

Abstract
Background and Purpose
Intrinsic and/or acquired resistance of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) commonly occurs in patients with non‐small‐cell lung cancer (NSCLC). Here, we developed a combined therapy of histone deacetylase inhibition by a novel HDAC inhibitor, YF454A, with erlotinib to overcome EGFR‐TKI resistance in NSCLC.
Experimental Approach
The sensitization of the effects of erlotinib by YF454A was examined in a panel of EGFR‐TKI‐resistant NSCLC cell lines in vitro and two different erlotinib‐resistant NSCLC xenograft mouse models in vivo. Western blotting and Affymetrix GeneChip expression analysis were further performed to determine the underlying mechanisms for the effects of the combination of erlotinib and YF454A.
Key Results
YF454A and erlotinib showed a strong synergy in the suppression of cell growth by blocking the cell cycle and triggering cell apoptosis in EGFR‐TKI‐resistant NSCLC cells. The combined treatment led to a significant decrease in tumour growth and tumour weight compared with single agents alone. Mechanistically, this combination therapy dramatically down‐regulated the expression of several crucial EGFR‐TKI resistance‐related receptor tyrosine kinases, such as Her2, c‐Met, IGF1R and AXL, at both the transcriptional and protein levels and consequently blocked the activation of downstream molecules Akt and ERK. Transcriptomic profiling analysis further revealed that YF454A and erlotinib synergistically suppressed the cell cycle pathway and decreased the transcription of cell‐cycle related genes, such as MSH6 and MCM7.
Conclusion and Implications
Our preclinical study of YF454A provides a rationale for combining erlotinib with a histone deacetylase inhibitor to treat NSCLC with EGFR‐TKI resistance.
Abbreviations
- CI
combination index
- EGFR
epidermal growth factor receptor
- HDAC
histone deacetylase
- NSCLC
non‐small‐cell lung cancer
- TKIs
tyrosine kinase inhibitors
Introduction
Lung cancer is the leading cause of cancer‐related mortalities in both sexes, accounting for more than one‐quarter of the total worldwide (Siegel et al., 2016). Non‐small‐cell‐lung cancer (NSCLC) accounts for approximately 87% of lung cancers. It is characterized by the accumulation of multiple genetic alterations and comprised of diverse histological subtypes (De Santis et al., 2014). Most patients with advanced NSCLC have poor diagnoses and have low survival rates (De Santis et al., 2014; Tan et al., 2015). Overexpression or mutation of epidermal growth factor receptors (EGFRs) plays an important role in the development, differentiation, survival and drug resistance of NSCLC (Chong and Janne, 2013; Hynes and Lane, 2005; Tan et al., 2015). Selective targeted EGFR tyrosine kinase inhibitors (TKIs), such as gefitinib, erlotinib and afatinib, have become the standard of care for first‐line treatment for advanced NSCLC with EGFR overexpression or mutations (Sequist et al., 2013; Tan et al., 2015). Various clinical trials have suggested that patients treated with EGFR‐TKIs show a great initial response and significant progression‐free survival in advanced NSCLC. However, the tumours eventually develop resistance, which becomes a key obstacle to successful therapy (Jackman et al., 2010).
The molecular basis of primary and acquired resistance to EGFR‐TKIs has been gradually identified but, as yet, has not been clarified(Chong and Janne, 2013; Lee et al., 2013; Soucheray et al., 2015; Webster, 2016). Activating KRAS mutations (Pao et al., 2005; Wheeler et al., 2010), overexpression of PIK3CA (Sartore‐Bianchi et al., 2009), loss‐of‐function of PTEN (Kokubo et al., 2005), IGF1R amplification and activation of the NF‐κB signalling pathway (Bivona et al., 2011) are all known to be involved in primary EGFR‐TKI resistance. Additionally, the T790M mutation of EGFR and activation of alternative receptor tyrosine kinases (amplification of MET and HER2 and overexpression of AXL) have been identified as contributing to acquired resistance of EGFR‐TKIs in NSCLC (Engelman et al., 2007; Gandara et al., 2014; Rho et al., 2014). Emerging data show that the down‐regulation of genes, such as BIM (Ng et al., 2012) involved in apoptosis and the epithelial–mesenchymal transition (Byers et al., 2013), contribute to both primary and acquired resistance to EGFR‐TKIs, markedly decreasing the therapeutic efficacy of EGFR‐TKIs. Therefore, the development of innovative strategies, such as a combined therapy that targets specific mutations or signalling pathways, is urgently needed to improve the therapeutic efficacy of EGFR‐TKIs in the treatment of NSCLC (Webster, 2016).
Histone deacetylase (HDAC) aberrations are identified as key drivers of several human diseases (e.g. cancer) (Falkenberg and Johnstone, 2014; Minucci and Pelicci, 2006). HDAC inhibitors are one of the major classes of epigenetic modulating agents for cancer therapy. They target tumour cell proliferation, cell cycle, apoptosis, senescence, cell migration and angiogenesis (Abend and Kehat, 2015; Falkenberg and Johnstone, 2014). Several HDAC inhibitors, including SAHA, FK228 and belinostat, were approved by the U.S. Food and Drug Administration for the treatment of cutaneous T‐cell lymphoma or/and peripheral T‐cell lymphoma from 2006 to 2015 (Abend and Kehat, 2015). In addition, multiple HDAC inhibitors have been reported to have therapeutic effects against various cancers in preclinical and clinical trials either as single agents or in combination therapies (Lakshmaiah et al., 2014). Recently, our group synthesized a novel class of hydroxamate‐based HDAC inhibitors with potent activities against breast cancer in vitro and in vivo (Yang et al., 2014). YF454A, the most potent candidate of this class, exhibited nanomolar half maximal inhibitory concentration (IC50) values toward class I and II b HDACs. However, the therapeutic effect of these novel HDAC inhibitors on NSCLC has not been defined, especially in primary or acquired EGFR‐TKI‐resistant NSCLC.
