

Abstract
Objective
To identify a genetic cause of early-onset systemic lupus erythematosus (SLE) in a large consanguineous family from Turkey and to study the mechanisms of disease.
Methods
We performed whole exome sequencing (WES) and SNP array genotyping in affected and unaffected family members. Protein studies, gene expression, cytokine profiling, neutrophil extracellular trap formation, and presence of low-density granulocytes were evaluated in patient primary cells and serum samples.
Results
We identified a novel homozygous loss-of-function mutation (p.Pro445Leufs*11) in the C1R gene. Sanger sequencing of 14 family members confirmed the homozygous mutation in four affected patients, and in an asymptomatic 9-year old female. Sera from patients with truncated C1r protein had low complement levels. Two affected siblings available for detailed evaluation exibited strong type I interferon inflammatory signatures despite being clinically inactive at the time of sampling. The type-I IFN transcriptional signature in patients’ blood correlated with disease expressivity, whereas the neutrophil signature in patients’ PBMCs was likely associated with disease severity. The affected female with the most severe phenotype, showed a stronger neutrophil signature, defined by enhanced neutrophil extracellular trap formation and the presence of low-density granulocytes. Analysis of exome data for modifying alleles suggested enrichment of common SLE-associated variants in the more severely affected patients. Lupus-associated HLA alleles or HLA haplotypes were not shared among the 4 affected subjects.
Conclusion
We report a novel high-penetrance mutation in C1R as the cause of monogenic SLE. Disease expressivity in this family appears to be influenced by additional common and rare genetic variants.
Keywords: Systemic Lupus Erythematosus, C1r, IFN-I signature, neutrophil extracellular trap (NETs), low-density granulocytes (LDGs)
Introduction
Systemic lupus erythematosus (SLE) is a complex autoimmune syndrome characterized by significant organ involvement. Most patients are diagnosed during adulthood and previous studies have suggested roles for genetic, epigenetic, environmental, and hormonal factors contributing to the pathogenesis of SLE. Genome-wide association studies (GWAS) revealed more than 40 SLE susceptibility loci(1). The disease-associated variants are generally common and have small but cumulative effects on disease susceptibility. Patients with early-onset symptoms, particularly during childhood, are more likely to have highly penetrant novel or rare variants in single genes(2).
Reduced serum levels of complement have been linked to SLE pathogenesis. A systematic analysis of all reported cases showed that the strength of the association between complement deficiency and SLE depends on the position of the missing protein in the cascade with the earlier complement components being associated with a higher incidence of SLE (C1>C4>C2)(3). Individuals with complete C1q deficiency have the highest risk of developing SLE while the deficiency of C1r/C1s has been associated with lower incidence of SLE (93% vs. 57% of deficient patients will develop SLE)(3). However, very few mutations in the complement genes have been reported. Biallelic loss-of-function (LOF) mutations in C1S have been found in lupus patients, but have also been found in asymptomatic individuals(4). Thus far, a LOF mutation causing complete C1r deficiency has only been identified in a single male patient of African-American ancestry who developed symptoms at the age of 3(5).
Because of the family history for SLE and consanguinity, we hypothesized that affected patients in the family should carry a homozygous high-penetrance mutation in a single gene. We investigated the effects of the C1r deficiency in innate and adaptive immune responses by analyzing whole blood cell transcriptional profiles, serum inflammatory cytokines, the presence of LDGs (a pathogenic subset described in SLE), and the formation of NETs. Finally, we analyzed the contribution of rare and common SLE-associated susceptibility alleles and HLA types to disease expressivity.
Materials and Methods
Patients
All patients in this family were followed at the Istanbul University, Cerrahpasa School of Medicine, Turkey. Patients P3 and P4 were also evaluated at the NIH Clinical Center. All patients and their healthy family members enrolled in the study provided written informed consent.
Whole exome sequencing
Genomic DNA samples were isolated from peripheral blood. WES and data analysis was performed as previously described(6). ExAC, 1000 genomes, dbSNP, NHLBI GO Exome Sequencing Project, Clinseq database, and an in-house exome database were used to filter for rare candidate variants.
