

Abstract
The field of Ni-catalysed cross-coupling has seen rapid recent growth because of the low cost of Ni, its earth abundance, and its ability to promote unique cross-coupling reactions. Whereas advances in the related field of Pd-catalysed cross-coupling have been driven by ligand design, the development of ligands specifically for Ni has received minimal attention. Here, we disclose a class of phosphines that enable the Ni-catalysed Csp3 Suzuki coupling of acetals with boronic acids to generate benzylic ethers, a reaction that failed with known ligands for Ni and designer phosphines for Pd. Using parameters to quantify phosphine steric and electronic properties together with regression statistical analysis, we identify a model for ligand success. The study suggests that effective phosphines feature remote steric hindrance, a concept that could guide future ligand design tailored to Ni. Our analysis also reveals that two classic descriptors for ligand steric environment—cone angle and % buried volume—are not equivalent, despite their treatment in the literature.
Over the past 50 years, Pd-catalysed cross-coupling has evolved to become one of the most useful strategies for carbon–carbon (C–C) and C–heteroatom bond formation1. The development of new ligands for Pd has been arguably the most important contributor to the advancement of these methods, enabling reactions with a broad range of substrates under mild and efficient conditions2–4. By contrast, the field of nickel-catalysed cross-coupling has witnessed tremendous activity over the past two decades5, but minimal effort has been dedicated to the identification of new ligand sets (Fig. 1a)6–9. Furthermore, phosphines developed for Pd catalysis have generally proven ineffective for Ni (refs 10–12). According to the example set by Pd, the design of new ligands for Ni should facilitate the refinement of existing methods and the identification of new chemical transformations. Here, we report the development of a new class of phosphines and demonstrate that these bench-stable ligands facilitate a Ni-catalysed Csp3 Suzuki coupling reaction that failed with known ligand architectures for Ni and Pd. Quantitative molecular parameterization of these ligands and multivariate correlation with reaction outcome are conducted13,14. These studies reveal that two of the most frequently used parameters to describe ligand steric effects—cone angle and buried volume—are not equivalent, despite their reported treatment in the literature15,16. Indeed, it is precisely their difference that provides an intuitive as well as quantitative rationale for their success and for the failure of ligands developed for Pd. We expect that the new phosphines in this study will find application in other Ni-catalysed coupling reactions. Moreover, the insight gained from molecular parameterization should serve as a general design principle for the identification of new ligands tailored for Ni.
Figure 1. Design of new ligands for Ni catalysis enables Suzuki coupling of benzylic acetals.
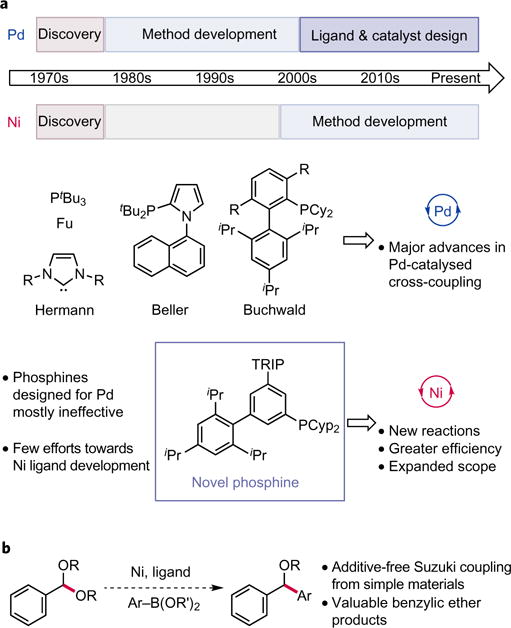
a, Ligand development in Pd versus Ni-catalysed cross-coupling. Ligands engineered for Pd catalysis have facilitated many advances in cross-coupling, including the discovery of altogether new transformations and the refinement of existing methods such that they can be used for large-scale synthesis. By contrast, minimal effort has been directed at ligand design for Ni-catalysed cross-coupling, despite the opportunity it presents for discovering novel reactions and for improving the efficiency of existing methods. b, Ligand design for Suzuki coupling of acetals. The development of a Ni-catalysed Suzuki coupling of acetals with readily available boronic acids would facilitate the preparation of valuable ether products by C–C bond formation. Application of known ligands for Ni and re-purposing ligands designed for Pd to this transformation were both unsuccessful in this regard, prompting the development and study of a new phosphine ligand class for Ni.
