

Abstract
Background
The dominant feature in neurodegenerative diseases is protein aggregations that lead to neuronal loss. Immunotherapies using antibodies or antibody fragments to target the aggregations are a highly perused approach. The molecular mechanisms underlying the amyloid-based immunotherapy are complex. Deciphering the properties of amyloidogenic proteins responsible for these diseases is essential to obtain insights into antibody recognition of the amyloid antigens.
Scope of Review
We systematically explore all available crystal structures of antibody-amyloid complexes related to neurodegenerative diseases, including antibodies that recognize the Aβ peptide, tau protein, prion protein, alpha-synuclein, huntingtin protein (mHTT), and polyglutamine.
Major Conclusions
We found that antibodies mostly use the conformational selection mechanism to recognize the highly flexible amyloid antigens. In particular, solanezumab bound to Aβ12–28 tripeptide motif conformation (F19F20A21), which is shared with the Aβ42 fibril. This motif, which is trapped by the antibody, may provide the missing link in amyloid formation. Water molecules often bridge between the antibody and amyloid, contributing to the recognition.
General Significance
This paper provides the structural basis for antibody recognition of amyloidogenic proteins. The analysis and discussion of known structures are expected to help in the design and optimization of antibodies in neurodegenerative diseases.
Keywords: Antibody, biological drug, amyloid, neurodegenerative diseases, Alzheimer’s disease, prion, antigen
Graphical Abstract
Introduction
Neurodegenerative diseases are caused by the progressive loss of structure and function of neurons, including neuronal death. Commonly studied NDs include Alzheimer (AD), Parkinson, Huntington, prion (“mad cow”) diseases, and Down’s syndrome (DS).[1–5] The dominant common feature is protein aggregation that leads to neuronal loss. For example, AD is characterized by the coexistence of the extracellular senile plaques of amyloid-β (Aβ) and the intracellular neurofibrillary tangles of tau protein[6]. Increasing evidence from studies in human, transgenic mice, cultured cells, wild type rodent and in vitro systems indicates that soluble oligomers of amyloidogenic proteins are responsible for amyloidosis[7, 8] and are the toxic agent.[9–11] Some data suggest that large aggregates can also lead to cytotoxicity [12, 13]. Protein aggregation leads to a complex, integrated pathophysiology in each of these diseases and is commensurate with loss of homeostatic regulation, including immune response, metabolic changes, synaptic loss, and neuronal death. Neurodegeneration is associated with a break-down of the blood-brain barrier (BBB) and/or blood spinal cord barrier (BSCB) which enables peripheral immune cells to infiltrate the central nervous system, further contributing to homeostatic dysregulation in the affected tissues.[14]
Immunotherapies using antibodies and antibody fragments to target the protein aggregation in neurodegenerative disease represent a highly used approach for the disease-modifying treatment.[15] Among these neurodegenerative diseases, the prion disease has long been recognized to have a protein only self-propagation infectious mechanism. Anti-prion antibodies and new vaccines have been tested over more than decade, aiming to break the immune tolerance to the prion protein, [16] including an antibody that can specifically neutralize PrPSc.[17] It now appears that similar prion-like mechanisms of self-propagation may underlie other neurodegenerative diseases as well. Blocking the self-propagation can support and guide vaccine development efforts. The identification of a pathological, self-promoting isoform offers a rational vaccine target.[18] Both active and passive anti-Aβ immunotherapies were shown to clear brain Aβ deposits. However, therapies aimed at reducing protein processing and clearance for AD have been unsuccessful in clinical trials, including an active anti-Aβ vaccine (AN1792) and two passive antibodies bapineuzumab and solanezumab[19]. These results suggest that targeting protein aggregation alone may be insufficient to treat NDs [14], with current approaches focusing on preventing downstream events of neuroinflammation and tau pathology by treating AD patients in the very early stages of the clinical symptoms.[20] Several drawbacks face the passive immunization approach; for example, the unfavorable drugs’ pharmacokinetics, the difficulty of the drugs to cross the blood brain barrier (BBB), and potential neurotoxicity. It has even been questioned “Is there still any hope for amyloid-based immunotherapy for Alzheimer’s disease”[19].
The molecular mechanisms underlying amyloid-based immunotherapy for neurodegenerative diseases are complex, and considerable work is needed to fully understand the properties of the amyloidogenic proteins responsible for the disease. Among these elucidating the features of the intrinsic disorder of monomeric protein and the landscape of polymorphic oligomer/fibril conformations may provide specific physico-chemical features for antibody recognition of the amyloid antigens.
Many proteins either contain at least one intrinsically disordered region (such as domain or linker in multidomain proteins)[21, 22] or are completely disordered.[23, 24] ‘Disordered’ or ‘intrinsically unstructured’ proteins lack a stable, well-defined structure under physiological conditions, existing in a continuum of conformations from the less to the more structured states.[25–28] IDPs are not only involved in a wide variety of physiological processes, but also involved in pathological aggregation processes associated with many human diseases such as neurodegenerative diseases.[29–32] Furthermore, amyloid formation involves highly polymorphic oligomer and fibril conformations.[33–37] Therefore, it is important to understand how antibodies respond and recognize the highly dynamic conformations in the monomeric, oligomeric, and fibrils states of the amyloid antigens. In this article, we systematically review all available crystal structures of antibody-amyloid protein complexes related to neurodegenerative diseases. We found that antibodies mostly use the conformational selection mechanism to recognize highly flexible amyloid antigens. The antigen conformations trapped in the antibody may provide important link to the conformational transitions in amyloid formation. Water molecules often bridge the interaction between antibodies and amyloid proteins, assisting in the conformation selection in antibody-antigen recognition.
1. Structural features of antibody recognition of Aβ peptide and tau protein
1.1 Three antibodies selected the N-terminal conformations of Aβ peptide
1.1.A. Highly populated extended conformation
The first antibody-Aβ peptide structures were solved in 2007.[38] The structures include the antigen binding domain fragments (Fabs) of two anti-protofibril antibodies (PFAs) PFA1 and PFA2 in complex with the Aβ(1–8) peptide DAEFRHDS and with the homolgous AKFRHD peptide (PDB code 2IPU, Figure 1, Table 1). PFA1-Fab and PFA2-Fab have identical L and similar H sequences. The key residues of the Aβ peptide for binding PFA1 and PFA2 consist of 3EFRHD7 at the Aβ N-terminus. The Aβ(1–8) peptide adopts nearly identical conformations (Cα RMS deviation for residues 2–8 is 0.48 Å) when bound to the two Fabs. The binding of the WWDDD motif in the H2 loop of both mAbs to the EFRHD sequence is mediated by a combination of salt bridges, hydrogen bonds, and hydrophobic contacts.[38]
Figure 1.

Overview of antibody-Aβ peptide complex structures. (A) The solid lines represent residues with visible electron density. The red dashed lines indicate the sequence range included in the complex structure. Each antibody-antigen complex is labeled by their PDB code and the Aβ sequence is in parenthesis. (B) The complex structure (pdb ID: 3IFN) of amyloid peptides (Aβ2–7) and antibody (12A11 Fabs) [40]. The heavy chain and light chain of 12A11 Fabs is colored in cyan and orange and the potential paratope is shown in stick model. The Aβ2–7 is shown in white surface model to represent its occupation in3D space and also stick model to represent the epitope. Water molecules associated with the antigen-antibody binding are shown in red dots.
Table 1.
