

A commercially available borate ester catalyzes amide formation from carboxylic acids and amines with very high efficiency.
Abstract
Chemical reactions for the formation of amide bonds are among the most commonly used transformations in organic chemistry, yet they are often highly inefficient. A novel protocol for amidation using a simple borate ester catalyst is reported. The process presents significant improvements over other catalytic amidation methods in terms of efficiency and safety, with an unprecedented substrate scope including functionalized heterocycles and even unprotected amino acids. The method was used to access a wide range of functionalized amide derivatives, including pharmaceutically relevant targets, important synthetic intermediates, a catalyst, and a natural product.
INTRODUCTION
Amide linkages are at the basis of all life processes, as the key connections in proteins. Although their importance is well recognized in chemistry as a common motif in pharmaceuticals and polymeric materials (1), their synthesis is often overlooked as a contemporary challenge. Improved methods for the synthesis of amide functionality are key to the sustainable future of chemical synthesis and manufacturing, especially if they can offer high efficiency and reduced environmental impact. This is a consequence of the fact that amide formation is typically achieved using inefficient and often hazardous reagents, which generate large quantities of waste products leading to high disposal costs (2). Accordingly, there have been recent calls from numerous major pharmaceutical companies for research into methods for “amide bond formation avoiding poor atom economy reagents” (3).
Despite recent reports of new strategies for amide bond formation from alcohols, aldehydes, or alkynes (4–6), direct condensation of a carboxylic acid and amine remains the most common approach to amide bond formation, owing to the ubiquity and stability of these functional groups. Conventional methods for direct amidation (Fig. 1A) formally proceed via a two-step sequence involving activated carboxylic acid derivatives, which then undergo aminolysis (7, 8). Indirect amide formations of this sort are expensive and waste-intensive and often suffer from functional group incompatibilities. The ideal approach would involve direct condensation of a carboxylic acid and amine in the presence of a catalyst because the only by-product of the reaction would be water. Direct thermal reaction without a catalyst has relatively limited scope and usually requires high temperatures (9, 10). In recent years, the use of group IV metal salts or boron compounds as catalysts has enabled amidation reactions to take place at lower temperatures (10–24). However, the use of these catalytic reactions in an industrial context is rare because of their limited reactivity with functionalized substrates and the inefficient procedures used [high dilution conditions and large quantities of waste-intensive molecular sieves (0.8 to 2.5 kg/mol)] (Fig. 1B) (10–24). Only boric acid has been applied as an amidation catalyst to any great extent on an industrial scale, but it is effective only for relatively reactive acid/amine combinations (25).
Fig. 1. Approaches to amide bond formation.
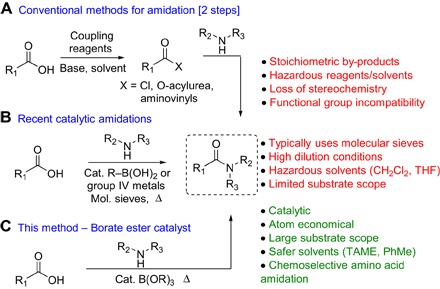
(A) Conventional methods for amidation proceeding via an activated carboxylic acid. (B) Recent catalytic amidations using group IV metal or boronic acid catalysts. THF, tetrahydrofuran. (C) Borate ester–catalyzed amide bond formation. TAME, tert-amyl methyl ether; PhMe, toluene.
To address this key issue of sustainability, we sought to develop an operationally simple catalytic method for direct amidation of the carboxylic acid/amine pair at high concentrations in “industrially preferred” solvents and without the need for any additives or dehydrating agents (26, 27). Our interest in the application of borate esters in stoichiometric amidation reactions led us to explore the use of these compounds as amidation catalysts, with a view to their suitability for application of preparing multigram quantities of amide. Here, we disclose a novel efficient protocol for amidation using a borate ester catalyst (Fig. 1C), with an unprecedented substrate scope. This method offers significant improvements in terms of safety and ease of setup and leads to a large reduction in the overall waste generated in an amidation reaction [process mass intensity (PMI)].
RESULTS AND DISCUSSION
Development of a highly versatile method for amidation
Borate esters have been demonstrated to mediate amidation reactions of functionalized carboxylic acids and amines (28, 29), yet a priori it was uncertain whether effective catalytic turnover could be achieved because the partially hydrolyzed borate ester 1 could readily undergo decomposition to leave a poorly reactive oligomeric boron oxide (Fig. 2A). Pleasingly, it was observed that effective turnover could be achieved using a Dean-Stark apparatus as an economical and efficient method for water removal, hence avoiding the need for wasteful molecular sieves. Following a solvent screen, tert-amyl methyl ether [TAME; boiling point (bp), 86°C] and PhMe (bp, 110°C) were identified as the most effective solvents. The former was preferable in most cases because it led to improved reactivity with functionalized substrates, and the lower bp reduces the energy requirements of the process. There was no background reaction under these conditions in the absence of a catalyst, and all boron-based catalysts examined gave measurably improved yields of amide 2 (Fig. 2B). Evaluation of a series of borate esters confirmed that a commercially available borate ester B(OCH2CF3)3 was the most effective catalyst for this transformation. As can be seen from the time-course plot (Fig. 2C), borate esters B(OMe)3 and B(OCH2CF3)3 significantly outperform boric acid, demonstrating that the alkoxy group on the boron atom substantially enhances the catalytic activity.
