

Abstract
Nine analogs of 1′S-1′-acetoxychavicol acetate (ACA) were hemi-synthesized and evaluated for their anticancer activities against seven human cancer cell lines. The aim of this study was to investigate the anti-proliferative, apoptotic, and anti-migration effects of these compounds and to explore the plausible underlying mechanisms of action. We found that ACA and all nine analogs were non toxic to human mammary epithelial cells (HMECs) used as normal control cells, and only ACA, 1′-acetoxyeugenol acetate (AEA), and 1′-acetoxy-3,5-dimethoxychavicol acetate (AMCA) inhibited the growth of MDA-MB-231 breast cancer cells with a half-maximal inhibitory concentration (IC50) value of <30.0 μM based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay results, and were selected for further investigation. DNA fragmentation assays showed that these three compounds markedly induced apoptosis of MDA-MB-231 cells. Western blot analysis revealed increased expression levels of cleaved PARP, p53, and Bax, while decreased expression levels of Bcl-2 and Bcl-xL were seen after treatment, indicating that apoptosis was induced via the mitochondrial pathway. Moreover, ACA, AEA, and AMCA effectively inhibited the migration of MDA-MB-231 cells. They also downregulated the expression levels of pFAK/FAK and pAkt/Akt via the integrin β1-mediated signaling pathway. Collectively, ACA and its hemi-synthetic analogs, AEA and AMCA are seen as potential anticancer agents following their abilities to suppress growth, induce apoptosis, and inhibit migration of breast cancer cells.
Keywords: 1′S-1′-acetoxychavicol acetate, ACA, ACA hemi-synthetic analogs, triple-negative breast cancer, MTT assay, DNA fragmentation, wound-healing assay, Western blot, integrin β1 signaling pathway
Introduction
Over the past half-century, natural products presented impressive achievements in drug discovery. However, major challenges, such as low production yields of natural products during scale-up efforts,1 inadequate natural resources,2 and their complex structures that impede improved structural modifications and synthesis of compounds, have hindered the development of these natural products as pharmaceutical drugs.3 In addition, screening of numerous extracts and purified compounds from a variety of sources involves substantial expenditure and time. Due to these limitations, it is imperative to perform structural modifications on potential compounds through organic hemi-synthesis, as this would allow discovery of analogs with higher efficacy.
1′S-1′-acetoxychavicol acetate (ACA) is a phenylpropanoid which can be found in the plant Alpinia conchigera (Zingiberaceae).4 It is known to exhibit a broad range of biological properties such as antiulcer,5 antifungal,6 inhibition of xanthine oxidase,7 inhibition of Epstein–Barr virus activation,8 and anticancer activity.9–12 Due to its wide range of biological functions, hemi-synthesis of different ACA analogs was warranted for enhancement purposes.13 However, various anticancer properties, such as anti-proliferative, apoptotic, and anti-migration effects of its analogs, are yet to be reported.
Breast cancer is the most common cancer in women in many countries, including Malaysia.14 Triple-negative breast cancer (TNBC) is one of the subgroups of breast cancer, which lacks expression of estrogen receptor (ER), progesterone receptor (PR), and HER-2.15 It constitutes roughly 15% of diagnosed breast cancers,16 and unfortunately, it does not respond to the most effective treatments for breast cancer such as targeted endocrine therapy.15 Moreover, the aggressiveness of TNBC leads to a high number of metastatic cases and deaths. Thus, the development of effective anticancer drugs for TNBC treatment is crucial.
In this study, the effects of ACA and its hemi-synthetic analogs on the proliferation of various cancer cell lines were assessed. We also investigated two other major anticancer properties, namely apoptosis and anti-migration effects, and discussed their underlying molecular mechanisms. We demonstrated that ACA, 1′-acetoxyeugenol acetate (AEA), and 1′-acetoxy-3,5-dimethoxychavicol acetate (AMCA) could inhibit MDA-MB-231 TNBC cell proliferation by inducing apoptosis through the regulation of vital proteins, namely PARP, p53, Bcl-2, Bcl-xL, and Bax. In addition, our results also revealed the potential of these three compounds against tumor metastasis by downregulating the expression of pFAK, focal adhesion kinase (FAK), pAkt, and Akt via the integrin β1-mediated pathway. These observations suggested that ACA, AEA, and AMCA may be potential agents in the treatment of TNBC.
Materials and methods
General chemistry procedure
Unless otherwise noted, all materials were obtained from commercial suppliers and were used without further purification. Reaction time and purity of products were monitored by thin-layer chromatography on Merck silica gel aluminum cards (0.2 mm thickness) with fluorescent indicator at 254 nm. Column chromatography was run on silica gel (200–300 mesh) obtained from EMD Millipore (Billerica, MA, USA). All spectral data were obtained using the following instruments: infrared (IR) on a Perkin Elmer RX1 FT-IR spectrometer, ultraviolet (UV) on a Shimadzu UV-160A UV–Visible Recording Spectrophotometer. One-dimensional (1D; 1H, 13C, distortionless enhancement by polarisation transfer) and two-dimensional (2D; correlation spectroscopy, nuclear overhauser effect spectroscopy, heteronuclear single-quantum coherence spectroscopy, heteronuclear multiple-bond connectivity) nuclear magnetic resonance (NMR) spectra using CDCl3 as solvent were recorded on Bruker AVN 400 (400 MHz for 1H NMR, 100 MHz for 13C NMR) spectrometer. Chemical shifts were internally referenced to the solvent signals in CDCl3 (1H, δ 7.26; 13C, δ 77.0), with tetramethylsilane as the internal standard and mass spectrometry (MS) on a Shimadzu gas chromatograph-MS spectrometer (HP 6890 Series Mass Selective Detector and HP 6890 Series GC System).
Isolation of ACA
The rhizomes of A. conchigera Griff. were collected from Jeli province of Kelantan, East Coast of Peninsular Malaysia.4 The sample was identified by Prof Dr Halijah Ibrahim from the Institute of Biological Science, Faculty of Science, University of Malaya. A voucher specimen (KL5049) was deposited in the Herbarium of Chemistry Department, Faculty of Science, University of Malaya. ACA was isolated following our previously published methods.4
General procedure to obtain compounds 2–8
4-Allyl-2,6-dimethoxyphenol (2), (S)-α-vinylbenzyl alcohol (3), and 4-allyl anisole (5) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Eugenol (4) was purchased from Merck & Co., Inc. (Whitehouse Station, NJ, USA). Compounds 6, 7, and 8 were synthesized from 2, 3, and 4, respectively, by reacting with acetic anhydride (Scheme 1).
Scheme 1.

Schematic preparation of ACA analogs.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; DMAP, 4-dimethylaminopyridine; RT, room temperature.
General procedure to synthesize compounds 17–20
Compounds 17–20 were synthesized by the modification of Lee and Ando’s17 procedure.
