

Abstract
Isolating small objects, such as particles, cells, and molecules, in individual aqueous droplets is useful for chemical and biological assays. We have developed a simple microfluidic platform to immobilize (park) microparticles at defined locations, and isolate particles in monodisperse droplets surrounded by immiscible oil. While conventional methods can only achieve stochastic encapsulation of objects within larger droplets, our in situ method ensures that a single particle is entrapped in a similar-sized droplet, with ~95% yield for parking and isolation. This enables time-lapse studies of reactions in confined volumes and can be used to perform enzymatic amplification of a desired signal to improve the sensitivity of diagnostic assays. To demonstrate the utility of our technique, we perform highly sensitive, multiplexed microRNA detection by isolating encoded, functional hydrogel microparticles in small aqueous droplets. Non-fouling hydrogel microparticles are attractive for microRNA detection due to favorable capture kinetics. By encapsulating these particles in droplets and employing a generalizable enzyme amplification scheme, we demonstrate an order of magnitude improvement in detection sensitivity compared to a non-amplified assay.
TOC
We demonstrate a microfluidic platform that immobilizes hydrogel microparticles at defined locations, and encapsulates particles in monodisperse droplets for sensitive bioassays.
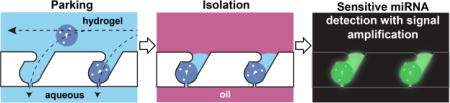
Introduction
Droplet microfluidics has emerged as a promising alternative to conventional multiwell plates for a variety of biological applications. Droplets can be used to isolate objects, such as cells, beads, viruses, and molecules1–8, and insulate one or multiple objects from the external environment, creating an appealing platform for high-throughput screening assays1–3. Droplets also enable accumulation of products from chemical or biological reactions within a small, confined volume, enhancing assay sensitivity for medical diagnostics4–8. Initially, many researchers focused on droplet production techniques such as T-junction breakup9 and flow focusing10. Such early production techniques relied on statistics in encapsulation, resulting in a Poisson distribution of isolated objects in droplets. Thus, droplet sorting11–13 and arrangement techniques2–4, 14–19 were developed to specifically monitor or collect droplets of interest. Static droplet arrays, a result of arrangement, enable repeated observation of droplets over time. Unlike sorting techniques, arrangement generally does not require complex external devices or protocols. Static droplet arrays can be generated in two ways: sequentially14–18 and in situ2–4, 19, 20. Sequential methods generate droplets upstream and arrange them downstream. In situ methods generate arranged droplets in a single step. Recently, novel droplet production techniques overcome stochastic encapsulation21–23, and these advances can be potentially adopted for sequential methods of droplet array production. However, in situ methods are generally advantageous over sequential methods because they do not require cumbersome operation procedures to ensure precise flow control during droplet generation. But challenges still exist for in situ platforms to overcome the statistics in encapsulation. This would require arranging objects before isolation, and matching object and droplet dimensions.
In this study, we developed a microfluidic platform to park microparticles in traps at pre-defined positions, and isolate them in water-in-oil droplets. Hydrodynamic force, associated with fluid flow through a pore in each trap, guides, squeezes, and parks particles with high yield (94.5 %). The microfluidic platform is designed to allocate only one particle per trap, overcoming Poisson statistics in encapsulation in the following isolation step. Based on a theoretical rationale, we back-flowed (flow direction from outlet to inlet) fluorinated oil to isolate a particle in a droplet at each trap, with homogeneous droplet size (CV = 7.9 %) and high isolation yield (96.4 %). Droplet production and arrangement were achieved simultaneously, avoiding cumbersome liquid handling and flow control.
The capabilities of this platform were demonstrated by detecting microRNA (miRNA) with high sensitivity. miRNA is a short non-coding RNA that post-transcriptionally regulates gene expression. miRNAs show distinct dysregulation patterns in disease contexts, such as cancer24–26, diabetes27, and cardiovascular diseases28, 29, and are considered potent biomarkers for disease diagnosis. Quantification of miRNA requires fulfilling crucial clinical needs: high sensitivity, specificity, reproducibility, wide dynamic range, and multiplexing capabilities with a short assay time30. Also, the quantification technique should be inexpensive, avoid target bias, and be compatible with various sample inputs (e.g. raw cells) with minimal required processing. However, existing approaches, such as quantitative real-time reverse-transcription polymerase chain reaction (qRT-PCR) and microarrays, cannot achieve all clinical needs at the same time30–32. qRT-PCR can achieve superior sensitivity, but is expensive and requires cumbersome sample extraction from raw cells. Microarrays have good multiplexing capabilities but require long assay times (overnight hybridization incubation).