Sanger sequencing and single base extension genotyping
We performed Sanger sequencing for C1R exons (BigDye Terminator v1.1 Cycle Sequencing kit,Applied Biosystems) on a 3130xl Genetic Analyzer (Applied Biosystems). Sequencher (Gene Codes) was used to analyze the sequencing data. We genotyped Turkish healthy controls and Behcet’s disease cases for the presence of the p.Pro445Leufs*11 mutation using the Sequenom iPLEX gold method (Sequenom). Genotypes were determined with Typer 4.0 software (Sequenom).
Targeted sequencing
Targeted sequencing of C1R was performed on pooled DNA samples from 300 patients with late-onset SLE. Nine amplicons covering the nine exons of C1R were generated using high-fidelity polymerase AccuPrime Pfx SuperMix (Invitrogen). Pooled PCR products were used to make a library and sequenced on a HiSeq instrument (Illumina).
HLA allele and haplotype analysis
Genomic DNA samples were analyzed using HumanOmni5Exome Bead array and an iScan array reader (Illumina). SNP genotypes were determined using GenomeStudio software (Illumina). SNPs residing within the major histocompatibility complex locus (chr6: 29Mbp - 34Mbp; hg19) were extracted and classical HLA alleles and haplotypes were determined by imputation using SNP2HLA (http://software.broadinstitute.org/mpg/snp2hla/) software and the Type 1 Diabetes Genetics Consortium dataset a reference.
Complement level measurement
The levels of complement components C1r, C1s, C1q, C2, C3, C4, and a total complement activity enzyme immunoassay were measured at National Jewish Health Advanced Diagnostic Laboratories, Denver.
NanoString assay
We extracted RNA using the PAXgene Blood RNA Kit (QIAGEN) and conducted gene expression analysis on the nCounter Analysis System (NanoString Technologies) using a codeset designed to target 32 IFN signature genes and/or 594 immunologically related genes. 200 ng total RNA was mixed with the capture and reporter probes and hybridized on the nCounter Prep Station. nSolver software was used for data analysis. The expression data was normalized by using a geometric mean of housekeeping genes.
Immunoblotting
IFN-γ (20ng/ml) or TNF (20ng/ml) was used to stimulate PBMCs overnight. Whole cell lysate from a patient and healthy control were prepared using cell lysis buffer. Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Immunoreactive proteins were detected with a C1r antibody Ab190800 (Abcam) and were visualized using ECL Plus western blotting substrate (ThermoFisher).
Cytokine measurement
Serum samples were tested for cytokine levels via a Bio-Plex Pro Human Cytokine 27-plex Assay (BioRad).
Cell isolation
Normal-density granulocytes were isolated from the red blood cell layer by dextran sedimentation for NET activity assays.
Neutrophil Extracellular Trap quantification by immunofluorescence microscopy
Donor normal-density granulocytes were plated at a density of 100,000 cells per one poly-L-lysine-coated (Sigma-Aldrich) coverslip and incubated for 15 minutes, then treated with or without lipopolysaccharide (LPS, 100 ng/mL, Sigma-Aldrich) (Life Technologies) for 3 hours. The coverslips were then fixed and stained for MPO and DNA. Images were acquired on a Zeiss LSM510 META confocal laser-scanning microscope (Carl Zeiss Microimaging).
Flow cytometry
Donor PBMCs were blocked in FACS solution (Biolegend) for 30 minutes. Anti-human surface markers were stained for 20 minutes: Pacific Blue CD3 & CD19, APC CD10, FITC CD14, PE CD15 (Biolegend). LDGs were identified as CD10+, CD14lo, CD15+.
qRT-PCR
RNA was isolated using a Direct-zol RNA MiniPrep kit (Zymo Research). Total RNA (1 μg) was reverse transcribed using iScript Reverse Transcriptase (BioRad) and qRT-PCR was performed using SSoAdvanced Universal SYBR Green (BioRad). Primer sequences are included in Supplementary Table 1. Samples were run in duplicate using a CFX96 C1000 Touch Real Time Thermal Cycler (Bio-Rad). Data was analyzed using Bio-Rad CFX Manager software.
Results
Clinical manifestations
The four affected patients (Figure 1A) shared a history of systemic inflammation, presence of anti-nuclear antibodies (ANA) and malar rash suggestive of SLE, while each patient presented with additional features of autoimmunity (Supplementary Table 2).
Figure 1.