The field of Ni catalysis has recently witnessed rapid growth owing to the base metal’s low cost and unique properties compared to its precious-metal sibling, Pd. One of the most significant contributions of Ni catalysis to cross-coupling methodology has been enabling the use of substrates bearing abundant and relatively inert Csp2–O and Csp3–O bonds as electrophilic coupling partners17. In this context, our group reported Ni-catalysed Suzuki couplings of N,O-acetals and O,O-acetals that deliver 2-arylated heterocycles using triphenylphosphine (PPh3) as a ligand18,19. A mechanistic study suggested a pathway involving boroxine-assisted ionization of the acetals followed by Ni-catalysed coupling of the resulting iminium or oxocarbenium ions20. Accordingly, we were able to develop an asymmetric variant of the N,O-acetal coupling using a chiral phosphonite ligand for Ni (ref. 21). We recently considered whether this strategy could be expanded to offer a general Csp3–C bond-forming approach to ether synthesis from a broad range of readily available and stable acyclic and cyclic acetals in combination with boronic acids (Fig. 1b). This reaction would offer an expedient synthesis of benzylic ethers—highly valuable pharmacophores in medicinal chemistry22—which are most commonly constructed by C–O bond formation.
The coupling of benzaldehyde dimethyl acetal 1 with para-fluorophenyl boroxine was chosen as the model system for optimization (Fig. 2a). This coupling was anticipated to be more challenging than our previously described Suzuki coupling of chromene acetals, because the Csp3–O bond of chromene acetals experiences significant bond weakening due to conjugation, and addition of Ni to the benzopyrylium delivers a stabilized π-allyl intermediate20, both not possible with simple acetals. Indeed, application of the previously identified conditions using PPh3 L1 as ligand only yielded trace ether 2 at elevated temperature. By contrast, use of tricyclohexylphosphine (PCy3, L2), one of the most common phosphines used for Ni-catalysed cross-coupling5,17, afforded modest amounts of product (29% yield). More hindered trialkylphosphines (tert-butylphosphine, PtBu3, L3) were ineffective, as were N-heterocyclic carbenes (L4). Alkyl arylphosphines, which possess intermediate steric hindrance and electronic character to trialkylphosphines and triarylphosphines, were also evaluated. Unsubstituted alkyl diarylphosphines (cyclohexyl diphenylphosphine, PCyPh2 L9 and cyclopentyl diphenylphosphine, PCypPh2 L11) were less effective than L2. However, PCy2Ph (L5) delivered similar yields to PCy3, as did the less hindered cyclopentyl variant PCyp2Ph (L8). More decorated versions of these ligand architectures have been pioneered by the Buchwald laboratory for Pd-catalysed couplings2. Unfortunately, members of this class, such as JohnPhos L6 and XPhos L7, provided no detectable product. The low reactivity observed with ligands developed for Pd is not unique to this example and highlights the limitations of these ligands when used with Ni (refs 10–12). Because Ni is smaller and more nucleophilic than Pd, we questioned whether a novel ligand framework designed specifically for this base metal could overcome the limitations of current ligands and achieve efficient cross-coupling. Furthermore, we sought to obtain a rationale for the effect of phosphine structure on reactivity to facilitate the development of other Ni-catalysed couplings and the discovery of additional ligand frameworks for Ni.
Figure 2. Ligand evaluation and timepoint studies.
