

Abstract
Experimental Lyme arthritis provides a mouse model for exploring the development of pathology following infection of C3H mice with Borrelia burgdorferi. Infected mice develop a reliable inflammatory arthritis of the ankle joint with severity that typically peaks around two to three weeks post-infection and then undergoes spontaneous resolution. This makes experimental Lyme arthritis an excellent model for investigating the mechanisms that drive both the development and resolution phases of inflammatory disease. Eicosanoids are powerful lipid mediators of inflammation and are known to regulate multiple aspects of inflammatory processes. While much is known about the role of eicosanoids in regulating immune responses during autoimmune disease and cancer, relatively little is known about their role during bacterial infection. In this review, we discuss the role of eicosanoid biosynthetic pathways in mediating inflammatory responses during bacterial infection using experimental Lyme arthritis as a model system. We point out the critical role eicosanoids play in disease development and highlight surprising differences between sterile autoimmune responses and those occurring in response to bacterial infection. These differences should be kept in mind when designing therapies and treatments for inflammatory diseases.
Keywords: Eicosanoids, Lyme Disease, Borrelia burgdorferi, COX-2, Lipidomics
1. Introduction
Lyme Disease is the most common arthropod-borne disease in the United States, with an estimated 300,000 new cases each year [1]. The disease is caused by infection with the spirochete, Borrelia burgdorferi, and is transmitted to the mammalian host via the bite of infected Ixodes ticks [2]. In humans, clinical diagnosis is typically aided by the development of an expanding bulls-eye rash (erythema migrans) at the site of the tick bite [3]. Prompt treatment of the infected individual with antibiotics at this stage will generally result in clearance of the spirochetes and cessation of symptoms [4]. However, a delay in treatment will often result in the manifestation of secondary disease symptoms, most commonly arthritis of the large joints, carditis, and neurological problems, which in some cases are quite difficult to treat [5].
Experimental Lyme borreliosis is a mouse model of Lyme Disease that mimics many of the disease symptoms (especially arthritis and carditis) seen in infected humans. Disease development is genetically regulated, with C3H mice being disease susceptible and C57BL/6 mice disease resistant [6]. Figure 1 provides an overview of immune parameters important during a typical infection in C3H mice. Following inoculation into the mammalian host, the spirochetes remain sequestered at the inoculation site for several days before systemically disseminating [7]. B. burgdorferi is a relatively slow growing bacterium, therefore the spirochetes gradually increase their numbers throughout the mammalian host for the next week or so, typically peaking two to three weeks post-infection [7]. The spirochetes have a tropism for joint tissue and colonize in the large joints, especially the knee and ankle, where they induce an inflammatory arthritis lasting several weeks. Antibody formation is thought to be the primary immune effector function responsible for control of B. burgdorferi numbers [8], therefore, as specific antibodies are generated the spirochetes are cleared from most tissues. However, sterile immunity is not achieved and the skin and urinary bladder remain infected most likely for the life of the host [9].
Figure 1. Parameters of experimental Lyme arthritis.
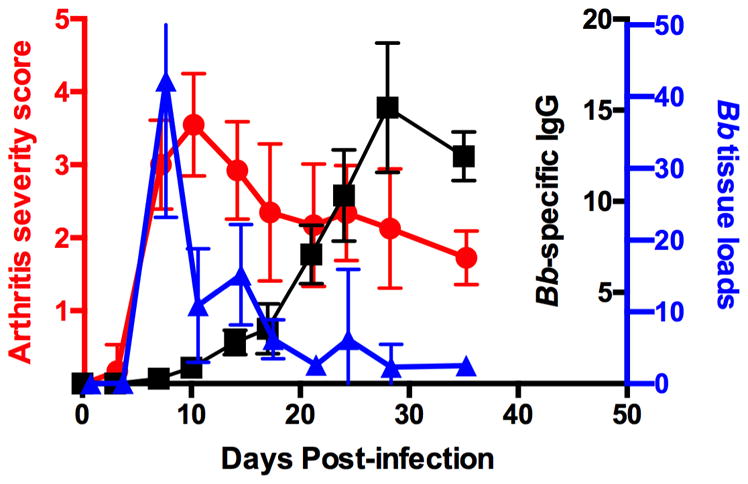
C3H mice were infected in the hind footpads with 1x105 B. burgdorferi and arthritis severity scores (red circles), Bb tissue loads (blue triangles), and Bb-specific IgG levels (black squares) were measured throughout the disease time course. Arthritis severity scores were determined from hematoxylin and eosin-stained tibiotarsal joints on a scale of 0–4. Bb tissue loads were determined by real-time PCR from knee tissue. Bb-specific IgG levels were determined by ELISA from serum. n=5 for each time point and the results are representative of two separate experiments. Figure was modified and used with permission from [15].