Our group has previously developed a hydrogel particle-based strategy for miRNA detection that overcomes many of the limitations of qRT-PCR and microarrays33. Non-fouling polyethylene glycol (PEG) particles enable quantification of miRNA in complex media, such as raw cells lysates34. Hydrogel particles can freely float to capture the majority of target miRNA in a sample, and the porous particles provide a solution-like environment, leading to high sensitivity and specificity in short assay times35. A unique post-hybridization labeling scheme eliminates target bias. Encoded hydrogel microparticles are synthesized in high-throughput by stop flow lithography (SFL)36, 37, a robust technique that enables multiplexing capabilities and high assay reproducibility. These features make this particle-based strategy a promising approach for point-of-care miRNA detection. However, although this technique demonstrates higher sensitivity than microarrays, the current limit of detection (LOD) is on the order of 1 amol33. Recent work has shown that this is comparable to the endogenous amount of one specific miRNA species from ~850 cells34. Thus, to improve the LOD of this strategy, we adopted the previously developed enzymatic amplification scheme8, 38 by using our microfluidic device for parking and isolation. Target-specific enzyme labeled particles were parked and isolated in aqueous droplets using our platform. Through an enzymatic reaction, fluorescent molecules were produced and accumulated in the droplets over time, resulting in signal amplification and one order of magnitude improved sensitivity compared to a direct labeling scheme, without compromising assay reproducibility, specificity, and multiplexing capabilities.
Methods
Microfluidic Device Fabrication
Microfluidic channels for both particle synthesis and parking/isolation were fabricated using previously reported processes33, 34, 36. PDMS (Sylgard 184, Dow Corning) was mixed in a 10:1 ratio and poured onto a SU-8 master (Microchem), fabricated by standard photolithography procedure, to make the top part of the PDMS channels. After curing overnight at 65 °C, PDMS channels were cut and punched to make an inlet and outlet. This PDMS channel was placed on top of a glass slide coated with half-cured PDMS (mixed in 10:1 ratio and cured ~37 min at 65 °C), and baked overnight for bonding. Heights of particle synthesis and parking/isolation channels were 42 and 38.5 µm, respectively.
Microparticle Synthesis
Microparticles were synthesized via stop flow lithography (SFL; schematic shown in Fig. S1)33, 34, 36. We flowed prepolymer solution through the synthesis channel using compressed air, stopped the flow, and exposed the channel with ultraviolet light (Thorlabs, 365 nm LED, 720 mW cm−2) patterned by a transparency mask (Fineline) designed in AUTOCAD. In this exposure step, the mask defined the shape of the 2D extruded, encoded microparticles, which were polymerized by photo-crosslinking. The three steps (flow, stop, exposure) were repeated to achieve semi-continuous particle synthesis. Synthesized particles were collected in a centrifuge tube and purified with PBST (phosphate buffered saline with 0.05 % Tween-20) by centrifugation. Particles used for the bioassay were oxidized after synthesis. Oxidation changes hydrophobic, non-reacted acrylate groups to carboxylic acid groups, reducing non-specific binding8. For oxidation, particles were incubated in 500 µM KMnO4 in 0.1 M Tris buffer (pH 8.8, filtered with 0.2 µM filter) for 5 min, purified, and stored in PBST (with 0.2 % Tween-20) containing 500 mM NaCl.
In this study, we synthesized soft, porous hydrogel particles, optimized for bioassays35, 38, 39. Prepolymer solution was prepared in two steps. First, a base solution was prepared by mixing 20 % (v/v) polyethylene glycol diacrylate (PEGDA, Sigma Aldrich, Mn = 700), 40 % (v/v) polyethylene glycol (PEG, Sigma Aldrich, Mn = 600; warmed to 35°C), 35% (v/v) 3× tris-EDTA buffer, and 5% (v/v) 2-hydroxy-2-methylpropiophenone (photoinitiator, Sigma Aldrich). This base solution was mixed with probe solution (Integrated DNA Technologies (IDT)) in a 9:1 ratio to make the prepolymer solution. The concentration and chemistry of each probe solution are described in supplementary table S1.
Particle Parking and Isolation
We used compressed air to generate pressure-driven flow for all steps. First, we flowed PBST (with 0.05 % Tween-20) in the parking/isolation channel until all air bubbles disappeared. Next, we flowed particle solution (PBST with 0.05 % Tween-20) at 2.0 psi from the inlet to park particles in traps (average particle height ~ 36 µm). For experiments where we monitored the enzymatic reaction, we then flowed substrate solution (chilled to 4 °C) from the outlet immediately before isolation. In this study, substrate solution was 200 µM fluorescein di-β-D-galactopyranoside (FDG, Thermo Fisher) dissolved in PBST (0.05 % Tween-20) containing 0.2% (v/v) DMSO. To isolate particles in droplets, we flowed FC-40 (Sigma-Aldrich) from the outlet until all particles were isolated. The oil/water interface moved through the channel at a speed of ~1 mm/s. We used a high speed camera (Phantom Miro M310, Vision Research) to image the parking and isolation processes. For calculation of parking and isolation yields, we flowed a total of ~75 particles into each channel.