AEA (17)
To a solution of vinylmagnesium bromide freshly prepared from magnesium turning (0.24 g, 9.87 mmol), 1 M vinyl bromide (6.6 mL, 6.57 mmol) and catalytic amount of iodine crystal in dry diethyl ether (20 mL), a solution of aldehyde 9 (0.50 g, 3.29 mmol) in diethyl ether (10 mL) under ice bath, were added. The ice bath was removed, and the mixture was allowed to warm to room temperature. After the reaction mixture was stored for 2 h at room temperature, saturated aqueous NH4Cl (10 mL) solution was added. The mixture was extracted with diethyl ether (2×20 mL). The resulting organic extracts were combined, and the solvent was removed under reduced pressure to yield a crude product. The oily product (intermediate 13) obtained by usual workup was acetylated with acetic anhydride (0.62 mL, 6.57 mmol) and 4-dimethylaminopyridine (DMAP; 0.81 g, 6.64 mmol) in dichloromethane (15 mL) in an ice bath. After consumption of starting material and product formation, the reaction was quenched with saturated aqueous NH4Cl (10 mL) solution and extracted with dichloromethane (3×15 mL), dried (NaSO4), and filtered. Evaporation of filtrate gave a yellow oil, which was purified by column chromatography on silica gel, and eluting with hexane/ethyl acetate (9:1) afforded the desired product as a yellowish oil 17 (0.53 g, 61%). The synthetic analog 17 is the same as the natural analog AEA that was previously studied.18,19
The spectral analysis of the yellowish oil: IR (neat) υmax, cm−1: 1,767, 1,742, 1,647, 1,607, 1,233, 1,198, 857. UV λmax, nm: 299.0. 1H NMR (CDCl3, 400 MHz), δ: 2.12 (3H, s), 2.31 (3H, s), 3.84 (3H, s), 5.25 (2H, dd, J =10.5), 5.99 (1H, m), 6.24 (1H, d, J =5.8), 6.95 (1H, s), 7.02 (2H, d, J =7.8). 13C NMR (CDCl3, 100 MHz), δ: 20.6 (CH3), 21.25 (CH3), 55.9 (OCH3), 75.7 (CH), 111.5 (CH), 117.0 (CH2), 119.6 (CH), 122.8 (CH), 135.9 (CH), 137.6 (C), 139.7 (C), 151.2 (C), 168.9 (C), 169.7 (C). MS m/z (electron ionization) 40, 45, 61, 77, 91, 107, 121, 264 (M+•). High resolution mass spectroscopy m/z (electron ionization): 264.0993 (M+• C14H16O5+• requires 264.0998).
AMCA (18)
To a solution of vinylmagnesium bromide freshly prepared from magnesium turning (0.17 g, 7.14 mmol), 1 M vinyl bromide (3.6 mL, 3.5 mmol) and catalytic amount of iodine crystal in dry diethyl ether (20 mL), a solution of aldehyde 10 (0.30 g, 1.65 mmol) in diethyl ether (10 mL) under ice bath, were added. The ice bath was removed, and the mixture was allowed to warm to room temperature. After the reaction mixture was stored for 1 h at room temperature, saturated aqueous NH4Cl (10 mL) solution was added. The mixture was extracted with diethyl ether (2×20 mL). The resulting organic extracts were combined, and the solvent was removed under reduced pressure to yield a crude product. The oily product (intermediate 14) obtained by usual workup was acetylated with acetic anhydride (0.33 mL, 3.30 mmol) and DMAP (0.41 g, 3.36 mmol) in dichloromethane (15 mL) in an ice bath. After consumption of starting material and product formation, the reaction was quenched with saturated aqueous NH4Cl (10 mL) solution and extracted with dichloromethane (3×15 mL), dried (NaSO4), and filtered. Evaporation of filtrate gave a yellow oil, which was purified by column chromatography on silica gel, and eluting with hexane/ethyl acetate (9:1) afforded the desired product as a yellowish oil 18 (0.30 g, 63%).
The spectral analysis of the yellowish oil: IR (neat) υmax, cm−1: 1,761, 1,645, 1,234. UV λmax, nm: 304.5. 1H NMR (CDCl3, 400 MHz), δ: 2.13 (3H, s), 2.33 (3H, s), 3.81 (6H, s), 5.31 (2H, dd, J =10.0 Hz), 5.98 (1H, m), 6.21 (1H, d, J =5.8 Hz), 6.61 (2H, s). 13C NMR (CDCl3, 100 MHz), δ: 20.4 (CH3), 21.2 (CH3), 56.1 (2OCH3), 76.0 (CH), 104.0 (2CH), 117.0 (CH2), 128.4 (C), 135.9 (CH), 137.2 (C), 152.1 (2C), 168.7 (C), 169.9 (C). HRMS m/z (EI): 294.1107 (M+• C15H18O6+• requires 294.1103).
1′-acetoxy-3,5-dimethoxychavicol (19)
To a solution of vinylmagnesium bromide freshly prepared from magnesium turning (0.92 g, 38.3 mmol), 1 M vinyl bromide (17 mL, 17.1 mmol) and catalytic amount of iodine crystal in dry diethyl ether (30 mL), a solution of aldehyde 11 (0.98 g, 5.89 mmol) in diethyl ether (10 mL) under ice bath, were added. The ice bath was removed, and the mixture was allowed to warm to room temperature. After the reaction mixture was stored for 2 h at room temperature, saturated aqueous NH4Cl (10 mL) solution was added. The mixture was extracted with diethyl ether (2×30 mL). The resulting organic extracts were combined, and the solvent was removed under reduced pressure to yield a crude product. The oily product (intermediate 15) obtained by usual workup was acetylated with acetic anhydride (0.60 mL, 6.35 mmol) and DMAP (1.52 g, 12.4 mmol) in dichloromethane (20 mL) in an ice bath. After consumption of starting material and product formation, the reaction was quenched with saturated aqueous NH4Cl (10 mL) solution and extracted with dichloromethane (3×15 mL), dried (NaSO4), and filtered. Evaporation of filtrate gave a yellow oil, which was purified by column chromatography on silica gel, and eluting with hexane/ethyl acetate (9:1) afforded the desired product as a yellowish oil 19 (0.90 g, 65%).
The spectral analysis of the yellowish oil: IR (CCl4) υmax, cm−1: 1,739, 1,602, 1,558, 1,519, 1,459, 1,232. UV λmax, nm: 304.5. 1H NMR (CDCl3, 400 MHz), δ: 2.11 (3H, s), 3.77 (6H, s), 5.31 (2H, dd, J =10.5 Hz), 5.97 (1H, m), 6.16 (1H, d, J =5.9 Hz), 6.38 (2H, s), 6.49 (1H, s). 13C NMR (CDCl3, 100 MHz), δ: 21.3 (CH3), 55.4 (2OCH3), 76.8 (CH), 100.0 (CH), 106.6 (2CH), 141.3 (C), 160.9 (2C), 170.0 (C). HRMS m/z (EI): 236.1045 (M+• C13H16O4+• requires 236.1049).
