

Abstract
Metastasis represents an end stage in the evolution of cancer progression and has been related to specific genetic pathways. Overexpression of mutant RAS in particular appears to promote invasion and metastasis, although exactly how this occurs has not been well characterized. We have previously shown that activation of the WASF3 protein regulates actin cytoskeleton dynamics that promote invasion. In this report we investigated how WASF3 overexpression interacts with mutant RAS to increased invasion and metastasis. The ability of RAS to promote invasion and metastasis was shown to be dependent on WASF3 activation in a PI3K and AKT dependent manner. Proteomics analysis demonstrates the presence of AKT in the WASF3 immunocomplex which is enhanced by overexpression of mutant RAS. During these processes activation of ERK1/2 is not affected by loss of WASF3 expression. Analysis of the relative involvement of p85 and p110 in the WASF3 complex demonstrates that mutant RAS promotes dissociation of p85 promoting activation of p110. These studies provide a deeper understanding of the critical role for WASF3 in facilitating increased invasion potential in cancer cells expressing mutant RAS and supports the idea that targeting WASF3 in metastatic cells overexpressing RAS may be used to suppress invasion and metastasis.
INTRODUCTION
The RAS genes are among the most commonly mutated oncogenes in human cancers particularly in certain types such as colon, pancreas and lung [Malumbres and Barbacid, 2003]. In addition to mutational activation, RAS genes are also often overexpressed in cancers such as breast and pancreas. In normal cells, growth factor stimulation leads to the activation of the RAS genes through switches between their GDP-bound inactive and GTP-bound active form, which promotes a variety of cellular responses. Normally this GDP-GTP transition is under tight regulation, but in cancer cells mutations in the RAS genes, largely at codons 12, 13 and 61, lead to constitutive activation of RAS by stabilizing the GTP-bound form [Castellano et al., 2011]. One of the main consequences of RAS activation is the promotion of invasion and metastasis, although the underlying mechanism is poorly understood [Castellano et al., 2011]. RAS activation leads to stimulation of the MAPK/ERK pathway leading to increased cell proliferation [Moodie et al., 1993; Vojtek et al., 1993] as well as the PI3 kinase (PI3K) pathway, which elicits a variety of cellular responses including invasion and metastasis [Cantley, 2002]. While the promotion of invasion and metastasis is a complex genetic process [Nguyen and Massagué, 2007], there are master proteins such as WASF3, which promote metastasis when overexpressed and, when inactivated, lead to suppression of invasion and metastasis, even in cells that overexpress mutant RAS [Sossey-Alaoui et al., 2005a, 2007a].
WASF3 is a member of the Wiskott-Aldridge family of proteins [Sossey-Alaoui et al., 2003] whose primary function is to regulate actin cytoskeleton dynamics leading to stimulation of membrane protrusion that lead to cell movement and invasion [Takenawa and Miki, 2001]. Knockdown of WASF3 in a variety of different cancer cell types leads to suppression of lamellipodia formation leading to suppression of invasion in vitro [Sossey-Alaoui et al., 2005b; Teng et al., 2010] and metastasis in vivo [Sossey-Alaoui et al., 2007a; Teng et al., 2010]. WASF3 also influences a series of pathways and genes that are related to metastasis [Sossey-Alaoui et al., 2005b; Teng et al., 2001, 2013] and its over expression is correlated with increased aggression and poor survival in primary breast cancers [Prat et al., 2010]. In model cell and animal studies, invasion and metastasis is correlated with high-level expression of WASF3, and cells that do not express WASF3 typically do not show the invasion/metastasis phenotype [Teng et al., 2011, 2014]. Re-expression of WASF3 in these cells promotes the invasive phenotype, further defining WASF3 as a metastasis-promoting gene. The invasion-promoting capacity of WASF3 is facilitated by its regulation of the KISS1 metastasis suppressor gene [Teng et al., 2011] which exerts control over the IkBa protein that normally suppresses NFkB. High level expression of WASF3 leads to increased NfKB levels in the nucleus through down regulation of IkBa activity. As a result, NFkB activates genes promoting metastasis such as IL6/8 and MMP3/9. Increased WASF3 levels have also been shown to occur in response to metastasis stimulating signals derived from hypoxia [Ghoshal et al., 2012] and cytokines [Teng et al., 2013], for example.
The fact that inactivating WASF3 in cells that are either mutant for KRAS, or overexpress KRAS, suppresses invasion suggested that the influence exerted over invasion/metastasis by mutant RAS is a WASF3 dependent event. Here we show that overexpression of mutant RAS promotes invasion in the presence of WASF3 protein and that WASF3 promotes invasion by influencing the association between PI3K and AKT which are activated downstream of RAS.
MATERIALS AND METHODS
Cell Lines and Standard Assays
All parental cell lines (MDA-MB-231, T47D, SKBR3, SW48, HCT116 and SW620) were purchased from the American Type Culture Collection (ATCC, Rockville, MD) and cultured as recommended. These cell lines have been routinely tested and authenticated by the ATCC. The nontransformed human breast epithelial MCF10A cell line and its derivatives were a generous gift of Dr. Ben Ho Park (Johns Hopkins University, Baltimore, MD). HCT116 derivatives were kindly provided by Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD). Standard cell culture, shRNA knockdown, lentiviral transduction, real-time RT-PCR, Western blot, immunoprecipitation (IP), immunofluorescence (IF), and cell proliferation assays were carried out as described previously [Sossey-Alaoui et al., 2005a].