In this study, we tested the hypothesis that the addition of YF454A may overcome erlotinib resistance in the EGFR‐TKI‐resistant NSCLC. We found that YF454A and erlotinib showed strong synergistic inhibitory effects on cell growth in a panel of EGFR‐TKI‐resistant NSCLC cells. Importantly, the synergy of the combined regimen was also observed in vivo. Our preclinical studies provide an applicable strategy that inhibition of histone deacetylases by YF454A may potentially improve the therapeutic efficacy of EGFR‐TKIs in the treatment of NSCLC.
Methods
Animal studies
Male BALB/cA nude mice (8‐week‐old) were obtained from National Rodent Laboratory Animal Resources (Shanghai, China). All mice were housed in rooms with appropriate temperature (20–22°C) and air humidity (60%) under a 12 h light/dark cycle (five mice per cage). Mice were given ad libitum water and food for 7 days prior to experimentation. All animal treatments were conducted according to Institutional Animal Care and Use Committee guidelines and under an institutional protocol approved by East China Normal University with respect to animal care and welfare assurance. Animal studies were reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
The EGFR‐TKI‐resistant NSCLC xenograft mouse model was established as described previously (Pang et al., 2009). A549 cells or PC9/ER cells (5 × 106 cells per mouse) were injected s.c. into the right hind flank of 6‐week‐old nude mice. Tumour volumes were evaluated by calipers and calculated using the standard formula: V = 0.52 × A × (B) 2, where A is the longest diameter of the tumour and B is the shortest diameter of the tumour. Once tumour volume exceeded approximately 200 mm3, mice were randomized into four groups: the control group (corn oil, p.o.; PBS, i.p.; daily); erlotinib (25 mg·kg−1; p.o.; daily); YF454A (25 mg·kg−1; i.p.; daily); or co‐treatment with erlotinib and YF454A. Solid tumour volume and mouse body weight were measured every 3 days. All mice were administered the different treatments for 27 consecutive days. Mice were killed, and solid tumours were extracted and weighed at the end of the experiments.
Cell culture and establishment of EGFR‐TKI‐resistant cell lines
The human NSCLC cell lines (A549, H1299, H1975 and PC9) were purchased from the American Type Culture Collection (Manassas, VA, USA). HCC827 was obtained from the Shanghai Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HCC827/ER (erlotinib‐resistant) was kindly provided by Dr Bin‐bing S. Zhou (Department of Haematology and Oncology, Shanghai Children's Medical Cenre, Shanghai Jiao Tong University School of Medicine, Shanghai, China). Cells were cultured in RPMI 1640 medium supplemented with 10% FBS (HyClone Laboratories, Logan, UT), 100 U·mL−1 of penicillin and streptomycin (HyClone) and maintained at 37°C under a humidified 95%:5% (v.v−1) mixture of air and CO2. All of the cell lines were authenticated by short tandem repeat analysis before use.
For the establishment of PC9/ER (erlotinib‐resistant) and HCC827/ER (erlotinib‐resistant) cell lines, parental PC9 and HCC827 cells were exposed to stepwise increasing concentrations of erlotinib from 10 nM up to 1 μM every 3 weeks for a period of 6 months. The established resistant cells were cultured in medium containing 1 μM of erlotinib and seeded into erlotinib‐free medium at least 2 weeks before the experiments.
Cell viability assays
Cells were incubated in 96‐well plates to attach overnight and then exposed to drug treatments for an additional 72 h. Cell viability was measured by CellTiter 96® AQueous One Solution (Promega, Madison, WI, USA). IC50 values were calculated using the GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). Combination index (CI) values were calculated by the CalcuSyn Version 2.0 software (Biosoft, Gambridge, UK). CI values <1, =1 and >1 indicate synergism, additive and antagonism between two drugs respectively (Chou, 2006; Chou and Talalay, 1984). Experiments were conducted three independent times (n = 3 per treatment group).
Cell cycle analysis
Cell cycle analysis was performed as described previously (Dai et al., 2012). PC9/ER cells were incubated with erlotinib (5 μM), YF454A (0.2 μM) or both drugs for 24 h. Cells were then fixed with 70% ethanol at −20°C overnight and stained with propidium iodide (Sigma) in the presence of ribonuclease A (Takara biotechnology, Dalian, China) for 30 min at room temperature. The cell cycle distribution was analysed by flow cytometry (FACS Calibur, BD Biosciences).
Apoptosis analysis
PC9/ER, H1229 and H1975 cells were exposed to different treatments for 48 h and then stained using BD Pharmingen Apoptosis Detection kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer's protocol. Cell apoptosis was assessed by flow cytometer (FACS Calibur, BD Biosciences) (Wang et al., 2014).
Clonogenic assay
Tumour cells were plated in 6‐well plates at a density of 800–1200 cells per well. After cells attached overnight, drugs at indicated concentrations were added to plates. DMSO served as the control vehicle. Medium with drugs was replaced every 3 days for a period of 7–14 days. The cells were then fixed with 4% paraformaldehyde and stained with 0.1% crystal violet. Colonies were counted by Image J software (NIH, Bethesda, MD, USA) (Zhang et al., 2014).
Western blotting assays
Western blotting analysis was performed as described previously (Wang et al., 2015). Briefly, treated cells were harvested and lysed in radio immunoprecipitation assay buffer. Cellular protein concentration was determined using a bicinchoninic acid assay (ThermoFisher Scientific, Waltham, MA, USA). Approximately 30–50 μg of total protein from whole cell lysates were run on 6–12% SDS‐polyacrylamide gels and transferred to nitrocellulose membranes (Millipore, Billerica, MA, USA) using the I‐Blot (Invitrogen, ThermoFisher Scientific). The membranes were then incubated with primary antibodies at 4°C overnight and sequentially incubated with appropriate anti‐rabbit, anti‐mouse or anti‐goat secondary antibodies for 1 h at room temperature. After several washes, visual signals were developed by the LI‐COR Odyssey Infrared Imaging system (LI‐COR Biosciences).
Real‐time PCR analysis
The total RNA was collected using RNAiso plus (TaKaRa Bio Inc., Shiga, Japan) from treated cells and then reverse‐transcribed to cDNA using the PrimeScript RT reagent kit (TaKaRa). Real‐time PCR was performed using the 96‐well Thermal iCycler (Biorad, Hercules, CA, USA). All amplified gene products were electrophoresed on agarose gels. Sequences of primers are listed in Supporting Information Table S1.