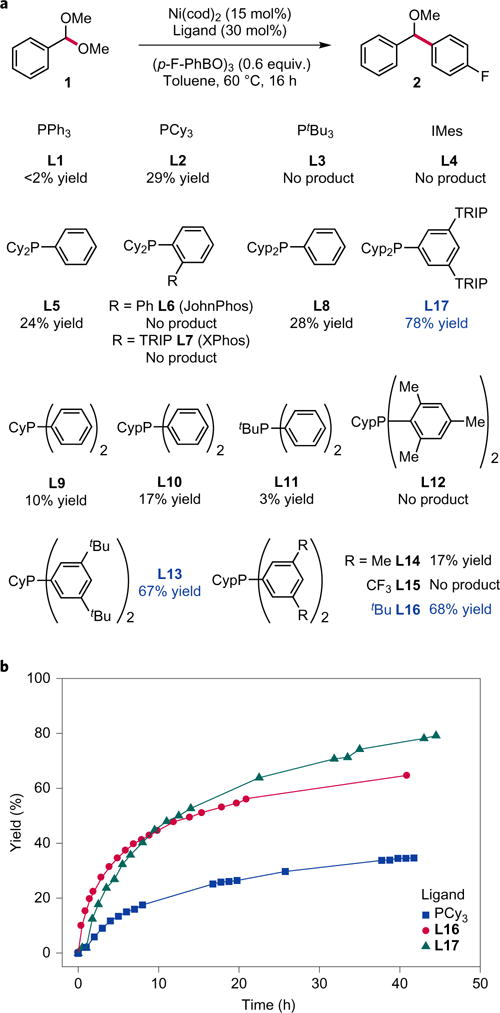
a, Screen of existing and novel phosphines for Ni. Ligand evaluation reveals that phosphines bearing tertiary alkyl groups or ortho-substituted aryl groups are completely ineffective. However, dialkylaryl [(alkyl)2PAr] and alkyldiaryl [(alkyl)PAr2] phosphines with secondary alkyl substituents and 3,5-substituted aryl groups are highly effective. IMes = 1,3-bis(2,4,6-trimethylphenyl)-imidazolium. Yields determined by 19F NMR with 2-fluorobiphenyl as a quantitative internal standard. b, Timepoint studies comparing the reaction profile with L16, L17 and PCy3 (L2). Ligands L16 and L17 afford highly active Ni catalysts in the Suzuki coupling; ligand L17 delivers the highest overall yields, despite a slower initial rate of reaction. Reactions were conducted at 60 °C in toluene (0.06 M), 15 mol% Ni(cod)2 and 30 mol% of ligand. cod = 1,5-cyclooctadiene.
We focused our efforts on modification of the aryl alkylphosphine architecture, because the parent members of this class provided modest reactivity and, unlike trialkylphosphines, this ligand scaffold is readily modifiable and tunable. A synthetic sequence that allowed facile access to these frameworks from readily available starting materials was developed (Supplementary Section C). Notably, many of the new phosphines shown in Fig. 2a were isolated on a gram scale as air-stable solids that could be stored on the bench for over a month without oxidation or decomposition.
We found that, within the alkyl diarylphosphine ((alkyl)PAr2) platform, 2-substitution on the aryl groups was detrimental to reactivity (L12), consistent with earlier observations using Buchwald ligands L6 and L7. Ligands with electron-withdrawing groups at the 3,5-positions were also ineffective (L15). In contrast, ligands bearing bulky, electron-donating substituents (tBu) at the 3,5-positions (L13, L16) afforded improved reactivity over the parent ligands (L9, L10), providing 67 and 68% yield of the desired ether product, respectively. We questioned whether inclusion of larger substituents at the 3,5-positions would further improve ligand performance. The 2,4,6-tri-isopropylphenyl (TRIP) group has proven of high utility in ligand and catalyst design as a very bulky substituent. For reasons of synthetic accessibility, we investigated modification of the (alkyl)2PAr framework with this substituent. Gratifyingly, the catalyst system using L17 demonstrated further improvement, delivering 2 in 78% yield.
The superior yields with L16 and L17 could be due to greater catalyst stability, improved rates of reaction, or both. To probe this question, NMR timepoint experiments were conducted to qualitatively assess reaction progress with various ligands (Fig. 2b). Ligands L16 and L17 exhibited greater rates than L2, thus suggesting these ligands generate more active Ni catalysts. Furthermore, these time-point studies illustrate that the catalyst system with L17 affords superior yields to L16, despite having a lower initial rate and an induction period.