DNA Constructs, Antibodies and Other Reagents
pLKO.1 lentiviral vectors harboring shRNA-targeting WASF3 were obtained from Open Biosystems (Huntsville, AL). The MISSION® shRNA against Kras was purchased from Sigma-Aldrich (St Louis, MO). The antibodies used for Western blot analysis were: WASF3, ERK1/2 and p-ERK1/2 (Thr202/Tyr204) (Cell Signaling Technology, Beverly, MA); AKT, p-AKT (Ser473), p85 and p110 (Abcam, Cambridge, MA); β-Actin, PY20 and KRAS (Sigma-Aldrich, St Louis, MO).
Immunoprecipitation
Cells were lysed in modified RIPA lysis buffer (Piece, IL). Lysate (1500 μg total protein) was incubated with the antibodies followed by addition of protein A/G agarose beads (Pierce) overnight at 4°C on a rotating platform. Beads were washed extensively in lysis buffer, and proteins were eluted with 2× Laemmli buffer (100 mM Tris-HCl pH 6.8, 20% glycerol, 4% SDS, 10% β-mercapto-ethanol) and heated to 95°C for 5 min before electrophoresis. For western blotting, equal amounts of protein were resolved by 8–10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes and probed with primary antibodies. Membranes were then incubated with species-specific horseradish peroxidase-conjugated secondary antibody (Pierce) followed by detection with ECL substrate (Pierce). For analysis of WASF3 phosphoactivation, IP protein was resolved by SDS-PAGE and probed with the PY20 antibody.
Invasion Assays
Invasion assays were performed in Matrigel-coated modified Boyden chambers with 8-μm pore size filters (BD Biosciences, MD). In brief, the serum-starved cells were added in the upper chamber (5 × 104 cells per insert) and RPMI 1640 medium with 5% FBS was used as a chemo attractant in the lower chamber. After 24 h incubation, non-invading cells that remained on the upper surface of the filter were removed and the cells that had passed through the filter and attached to the bottom of the membrane were stained with crystal violet cell stain solution (Millipore, Billerica, MA). The number of cells/mm2 were then counted and expressed as a percentage of cells in the control groups.
In Vivo Metastasis Assays
All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Augusta University. Six-week-old SCID mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and were injected with 1×106 cells via lateral tail veins to establish tumor metastasis models as described previously [Sossey-Alaoui et al., 2007a]. The mice were sacrificed on day 56 and metastatic nodules on the surface of the lung and liver were counted. The lungs and livers were then fixed and processed for histological analyses. Hematoxylin and eosin (H&E) staining was used to examine tumor metastasis in the lung and liver.
RESULTS
Mutant KRAS Enhances WASF3 Phosphoactivation and Invasion in MCF10A Cells
To address fundamental relationships between WASF3 and RAS, we first used a normal cell system that expresses both genes at endogenous levels. MCF10A cells are immortalized, non-transformed cells derived from primary epithelial breast cells that are non-invasive in vitro and non-tumorigenic in vivo [Soule et al., 1990]. Although our previous studies show that WASF3 is typically not expressed in non-invasive cells [Teng et al., 2014], relatively high levels of WASF3 protein were surprisingly detected in MCF10A cells, albeit still lower than in the invasive breast cancer cells such as MDA-MB-231 (Figure 1A). Beyond protein expression, however, phosphoactivation of WASF3 is required for its ability to promote invasion/metastasis [Sossey-Alaoui et al., 2007b; Teng et al., 2014], and analysis of the WASF3 phosphorylation status in MCF10A cells showed only a low-level, steady state activation of WASF3 (Fig. 1A), consistent with that seen in other non invasive cells.
Figure 1.
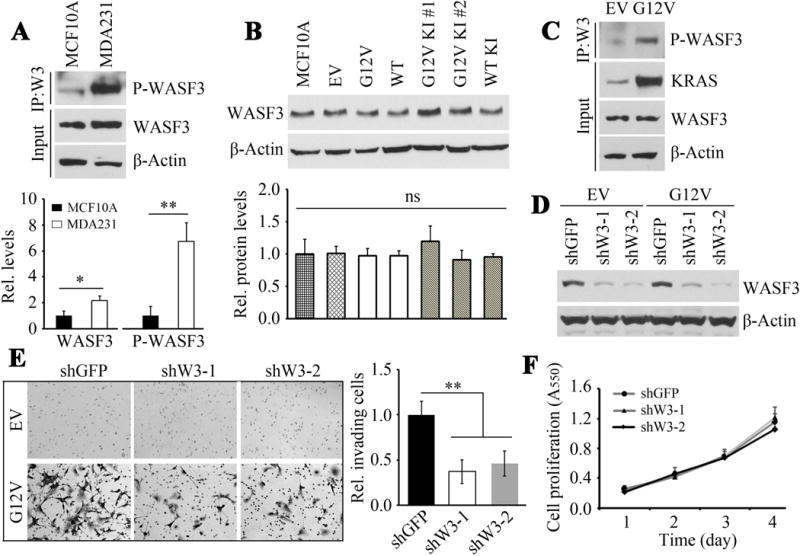
MCF10A cells express relatively high levels of WASF3 (input) compared with MDA231 cells (A) but significantly lower levels of phosphoactivated WASF3. WASF3 levels are not significantly different in derivatives of MCF10A cells overexpressing either the mutant (G12V) or wild type (WT) RAS gene (B) or in two isogenic MCF10A clones with G12V (KI#, KI#2) or wild type knock-in (WT KI). In contrast, phosphoactivated WASF3 is significantly increased when mutant KRAS (G12V) is overexpressed in MCF10A, while WASF3 protein levels are unaffected (C). Knockdown of WASF3 (D) using two different shRNAs (ShW3-1, -2) leads to a significant reduction in invasion potential in the mutant (G12V) KRAS-overexpressing cells (E) while cell proliferation is not affected (F).