Fig. 2. Towards a borate-catalyzed amide coupling.
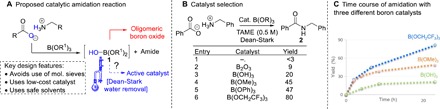
(A) Proposed catalytic cycle for amidation. (B) Catalyst selection. (C) Time course of borate amidation with different boron catalysts.
The efficiency of the amidation process was further enhanced through the use of an operationally simple method for purification of the amide products using scavenger resins (Fig. 3), which remove unreacted acid and amine, as well as boron-containing impurities. This significantly reduces the solvent requirements of the process by removing the need for aqueous/organic separations and/or chromatographic purification in most cases.
Fig. 3. Scope of borate-catalyzed amide bond formation.
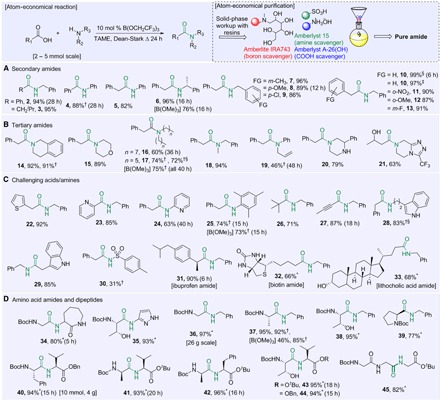
(A) Secondary amides. (B) Tertiary amides. (C) Challenging amides. (D) Amino acid amides. mol %, mole percent; Boc, tert-butoxycarbonyl. Reactions run according to general procedure A for 24 hours unless stated otherwise. *Amide synthesized using 20 mole percent (mol %) B(OCH2CF3)3. †Reaction performed in PhMe instead of TAME. ‡Amide synthesized using 1 mol % B(OCH2CF3)3. §Amide synthesized using 5 mol % B(OCH2CF3)3.
Scope of the reaction
To evaluate the reaction scope, we explored the preparation of a selection of amides (Fig. 3). A variety of primary amines were successfully coupled with simple carboxylic acids to give secondary amides 2 to 13 in excellent yields. In contrast to existing catalytic amidation reactions, a range of tertiary amides could also be prepared, including examples derived from both cyclic (14, 15, 20, and 21) and acylic (16 to 19) secondary amines, the latter being particularly difficult compounds to prepare via catalytic amidation. Furthermore, challenging amides were prepared from functionalized/heterocyclic carboxylic acids or amines (22 to 33), demonstrating the unprecedented scope of this catalytic amidation reaction. It was even possible to acylate a poorly nucleophilic sulfonamide to give the corresponding derivative 30, albeit in moderate yield. Naturally occurring carboxylic acids such as biotin (32) and lithocholic acid (33) were also amenable to amide bond formation. Nonsteroidal anti-inflammatory ibuprofen also smoothly reacted to form the corresponding amide 31. Our studies also suggested that B(OMe)3 was a reasonably effective and very low-cost catalyst for synthesizing relatively unfunctionalized amides when PhMe is used as solvent (6, 17, 25, and 37).
We next directed our efforts to amidation reactions of amino acids due to their importance as low-cost renewable raw materials that can be applied to the synthesis of many biologically active targets. As such, the compatibility of our methodology with this type of building block was an important aspect to examine because many of the existing catalytic amidation methods are unsuccessful with these compounds (11–24). Coupling of N-Boc–protected amino acids with a range of amines, including both simple aliphatic amines (36 to 39) and functionalized examples (34 and 35), gave the corresponding amides in excellent yields. The synthesis of dipeptides (40 to 44) and even a tripeptide (45) was also demonstrated successfully. Pleasingly, the free hydroxyl functionality of threonine did not require protection, and the corresponding amino amides (35 and 38) and dipeptides (43 and 44) were obtained in excellent yields. No detectable racemization was observed for any of the amino acid derivatives.
Amidation with unprotected amino acids
In an effort to address the unmet need for novel chemoselective strategies in chemical synthesis and thereby further prevent the generation of unnecessary waste by-products, we sought to test the application of our catalytic amidation reaction with unprotected amino acids. Protecting group manipulations and amide bond formation account for about a third of the reactions carried out in the synthesis of pharmaceutical intermediates (30, 31), so overcoming these synthetic hurdles could pave the way toward greater sustainability in the chemical industry. Perhaps the most impressive feat of this methodology, catalytic amidation of unprotected amino acids, could be achieved in a chemoselective manner, thereby circumventing the need for protection/deprotection of the amino group (Fig. 4) (32). This represents a novel, practical, and more atom-economical route to primary amino acid derivatives, which are a well-documented class of potent anticonvulsants and agents for neuropathic pain treatment (33, 34). Owing to the unreactive/insoluble nature of free amino acids, a higher catalyst loading and a slight excess of amine were necessary for this type of reaction to go to completion. This unique catalytic and chemoselective amide bond formation displayed a broad substrate scope with a variety of functionalized free amino acids and amines. Simple unfunctionalized amino amides (47, 48, 50, 51, and 53 to 59) were obtained in high yields. Diverse functional groups, including hydroxyl groups (52), heterocycles (46), and sulfides (49), were well tolerated. A β-amino acid could also be converted to the corresponding amide, albeit in lower yield (55). Pleasingly, glutamic acid underwent a tandem cyclization/amidation to give the corresponding pyroglutamide (60) in good yield. This method provides a highly efficient route for the catalytic synthesis of amino amides directly in one step from readily available free amino acids.