Summary of the complex structure of Aβ-antibodies
| PDB code | antigen | antibody | epitope | paratope | refs | |
|---|---|---|---|---|---|---|
|
| ||||||
| 2IPU | Aβ(1–8)peptide DAEFRHDS | Fabs of PFA1 | DAEFRHDS | WWDDD | [38] | |
|
| ||||||
| 2R0W | Aβ(1–8)peptide DAEFRHDS | Fabs of PFA2 | DAEFRHDS | WWDDD | [38] | |
|
| ||||||
| 2R0Z | GRIP1 | Fabs of PFA1 | AKFRHD | WWDDD | [38] | |
|
| ||||||
| 2IPT | N/A | PFA1 Fab | N/A | N/A | [38] | |
|
| ||||||
| 2IQ9 | N/A | PFA2 FAB fragment, triclinic apo form | N/A | N/A | [38] | |
|
| ||||||
| 2IQA | N/A | PFA2 FAB fragment, monoclinic apo form | N/A | N/A | [38] | |
|
| ||||||
| 3BAE | Aβ(1–28) | WO2 | Aβ(2–8) | [39] | ||
|
| ||||||
| 3BKC | N/A | WO2 Fab (Form B) | N/A | N/A | [39] | |
|
| ||||||
| 3BKM | N/A | WO2 Fab (Form A) | N/A | N/A | [39] | |
|
| ||||||
| 3BKJ | Aβ (1–16) | WO2 Fab | Aβ(2–8) | [39] | ||
|
| ||||||
| 3EYS | pyro-Glu3-A pEFRHDS | PFA1 | [44] | |||
|
| ||||||
| 3EYS | Ror2 REEFRHEA | PFA1 | [44] | |||
|
| ||||||
| 3IFO | Aβ 1–7 | 10D5 Fabs | 3EFRHD7 | [40] | ||
|
| ||||||
| 3IFL | Aβ 1–7 | 12A11 Fabs | 3EFRHD7 | [40] | ||
|
| ||||||
| 3IFN | Aβ 1–40 | 12A11 Fabs | 3EFRHD7 | [40] | ||
|
| ||||||
| 3IFP | Aβ 1–7 | 12B4 Fabs | 3EFRHD7 | [40] | ||
|
| ||||||
| 3LN9 | Amyloid Fibril | B10 | ? | positively charged residues of CDRs1–3 | [47] | |
|
| ||||||
| 4OJF | Aβ 1–8 | 3D6 (Bapineuzumab) | 1DAEFR5 | L1 24KSSQSLLDSDGKTYLN34 | [48] | |
| L2 50LVSKLDS56 | ||||||
| L3 89WQGTHFPRT97 | ||||||
| H1 26GFTFSNYGMS35 | ||||||
| H2 50SIRSGGGRTYYSDNVKG65 | ||||||
| H3 95YDHYSGSSDY102 | ||||||
|
| ||||||
| 4HIX | Aβ 1–40 | Bapineuzumab | 1DAEFRH6 | [48] | ||
|
| ||||||
| 4IFO | IDE | Fab(IDE) | K324/Y325/402/459/R 460/D462/E465/ | Y54/Y55/102/Y114/S108/109/Y1 12/Y110 | [79] | |
|
| ||||||
| 3MCL | N/A | C706 Fab | N/A | N/A | [80] | |
|
| ||||||
| 3O11 | N/A | C706 Fab | N/A | N/A | [80] | |
|
| ||||||
| 3TPK | N/A | KW1 | [46] | |||
|
| ||||||
| 3U0T | Aβ 40 | Ponezumab | Aβ30–40 | [53] | ||
|
| ||||||
| 4M1C | IDE/ Aβ16–20 complex | Fab | ||||
|
| ||||||
| 4ONF | Aβ 1–7 | 3D6 | 1DAEFRHD7 | [49] | ||
|
| ||||||
| 4ONG | Aβ 1–40 | 3D6 | 1DAEFRHD7 | [49] | ||
|
| ||||||
| 5CSZ | Aβ 1–11 | Gantenerumab | [45] | |||
|
| ||||||
| 4XXD | Aβ 12–28 | solanezumab Fab | Aβ 16–26 | [51] | ||
One year later (2008), the crystal structures of antibody WO2 in complex with Aβ 1–16 and 1–28 were solved.[39] In both cases, only residues 2–7 have stable structure, and all other residues have no electron density. The Aβ peptide adopts an extended, coil-like conformation across its major immunodominant B-cell epitope between residues 2 and 8. Residues 2–8 in the Aβ structures derived from the WO2:Aβ(1–16) and WO2:Aβ(1–28) crystals have almost identical conformations (RMSD of 0.08 Å for all Cα atoms) apart from minor deviations at both ends of the peptide. All residues from Phe4 to Ser8, except Asp7, make close contact with the WO2 heavy-chain H3.[39]
In 2010, the structures of three other antibodies (12A11, 10D5, and 12B4) recognizing Aβ residues 3–7 were revealed, with Aβ structures surprisingly similar to these in PFA1-2 and WO2 binding.[40] Residues 2–6 of the peptide could be visualized unambiguously in all four structures (antibody12A11+Aβ(1–7), antibody12A11+Aβ(1–40), antibody10D5+Aβ(1–7), and antibod12B4+Aβ(1–7)). Aβ D1 could be modeled only in the 12A11+Aβ(1–7) structure. In all three antibodies, Aβ residues Ala2 and Glu3 interact principally with the light chain CDRs L3 and L1, whereas Arg5 and Asp7 interact with CDRs H2 and H3, respectively. Aβ Phe4 and His6 are the most deeply buried in the groove, with their side chains interacting with both the light and the heavy chain. Both Phe4 and His6 are in direct contact with CDRs L1 and L3. Phe4 is also in direct contact with the heavy chain CDR H2 and residue His6 contacts CDR H3.
It is interesting to see that six independently derived antibodies mentioned above (12A11, 10D5, 12B4, PFA1, PFA2, and WO2) recognize the same N-terminal conformation of the Aβ peptide. Superposition of the peptide in the complexes gives an average RMSD of 0.68 Å for all atoms of residues 3–6.[40] While the similarity in Aβ conformation is reflected in the strong sequence conservation in the CDR loops L1, L2, L3, H1, and H2, there are notable differences in these regions and in the H3 loop.[40] Antibodies are not static or rigid, including when fitting the Aβ conformation. PFA1 and PFA2 undergo conformational changes in order to bind Aβ (> 1 Å), most notably in the CDR H3. This region is located near the C terminus of the Aβ(1–7) peptide, and the different antibodies show some variation in their interactions with this portion of Aβ. It has been postulated that the differences in sequence and structure of CDR-H3 contribute to the different in vivo activities observed in the CFC assay between 12A11, 10D4, and 12B4. These three antibodies targeting the same linear epitope of Aβ, Aβ(3–7), differ in their ability to reverse contextual fear conditioning deficits in Tg2576 mice in an acute testing paradigm.[40]
Since Aβ peptide fragments are disordered in solution and there is no well-defined stable conformation, the invariance of this selected conformation in short peptide as well as in longer Aβ(1–16), Aβ(1–28), and Aβ(1–40) does not indicate that the bound conformation is the most stable in solution. Additional evidence has shown that even with perturbations from mutations and ion coordination, the binding mode and conformation of the N-terminal residues are surprisingly stable. For example, it is known that Zn2+ ion binds the N-terminal 1–16 region and forms very stable structures[37, 41, 42], and that the presence of metals affects the Aβ aggregation state[43]. However, crystal soaks and co-crystallization experiments for WO2:Aβ with Cu2+ and Zn2+ ions revealed that even with the extended Aβ(1–28) peptide present, no change of Aβ-antibody interaction was observed, and no tertiary structures were induced such as those observed in metal-binding soluble Aβ peptides in NMR studies.[39]
Alanine scan of the contribution of positions 1–8 in the Aβ(1–40) peptide to the binding of PFA1-2 indicates that except D1, all other positions are sensitive to the Ala mutations. The binding is significantly impaired or eliminated in Aβ(1–40) mutants for Ala mutations at positions 3, 4, 5, and 6. The D7A mutant peptide exhibits significantly reduced binding to both mAbs. Position 2 appears optimized and acts to increase binding affinity. For example, the peptide sequence REEFRHEA, derived from the human receptor-related neurotrophic tyrosine kinase (ROR2), actually binds to PFA1 and PFA2 with approximately twice the affinity of the WT Aβ(1–7) peptide AEFRHD.[38]
Since the E3A mutant does not bind the antibodies, it is surprising to see that a similar short AKFRHD peptide is able to bind the antibody PFA1-2. The binding of AEFRHD to the antibodies PFA1 and 2 are within expectation. In contrast, the E3K substitution results in replacing the favorable ion-pair between E3 to His by a less favorable van der Waals contact formed between two basic residues. Thus the charge reversion by the E3K mutation should completely eliminate the peptide-antibody binding. However, the affinities of PFA1 and 2 Fabs to AKFRHD are only 28 and 35 times lower than binding affinity to AEFRHD.[38] The binding structure of the AKFRHD (“E3K”) peptide to PFA1 is nearly identical (Cα RMSD is 0.23 Å) to that of Aβ(1–8). This indicates that the antibody is able to adjust its interaction to select a matched peptide conformation.[38]
The pyroglutaminyl (pyro-Glu) forms at residues 3 and 11 are implicated in the onset of AD, since AD patients have abundant pyro-Glu-Aβ in their neuritic amyloid deposits. The Glu to pyro-Glu substitution at position 3 has a drastic effect on mAb binding. PFA1 Fab binds pyro-Glu3-Aβ with a greatly reduced affinity (77-fold difference in Kd) when compared to Aβ(1–8).[44] However, the Cα RMSD for residues 3–8 between the WT Aβ and pyro-Glu3-Aβ(3–8) is only 0.24 Å, with some structural adjustment at E3 position. Interestingly, the pyroglutaminyl ring occupies space overlapping that of the E3 side chain of WT Aβ, indicating the robustness of mutual conformational selection between the peptide and antibody. Comparison of the structures of the peptides in complex with PFA1 shows that the greatest conformational flexibility occurs at residues 2 to 3 and 8 of the peptide. These structures provide the molecular basis of the specificity tolerance of PFA1 and its ability to recognize Aβ N-terminal heterogeneity.[44]
1.1B. Extended binding mode of gantenerumab and the implication of fibril binding
The crystal structure obtained from the gantenerumab Fab with Aβ(1–11) or Aβ(3–11) peptides revealed a unique binding motif of the Aβ peptides to CDRs 1, 2, and 3 of the heavy chain and CDR3 of the light chain.[45] Residues 1–11 of Aβ peptide in the gantenerumab structure were observed bound in an extended conformation, with the N-terminal to C-terminal orientation of Aβ reversed by 180° relative to the published structures discussed in section A.