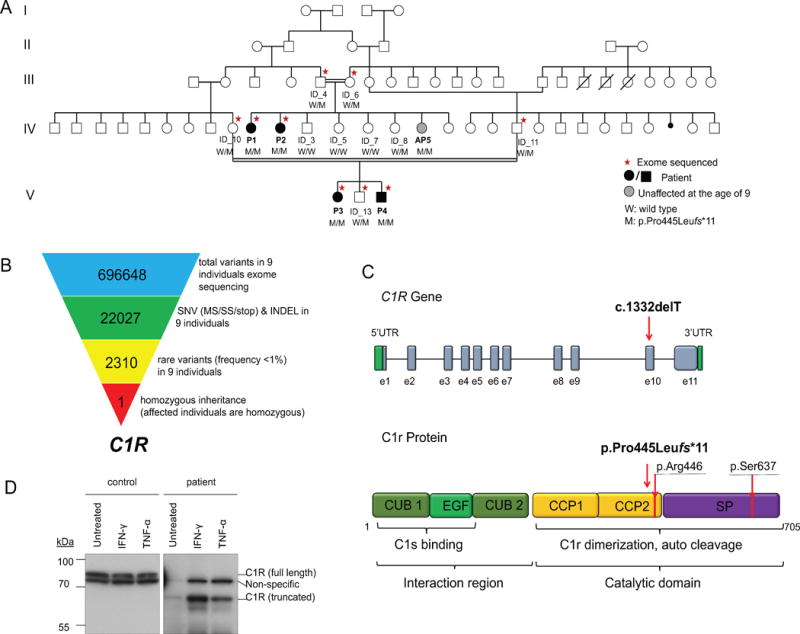
C1R mutation causes recessively inherited systemic lupus erythematosus.
(A) Pedigree of the large consanguineous family with recessively inherited homozygous mutation in the C1R gene. The individuals selected for WES are marked with asterisks. All the individual genotypes were confirmed by Sanger sequencing. Patients diagnosed with SLE are indicated by black filled symbols. The unaffected 9-year old patient with homozygous mutation is indicated by the grey filled circle.
(B) Schematic representation of the exome data-filtering approach used to identify homozygous recessively inherited exonic variants. Selection for rare exonic variants with frequency less than 1% resulted in 2310 variants. The assumption of homozygous inheritance in the affected patients led to identification of a single candidate exonic variant in C1R. (SNV: single nucleotide variants including missense, splice site variants, and stop codon variants; INDEL, frame shift and non-frame shift insertions and deletions)
(C) Schematic representations of the C1R gene and C1r protein domains show the position of novel c.1332delT, p.Pro445Leufs*11 mutation. Arginine residue 446 is the C1r cleavage site, Serine residue 637 is the active site of the protein.
(D)C1r protein expression in a patient’s PBMCs compared to a healthy control at basal level and after IFN-γ or TNF stimulation. Whole-cell lysates from the C1r mutation homozygous patient AP5 and one healthy donor were immunoblotted with antibodies for C1r.
The first pair of female affected siblings, patients P1 and P2, developed symptoms at age of 10 and 2 years, respectively, and had a milder course of disease than the second pair of affected siblings, patients P3 (female) and P4 (male). At 13 years old, coinciding with puberty, patient P3 developed frequent severe headaches accompanied by long-lasting fevers (40°C) and bilateral painful cervical lymphadenopathy. She gradually progressed with other symptoms including malar rash, hemorrhagic vesicular eruptions, and proteinuria. Her neurological symptoms included delirium, hallucinations, generalized tonic-clonic and absence seizures. Brain MRI did not show evidence of focal lesions, however, lumbar puncture revealed increased intracranial pressure. Renal biopsy showed evidence of mesangial proliferative nephritis. Later in her disease course she developed diffuse hair loss, photosensitivity, and weight loss. She has residual neurological and cognitive impairments, sicca syndrome and aphthous lesions. Her disease is controlled with monthly IV methylprednisolone, cyclophosphamide, azathioprine, and hydroxychloroquine.