1′-acetoxy-4-methoxychavicol (20)
To a solution of vinylmagnesium bromide freshly prepared from magnesium turning (2.14 g, 89.2 mmol), 1 M vinyl bromide (37 mL, 37.0 mmol) and catalytic amount of iodine crystal in dry diethyl ether (50 mL), a solution of aldehyde 12 (1.68 g, 12.3 mmol) in diethyl ether (10 mL) under ice bath, were added. The ice bath was removed, and the mixture was allowed to warm to room temperature. After the reaction mixture was stored for 3 h at room temperature, saturated aqueous NH4Cl (10 mL) solution was added. The mixture was extracted with diethyl ether (2×30 mL). The resulting organic extracts were combined, and the solvent was removed under reduced pressure to yield a crude product. The oily product (intermediate 16) obtained by usual workup was acetylated with acetic anhydride (1.51 mL, 16.0 mmol) and DMAP (3.09 g, 25.3 mmol) in dichloromethane (25 mL) in an ice bath. After consumption of starting material and product formation, the reaction was quenched with saturated aqueous NH4Cl (10 mL) solution and extracted with dichloromethane (3×15 mL), dried (NaSO4), and filtered. Evaporation of filtrate gave a yellow oil, which was purified by column chromatography on silica gel, eluting with hexane/ethyl acetate (95:5) afforded the desired product as a yellowish oil 20 (1.57 g, 62%).
The spectral analysis of the yellowish oil: IR (neat) υmax, cm−1: 1,737, 1,515, 1,236. UV λmax, nm: 304.5. 1 HNMR (CDCl3, 400 MHz), δ: 2.07 (3H, s), 3.78 (3H, s), 5.28 (2H, dd, J =10.4 Hz), 5.99 (1H, m), 6.22 (1H, d, J =5.8 Hz), 6.88 (2H, d, J =6.8 Hz), 7.28 (2H, d, J =6.8 Hz). 13C NMR (CDCl3, 100 MHz), δ: 21.3 (CH3), 55.3 (OCH3), 76.3 (CH), 113.9 (2CH), 116.5 (CH2), 128.8 (2CH), 136.4 (CH), 159.5 (C), 169.1 (C). MS m/z (EI) 43, 65, 77, 91, 104, 121, 131,149, 164, 206 (M+•). HRMS m/z (EI): 206.0948 (M+• C12H14O3+• requires 206.0943).
Cell lines and culture conditions
A total of seven human cancer cell lines and one normal cell line were used in this study. Five cell lines (PC-3 [prostate cancer cells], MCF-7 and MDA-MB-231 [breast cancer cells], EJ-28 and RT-112 [urinary bladder cancer cells]) were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium, while two cell lines (HSC-4 [oral squamous cancer cells] and HepG2 [liver cancer cells]) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. The human mammary epithelial cell line (HMEC) used as the normal cell control was cultured in mammary epithelial growth medium. The cell lines such as PC-3, MCF-7, MDA-MB-231, EJ-28, HepG2 were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), RT-112 from German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany), and HSC-4 from Malaysian Cancer Research Initiative Foundation (CARIF, Selangor, Malaysia), while HMEC was obtained from Lonza Inc. (Allendale, NJ, USA). All cultures were maintained in an incubator with humidified atmosphere (5% CO2) at 37°C.
Cell viability assay
Cytotoxic effects of ACA and its analogs (Figure 1) on the seven cancer cell lines were determined by using the in vitro 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. All tested compounds were dissolved in dimethyl sulfoxide (DMSO) to a final concentration of 10.0 mM. In brief, cells were seeded in triplicates on 96-well plates at 1.0×104 cells/well. After 24 h, cells were treated with increasing concentrations (5.0–50.0 μM) of ACA and its analogs for 24 h. A total of 10.0 μL of MTT reagent (5.0 mg/mL) was added to each well. After incubation in the dark at 37°C for 2 h, the unreacted MTT dye was removed by aspiration and 200.0 μL of DMSO was added to dissolve MTT purple formazan. The absorbance was determined at 579 nm by microtiter plate reader (Tecan Sunrise®, Männedorf, Switzerland), with a reference wavelength of 650 nm.
Figure 1.

The structures of ACA and its analogs (2, 5–8, and 17–20).
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate.
DNA fragmentation assay
MDA-MB-231 breast cancer cells were treated with ACA, AEA, and AMCA at their half-maximal inhibitory concentrations (IC50; 4.8, 9.5 and 29.6 μM, respectively) for 24 h. Total DNA was extracted from both untreated and treated cells using the Suicide Track™ DNA Ladder Isolation Kit (Calbiochem, La Jolla, CA, USA) according to the manufacturer’s protocol. Analysis of extracted DNA was carried out on a 1.0% (w/v) agarose gel electrophoresis and stained with ethidium bromide. Observation of DNA fragmentation was performed under UV illumination and visualized using a gel documentation system (Alpha Innotech, San Leandro, CA, USA).
Anti-migration assay
The anti-migration effects of ACA and its analogs were determined using the wound-healing assay. MDA-MB-231 cells were seeded in six-well plates and cultured until 80% confluent. After an overnight cultivation, complete medium was then replaced by serum-free medium treated with 1 μg/mL mitomycin-C, and the cells were further incubated at 37°C for 2 h to stop cell proliferation. A vertical scratch was made using a sterile pipette tip across the well, and cell debris was removed by washing with 1× phosphate-buffered saline. Cells were treated with control solvent or ACA and its analogs at their 20% inhibitory concentrations (IC20; Table S1) in serum-free medium. IC20 were used instead of IC50, because IC20 were less toxic on the cells. Images of wounded cells were captured using an inverted fluorescence microscope, Nikon Eclipse TS-100 (Nikon Corporation, Tokyo, Japan), at 0 and 24 h after wounding. The distance between two sides of the wound was analyzed using Tscratch software, version 1.0 (The Mathworks, Natick, MA, USA).
Western blot assay
MDA-MB-231 cells were treated with ACA, AEA, and AMCA and incubated for 24 h. Total proteins were extracted using the NE-PER nuclear and cytoplasmic extraction kit (Pierce, Rockford, IL, USA) according to the manufacturer’s protocol. Protein concentrations were determined using the Bio-Rad DC protein assay (Bio-Rad Laboratories Inc., Hercules, CA, USA) according to the manufacturer’s protocol. Equal amounts of protein were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes by semi-dry transfer system (Bio-Rad Laboratories Inc.). Membranes were blocked with 5% (w/v) bovine serum albumin and incubated with primary antibodies against GAPDH, Bcl-2, Bcl-xL, Bax, p53, PARP, β1 integrin, FAK, pFAK, Akt, and pAkt (1:1,000 dilutions; Cell Signaling Technology, Danvers, MA, USA). Horseradish peroxidase-linked secondary antibodies were added, and bound proteins were detected through enhanced chemiluminescence reagent (Advan-sta, Menlo Park, CA, USA) and visualized on a Fusion FX7 imaging system (Vilbert Lonmart, Eberhardzell, Germany).
Statistical analysis
The results were presented as mean ± standard error of the mean for triplicates of all experiments. One-way ANOVA was used to analyze the statistically significant differences with a confidence level of p≤0.05.