Experimental Lyme arthritis is an inflammatory arthritis and is dependent on the presence of the spirochetes in the joint tissue. As spirochete numbers increase systemically the microbes colonize the synovium and areas around the tendon sheaths, and in susceptible strains of mice this causes inflammation and arthritis development [6]. The development of disease is mediated by an influx of neutrophils and macrophages and is independent of participation by T and B cells [10]. This is mechanistically different than other commonly used murine models of arthritis, such as collagen-induced arthritis (CIA) or the K/BxN arthritis model, which depend upon the presence of auto-reactive antibodies and the activation of the adaptive immune response for development of disease [11]. Conversely, experimental Lyme arthritis is mediated solely by innate immunity [12]. However, B cells are thought to play an important role in Lyme arthritis resolution, as passive transfer of immune serum into B. burgdorferi-infected scid mice, which are devoid of T and B cells, causes spirochete clearance and disease resolution [8].
2. Eicosanoids in experimental Lyme arthritis
Eicosanoids are 20-carbon fatty acid metabolites of arachidonic acid (AA) that play numerous regulatory roles throughout the body (Figure 2), but are especially important during inflammatory responses [13]. Following tissue injury or infection AA is released from membrane stores primarily through the action of cytosolic phospholipase A2 (cPLA2). The free AA can then be used by several cell-specific enzymes of the cyclooxygenase (COX), lipoxygenase (LOX), or cytochrome P450 (CYP450) metabolic pathways as the substrate for the production of numerous bioactive lipids. These lipids have a wide range of biological activities and play a role in regulating many pathways, especially vascular tone and inflammation. In addition to AA, other fatty acids, such as the omega-3 fatty acids docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), can be metabolized by these same enzymes to produce other bioactive lipid metabolites, such as the protectins and resolvins [14].
Figure 2. Simplified schematic of the eicosanoid biosynthetic pathway.

Upon injury or infection arachidonic acid (AA) is released from membrane stores through the action of cytosolic phospholipase A2. AA is then acted upon by various enzymes (blue boxes) and converted into numerous bioactive lipid compounds (ovals) with wide ranging pharmacological effects.
Since experimental Lyme arthritis is an inflammatory arthritis it was anticipated that eicosanoids would be involved in regulating its development and resolution: however; which eicosanoids were involved and the timing of their production was unknown. To answer these questions we pioneered a new experimental technique to comprehensively analyze nearly the entire known eicosanoid metabolome (along with a few other bioactive lipids) from B. burgdorferi-infected joints over the complete time-course of infection [15]. We harvested ankle joints from B. burgdorferi-infected and sham-infected arthritis-susceptible C3H and arthritis-resistant DBA/2 mice at nine different time points during the course of disease. Infection of C3H mice with B. burgdorferi induced significant eicosanoid production across multiple metabolomic pathways; including the COX, LOX and CYP450 pathways, in addition to other non-enzymatic metabolic breakdown products (Figure 3). In general, most of the metabolic pathways in the susceptible C3H mice were activated at time points before and near the peak of inflammation. In contrast, several of these same pathways appeared to be actively suppressed in the disease-resistant DBA/2 mice [15]. This demonstrates a fundamentally different response to infection with B. burgdorferi in disease-resistant and –susceptible mouse strains and has important implications for disease development in humans. However, the mechanisms responsible for these differences remain to be elucidated.
Figure 3. Lipidomic profiling of temporal changes in eicosanoids and other related bioactive lipid biosynthetic pathways.

Heat map depicting fold changes in lipid mediators from B. burgdorferi-infected joints compared to uninfected (day 0). In addition, joint levels of selected lipid mediators are shown over time for n = 8–10 mice per time point. Figure was adapted with permission from [15].