Microarray analysis
PC9/ER cells were treated with YF454A (0.2 μM), erlotinib (5 μM) or YF454A/erlotinib for 12 h. The RNA of treated cells was extracted using RNAiso plus (TaKaRa) according to the manufacturer's protocol. Microarray analysis was profiled by the Affymetrix GeneChip® Human Transcriptome Array 2.0 (Affymetrix, Santa Clara, CA, USA). The microarray data discussed in this study have been deposited in the National Centre for Biotechnology Information Gene Expression Omnibus and is accessible through series accession number GSE80316 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc= GSE80316).
Raw CEL files were normalized using the Affymetrix Expression Console Software 1.30 with the Robust Multi‐array Average algorithm. Differential gene expression analysis among groups was performed using Bayesian Analysis of Variance for Microarrays (BAMarray) 3.0. The resulting gene lists were further narrowed down using STEM v. 1.3.8 (Short Time‐series Expression Miner) into genes whose expression showed more than twofold change compared with the control (FC > =2 or FC < = −2). P values less than 0.05 were corrected for multiple testing by the BH method.
Analysis of differentially expressed genes using patient data
Read count data for primary lung adenocarcinoma and matched normal lung tissues were downloaded from The Cancer Genome Atlas (January, 2015) (Cancer Genome Atlas Research N, 2014). The edgeR package (Robinson et al., 2010) was used for differential expression gene analysis on RNA‐seq read count data.
Kaplan–Meier survival analysis
Kaplan–Meier survival analysis was conducted using gene microarray data and clinical data (Gyorffy et al., 2013). All P‐values were performed using the Log‐rank test (Gyorffy et al., 2013).
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Results are presented as mean ± SEM. Data analysis was carried out using Microsoft Excel and GraphPad Prism Software version 5.0. Groups which contained five or more independent repeats were subjected to statistical analysis. For comparisons of two groups, a two‐tailed unpaired t‐test was used; for comparisons of multiple groups, one‐way ANOVA analysis followed by Tukey post hoc tests was applied. Post hoc tests were run only if F achieved P < 0.05, and there was no significant inhomogeneity. Statistical significance was set as P < 0.05.
Randomization and blinding
All experiments in this study were conducted in a randomized manner. The in vivo experiments were performed by investigators blinded to the treatment groups. The in vitro experiments were not performed with blinding because the assays were carried out under standardized procedures and revealed strictly quantitative data.
Materials
Erlotinib and SAHA were purchased from Selleck Chemicals (Houston, TX, USA). YF454A [N1‐((5‐(5‐pyrimidinyl)‐2‐thiopheneyl) methyl)‐N7‐hydroxyN1‐(4‐methoxyphenyl) heptane‐diamide] and other lead compounds were synthesized in house (Yang et al., 2014). All compounds were dissolved in DMSO and stored at −80°C. The p‐Akt (Ser473), Akt, p‐ERK1/2 (Thr202/Tyr204), ERK1/2, p‐EGFR (pTyr1068), EGFR, p‐Her2 (Tyr1221/1222), Her2, p‐c‐Met(Tyr1234/1235), c‐Met, p‐IGF1R (pTyr1316), IGF1R, Chk1, cyclin D1 and E2F1 antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA). Antibodies against Cdc25A, p‐Rb (Ser807/811) and Rb were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The p‐AXL (Tyr779) and AXL antibodies were obtained from R&D System (Minneapolis, MN, USA), and β‐actin antibody was purchased from Sigma (St. Louis, MO, USA). Secondary antibodies including IRDye 800CW Goat anti‐mouse IgG (H + L), IRDye 800CW goat anti‐rabbit IgG (H + L) and IRDye 800CW donkey anti‐goat IgG (H + L) were obtained from LI‐COR Biosciences (Lincoln, NE, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Cytotoxic synergy of YF454A and erlotinib in EGFR‐TKI‐resistant NSCLC cells
Our group recently synthesized a series of novel hydroxamate‐based pan‐HDAC inhibitors that exhibited nanomolar IC50 values toward class I and II b HDACs (Yang et al., 2014). The lead compound, YF454A, exhibited anti‐proliferative activity against human breast cancer in vitro and in vivo (Nakagawa et al., 2013; Yang et al., 2014). However, the therapeutic effect of these novel HDAC inhibitors on NSCLC has not been defined, especially in primary or acquired EGFR‐TKI‐resistant NSCLC. We first chose a panel of EGFR‐TKI‐resistant NSCLC cell lines with diverse EGFR or RAS genotypes for experimental use: A549 (EGFR wild‐type, KRAS mutant), H1299 (EGFR wild‐type, NRAS mutant) and H1975 (L858R and T790M EGFR mutations) (Yeh et al., 2011). As expected, IC50 of erlotinib was over 10 μM against these resistant cell lines (Supporting Information Table S2). The acquired EGFR‐TKI‐resistant cell lines PC9/ER (IC50 = 5.05 μM) exhibited 250‐fold more resistance to erlotinib than that of parental PC9 cells (IC50 = 0.02 μM; Figure 1A). In addition, HCC827/ER (IC50 > 10 μM) showed approximate 400‐fold more resistance to erlotinib than that of parental HCC827 cells (IC50 = 0.02 μM; Figure 1A). Given the significance of the receptor tyrosine kinase signalling pathways involved in EGFR‐TKI resistance in NSCLC, we assessed basal expression and phosphorylation levels of several receptor tyrosine kinases (Her2, EGFR, c‐Met, IGF1R and AXL) and their downstream molecules in PC9/ER and HCC827/ER cells by Western blotting assays. In agreement with previous studies (Chong and Janne, 2013; Rho et al., 2014), increased activity of Her2, AXL and AKT was observed in PC9/ER cells, and hyper‐phosphorylation of c‐Met and AXL was observed in HCC827/ER cells (Figure 1B). Notably, down‐regulation of EGFR occurred in both cell lines with required resistance to EGFR‐TKIs.
Figure 1.
