

Abstract
Objectives
The diagnosis of chronic intestinal pseudo-obstruction (CIPO) has depended on clinical features, manometry, and imaging. This report aimed to determine the efficacy of sequencing the ACTG2 gene for diagnosis. In addition, the goal was to determine how often a mutation would be found in our randomly collected cohort of probands and those probands published previously.
Methods
Whole exome sequencing was performed in 4 probands with CIPO. Subsequently, only the ACTG2 gene was sequenced in another 24 probands (total 28). We analyzed published data of 83 probands plus our 28 [total 111] and determined how many had pathogenic variants and the precise genotype.
Results
Whole exome and Sanger sequencing revealed a pathogenic variant in the ACTG2 gene in 4/28 of our probands and in 45 out of 83 published probands [49/111(44.1%)]. Moreover, a mutational hotspot in the ACTG2 gene was recognized. Genetic heterogeneity is evident.
Conclusion
Pooled gene sequencing results from one individual in each of 111 families enabled a precise diagnosis of an ACTG2 mutation in 49 (44%). The benefit to patients and families of early confirmation of a motility disorder not only helps avoid unnecessary intervention, but also enables institution of appropriate treatments and avoidance of secondary disorders such as malnutrition and poor growth. Knowledge of a pathogenic variant in a parent, with a 50% risk of recurrence, provides an opportunity for genetic counseling.
Keywords: Visceral myopathy, gastroparesis, gastrointestinal transplantation, mega-ureter
INTRODUCTION
Chronic Intestinal Pseudo-Obstruction [CIPO], the nomenclature for which has included megacystis-microcolon-intestinal hypoperistalsis syndrome and hollow visceral myopathy.1–4 [MIM 155310] is a mostly severely debilitating disorder of enteric smooth muscle. Other forms of CIPO may occur as a consequence of mitochondrial disease.5 This is a disorder that frequently demands total parenteral nutrition (TPN) for decades and which may lead eventually to total intestinal, liver, pancreas and spleen transplantation, or death before such dramatic interventions. The clinical features of this progressive disorder encompass a variable phenotype. Gastro-intestinal symptoms and signs due to gastroparesis typically include abdominal distension and pain, nausea, and vomiting. Often, there is a need for abdominal surgery, which may include gastrostomy, ileostomy, intestinal resection, or colectomy. Survivors with intestinal failure may need total gastro-intestinal transplantation. Megacystis and megaureter, often evident at birth, may also be detected prenatally by ultrasound imaging. Bladder involvement is almost invariable in all and frequently severe. Lifetime catheterization of the bladder is almost invariable in all who have megacystis despite surgical efforts to limit the size of the bladder. A high mortality rate attends this disorder 6–9 with one report indicating a 19.7% survival rate.6
The genetic basis for CIPO was uncertain until the first Finish report3 of a family in which an autosomal dominant pathogenic variant in the enteric smooth muscle actin γ-2 (ACTG2) gene was determined in adult onset visceral myopathy. Additional families with pathogenic variants in this gene have subsequently been identified, with 45 probands reported3,4,10–16 from randomly collected families. However, some families manifest autosomal dominant inheritance but do not have an ACTG2 mutation, implying that one or more genes remain to be discovered.
This report focuses on four probands with ACTG2 pathogenic variants from 4 families with severe CIPO and megacystis, out of a randomly collected cohort of 28 probands.
METHODS
Four of our probands had whole exome sequencing by the University of Washington Center for Mendelian Genomics, with us analyzing the data provided. After a single pathogenic variant was found in one proband, we proceeded with Sanger sequencing of our other 24 probands with their consent. This study received IRB approval (#H-23846/1797B) at Boston University School of Medicine.