In the C3H mice, many of the metabolites displayed a biphasic expression pattern, with an initial peak corresponding to the peak of inflammation and then a second peak around day 28 post-infection, which is typically the onset of resolution in this model. This correlates well with the hypothesis that some eicosanoids, for example the prostaglandin PGE2, may play a role in both the induction and resolution phases of the inflammatory response [16]. PGE2 and PGD2 were highly expressed COX-2 metabolites, and leukotrienes LTE4 and 5-HETE (5-hydroxyeicosatetraenoic acid) were the most highly expressed 5-LOX metabolites in the joints of infected C3H mice (Figure 3). These are known mediators of inflammatory responses so it is perhaps not surprising to find high levels in the infected tissue. We also found high levels of many of the 12/15-LOX metabolites, including the docosanoid protectin, 15-HETE, 12-HETE, and the hepoxilin HXA3. Production of these metabolites may reflect the tight control needed over inflammatory processes. HXA3 is known to recruit neutrophils [17] while 12-HETE can recruit monocytes by promoting the expression of MCP-1 [18]. In contrast, protectin D1 has been shown to inhibit neutrophil recruitment in vivo [19]. In addition, 15-HETE can be converted to the pro-resolution molecule, lipoxin LXA4, by 5-LOX and thus may be important for the induction of disease resolution [20]. We also found significantly higher levels of the docosanoid 17-HdoHE, a stable form of the protectin and resolvin biosynthetic intermediate, 17-HpDoHE, in infected C3H mouse joints. Finally, several metabolites from the CYP metabolic pathway were also highly expressed. The epoxyeicosatrienoic acids (EET), 14,15-EET and 11,12-EET can modulate inflammation by promoting vasodilation allowing inflammatory cell transmigration from the vasculature into infected tissue [21]. These results demonstrate the complex and cooperative nature of eicosanoid biosynthesis in regulating inflammation in response to infection.
By using this comprehensive lipidomics approach an overview model can be constructed of how activation of the eicosanoid metabolic pathway might regulate the development and resolution of inflammatory responses (Figure 4). Following recognition of infection by innate immune cells, cPLA2 and other specific eicosanoid metabolic enzymes are activated. cPLA2 releases AA from membrane stores and this is converted to specific eicosanoid metabolic products in various cell types, such as tissue macrophages and endothelial cells. PGE2 and PGD2 likely promote the onset of inflammation, along with specific cytokines and chemokines, by activating the local vasculature and increasing vascular permeability. Anti-inflammatory products, such as the PGD2 dehydration product 15d-PGJ2, are produced at the same time to modulate the response so that it is not too damaging to host tissue. 12-HETE and various EETs may help to control vascular tone during inflammatory responses and counterbalance pro-inflammatory signals. In cooperation with locally released cytokine and chemokine signals 12-HETE and HXA3 may participate in the recruitment of monocytes and neutrophils from the vasculature [18, 22]. These signals induce inflammation and result in arthritis development in the joints of infected mice. As Borrelia-specific antibody responses rise, the spirochetes are cleared from the joints inducing the production of pro-resolution mediators to decrease arthritis severity. Once the microbial threat is removed, alternatively activated macrophages can be recruited to remove tissue debris and release pro-resolving molecules, like the protectins, to restore the tissue to homeostasis and full functionality of the joint.
Figure 4. Model of lipid mediator regulation of experimental Lyme arthritis.