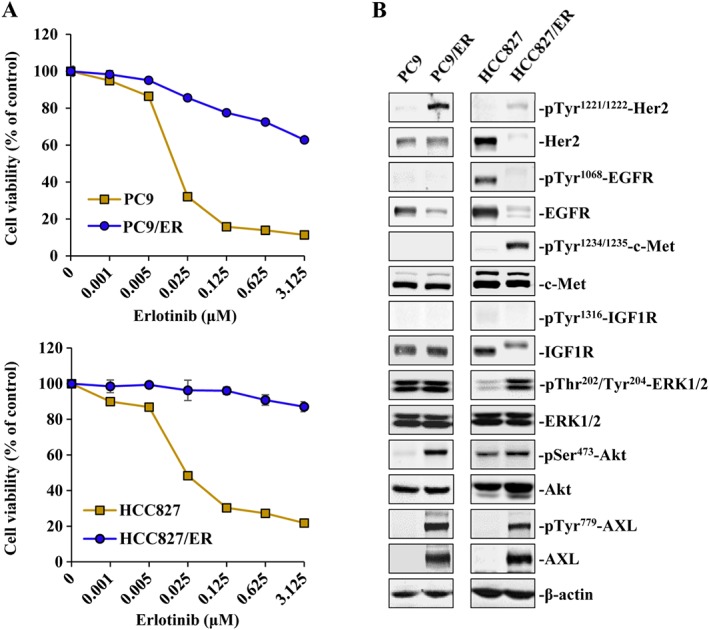
Molecular characterization of EGFR‐TKI‐resistant NSCLC cells. (A) Cytotoxicity of erlotinib in PC9, PC9/ER, HCC827 and HCC827/ER cells was determined using the cell viability assays. Means from three independent experiments are shown with bars representing SEM (n = 3 per group). (B) Characterization of phosphorylation status of tyrosine kinases including Her2, EGFR, Met, IGF1R, AXL and their downstream molecules Akt and ERK in PC9, PC9/ER, HCC827 and HCC827/ER cells respectively.
Next, we examined the cytotoxicity of our novel HDAC inhibitors (YF454A, YF513 and YF441B; their chemical structures are shown in Supporting Information Figure S1) in the above EGFR‐TKI‐resistant NSCLC cell lines. The classic HDAC inhibitor SAHA was used as the control. As shown in Supporting Information Table S2, three of our HDAC inhibitors (YF454A, YF513 and YF441B) showed comparable or better anti‐proliferative effects than that of SAHA in all five EGFR‐TKI‐resistant NSCLC cell lines. Notably, the IC50 values of YF454A ranged from 0.15 to 3.50 μM, which were almost 5–10‐fold lower than those of SAHA. To identify whether YF454A was synergistic with erlotinib in EGFR‐TKI‐resistant NSCLC, we treated cells with increasing concentrations of drug pairs. Compared with YF454A or erlotinib single agent alone, the combination of YF454A and erlotinib augmented cytotoxicity in all cell lines tested (Figure 2A–E). Most of the CI values at various combinations were less than 0.7, suggesting a strong synergy of YF454A and erlotinib in both primary and acquired EGFR‐TKI‐resistant NSCLC cells. To test whether YF454A and erlotinib also hold synergistic effects in EGFR‐TKI‐sensitive cells, we treated parental PC9 and HCC827 cells with increasing concentrations of YF454A alone or in combination with erlotinib. Our results showed that YF454A dose‐dependently blocked the growth of EGFR‐TKI‐sensitive cells (Supporting Information Figure S2A(i), B(i); IC50 = 0.08 μM in PC9 cells and IC50 = 0.15 μM in HCC827 cells). However, the CI values of this drug pair at different combinations in EGFR‐TKI‐sensitive cells were all close to 1 (Supporting Information Figure S2A(ii), B(ii)), which were much higher than that in EGFR‐TKI‐resistant NSCLC cells, indicating that EGFR‐TKI‐resistant NSCLC cells might be more fragile to the combination treatment.
Figure 2.
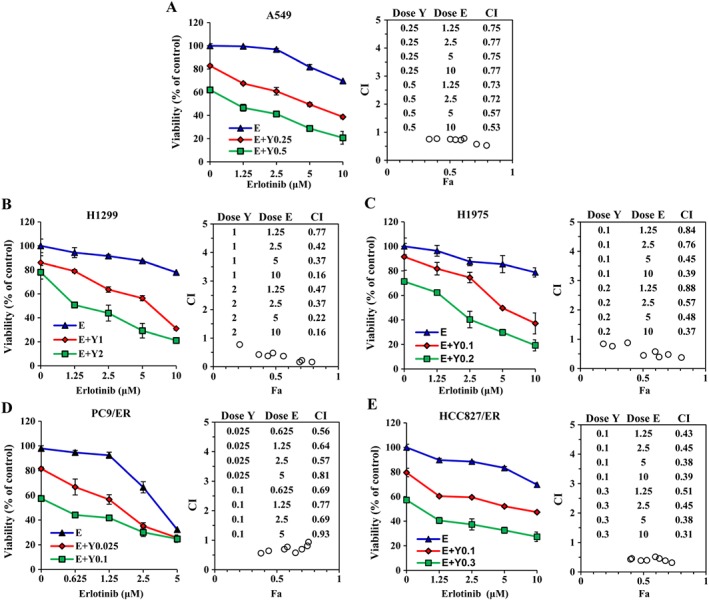
YF454A enhances erlotinib‐induced cytotoxicity in EGFR‐TKI‐resistant NSCLC cells. A549 (A), H1299 (B), H1975 (C), PC9/ER (D) and HCC827/ER (E) cells were treated with YF454A (Y) alone or in combination with erlotinib (E) for 72 h. Cell viability was measured. The combined interaction of dual treatment was analysed using the CI equation and presented as Fa (fraction affected by the dose) in the Fa–CI plots. CI values were calculated using the Calcusyn software package (Biosoft, Cambridge, UK). Means from three independent experiments are shown with bars representing SEM (n = 3 per group).