Next, we sought to understand what structural features of the new ligands are responsible for their significantly improved rate and catalyst stability. In particular, we sought to quantify the steric and electronic properties of these ligands using numerical parameters and search for statistical correlations with reaction yield. The electronic properties of the ligands were evaluated using the computationally derived minimum electrostatic potential (Vmin) parameter, which correlates with the Tolman electronic parameter23. No strong correlation with yield was observed using Vmin alone (r2 = 0.18, Supplementary Fig. 2); notably, L16 and L17 have only slightly greater Vmin values than the parent scaffolds L6 and L4, respectively. The steric properties of the ligands were also quantified and analysed. To achieve internal consistency, all steric parameters were computed from molecular mechanics models as opposed to using literature values (Supplementary Section H). The first steric parameter examined was Tolman’s cone angle metric15,24. Although the cone angle correlated strongly with yield for a range of ligands (circles in Fig. 3a), JohnPhos, XPhos, PtBu3 and L12 were notable outliers (triangles). Another parameter often used to quantify ligand steric effects is buried volume (%Vbur)16,25. A qualitative trend readily became apparent with this parameter: ligands with large buried volumes are ineffective. The plot of %Vbur in Fig. 3a illustrates that a ligand possessing a high buried volume is inactive, regardless of its cone angle (for example, JohnPhos, L12). Notably, cone angle and %Vbur have been considered as equivalent measures of ligand steric size and used interchangeably in the literature16. The divergence in the parameter–reactivity relationships shown in Fig. 3a provides a striking example showing how these two metrics are far from equivalent (Fig. 3b). We sought to understand the origin of this discrepancy and how it translates to a structure-based insight into ligand effectiveness.
Figure 3. Steric parameterization and analysis.
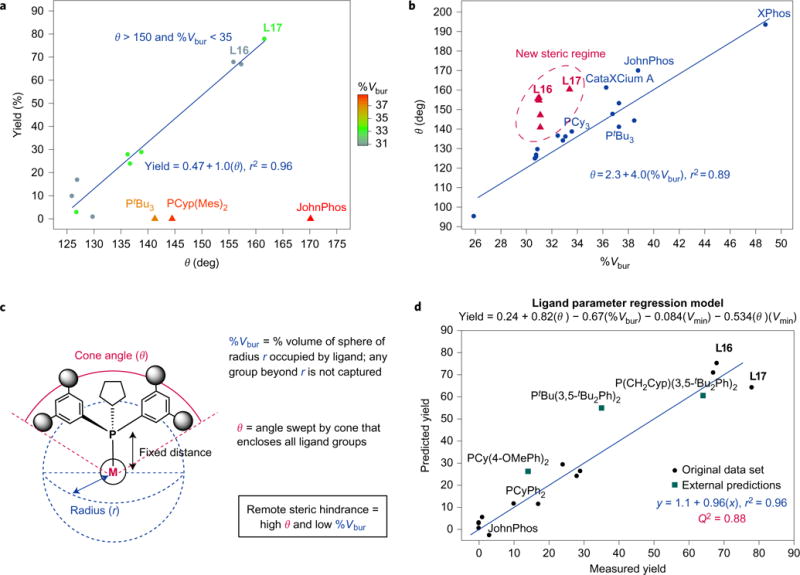
a, Plot of yield versus cone angle (θ) and buried volume (%Vbur). A comparison of the ligand steric parameters against reaction yield reveals that larger cone angle correlates with higher yield (circles), with the exception of ligands featuring high buried volume (triangles). The regression equation only includes the circle data points. b, Plot of cone angle versus buried volume. Cone angle and buried volume have previously been treated as equivalent measures of ligand steric environment, as illustrated by a 1:1 mapping of the two parameters (circle data points). The regression equation only includes circle data points. The new phosphines prepared in this study (triangles) fall outside this 1:1 mapping, highlighting that these two terms are not equivalent, by definition. c, Definition of cone angle and buried volume. Both measure steric encumbrance but with different distance dependencies. Buried volume only accounts for steric hindrance proximal to the metal whereas cone angle is sensitive to ligand size at a distance. A ligand with a large cone angle and low %Vbur, such as those most successful in this new method, therefore possesses remote steric hindrance. d, Plot of predicted yield versus actual yield. A quantitative model to relate cone angle, buried volume and Vmin to yield was obtained by regression analysis. This model accounts for the entire scope of ligands tested in the study and features a mathematical representation of the remote steric effect concept. We show that the model can predict the performance of ligands (green squares) that feature unique structural motifs to those included in the ligand training set.