MCF10A, unlike transformed cells such as MDA-MB-231 that harbor a G12V mutation in KRAS (KRASG12V), express a wild type KRAS gene (KRASWT) and have been used to investigate the role RAS in early stages of the transformation process [Konishi et al., 2007]. When MCF10A cells are forced to overexpress KRASG12V, they become invasive in vitro [Teng et al., 2012]. Many cancer cells, however, express mutant RAS at physiologically normally regulated levels [Gustafson et al., 2009]. To investigate how WASF3 is affected under these circumstances, we used the isogenic pairs of MCF10A cells described by Konishi et al [2007], which have been engineered to express endogenously either KRASG12V or KRASWT monoallelically. WASF3 protein levels in these isogenic pairs were identical (Fig. 1B). In contrast, forced overexpression of KRASG12V in MCF10A cells, while not altering WASF3 protein levels (Fig. 1B), dramatically increased WASF3 phosphorylation levels (Fig. 1C).
While knockdown of WASF3 in aggressive breast cancer cells leads to loss of invasion [Sossey-Alaoui et al., 2005a], knockdown of WASF3 in MCF10A cells (Fig. 1D) did not affect endogenous invasion levels (Fig. 1E). In contrast, when WASF3 was knocked down in MCF10A cells overexpressing KRASG12V, invasion was remarkably reduced (Fig. 1E), without significant changes in cell proliferation (Fig. 1F), compared with the knockdown control cells. These observations demonstrate that overexpression of KRASG12V in non transformed MCF10A cells promotes invasion that correlates with phosphoactivation of WASF3 in this cell system.
WASF3 Phosphoactivation Promotes Mutant Kras-Dependent AKT Activation
PI3K/AKT and MAPK/ERK are the two main downstream effector pathways of KRAS [Castellano and Downward, 2011]. The p85 component of PI3K binds directly to WASF3 and PI3K activity is required for WASF3 activation [Sossey-Alaoui et al., 2005b]. To understand better the impact of WASF3 on KRASG12V-mediated PI3K and MAPK signaling in the MCF10A model system, we analyzed the relationship between WASF3 activation and AKT and ERK1/2 activation in KRASG12V expressing cells. In KRASG12V overexpressing cells, knockdown of WASF3 led to an almost complete suppression of AKT activation, but not pERK1/2 activation (Fig. 2A). In contrast, only a small change in pAKT levels was observed when WASF3 was knocked down in the cells expressing KRASG12V at endogenous levels (Fig. 2A). pAKT levels in MCF10A were comparable between cells expressing either KRASWT or KRASG12V, but higher in cells overexpressing KRASG12V (Fig. 2B). Analysis of WASF3 phosphorylation levels in these MCF10A cells showed dramatically higher levels in cells overexpressing KRASG12V compared to those expressing monoallelic KRASWT or KRASG12V (Fig. 2B). These data indicate that high-level expression of mutant KRAS enhances WASF3 activation, and activated WASF3 may participate in facilitating AKT phosphoactivation.
Figure 2.
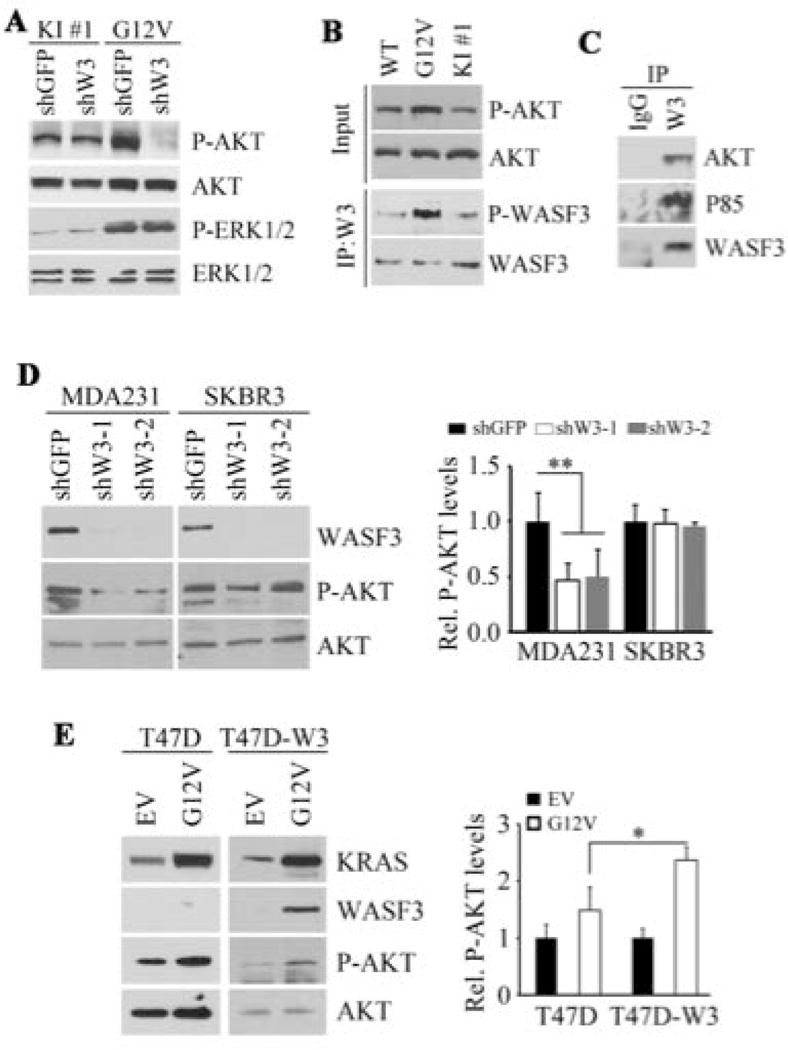
Knockdown of WASF3 in MCF10A cells expressing either endogenous levels (KI#1) of mutant RAS, or overexpressing (G12V) mutant KRAS shows suppression of AKT activation only in the cells overexpressing the mutant RAS (A). ERK1/2 levels were not affected in either cell type. Increases in phosphoactivated AKT (B) was seen in MCF10A cells overexpressing the mutant RAS which was accompanied by increased activation of WASF3. Immunoprecipitation of WASF3 from MDA-MB-231 cells shows the presence of p85 and AKT in the immunocomplex (C). When WASF3 was knocked down in MDA-MB-231 cells, phospho-AKT levels were reduced whereas there was no effect on pAKT when WASF3 was knocked down in SKBR3 cells (D). Overexpression of mutant KRAS (G12V) in T47D cells leads to increased pAKT levels which was more significant when WASF3 was also overexpressed (D).