Combining YF454A with erlotinib leads to cell‐cycle arrest, apoptosis and irreversible growth arrest in vitro
Cell cycle analysis showed that YF454A alone or in combination with erlotinib dramatically arrested the cell cycle at G1 phase with a concomitant decrease in G2/M phase compared with the vehicle control group (Figure 3A). Apoptosis assays further validated the cell‐killing effects of the co‐treatment of YF454A and erlotinib. As shown in Figure 3B, dual treatments for 48 h significantly increased cell apoptosis compared with each drug alone (29.75% in PC9/ER cells, 28.25% in H1299 cells and 65.36% in H1975 cells; P < 0.05). A two‐dimensional‐clonogenic assay was also carried out to test the inhibitory effect of the combined treatment in PC9/ER, H1299 and A549 cells respectively. The results showed that YF454A and erlotinib synergistically blocked the colony formation of all three cell lines (P < 0.05; Figure 3C, D). Taken together, these results suggest that the synergy of YF454A and erlotinib apparently reduces the growth and survival of EGFR‐TKI‐resistant NSCLC in vitro.
Figure 3.
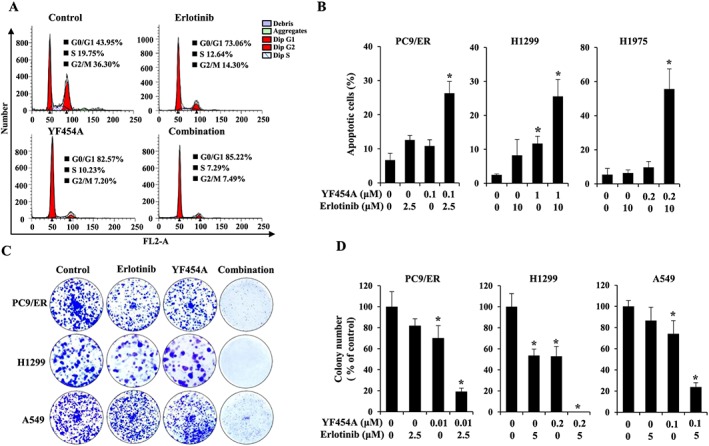
The addition of YF454A potentiates erlotinib‐mediated inhibition of the growth of EGFR‐TKI‐resistant NSCLC in vitro. (A, B) YF454A and erlotinib showed a strong synergy in the suppression of cell growth by blocking the cell cycle (A) and triggering cell apoptosis (B). PC9/ER cells were treated with 0.2 μM of YF454A, 5 μM of erlotinib or YF454A/erlotinib for 24 h. Treated cells was stained with propidium iodide, and cell cycle distribution was assessed by flow cytometry. Apoptotic cells were stained with propidium iodide/Annexin V‐FITC and analysed by flow cytometry. (C) The combination of YF454A and erlotinib dramatically suppressed colony formation of EGFR‐TKI‐resistant NSCLC cells. PC9/ER, H1299 and A549 cells were treated with indicated concentrations of single drugs or drug combinations for 7–14 days. Colonies were stained with 0.1% crystal violet and photographed (magnification; 40×). (D) The relative colony number. The colony number was counted by Image J software (NIH, Bethesda, MD), and results represent the mean of three independent experiments. Error bars represent SEM (n = 6 per group). One‐way ANOVA with Turkey post hoc tests were performed; *P < 0.05 versus the vehicle control group.
Enhanced therapeutic efficacy of erlotinib by YF454A in vivo
To evaluate whether YF454A potentiated the efficacy of erlotinib in vivo, we established primary and acquired EGFR‐TKI‐resistant NSCLC xenograft mouse models. Primary EGFR‐TKI‐resistant NSCLC A549 cells (EGFR wild‐type; KRAS mutant) and acquired EGFR‐TKI‐resistant NSCLC PC9/ER cells were used respectively. In the A549 tumour cell xenograft mouse model, the co‐treatment with YF454A and erlotinib produced a significant tumour regression as compared with the control group (P < 0.05) or each drug alone group (P < 0.05) (Figure 4A). Moreover, YF454A and erlotinib synergistically decreased tumour weight compared with the single agent groups (Figure 4B), with no obvious effect of systematic toxicity on mouse body weight (Supporting Information Figure S3A).
Figure 4.
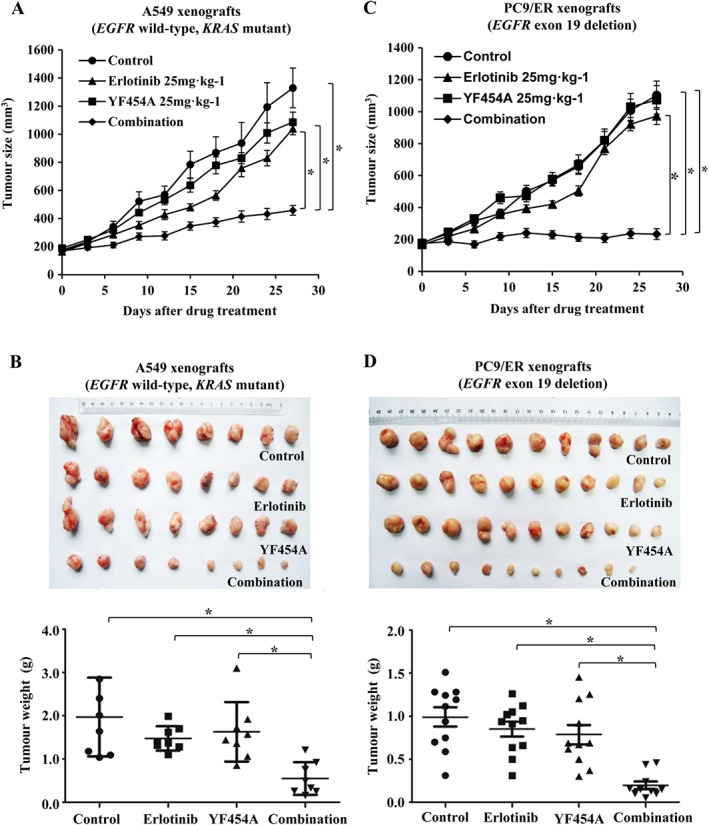
The combination of YF454A and erlotinib synergistically suppresses the growth of EGFR‐TKI resistant tumour xenografts in vivo. (A, B) The combination of YF454A and erlotinib inhibited the growth of primary EGFR‐TKI‐resistant A549 xenografts (n = 8). Nude mice bearing A549 tumours were treated with different drugs or drug combinations. The tumour volume in mice was plotted every 3 days (A), and tumour weights were measured when the mice were killed on day 28 (B). (C, D) The tumour growth of acquired EGFR‐TKI‐resistant PC9/ER xenografts (n = 11 in the vehicle control group, erlotinib group and YF454A group; n = 10 in the combination group) was dramatically suppressed by the co‐treatment with YF454A and erlotinib, as indicated by tumour volume curves (C) and tumour weights in different groups on day 28 (D). Means are shown with bars representing SEM. One‐way ANOVA with Turkey's post hoc tests were performed; *P < 0.05 significantly different as indicated. See also Supporting Information Figure S3.