The difference between these two steric parameters can be appreciated through an understanding of the distinct methods each uses to describe ligand size. The buried volume metric emphasizes steric hindrance proximal to the metal by measuring the volume occupied by the ligand in a sphere of defined radius (r = 3.5 Å, Fig. 3c). By contrast, the cone angle parameter measures a cone that encloses all groups of the ligand, regardless of how far they are from the metal, and is therefore sensitive to ligand size, even at a distance. Because most ligands have operated in the first coordination sphere, a correlation between the two parameters has been observed; however, this correlation is not required by definition. Taken together, ligands with small buried volume and large cone angle are the ones that feature remote steric hindrance; they are relatively unencumbered near the metal binding site but have large groups distant from the metal. Given the smaller size of Ni relative to Pd and the shorter Ni–P bond distance (2.05 versus 2.28 Å), it makes intuitive sense that sterically hindered phosphines designed for Pd catalysis would crowd a Ni centre, preventing critical substrate or even ligand-binding events. In contrast, new ligands L16 and L17 probably permit coordination of reaction components to Ni while still discouraging the binding of multiple ligand equivalents and preventing bimetallic deactivation mechanisms26. Additional studies are currently ongoing to investigate the specific influence ligands L16 and L17 impart on catalyst structure and activity.
Going further, a quantitative model to relate the cone angle and %Vbur with yield was developed to explain the outliers and provide quantitative support for the remote steric effect hypothesis (Supplementary Section G). A positive correlation with cone angle was observed, as well as an inverse correlation with the buried volume (Fig. 3d). Taken together, these two terms are a mathematical representation of the remote steric effect concept. The function can be visualized by plotting predicted versus measured yield (Fig. 3d). A slope approaching one and a small intercept indicate the model is accurate, while an r2 value of 0.96 demonstrates the model’s precision. Notably, the model correctly accounts for the entire scope of ligands tested in the study, including ligands with high buried volume (JohnPhos, PtBu3), whereas they fell as outliers in the simple yield versus cone angle correlation (Fig. 3a). Leave-one-out cross-validation of the model was also performed. A Q2 value of 0.88 indicates that the model has potential predictive ability on a new data set, a feature made more attractive given that all of the ligand parameters are generated computationally rather than obtained from experiment or literature. Indeed, a preliminary evaluation suggests that the model can predict the performance of ligands (Fig. 3d, squares) across a range of structures, including two ligands possessing substituents not present in the training set and a third ligand predicted and found to be active despite its bearing a tert-butyl substituent that rendered all ligands in the training set inactive.
Having arrived at an enabling ligand, the scope of boroxine nucleophiles was then evaluated (Table 1). Both electron-deficient (2,3) and electron-neutral (5) phenylboroxines afforded ethers in good to excellent yields, whereas electron-rich phenylboroxines (4) delivered modest reaction efficiency, presumably due to their attenuated Lewis acidity20. Carbonyl-functionalized (6) and sterically hindered (8) boroxines are competent coupling partners. Furthermore, ligand L17 induces good reaction efficiency with heterocyclic boroxines (9), as long as the heterocycle is not strongly Lewis basic. Acetal scope investigations also revealed tolerance for electronic and steric variation. Ortho-substitution on the benzaldehyde was well tolerated under the reaction conditions (10). Both electron-rich (11) and electron-neutral (12) acetals were also compatible; the former affords a synthetic alternative to the limitation of using electron-rich aryl boroxines described above. Unfortunately, electron-deficient acetals did not undergo coupling. This result, taken together with the ineffectiveness of electron-rich boroxines, is consistent with a mechanism involving boronic acid-assisted ionization of the acetal followed by oxidative addition to the resulting oxocarbenium intermediate20. Notably, no over-arylation of the ether products to give triarylalkanes is observed, even though Jarvo and Shi have reported Ni-catalysed couplings of benzylic ethers related to the products in Table 1 (refs 27,28).