miRNA Detection Assay
The bioassay was performed following previously reported protocols with some modifications8, 33, 34. In this work, samples were processed in a 0.65 ml microcentrifuge tube for all steps before the parking process. All solvents were filtered with a 0.2 µm sterile filter. All purification steps were repeated three times and performed by centrifugation with 500 µl of buffer. All incubation steps were conducted in a thermoshaker (Thomas Scientific) at 1500 rpm. Unless specified, all PBST solutions contained 0.2 % (v/v) Tween-20 to prevent particle-particle adhesion. Fluorescence signal was detected by an interline CCD camera (Clara, Andor) with 0.2 second exposure time. For hybridization, target miRNA was diluted in TET buffer (1× Tris-EDTA buffer with 0.05 % Tween-20) containing 500 mM NaCl, and 25 microparticles in the same buffer were added to make a total volume of 50 µl. The sample was incubated at 65 °C for 90 min. After hybridization, samples were purified with TET buffer containing 50 mM NaCl. For ligation, we added 245 µl of pre-mixed ligation solution (100 µl 10x NEBuffer 2 (New England Biolabs), 900 µl TET, 250 nM ATP, 40 nM biotinylated universal linker (IDT), and 800 U/ml T4 DNA ligase) into the purified particle sample. This mixture was incubated at 21.5 °C for 30 min.
For direct tagging with a fluorescent reporter, we purified the sample with TET containing 50 mM NaCl, and added 5 µl of 20 µg/ml streptavidin-R-phycoerythrin (SAPE, Invitrogen) dissolved in TET. Samples were incubated at 21.5 °C for 45 min. Then, the sample was purified with TET containing 50 mM NaCl, and dispersed in PTET (5x TE with 25% (v/v) PEG 400 and 0.05% Tween-20). 10 µl of particle solution was placed on top of a glass slide, covered by a cover glass, and imaged under excitation (20X objective, X-Cite 120LED, Omega XF101-2 filter set: λex/λem = 525/565 nm, 2.0 W cm−2). For enzyme labeling and amplification, we purified the sample with PBST and added 2.5 µl of 20 µg/ml streptavidin-β-galactosidase (SAB (Invitrogen), in PBST, filtered with 0.45 µm filter) into the 50 µl sample. Samples were incubated at 21.5 °C for 30 min. Then, the sample was purified with PBST and incubated at 21.5 °C for 60 min to enable the outward diffusion of physically trapped enzyme inside the particles. Next, we purified the sample with PBST (with 0.05 % (v/v) Tween-20) and followed the described parking/isolation procedure. After isolation, the whole channel was stored in a sealed humid chamber until imaging (1 hr incubation, unless otherwise specified). Fluorescence signal was detected under excitation using the same Andor CCD camera (20x objective, X-Cite 120LED, Omega XF100-3 filter set: λex/λem = 470/545 nm, 0.11 W cm−2). For multiplexed assays, 25 particles for each miRNA (i.e. 75 total particles for 3 distinct miRNA species) and all miRNA targets were mixed together during hybridization. The rest of the assay followed the same described protocol, and all particles were parked and isolated in the same channel. For all experiments, we imaged at least 5 particles for every condition.
Results and Discussion
Design Principles for Particle Parking and Isolation Device
The driving force for particle arrangement should satisfy three requirements; it should be controllable, be directed towards the desired location, and act on all objects in the same manner regardless of chemical composition. Our microfluidic device comprises an inlet, main channels, and an outlet (Fig. 1a). The inlet has a dust filter to block debris that can cause potential problems in a linear channel. The main channel has 95 traps (i.e. parking lots) for microparticle parking. Once the particle solution flows into the channel, the liquid flow bifurcates; one branch follows the main channel, and the other passes through the pores in each trap (Fig. 1, with dimensions specified in figure S2). The second branch of liquid flow generates a hydrodynamic force, which guides and parks a microparticle into each trap. This hydrodynamic force can be controlled by the applied pressure, points towards the trap, and acts on all types of objects in the same manner. Thus, it satisfies all desired characteristics for particle arrangement, and enables high parking yield (94.5 ± 5.3 % of all particles introduced into the channel are successfully parked, Fig. 1c). Such high parking yield can be achieved at various pressures (Fig. S3), and all particles are parked regardless of initial position within the channel (Fig. S4). Guided microparticles are squeezed into traps (Fig. 2a, Movie S1) with entrance widths slightly smaller than particle diameter, and the parked particles do not pass through the traps due to geometric constraints, resulting in a fixed particle position. Once a particle occupies a trap, the hydrodynamic resistance through the trap increases significantly, reducing the driving force toward the same trap and forcing other particles to park in empty traps. This ensures that each trap always contains a single particle. This design principle has been used to trap cells40–42, beads43, particles44, 45, and embryos46, 47 in microfluidic devices. In this work, we synthesized microparticles of 100 µm diameter via stop flow lithography (SFL)33–37, with 105 particles/hr throughput, to demonstrate parking and isolation in our platform. Particles in aqueous solution (PBST) were first parked within the channel, then isolated in water-in-oil droplets by flowing fluorinated oil (FC-40) from the outlet of the device. We were able to achieve high isolation yield (96.4 ± 1.7 % of parked particles are isolated, Fig. 1d, Movie S2) with homogeneous droplet sizes less than 500 pl in volume (CV = 7.9 %). It is possible to precisely control the volume of the droplets by tailoring device geometry, and droplets can be fabricated with minimal excess volume compared to the encapsulated object. These characteristics, along with simple device setup and operation, make this an ideal system for encapsulating single objects in isolated aqueous droplets with high yield. While we demonstrate the utility of this platform for miRNA detection by parking functionalized hydrogel particles, this technique can also be adapted for parking other soft, biological objects, such as cells, spheroids, and embryos.