Similarly, addition of YF454A to erlotinib also remarkably slowed the progression of tumours without apparently inhibitory effect on mouse weight in the acquired EGFR‐TKI‐resistant NSCLC PC9/ER xenograft mouse model (P < 0.05 vs. the control group or each single agent group; Figure 4C, D and Supporting Information Figure S3B). The tumour volume and weight of PC9/ER xenografts were strikingly decreased after a 4 week treatment. Collectively, these preclinical data reveal that YF454A might be an effective anti‐tumour candidate in combination with erlotinib to overcome EGFR‐TKI resistance in vivo.
The combination of YF454A and erlotinib significantly suppresses the receptor tyrosine kinase signalling pathways in EGFR‐TKI‐resistant NSCLC cells
The EGFR T790M mutation and ‘bypass’ receptor tyrosine kinases (HER2, IGF1R, MET and AXL) amplification are well‐documented mechanisms of EGFR‐TKI resistance in lung cancer (Jackman et al., 2010). To further explore the molecular basis of synergistically anti‐proliferative activity of YF454A and erlotinib in EGFR‐TKI‐resistant NSCLC cells, the protein expression and phosphorylation levels of reported receptor tyrosine kinases were further examined by Western blotting and real‐time PCR assays. As shown in Figure 5A(i), PC9/ER cells treated with either YF454A alone or in combination with erlotinib for 24 h showed apparent down‐regulation of total protein levels of EGFR, Her2, IGFR, c‐Met and AXL. Time‐dependent down‐regulation of receptor tyrosine kinases was also observed in the combined regimen (Figure 5B(i)). To test whether the decreased expression of receptor tyrosine kinases mediated by the drugs was due to transcription inhibition, we further performed the PCR analysis, and the results showed that the inhibitory actions on ‘bypass’ receptor tyrosine kinases mediated by YF454A or YF454A/erlotinib occurred at the transcriptional level (Figure 5A(ii), B(ii)). Collectively, all these results suggest that the combined therapy of YF454A and erlotinib dramatically inhibit multiple ‘bypass’ receptor tyrosine kinase pathways in EGFR‐TKI resistance at both the transcriptional and protein levels.
Figure 5.
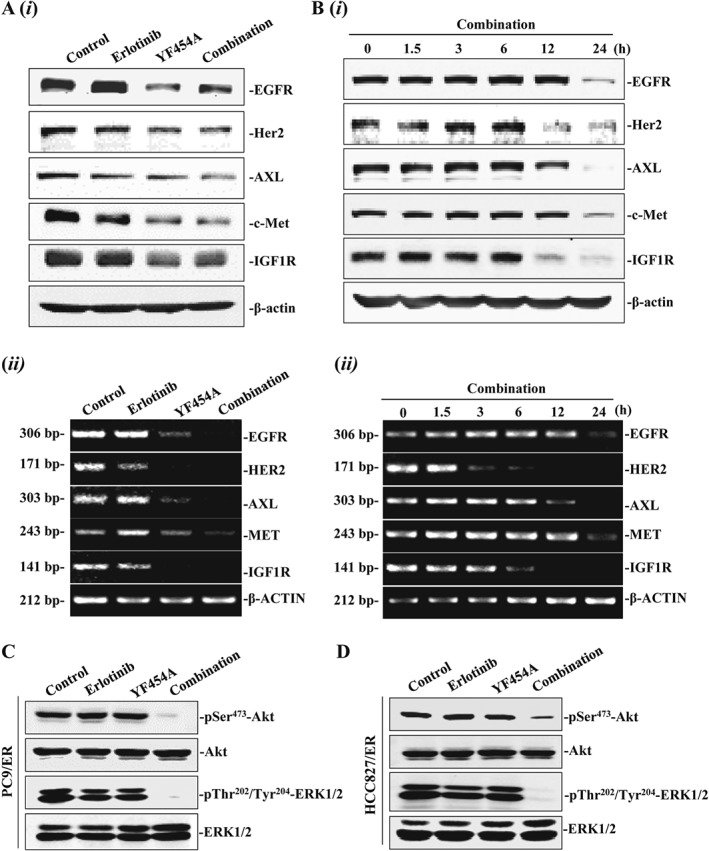
YF454A plus erlotinib significantly suppresses the receptor tyrosine kinase signalling pathways in EGFR‐TKI‐resistant NSCLC cells. (A) Effects of either YF454A or erlotinib alone or in combination on the protein and mRNA expression of EGFR, Her2, AXL, c‐Met and IGF‐1R. PC9/ER cells were treated with YF454A (0.2 μM), erlotinib (5 μM) or YF454A/erlotinib for 24 h. Cell lysates and total mRNA were prepared, and Western blotting assays (i) and real‐time PCR analysis (ii) were further carried out. (B) The combination of YF454A and erlotinib time‐dependently suppressed protein expression (i) and mRNA expression (ii) of EGFR, HER2, AXL, MET and IGF‐1R. (C, D) YF454A/erlotinib down‐regulated the phosphorylation of Akt and ERK in PC9/ER cells (C) and in HCC827/ER cells (D). PC9/ER and HCC827/ER cells were treated with YF454A (0.2 μM) and erlotinib (5 μM) as a single agent alone or in combination for 24 h. The whole‐cell lysates were prepared and probed with specific Akt and ERK antibodies. Western blotting and real‐time PCR assays were performed three independent times, and representative images are shown.