Results
Effect of hemi-synthetic ACA analogs on the viability of various cancer cell lines
Cytotoxicity evaluation of ACA and its analogs (Figure 1) in various human cancer cell lines was performed using the MTT assay method with results expressed as mean IC50 values. Seven cancer cell lines were used in this study to explore whether different ACA analogs could exhibit a similar broad spectrum cytotoxic effect as was reported for ACA on various cancer types.9–12 All tested analogs indicated weak cytotoxicity except for compounds ACA, AEA, and AMCA with potencies ≤30.0 μM (Table 1). Meanwhile, all compounds were shown to have minimal cytotoxic activity toward HMECs used as normal human breast cell control, indicating nontoxic effects against normal cells at therapeutic doses. Solvent control with DMSO at ≤1.0% (v/v) showed insignificant effects on cancer cell viability (data not shown), also indicating that cytotoxicity was induced as a result of treatment with tested compounds instead of DMSO, which is known to be cytotoxic at high concentrations.20 ACA induced cytotoxicity on all cancer cell lines tested, with highest levels of efficacy being observed in breast adenocarcinoma (MDA-MB-231) and oral squamous carcinoma cells (HSC-4) at 24 h post treatment time. Although AEA and AMCA also induced cytotoxicity, their effects were found to be selective toward specific cancer cell lines. AEA was found to induce high levels of cytotoxicity in bladder adenocarcinoma (EJ-28), hepatocarcinoma (HepG2), and oral SCC (HSC-4), while AMCA only showed cytotoxic effects against MDA-MB-231 cells (Table 1).
Table 1.
IC50 values for ACA and its analogs on various cancer cell lines and normal cells
| Cell lines | IC50 (μM)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ACA | 2 | 5 | 6 | 7 | 8 | AEA | AMCA | 19 | 20 | |
| Breast adenocarcinoma (MDA-MB-231) | 4.8±0.4 | N/A | N/A | N/A | N/A | N/A | 9.5±0.3 | 29.6±5.6 | N/A | N/A |
| Breast adenocarcinoma (MCF-7) | 30.0±0.3 | N/A | N/A | N/A | N/A | N/A | 25.2±0.6 | N/A | N/A | N/A |
| Bladder carcinoma (RT-112) | 14.1±3.8 | N/A | N/A | N/A | N/A | N/A | 10.9±2.4 | N/A | N/A | N/A |
| Bladder carcinoma (EJ-28) | 8.2±0.9 | N/A | N/A | N/A | N/A | N/A | 4.2±2.2 | N/A | N/A | N/A |
| Prostate carcinoma (PC-3) | 26.7±2.3 | N/A | N/A | N/A | N/A | N/A | 13.8±0.8 | N/A | N/A | N/A |
| Oral squamous carcinoma (HSC-4) | 5.5±0.5 | N/A | N/A | N/A | N/A | N/A | 5.1±0.2 | N/A | N/A | N/A |
| Hepatocyte carcinoma (HepG2) | 18.0±0.8 | N/A | N/A | N/A | N/A | N/A | 4.3±0.5 | N/A | N/A | N/A |
| HMECs | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
Notes: Cells were treated with the test compounds for 24 h. Data are reported as mean ± SEM (n=3). N/A denotes that cell viability levels were maintained above 50% at maximum concentration of test compound (50 μM) at 24 h of incubation.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; HMECs, human mammary epithelial cells; IC50, half-maximal inhibitory concentration; SEM, standard error of the mean; N/A, not applicable.
Effect of ACA, AEA, and AMCA on DNA fragmentation
Next, to determine whether their cytotoxic effects are mediated through apoptosis, we performed DNA fragmentation assays. As shown in Figure 2, DNA from the untreated group showed no degradation, whereas DNA laddering was observed in MDA-MB-231 cells that were treated with ACA, AEA, and AMCA at their IC50. This typical oligonucleosomal DNA degradation is one of the hallmarks of apoptotic cell death, thus suggesting that ACA, AEA, and AMCA induce cell death through apoptosis.
Figure 2.
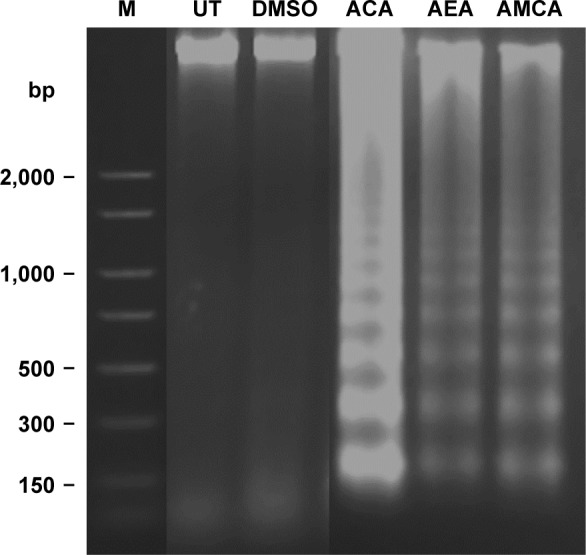
Agarose gel electrophoresis of DNA in MDA-MB-231 cells treated with ACA, AEA, and AMCA.
Notes: Cells were untreated (lane UT) or treated with DMSO 0.08% (v/v), ACA, AEA, AMCA at their IC50 (4.8, 9.5, and 29.6 μM, respectively) for 24 h. Left lane (M) shows DNA size markers. Cellular DNA was extracted and subjected to agarose gel electrophoresis.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; DMSO, dimethyl sulfoxide; DNA, deoxyribonucleic acid; IC50, half-maximal inhibitory concentration; UT, untreated.
Effect of ACA, AEA, and AMCAon PARP, p53, and Bcl-2 family members
To further verify the occurrence of apoptosis induced by ACA, AEA, and AMCA on MDA-MB-231 cells, we performed PARP cleavage analysis, a common marker for induction of apoptosis.21 As shown in Figure 3A, the cleavage of full-length PARP enzymes (116 kDa) into an 89 kDa subunit protein was observed in the MDA-MB-231 cells that had been treated with ACA, AEA, and AMCA at their IC50. Based on densitometry analysis, these three compounds increased the cleavage of PARP by 7.8-, 4.8-, and 1.8-fold compared to untreated cells, respectively (Figure 3B). ACA, AEA, and AMCA were also found to decrease Bcl-2 and Bcl-xL anti-apoptotic protein levels and increase the expression levels of p53 and pro-apoptotic Bax. As shown in Figure 3C and D, treatment with ACA, AEA, and AMCA at their IC50 for 24 h significantly decreased the expression level of Bcl-2 by ~40%–50% and Bcl-xL by ~30%–50% when compared to untreated cells. The p53 levels in MDA-MB-231 cells increased by 3.3-, 3.4-, and 2.3-fold after treatment with ACA, AEA, and AMCA, respectively (Figure 3E). The three compounds upregulated the expression of pro-apoptotic Bax by 6.2-, 9.4-, and 2.9-fold, respectively (Figure 3F). Collectively, this suggests that PARP, p53, Bcl-2, Bcl-xL, and Bax were involved in inducing apoptosis via the intrinsic pathway when treated with ACA, AEA, and AMCA.
Figure 3.

Effects of ACA, AEA, and AMCA on the apoptotic pathway.
Notes: Expression levels of PARP, p53, Bcl-2, Bcl-xL, and Bax proteins from MDA-MB-231 cells were analyzed via Western blot analysis (A). GAPDH was used as a loading control. The quantitative data (B–F) are presented as mean ± SEM from three independent experiments (*p≤0.05 by two-tailed unpaired t-test).