Fig. 4. Chemoselective amide bond formation from amino acids.
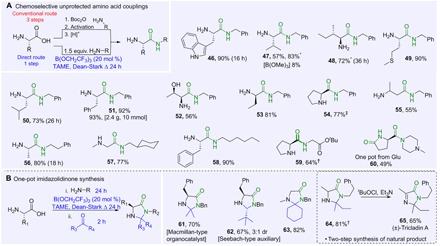
(A) Chemoselective amino acid couplings. (B) Sequential amidation/condensation to form imidazolidinones. dr, diastereomeric ratio. Reactions run according to general procedures B and C for 24 hours unless stated otherwise. *Using 30 mol % B(OCH2CF3)3. †Using 2 equiv. of amine. ‡Using 1.2 equiv. of benzylamine.
One-pot sequential condensations
Given that B(OCH2CF3)3 has previously been shown to promote imine formation when used stoichiometrically (35), we also explored a one-pot unprotected amino acid amidation/condensation reaction to provide access to imidazolidinones in a single step. Using this approach, we were able to prepare a Macmillan-type organocatalyst (61) and a Seebach-type auxiliary (62), as well as cyclohexanone derivative 63, in one-pot procedures starting from the unprotected amino acid. Furthermore, we were also able to synthesize the natural product (±)-Tricladin A (65) from alanine in a two-step, rather than a five-step, sequence (36): The one-pot sequential direct amidation/condensation with 2-butanone provided 64 in 81% yield, which could then be converted into the racemic natural product via a literature oxidation method.
Synthetic applications of the reaction
We then went on to explore the application of our methodology to the synthesis of active pharmaceutical ingredients (APIs) (Fig. 5). The amide bond formation steps within the syntheses of several top-selling pharmaceuticals, including Valsartan (Diovan) (66), Bunazosin (Andante) (67), GVS-111 (Noopept) (68), Atorvastatin (Lipitor) (69), Granisetron (Kytril) (70), and Sitagliptin (Januvia) (71 and 72), and all, proceeded well using our new amidation procedure. Similarly, Fasoracetam (73) was synthesized in one step from glutamic acid, a cheap and readily available chemical feedstock.
Fig. 5. Application to the synthesis of APIs.
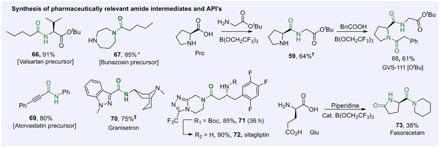
Reactions run according to general procedures A or B for 24 hours unless stated otherwise. *Using 1.5 equiv. of homopiperazine. †Using 2 equiv. of H-Gly-OttBu. ‡Using 1.0 equiv. of B(OCH2CF3)3.
Because our goal was to develop a highly efficient and scalable amidation protocol, we sought to demonstrate the efficiency of our method by benchmarking it against other known catalytic amide bond formation processes. The PMI [PMI = (raw material input)/(bulk product output)] provides a widely used measure for the efficiency of a chemical process and was used to compare a selection of recently reported catalytic amidation reactions with the present method (Fig. 6) (37). The synthesis of phenylacetamide 10 is ubiquitous in the amidation literature, so this compound was selected for comparison (12–24, 38). Pleasingly, our procedure showed clear improvements over existing catalytic amidation methods with regard to the PMI for both (i) the reaction conditions (more concentrated; no additives or molecular sieves) and (ii) the workup (no liquid-phase extraction). The PMI values were also calculated for two further large-scale borate-catalyzed amide syntheses. Although the solid-phase workup procedure is still highly efficient on a multigram scale (36), direct crystallization of the amide product from the reaction mixture leads to a further improvement in the PMI (74).
Fig. 6. Environmental metrics for catalytic amidation reactions.
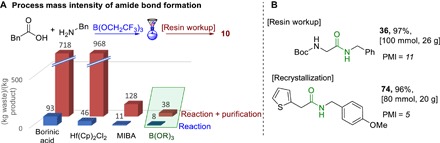
(A) PMI calculations for a selection of catalytic amide bond formation processes. MIBA, 5-methoxy-2-iodophenylboronic acid. (B) Improved PMIs of large-scale borate-catalyzed amidation procedures.