Even though the crystal structure only revealed the binding of Aβ(1–11) region, the antibody is able to recognize the middle hydrophobic region of Aβ. The central region of Aβ that was most pronounced is the decapeptide VFFAEDVGSN. With the inclusion of 8–11 and the 18–27, gantenerumab is able to bind the Aβ monomer, oligomer, as well as fibril. The affinities of gantenerumab to different aggregation states of the Aβ42 peptide are 0.6 nM, 1.2 nM, and 17nM for fibrils, oligomers, and monomers, respectively.[45] The high avidity binding to aggregated A42 and oligomeric Aβ42 contribute to the ability of restoring synaptic deficits in the transgenic mice study. In functional assays, gantenerumab induced cellular phagocytosis of human amyloid-β deposits in AD brain slices and neutralized oligomeric Aβ42 in rat brain. However, gantenerumab did not alter plasma Aβ suggesting undisturbed systemic clearance of soluble Aβ. These studies demonstrated that gantenerumab preferentially interacts with aggregated Aβ in the brain and lowers amyloid-β by eliciting effector cell-mediated clearance.[45] It is therefore interesting to check possible binding modes of gantenerumab and the Aβ fibril as we know that both the N-terminal and central portions of Aβ are recognized by gantenerumab. The N-terminal region is often modeled as random conformation in the fibril state. However, it is likely that the gantenerumab-locked Aβ(1–11) conformation will be enriched in the oligomer and fibril state, leading to increased binding affinity of gantenerumab to the Aβ oligomer.
Several other structures of the antibodies that recognize the oligomer or fibril were solved in the apo form only. Thus, it is still unknown what are the structural features of antibody-oligomer or antibody-fibril recognition. KW1 is an oligomer-specific antibody fragment. KW1 not only discriminates between oligomers and other Aβ conformations, such as fibrils or disaggregated peptides; it also differentiates between different types of Aβ oligomers, such as those formed by Aβ(1–40) and Aβ(1–42) peptide. X-ray crystallography, NMR spectroscopy, and peptide array measurements imply that KW1 recognizes oligomers through a hydrophobic and aromatic surface motif that includes Aβ residues 18–20. [46] By altering a specific step of the fibrillogenic cascade, it prevents the formation of mature Aβ fibrils and induces the accumulation of nonfibrillar aggregates.[46] Antibody B10 only binds the Aβ(1–40) fibril but not the monomer or oligomers. B10 presents poly-amyloid specific binding and interacts with fibrillar structures consisting of different polypeptide chains; not only Aβ(1–40). While the antigen surface is characterized by highly acidic properties, the antigen-binding site of B10 is strongly basic. Mutations of these basic residues into alanine potently impair fibril binding, and reduce B10-fibril interactions which is also observed when the fibril carboxyl groups are covalently masked by a chemical modification. These data imply that the B10 conformational specificity for amyloid fibrils depends upon specific electrostatic interactions with an acidic moiety, which is common to different amyloid fibrils.[47]
1.1C. Bapineuzumab family antibodies select helical conformation of Aβ N-terminus
Bapineuzumab (AAB-001) and its derivative (AAB-003) are humanized versions of the anti-Aβ murine antibody 3D6 which targets amyloid Aβ plaques. The common Fab fragment of these immunotherapies has been expressed, purified and crystallized in complex with Aβ peptides (residues 1–8 and 1–28).[48] The amino-terminal conformation of Aβ recognized by the Bapineuzumab family has a helical conformation,[48, 49] which is very similar to that of TFE (2,2,2-trifluoroethylalcohol)-stabilized solution structures of Aβ (RMSD 0.8 Å for Cα atoms when the lowest energy NMR structure is superimposed). The structure of 3D6 with Aβ(1–7) reveals excellent agreement (RMSD 0.56 Å) with the Bapineuzumab (PDB code 4HIX) structure. The helical conformation is stabilized by five putative intramolecular hydrogen bonds. Three of the five intra-Aβ hydrogen bonds involve Glu3, the first between its backbone amide and a side-chain carboxyl, and two bonds between the side-chain carboxyls and the free N-terminal amine of Asp1. The key role of Glu3 in maintaining the helical structure partially explains the dominant loop conformation of E3K and pyro-Glu-Aβ in antibody PFA recognition, since these mutations likely destroy the helical hydrogen bonds. The helical structure explains the Bapineuzumab antibody’s exquisite selectivity for particular Aβ species and why it cannot recognize N-terminally modified or truncated Aβ peptides.[50] Preferential binding of Bapineuzumab for plaque deposited Aβ(1–11) suggests that this helical conformation at the N-terminus is either enriched or exists in an equilibrium of conformational states in dense plaque deposits, which also have polymorphic structures. This is similar to Gantenerumab’s preference of Aβ oligomers and fibrils.
Clearly, Aβ does not adopt a single conformation in solution. The conformational ensemble of the amino terminus has a helical population, which is recognized by the Bapineuzumab family antibodies. The reason that Bapineuzumab and 3D6 captured the helical conformation could be that their binding motifs are more hydrophobic. For example, the WWDDD motif conserved between PFA1 and PFA2, and YWDDD in WO2, is not found in the Bapineuzumab CDR-H2, where the corresponding sequence is RSGGG.[49] Such a binding pocket environment may have some similarity as the TFE, which stabilizes helical structures in solution.
1.2 Structures of the antibodies in complex with the central and C-terminal motifs of the Aβ peptide
Solanezumab developed in Eli Lilly is one of the leading antibodies targeting Aβ in clinical trials. The solanezumab structure has been solved in complex with Aβ(12–28), and residues 16–26 are visible.[51] The antibody interacts with the Aβ peptide backbone and side-chains of K16, F19, F20, and D23. While Aβ residues 16–18 are in an extended coil conformation, residues 20 to 26 adopt a helical conformation which is similar to the helical region in the HFIP-induced solution state helical Aβ structure.
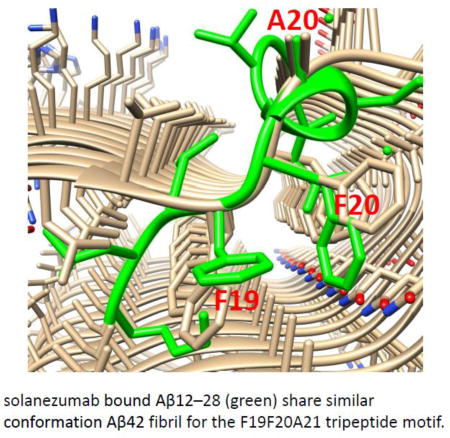
The conformation of the central F19-F20-A21 tri-peptide segment is interesting. The Phe19-Phe20 dipeptide provides dominant hydrophobic interactions with solanezumab, contributing 42% of the 960 Å2 interface area of the Aβ contacting solanezumab. The side-chains of two phenylalanines are deeply buried in the antibody, whose pocket is mostly hydrophobic. Surprisingly, we found that the F19-F20-A21 tri-peptide conformation matches the F19-F20-A21 tri-peptide motif in our recently solved Aβ1–42 fibril structure[52] (Figure 2). This unexpected connection of the antibody-recognized Aβ conformation and its definite role in the core region of Aβ(1–42) fibril has two implications. First, the connection provides insights into the mechanism of Aβ(1–42) formation. This F19-F20-A21 tri-peptide conformation should have sufficient population time to be recognized by solanezumab. The conservation of this tri-peptide conformation in the Aβ(1–42) fibril structure indicates that it is likely to be a nucleation core for oligomer and fibril formation. Second, this tri-peptide conformation is also likely to be important for developing antibodies that recognize the monomer, oligomer, and fibril in the central region of the Aβ peptide. While solanezumab (and the parent antibody) are known to only bind monomeric soluble Aβ, the closely related crenezumab has been described as having high affinity for monomeric, oligomeric and fibrillar forms. It has been shown that all CDRs are identical in length to their counterpart in solanezumab and crenezumab. Three Aβ binding loops, L2, L3, and the highly variable H3 are identical in composition for these two antibodies. The remarkable conservation of Aβ-binding residues in crenezumab explains the observed shared cross reactivity of solanezumab and crenezumab with proteins abundant in plasma that exhibit this Phe-Phe dipeptide.[51]
Figure 2.