To extend these observations to metastatic tumor cells, we investigated the relationship between WASF3 and RAS expression in MDA-MB-231 breast cancer cells which express KRASG12V. In our previous studies of WASF3-associated proteins in MDA-MB-231 cells using mass spectrometry analysis [Teng et al., 2013; Zhou et al., 2016], we identified the AKT Interacting Protein (AKTIP) in the WASF3 immunocomplex, supporting the possibility that WASF3 interacts with AKT (Supplementary Fig. S1). To confirm this interaction, we performed IP from MDA-MB-231 cells and demonstrated the presence of AKT in the WASF3 immunocomplex (Fig. 2C). The p85 regulatory component of PI3K was also present (Fig. 2C), as we have described previously [Sossey-Alaoui et al., 2005b]. When WASF3 was knocked down in MDA-MB-231 cells, levels of activated pAKT were suppressed (Fig. 2D), suggesting a functional interaction between WASF3 and AKT. In contrast, when WASF3 was knocked down in SKBR3 cells, which harbor wild type KRAS, pAKT levels were not affected (Fig. 2D). When KRASG12V was overexpressed in parental T47D breast cancer cells, which do not normally express WASF3 [Teng et al., 2014] and also harbor a wild-type KRAS gene, pAKT levels increased (Figure 2E) and when WASF3 was also overexpressed in these cells (Fig., 2E) pAKT levels were further increased. These studies establish that the ability of mutant RAS to mediate activation of AKT is dependent on WASF3.
Knockdown of WASF3 Leads to Reduced pAKT Levels, Invasion and Metastasis in Colon Cancer Cells
We next extended our studies relating RAS and WASF3 expression from non-transformed cells to cancer cells. Although KRAS mutations are seen in many different cancers, they are particularly prevalent in colon cancers [Malumbres and Barbacid, 2003], and have been shown to play a critical role in the oncogenic progression of colon cancer [Castellano and Downward, 2011]. We therefore investigated the relationship between WASF3 and KRAS in colon cancer cell lines. Here, the positive relationship between WASF3 expression levels and invasion is maintained (Fig. 3A). Non-invasive SW48 cells express KRASWT, poorly invasive HCT116 cells express KRASG12V at low levels and metastatic SW620 cells express KRASG12V at high levels [Teng et al., 2012]. When WASF3 was knocked down in these three colon cancer cell lines, there was a proportional reduction in both pAKT levels (Fig. 3A) and invasion (Fig. 3B) in HCT116 and SW620 cells, but not in SW48 cells. Thus, as in the breast cell systems described above, WASF3 expression is also associated with AKT phosphorylation in the presence of mutant KRAS in colon cancer cells. Since suppression of invasion as a result of WASF3 knockdown was more significant in SW620 cells than in HCT116 cells, we investigated the metastatic potential of these cells using intravenous xenografts in SCID mice. In this experimental model of metastasis, mice injected with WASF3 knockdown SW620 cells showed significantly lower numbers of surface nodules on the liver (Fig. 3C) and lungs (Fig. 3D) of these mice compared with mice injected with parental cells. Histological analysis further confirmed these observations (Figs. 3C and D), demonstrating that WASF3 promotes metastasis in colon cancer cells.
Figure 3.
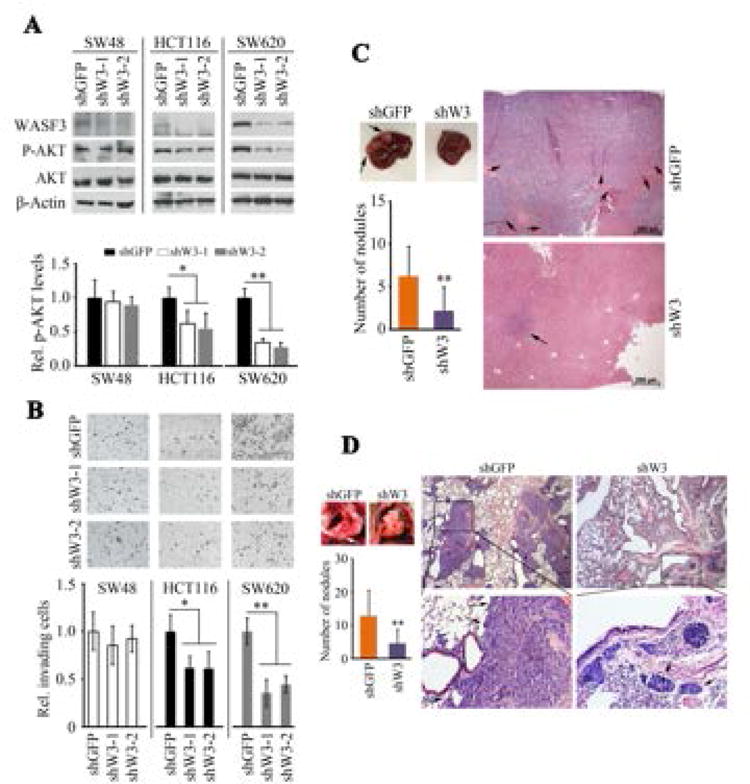
Expression levels of WASF3 are increased in highly invasive SW620 cells compared with less invasive HCT116 and noninvasive SW48 cells (A). pAKT levels are also reduced proportional to the endogenous levels of WASF3 expression. Knockdown of WASF3 in these three cell lines shows no affect on invasion for SW48 cells, a significant suppression of invasion in HCT116 cells and a highly significant suppression of invasion in SW620 cells (B). Metastasis assays for SW620 cells in SCID mice shows a reduced number of surface nodules on the livers from cells in which WASF3 was knocked down (C), which was also seen in the lungs from the same animals (D).