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; Bax, Bcl-2-associated X protein; Bcl-2, B-cell lymphoma 2; Bcl-xL, B-cell lymphoma-extra large; DMSO, dimethyl sulfoxide; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PARP, poly(ADP-ribose) polymerase; p53, tumor suppressor p53; SEM, standard error of the mean.
Effect of hemi-synthetic ACA analogs on anti-migration of MDA-MB-231 cells
Cell migration is an important step in the metastatic process.22 Therefore, the effects of ACA and its analogs on cell migration were also examined on breast cancer MDA-MB-231 cells (Figures 4 and S1). This cell line was chosen for anti-migration study due to its highly aggressive and strongly metastatic properties among the selected panel of cell lines.23,24 Our data showed that ACA, AEA, and AMCA and compounds 5–8 and 19–20 significantly inhibited the migration rate of MDA-MB-231 cells compared with untreated cells. Furthermore, results revealed that the difference in inhibition of cell migration between untreated cells and cells treated with DMSO was insignificant (p-value >0.05), suggesting that the anti-migration effects seen were not due to the solvent.
Figure 4.

Effects of ACA and its analogs on the cell migration of MDA-MB-231 cells.
Notes: Cells were wounded and treated with DMSO 0.08% (v/v), ACA, and nine analogs at IC20 for 24 h. The open wound area at 24 h was quantified using TScratch software relative to wound area at 0 h. The mean percentage of area migrated ± SEM from three separate experiments is illustrated. Statistically significant differences were compared between untreated conditions versus treatment groups with (*) denoting p-values ≤0.05.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; DMSO, dimethyl sulfoxide; IC20, 20% inhibitory concentration; SEM, standard error of the mean.
Effect of ACA, AEA, and AMCA on downregulators of integrin β1
In addition to anti-proliferative effects and induction of apoptosis, ACA, AEA, and AMCA also showed significant anti-migration effects on MDA-MB-231 breast cancer cells. Thus, the molecular mechanism related to the anti-migration effects of these compounds was also investigated. The effect of these compounds on the expression of integrin β1, which is a major player in tumor metastasis that mediate the adhesion of cells on extracellular matrix (ECM),25 and some of its downstream molecules were determined. We examined the integrin-mediated pathway on MDA-MB-231 breast cancer cell line by Western blot analysis and found that the level of integrin β1 decreased following ACA, AEA, and AMCA treatment (Figure 5A). Upon integrin β1 binding to ECM, the downstream signaling molecules such as FAK and Akt were phosphorylated26 and participated in the regulation of cell migration. In analyzing the integrin-induced signaling pathway, phosphorylation of FAK and Akt was significantly reduced by ACA, AEA, and AMCA after 24 h (Figure 5A). As shown in Figure 5B, the integrin β1 levels of MDA-MB-231 cells decreased by 15.5%, 37.1%, and 93.3% after treatment with ACA, AEA, and AMCA, respectively. The reduction level of phosphorylated FAK and Akt by ACA, AEA, and AMCA was about 70%–94% (Figure 5C and D).
Figure 5.

Effects of ACA, AEA, and AMCA on the migration pathway.
Notes: Expression levels of integrin β1, FAK, pFAK, Akt, and pAkt proteins from MDA-MB-231 cells were analyzed via Western blot analysis (A). GAPDH was used as a loading control. The quantitative data (B–D) are presented as mean ± SEM from three independent experiments (*p≤0.05 by two-tailed unpaired t-test).
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; Akt, protein kinase B; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; DMSO, dimethyl sulfoxide; FAK, focal adhesion kinase; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; integrin β1, integrin subunit beta 1; pAkt, phosphorylated protein kinase B; pFAK, phosphorylated focal adhesion kinase; SEM, standard error of the mean.
Discussion
ACA is a natural product that exerts anticancer effects by inducing apoptosis27 and cell cycle arrest4 and inhibiting angiogenesis28 and metastasis.29 Wang et al29 showed that ACA exerted cytotoxic effects and inhibited the signal transduction pathway related to tumor metastasis, such as signal transducer and activator of transcription 3 (STAT3), Src homology region 2 domain-containing phosphatase 1 (SHP-1), and matrix metalloproteinase (MMP)-mediated pathway. In this study, ACA inhibited the growth of MDA-MB-231 cells with IC50 of 4.8 μM on the breast cancer cell line, which is about 10 times lower than Wang et al’s study. The significant difference of the results may be due to the heterogeneity of cancer cells30 and the changes that occurred in cells during cultivation in different laboratory conditions.
Even though ACA was seen to induce apoptosis and inhibit migration on some cancer cell types, the apoptotic induction and anti-migration mechanisms of hemi-synthetic ACA analogs on breast cancer cells have never been reported. In this study, we revealed that not only ACA but also two of its analogs were able to induce apoptosis and suppress the migration of MDA-MB-231 breast cancer cells.
In this study, three compounds (ACA, AEA, and AMCA) possessed 1′-acetoxyl group and showed better cytotoxicity than other compounds. This highlighted the importance of 1′-acetoxyl group in governing anti-proliferative activity. The absence of the 1′-acetoxyl group (compounds 2, 5, 6, 8) could lead to a reduction in inhibitory effects on cancer cell growth. These findings are consistent with the study by Murakami et al.13 On the other hand, AMCA, which possesses an additional methoxyl group at the 5-position compared to AEA, exerted a weaker effect against MDA-MB-231 cells, suggesting that the presence of an additional methoxyl group at 5-position may reduce cytotoxic activity. In a study by Xu et al,31 the authors reported the structural factors of ACA that regulate tumor cell viability, intracellular glutathione level, and glutathione reductase activity in Ehrlich ascites tumor cells. They suggested that the acetoxyl group substituted at para position of the benzene ring was essential, and similarly we found that analog without an acetoxyl group at the 4-position (such as compounds 2 and 20) showed insignificant cytotoxic effects.
MDA-MB-231 was reported as the most sensitive cell line toward the compounds tested in terms of cytotoxicity and was thus selected as a model for further investigation. Another type of breast cancer cell, MCF-7 cell line, was only sensitive to ACA and AEA. Despite both cell lines being breast cancer cell lines, MDA-MB-231 is a triple-negative (lack of ER, PR, and HER-2 expression) breast cancer cell line, while MCF-7 is an ER- and PR-positive breast cancer cell line. Thus, these differences could be the underlying cause toward response variations and drug sensitivity.32
DNA fragmentation is a cellular hallmark of apoptosis. In apoptotic cells, endonucleases cleave DNA into internucleosomal fragments of roughly 180–200 bp.33 Since ACA, AEA, and AMCA showed active anti-proliferative effects and induced internucleosomal DNA fragmentation on MDA-MB-231 cells, induction of apoptosis was confirmed. During the apoptotic process, proteolytic cleavage of PARP due to the activation of caspase-334 was seen in this study. Previous study showed that ACA induced apoptosis via the activation of caspase-3-like activity.35 Thus, the alteration of PARP by ACA, AEA, and AMCA was in agreement with the induction of apoptosis via the activation of caspase-3. The activation of the p53 protein is also related to induction of apoptosis36 and regulation of Bcl-2 family members’ expression.37 Anti-apoptotic proteins, Bcl-2 and Bcl-xL, act as inhibitors of the mitochondrial intrinsic pathway that block the release of cytochromes and counteract the effects of pro-apoptotic proteins such as Bax.38 Pro-apoptotic protein, Bax, redistributes from the cytosol to mitochondria during apoptotic events39 to cause dysfunction of the mitochondrial membrane.40 In this study, the expression of Bax was increased by treatment with ACA, AEA, and AMCA, while the expression of Bcl-2 and Bcl-xL was decreased. These observations suggested that the induction of apoptosis by ACA, AEA, and AMCA was triggered by the upregulation of pro-apoptotic proteins and downregulation of anti-apoptotic proteins related to the mitochondrial intrinsic pathway. Furthermore, previous studies showed that ACA and AEA suppressed the growth of cancer cells by inhibiting nuclear factor-κB (NF-κB) pathway.19,41–43 The expression of the anti-apoptotic proteins, such as Bcl-244 and Bcl-xL,45 was regulated by NF-κB. Hence, the induction of apoptosis by ACA, AEA, and AMCA may also be linked to the inactivation of the NF-κB.