Comparison of the F19F20A21 tripeptide conformations in Aβ42 fibril and in solanezumab bound Aβ(12–28) (green). A. The overall structure of the Aβ42 fibril and Aβ(12–28) conformation. B. The match with the F19F20A21 tripeptide.
The structural features of the C-terminal region of the Aβ peptide that interact with the antibody are also known. Ponezumab, a humanized monoclonal antibody, binds specifically to the C-terminus of Aβ40. The X-ray crystal structure of ponezumab in complex with Aβ40 reveal that the Aβ40 carboxyl moiety makes extensive contacts with ponezumab.[53] Within the ponezumab-Aβ40 complex structure, 11 Aβ residues from A30 to V40 are visible in the electron density maps. The conformation of the C-terminal region is characterized by four β-turns (31–34, 33–36, 35–38, and 36–39). V40 interacts most extensively with ponezumab; 35% of the total binding interface buries the Aβ V40 residue including the negatively charged carboxylic acid (COO-) moiety. No binding was detectable for the Aβ(17–40) amide or Aβ(17–42), explaining the critical role of the C-terminal carboxylic acid for ponezumab activity.[53] Following similar comparison of the amyloid structure and the F19-F20-A21 tripeptide conformation in solanezumab-Aβ(12–28) complex, it is very likely that some turn conformation in the ponezumab-Aβ40 complex structure will be similar to that in the Aβ40 oligomer or fibril structures.
1.3 Structures of the Antibodies in recognizing peptides from the tau protein
The monoclonal antibody MN423 is specific to the tertiary structure of the tau PHF core fragment 386TDHGAE391, where the crystal structure of the complex has been solved by refining a computational docking model of the peptide with MN423 (Table 2).[54, 55] The hexapeptide 386TDHGAE391 adopts a loop form without any direct intra-chain hydrogen bonds. The core fragment makes seven direct and five water-mediated hydrogen bonds with MN423. Comparison of the apo and holo MN423 structure indicates that MN423 combining site in both structures is identical. Thus the antibody-tau peptide recognition may need small activation energy.[54] The loop conformation of the hexapeptide 386TDHGAE391 could represent the genuine structure of the core PHF tau C-terminus. Interestingly, the hexapeptide lies outside the repeat region known to convert to β-structure during PHF assembly, and its close association with the compact PHF core suggests a potential role of the C-terminal core peptide in the PHF assembly.[55] Interestingly, the data from in silico docking of tau peptide into the antibody mold indicated that computational approaches provide results comparable to X-ray crystallography for this antibody-tau complex,[54] helped by very small antibody conformation change upon antigen recognition.
Table 2.
Summary of the complex structure of tau-antibodies
One of the driving forces in tau aggregation is hyperphosphoylation. Specific recombinant chicken antibodies to three phosphoepitopes in Alzheimer disease-associated tau protein have been developed. Each antibody shows full specificity for a single phosphopeptide. The Fab fragment forms complex with the tau peptide 224KKVAVVR(pT231)PPK-(pS235)PSSAKC241, with two phosphorylation sites at Thr-231 and Ser-235. The crystal structure of the Fab-tau peptide represents the first antiphospho antibody in complex with its cognate phosphoepitope.[56]
In the Fab-peptide co-crystal structure, 10 amino acids (225KVAVVR(pT)PPK234) are visible, of which six (225KVAVVR(pT231)) interact directly with the Fab fragment. The remaining eight residues of the peptide are disordered. A long, disulfide-constrained CDR-H3 mediates the peptide recognition. The segment 224–241 is in the N-terminal side of the tau repeat domains, which starts at residue 243. Immunohistochemistry data show that the antibody does recognize the phosphorylated epitope in the intact molecule.[56]Therefore, the segment 224–241 should be exposed in the full-length tau and the conformation represents a populated state in the full-length tau. The critical phosphorylation site (pThr-231) is exclusively recognized by CDR-H2, which forms a positively charged pocket to accommodate the phosphate. The highly specific phosphate recognition explains why the antibody does not bind to non-phosphorylated peptides with the same sequence.[56]
Monoclonal antibody DC8E8 is able to recognize the PGGG motif, four of which separate the four repeats in tau core domain. It has been shown that the DC8E8 neutralizes tau in a murine model of tauopathy.[57] The X-ray structure of the DC8E8 Fab apo form suggested that the four DC8E8 epitopes form protruding structures on the tau molecule. The flexibility of the binding site allows DC8E8 to adapt to four homologous, albeit not identical antigens. DC8E8 is able to discriminate between the healthy and diseased tau proteome, making its epitopes suitable targets, and DC8E8 a suitable candidate molecule, for AD immunotherapy.[57] Figure 3 shows the location of the four motifs in K18 monomer[58] and K18 fibril[36, 59]. With the addition of the C- and N-terminal residues, the PGGG motifs could be buried inside. The PGGG motifs in full length fibrils are also partially covered by the C- and N-terminal brush shells. However, phosphorylation will help to expose the HXPGGG motifs[60] and allow FC8E8 recognition.
Figure 3.

The locations of the HXPGGG motifs in the K18 monomer[58] (A) and K18 fibril[36, 59] (B). C. A model of full-length tau fibril structure[60].
2. Structural features of antibody recognition of other amyloid proteins
2.1 Prion proteins
The X-ray crystallographic structures of the anti-Syrian hamster prion protein (SHaPrP) monoclonal Fab 3F4 alone, as well as the complex with its cognate peptide epitope (SHaPrP 104–113), have been determined (Table 3).[61] The conformation of the decapeptide is an Omega-loop, stabilized by the presence of at least two intramolecular hydrogen bonds between Thr107/M109 and His111/Lys106. The importance of the hydrogen bond in stabilizing the Omega-loop can be inferred from the absolute conservation of Thr107 and His111 in all PrP species. The peptide interacts in a U-shaped groove on the Fab, with deep penetration of two side-chains, Met109 and Met112 into the Fab surface. The deep pocket that warps the peptide caused substantial alterations in the antibody region upon epitope binding. The antibody conformation can also be caused by the need to accommodate the peptide conformational change. The loop is disordered or flexible in the three NMR structures of PrP. The conformational change may be the result of the pH differences between the crystallization conditions (pH 7.0) and the NMR studies (acidic conditions). Alternatively, this region of PrP may indeed be flexible, existing in an ensemble of states. It is also interesting to note that this Omega-loop structure could form a p-hairpin in PrPSc, indicating that this flexible region undergoes conformational rearrangement that is an essential feature of prion disease.[61]
Table 3.