Figure 1. Microfluidic device and protocol for microparticle parking and isolation.

(a) Schematics of microparticle parking and isolation. The device is shown before parking, after parking, and after isolation with fluorinated oil (FC-40). The top row shows close-up images of the outlined area, and the bottom row shows the whole device during each of the three steps. (b-d) Bright field images of the microfluidic channel before parking (b), after parking (c), and after isolation (d). Only one particle occupies each trap, and trapped particles are isolated in individual homogeneous-sized droplets (volume < 500 pl; CV = 7.9%) with high yield. Yields of parking and isolation are 94.5 (± 5.3) and 96.4 (± 1.7) %, respectively. Scale bar is 300 µm.
Figure 2. Analysis of parking and isolation process.

(a-b) Time-lapse images of particle parking (a) and isolation (b) captured by a high speed camera. Microparticles are guided, squeezed, and parked in a trap by a hydrodynamic force associated with flow through the pore in each trap. Trapped particles are isolated by back-flow of fluorinated oil. (c-f) Schematics for scaling analysis: forward-flow (c-d) and back-flow cases (e-f) in wide (c, e) and close-up views (e, f). Back-flow prevents penetration of the oil phase into the trap, ensuring homogeneous droplet size and preventing particle deformation (see Fig. S5-S7 for low magnification images of forward-flow and back-flow cases). Scale bar is 100 µm.
Rationale of Back-flow for Isolation
During particle isolation, back-flow of FC-40 from the device outlet towards the inlet (opposite to the direction used for particle parking) holds significant advantage over forward-flow (from inlet to outlet). As the oil moves through the channel, the oil/water interface should not trespass (enter into) the traps because the interfacial tension between FC-40 and PBST (20.9 ± 0.3 mN/m, measured using a goniometer from Ramé-hart Instrument Co.) can induce deformation of the soft objects parked in each trap. In addition, because the interface continues to trespass the trap until the oil in the main channel passes the opposite side of the trap, this phenomenon can cause variation in droplet size, depending on the time required for isolation. Backflow of FC-40 prevents the penetration of the interface into the trap while forward-flow does not (Fig. 2b, Fig. S5 and S6 show wide-view images displaying the behavior of the interface in the case of forward-flow and back-flow, respectively).
Schematics of each case are shown in figure 2c-f (Fig. 2c, d for forward-flow, and Fig. 2e, f for back-flow). Consider the point when the oil/water interface is near one side of the trap (Fig. 2c, e). P1 is the pressure of the oil phase near the interface, P2 is the pressure of the water phase near the interface in the trap, and P3 is the pressure of the water phase downstream, near the opposite side of the trap. κL and κS are the mean curvatures of the interface in the main channel and the trap, respectively. The applied pressure (ΔP ≡ P1 − P3) is the force driving the interface to trespass the trap. The opposing force that inhibits trespassing is the interfacial tension related to κS. Therefore, we can define a dimensionless number, Ψ, that provides a criterion for trespassing.
| (1) |
Ψ is the ratio between applied pressure and maximum interfacial tension. γ is the surface tension between oil and water, D is the width of the trap entrance/pore, and H is the height of the channel. The maximum interfacial tension is determined by channel geometry.
| (2) |
DE and DP are the widths of the trap entrance and trap pore, respectively. In our channel design (Fig. S2), DP ~ 0.2 DE; DE is larger to allow particles to park, and DP is smaller to prevent particles from passing through. To achieve maximal interfacial tension and prevent trespassing (Ψ≤1), we should minimize D by allowing the oil/water interface to pass by DP first. This is achieved by the back-flow of FC40, which prevents trespassing and enables the formation of uniform-sized droplets. It is worth noting that we were able to isolate objects without any deformation, regardless of object softness (Fig. S7 shows the generation of empty droplets). However, back-flow requires the application of lower pressure than during parking in order to retain particles in traps, resulting in a longer process time (Fig. 2b). The isolation process can be accelerated when particles are stiffer, or when the trap entrance and pore are smaller, ensuring that no particles escape and that the oil/water interface does not trespass the trap.