ACA has also been reported to inhibit metastasis and invasion on different cancer cell types, such as breast cancers,29 oral cancers,43 prostate cancers,28 and lung cancers.46 These investigations point out that ACA may generally suppress cell migration and invasion in various cancer cell types. In this study, we reported that ACA and its analogs exhibit anti-migration effects on MDA-MB-231 cells. Although some of the ACA analogs showed poor potency against cancer cell proliferation, they exhibited strong effects against migration and thus reignite interest for further studies on the molecular mechanisms governing anti-migration activity of ACA and its analogs. In this study, most of the hemi-synthetic ACA analogs were not cytotoxic within the 5.0–50.0 μM dose range, but many of them were able to inhibit cancer cell migration with nontoxic concentrations. Hence, the anti-migration activity of the ACA analogs is not linked to their direct cytotoxic effects on cancer cells. The inhibition of cell migration by the compounds occurred at concentrations lower than those that inhibited cell proliferation. The difference in effective concentrations suggested that different mechanisms may be involved to promote ACA analogs’ anti-migration effects versus cytotoxic ability.
Cancer cell motility by metastasis is a main step for the progression of malignancy.47 Tumor cells take multisteps to metastasize from their initial site to secondary organs by migration through basement membranes and extracellular matrices.48 The migration process is stimulated extracellularly and initiated by integrins and intracellular signaling proteins located at the sites of focal adhesion assembly and disassembly.49 Regulating integrin β1 signaling of FAK through modulation of Akt activity could lead to inhibition of cell migration.50 Many human malignant cancers show increased FAK expression promoted by integrin β1 to result in increased metastasis.51 In this study, breast cancer cells treated with ACA, AEA, and AMCA suppressed integrin β1 after 24 h (Figure 5), which indicated their ability to interrupt the integrin signaling pathway. Since integrin acts as the activator of FAK, we examined the effect of these compounds on the activation of pFAK. Upon treatment, FAK phosphorylation was significantly reduced in MDA-MB-231 cells. ACA, AEA, and AMCA also decreased Akt phosphorylation, showing that the activity of pAkt was correlated with FAK phosphorylation. Thus, relative intensities of integrin β1, pFAK/FAK, and pAkt/Akt showed that ACA, AEA, and AMCA showed anti-migration potency and is dependent, at least in part, on the integrin β1-mediated signaling pathway. Wang et al29 showed that inhibition of metastasis by ACA is through interference with the STAT3 signaling pathway. Their findings support our current result as STAT3 proteins are known to be interrelated to integrin β1. Nevertheless, the mechanism(s) in which ACA and its analogs participate in to achieve their anticancer role on TNBCs will require further in-depth investigation, such as the evaluation of key proteins involved in other metastatic signaling pathways.
In addition, the efficacy of ACA is also being studied for its use in combination studies with other drugs/biologics. For example, ACA reduced tumor volume and side effects in vivo when used in combination with alpha-fetoprotein (AFP).52
While stand-alone doses of the analogs may not seem clinically relevant at this stage, such structural modifications of ACA could potentially serve as a platform to find better candidates which can be used in combination regimens during preclinical developmental studies. This study demonstrates that there is still room for improvement in designing modified ACA analogs with increased anticancer activities.
Conclusion
We have identified that ACA, AEA, and AMCA significantly inhibited growth, induced apoptosis, and suppressed migration of human breast cancer cells. Overall, these preliminary results revealed that the anti-proliferative activities are not absolutely required for the suppression of migration in cancer cells by ACA and its analogs. In addition, analogs active in anti-proliferation against selected cancer cells were seen to possess better anti-migration ability against breast cancer cells compared to the natural product itself. Thus, this study demonstrates that there is still room for improvement in designing new analogs with increased anticancer activities. However, more investigations are required to identify the different targets of such ACA analogs at the molecular level and validate the molecular mechanisms in animal models for effective treatment of breast cancer. Overall, this study suggested that ACA, along with its hemi-synthetic analogs, AEA and AMCA, are potential combinatory therapeutic agents for the treatment of TNBCs.
Supplementary materials
Representative pictures of wound-healing assay of MDA-MB-231 cells after various treatments of ACA and its analogs at IC20.
Notes: The open wound area at 24 h was quantified using TScratch software relative to wound area at 0 h. All data are presented as mean ± SEM from three independent replicates, with representative images at 40× magnification shown from each treatment group.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; DMSO, dimethyl sulfoxide; IC20, 20% inhibitory concentration; SEM, standard error of the mean.
Table S1.
IC20 values for ACA and its analogs on MDA-MB-231 cell line
| IC20 (μM)
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ACA | 2 | 5 | 6 | 7 | 8 | AEA | AMCA | 19 | 20 |
| 2.0 | 48.0 | 32.0 | 50.0 | 31.0 | 50.0 | 4.0 | 12.0 | 28.0 | 45.0 |
Note: IC20 for each analog was used to treat MDA-MB-231 cells for wound-healing assay.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; IC20, 20% inhibitory concentration.
Acknowledgments
ACA and analogs were prepared by Chin Hui Kee. This study was supported by the University of Malaya Postgraduate Research Grant (PV050-2012A, PG100-2012B), the Centre for Research in Biotechnology for Agriculture (CEBAR) Flagship Grant (RU005C-2014), Chemistry-HIR Grant UM.0000091/HIR.C3, RP001-2012A/B, Malaysian Ministry of Higher Education (MOHE) Fundamental Research Grant Scheme (FRGS) (FRGS/1/2014/SG05/UCSI/03/1), and the Biosecurity and Biosafety for Tropical Agricultural Bioeconomy Grant (RU015-2016).