Whole Exome Sequencing
Initial quality control entailed DNA quantification, sex typing and molecular “fingerprinting” using a high frequency genotyping assay. Library construction and exome capture were automated (Perkin-Elmer Janus II) in a 96-well plate format. 1 ug of genomic DNA was subjected to a series of shotgun library construction steps, including fragmentation through acoustic sonication (Covaris), end-polishing and A-tailing, ligation of sequencing adaptors, and PCR amplification with 8 bp barcodes for multiplexing. Libraries undergo exome capture using the Roche/Nimblegen SeqCap EZ v2.0 (~36.5 MB target). Prior to sequencing, the library concentration is determined by triplicate qPCR and molecular weight distributions verified on the Agilent Bioanalyzer (150 ± 15 bp). Barcoded exome libraries were pooled using liquid handling robotics prior to clustering (Illumina cBot) and loading. Massively parallel sequencing-by-synthesis with fluorescently labeled, reversibly terminating nucleotides was carried out on the HiSeq sequencer.
The sequencing analysis pipeline consists of base calling, alignment, local realignment, duplicate removal, quality recalibration, data merging, variant detection, genotyping and annotation using a combined suite of Illumina software, other software packages (Genome Analysis ToolKit [GATK], Picard, BWA-MEM, SAMTools, and in-house custom scripts). Variant detection and genotyping are performed using the HaplotypeCaller (HC) tool from GATK (3.2). A variant quality score recalibration (VQSR) on the “raw” variant call format (VCF) is used to generate a filtered VCF call set.
Data quality control included an assessment of: (1) total reads (minimum of 50 million PE50 reads); (2) library complexity (3) capture efficiency (4) coverage distribution: 90% at 8X required for completion; (5) capture uniformity; (6) raw error rates; (7) Transition/Transversion ratio (Ti/Tv); (8) distribution of known and novel variants relative to dbSNP is typically < 7% (9) fingerprint concordance > 99%; (10) sample homozygosity and heterozygosity and (11) sample contamination validation. Exome completion is defined as having > 90% of the exome target at > 8X coverage and >80% of the exome target at > 20X coverage. Typically this requires mean coverage of the target at 50–60X. The SeattleSeq Annotation Server (http://gvs.gs.washington.edu/SeattleSeqAnnotation/) was used to annotate the final VCF file.
Sanger Sequencing
Primers flanking each of the 8 coding exons and flanking intronic regions of ACTG2 were designed using the Primer3 program (http://bioinfo.ut.ee/primer3/). After treatment with ExoSAP-IT, the PCR product was sequenced with the BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems) followed by capillary electrophoresis on an ABI 3730 sequencer.
RESULTS
Our patients were randomly recruited over at least fifteen years. All had the typical symptoms and signs of CIPO with or without megacystis. These included bloating, abdominal distention and pain, pseudo-obstruction, nausea, vomiting and failure to thrive, as well as weight loss.
The clinical data for our four probands (Cases #1, 3, 4, and 5) and their family members (cases #2, 6, and 7) with ACTG2 pathogenic variants in this report (Table 1) uniformly reflect severe CIPO and megacystis. Megacystis was present prenatally and evident at birth in all 7. Three died at 6 months, 2 years, and 11.5 years of age. One mother at 38 years of age suffering intestinal failure had total visceral exenteration that included her stomach, intestine, liver, pancreas, gall-bladder and spleen followed by multi-organ transplantation. She had required TPN for 35 years, from the age of 3. Four of the 7 needed long-term TPN. Three each had gastrostomy, colectomy, and ileostomy. Because of the megacystis, 6 have endured life-time bladder catheterization.
Table 1.