Static Particle-Containing Droplet Arrays: Product Accumulation and Signal Amplification
Droplets at fixed locations can accumulate product from chemical reactions and enable observation over time. Product accumulation can amplify a desired signal and enable the detection of previously non-detectable signals, a useful feature for diagnostics4–8. We synthesized biotin-functionalized microparticles with various biotin probe concentrations in the prepolymer solution. These particles were labeled with streptavidin-conjugated enzyme (streptavidin-β-galactosidase, SAB), washed, and parked in our microfluidic platform (Fig. 3a). After parking, non-fluorescent substrate (fluorescein di-β-D-galactopyranoside, FDG) solution was back-flowed at low temperature (4 °C). The low temperature quenches the reaction before isolation takes place, and back-flow minimizes the dilution of the substrate solution. Substrate-soaked particles were then isolated in water-in-oil droplets. Fluorescein, a fluorescent product generated from the enzymatic reaction, was accumulated in the droplet, amplifying the detected signal (Fig. 3b, c). The streptavidin-conjugated enzyme (SAB) specifically labels the biotin probes conjugated to the particles, and continually turns over excess substrate into fluorescent product. Thus, the signal difference between particles with and without biotin becomes more distinct over time. In addition, only droplets containing particles showed an increase in signal, while empty droplets did not. This is evidence that each droplet was isolated from the others, and that diffusion of fluorescein outside of the aqueous phase is suppressed due to low solubility in FC-40.
Figure 3. Signal amplification in static droplet array.

(a) Schematic of amplification demonstration. Biotinylated particles are labeled by streptavidin-conjugated enzyme (SAB), parked, soaked by substrate (FDG), and isolated in an aqueous droplet. (b, c) Signal plot (b) and fluorescent time-lapse images (c) of particles in an isolated droplet with/without a biotinylated probe. Over time, fluorescein is generated by the enzymatic reaction and accumulated within the droplet, amplifying the signal in the biotinylated particle. Scale bars are 100 µm.
Highly Sensitive, Multiplexed miRNA Detection
We demonstrate our platform’s capabilities by improving the LOD of a hydrogel particle-based microRNA detection strategy. As discussed, non-fouling hydrogel particles present an appealing motif for miRNA detection with multiplexing capabilities, high sensitivity, specificity, and reproducibility34, 39. Prior work employing a direct tagging strategy to label hydrogel particles showed a detection limit on the order of 1 amol for various miRNA species33. For a different, non-particle based assay with lower overall sensitivity, use of an enzymatic amplification scheme improved the LOD one order of magnitude compared to direct tagging8. In this work, by isolating hydrogel particles, we adopt the enzymatic amplification scheme to particle-based assays to achieve improved miRNA detection sensitivity.
We synthesized microparticles incorporating an acrylated synthetic DNA probe. The acrylate group on the probe ensures that the molecule is covalently bound to the PEGDA microparticles during photo-crosslinking, and the DNA chain has a complementary sequence to the miRNA target to be detected. In addition, the end of the DNA chain also contains a sequence that is complementary to a universal linker functionalized with biotin; this enables subsequent labeling by streptavidin-conjugated moieties without sequence bias33. During hybridization under carefully optimized conditions, the target miRNA specifically bound to complementary probes in the hydrogel particles. After a 90 min hybridization step, the particles were sequentially labeled with a biotinylated universal linker (30 min) ligated to bound target miRNA, and a streptavidin-conjugated reporter. In order to show the effect of amplification, we used two different reporters, a fluorescent molecule (SAPE, 45 min) for direct tagging, and an enzyme (SAB, 30 min) for the amplification strategy. For the amplification strategy, enzyme-labeled particles were then parked within the microfluidic channel, soaked in substrate solution (FDG) at low temperature, and isolated in an aqueous droplet (Fig. 4a). After isolation, the enzymatic reaction was allowed to run for 60 min before imaging. We note that the amplification strategy protocol (total assay time: 4hr 30 min) is slightly longer than direct tagging (2hr 45 min), mainly due to the enzymatic reaction time. For future point-of-care applications, the reaction time (1hr) can be reduced to 10 min without harming the LOD, as demonstrated by figure S10. However, the current total assay time is still short enough for point-of-care use, and is significantly shorter than microarray hybridization techniques (10-20 hr31, 48). For both direct tagging and amplification strategies, we measured the signals corresponding to different target amounts to plot a calibration curve, and calculated the LOD based on the trend line. Here, LOD is defined as the target miRNA amount which shows a net signal equal to three times the signal-to-noise ratio, and the amplification factor is defined as the ratio of the LOD of direct tagging compared to the amplification strategy. Both amplification and direct-tagging strategies showed linear responses for three different miRNA species, ensuring precise quantification. For the amplification strategy, we observed a linear response over a 3-log concentration range; the assay’s high dynamic range is advantageous for quantification of miRNAs, which are known to show a wide range of abundance30, 49.
Figure 4. Application to highly sensitive miRNA detection.

(a) Schematic of miRNA detection using direct tagging or amplification strategies. (b-d) Calibration curves for quantification of three different miRNA species. Orange and blue dotted lines represent the limit of detection (LOD) of direct tagging (SAPE) and amplification (enzyme) schemes, respectively. Solid lines show best fits for the data (least-squares linear fit of log-transformed values). Amplification factors are 12 for let7a (b), 11 for miR145 (c), and 8 for miR221 (d). Our platform for particle isolation within droplets enables the use of the amplification strategy, resulting in one order of magnitude improved LOD compared to direct tagging.