Unlike SW620, parental HCT116 cells express a wild type KRAS gene, are weakly invasive and express lower levels of WASF3. These cells have also been engineered to express either KRASWT or KRASG13D monoallelically [Teng et al., 2012], which allowed analysis of the relationship between WASF3 expression and mutant KRAS status in these isogenic pairs of cells. No difference in cell proliferation was observed whether the KRASWT or KRASG13D allele was expressed (Fig. 4A). When only the KRASG13D was expressed, there was a highly significant increase in cell invasion potential compared with either parental HCT116 cells or those expressing only KRASWT (Fig. 4B). Cells expressing only KRASWT showed a significant reduction in invasion compared with the parental cells, but invasion in the parental cells was significantly lower than in KRASWT expressing cells. These data suggest that the presence of KRASWT can influence the ability of KRASG13D to promote invasion. Consistent with these observations, levels of phosphoactivated WASF3 were higher in the cells expressing KRASG13D compared with those expressing KRASWT (Fig. 4C). When WASF3 was knocked down in the HCT116 isogenic derivatives (Fig. 4D), invasion was significantly reduced in a KRASG13D expressing cells compared with KRASWT expressing cells (Fig. 4E) confirming the importance of WASF3 for KRASG13D -induced invasion.
Figure 4.
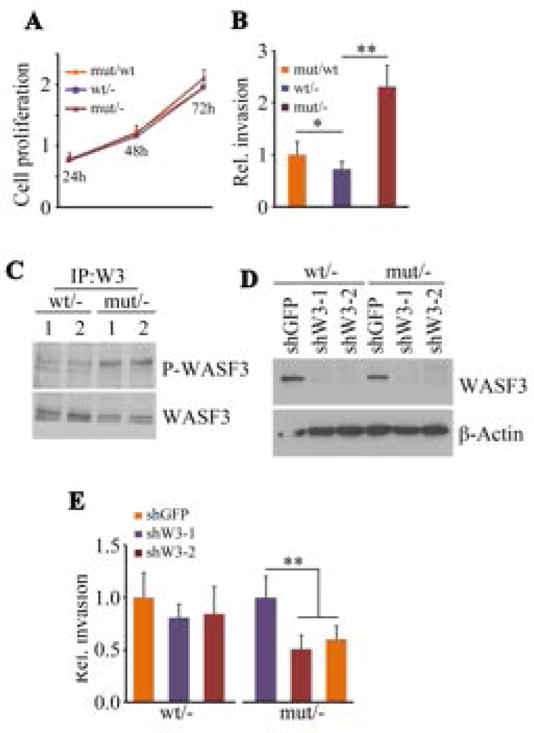
HCT116 cells show no difference in cell proliferation whether the monoallelic wild-type (wt/-) or mutant (mut/-) KRAS was expressed (A) and expression of the mutant KRAS allele on a wild-type background (mut/wt) similarly showed no change in proliferation. In contrast, invasion potential was significantly increased in cells expressing the monoallelic mutant KRAS compared with cells expressing monoallelic, wild type KRAS (B). Cells heterozygous for mutant and wild type RAS showed an intermediate increase in invasion potential. Immunoprecipitation of WASF3 from wt/- or mut/- HCT116 cells showed increased levels of activated phospho-WASF3 in the mut/- cells (C). Knockdown of WASF3 (D), using two different shRNAs, in the monoalleleic, isogenic HCT116 pairs showed reduced invasion only in the mut/- cells (E).
WASF3 Activation Influences the Interaction Between p85 and p110
The catalytic activity of the p110 subunit of PI3K, which activates AKT, is suppressed through association with the p85 subunit [Castellano and Downward, 2011]. When the endogenous p85 protein was immunoprecipitated from MCF10A isogenic pairs, higher levels of WASF3 protein were seen in the p85 immunocomplex from cells expressing KRASG12V compared with the cells expressing KRASWT (Fig. 5A). Together with the results shown in Figure 2B, these observations suggest that this association depends on WASF3 phosphorylation. We have previously shown that overexpression of WASF3 in the non-metastatic T47D breast cancer cells increases motility [Teng et al., 2014]. When KRASG12V was introduced into T47D cells overexpressing exogenous WASF3, there was a reduction in the levels of p110 associated with p85, supporting increased PI3K activity (Fig. 5B). In contrast, knockdown of WASF3 in either breast cancer MDA-MB-231 cells (Fig. 5C) or colon cancer SW620 cells (Fig. 5D), led to an enhanced interaction between p85 and p110. It appears, therefore, that WASF3 influences increased dissociation between p85 and p110, accounting for increased AKT activation (Fig. 5E).