The PI3K/AKT and MAPK pathways are critical signalling cascades downstream of receptor tyrosine kinases, controlling multiple biological actions of cancer cells, such as cell proliferation, survival, invasiveness and drug resistance. Therefore, we tested the inhibitory effect of the combined regimen on downstream pathways of receptor tyrosine kinases in EGFR‐TKI‐resistant NSCLC cells. As expected, erlotinib or YF454A alone did not affect the active forms of AKT and ERK, whereas the combined treatment potently decreased the levels of phospho‐AKT and phospho‐ERK in treated PC9/ER cells (Figure 5C) and HCC827/ER cells (Figure 5D).
YF454A and erlotinib synergistically inhibit the expression of cell cycle related genes
Up to 20% of well‐known genes are affected by HDAC inhibitors, among which cell cycle regulators are primary targets of HDAC inhibition (Peart et al., 2005). To further understand the anti‐proliferative and pro‐apoptotic effect of the combined regimen, we further examined several key cell‐cycle regulators controlling G1/S cell‐cycle progression, including Chk1, Cdc25A, Cyclin D1, retinoblastoma protein (Rb) and E2F1. The E2F family of transcription factors regulate target genes that are crucial for S‐phase progression and the onco‐suppressor Rb recruits HDACs to E2F target genes (Minucci and Pelicci, 2006). We found that all of the above cell‐cycle regulators were substantially suppressed by erlotinib when YF454A was added, suggesting that HDAC inhibition greatly contributes to the anti‐proliferative and pro‐apoptotic effect of the combined treatment (Figure 6A). Inhibition of these proteins was a consequence of transcriptional regulation, as their mRNA expression was decreased under the same conditions (Figure 6B). The protein and mRNA levels of Chk1, Cdc25A and E2F1 were also found to be dramatically suppressed by the co‐treatment in a time‐dependent manner (Figure 6C, D). It has been reported that HDAC inhibitors caused cell cycle arrest by p53‐independent induction of p21WAF1/CIP1 (Sandor et al., 2000). P21 inhibits G1‐cyclin/CDK complexes and decreases the activity of Rb, thereby regulating G1/S cell cycle transition (Vaziri et al., 1998). In our study, p53‐independent induction of p21 was observed by either YF454A alone or in combination with erlotinib (Figure 6E, F). Collectively, these data indicate that potentiation of erlotinib by YF454A effectively inhibits cell proliferation and induces cell apoptosis primarily by targeting cell‐cycle progression.
Figure 6.
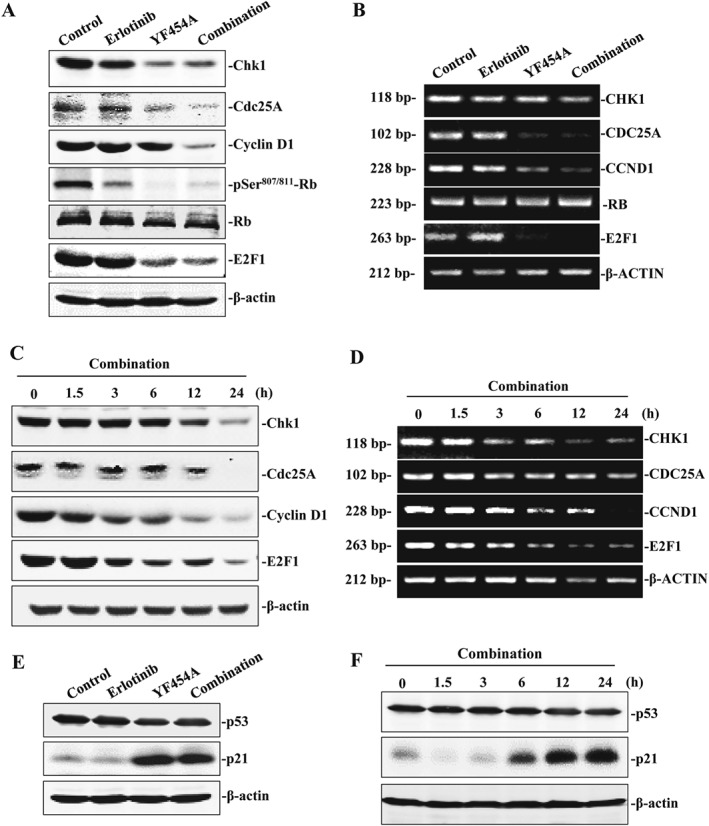
The cell cycle pathway is involved in the synergy of YF454A and erlotinib in EGFR‐TKI‐resistant NSCLC. (A, B) Effect of YF454A and erlotinib as a single agent alone or in combination on the cyclin D1/Rb/E2F pathway in PC9/ER cells. Protein expression was detected by Western blotting assays with specific antibodies (A), and gene expression was examined by real‐time PCR analysis (B). (C, D) A time‐dependent effect of the combined treatment on the molecules involved in the regulation of G1/S cell‐cycle progression. PC9/ER cells were treated with the two drugs for the indicated time intervals. Protein levels and transcriptional levels of Chk1, Cdc25A, cyclin D1 and E2F1 were examined by Western blotting assays (C) and real‐time PCR analysis (D) respectively. (E, F) YF454A and erlotinib as a single agent alone or in combination led to induction of p21, independent of p53 regulation. PC9/ER cells were treated with YF454A (0.2 μM), erlotinib (5 μM) or both drugs (1:1) for 24 h (E) or with a combined treatment of YF454A and erlotinib for 1.5, 3, 6, 12 and 24 h (F). The protein level of p21 and p53 was probed. Western blotting and real‐time PCR assays were performed three independent times, and representative images are shown.
Transcriptome alteration mediated by the synergy of YF454A and elotinib
To further elucidate the possible mechanism of the enhanced activity of elotinib by YF454A in EGFR‐TKI‐resistant NSCLC cells, we performed a microarray analysis to examine the global transcriptome profile using an Affymetrix microarray platform in PC9/ER cells. We identified genes whose mRNA levels were expressed differentially among different treatment groups using differential expression genes analysis and found that several differentially expressed genes were highly regulated by the co‐treatment compared with either erlotinib or YF454A single agents alone (fold change ≥2 or fold change ≤−2 and P ≤0.05) (Figure 7A). Pathway enrichment analysis revealed that the cell cycle pathway showed the highest rank according to enrichment score [−log10 (P‐value)] (Figure 7B).