Clinical Data of 4 probands (#1, 3, 4, 5) and 3 affected family members (2-A, 6-B, and 7-B) with CIPO and Megacystis
| Clinical Data | Case 1-A | Case 2-A | Case 3 | Case 4 | Case 5-B | Case 6-B | Case 7-B |
|---|---|---|---|---|---|---|---|
| ACTG2 Mutation | R257C | R257C | R257C | R257C | R40H | R40H* | R40H* |
| Current Age | 39 | 6 | Died | Died | 3 | Died | 28 |
| Age of Onset | Birth | Prenatal | Prenatal | Birth | Birth | Prenatal | Birth |
| Age of Death | − | − | 2 | 11.5 | − | 6 mths | − |
| Gender | F | F | F | M | M | M | M |
| Parent or Sib affected | − | + | − | − | + | + | − |
| Pseudo-Obstruction | + | + | + | + | + | + | + |
| Colectomy | + | + | − | − | − | − | + |
| Ileostomy | + | + | − | − | − | − | + |
| Cholecystectomy | + | − | − | − | − | − | − |
| Cholelithiasis | + | − | − | − | − | − | − |
| Microcolon | − | − | − | − | − | − | − |
| Gastrostomy | + | + | − | + | − | − | − |
| Nissen Fundoplication | + | + | − | − | − | − | − |
| Small bowel resection | + | + | − | − | − | − | − |
| Malrotation/Volvulus | − | − | + | + | − | − | − |
| GE Reflux | + | + | + | + | − | − | − |
| Cirrhosis/Liver Failure | + | − | − | + | − | − | − |
| Long-term TPN dependence | 35 years | 6 years | + | + | − | − | − |
| Organ Transplantation | + | − | − | − | − | − | − |
| Megacystis | + | + | + | + | + | + | + |
| Long-term Bladder catheterization | − | + | + | + | + | + | + |
| Hydronephrosis | + | − | − | − | + | + | − |
| Premature labor | + | − | N/K | + | − | + | + |
| Other medical disorders | Thrombosis inferior vena cava/Renal vein | VSD | − | Pancreatitis Seizures | − | − | N/K |
N/K = not known
By inference
+ = Yes
A = Family A
B = Family B
− = No
The parents of two of our four probands had no ACTG2 pathogenic variants, in all likelihood reflecting de novo variants.
In those without an ACTG2 mutation, the noted details reflect only information from the last contact which, for many, was at least ten years. The onset was apparent prenatally or by two years of age in 18/24. 21 were female. Two children died. 8/24 had colectomies, 2/24 had malrotation or volvulus, 5/24 had ileostomy or jejunostomy or cecostomy, 10/24 needed TPN [one for 29 years], and 11/24 had megacystis or urinary retention. All were Caucasians. All had manometry (dysmotility patterns not known by us) and/or endoscopy.
DISCUSSION
An autosomal dominant mode of inheritance with complete penetrance is clear in the familial cases with ACTG2 pathogenic variants. Autosomal recessive inheritance of CIPO is highly likely in children of consanguineous unions 17,18 or in affected siblings with healthy parents (bearing in mind gonadal mosaicism). A homozygous pathogenic variant in the RAD21 gene was reported in one consanguineous family.18 4/28 of our cohort and 15/27 in the report by Wangler et al.4 were found to harbor an ACTG2 pathogenic variant. The difference between these two randomly collected cohorts remains unexplained, but may reflect inaccurate clinical diagnoses.
Of the reported 45 probands plus our 4 with CIPO, 33/49 (73.3%) have pathogenic variants at either amino acid R178 or R257. The most common pathogenic variants observed, R178C and R257C, involve a C>T transition at CpG dinucleotides (Figure 1). Methylated CpG (mCpG) sequences frequently undergo mutation caused by random deamination of 5-methycytosines leading to a C>T transition. Thus, the two amino acids, R178 and R257, are likely mutational hotspots in ACTG2.
Figure 1.