Figure 5.
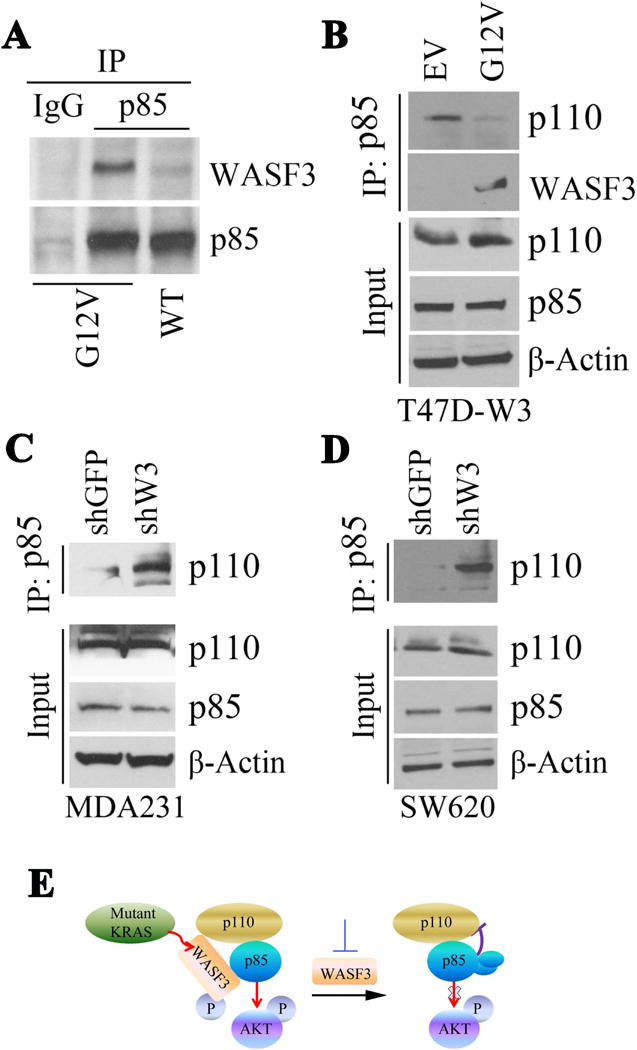
Immunoprecipitation of p85 from MCF10A cells expressing either the G12V mutation or wild-type (WT) RAS shows increased levels of WASF3 in the immunocomplex in the presence of mutant RAS (A). In T47D cells expressing exogenous WASF3, the p110 component of PI3K is reduced in the p85 immunocomplex (B). Knockdown of WASF3 in MDA231 cells (C) or SW620 cells (D) shows increased levels of p110 in the p85 immunocomplex. (E) Model describing potential involvement of WASF3 control of AKT phosphorylation.
WASF3 is an Important Mediator in KRAS-AKT Signaling
When KRASG12V was overexpressed in HCT116 cells showing monoallelic expression of KRASWT, both pWASF3 and pAKT levels were increased (Fig. 6A). When mutant KRAS was knocked down in the KRASG13D expressing HCT116 cells, pAKT and pWASF3 levels were dramatically decreased (Fig. 6B). Analysis of the relative engagement of p110 with p85 in the WASF3 immunocomplex in HCT116 isogenic cell lines (Fig. 6C) shows increased WASF3 levels in the p85 immunocomplex in KRASG12V cells compared with those expressing KRASWT. This change was accompanied by a concomitant decrease of p110 levels in the complex. Since activation of AKT can be initiated by various extracellular signals promoting PI3K signaling [Castellano and Downward, 2011], we explored the role of WASF3 in KRAS-mediated AKT activation using WASF3 knockdown in the isogenic HCT116. As shown in Figure 6D, loss of WASF3 function led to a significant decrease in pAKT levels in the KRASG13D-expressing cells compared with those expressing KRASWT. These observations support the mechanism where WASF3 promotes AKT activation by suppressing the inhibitory effect of p85 on p110, and that this event is enhanced by mutant KRAS.
Figure 6.
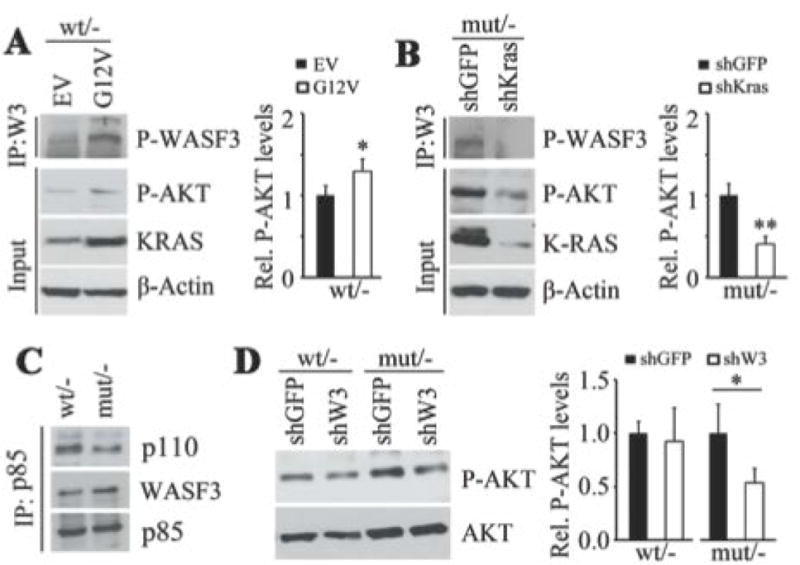
When exogenous mutant KRAS (G12V) was over expressed in HCT116 cells showing monoallelic expression of wild-type KRAS, increased levels of both pAKT and pWASF3 levels are seen in the WASF3 immunocomplex (A). When KRAS is knocked down in the monoallelic mutant RAS cells (mut/-), pAKT and pWASF3 levels in the WASF3 immunocomplex are decreased (B). IP of p85 in the monoallelic wild-type and mutant RAS HCT116 cells shows decreased levels of p110 associated with p85 (C). In the same isogenic pairs (D) knockdown of WASF3 only leads to reduced pAKT levels in cells expressing the mutant RAS allele.