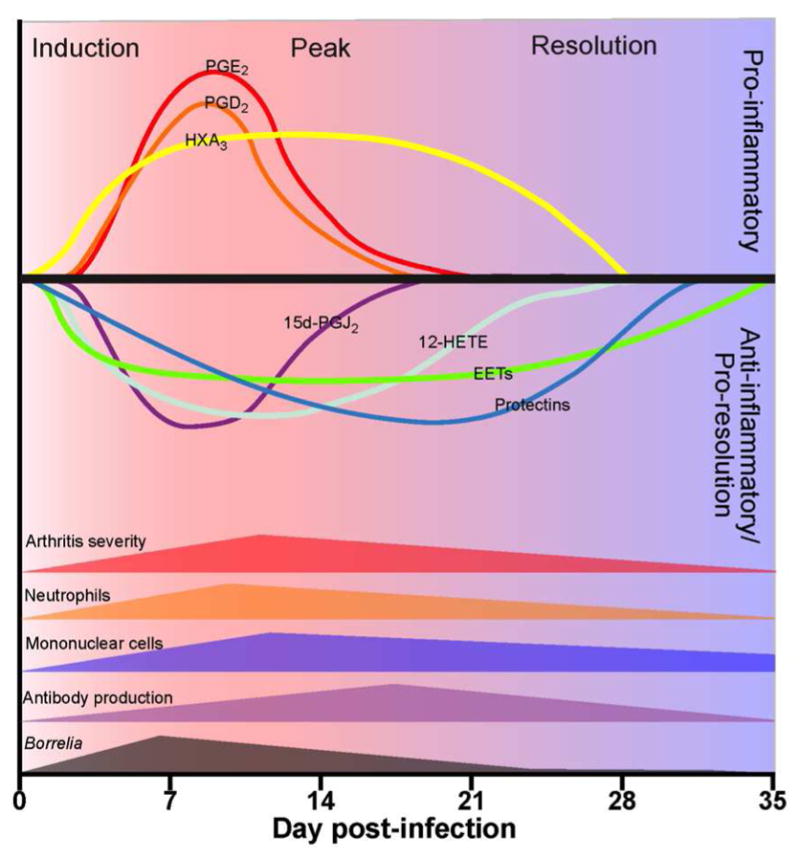
Following infection both pro-and anti-inflammatory mediators are produced and inflammation ensues. As the spirochetes are cleared by antibody and innate immune cells, other pro-resolution lipid mediators are produced which stimulate resolution and a return of the tissue to homeostasis. Figure is adapted with permission from [15].
3. Impact of eicosanoids on experimental Lyme arthritis
3.1 COX-2 inhibition
As demonstrated above, infection of C3H mice with B. burgdorferi induces the production of numerous eicosanoids in infected joint tissue. But what impact does the production of these compounds have on disease pathogenesis? Administration of prostaglandins into tissue is known to induce several of the cardinal signs of inflammation, such as pain, swelling and redness [23]. Inhibition of prostaglandin production via COX inhibition has been demonstrated to ameliorate arthritis development in several autoimmune models of arthritis, including CIA, K/BxN, and adjuvant-induced arthritis [24–26], but the effect this might have on an inflammatory arthritis was unclear. We infected C3H mice treated with a COX-2-specific inhibitor, celecoxib, or C3H mice deficient in COX-2 (COX-2−/−) with B. burgdorferi and followed arthritis development for 35 days [27]. We found that COX-2 inhibition or deletion did not have a significant impact on arthritis development, but rather inhibited arthritis resolution. This effect was not due to an alteration in development of Borrelia-specific antibody responses or failure of spirochete clearance from joint tissue as these were similar to control animals. Thus, by inhibiting prostaglandin synthesis via COX-2, we converted a transient inflammatory response into a chronic inflammatory disease. The mechanisms behind this phenomenon are not clear. COX-2-deficient mice infected with B. burgdorferi do not develop the typical joint swelling seen in wild-type mice, but the underlying recruitment of inflammatory cells, such as neutrophils and monocytes, appears to be unaffected [27]. The loss of COX-2 activity decreases the production of anti-inflammatory prostaglandins, like PGD2 and 15d-PGJ2, and thus may impact disease resolution. In addition, the COX-2 derived product, 15-HETE, can be converted to LXA4 by 5-LOX activity [20], and PGE2 can induce PMN to switch from producing LTB4 to LXA4 [16] and thus primarily impact disease resolution. In contrast, inhibition of COX-2 activity prevented the development of CIA or adjuvant-induced arthritis, while inhibition of COX-1 inhibited K/BxN arthritis via PGI2 inhibition [24–26]. The reasons for these differences in arthritis outcome when blocking COX are not clear; however, NSAIDs are a common treatment for arthritis patients. While the treatment is effective in alleviating pain and swelling associated with arthritis it does not slow down the underlying joint inflammation, and it may actually make it worse.