Table 1.
Scope investigation.
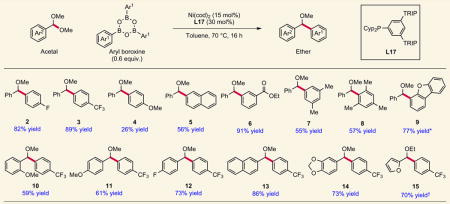
|
A range of aryl boroxines undergo cross-coupling with dimethyl acetals derived from benzaldehydes. Yields shown are isolated yields. *Run at 60 °C. †2-furan diethyl acetal used.
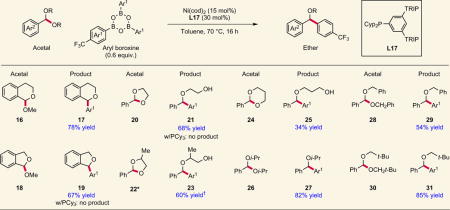
More complex substrates, including exo- and endocyclic, bidentate and hindered acetals, also underwent coupling to provide several important structural motifs (Table 2). Products 17 and 19 are of particular interest because they represent the structural cores of important pharmaceutical compounds29. Notably, phthalan and dioxolane acetals 18 and 20, which were completely unreactive with PCy3, undergo C–C bond formation using L17 to afford α-arylated ethers 19 and 21 in good yield. The products 21 and 23 possess an alcohol tether capable of undergoing further functionalization, including serving as a directing group for additional Ni-catalysed coupling to give bioactive triarylmethane products30,31. Acetal 22 (1.3:1 d.r.) reacts to generate 23 in a 2:1 mixture of regioisomers, with the major regioisomer (3.8:1 d.r.) derived from functionalization of the less substituted C–O bond. Compared with the five-membered cyclic acetals 20 and 22, dioxane acetal 24 was less reactive. Perhaps most notably, acetals derived from sterically hindered alcohols (22, 26, 30) deliver products in excellent yield that could not be easily obtained through most C–O bond-forming methods32. Although acid-catalysed approaches to these products exist, they are typically limited to methyl ether formation from electron-rich or neutral benzhydrols and require strong acid and forcing conditions33. Furthermore, our laboratory previously described a reductive cross-coupling method for the synthesis of this same product class from acetals and aryl iodides using a stoichiometric reductant and a potent Lewis acid34. This reductive approach failed with substituted acetals such as 22, 26, 28 and 30, highlighting a major strategic advantage of this new method.
Table 2.
Scope investigation with more complex acetal substrates.
|
Several exocyclic and endocyclic acetals undergo C–C bond formation to afford valuable cyclic and acyclic ethers. Access to sterically hindered ethers is also possible using this method. In the cases examined, the new phosphine framework is exclusively effective. PCy3, a ligand that delivered low but measurable yield in the model reaction, is completely inactive, highlighting the impact of ligand development on reaction discovery. Yields shown are isolated yields. *1.3:1 mixture of diastereomers. †Run at 85 °C; isolated as a 2:1 mixture of regioisomers (major regioisomer shown).
In conclusion, we have developed a novel class of aryl alkylphosphines containing bulky groups at the 3,5-positions (L17) that confer high activity upon Ni catalysts for the Suzuki coupling of benzylic acetals. Parameterization and modelling studies reveal that the effectiveness of these ligands is a function of remote steric hindrance, a structural concept relatively unexplored in ligand design. We reveal a divergence between the cone angle and buried volume (%Vbur) parameters, two measures of steric size previously considered to be equivalent. Whereas %Vbur only describes steric hindrance in the metal’s first coordination sphere, cone angle captures it beyond the immediate proximity of the metal. We show that the two can be used in conjunction to develop a quantitative model for predicting ligand reactivity. We believe this new ligand architecture and the concept of remote steric hindrance will lead to significant advances in both Ni catalysis and ligand design.