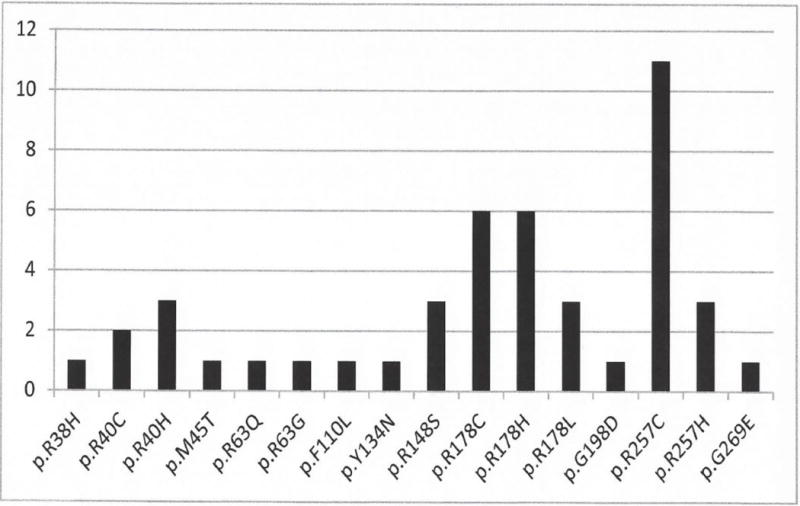
Shown are the 16 pathogenic variants reported in the ACTG2 gene and the number of times each mutation was observed in 45 probands.
Thus far, all pathogenic variants detected in the ACTG2 gene have been missense variants with no exon or whole gene deletions/duplications being reported. These variants lead to changes in protein function, impair ACTG2 polymerization, and contribute to reduced smooth muscle cell contractility10. While pathogenic variants in recessive and mitochondrial genes that cause CIPO and or bladder involvement are known, they are invariably associated with disorders or defects in other organ systems.19 From our 4 probands, together with 45 other published molecularly confirmed probands, a few early insights are emerging, but further gene discovery is awaited.
The highly variable phenotype and the few that have had molecular studies have made a reliable estimate of the incidence or prevalence of CIPO with or without megacystis20 difficult to determine. The realization that a proven affected parent may have only minor gastrointestinal symptoms [bloating, constipation or diarrhea or irritable bowel syndrome] complicates ascertainment. It is clear, however, that females are much more often affected than males.6 Compilation of our probands with those published3,4,10–16 thus far show 49/111 (44.1%) with ACTG2 pathogenic variants. No affected siblings with ACTG2 pathogenic variants with parents shown not to harbor the culprit pathogenic variant have been reported. Hence, germline mosaicism remains as a possibility in such cases. While genotype-phenotype correlations are still unclear, de novo pathogenic variants in ACTG2 may convey more severe disease than when inherited.21
The spectrum of severity of CIPO with or without megacystis is wide, ranging from profound prenatal or neonatal megacystis with or without prune belly syndrome22 and CIPO, to non-specific constipation and abdominal bloating without bladder involvement. Our patients and the majority of those published with ACTG2 pathogenic variants, have or had severe gastroparesis or bladder problems. Repeated surgical interventions have included intestinal decompression, gastrostomy, colectomy, small gut resection, ileostomy, colostomy and Nissen fundoplication. One of our patients at 38 years of age with intestinal failure, underwent transplantation of her entire intestine, stomach, liver, pancreas, and spleen.
Affected children have invariably been subject to multiple and repeated diagnostic efforts that included intestinal biopsy, radiologic studies, manometry and cystoscopy. Now from a blood sample a precise diagnosis can be made in a week, by sequencing the ACTG2 gene. The benefit to patients and families of early confirmation of a motility disorder not only helps avoid unnecessary intervention, but also enables institution of appropriate treatments and avoidance of secondary disorders such as malnutrition and poor growth. Moreover, knowledge of a familial ACTG2 pathogenic variant in a parent provides an opportunity for avoidance or prevention of a recurrence via prenatal genetic diagnosis or preimplantation genetic diagnosis.23
What is Known
Chronic Intestinal Pseudo-Obstruction with or without megacystis is a debilitating dominant or recessive disorder with a high mortality rate and a variable phenotype.
Diagnosis has depended on clinical signs, manometry, and radiology.
Pathogenic mutations in a single gene have been recognized recently.
Very few families have had gene sequencing.
What is New
4/28 of our probands had an ACTG2 gene mutation, enabling a precise diagnosis.
Sequencing of this gene in our 28 probands and 83 published probands shows for the first time that 49/111 (44.1%) have an ACTG2 mutation.