3.2 5-LOX inhibition
Similar results were found when we infected C3H mice deficient in 5-LOX with B. burgdorferi [28]. Mice deficient in 5-LOX products had also been shown to be resistant to the development of CIA and other models of autoimmune arthritis primarily by blocking LTB4-mediated recruitment of neutrophils into the joint [29, 30]. In experimental Lyme arthritis, infection of 5-LOX−/− mice resulted in normal development of Lyme arthritis, but a defect in arthritis resolution. These results demonstrate the complexity of the lipid mediator network and their cooperativity in regulating the inflammatory process. Interestingly, during our lipidomics study, we found that not only did COX-2−/− mice have an expected defect in prostaglandin production, but also an unexpected defect in production of some 5-LOX metabolites [15]. These results demonstrate we have much to learn before the tremendous potential of this regulatory network can be harnessed for the treatment of inflammatory diseases. For now, we must be cognizant of the differences in responses between model systems, especially between autoimmune and infectious disease models, when designing therapies to impact inflammatory disease.
Highlights.
C3H mice infected with Borrelia burgdorferi develop an inflammatory arthritis.
Select bioactive lipids from infected ankles were quantified using lipidomics.
COX-2 inhibition or deletion results in non-resolution of arthritis.
Lipidomics revealed that COX-2 deletion leads to loss of some 5-LOX metabolites.
5-LOX inhibition or deletion also resulted in non-resolution of arthritis.
Acknowledgments
We thank NIH grants R01 GM20501 (E.A.D.) and R01 AR052748 (C.R.B.) for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kuehn BM. CDC estimates 300,000 US cases of Lyme disease annually. JAMA. 2013;310(11):1110. doi: 10.1001/jama.2013.278331. [DOI] [PubMed] [Google Scholar]
- 2.Burgdorfer W, Barbour AG, Hayes SF, Benach JL, Grunwaldt E, Davis JP. Lyme disease-a tick-borne spirochetosis? Science. 1982;216(4552):1317–1319. doi: 10.1126/science.7043737. [DOI] [PubMed] [Google Scholar]
- 3.Steere AC, Hardin JA, Malawista SE. Erythema chronicum migrans and Lyme arthritis: cryoimmunoglobulins and clinical activity of skin and joints. Science. 1977;196(4294):1121–1122. doi: 10.1126/science.870973. [DOI] [PubMed] [Google Scholar]
- 4.Steere AC. Lyme disease: a growing threat to urban populations. Proc Nat Acad Sci, USA. 1994;91(7):2378–2383. doi: 10.1073/pnas.91.7.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steere AC, Schoen RT, Taylor E. The clinical evolution of Lyme arthritis. Ann Intern Med. 1987;107(5):725–731. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- 6.Barthold SW, Beck DS, Hansen GM, Terwilliger GA, Moody KD. Lyme borreliosis in selected strains and ages of laboratory mice. J Infect Dis. 1990;162(1):133–138. doi: 10.1093/infdis/162.1.133. [DOI] [PubMed] [Google Scholar]
- 7.Barthold SW, Persing DH, Armstrong AL, Peeples RA. Kinetics of Borrelia burgdorferi dissemination and evolution of disease after intradermal inoculation of mice. Am J Path. 1991;139(2):263–273. [PMC free article] [PubMed] [Google Scholar]
- 8.Barthold SW, deSouza M, Feng S. Serum-mediated resolution of Lyme arthritis in mice. Lab Invest. 1996;74(1):57–67. [PubMed] [Google Scholar]
- 9.Barthold SW, Janotka JL, Smith AL, Persing DH. Chronic Lyme borreliosis in the laboratory mouse. Am J Path. 1993;143(3):959–971. [PMC free article] [PubMed] [Google Scholar]
- 10.Schaible UE, Kramer MD, Museteanu C, Zimmer G, Mossmann H, Simon MM. The severe combined immunodeficiency (scid) mouse. A laboratory model for the analysis of Lyme arthritis and carditis. J Exp Med. 1989;170(4):1427–1432. doi: 10.1084/jem.170.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nandakumar KS, Holmdahl R. Antibody-induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis ResTher. 2006;8(6):223–233. doi: 10.1186/ar2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown CR, Reiner SL. Genetic control of experimental Lyme arthritis in the absence of specific immunity. Infect Immun. 1999;67(4):1967–1973. doi: 10.1128/iai.67.4.1967-1973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol. 2015;15(8):511–523. doi: 10.1038/nri3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. 