Her brother, P4, first developed symptoms at age 7, in the form of axillary lymphadenopathy and fevers (39°C). He was found to have proteinuria and a renal biopsy revealed acute proliferative glomerulonephritis. Immunofluorescence microscopy showed diffuse deposition of IgG, C3, and C1q. A year later, he developed a malar rash, photosensitivity, and a scaly erythematous patchy rash consistent with discoid lupus. He has also complained of photophobia, xerophtalmia and aphthous mouth lesions. His symptoms have stabilized with a combination of immunosuppressive therapy of prednisone and azathioprine.
Genetic analysis
We performed WES in the four patients, four unaffected parents and one unaffected sibling (Figure 1A). We selected variants with a frequency of less than 1% in various public databases and in our in-house database of 839 exomes (Figure 1B). Assuming homozygous inheritance in affected patients allowed to identify 6 candidate variants, 5 of which were non-coding and thus less likely to be pathogenic. Four rare/+novel homozygous non-coding variants were inherited on the same haplotype co-segregating with the novel coding variant in C1R (Supplementary Table 3).
All affected patients were homozygous for a 1bp deletion (c.1332delT) in the C1R gene, corresponding to the p.Pro445Leufs*11 protein mutation (Figure 1C, Supplementary Figure 1). The C1R gene encodes complement component 1, r subcomponent. Sanger sequencing of 14 family members confirmed the proper segregation of the p.Pro445Leufs*11 frameshift mutation, and identified a 9-year old asymptomatic female (AP5) to carry the same homozygous mutation (Figure 1A). We genotyped 1706 Turkish healthy controls and 1618 Turkish patients with Behcet’s disease and did not identify any carriers. Targeted sequencing on 300 sporadic SLE patients with late-onset symptoms also did not identify any patients with biallelic LOF mutations in C1R. Western blotting of mutant PBMCs confirmed that the p.Pro445Leufs*11 variant creates truncated proteins (Figure 1D). The truncated C1R protein lacks the light chain domain, which contains the catalytic domain of the activated (cleaved) C1r protein. Therefore the mutant protein is expected to be unable to cleave and activate C1s (Figure 1C).
We attempted to explain the difference in the disease severity in patients P1, P2, P3, and P4 by looking for modifying alleles. We analyzed the exome data for rare exonic predicted damaging variants and common SLE-associated variants, present in one or more patients. We found 3 rare coding variants in CD44, DHCR7, and PXK genes have been implicated in the pathogenesis of SLE. Patient P1 and P2 had no variants, P3 and P4 carried 3 and 2 of these variants, respectively (Supplementary Table 4). We then looked for presence of common SLE-associated variants (SNPs) that were polymorphic in this family. We found eleven variants both in affected and unaffected family members, as expected, however they were enriched in the more severly affected patients P3 and P4 (Table 1). We determined classical HLA alleles in four affected patients (P1, P2, P3, P4) and two unaffected family members. No single HLA allele was shared among the 4 affected subjects. Finally, none of the previously reported SLE-associated extended MHC haplotypes were observed in the family (Supplementary Table 5 and 6).
Table 1.
Common SLE-associated variants identified in the family by whole-exome sequencing*
| Chr. | Position | Ref. | Var. | Type | Gene | Consequence | dbID | HGMDid | ExAC_freq | P1 | P2 | P3 | P4 | ID_10 | ID_11 | ID_13 | ID_4 | ID_6 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 16 | 3707746 | G | A | Nonsynonymous_SNV | DNASE1 | c.731G>A:p.R244Q | rs1053874 | CM042698 | 0.371 | GA | AA | GA | AA | GA | GA | AA | AA | GA |