Footnotes
Author contributions
SKL carried out the biological experiments and wrote the manuscript. MNA isolated ACA, synthesized the analogs, and contributed to drafting the manuscript. LLAI helped in solving technical experiment problems and editing the manuscript. KA participated in the design of the analogs. NHN conceived the entire study and helped in editing the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Fett-Neto AG, DiCosmo F, Reynolds WF, Sakata K. Cell culture of Taxus as a source of the antineoplastic drug taxol and related taxanes. Biotechnology. 1992;10(12):1572–1575. doi: 10.1038/nbt1292-1572. [DOI] [PubMed] [Google Scholar]
- 2.Datta A, Srivastava PS. Variation in vinblastine production by Catharanthus roseus, during in vivo and in vitro differentiation. Phytochemistry. 1997;46(1):135–137. [Google Scholar]
- 3.Morrison KC, Hergenrother PJ. Natural products as starting points for the synthesis of complex and diverse compounds. Nat Prod Rep. 2014;31(1):6–14. doi: 10.1039/c3np70063a. [DOI] [PubMed] [Google Scholar]
- 4.Awang K, Azmi MN, Aun LI, Aziz AN, Ibrahim H, Nagoor NH. The apoptotic effect of 1′s-1′-acetoxychavicol acetate from Alpinia conchigera on human cancer cells. Molecules. 2010;15(11):8048–8059. doi: 10.3390/molecules15118048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitsui S, Kobayashi S, Nagahori H, Ogiso A. Constituents from seeds of Alpinia galanga Wild, and their anti-ulcer activities. Chem Pharm Bull. 1976;24(10):2377–2382. doi: 10.1248/cpb.24.2377. [DOI] [PubMed] [Google Scholar]
- 6.Janssen AM, Scheffer JJ. Acetoxychavicol acetate, an antifungal component of Alpinia galanga. Planta Med. 1985;51(6):507–511. [PubMed] [Google Scholar]
- 7.Noro T, Sekiya T, Katoh M, et al. Inhibitors of xanthine oxidase from Alpinia galanga. Chem Pharm Bull. 1988;36(1):244–248. [Google Scholar]
- 8.Kondo A, Ohigashi H, Murakami A, Suratwadee J, Koshimizu K. 1′-Acetoxychavicol acetate as a potent inhibitor of tumor promoter-induced Epstein-Barr virus activation from Languas galanga, a traditional Thai Condiment. Biosci Biotechnol Biochem. 1993;57(8):1344–1345. [Google Scholar]
- 9.Murakami A, Ohura S, Nakamura Y, Koshimizu K, Ohigashi H. 1′-Acetoxychavicol acetate, a superoxide anion generation inhibitor, potently inhibits tumor promotion by 12-O-tetradecanoylphorbol-13-acetate in ICR mouse skin. Oncology. 1996;53(5):386–391. doi: 10.1159/000227593. [DOI] [PubMed] [Google Scholar]
- 10.Ohnishi M, Tanaka T, Makita H, et al. Chemopreventive effect of a xanthine oxidase inhibitor, 1′-acetoxychavicol acetate, on rat oral carcinogenesis. Jpn J Cancer Res. 1996;87(4):349–356. doi: 10.1111/j.1349-7006.1996.tb00229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka T, Makita H, Kawamori T, et al. A xanthine oxidase inhibitor 1′-acetoxychavicol acetate inhibits azoxymethane-induced colonic aberrant crypt foci in rats. Carcinogenesis. 1997;18(5):1113–1118. doi: 10.1093/carcin/18.5.1113. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi Y, Nakae D, Akai H, et al. Prevention by 1′-acetoxychavicol acetate of the induction but not growth of putative preneoplastic, glutathione S-transferase placental form-positive, focal lesions in the livers of rats fed a choline-deficient, L-amino acid-defined diet. Carcinogenesis. 1998;19(10):1809–1814. doi: 10.1093/carcin/19.10.1809. [DOI] [PubMed] [Google Scholar]
- 13.Murakami A, Toyota K, Ohura S, Koshimizu K, Ohigashi H. Structure-activity relationships of (1′S)-1′-acetoxychavicol acetate, a major constituent of a Southeast Asian condiment plant Languas galanga, on the inhibition of tumor-promoter-induced Epstein-Barr virus activation. J Agric Food Chem. 2000;48(5):1518–1523. doi: 10.1021/jf990528r. [DOI] [PubMed] [Google Scholar]
- 14.Dahlui M, Ramli S, Bulgiba AM. Breast cancer prevention and control programs in Malaysia. Asian Pac J Cancer Prev. 2011;12(6):1631–1634. [PubMed] [Google Scholar]
- 15.Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011;16(1):1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 16.Chacón RD, Costanzo MV. Triple-negative breast cancer. Breast Cancer Res. 2010;12(2):1–9. doi: 10.1186/bcr2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S-J, Ando T. Optically active 1-acetoxychavicol acetate and its positional isomers: synthesis and repellent effect against adzuki bean weevil. J Pestic Sci. 2001;26:76–81. [Google Scholar]
- 18.Hasima N, Aun LI, Azmi MN, et al. 1′S-1′-acetoxyeugenol acetate: a new chemotherapeutic natural compound against MCF-7 human breast cancer cells. Phytomedicine. 2010;17(12):935–939. doi: 10.1016/j.phymed.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 19.In LL, Azmi MN, Ibrahim H, Awang K, Nagoor NH. 1′S-1′-acetoxyeugenol acetate: a novel phenylpropanoid from Alpinia conchigera enhances the apoptotic effects of paclitaxel in MCF-7 cells through NF-kappaB inactivation. Anticancer Drugs. 2011;22(5):424–434. doi: 10.1097/CAD.0b013e328343cbe6. [DOI] [PubMed] [Google Scholar]
- 20.Wang C-C, Lin S-Y, Lai Y-H, Liu Y-J, Hsu Y-L, Chen JJW. Dimethyl sulfoxide promotes the multiple functions of the tumor suppressor HLJ1 through activator protein-1 activation in NSCLC cells. PLoS One. 2012;7(4):e33772. doi: 10.1371/journal.pone.0033772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diefenbach J, Burkle A. Introduction to poly(ADP-ribose) metabolism. Cell Mol Life Sci. 2005;62(7–8):721–730. doi: 10.1007/s00018-004-4503-3. [DOI] [PubMed] [Google Scholar]
- 22.Harlozinska A. Progress in molecular mechanisms of tumor metastasis and angiogenesis. Anticancer Res. 2005;25(5):3327–3333. [PubMed] [Google Scholar]
- 23.Price JE, Polyzos A, Zhang RD, Daniels LM. Tumorigenicity and metastasis of human breast carcinoma cell lines in nude mice. Cancer Res. 1990;50(3):717–721. [PubMed] [Google Scholar]
- 24.Nair HK, Rao KVK, Aalinkeel R, Mahajan S, Chawda R, Schwartz SA. Inhibition of prostate cancer cell colony formation by the flavonoid quercetin correlates with modulation of specific regulatory genes. Clin Diagn Lab Immunol. 2004;11(1):63–69. doi: 10.1128/CDLI.11.1.63-69.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin H, Varner J. Integrins: roles in cancer development and as treatment targets. Br J Cancer. 2004;90(3):561–565. doi: 10.1038/sj.bjc.6601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vachon PH. Integrin signaling, cell survival, and anoikis: distinctions, differences, and differentiation. J Sig Transduction. 2011;2011:18. doi: 10.1155/2011/738137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito K, Nakazato T, Murakami A. Induction of apoptosis in human myeloid leukemic cells by 1′-acetoxychavicol acetate through a mitochondrial-and Fas mediated dual mechanism. Clin Cancer Res. 2004;10:2120–2130. doi: 10.1158/1078-0432.ccr-1142-03. [DOI] [PubMed] [Google Scholar]