2011;111(10):5922–5943. doi: 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blaho VA, Buczynski MW, Brown CR, Dennis EA. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J Biol Chem. 2009;284(32):21599–21612. doi: 10.1074/jbc.M109.003822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol. 2001;2(7):612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 17.Mrsny RJ, Gewirtz AT, Siccardi D, Savidge T, Hurley BP, Madara JL, McCormick BA. Identification of hepoxilin A3 in inflammatory events: A required role in neutrophil migration across intestinal epithelia. Proc Nat Acad Sci, USA. 2004;101(19):7421–7426. doi: 10.1073/pnas.0400832101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wen Y, Gu J, Vandenhoff GE, Liu X, Nadler JL. Role of 12/15-lipoxygenase in the expression of MCP-1 in mouse macrophages. American journal of physiology Heart and circulatory physiology. 2008;294(4):H1933–1938. doi: 10.1152/ajpheart.00260.2007. [DOI] [PubMed] [Google Scholar]
- 19.Serhan CN, Gotlinger K, Hong S, Lu Y, Siegelman J, Baer T, Yang R, Colgan SP, Petasis NA. Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: assignments of dihydroxy-containing docosatrienes. J Immunol. 2006;176(3):1848–1859. doi: 10.4049/jimmunol.176.3.1848. [DOI] [PubMed] [Google Scholar]
- 20.Norris PC, Gosselin D, Reichart D, Glass CK, Dennis EA. Phospholipase A2 regulates eicosanoid class switching during inflammasome activation. Proc Nat Acad Sci, USA. 2014 doi: 10.1073/pnas.1404372111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaelis UR, Fleming I. From endothelium-derived hyperpolarizing factor (EDHF) to angiogenesis: Epoxyeicosatrienoic acids (EETs) and cell signaling. Pharmacol Ther. 2006;111(3):584–595. doi: 10.1016/j.pharmthera.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 22.Tamang DL, Pirzai W, Priebe GP, Traficante DC, Pier GB, Falck JR, Morisseau C, Hammock BD, McCormick BA, Gronert K, et al. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J Immunol. 2012;189(10):4960–4969. doi: 10.4049/jimmunol.1201922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochimica et biophysica acta. 2015;1851(4):414–421. doi: 10.1016/j.bbalip.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 24.Myers LK, Kang AH, Postlethwaite AE, Rosloniec EF, Morham SG, Shlopov BV, Goorha S, Ballou LR. The genetic ablation of cyclooxygenase 2 prevents the development of autoimmune arthritis. Arthritis Rheum. 2000;43(12):2687–2693. doi: 10.1002/1529-0131(200012)43:12<2687::AID-ANR8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, Boilard E, Nigrovic PA, Clark P, Xu D, FitzGerald GA, Audoly LP, Lee DM. Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum–transfer arthritis. Arthritis Rheum. 2008;58(5):1354–1365. doi: 10.1002/art.23453. [DOI] [PubMed] [Google Scholar]
- 26.Anderson GD, Hauser SD, McGarity KL, Bremer ME, Isakson PC, Gregory SA. Selective inhibition of cyclooxygenase (COX)-2 reverses inflammation and expression of COX-2 and interleukin 6 in rat adjuvant arthritis. J Clin Invest. 1996;97(11):2672. doi: 10.1172/JCI118717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blaho VA, Mitchell WJ, Brown CR. Arthritis develops but fails to resolve during inhibition of cyclooxygenase 2 in a murine model of Lyme disease. Arthritis Rheum. 2008;58(5):1485–1495. doi: 10.1002/art.23371. [DOI] [PubMed] [Google Scholar]
- 28.Blaho VA, Zhang Y, Hughes-Hanks JM, Brown CR. 5-Lipoxygenase-deficient mice infected with Borrelia burgdorferi develop persistent arthritis. J Immunol. 2011;186(5):3076–3084. doi: 10.4049/jimmunol.1003473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffiths RJ, Pettipher ER, Koch K, Farrell CA, Breslow R, Conklyn MJ, Smith MA, Hackman BC, Wimberly DJ, Milici AJ, et al. Leukotriene B 4 plays a critical role in the progression of collagen- induced arthritis. Proc Nat Acad Sci, USA. 1995;92(2):517–521. doi: 10.1073/pnas.92.2.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim ND, Chou RC, Seung E, Tager AM, Luster AD. A unique requirement for the leukotriene B4 receptor BLT1 for neutrophil recruitment in inflammatory arthritis. Journal of Experimental Medicine. 2006;203(4):829–835. doi: 10.1084/jem.20052349. [DOI] [PMC free article] [PubMed] [Google Scholar]