| 19 | 10472932 | A | G | Intronic | TYK2 | – | rs280519 | – | 0.508 | AA | AA | GG | AG | GG | AG | GG | AA | AG |
| 19 | 10475651 | C | A | Nonsynonymous_SNV | TYK2 | c.1084G>T:p.V362F | rs2304256 | CM050351 | 0.275 | CC | CC | CA | CC | CA | CC | CA | CC | CA |
| 12 | 10532325 | T | C | Nonsynonymous_SNV | KLRK1 | c.214A>G:p.T72A | rs2255336 | CM099345 | 0.803 | TC | CC | TC | TC | CC | TC | TC | CC | TC |
| 1 | 12252954 | T | G | Nonsynonymous_SNV | TNFRSF1B | c.587T>G:p.M196R | rs1061622 | CM022071 | 0.223 | TT | TT | TG | TT | TG | TT | TT | TT | TT |
| 16 | 31276810 | G | A | Nonsynonymous_SNV | ITGAM | c.230G>A:p.R77H | rs1143679 | CM080418 | 0.092 | GG | GG | GA | GA | GA | GG | GG | GG | GG |
| 2 | 33701889 | T | C | Intronic | RASGRP3 | – | rs13385731 | – | NA | TT | TT | TT | TC | TT | TC | TC | TT | TT |
| 15 | 89450586 | G | T | Nonsynonymous_SNV | MFGE8 | c.226C>A:p.L76M | rs1878326 | CM094608 | 0.622 | TT | TT | GT | TT | TT | GT | TT | TT | GT |
| 4 | 102751075 | G | A | Nonsynonymous_SNV | BANK1 | c.182G>A:p.R61H | rs10516487 | CM080095 | 0.255 | GG | GG | GA | GG | GG | GA | GG | GG | GG |
| 4 | 102839286 | G | A | Nonsynonymous_SNV | BANK1 | c.1147G>A:p.A383T | rs3733197 | CM080096 | 0.303 | GG | GG | AA | GG | GA | GA | GA | GG | GG |
| 1 | 161479744 | A | G | Nonsynonymous_SNV | FCGR2A | c.500A>G:p.H167R | rs1801274 | – | 0.479 | GG | GG | AG | AG | AA | GG | AG | AG | GG |
Whole-exome sequencing was performed in the affected family members (P1–P4) and 5 additional family members (ID_10, ID_11, ID_13, ID_4, and ID_6) (Figure 1). Boldface letters indicate systemic lupus erythematosus (SLE) susceptibility alleles. Chr. = chromosome; Ref. = reference; Var. = variant; HGMD = Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php); ExAC = Exon Aggregation Consortium; SNV = single-nucleotide variant.
Low complement levels
We measured patients serum complement levels to determine the functional effect of this mutation (Supplementary Table 7). Patients P3 and P4 had noticeably low C1r and C1s levels, while C1q and C2-4 levels were normal to high. The total complement activity level was <10 CAE units, which is far below the normal level of 55-145 CAE units. Samples from other patients were not available for testing.
Type-I Interferon (IFN-I) signature and cytokine levels
We investigated the presence of IFN gene expression signature in whole blood samples from patients P3, P4 and from the asymptomatic mutation-positive patient (AP5). Both affected patients showed significant up regulation of IFN response genes (Figure 2A, Supplementary Figure 2A). The male patient P4 had more pronounced IFN-I signature than the female patient P3, while the gene expression profile of the asymptomatic patient AP5 was similar to a healthy control (Figure 2A). The strong inflammatory signature in the male patient P4 correlated with significantly higher levels of serum proinflammatory, anti-inflammatory cytokines and chemokines than in female patients (Figure 2B). Gene expression profililing of 594 immuno-related genes demonstrated that patients P3 and P4 have increased expression of NF-κB and Jak-Stat pathway genes, while the asymptomatic patient AP5 appeared more similar to 4 healthy controls (Supplementary Figure 2B).
Figure 2.
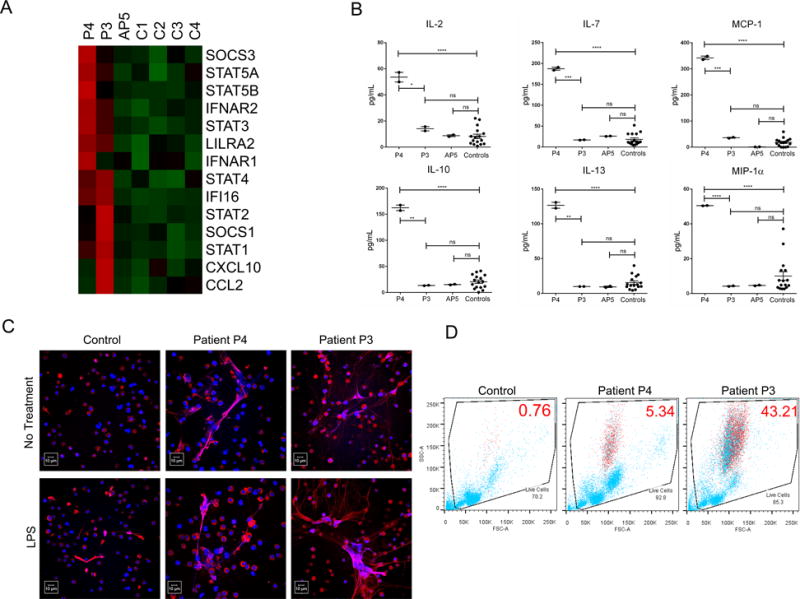
Patients with the C1R mutation exhibit enhanced IFN-I signature, inflammatory cytokine profile, high levels of NETosis and a low-density granulocyte signature.