Mechanism of the reaction
Because the alkoxy group present on the borate ester exerts a significant effect on the catalytic activity (Fig. 2B), at least one of these groups must remain attached to the boron atom during the catalytic cycle. Analysis of the contents of the Dean-Stark trap by 19F nuclear magnetic resonance (NMR) showed that less than 1 equivalent of trifluoroethanol was removed from the reaction mixture over the course of the amidation reaction, suggesting that the active catalyst has a general structure XB(OCH2CF3)2.
Using a graphical method involving variable time normalization developed by Burés (39, 40), we were able to elucidate the reaction orders from concentration profiles of the reaction. As expected, analysis of the reaction kinetics for the formation of amide 6 suggests a positive dependence on the concentration of borate ester catalyst (0.8th order). This is consistent with a reaction that is first-order in catalyst, but with some competitive decomposition of the active species (41). The reaction rate was independent of amine concentration (0th order) but displayed a positive correlation with acid concentration (0.5th order). The noninteger reaction order with respect to the carboxylic acid could be explained by off-cycle equilibria, for example, reversible formation of the amine carboxylate salt 75 (equil. 1) (Fig. 7A) (41).
Fig. 7. Mechanism of the amidation reaction.
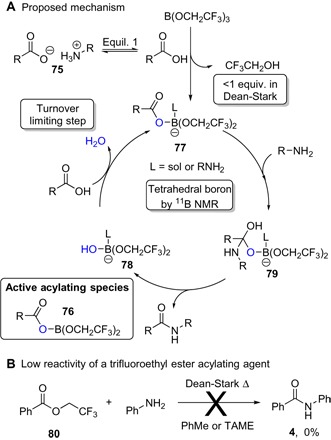
(A) Proposed mechanism. (B) Low reactivity of a trifluoroethyl ester acylating agent. Sol, solvent.
As a consequence of these observations, we propose that the active acylating agent is likely to be an acyloxyboron compound (76) bearing two trifluoroethoxy groups, related to the intermediates previously suggested for stoichiometric amidation reactions mediated by alkoxyboron compounds (42, 43). The 11B NMR of the reaction mixture only showed that tetrahedral boron species are present, so 76 is likely present as a Lewis base adduct 77 with the amine or solvent (L = amine, trifluoroethanol, or TAME). The former is more likely because the amine is a stronger Lewis base. Note that the first-order dependence on catalyst does not preclude an active species containing two or more boron atoms with bridging carboxylate ligands (for example, [77]2) (41, 44). On the basis of the kinetic data, we propose a catalytic cycle as shown (Fig. 7A), in which condensation of the carboxylic acid with the monohydroxyboron species 78, with concomitant water removal, is the turnover limiting step.
Finally, an alternative pathway in which a trifluoroethyl ester, such as 80, acts as the acylating species (45, 46) was excluded on the basis that ester 80 is not a competent acylating agent for poorly nucleophilic amines such as aniline (Fig. 7B). Further work is under way to fully elucidate the mechanism of this amidation reaction to facilitate the design of more active catalysts.
Conclusions
Our new borate-catalyzed amide coupling reaction has many advantages over existing methods for amidation: It uses a simple, commercially available catalyst, and the protocol proceeds with high efficiency (low PMI value), with a remarkably broad substrate scope, including application to the synthesis of many pharmaceutically relevant compounds. The procedure can easily be performed on a multigram scale, and the products can be isolated either via a filtration workup or by direct crystallization from the reaction mixture. We anticipate that this method will find many applications in the synthesis of amides in a wide range of fields.
MATERIALS AND METHODS
General amidation procedure A
A stirred suspension of an amine (5.0 to 5.5 mmol) and carboxylic acid (5.0 mmol) in TAME (5 ml) was heated to reflux (bp, 86°C) in Dean-Stark apparatus (side arm filled with TAME), and B(OCH2CF3)3 (0.5 mmol; 5 ml of a 0.1 M solution in TAME) was added into the reaction mixture through the Dean-Stark apparatus. An air condenser was fitted, and the reaction mixture was stirred for 2 to 36 hours. Upon completion, the reaction mixture was cooled down to room temperature and concentrated in vacuo. The crude mixture was dissolved in dimethyl carbonate (10 ml) and H2O (0.5 ml); Amberlite IRA743 (0.25 g), Amberlyst A15 (0.5 g), and Amberlyst A-26(OH) (0.5 g) resins were added; the resulting suspension was stirred for 30 min. After the disappearance of any remaining starting materials [monitored by thin-layer chromatography (TLC)], MgSO4 (~0.5 g) was added. The reaction was filtered, the reaction flask was washed with dimethyl carbonate (2 × 10 ml), and the combined filtrates were concentrated in vacuo to give pure amide.
General amidation procedure B for unprotected amino acids
A stirred suspension of an amine (7.5 mmol) and unprotected amino acid (5 mmol) in TAME (2.5 ml) was heated to reflux (bp, 86°C) in a Dean-Stark apparatus (side arm filled with TAME), and B(OCH2CF3)3 (1 mmol; 2.5 ml of a 0.4 M solution in TAME) was added through the Dean-Stark apparatus. An air condenser was fitted, and the reaction mixture was stirred for 24 hours. Upon completion, the reaction mixture was concentrated in vacuo and dry-loaded onto silica gel for column chromatography.