Depending on the miRNA species, the amplification factor showed some variation (Fig. 4b-d, amplification factor = 12 for let-7a, 11 for miR145, and 8 for miR221). For the three different miRNA species tested, we could conclude that enzymatic amplification improved the LOD one order of magnitude compared to direct tagging. The LODs for the enzyme amplified assays were on the order of 0.1 amol for the three miRNAs tested. In the case of let-7a, the amplification strategy’s LOD of 0.14 amol is equivalent to the endogenous amount in ~70 MCF10A breast cells, or ~40 MCF7 breast cancer cells50. In the future, to approach single cell level detection, further improvement in LOD can be achieved by decreasing the total surface area of the microparticles35. For instance, as the particle number and the length scale of the particles decreases five and three times respectively, it should be possible to achieve single cell analysis.
To demonstrate multiplexing capabilities, we encoded particles by using mask-patterned ultraviolet light during particle synthesis by SFL. At the center of each particle, there is a right, non-isosceles triangle, which has chirality, indicating the direction of decoding. Eight bit-coding spots surround this triangle, providing 28 encoding capacity (Fig. 5a). It is worth noting that these codes could be successfully identified because particles were not deformed after isolation by using back-flow of FC-40. Three distinctive dysregulation patterns were detected using three encoded particles containing different miRNA probes (Fig. 5b, enlarged image is shown in figure S11; figure 5c shows low magnification images of the first case of figure 5b). Each dysregulation pattern demonstrated here represents a potential disease profile. For example, the first dysregulation pattern shown in figure 5b (increased let-7a and miR145 expression, decreased miR221 expression) has been observed in metabolic diseases including type 2 diabetes mellitus and obesity51–54. Although the relative expression of different miRNAs may vary between studies due to differences in cell type, patient cohort, and disease stage or subtype, the three miRNA species we studied (let-7a, miR145, and miR221) have also been shown to have diagnostic value for prostate cancer55 and triple negative breast cancer56. In figure 5, + sign represents 1 amol of spiked-in target, an amount which is nearly undetectable by direct tagging, but is clearly distinguishable from negative controls (- sign) in our demonstration. In this demonstration, we take advantage of the excellent target-capturing capabilities of hydrogel microparticles, and the product-accumulating capabilities of isolated droplets. We showcase how our platform can be used as a general strategy to improve the sensitivity of bioassays through enzymatic amplification without compromising target specificity, reproducibility, multiplexing capabilities, and dynamic range.
Figure 5. Multiplexed miRNA assay.

(a) Schematic of decoding of shape-encoded microparticles. Particles used for detection of each target miRNA have distinct codes. (b) Multiplexed miRNA assays for three distinctive dysregulation patterns. + and – signs represent 1 amol of spiked-in target and the negative control, respectively. (c) Bright field and fluorescent images of a microfluidic channel for the dysregulation pattern presented in the first row of figure 4b (let7a: +, miR145: +, miR221: -). Scale bars are 50 (b) and 200 µm (c).
Conclusion
Our microfluidic platform enables one to park particles in traps and isolate them in droplets with high yield for both parking and isolation, while eliminating droplet size heterogeneity and particle deformation. The hydrodynamic driving force is designed to fulfill the optimal requirements for particle arrangement, resulting in high parking yield and a single particle in each trap. Since we first allocate one particle per trap, we can overcome the statistics in encapsulation in the following isolation step. Scaling theory rationalizes the back-flow of oil, which ensures homogeneous droplet size and no deformation of encapsulated particles. The setup and operation of the entire process is simple and robust, and does not require cumbersome flow control in contrast to conventional droplet production methods. Our platform provides a method to integrate particle-based miRNA detection with an enzyme amplification scheme, to create a promising candidate for point-of-care diagnostics. Our system achieves one order of magnitude improved sensitivity compared to the competing direct tagging, particle-based strategy, without compromising other clinical requirements, such as multiplexing capabilities, specificity, reproducibility, and dynamic range. The same amplification strategy can be also be applied to other particle-based assays for protein57 or DNA detection37. Through integration with an automated microscope stage, our platform can also achieve fully automated interrogation as a result of fixed particle locations, and particle shape-based encoding. The design principle of our platform is universal and can be applied to objects of any length scale, chemical composition, or elastic modulus. In addition, devices are fabricated by simple molding methods with inexpensive materials (PDMS). Thus, we envision that this universal platform can be used with other soft, biological entities42 and find utility in diverse biomedical applications.
Supplementary Material
Acknowledgments
We gratefully acknowledge funding from NIH-NCI Grant 5R21CA177393-02, a Samsung Scholarship to J.J.K., and an NSERC fellowship to L.C. Microfabrication was performed at MTL, MIT. We also thank A. Tentori, S. Shapiro, M. Nagarajan, and A. Gupta at MIT for insightful discussions.