DISCUSSION
The association between mutant RAS and increased invasion and metastatic potential of cancer cells is well established, although the mechanisms behind these observations are poorly understood [Castellano and Downward, 2011]. Promotion of the metastatic phenotype occurs through signaling cascades initiated by a variety of extracellular signals such as hypoxia, growth factors and cytokines. We have shown in many cancer types, that WASF3 is required to transmit these extracellular stimuli to promote invasion and metastasis, specifically by acting as the nexus to assemble protein complexes involved in this process. Thus, without WASF3, metastatic signals from PDGF [Sossey-Alaoui et al., 2005b], IL6 [Teng et al., 2013) and ERBB2/3 [Yun et al.,2005], for example, cannot promote invasion, even though their effect on proliferation is unaffected, presumably because a different set of intermediates is involved in these processes. Activation of receptors can promote invasion and metastasis by activating RAS [Castellano and Downward, 2011] but mutational activation of RAS bypasses the need for receptor mediated signaling. This has been shown in studies targeting the EGFR receptor in lung cancer, for example, which is only effective in cells with wild type RAS [Teng et al 2016a]. Part of the pro-metastatic signaling cascade through RAS activation, involves AKT activation in a PI3K-dependent manner [Castellano and Downward, 2011]. Activation of AKT has also been shown independently to promote metastasis [Linardou et al., 2008]. WASF3 assembles different protein complexes [Chin et al 2009] including a variety of kinases such as ABL [Sossey-Alaoui et al., 2007b], JAK2 [Teng et al., 2013] as well as the p85 regulatory subunit of PI3K [Sossey-Alaoui et al., 2005b]. We now also show the presence of the p110 subunit in the WASF3 complex which mediates a competitive interaction between WASF3 and p85/p110 subunits of PI3K, in which the catalytic subunit is release from its repression by p85 in response to RAS signaling, leading to AKT activation. Evidence presented here demonstrates that WASF3 is a critical downstream intermediate effector for the ability of mutant RAS to promote invasion through regulation of the interaction between PI3K and AKT.
RAS activates RAC through PI3K [Castellano and Downward, 2011] and WASF3 is also activated by RAC1/2 [Teng et al., 2016b], which is required for its ability to promote invasion and metastasis. RAC performs this function by interacting with proteins associated with the N-terminus of the WASF3 complex that normally hold WASF3 in an autorepressed, inactive conformation [Teng et al., 2016c]. We showed recently that disrupting this complex using stapled peptide mimics leads to suppression of WASF3 activation and destabilization of the complex leading to loss of invasion and metastasis [Teng et al., 2016c, d]. This effect was seen in breast, prostate and colon cancer cells expressing mutant RAS, further supporting the idea that WASF3 is required to transmit the RAS signal to effect metastasis in different cancer types. In fact, WASF3 is important as an intermediate for other signaling molecules that promote invasion and metastasis such as HIF1 [Ghoshal et al., 2012], ERBB2 [Yun et al., 2009], PDGF [Sossey-Alaoui et al., 2005b] and JAK2/STAT3 [Teng et al., 2013], supporting the contention that WASF3 is at the nexus of multiple signaling cascades promoting metastasis. As such, developing a strategy to disrupt WASF3 function directly, rather that targeting upstream components of the pathway may be a more effective therapeutic strategy to control invasion and metastasis that has widespread implications, especially for tumors expressing mutant RAS and where therapies targeting RAS have not been successful [Malumbres and Barbacid, 2003].
Although there is evidence that mutated RAS can promote invasion, this effect is more pronounced when it is also overexpressed as seen in the MCF10A model presented here. Furthermore, although isolated studies suggest a wild type allele has a neutral or positive effect on the malignant phenotype, the majority of studies suggest a wild type allele has a suppressive effect over a mutant allele when expressed at physiologically regulated levels [Gustafson et al., 2009]. Our studies using isogenic HCT 116 cancer cells show this suppressive effect on invasion in cells heterozygous for mutant RAS but loss of the WT allele in these cells enhances the effect of the mutant allele on invasion in a WASF3-dependent manner. This is the proposed underlying basis for loss of heterozygosity at the RAS loci which eliminates the wild type allele [Zhou et al., 2016].
Since the levels of activated WASF3 seem to be proportional to the invasion potential in cancer cells it is consistent that higher levels of mutant RAS leads to increased WASF3 activation not seen when RAS is expressed at physiological levels. This is supported by the activation of AKT which is again not increased in the isogenic pairs described by Konishi et al., [2007] but is increased when RAS is overexpressed and is lost when WASF3 is knocked down. These studies, therefore, provide an explanation for the role of mutant RAS in promoting invasion and metastasis which involve activation of signaling pathways in a WASF3-dependent manner.
Supplementary Material
Acknowledgments
We are grateful to Yun Mei and Xiaoou Ren for technical help.