Summary of the complex structure of prion-antibodies
| code | antigen | antibody | epitope | refs |
|---|---|---|---|---|
|
| ||||
| 2w9e | huPrP (119–231) | ICSM 18 Fab | (H1, 143–156) | [63] |
|
| ||||
| 1tpx | OvPrPC and OvPrPSC | VRQ14 Fab | 188–199 | [64] |
| (114–234) | ||||
| 1. A136-R154-R171 | ||||
| 2. V136-R154-Q171 | ||||
| 3. A136-R154-Q171 | ||||
|
| ||||
| 4DGI | huPrPc (120–230) | POM1 Fab | Primary: (residues 140–147) | [65] |
| Secondary: Lys204, Arg208 and Gln212 | ||||
|
| ||||
| 4YX2 | boPrP(103–242), | POM1 Fab | The same with huPrP | [66] |
| 4YXH | dePrP (123–231), | |||
| 4YXK | ekPrP (124–231) , | |||
| 4YXL | shPrP (90–230) | |||
|
| ||||
| 4j8r | Peptide OR2 | POM2 Fab | PHGGSWGQPHGGSWGQ | [62] |
| PHGGSWGQPHGGS WGQ | ||||
|
| ||||
| 1cr9 | peptide epitope (SHaPrP 104–113) | 3F4 Fab | KPKTNMKHMA | [61] |
|
| ||||
| 4kml | full-length HuPrP | nanobody (Nb484) | discontinuous epitope including residues | [67] |
| 123–125 in the β0-β1 loop, residues 164–170 in the β2-α2 | ||||
| loop, and residues | ||||
| 174–185 in the α2-helix. | ||||
Prion protein (PrP(c)) has a highly flexible N-terminal domain, which contains 4–5 repeats of eight amino acids long peptide known as the octapeptide repeat (OR) domain. [62] The repeats are rich in glycine residues, and the OR domain is intrinsically disordered in solution. The structure of a tandem OR repeat, PHGGSWGQPHGGSWGQ of PrPc in complex with the POM2 antibody Fab molecule OR, was solved. There are two copies of OR molecules in unit cell, and the overall RMSD between the Cα atoms of the OR2 peptide in Mol1 and Mol2 is 1.28 Å. Consistent with the large RMSD, there are also some differences observed in the hydrogen bonding of the OR2 peptides in Mol1 and Mol2. The Trp6 residue is better positioned in the binding cleft of the POM2 Fab in Mol2 than in Mol1 facilitating the hydrogen bonding to the backbone carbonyl oxygen of POM2. Both copies of the OR2 adopted an extended conformation, indicating that the bound Fab does not select any putative native β-turn conformation of the ORs.[62]
The structures of several antibodies in complex with the C-terminal of PrPC have been well studied. The monoclonal antibody ICSM18 recognizes huPrP(119–231) [63], VRQ14 Fab binds to ovine PrPC and to PrPSc, [64] POM1 Fab binds PrPC from many species[65, 66], and nanobody Nb484 binds to full length human prion protein[67]. These antibodies recognize different epitopes, residues 143–156 for ICSM18, residues 140–147 and 204–212 for POM1, and 188–199 for VRQ14. Structural superpositions of the bound PrPc in the different complexes indicate limited conformational variations among the PrPc in different complexes. In all cases, many disordered structures in the free PrPC conformations have been stabilized and provided important information for the conformational dynamics of PrPC and its conversion to PrPSc. Molecular dynamics simulation studies on six prion proteins (cow, deer, elk, Syrian hamster, mouse and human) revealed differences in the local fluctuations and imply that these differences have possible roles in the unfolding of the globular domains and the strain selection and the preferred conformations adopted upon binding. [66]
POM1 Fab and ICSM18 Fab, recognize adjacent PrPc epitopes in the 143–156 region with completely different binding modes. The two Fabs bind the common huPrPc epitope in an opposite manner. Superimposing the Fab fragments of the POM1 and ICSM18 antibodies reveals that the bound huPrPc molecules rotate 180 degree in the respective CDRs. The reason for this large difference is that the antibody-antigen interactions of these two Fabs are largely electrostatic, and the two Fabs exploit different binding residues to satisfy salt bridge formation and other polar interactions.
PrP residues 188–199 are in the C-terminal part of helix H2 and the N-terminal part of the H2-H3 loop and they are mostly hydrophobic. Only one face of helix H2 is buried in the VRQ14 Fab-combining site. The combined structural and immunochemical data provide strong evidence that the C-terminal end of helix H2 and the N-terminal part of the H2-H3 loop are conserved in PrPSc. The hinge region of end of helix H2 and of the H2-H3 loop is also the region which characterizes the only secondary structure difference between the huPrP monomer in the dimer and ovine PrP. This hinge forms a small antiparallel-sheet in the covalent dimer in which four main-chain hydrogen bonds compensate for the three main-chain H2 helix hydrogen bonds disrupted in the dimer by the H3 exchange. Based on the structure of VRQ14 ovine PrPC complex and recognition of both PrPC and to PrPSc, it was proposed that the two PrPC C-terminal alpha-helices are conserved in PrPSc, whereas secondary structure changes are located in the N-terminal alpha-helix. [64]
The interaction of ICSM18 with huPrP (119–231) indicated a clear correlation between the ability to inhibit PrPSc propagation and binding affinity for a PrPC-type conformation for therapeutic antibodies. In the crystal structure of ICSM18 binding with huPrP (119–231), interactions between two neighboring PrP molecules are mediated by close homotypic contacts between residues at position 129 that lead to the formation of a 4-stranded intermolecular β-sheet. This region harbors the strongest common susceptibility polymorphisms in the human prion disease. The importance of this residue in mediating protein-protein contact could explain the genetic susceptibility. [63] The importance of a nearby region in PrPSc propagation can be inferred from the nanobody-stabilized prion protein structure. The nanobody (Nb484) binds to full-length human prion protein and revealed unprecedented structural features at the N-terminal palindromic sequence AGAAAAGA (residues 113–120). The palindromic motif adopts a stable and fully extended configuration to form a three-stranded antiparallel β-sheet. Thus the structure supports the long standing hypothesis that the palindromic sequence AGAAAAGA mediates β-sheet formation in early stage of the PrPC conversion to PrPSc. [67]
2.2 alpha-synuclein and polyglutamine
Several structures of antibody-bound peptides from alpha-synuclein and polyglutamine are known (Table 4). Huntington’s disease is triggered by misfolding of fragments of mutant forms of the huntingtin protein (mHTT) with aberrant polyglutamine expansions. Three anti-polyQ antibodies-MW1, 1C2 and 3B5H10-have been extensively used to probe the conformation of polyQ. The crystal structure of the MW1 epitope reveals a linear, non-pathogenic polyQ. The MW1 and 1C2 antibodies have similar sequences and structures, consistent with their binding to short polyQ and their polyQ length-discrimination properties. The 3B5H10 epitope is actually a short, non-pathogenic polyQ. All three antibodies MW1, 1C2 and 3B5H10 interact similarly with polyQ of various lengths, and bind small polyQ epitopes in similar linear and extended conformations. [68]
Table 4.