For the first time, a mutational hotspot is recognized.
Acknowledgments
Whole exome sequencing of 4 families was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant U54HG006493 to Drs. Debbie Nickerson, Michael Bamshad, and Suzanne Leal. The bioinformatic analyses were done at The Center for Human Genetics.
We thank Malgorzata Nowaczyk, M.D. and Gina Cowing, MS, CGC for referring one family to us.
Grant Support: National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant U54HG006493
Footnotes
Disclosures: None of the authors have any disclosures to make. The authors have no conflicts of interests relevant to this article to disclose.
AUTHOR CONTRIBUTIONS
Dr. A. Milunsky conceptualized and designed the study, reviewed the literature, analyzed the data, drafted the initial manuscript, and approved the final manuscript as submitted.
Dr. Baldwin did the bioinformatics analysis of the whole exome sequencing and the analysis of the Sanger sequencing data.
Dr. Zhang performed the Sanger sequencing.
Mr. Primack assisted in the Sanger sequencing and data analysis.
Dr. Curnow provided detailed clinical data on one of our four families.
Dr. J. Milunsky assisted in the analysis of the molecular data, reviewed and revised the manuscript and approved the final manuscript as submitted.
References
- 1.Berdon W, Baker D, Blanc W, et al. Megacystis-microcolon-intestinal hypoperistalsis syndrome: a new cause of intestinal obstruction in the newborn. Report of radiologic findings in five newborn girls. AJR Am J Roentgenol. 1976;126:957–964. doi: 10.2214/ajr.126.5.957. [DOI] [PubMed] [Google Scholar]
- 2.Puri P, Shinkai M. Megacystis microcolon intestinal hypoperistalsis syndrome. Semin Pediatr Surg. 2005;14:58–63. doi: 10.1053/j.sempedsurg.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 3.Lehtonen HJ, Sipponen T, Tojkander S, et al. Segregation of a missense variant in enteric smooth muscle actin gamma-2 with autosomal dominant familial visceral myopathy. Gastroenterology. 2012;143:1482–1491. doi: 10.1053/j.gastro.2012.08.045. [DOI] [PubMed] [Google Scholar]
- 4.Wangler MF, Gonzaga-Jauregui C, Gambin T, et al. Heterozygous de novo and inherited mutations in the smooth muscle actin (ACTG2) gene underlie megacystis-microcolon-intestinal hypoperistalsis syndrome. PLoS Genet. 2014;10:e1004258. doi: 10.1371/journal.pgen.1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amoit A, Tchikviladzé M, Joly F, et al. Frequency of mitochondrial defects in patients with chronic intestinal pseudo-obstruction. Gastroenterology. 2009;137:101–9. doi: 10.1053/j.gastro.2009.03.054. [DOI] [PubMed] [Google Scholar]
- 6.Gosemann JH, Puri P. Megacystis microcolon intestinal hypoperistalsis syndrome: systematic review of outcome. Pediatr Surg Int. 2011;27:1041–1046. doi: 10.1007/s00383-011-2954-9. [DOI] [PubMed] [Google Scholar]
- 7.Sabbagh C, Amiot A, Maggiori L, et al. Non-transplantation surgical approach for chronic intestinal pseudo-obstruction: analysis of 63 adult consecutive cases. Neurogastroenterol Motil. 2013;25:680–686. doi: 10.1111/nmo.12191. [DOI] [PubMed] [Google Scholar]
- 8.Ueno T, Wada M, Hoshino K, et al. A national survey of patients with intestinal motility disorder who are potential candidates for intestinal transplantation in Japan. Transplant Proc. 2013;45:2029–2031. doi: 10.1016/j.transproceed.2013.01.092. [DOI] [PubMed] [Google Scholar]