Supported by: CA120510 from the National Institutes of Health
References
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- Castellano E, Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes & Cancer. 2011;2:261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin YR, Toker A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009;21:470–476. doi: 10.1016/j.cellsig.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- Ghoshal P, Teng Y, Lesoon L, Cowell JK. HIF1A induces expression of the WASF3 Metastasis Associated Gene under hypoxic conditions. Int J Cancer. 2012;131:E905–15. doi: 10.1002/ijc.27631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson TL, Wellberg E, Laffin B, Schilling L, Metz RP, Zahnow CA, Porter WW. Ha-Ras transformation of MCF10A cells leads to repression of Singleminded-2s through NOTCH and C/EBPbeta. Oncogene. 2009;28:1561–1568. doi: 10.1038/onc.2008.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi H, Karakas B, Abukhdeir AM, Lauring J, Gustin JP, Garay JP, Konishi Y, Gallmeier E, Bachman KE, Park BH. Knock-in of mutant K-ras in nontumorigenic human epithelial cells as a new model for studying K-ras mediated transformation. Cancer Res. 2007;67:8460–8467. doi: 10.1158/0008-5472.CAN-07-0108. [DOI] [PubMed] [Google Scholar]
- Linardou H, Dahabreh IJ, Kanaloupiti D, Siannis F, Bafaloukos D, Kosmidis P, Papadimitriou CA, Murray S. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9:962–972. doi: 10.1016/S1470-2045(08)70206-7. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;2003:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- Moodie SA1, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Massagué J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8:341–52. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Head K, Nowak N, Cowell JK. Characterization of the genomic organization and expression profile of the human and mouse WAVE gene family. Mamm Genome. 2003;14:314–322. doi: 10.1007/s00335-002-2247-7. [DOI] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Ranalli TA, Li X, Cowell JK. WAVE3 promotes cell motility and invasion through the regulation of MMP-1, MMP-3 and MMP-9- expression. Exp Cell Res. 2005a;308:135–145. doi: 10.1016/j.yexcr.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Li X, Ranalli TA, Cowell JK. WAVE3-mediated cell migration and lamellipodia formation are regulated downstream of phosphatidylinositol 3-kinase. J Biol Chem. 2005b;280:21748–21755. doi: 10.1074/jbc.M500503200. [DOI] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Safina A, Li X, Vaughan MM, Hicks DG, Bakin AV, Cowell JK. Down-regulation of WAVE3, a metastasis promoter gene, inhibits invasion and metastasis of breast cancer cells. Am J Pathol. 2007a;170:2112–2121. doi: 10.2353/ajpath.2007.060975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossey-Alaoui K, Li X, Cowell JK. c-Abl-mediated phosphorylation of WAVE3 is required for lamellipodia formation and cell migration. J Biol Chem. 2007b;282:26257–16265. doi: 10.1074/jbc.M701484200. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD, Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–6086. [PubMed] [Google Scholar]
- Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001;114:1801–1809. doi: 10.1242/jcs.114.10.1801. [DOI] [PubMed] [Google Scholar]
- Teng Y, Ren MQ, Cheney R, Sharma S, Cowell JK. Inactivation of the WASF3 gene in prostate cancer cells leads to suppression of tumorigenicity and metastases. Br J Cancer. 2010;103:1066–1075. doi: 10.1038/sj.bjc.6605850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Liu M, Cowell JK. Functional interrelationship between the WASF3 and KISS1 metastasis-associated genes in breast cancer cells. Int J Cancer. 2011;129:2825–2835. doi: 10.1002/ijc.25964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Ngoka L, Mei Y, Lesoon L, Cowell JK. HSP90 and HSP70 are essential for stabilization and activation of the WASF3 metastasis promoting protein. J Biol Chem. 2012;287:10051–10059. doi: 10.1074/jbc.M111.335000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Ghoshal P, Ngoka L, Mei Y, Cowell JK. Critical role of the WASF3 gene in JAK2/STAT3 regulation of cancer cell invasion. Carcinogenesis. 2013;34:1994–1999. doi: 10.1093/carcin/bgt167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Mei Y, Hawthorn L, Cowell JK. WASF3 regulates miR-200 inactivation by ZEB1 through suppression of KISS1 leading to increased invasiveness in breast cancer cells. Oncogene. 2014;33:203–211. doi: 10.1038/onc.2012.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Pi W, Wang Y, Cowell JK. WASF3 provides the conduit to facilitate invasion and metastasis in breast cancer cells through HER2/HER3 signaling. Oncogene. 2016a;35:4633–4640. doi: 10.1038/onc.2015.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Ren X, Li H, Shull A, Kim J, Cowell JK. Mitochondrial ATAD3A combines with GRP78 to regulate the WASF3 metastasis-promoting protein. Oncogene. 2016b;35:333–343. doi: 10.1038/onc.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Bahassan A, Dong D, Hanold LE, Ren X, Kennedy EJ, Cowell JK. Targeting the WASF3-CYFIP1 complex using stapled peptides suppresses cancer cell invasion. Cancer Res. 2016c;276:965–973. doi: 10.1158/0008-5472.CAN-15-1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng Y, Qin H, Bahassan A, Bendzunas NG, Kennedy EJ, Cowell JK. The WASF3-NCKAP1-CYFIP1 complex is essential for breast cancer metastasis. Cancer Research. 2016d;76:5133–142. doi: 10.1158/0008-5472.CAN-16-0562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojtek AB, Hollenberg SM, Cooper JA. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA, Jr, Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Der CJ, Cox AD. The role of wild type RAS isoforms in cancer. Semin Cell Dev Biol. 2016;58:60–69. doi: 10.1016/j.semcdb.2016.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.