(A) NanoString analysis for IFN-I signature genes in symptomatic patients (P3 and P4), an asymptomatic patient (AP5), and a healthy control.
(B) Cytokine profiling comparing serum from patient P4, patient P3, and asymptomatic patient AP5 with healthy controls. Data represent mean ± S.E.M from analysis of duplicates of each individual. * indicates P < 0.05; ** indicates P < 0.01; *** indicates P < 0.001; **** indicates P < 0.0001.
(C) Spontaneous (without LPS) and induced (with LPS) NET production was analyzed by immunofluorescence (MPO in red and DNA in blue, scale bar = 10 μm).
(D) The percentage of low-density granulocytes (CD10+, CD14lo, CD15+) identified with red symbols was determined by flow cytometry of each individual’s PBMCs. Forward-scattered light (FSC) is proportional to cell-surface area or size. Side-scattered light (SSC) is proportional to cell granularity or internal complexity.
Neutrophil phenotype
NETs have been shown to play important role in modulating immune responses in a variety of inflammatory and autoimmune diseases including SLE(7). We examined NET formation of normal-density granulocytes from P3, P4 and a control donor by immunofluorescence. The female patient P3 showed higher levels of spontaneous NETosis and LPS-induced NETosis compared with the P4 male patient and control (Figure 2C). The male patient P4 exhibited some enhanced NET production when compared to the control, but the results were not as dramatic as in the female patient.
A unique population of granulocytes known as LDGs has been associated with increased propensity for spontaneous NET formation (8). The frequency of LDGs in PBMC fractions was quantified in patients P3, P4 and a healthy control. The female P3 patient showed a markedly higher percentage of LDGs (43.21%) compared to the male P4 patient (5.34%) and control donor (0.76%) (Figure 2D). Additionally, qRT-PCR analysis of the PBMCs for granulocyte-expressed proteins (myeloperoxidase [MPO] and cathepsin G [Cath G]) showed a higher granulocyte signature in the PBMCs from patient P3 than the P4 patient (Supplementary Figure 2C). These results suggest that the deficiency of C1r is associated with a pro-inflammatory granulocyte phenotype, including enhanced NETosis and higher percentage of LDGs.
Discussion
C1r is a serine protease that combines with C1q to form the C1 complex of the classical complement activation pathway. Loss-of-function mutations at the beginning steps of complement activation can have major effects on all downstream complement proteins. This is the second report of a novel high-penetrance mutation in C1R as the cause of monogenic and familial type of SLE. The C1r deficient patients in this study are phenotypically similar to the previously reported patient with the C1r deficiency (Supplementary Table 2).
This consanguineous family with multiple affected patients provided a platform for the analysis of role for modifying alleles in disease expressivity. Despite the common genetic and enviromental background there are phenotypic variations even amongst patients of the same gender, which cannot be fully explained by the novel deleterious mutation in C1R. Our data suggest that in addition to the deleterious C1R mutation, other rare and common gene variants may have contributed to the disease severity (Supplementary Figure 3). In particular, patient P3, who had CNS and renal disease, carried more SLE risk alleles than the other affected members of the family. Interestingly, classical SLE-associated HLA alleles were not identified in this family.
Until recently, dysregulated lymphocytes and aberrant antigen presentation have been considered major factors contributing to inflammation in SLE patients. Current models of lupus pathogenesis include a role for activated granulocytes, enhanced NET formation and type-I interferon producing cells(9). Enhanced NETosis in SLE patients may provide a potent stimulus for IFNα release by plasmacytoid dendritic cells (pDCs), inflammasome activation, and can also directly lead to tissue damage(10, 11). One interesting finding of this study was the difference in inflammatory signatures between the male and female patients despite the fact that neither had active disease at time of sampling. The male patient P4 showed a higher inflammatory cytokine signature than the female P3 patient. The female patient P3 had most severe disease and a stronger neutrophil signature, which might be related to the role of estrogen signaling in the regulation of immune pathways or a gender-attributed difference in microbiota (12).