- 9.Lauro A, Zanfi C, Pellegrini S, et al. Isolated intestinal transplant for chronic intestinal pseudo-obstruction in adults: Long-term outcome. Transplant Proc. 2013;45:3351–3355. doi: 10.1016/j.transproceed.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 10.Halim D, Hofstra RM, Signorile L, et al. ACTG2 variants impair actin polymerization in sporadic megacystis microcolon intestinal hypoperistalsis syndrome. Hum Mol Genet. 2015;25:571–583. doi: 10.1093/hmg/ddv497. [DOI] [PubMed] [Google Scholar]
- 11.Klar J, Raykova D, Gustafson E, et al. Phenotypic expansion of visceral myopathy associated with ACTG2 tandem base substitution. Eur J Hum Genet. 2015;23:1679–1683. doi: 10.1038/ejhg.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matera I, Rusmini M, Guo Y, et al. Variants of the ACTG2 gene correlate with degree of severity and presence of megacystis in chronic intestinal pseudo-obstruction. Eur J Hum Genet. 2016:1–5. doi: 10.1038/ejhg.2015.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holla Ø, Bock G, Busk Ø, et al. Familial visceral myopathy diagnosed by exome sequencing of a patient with chronic intestinal pseudo-obstruction. Endoscopy. 2014;46:533–537. doi: 10.1055/s-0034-1365142. [DOI] [PubMed] [Google Scholar]
- 14.Tuzovic L, Tang S, Miller RS, et al. New insights into the genetics of fetal megacystis: ACTG2 mutations, encoding x03B3;-2 smooth muscle actin in megacystis microcolon intestinal hypoperistalsis syndrome (Berdon Syndrome) Fetal Diagn Ther. 2015;38:296–306. doi: 10.1159/000381638. [DOI] [PubMed] [Google Scholar]
- 15.Thorson W, Diaz-Horta O, Foster J, et al. De novo ACTG2 mutations cause congenital distended bladder, microcolon, and intestinal hypoperistalsis. Hum Genet. 2013;133:737–742. doi: 10.1007/s00439-013-1406-0. [DOI] [PubMed] [Google Scholar]
- 16.Annerén G, Meurling S, Olsen L. Megacystis-microcolon-intestinal hypoperistalsis syndrome(MMIHS), an autosomal recessive disorder: Clinical reports and review of the literature. Am J Med Genet. 1991;41:251–254. doi: 10.1002/ajmg.1320410224. [DOI] [PubMed] [Google Scholar]
- 17.Xiao Lu W, Wang YJ, et al. Mutation in actin [gamma]-2 responsible for megacystis microcolon intestinal hypoperistalis syndrome in four Chinese patients. J Pediatr Gastroenterol Nutr. 2016 doi: 10.1097/MPG.0000000000001204. [DOI] [PubMed] [Google Scholar]
- 18.Bonora E, Bianco F, Cordeddu L, et al. Mutations in RAD21 disrupt regulation of APOB in patients with chronic intestinal pseudo-obstruction. Gastroenterology. 2015;148:771–782. doi: 10.1053/j.gastro.2014.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antonucci A, Fronzoni L, Cogliandro L, et al. Chronic intestinal pseudo-obstruction. World J Gastroenterol. 2008;14:2953–2961. doi: 10.3748/wjg.14.2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higman D, Peters P, Stewart M. Familial hollow visceral myopathy with varying urological manifestations. Br J Urol. 1992;70:435–438. doi: 10.1111/j.1464-410x.1992.tb15804.x. [DOI] [PubMed] [Google Scholar]
- 21.Wangler MF, Beaudet AL. ACTG2-Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2015. [Google Scholar]
- 22.Richer J, Milewicz DM, Gow R, et al. R179H mutation in ACTA2 expanding the phenotype to include prune-belly sequence and skin manifestations. Am J Med Genet Part A. 2012;158A:664–668. doi: 10.1002/ajmg.a.35206. [DOI] [PubMed] [Google Scholar]
- 23.Milunsky A, Milunsky JM, editors. Genetic disorders and the fetus: Diagnosis, prevention and treatment. 7th. Hoboken: Wiley & Sons; 2016. [Google Scholar]