Summary of the complex structure of antibodies with other amyloids
The C4 single-chain Fv antibody (scFv) binds to the first 17 residues of huntingtin [HTT(1–17)] and generates substantial protection against multiple phenotypic pathologies in situ and in vivo. Residues 3–11 of the bound HTT(1–17) peptide fold into a right-handed α-helix, while residues 12–17 of HTT(1–17) are in an extended conformation, of which residues 12–15 adopt a β-sheet structure and interact with the same region of another C4 scFv:HTT(1–17) complex in the asymmetric unit, resulting in a β-sheet interface within a dimeric C4 scFv:HTT(1–17) complex. [69]
Human α-synuclein is a 140-residue intrinsically disordered protein. The crystal structure of the C-terminal fragment of alpha-synuclein 132GYQDYEPEA140 in complex with single-domain camelid antibody, NbSyn2 is available.[70] The position of Tyr136, Glu137, Pro138, Glu139, and Ala140 of α-synuclein can be identified, and their interactions with NbSyn2 are mostly electrostatic, with water molecules bridging the main-chain atoms of alpha-synuclein to atoms of NbSyn2. However, since this region is not involved in amyloid formation[37], NbSyn2 binding has no effects on the fibril formation of alpha-synuclein. [70]
3. Water-assisted conformational recognition in antibody-antigen interactions
Water molecules play an important role in protein folding and protein-protein interactions through their association with proteins.[71] Among all the protein-protein complex structures in the pdb, about 21% of the water molecules were found to be involved in bridging interactions between the proteins.[72] Water molecules are an integral part of the antibody NbSyn2 recognition of alpha-synuclein, with most of them located in the binding regions bridging the protein contacts. The water-mediated interactions likely persist in solution as well, as suggested by the NMR observation of the broadening of the amide and carbonyl resonances of α-synuclein residues involved in binding. [70]
The strong involvement of water molecules has been found in many antibody-protein complexes discussed in this paper. [70] In the PFA1-WT Aβ complex (PDB code 2IPU), water molecules bridge the E3 interaction with PFA1. However, in the PFA1- Ror2(518–525) complex structure (PDB code 3EYS), the absence of the water molecule prevents the formation of the hydrogen bond between the water and E3, resulting in side-chain conformational flexibility of Ror2(518–525)’s E3 which finds a new hydrogen bond partner in LC H93, and the water-mediated interaction around the carboxyl terminus also changes. [44] In the humanized Bapineuzumab 3D6-Aβ peptide complex, there are five water-mediated hydrogen bonds. [50] Several interactions between the peptide and the antibody involve bridging water molecules. Similar water-bridged antibody interactions are also observed in the 4HIX crystal structure.[48, 50] Water molecules are inside the deep pocket of the antibody binding site, and interact with residues from the N-terminus of the Aβ peptide.[49]
The antibody-tau fragment structure contains a large number of water molecules. The structure of the complex revealed that the PHF core C-terminus binds to MN423 combining site with five water-mediated hydrogen bonds.[55] The water molecule tightly bound in the combining site by hypervariable loops L3 and H3 is not displaced by PHF tau peptide binding and plays an important role in its recognition. In the antibody recognition of the phosphorylated tau, there is a water-mediated hydrogen-bonding network between phosphate oxygen and side chain nitrogen of Arg-230. [56]
Prion-antibody recognition is also water-mediated. In the Fab 3F4 SHaPrP 104–113 complex, two water molecules are present at the interface between the peptide and the antibody. One water molecule links the backbone nitrogen atom of Lys-P106 with Tyr-H33 of the antibody. The second water molecule binds in a structurally conserved site on the light chain, forming a hydrogen bond with the carbonyl oxygen atom of Lys-P110 of the peptide as well as a long hydrogen bond with His-L95A. Interestingly, the uncomplexed structure contains a solvent molecule bound to His-L95A as well. Peptide binding shifts, rather than replaces, the water molecule upon complexation. [61] In the POM2 Fab OR repeat peptide interaction, The amide nitrogen of Pro P1 residue forms a hydrogen bond with a water molecule situated in this pocket which in turn hydrogen bonds with the Glu H35 residue of the CDR H1 of the POM2 Fab. [62]
Conclusions
The molecular details of the recognition of disordered antigens by their cognate antibodies are less studied and known than ‘conventional’ folded protein antigens. It has been suggested that antibodies only bind disordered antigens weakly. However, since IDPs are not random and there are dominant conformations in the ensemble that can be stabilized by selected conformations. Using a large dataset of protein antigens, a recent study [73] showed that disordered epitopes are as likely to be recognized by antibodies as ordered epitopes. They pointed out that (1) disordered proteins are common targets of antibody responses, they do not affect affinity maturation, and the antibody affinity is only weakly influenced by epitope disorder; (2) disordered epitopes are smaller than ordered epitopes, and disordered epitopes are enveloped by concave paratopes. The structural basis for the unexpectedly high affinity of antibodies to disordered epitopes is that concave paratopes maximize the extent and complementarity of the interaction.[73]
Indeed, our examination of the known structures of the antibody complexes with flexible amyloid proteins agrees that antibody affinity is only weakly influenced by epitope disorder. It is interesting to note that most antibodies surveyed here selected unordered epitopes. One of reason could be conformation polymorphism for oligomeric and fibrillar amyloid, which are more difficult than monomer to change conformation. Thus, even though an antibody can recognize oligomeric or fibrillary amyloid, the polymorphism prevent crystallization of the antibody-antigen complex. Antibodies can select and stabilize biologically significant conformations of the monomeric amyloid proteins. Antibody-antigen interactions optimize the coupling of rigid and flexible protein binding sites for specific antigen binding. Antibody variable regions are necessarily flexible to enable recognition of extremely diversified targets. Clearly, nature utilized conformational selection to fit specific targets. The ‘conformational selection and population shift’ model[74–77] provides a realistic molecular recognition mechanism considering the protein conformational ensemble, [78] since both binding partners are flexible and have conformational distributions. During binding, the conformers that are most complementary to some pre-existing ligand conformations can be selected and the equilibrium shifts toward these conformers to form complexes. [74–77]
We found that antibodies mostly use the conformational selection mechanism to recognize highly flexible amyloid antigens. The antigen conformations trapped in the antibody bound form may provide a link - thereby helping to understand the conformational transitions cascade in amyloid formation. In particular, we discovered that solanezumab-bound Aβ(12–28) shares a similar conformation in the Aβ42 fibril, involving the F19F20A21 tripeptide, pointing to a new recognition motif. Water molecules often bridge the interaction between antibody and amyloid proteins, assisting in the conformational selection process in antibody-antigen recognition. In future designs, it should be possible to explicitly include water molecules to optimize the antibody-antigen interaction. Currently, there is still no available structure for an antibody in complex with amyloid protein in oligomer or fibril state. Computational modeling may help by exploring the protein conformational ensemble.
Highlights.
Antibody drugs targeting protein aggregation are difficult to develop
Available crystal structures of antibody-amyloid complexes are reviewed
The antibody selects antigen conformation which is not necessarily the most stable
Solanezumab bound Aβ12–28 F19F20A21 motif as shared with Aβ42 fibril
This motif may be important in amyloid formation thus serve as a new target
Acknowledgments
This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: None
References
- 1.Sipe JD. Amyloidosis. Annual Review of Biochemistry. 1992;61:947–975. doi: 10.1146/annurev.bi.61.070192.004503. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nature Cell Biology. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 3.Sunde M, Blake CCF. From the globular to the fibrous state: protein structure and structural conversion in amyloid formation. Quarterly reviews of biophysics. 1998;31:1. doi: 10.1017/s0033583598003400. [DOI] [PubMed] [Google Scholar]
- 4.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annual Review of Neuroscience. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 5.Lalowski M, Golabek A, Lemere CA, Selkoe DJ, Wisniewski HM, Beavis RC, Frangione B, Wisniewski T. The “nonamyloidogenic” p3 fragment (amyloid beta 17–42) is a major constituent of Down’s syndrome cerebellar preamyloid. Journal of Biological Chemistry. 1996;271:33623–33631. doi: 10.1074/jbc.271.52.33623. [DOI] [PubMed] [Google Scholar]