- 28.Pang X, Zhang L, Lai L, et al. 1′-Acetoxychavicol acetate suppresses angiogenesis-mediated human prostate tumor growth by targeting VEGF-mediated Src-FAK-Rho GTPase-signaling pathway. Carcinogenesis. 2011;32(6):904–912. doi: 10.1093/carcin/bgr052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang J, Zhang L, Chen G, et al. Small molecule 1′-acetoxychavicol acetate suppresses breast tumor metastasis by regulating the SHP-1/STAT3/MMPs signaling pathway. Breast Cancer Res Treat. 2014;148(2):279–289. doi: 10.1007/s10549-014-3165-6. [DOI] [PubMed] [Google Scholar]
- 30.Nugoli M, Chuchana P, Vendrell J, et al. Genetic variability in MCF-7 sublines: evidence of rapid genomic and RNA expression profile modifications. BMC Cancer. 2003;3(1):13. doi: 10.1186/1471-2407-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu S, Kojima-Yuasa A, Azuma H, Kennedy DO, Konishi Y, Matsui-Yuasa I. Comparison of glutathione reductase activity and the intracellular glutathione reducing effects of 13 derivatives of 1′-acetoxychavicol acetate in Ehrlich ascites tumor cells. Chem Biol Interact. 2010;185(3):235–240. doi: 10.1016/j.cbi.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 32.Rouzier R, Perou CM, Symmans WF, et al. Breast cancer molecular subtypes respond differently to preoperative chemotherapy. Clin Cancer Res. 2005;11(16):5678–5685. doi: 10.1158/1078-0432.CCR-04-2421. [DOI] [PubMed] [Google Scholar]
- 33.Zhang JH, Xu M. DNA fragmentation in apoptosis. Cell Res. 2000;10(3):205–211. doi: 10.1038/sj.cr.7290049. [DOI] [PubMed] [Google Scholar]
- 34.Los M, Mozoluk M, Ferrari D, et al. Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol Biol Cell. 2002;13(3):978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moffatt J, Hashimoto M, Kojima A, et al. Apoptosis induced by 1′-acetoxychavicol acetate in Ehrlich ascites tumor cells is associated with modulation of polyamine metabolism and caspase-3 activation. Carcinogenesis. 2000;21(12):2151–2157. doi: 10.1093/carcin/21.12.2151. [DOI] [PubMed] [Google Scholar]
- 36.Geng QQ, Dong DF, Chen NZ, et al. Induction of p53 expression and apoptosis by a recombinant dual-target MDM2/MDMX inhibitory protein in wild-type p53 breast cancer cells. Int J Oncol. 2013;43(6):1935–1942. doi: 10.3892/ijo.2013.2138. [DOI] [PubMed] [Google Scholar]
- 37.Sax JK, El-Deiry WS. p53 downstream targets and chemosensitivity. Cell Death Differ. 2003;10(4):413–417. doi: 10.1038/sj.cdd.4401227. [DOI] [PubMed] [Google Scholar]
- 38.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15(4):1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000;7(1):102–111. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- 40.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Misawa T, Dodo K, Ishikawa M, et al. Structure-activity relationships of benzhydrol derivatives based on 1′-acetoxychavicol acetate (ACA) and their inhibitory activities on multiple myeloma cell growth via inactivation of the NF-kappaB pathway. Bioorg Med Chem. 2015;23(9):2241–2246. doi: 10.1016/j.bmc.2015.02.039. [DOI] [PubMed] [Google Scholar]
- 42.Ito K, Nakazato T, Xian MJ, et al. 1′-Acetoxychavicol acetate is a novel nuclear factor kappaB inhibitor with significant activity against multiple myeloma in vitro and in vivo. Cancer Res. 2005;65(10):4417–4424. doi: 10.1158/0008-5472.CAN-05-0072. [DOI] [PubMed] [Google Scholar]
- 43.In LL, Arshad NM, Ibrahim H, Azmi MN, Awang K, Nagoor NH. 1′-Acetoxychavicol acetate inhibits growth of human oral carcinoma xenograft in mice and potentiates cisplatin effect via proinflammatory microenvironment alterations. BMC Complement Altern Med. 2012;12:179. doi: 10.1186/1472-6882-12-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20(50):7342–7351. doi: 10.1038/sj.onc.1204926. [DOI] [PubMed] [Google Scholar]
- 45.Tamatani M, Che YH, Matsuzaki H, et al. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFκB activation in primary hippocampal neurons. J Biol Chem. 1999;274(13):8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- 46.Ichikawa H, Takada Y, Murakami A, Aggarwal BB. Identification of a novel blocker of IκBα kinase that enhances cellular apoptosis and inhibits cellular invasion through suppression of NF-κB-regulated gene products. J Immunol. 2005;174(11):7383–7392. doi: 10.4049/jimmunol.174.11.7383. [DOI] [PubMed] [Google Scholar]
- 47.Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 48.van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728(1–2):23–34. doi: 10.1016/j.mrrev.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Golubovskaya VM, Kweh FA, Cance WG. Focal adhesion kinase and cancer. Histol Histopathol. 2009;24(4):503–510. doi: 10.14670/HH-24.503. [DOI] [PubMed] [Google Scholar]
- 50.Choi Y-A, Kim D-K, Kang S-S, Sonn J-K, Jin E-J. Integrin signaling and cell spreading alterations by rottlerin treatment of chick limb bud mesenchymal cells. Biochimie. 2009;91(5):624–631. doi: 10.1016/j.biochi.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 51.Zhao J, Guan JL. Signal transduction by focal adhesion kinase in cancer. Cancer Metastasis Rev. 2009;28(1–2):35–49. doi: 10.1007/s10555-008-9165-4. [DOI] [PubMed] [Google Scholar]
- 52.Arshad NM, In LLA, Soh TL, et al. Recombinant human alpha fetoprotein synergistically potentiates the anti-cancer effects of 1′-S-1′-acetoxychavicol acetate when used as a complex against human tumours harbouring AFP-receptors. Oncotarget. 2015;6(18):16151–16167. doi: 10.18632/oncotarget.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative pictures of wound-healing assay of MDA-MB-231 cells after various treatments of ACA and its analogs at IC20.
Notes: The open wound area at 24 h was quantified using TScratch software relative to wound area at 0 h. All data are presented as mean ± SEM from three independent replicates, with representative images at 40× magnification shown from each treatment group.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; DMSO, dimethyl sulfoxide; IC20, 20% inhibitory concentration; SEM, standard error of the mean.
Table S1.
IC20 values for ACA and its analogs on MDA-MB-231 cell line
| IC20 (μM)
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ACA | 2 | 5 | 6 | 7 | 8 | AEA | AMCA | 19 | 20 |
| 2.0 | 48.0 | 32.0 | 50.0 | 31.0 | 50.0 | 4.0 | 12.0 | 28.0 | 45.0 |
Note: IC20 for each analog was used to treat MDA-MB-231 cells for wound-healing assay.
Abbreviations: ACA, 1′S-1′-acetoxychavicol acetate; AEA, 1′-acetoxyeugenol acetate; AMCA, 1′-acetoxy-3,5-dimethoxychavicol acetate; IC20, 20% inhibitory concentration.