General procedure C for synthesis of imidazolidinones
Following general procedure B, after heating to reflux for 24 hours, a solution of aldehyde or ketone (10 mmol) in TAME (5 ml) was added dropwise over 10 min into the reaction mixture (fig. S2). The reaction was left to stir for 1 hour. If the reaction was not complete, as seen by the disappearance of the intermediate amino amide by TLC (revealed with ninhydrin stain) or high-performance liquid chromatography (HPLC), a further portion of aldehyde or ketone (5 mmol) in TAME (2 ml) was added dropwise over 5 min into the reaction mixture, which was left to stir for another hour. Once complete, the reaction was cooled to room temperature and concentrated in vacuo. The product was purified by flash column chromatography.
Supplementary Material
Acknowledgments
We acknowledge A. Aliev [University College London (UCL)] for the help with 11B NMR and the Engineering and Physical Sciences Research Council (EPSRC) UK National Mass Spectrometry Facility at Swansea University. Funding: We would like to thank GlaxoSmithKline and UCL Chemistry/EPSRC for providing funding to support a PhD studentship. Author contributions: M.T.S. performed all the experimental work. M.T.S., L.T.B., and T.D.S. conceived the project, designed the experiments, analyzed the data, and contributed to the preparation of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Experimental procedures, reaction troubleshooting, extensive optimization data, NMR spectra, and mass spectrometry data are available in the Supplementary Materials. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1701028/DC1
General methods
Optimization of reaction parameters
General procedures
Resin capacity in varying solvents
PMI calculations
Mechanistic studies
Spectroscopic data
1H and 13C NMR spectra
Chiral HPLC traces for enantiopurity measurements
1H and 13C NMR spectra for enantiopurity measurements
table S1. Solvent screen for general amidation.
table S2. Screening of borate reagents in amino acid amidation.
table S3. Varying time, catalyst loading, and equivalents of amine.
table S4. Varying concentration.
table S5. Reaction troubleshooting.
table S6. Solvent screen for resin workup.
table S7. Raw data for PMI calculations for amidation product 10.
table S8. PMI calculations for amidation product 10.
table S9. Raw data for PMI calculations.
table S10. PMI calculations.
table S11. Raw data for determination of order in catalyst.
table S12. Raw data for determination of order in catalyst.
table S13. Raw data for determination of order in catalyst.
table S14. Raw data for determination of order in amine.
table S15. Raw data for determination of order in amine.
table S16. Raw data for determination of order in amine.
table S17. Raw data for determination of order in acid.
table S18. Raw data for determination of order in acid.
table S19. Raw data for determination of order in acid.
table S20. Raw data for determination of order in acid.
table S21. Raw data for determination of order in acid.
fig. S1. Representative example of a Dean-Stark reaction setup.
fig. S2. Representative examples of a Dean-Stark setup with adaptor for the addition of ketone/aldehyde.
fig. S3. Representative example of a resin workup.
fig. S4. Green metrics for catalytic amidation protocols.
fig. S5. 19F NMR spectra of the crude reaction mixture (top) and the Dean-Stark (bottom) with fluorobenzene as an internal standard.
fig. S6. 11B NMR spectra of B(OCH2CF3)3 (top) and reaction mixture (bottom two) at 4- and 24-hour intervals.
REFERENCES AND NOTES
- 1.Pattabiraman V. R., Bode J. W., Rethinking amide bond synthesis. Nature 480, 471–479 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Dunetz J. R., Magano J., Weisenburger G. A., Large-scale applications of amide coupling reagents for the synthesis of pharmaceuticals. Org. Process Res. Dev. 20, 140–177 (2016). [Google Scholar]
- 3.Constable D. J. C., Dunn P. J., Hayler J. D., Humphrey G. R., Leazer J. L. Jr, Linderman R. J., Lorenz K., Manley J., Pearlman B. A., Wells A., Zaks A.. Zhang T. Y., Key green chemistry research areas—A perspective from pharmaceutical manufacturers. Green Chem. 9, 411–420 (2007). [Google Scholar]
- 4.Chan W.-K., Ho C.-M., Wong M.-K., Che C.-M., Oxidative amide synthesis and N-terminal α-amino group ligation of peptides in aqueous medium. J. Am. Chem. Soc. 128, 14796–14797 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Gunanathan C., Ben-David Y., Milstein D., Direct synthesis of amides from alcohols and amines with liberation of H2. Science 317, 790–792 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Vora H. U., Rovis T., Nucleophilic carbene and HOAt relay catalysis in an amide bond coupling: An orthogonal peptide bond forming reaction. J. Am. Chem. Soc. 129, 13796–13797 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valeur E., Bradley M., Amide bond formation: Beyond the myth of coupling reagents. Chem. Soc. Rev. 38, 606–631 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Krause T., Baader S., Erb B., Gooßen L. J., Atom-economic catalytic amide synthesis from amines and carboxylic acids activated in situ with acetylenes. Nat. Commun. 7, 11732 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gooßen L. J., Ohlmann D. M., Lange P. P., The thermal amidation of carboxylic acids revisited. Synthesis 2009, 160–164 (2009). [Google Scholar]