Supplementary Material
Acknowledgments
The authors acknowledge financial support from NIGMS (R01 GM100985). The authors also thank M.S. Sigman (University of Utah) for helpful suggestions on this manuscript and B. Shields, T. Graham (Merck) and D. Ahneman for discussions.
Footnotes
Data availability
Crystallographic data have been deposited at the Cambridge Crystallographic Data Centre (CCDC) as CCDC 1520891 (S-1) and can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/getstructures. Procedures, code and data for statistical analysis are available in the Supplementary Information at http://www.nature.com/nchem.
Author contributions
K.W. and A.G.D. conceived the work. K.W. performed and analysed the experiments and calculations. K.W. and A.G.D. designed the experiments, analysed the data and co-wrote the paper.
Additional information
Supplementary information and chemical compound information are available in the online version of the paper. Correspondence and requests for materials should be addressed to A.G.D.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Johansson Seechurn CCC, Kitching MO, Colacot TJ, Snieckus V. Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 nobel prize. Angew Chem Int Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 2.Martin R, Buchwald SL. Palladium-catalyzed Suzuki–Miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fu GC. The development of versatile methods for palladium-catalyzed coupling reactions of aryl electrophiles through the use of P(t- Bu)3 and PCy3 as ligands. Acc Chem Res. 2008;41:1555–1564. doi: 10.1021/ar800148f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marion N, Nolan SP. Well-defined N-heterocyclic carbenes–palladium(II) precatalysts for cross-coupling reactions. Acc Chem Res. 2008;41:1440–1449. doi: 10.1021/ar800020y. [DOI] [PubMed] [Google Scholar]
- 5.Tasker SZ, Standley EA, Jamison TF. Recent advances in homogeneous nickel catalysis. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zultanski SL, Fu GC. Catalytic asymmetric γ-alkylation of carbonyl compounds via stereoconvergent Suzuki cross-couplings. J Am Chem Soc. 2011;133:15362–15364. doi: 10.1021/ja2079515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tobisu M, Yasutome A, Kinuta H, Nakamura K, Chatani N. 1,3-Dicyclohexylimidazol-2-ylidene as a superior ligand for the nickel-catalyzed cross-couplings of aryl and benzyl methyl ethers with organoboron reagents. Org Lett. 2014;16:5572–5575. doi: 10.1021/ol502583h. [DOI] [PubMed] [Google Scholar]
- 8.Lavoie CM, et al. Challenging nickel-catalysed amine arylations enabled by tailored ancillary ligand design. Nat Commun. 2016;7:11073. doi: 10.1038/ncomms11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hansen EC, et al. New ligands for nickel catalysis from diverse pharmaceutical heterocycle libraries. Nat Chem. 2016;8:1126–1130. doi: 10.1038/nchem.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nielsen DK, Doyle AG. Nickel-catalyzed cross-coupling of styrenyl epoxides with boronic acids. Angew Chem Int Ed. 2011;50:6056–6059. doi: 10.1002/anie.201101191. [DOI] [PubMed] [Google Scholar]
- 11.Hie L, et al. Conversion of amides to esters by the nickel-catalysed activation of amide C–N bonds. Nature. 2015;524:79–83. doi: 10.1038/nature14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao X. Nickel-catalyzed arylation of heteroaryl-containing diarylmethanes: exceptional reactivity of the Ni IX-based catalyst. Chem Sci. 2016;7:611–618. doi: 10.1039/c5sc03704b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harper KC, Sigman MS. Three-dimensional correlation of steric and electronic free energy relationships guides asymmetric propargylation. Science. 2011;333:1875–1878. doi: 10.1126/science.1206997. [DOI] [PubMed] [Google Scholar]
- 14.Milo A, Bess EN, Sigman MS. Interrogating selectivity in catalysis using molecular vibrations. Nature. 2014;507:210–214. doi: 10.1038/nature13019. [DOI] [PubMed] [Google Scholar]