- 6.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 7.Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer’s disease and other neuro degenerative disorders: The emerging role of oligomeric assemblies. Journal of Neuroscience Research. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 8.Walsh DM, Selkoe DJ. Oligomers on the brain: the emerging role of soluble protein aggregates in neurodegeneration. Protein Pept Lett. 2004;11:213–228. doi: 10.2174/0929866043407174. [DOI] [PubMed] [Google Scholar]
- 9.Hardy J, Selkoe DJ. Science. Vol. 297. New York, N.Y: 2002. Medicine - The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics; pp. 353–356. [DOI] [PubMed] [Google Scholar]
- 10.Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- 11.Baumketner A, Bernstein SL, Wyttenbach T, Bitan G, Teplow DB, Bowers MT, Shea JE. Amyloid beta-protein monomer structure: a computational and experimental study. Protein Sci. 2006;15:420–428. doi: 10.1110/ps.051762406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshiike Y, Akagi T, Takashima A. Surface structure of amyloid-beta fibrils contributes to cytotoxicity. Biochemistry. 2007;46:9805–9812. doi: 10.1021/bi700455c. [DOI] [PubMed] [Google Scholar]
- 14.Wood LB, Winslow AR, Strasser SD. Systems biology of neurodegenerative diseases. Integrative Biology. 2015;7:758–775. doi: 10.1039/c5ib00031a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Genst E, Messer A, Dobson CM. Antibodies and protein misfolding: From structural research tools to therapeutic strategies. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2014;1844:1907–1919. doi: 10.1016/j.bbapap.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 16.Rovis TL, Legname G. Prion protein-specific antibodies-development, modes of action and therapeutics application. Viruses. 2014;6:3719–3737. doi: 10.3390/v6103719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taschuk R, Van der Merwe J, Marciniuk K, Potter A, Cashman N, Griebel P, Napper S. In vitro neutralization of prions with PrP(Sc)-specific antibodies. Prion. 2015;9:292–303. doi: 10.1080/19336896.2015.1071761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marciniuk K, Taschuk R, Napper S. Evidence for prion-like mechanisms in several neurodegenerative diseases: potential implications for immunotherapy. Clin Dev Immunol. 2013;2013:473706. doi: 10.1155/2013/473706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panza F, Logroscino G, Imbimbo BP, Solfrizzi V. Is there still any hope for amyloid-based immunotherapy for Alzheimer’s disease? Curr Opin Psychiatry. 2014;27:128–137. doi: 10.1097/YCO.0000000000000041. [DOI] [PubMed] [Google Scholar]
- 20.Lemere CA. Immunotherapy for Alzheimer’s disease: hoops and hurdles. Mol Neurodegener. 2013;8:36. doi: 10.1186/1750-1326-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maciejewski M, Tjandra N, Barlow PN. Estimation of interdomain flexibility of N-terminus of factor H using residual dipolar couplings. Biochemistry. 2011;50:8138–8149. doi: 10.1021/bi200575b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kukic P, Camilloni C, Cavalli A, Vendruscolo M. Determination of the individual roles of the linker residues in the interdomain motions of calmodulin using NMR chemical shifts. J. Mol. Biol. 2014;426:1826–1838. doi: 10.1016/j.jmb.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Chouard T. Structural biology: Breaking the protein rules. Nature. 2011;471:151–153. doi: 10.1038/471151a. [DOI] [PubMed] [Google Scholar]
- 24.Uversky VN. Introduction to intrinsically disordered proteins (IDPs) Chem. Rev. 2014;114:6557–6560. doi: 10.1021/cr500288y. [DOI] [PubMed] [Google Scholar]
- 25.Gunasekaran K, Tsai CJ, Kumar S, Zanuy D, Nussinov R. Extended disordered proteins: targeting function with less scaffold. Trends Biochem. Sci. 2003;28:81–85. doi: 10.1016/S0968-0004(03)00003-3. [DOI] [PubMed] [Google Scholar]
- 26.Kortemme T, Kelly MJ, Kay LE, Forman-Kay J, Serrano L. Similarities between the spectrin SH3 domain denatured state and its folding transition state. J. Mol. Biol. 2000;297:1217–1229. doi: 10.1006/jmbi.2000.3618. [DOI] [PubMed] [Google Scholar]
- 27.Mittag T, Forman-Kay JD. Atomic-level characterization of disordered protein ensembles. Curr. Opin. Struct. Biol. 2007;17:3–14. doi: 10.1016/j.sbi.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Shortle D, Ackerman MS. Persistence of native-like topology in a denatured protein in 8 M urea. Science. 2001;293:487–489. doi: 10.1126/science.1060438. [DOI] [PubMed] [Google Scholar]
- 29.Carrell RW, Lomas DA. Conformational disease. Lancet. 1997;350:134–138. doi: 10.1016/S0140-6736(97)02073-4. [DOI] [PubMed] [Google Scholar]
- 30.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 31.Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- 32.Uversky VN, Dave V, Iakoucheva LM, Malaney P, Metallo SJ, Pathak RR, Joerger AC. Pathological unfoldomics of uncontrolled chaos: intrinsically disordered proteins and human diseases. Chem. Rev. 2014;114:6844–6879. doi: 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller Y, Ma B, Nussinov R. Polymorphism in Alzheimer Abeta amyloid organization reflects conformational selection in a rugged energy landscape. Chem Rev. 2010;110:4820–4838. doi: 10.1021/cr900377t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma B, Nussinov R. Polymorphic C-terminal beta-sheet interactions determine the formation of fibril or amyloid beta-derived diffusible ligand-like globulomer for the Alzheimer Abeta42 dodecamer. J Biol Chem. 2010;285:37102–37110. doi: 10.1074/jbc.M110.133488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ma B, Nussinov R. Polymorphic triple beta-sheet structures contribute to amide hydrogen/deuterium (H/D) exchange protection in the Alzheimer amyloid beta42 peptide. J Biol Chem. 2011;286:34244–34253. doi: 10.1074/jbc.M111.241141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu X, Luo Y, Dinkel P, Zheng J, Wei G, Margittai M, Nussinov R, Ma B. Cross-seeding and conformational selection between three- and four-repeat human Tau proteins. J Biol Chem. 2012;287:14950–14959. doi: 10.1074/jbc.M112.340794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.L Xu, Nussinov R, Ma B. Coupling of the non-amyloid-component (NAC) domain and the KTK(E/Q)GV repeats stabilize the alpha-synuclein fibrils. European journal of medicinal chemistry. 2016 doi: 10.1016/j.ejmech.2016.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardberg AS, Dice LT, Ou S, Rich RL, Helmbrecht E, Ko J, Wetzel R, Myszka DG, Patterson PH, Dealwis C. Molecular basis for passive immunotherapy of Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2007;104:15659–15664. doi: 10.1073/pnas.0705888104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miles LA, Wun KS, Crespi GA, Fodero-Tavoletti MT, Galatis D, Bagley CJ, Beyreuther K, Masters CL, Cappai R, McKinstry WJ. Amyloid-β-anti-amyloid-β complex structure reveals an extended conformation in the immunodominant B-cell epitope. Journal of molecular biology. 2008;377:181–192. doi: 10.1016/j.jmb.2007.12.036. [DOI] [PubMed] [Google Scholar]
- 40.Basi GS, Feinberg H, Oshidari F, Anderson J, Barbour R, Baker J, Comery TA, Diep L, Gill D, Johnson-Wood K. Structural correlates of antibodies associated with acute reversal of amyloid β-related behavioral deficits in a mouse model of Alzheimer disease. Journal of Biological Chemistry. 2010;285:3417–3427. doi: 10.1074/jbc.M109.045187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zirah S, Kozin SA, Mazur AK, Blond A, Cheminant M, Segalas-Milazzo I, Debey P, Rebuffat S. Structural Changes of Region 1–16 of the Alzheimer Disease Amyloid beta-Peptide upon Zinc Binding and in Vitro Aging. Journal of Biological Chemistry. 2005;281:2151–2161. doi: 10.1074/jbc.M504454200. [DOI] [PubMed] [Google Scholar]
- 42.Tsvetkov PO, Kulikova AA, Golovin AV, Tkachev YV, Archakov AI, Kozin SA, Makarov AA. Minimal Zn2+ Binding Site of Amyloid-β. Biophysical Journal. 2010;99:L84–L86. doi: 10.1016/j.bpj.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller Y, Ma B, Nussinov R. Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc Natl Acad Sci U S A. 2010;107:9490–9495. doi: 10.1073/pnas.0913114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gardberg A, Dice L, Pridgen K, Ko J, Patterson P, Ou S, Wetzel R, Dealwis C. Structures of Aβ- Related Peptide- Monoclonal Antibody Complexes. Biochemistry. 2009;48:5210–5217. doi: 10.1021/bi9001216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, Rothe C, Urban M, Bardroff M, Winter M, Nordstedt C, Loetscher H. Gantenerumab: a novel human anti-Abeta antibody demonstrates sustained cerebral amyloid-beta binding and elicits cell-mediated removal of human amyloid-beta. J Alzheimers Dis. 2012;28:49–69. doi: 10.3233/JAD-2011-110977. [DOI] [PubMed] [Google Scholar]
- 46.Morgado I, Wieligmann K, Bereza M, Rönicke R, Meinhardt K, Annamalai K, Baumann M, Wacker J, Hortschansky P, Maleševid M. Molecular basis of β-amyloid oligomer recognition with a conformational antibody fragment. Proceedings of the National Academy of Sciences. 2012;109:12503–12508. doi: 10.1073/pnas.1206433109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haupt C, Morgado I, Kumar ST, Parthier C, Bereza M, Hortschansky P, Stubbs MT, Horn U, Fändrich M. Amyloid fibril recognition with the conformational B10 antibody fragment depends on electrostatic interactions. Journal of molecular biology. 2011;405:341–348. doi: 10.1016/j.jmb.2010.10.059. [DOI] [PubMed] [Google Scholar]