- 10.Allen C. L., Chhatwal A. R., Williams J. M. J., Direct amide formation from unactivated carboxylic acids and amines. Chem. Commun. 48, 666–668 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Lanigan R. M., Sheppard T. D., Recent developments in amide synthesis: Direct amidation of carboxylic acids and transamidation reactions. Eur. J. Org. Chem. 2013, 7453–7465 (2013). [Google Scholar]
- 12.Ishihara K., Ohara S., Yamamoto H., 3,4,5-Trifluorobenzeneboronic acid as an extremely active amidation catalyst. J. Org. Chem. 61, 4196–4197 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Maki T., Ishihara, K.. Yamamoto H., 4,5,6,7-Tetrachlorobenzo[d][1,3,2]dioxaborol-2-ol as an effective catalyst for the amide condensation of sterically demanding carboxylic acids. Org. Lett. 8, 1431–1434 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Lundberg H., Tinnis F., Adolfsson H., Direct amide coupling of non-activated carboxylic acids and amines catalysed by zirconium(IV) chloride. Chem. Eur. J. 18, 3822–3826 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Noda H., Furutachi M., Asada Y., Shibasaki M., Kumagai N., Unique physicochemical and catalytic properties dictated by the B3NO2 ring system. Nat. Chem. 9, 571–577 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Lundberg H., Adolfsson H., Hafnium-catalyzed direct amide formation at room temperature. ACS Catal. 5, 3271–3277 (2015). [Google Scholar]
- 17.Fatemi S., Gernigon N., Hall D. G., A multigram-scale lower E-factor procedure for MIBA-catalyzed direct amidation and its application to the coupling of alpha and beta aminoacids. Green Chem. 17, 4016–4028 (2015). [Google Scholar]
- 18.Mohy El Dine T., Erb W., Berhault Y., Rouden J., Blanchet J., Catalytic chemical amide synthesis at room temperature: One more step toward peptide synthesis. J. Org. Chem. 80, 4532–4544 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Arnold K., Davies B., Hérault D., Whiting A., Asymmetric direct amide synthesis by kinetic amine resolution: A chiral bifunctional aminoboronic acid catalyzed reaction between a racemic amine and an achiral carboxylic acid. Angew. Chem. Int. Ed. 47, 2673–2676 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Al-Zoubi R. M., Marion O., Hall D. G., Direct and waste-free amidations and cycloadditions by organocatalytic activation of carboxylic acids at room temperature. Angew. Chem. Int. Ed. 47, 2876–2879 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Arnold K., Davies B., Giles R. L., Grosjean C., Smith G. E., Whiting A., To catalyze or not to catalyze? Insight into direct amide bond formation from amines and carboxylic acids under thermal and catalyzed conditions. Adv. Synth. Catal. 348, 813–820 (2006). [Google Scholar]
- 22.Arnold K., Batsanov A. S., Davies B., Whiting A., Synthesis, evaluation and application of novel bifunctional N,N-di-isopropylbenzylamineboronic acid catalysts for direct amide formation between carboxylic acids and amines. Green Chem. 10, 124–134 (2008). [Google Scholar]
- 23.Gernigon N., Al-Zoubi R. M., Hall D. G., Direct amidation of carboxylic acids catalyzed by ortho-iodo arylboronic acids: Catalyst optimization, scope, and preliminary mechanistic study supporting a peculiar halogen acceleration effect. J. Org. Chem. 77, 8386–8400 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Ishihara K., Lu Y., Boronic acid–DMAPO cooperative catalysis for dehydrative condensation between carboxylic acids and amines. Chem. Sci. 7, 1276–1280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mylavarapu R. K., Kondaiah G. C. M., Kolla N., Veeramalla R., Koilkonda P., Bhattacharya A., Bandichhor R., Boric acid catalyzed amidation in the synthesis of active pharmaceutical ingredients. Org. Process Res. Dev. 11, 1065–1068 (2007). [Google Scholar]
- 26.Prat D., Wells A., Hayler J., Sneddon H., McElroy C. R., Abou-Shehada S., Dunn P. J., CHEM21 selection guide of classical- and less classical-solvents. Green Chem. 18, 288–296 (2016). [Google Scholar]
- 27.P. J. Dunn, K. K. Hii, M. J. Krische, M. T. Williams, in Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries (Wiley, 2013), pp. 89–120. [Google Scholar]