References
- 1.Brouzes E, Medkova M, Savenelli N, Marran D, Twardowski M, Hutchison JB, Rothberg JM, Link DR, Perrimon N, Samuels ML. P Natl Acad Sci USA. 2009;106:14195–14200. doi: 10.1073/pnas.0903542106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amselem G, Guermonprez C, Drogue B, Michelin S, Baroud CN. Lab Chip. 2016;16:4200–4211. doi: 10.1039/c6lc00968a. [DOI] [PubMed] [Google Scholar]
- 3.Bithi SS, Vanapalli SA. Sci Rep-Uk. 2017;7 doi: 10.1038/srep41707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bose S, Wan Z, Carr A, Rizvi AH, Vieira G, Pe'er D, Sims PA. Genome biology. 2015;16:120. doi: 10.1186/s13059-015-0684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han HS, Cantalupo PG, Rotem A, Cockrell SK, Carbonnaux M, Pipas JM, Weitz DA. Angew Chem Int Edit. 2015;54:13985–13988. doi: 10.1002/anie.201507047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell EP, Randall JD, Provuncher GK, Walt DR, Duffy DC. Nature biotechnology. 2010;28:595–599. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H, Srinivas RL, Gupta A, Doyle PS. Angewandte Chemie. 2015;54:2477–2481. doi: 10.1002/anie.201409489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thorsen T, Roberts RW, Arnold FH, Quake SR. Physical review letters. 2001;86:4163–4166. doi: 10.1103/PhysRevLett.86.4163. [DOI] [PubMed] [Google Scholar]
- 10.Anna SL, Bontoux N, Stone HA. Appl Phys Lett. 2003;82:364–366. [Google Scholar]
- 11.Franke T, Abate AR, Weitz DA, Wixforth A. Lab Chip. 2009;9:2625–2627. doi: 10.1039/b906819h. [DOI] [PubMed] [Google Scholar]
- 12.Ahn K, Kerbage C, Hunt TP, Westervelt RM, Link DR, Weitz DA. Appl Phys Lett. 2006;88 [Google Scholar]
- 13.Chabert M, Viovy JL. Proc Natl Acad Sci U S A. 2008;105:3191–3196. doi: 10.1073/pnas.0708321105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi WW, Qin JH, Ye NN, Lin BC. Lab Chip. 2008;8:1432–1435. doi: 10.1039/b808753a. [DOI] [PubMed] [Google Scholar]
- 15.Schmitz CHJ, Rowat AC, Koster S, Weitz DA. Lab Chip. 2009;9:44–49. doi: 10.1039/b809670h. [DOI] [PubMed] [Google Scholar]
- 16.Huebner A, Bratton D, Whyte G, Yang M, deMello AJ, Abell C, Hollfelder F. Lab Chip. 2009;9:692–698. doi: 10.1039/b813709a. [DOI] [PubMed] [Google Scholar]
- 17.Korczyk PM, Derzsi L, Jakiela S, Garstecki P. Lab Chip. 2013;13:4096–4102. doi: 10.1039/c3lc50347j. [DOI] [PubMed] [Google Scholar]
- 18.Abbyad P, Dangla R, Alexandrou A, Baroud CN. Lab Chip. 2011;11:813–821. doi: 10.1039/c0lc00104j. [DOI] [PubMed] [Google Scholar]
- 19.Sun M, Bithi SS, Vanapalli SA. Lab Chip. 2011;11:3949–3952. doi: 10.1039/c1lc20709a. [DOI] [PubMed] [Google Scholar]
- 20.Gumuscu B, Bomer JG, van den Berg A, Eijkel JCT. Lab Chip. 2015;15:664–667. doi: 10.1039/c4lc01350f. [DOI] [PubMed] [Google Scholar]
- 21.Abate AR, Chen CH, Agresti JJ, Weitz DA. Lab Chip. 2009;9:2628–2631. doi: 10.1039/b909386a. [DOI] [PubMed] [Google Scholar]
- 22.Edd JF, Di Carlo D, Humphry KJ, Koster S, Irimia D, Weitz DA, Toner M. Lab Chip. 2008;8:1262–1264. doi: 10.1039/b805456h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kemna EWM, Schoeman RM, Wolbers F, Vermes I, Weitz DA, van den Berg A. Lab Chip. 2012;12:2881–2887. doi: 10.1039/c2lc00013j. [DOI] [PubMed] [Google Scholar]
- 24.Calin GA, Croce CM. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 25.Melo SA, Esteller M. Febs Lett. 2011;585:2087–2099. doi: 10.1016/j.febslet.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 26.Chen PS, Su JL, Hung MC. J Biomed Sci. 2012;19 doi: 10.1186/1423-0127-19-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pandey AK, Agarwal P, Kaur K, Datta M. Cell Physiol Biochem. 2009;23:221–232. doi: 10.1159/000218169. [DOI] [PubMed] [Google Scholar]
- 28.Small EM, Olson EN. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thum T, Mayr M. Cardiovasc Res. 2012;93:543–544. doi: 10.1093/cvr/cvs085. [DOI] [PubMed] [Google Scholar]
- 30.Pritchard CC, Cheng HH, Tewari M. Nat Rev Genet. 2012;13:358–369. doi: 10.1038/nrg3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chugh P, Dittmer DP. Wires Rna. 2012;3:601–616. doi: 10.1002/wrna.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker M. Nat Methods. 2010;7:687–692. doi: 10.1038/nmeth0910-687. [DOI] [PubMed] [Google Scholar]