- 15.Tolman CA. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem Rev. 1977;77:313–348. [Google Scholar]
- 16.Clavier H, Nolan SP. Percent buried volume for phosphine and N- heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem Commun. 2010;46:841–861. doi: 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]
- 17.Rosen BM, et al. Nickel-catalyzed cross-couplings involving carbon–oxygen bonds. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graham TJA, Shields JD, Doyle AG. Transition metal-catalyzed cross coupling with N-acyliminium ions derived from quinolines and isoquinolines. Chem Sci. 2011;2:980–984. [Google Scholar]
- 19.Graham TJA, Doyle AG. Nickel-catalyzed cross-coupling of chromene acetals and boronic acids. Org Lett. 2012;14:1616–1619. doi: 10.1021/ol300364s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sylvester KT, Wu K, Doyle AG. Mechanistic investigation of the nickel-catalyzed Suzuki reaction of N,O-acetals: evidence for boronic acid assisted oxidative addition and an iminium activation pathway. J Am Chem Soc. 2012;134:16967–16970. doi: 10.1021/ja3079362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shields JD, Ahneman DT, Doyle AG. Enantioselective, nickel-catalyzed Suzuki cross-coupling of quinolinium ions. Org Lett. 2014;16:142–145. doi: 10.1021/ol4031364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ameen D, Snape TJ. Chiral 11-diaryl compounds as important pharmacophores. Med Chem Commun. 2013;4:893–907. [Google Scholar]
- 23.Suresh CH, Koga N. Quantifying the electronic effect of substituted phosphine ligands via molecular electrostatic potential. Inorg Chem. 2002;41:1573–1578. doi: 10.1021/ic0109400. [DOI] [PubMed] [Google Scholar]
- 24.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. University Science: 2010. [Google Scholar]
- 25.Poater A, et al. Sambvca: a Web application for the calculation of the buried volume of N-heterocyclic carbene ligands. Eur J Inorg Chem. 2009;2009:1759–1766. [Google Scholar]
- 26.Barder TE, Walker SD, Martinelli JR, Buchwald SL. Catalysts for Suzuki–Miyaura coupling processes: scope and studies of the effect of ligand structure. J Am Chem Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 27.Taylor BLH, Harris MR, Jarvo ER. Synthesis of enantioenriched triarylmethanes by stereospecific cross-coupling reactions. Angew Chem Int Ed. 2012;51:7790–7793. doi: 10.1002/anie.201202527. [DOI] [PubMed] [Google Scholar]
- 28.Guan BT, et al. Direct benzylic alkylation via Ni-catalyzed selective benzylic sp3 C−O activation. J Am Chem Soc. 2008;130:3268–3269. doi: 10.1021/ja710944j. [DOI] [PubMed] [Google Scholar]
- 29.Eildal JNN, et al. From the selective serotonin transporter inhibitor citalopram to the selective norepinephrine transporter inhibitor talopram: synthesis and structure–activity relationship studies. J Med Chem. 2008;51:3045–3048. doi: 10.1021/jm701602g. [DOI] [PubMed] [Google Scholar]
- 30.Greene MA, Yonova IM, Williams FJ, Jarvo ER. Traceless directing group for stereospecific nickel-catalyzed alkyl–alkyl cross-coupling reactions. Org Lett. 2012;14:4293–4296. doi: 10.1021/ol300891k. [DOI] [PubMed] [Google Scholar]
- 31.Tollefson EJ, Hanna LE, Jarvo ER. Stereospecific nickel-catalyzed cross-coupling reactions of benzylic ethers and esters. Acc Chem Res. 2015;48:2344–2353. doi: 10.1021/acs.accounts.5b00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williamson AXLV. Theory of ætherification. Philos Mag Series 3. 1850;37:350–356. [Google Scholar]
- 33.Fuson RC, Jackson HL. A comparison of certain dimesitylmethyl derivatives with the corresponding triarylmethyl compounds. J Am Chem Soc. 1950;72:351–353. [Google Scholar]
- 34.Arendt KM, Doyle AG. Dialkyl ether formation by nickel-catalyzed cross-coupling of acetals and aryl iodides. Angew Chem Int Ed. 2015;54:9876–9880. doi: 10.1002/anie.201503936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.