- 28.Starkov P., Sheppard T. D., Borate esters as convenient reagents for direct amidation of carboxylic acids and transamidation of primary amides. Org. Biomol. Chem. 9, 1320–1323 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Lanigan R. M., Starkov P., Sheppard T. D., Direct synthesis of amides from carboxylic acids and amines using B(OCH2CF3)3. J. Org. Chem. 78, 4512–4523 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cooper T. W. J., Campbell I. B., Macdonald S. J. F., Factors determining the selection of organic reactions by medicinal chemists and the use of these reactions in arrays (small focused libraries). Angew. Chem. Int. Ed. 49, 8082–8091 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Carey J. S., Laffan D., Thomson C., Williams M. T., Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 4, 2337–2347 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Lanigan R. M., Karaluka V., Sabatini M. T., Starkov P., Badland M., Boulton L., Sheppard T. D., Direct amidation of unprotected amino acids using B(OCH2CF3)3. Chem. Commun. 52, 8846–8849 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Simoens S., Lacosamide as adjunctive therapy for partial-onset epileptic seizures: A review of the clinical and economic literature. Curr. Med. Res. Opin. 27, 1329–1338 (2011). [DOI] [PubMed] [Google Scholar]
- 34.King A. M., Salomé C., Dinsmore J., Salomé-Grosjean E., De Ryck M., Kaminski R., Valade A., Kohn H., Primary amino acid derivatives: Compounds with anticonvulsant and neuropathic pain protection activities. J. Med. Chem. 54, 4815–4830 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Reeves J. T., Visco M. D., Marsini M. A., Grinberg N., Busacca C. A., Mattson A. E., Senanayake C. H., A general method for imine formation using B(OCH2CF3)3. Org. Lett. 17, 2442–2445 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Zhao H., Huang Z., Chen W., Total synthesis of tricladins A and B and identification of their absolute configuration. J. Org. Chem. 79, 11290–11294 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Fennie M. W., Roth J. M., Comparing amide-forming reactions using green chemistry metrics in an undergraduate organic laboratory. J. Chem. Educ. 93, 1788–1793 (2016). [Google Scholar]
- 38.Mohy El Dine T., Rouden J., Blanchet J., Borinic acid catalysed peptide synthesis. Chem. Commun. 51, 16084–16087 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Burés J., Variable time normalization analysis: General graphical elucidation of reaction orders from concentration profiles. Angew. Chem. Int. Ed. 55, 16084–16087 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Burés J., A simple graphical method to determine the order in catalyst. Angew. Chem. Int. Ed. 55, 2028–2031 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lundberg H., Tinnis F., Zhang J., Algarra A. G., Himo F., Adolfsson H., Mechanistic elucidation of zirconium-catalyzed direct amidation. J. Am. Chem. Soc. 139, 2286–2295 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Pelter A., Levitt T. E., Nelsoni P., Some amide forming reactions involving boron reagents. Tetrahedron 26, 1539–1544 (1970). [Google Scholar]
- 43.Collum D. B., Chen S.-C., Ganem B., A new synthesis of amides and macrocyclic lactams. J. Org. Chem. 43, 4393–4394 (1978). [Google Scholar]
- 44.Burés J., What is the order of a reaction? Top. Catal. 60, 631–633 (2017). [Google Scholar]
- 45.Caldwell N., Jamieson C., Simpson I., Watson A. J. B., Catalytic amidation of unactivated ester derivatives mediated by trifluoroethanol. Chem. Commun. 51, 9495–9498 (2015). [DOI] [PubMed] [Google Scholar]
- 46.McPherson C. G., Caldwell N., Jamieson C., Simpson I., Watson A. J. B., Amidation of unactivated ester derivatives mediated by trifluoroethanol. Org. Biomol. Chem. 15, 3507–3518 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Marfey P., Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 49, 591–596 (1984). [Google Scholar]
- 48.Jimenez-Gonzalez C., Ponder C. S., Broxterman Q. B., Manley J. B., Using the right green yardstick: Why process mass intensity is used in the pharmaceutical industry to drive more sustainable processes. Org. Process Res. Dev. 15, 912–917 (2011). [Google Scholar]
- 49.Sharma N., Sekar G., Stable and reusable binaphthyl-supported palladium catalyst for aminocarbonylation of aryl iodides. Adv. Synth. Catal. 358, 314–320 (2016). [Google Scholar]
- 50.Salehi P., Motlagh A. R., Silica gel supported ferric perchlorate: A new and efficient reagent for one pot synthesis of amides from benzylic alcohols. Synth. Commun. 30, 671–675 (2000). [Google Scholar]
- 51.Karaluka V., Lanigan R. M., Murray P. M., Badland M., Sheppard T. D., B(OCH2CF3)3-mediated direct amidation of pharmaceutically relevant building blocks in cyclopentyl methyl ether. Org. Biomol. Chem. 13, 10888–10894 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Sudta P., Kirk N., Bezos A., Gurlica A., Mitchell R., Weber T., Willis A. C., Prabpai S., Kongsaeree P., Parish C. R., Suksamran S., Kelso M. J., Synthesis, structural characterisation, and preliminary evaluation of non-indolin-2-one-based angiogenesis inhibitors related to sunitinib (Sutent®). Aust. J. Chem. 66, 864–873 (2013). [Google Scholar]