Hypotheses on the mechanism of interaction between NETs and complement are rather mixed in the literature (13)(14). The origin of LDGs remains unclear and whether these cells are immature granulocytes or represent a distinct mature subset needs to be further characterized(10). It is possible that the immune dysregulation that follows C1r deficiency or the distinct cytokine milieu may have led to the generation of these cells. Hormonal or genetic factors may also play a role, given that the female patient had a significantly higher number of LDGs. The specific role of gender in the regulation of adaptive and innate immune responses in SLE and its association with response to therapy need to be systematically studied in large cohorts of patients. Analyzing the combined effect of rare and common gene variants in SLE may shed a light on interactions between signaling pathways and may further reveal potential therapeutic targets.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institute of Arthritis and Musculoskeletal and Skin Diseases and by the NIH Clinical Center. Dr. Erkan Demirkaya was recipient of the Research Fellowship Program for International Researchers, which is supported by The Scientific and Technological Research Council of Turkey (TUBITAK; B.14.2.TBT.0.06.01-219-84). We also thank all the patients and the family members for their enthusiastic support during this research study.
Footnotes
Author Contributions
E.D., Q.Z., and I.A. designed the study. Q.Z., C.K.S., N.D., W.T., E.F.R., M.T. Y.H.P., J.C., and S.C.C. performed experiments. Q.Z., C.K.S., M.J.O., M.G. and I.A. analyzed and interpreted the data. E.D., P.H., K.B., D.S., A.A., S.S., S.C., S.A.H, A.K.O., D.L.K., and O.K. enrolled the patients and collected and interpreted clinical information. Q.Z., C.K.S., M.J.K, and I.A. wrote the manuscript. I.A. directed and supervised the research. All authors contributed to the review and approval of the manuscript.
Competing Interests
The authors declare no competing financial interests.
References
- 1.Cui Y, Sheng Y, Zhang X. Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun. 2013;41:25–33. doi: 10.1016/j.jaut.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Niewold TB. Advances in lupus genetics. Curr Opin Rheumatol. 2015;27(5):440–7. doi: 10.1097/BOR.0000000000000205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis MJ, Botto M. Complement deficiencies in humans and animals: links to autoimmunity. Autoimmunity. 2006;39(5):367–78. [Google Scholar]
- 4.Bienaime F, Quartier P, Dragon-Durey MA, Fremeaux-Bacchi V, Bader-Meunier B, Patey N, et al. Lupus nephritis associated with complete C1s deficiency efficiently treated with rituximab: a case report. Arthritis Care Res (Hoboken) 2010;62(9):1346–50. doi: 10.1002/acr.20163. [DOI] [PubMed] [Google Scholar]
- 5.Wu YL, Brookshire BP, Verani RR, Arnett FC, Yu CY. Clinical presentations and molecular basis of complement C1r deficiency in a male African-American patient with systemic lupus erythematosus. Lupus. 2011;20(11):1126–34. doi: 10.1177/0961203311404914. [DOI] [PubMed] [Google Scholar]
- 6.Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. 2015 doi: 10.1038/ng.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith CK, Kaplan MJ. The role of neutrophils in the pathogenesis of systemic lupus erythematosus. Curr Opin Rheumatol. 2015;27(5):448–53. doi: 10.1097/BOR.0000000000000197. [DOI] [PubMed] [Google Scholar]
- 8.Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. 2011;187(1):538–52. doi: 10.4049/jimmunol.1100450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu Y, Su K. Neutrophil Extracellular Traps and Systemic Lupus Erythematosus. J Clin Cell Immunol. 2013;4 doi: 10.4172/2155-9899.1000139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–53. doi: 10.1038/nm.4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3(73):73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, et al. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339(6123):1084–8. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Wang C, Zhao MH, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol. 2015;181(3):518–27.14. doi: 10.1111/cei.12654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leffler J, Martin M, Gullstrand B, Tyden H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. 2012;188(7):3522–31. doi: 10.4049/jimmunol.1102404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.