- 48.Miles LA, Crespi GA, Doughty L, Parker MW. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Scientific reports. 2013;3 doi: 10.1038/srep01302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feinberg H, Saldanha JW, Diep L, Goel A, Widom A, Veldman GM, Weis WI, Schenk D, Basi GS. Crystal structure reveals conservation of amyloid-beta conformation recognized by 3D6 following humanization to bapineuzumab. Alzheimers Res Ther. 2014;6:31. doi: 10.1186/alzrt261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miles LA, Crespi GA, Doughty L, Parker MW. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Sci Rep. 2013;3:1302. doi: 10.1038/srep01302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Crespi GA, Hermans SJ, Parker MW, Miles LA. Molecular basis for mid-region amyloid-β capture by leading Alzheimer’s disease immunotherapies. Scientific reports. 2015;5 doi: 10.1038/srep09649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Abeta(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol. 2015;22:499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.La Porte SL, Bollini SS, Lanz TA, Abdiche YN, Rusnak AS, Ho W-H, Kobayashi D, Harrabi O, Pappas D, Mina EW. Structural basis of C-terminal β-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer’s disease. Journal of molecular biology. 2012;421:525–536. doi: 10.1016/j.jmb.2011.11.047. [DOI] [PubMed] [Google Scholar]
- 54.Skrabana R, Dvorsky R, Sevcik J, Novak M. Monoclonal antibody MN423 as a stable mold facilitates structure determination of disordered tau protein. Journal of structural biology. 2010;171:74–81. doi: 10.1016/j.jsb.2010.02.016. [DOI] [PubMed] [Google Scholar]
- 55.Sevcik J, Skrabana R, Dvorsky R, Csokova N, Iqbal K, Novak M. X-ray structure of the PHF core C- terminus: insight into the folding of the intrinsically disordered protein tau in Alzheimer’s disease. FEBS Lett. 2007;581:5872–5878. doi: 10.1016/j.febslet.2007.11.067. [DOI] [PubMed] [Google Scholar]
- 56.Shih HH, Tu C, Cao W, Klein A, Ramsey R, Fennell BJ, Lambert M, Shúilleabháin DN, Autin B, Kouranova E. An ultra-specific avian antibody to phosphorylated tau protein reveals a unique mechanism for phosphoepitope recognition. Journal of Biological Chemistry. 2012;287:44425–44434. doi: 10.1074/jbc.M112.415935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kontsekova E, Zilka N, Kovacech B, Skrabana R, Novak M. Identification of structural determinants on tau protein essential for its pathological function: novel therapeutic target for tau immunotherapy in Alzheimer’s disease. Alzheimers Res Ther. 2014;6:45. doi: 10.1186/alzrt277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo Y, Ma B, Nussinov R, Wei G. Structural Insight into Tau Protein’s Paradox of Intrinsically Disordered Behavior, Self-Acetylation Activity, and Aggregation. J Phys Chem Lett. 2014;5:3026–3031. doi: 10.1021/jz501457f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siddiqua A, Luo Y, Meyer V, Swanson MA, Yu X, Wei G, Zheng J, Eaton GR, Ma B, Nussinov R, Eaton SS, Margittai M. Conformational basis for asymmetric seeding barrier in filaments of three- and four-repeat tau. J Am Chem Soc. 2012;134:10271–10278. doi: 10.1021/ja303498q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu L, Zheng J, Margittai M, Nussinov R, Ma B. Hyperphopsphorylation Promotes Tau Aggregation by Modulating Filament Structure and Stability. ACS Chem Neurosci. 2016 doi: 10.1021/acschemneuro.5b00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanyo ZF, Pan K-M, Williamson RA, Burton DR, Prusiner SB, Fletterick RJ, Cohen FE. Antibody binding defines a structure for an epitope that participates in the PrP C→ PrP Sc conformational change. Journal of molecular biology. 1999;293:855–863. doi: 10.1006/jmbi.1999.3193. [DOI] [PubMed] [Google Scholar]
- 62.Swayampakula M, Baral PK, Aguzzi A, Kav NN, James MN. The crystal structure of an octapeptide repeat of the Prion protein in complex with a Fab fragment of the POM2 antibody. Protein Science. 2013;22:893–903. doi: 10.1002/pro.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Antonyuk S, Trevitt C, Strange R, Jackson G, Sangar D, Batchelor M, Cooper S, Fraser C, Jones S, Georgiou T. Crystal structure of human prion protein bound to a therapeutic antibody. Proceedings of the National Academy of Sciences. 2009;106:2554–2558. doi: 10.1073/pnas.0809170106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eghiaian F, Grosclaude J, Lesceu S, Debey P, Doublet B, Tréguer E, Rezaei H, Knossow M. Insight into the PrPC→ PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10254–10259. doi: 10.1073/pnas.0400014101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baral PK, Wieland B, Swayampakula M, Polymenidou M, Rahman MH, Kav NN, Aguzzi A, James MN. Structural studies on the folded domain of the human prion protein bound to the Fab fragment of the antibody POM1. Acta Crystallographica Section D: Biological Crystallography. 2012;68:1501–1512. doi: 10.1107/S0907444912037328. [DOI] [PubMed] [Google Scholar]
- 66.Baral PK, Swayampakula M, Aguzzi A, James MN. X-ray structural and molecular dynamical studies of the globular domains of cow, deer, elk and Syrian hamster prion proteins. Journal of structural biology. 2015;192:37–47. doi: 10.1016/j.jsb.2015.08.014. [DOI] [PubMed] [Google Scholar]
- 67.Abskharon RN, Giachin G, Wohlkonig A, Soror SH, Pardon E, Legname G, Steyaert J. Probing the N-terminal β-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. Journal of the American Chemical Society. 2014;136:937–944. doi: 10.1021/ja407527p. [DOI] [PubMed] [Google Scholar]
- 68.Klein FA, Zeder-Lutz G, Cousido-Siah A, Mitschler A, Katz A, Eberling P, Mandel J-L, Podjarny A, Trottier Y. Linear and extended: a common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Human molecular genetics. 2013;22:4215–4223. doi: 10.1093/hmg/ddt273. [DOI] [PubMed] [Google Scholar]
- 69.De Genst E, Chirgadze DY, Klein FA, Butler DC, Matak-Vinkovid D, Trottier Y, Huston JS, Messer A, Dobson CM. Structure of a single-chain fv bound to the 17 N-terminal residues of huntingtin provides insights into pathogenic amyloid formation and suppression. Journal of molecular biology. 2015;427:2166–2178. doi: 10.1016/j.jmb.2015.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Genst EJ, Guilliams T, Wellens J, O’Day EM, Waudby CA, Meehan S, Dumoulin M, Hsu S-TD, Cremades N, Verschueren KH. Structure and properties of a complex of α-synuclein and a single-domain camelid antibody. Journal of molecular biology. 2010;402:326–343. doi: 10.1016/j.jmb.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 71.Hong S, Kim D. Interaction between bound water molecules and local protein structures: A statistical analysis of the hydrogen bond structures around bound water molecules. Proteins. 2016;84:43–51. doi: 10.1002/prot.24953. [DOI] [PubMed] [Google Scholar]
- 72.Ahmed MH, Spyrakis F, Cozzini P, Tripathi PK, Mozzarelli A, Scarsdale JN, Safo MA, Kellogg GE. Bound water at protein-protein interfaces: partners, roles and hydrophobic bubbles as a conserved motif. PLoS One. 2011;6:e24712. doi: 10.1371/journal.pone.0024712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.MacRaild Christopher A, Richards Jack S, Anders Robin F, Norton Raymond S. Antibody Recognition of Disordered Antigens. Structure. 2016;24:148–157. doi: 10.1016/j.str.2015.10.028. [DOI] [PubMed] [Google Scholar]
- 74.Ma B, Kumar S, Tsai CJ, Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 75.Tsai CJ, Kumar S, Ma B, Nussinov R. Folding funnels, binding funnels, and protein function. Protein Sci. 1999;8:1181–1190. doi: 10.1110/ps.8.6.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsai CJ, Ma B, Nussinov R. Folding and binding cascades: shifts in energy landscapes. Proc Natl Acad Sci U S A. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumar S, Ma B, Tsai CJ, Sinha N, Nussinov R. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci. 2000;9:10–19. doi: 10.1110/ps.9.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei G, Xi W, Nussinov R, Ma B. Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem Rev. 2016 doi: 10.1021/acs.chemrev.5b00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McCord LA, Liang WG, Dowdell E, Kalas V, Hoey RJ, Koide A, Koide S, Tang W-J. Conformational states and recognition of amyloidogenic peptides of human insulin-degrading enzyme. Proceedings of the National Academy of Sciences. 2013;110:13827–13832. doi: 10.1073/pnas.1304575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Teplyakov A, Obmolova G, Canziani G, Zhao Y, Gutshall L, Jung SS, Gilliland GL. His-tag binding by antibody C706 mimics β-amyloid recognition. Journal of Molecular Recognition. 2011;24:570–575. doi: 10.1002/jmr.1069. [DOI] [PubMed] [Google Scholar]
- 81.Sevcik J, Skrabana R, Dvorsky R, Csokova N, Iqbal K, Novak M. X-ray structure of the PHF core C-terminus: insight into the folding of the intrinsically disordered protein tau in Alzheimer’s disease. FEBS letters. 2007;581:5872–5878. doi: 10.1016/j.febslet.2007.11.067. [DOI] [PubMed] [Google Scholar]