- 33.Chapin SC, Appleyard DC, Pregibon DC, Doyle PS. Angew Chem Int Edit. 2011;50:2289–2293. doi: 10.1002/anie.201006523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee H, Shapiro SJ, Chapin SC, Doyle PS. Anal Chem. 2016;88:3075–3081. doi: 10.1021/acs.analchem.5b03902. [DOI] [PubMed] [Google Scholar]
- 35.Pregibon DC, Doyle PS. Anal Chem. 2009;81:4873–4881. doi: 10.1021/ac9005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dendukuri D, Pregibon DC, Collins J, Hatton TA, Doyle PS. Nat Mater. 2006;5:365–369. doi: 10.1038/nmat1617. [DOI] [PubMed] [Google Scholar]
- 37.Pregibon DC, Toner M, Doyle PS. Science. 2007;315:1393–1396. doi: 10.1126/science.1134929. [DOI] [PubMed] [Google Scholar]
- 38.Srinivas RL, Johnson SD, Doyle PS. Anal Chem. 2013;85:12099–12107. doi: 10.1021/ac403201p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chapin SC, Appleyard DC, Pregibon DC, Doyle PS. Angewandte Chemie. 2011;50:2289–2293. doi: 10.1002/anie.201006523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chung K, Rivet CA, Kemp ML, Lu H. Anal Chem. 2011;83:7044–7052. doi: 10.1021/ac2011153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sochol RD, Dueck ME, Li S, Lee LP, Lin L. Lab Chip. 2012;12:5051–5056. doi: 10.1039/c2lc40704c. [DOI] [PubMed] [Google Scholar]
- 42.Sauzade M, Brouzes E. Lab Chip. 2017;17:2186–2192. doi: 10.1039/c7lc00283a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teshima T, Ishihara H, Iwai K, Adachi A, Takeuchi S. Lab Chip. 2010;10:2443–2448. doi: 10.1039/c004986g. [DOI] [PubMed] [Google Scholar]
- 44.Yeom SY, Son CH, Kim BS, Tag SH, Nam E, Shin H, Kim SH, Gang H, Lee HJ, Choi J, Im HI, Cho IJ, Choi N. Anal Chem. 2016;88:4259–4268. doi: 10.1021/acs.analchem.5b04190. [DOI] [PubMed] [Google Scholar]
- 45.Kim JJ, Bong KW, Reategui E, Irimia D, Doyle PS. Nat Mater. 2017;16:139–146. doi: 10.1038/nmat4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Akagi J, Khoshmanesh K, Evans B, Hall CJ, Crosier KE, Cooper JM, Crosier PS, Wlodkowic D. PloS one. 2012;7:e36630. doi: 10.1371/journal.pone.0036630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung K, Kim Y, Kanodia JS, Gong E, Shvartsman SY, Lu H. Nat Methods. 2011;8:171–176. doi: 10.1038/nmeth.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yauk CL, Rowan-Carroll A, Stead JDH, Williams A. Bmc Genomics. 2010;11 doi: 10.1186/1471-2164-11-330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liang Y, Ridzon D, Wong L, Chen CF. Bmc Genomics. 2007;8 doi: 10.1186/1471-2164-8-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guzman N, Agarwal K, Asthagiri D, Yu LB, Saji M, Ringel MD, Paulaitis ME. Mol Cancer Res. 2015;13:891–901. doi: 10.1158/1541-7786.MCR-14-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Deiuliis JA. Int J Obesity. 2016;40:88–101. doi: 10.1038/ijo.2015.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lorente-Cebrian S, Mejhert N, Kulyte A, Laurencikiene J, Astrom G, Heden P, Ryden M, Arner P. PloS one. 2014;9 doi: 10.1371/journal.pone.0086800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jiang LQ, Franck N, Egan B, Sjogren RJO, Katayama M, Duque-Guimaraes D, Arner P, Zierath JR, Krook A. Am J Physiol-Endoc M. 2013;305:E1359–E1366. doi: 10.1152/ajpendo.00236.2013. [DOI] [PubMed] [Google Scholar]
- 54.Chou WW, Wang YT, Liao YC, Chuang SC, Wang SN, Juo SHH. Cell Physiol Biochem. 2013;32:127–137. doi: 10.1159/000350131. [DOI] [PubMed] [Google Scholar]
- 55.Bertoli G, Cava C, Castiglioni I. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17030421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thakur S, Grover RK, Gupta S, Yadav AK, Das BC. PloS one. 2016;11 doi: 10.1371/journal.pone.0158946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Srinivas RL, Chapin SC, Doyle PS. Anal Chem. 2011;83:9138–9145. doi: 10.1021/ac202335u. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.