- 53.Agwada V. C., Potential central nervous system active agents. 2. Synthesis of N-benzylphenylacetamides. J. Chem. Eng. Data 27, 481–483 (1982). [Google Scholar]
- 54.Morimoto H., Fujiwara R., Shimizu Y., Morisaki K., Ohshima T., Lanthanum(III) triflate catalyzed direct amidation of esters. Org. Lett. 16, 2018–2021 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Aspin S. J., Taillemaud S., Cyr P., Charette A. B., 9-Silafluorenyl dichlorides as chemically ligating coupling agents and their application in peptide synthesis. Angew. Chem. Int. Ed. 55, 13833–13837 (2016). [DOI] [PubMed] [Google Scholar]
- 56.Mamillapalli N. C., Sekar G., Chemoselective reductive deoxygenation and reduction of α-keto amides by using a palladium catalyst. Adv. Synth. Catal. 357, 3273–3283 (2015). [Google Scholar]
- 57.Watson A. J. A., Maxwell A. C., Williams J. M. J., Ruthenium-catalyzed oxidation of alcohols into amides. Org. Lett. 11, 2667–2670 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Sarode P. B., Bahekar S. P., Chandak H. S., DABCO/AcOH jointly accelerated copper(I)-catalysed cycloaddition of azides and alkynes on water at room temperature. Synlett 27, 2681–2684 (2016). [Google Scholar]
- 59.Yasuda S., Kumagai N., Shibasaki M., Direct asymmetric α-allylation of ketones with allylic alcohols via Pd/enamine cooperative function. Heterocycles 86, 745–757 (2012). [Google Scholar]
- 60.Selkälä S. A., Koskinen A. M. P., Preparation of bicyclo[4.3.0]nonanes by an organocatalytic intramolecular Diels–Alder reaction. Eur. J. Org. Chem. 2005, 1620–1624 (2005). [Google Scholar]
- 61.Mintz M. J., Walling C., t-Butyl hypochlorite. Org. Synth. 49, 9 (1969). [Google Scholar]
- 62.Chou W.-C., Tan C.-W., Chen S.-F., Ku H., One-pot neat reactions of carboxylic esters and alkylenediamines for efficient preparation of N-acylalkylenediamines. J. Org. Chem. 63, 10015–10017 (1998). [Google Scholar]
- 63.Schäfera I., Opatz T., Pyrroles and indolizidines from deprotonated α-(alkylideneamino)nitriles. Synthesis 2011, 1691–1704 (2011). [Google Scholar]
- 64.Bermudez J., Fake C. S., Joiner G. F., Joiner K. A., King F. D., Miner W. D., Sanger G. J., 5-Hydroxytryptamine (5-HT3) receptor antagonists. 1. Indazole and indolizine-3-carboxylic acid derivatives. J. Med. Chem. 33, 1924–1929 (1990). [DOI] [PubMed] [Google Scholar]
- 65.Zhou S., Wang J., Chen X., Aceña J. L., Soloshonok V. A., Liu H., Chemical kinetic resolution of unprotected β-substituted β-amino acids using recyclable chiral ligands. Angew. Chem. Int. Ed. 53, 7883–7886 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Amedjkouh M., Ahlberg P., Synthesis of chiral diamines using novel 2-trichloromethyloxazolidin-4-one precursors derived from 5-oxo-proline and proline. Tetrahedron Asymmetry 13, 2229–2234 (2002). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1701028/DC1
General methods
Optimization of reaction parameters
General procedures
Resin capacity in varying solvents
PMI calculations
Mechanistic studies
Spectroscopic data
1H and 13C NMR spectra
Chiral HPLC traces for enantiopurity measurements
1H and 13C NMR spectra for enantiopurity measurements
table S1. Solvent screen for general amidation.
table S2. Screening of borate reagents in amino acid amidation.
table S3. Varying time, catalyst loading, and equivalents of amine.
table S4. Varying concentration.
table S5. Reaction troubleshooting.
table S6. Solvent screen for resin workup.
table S7. Raw data for PMI calculations for amidation product 10.
table S8. PMI calculations for amidation product 10.
table S9. Raw data for PMI calculations.
table S10. PMI calculations.
table S11. Raw data for determination of order in catalyst.
table S12. Raw data for determination of order in catalyst.
table S13. Raw data for determination of order in catalyst.
table S14. Raw data for determination of order in amine.
table S15. Raw data for determination of order in amine.
table S16. Raw data for determination of order in amine.
table S17. Raw data for determination of order in acid.
table S18. Raw data for determination of order in acid.
table S19. Raw data for determination of order in acid.
table S20. Raw data for determination of order in acid.
table S21. Raw data for determination of order in acid.
fig. S1. Representative example of a Dean-Stark reaction setup.
fig. S2. Representative examples of a Dean-Stark setup with adaptor for the addition of ketone/aldehyde.
fig. S3. Representative example of a resin workup.
fig. S4. Green metrics for catalytic amidation protocols.
fig. S5. 19F NMR spectra of the crude reaction mixture (top) and the Dean-Stark (bottom) with fluorobenzene as an internal standard.
fig. S6. 11B NMR spectra of B(OCH2CF3)3 (top) and reaction mixture (bottom two) at 4- and 24-hour intervals.