Figure 7.
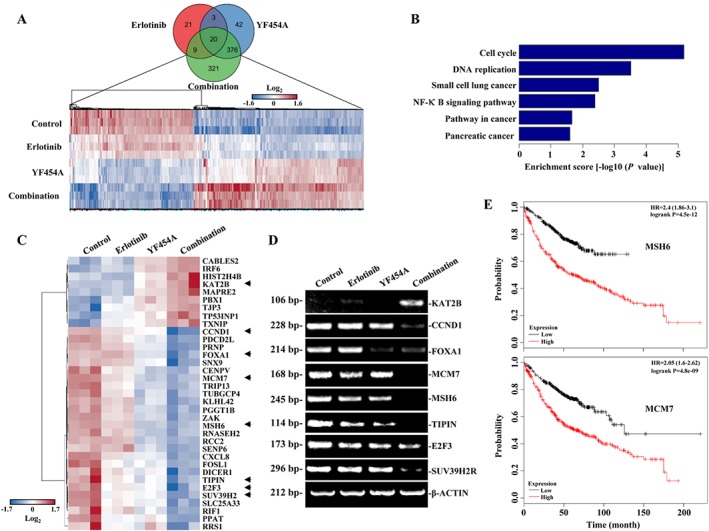
Transcriptome alteration mediated by the synergy of YF454A and elotinib. (A) Venn diagrams showed the gene profile of different groups. PC9/ER cells were treated withYF454A (0.2 μM), erlotinib (5 μM) or YF454A/erlotinib for 12 h. Total mRNA of different groups was prepared, and microarray analysis was carried out by using Human Transcriptome Array 2.0 (n = 3 per group). (B) The top six enrichment pathways. Pathway enrichment was analysed based on the subsets of down‐regulated differentially expressed genes. (C) Cell‐cycle‐related genes that were significantly suppressed by the co‐treatment of YF454A and erlotinib. Heat map is coloured according to the Z‐scores of expression values. Arrowheads indicate the differentially expressed genes that were involved in the G1/S phase cell‐cycle progression. (D) Real‐time PCR assays validated the effects of the indicated treatments on genes that regulated the G1/S phase progression. (E) Cell‐cycle‐related genes MSH6 and MCM7 were significantly associated with patient survival of lung adenocarcinoma. P‐values were performed using the Log‐rank test.
The gene ontology analysis further showed that genes prominently affected by the combination of erlotinib and YF454A were highly enriched in cell‐cycle and DNA replication progression (Supporting Information Table S3). The expression pattern of several important genes regulating cell‐cycle progression was remarkably activated or suppressed by the combined treatment as compared with each drug alone in PC/ER cells, including MCM7, MSH6, KAT2B, CCND1, TIPIN, E2F3, SUV39H2 and others (Figure 7C). The potent suppression of these genes mediated by the co‐treatment was further validated by the real‐time PCR assays (Figure 7D). Notably, we found that MSH6, MCM7, E2F3, TIPIN and SUV39H2 were significantly up‐regulated in patients with lung adenocarcinoma compared to the matched normal lung tissues based on the Cancer Genome Atlas database (Supporting Information Table S4). It has been reported that Cyclin D1 and E2F3 are essential components in the HER2/RAS oncogenic pathway (Wu et al., 2015; Yu et al., 2001), and down‐regulation of Cyclin D1 and E2F3 leads to significant delay of mammary tumour onset (Wu et al., 2015; Yu et al., 2001). We further found that increased expression of MSH6 and MCM7 was associated with poor survival in lung adenocarcinoma patients (Figure 7E and Supporting Information Table S4), suggesting that decreased expression of MSH6 and MCM7 by the synergy of YF454A and erlotinib might increase the clinical benefit for patients with lung adenocarcinoma. Taken together, these results show that erlotinib and YF454A synergistically regulate the transcription of cell‐cycle‐related genes involved in G1/S phase progression (e.g. MSH6 and MCM7), suggesting a major molecular basis for overcoming EGFR‐TKI resistance in NSCLC.
Discussion
Targeted cancer therapy has been significantly advanced since the last decade. Erlotinib was approved by the U.S. Food and Drug Administration for first‐line treatment of metastatic EGFR‐mutant lung cancer in 2013, and gefitinib was approved as a first‐line treatment for NSCLC in 2015 (Jackson and Chester, 2015). However, progression‐free survival with these therapies is rather disappointing due to the development of various types of resistance (Chong and Janne, 2013). Two main resistance mechanisms have been revealed in patients: mutation of EGFR to a drug‐resistant state (T790M or S492R mutations) in which cells are insensitive to gefitinib or erlotinib at the enzyme level and activation or up‐regulation of ‘bypass’ receptor tyrosine kinases (such as MET, HER2 and IGF1R) (Chong and Janne, 2013; Wheeler et al., 2010). Increasing knowledge concerning the resistance mechanism provides a chance to exploit new mechanism‐based inhibitors or combination therapies to prevent or overcome EGFR‐TKI resistance in clinical settings.
Previous studies indicated that HDAC inhibitors, a group of drugs targeting epigenetics, have been evaluated for their synergist effects when administered in combination with various conventional chemotherapeutic drugs for different types of solid tumours (Bose et al., 2014; Minucci and Pelicci, 2006). Notably, SAHA was shown to circumvent the EGFR‐TKI resistance caused by the BIM polymorphism (Nakagawa et al., 2013; Ng et al., 2012). CUDC‐101, a multi‐targeted HDAC, EGFR and HER2 inhibitor, decreased the MET and AXL signalling pathways, restored E‐cadherin expression and reduced cell migration in EGFR‐TKI‐resistant NSCLC (Wang et al., 2013). The HDAC inhibitor, MPTOE028, yielded significant growth inhibition in erlotinib‐resistant lung adenocarcinoma by blocking the EGFR/HER2 signalling and many other compensatory pathways (Chen et al., 2013). Furthermore, the addition of an HDAC inhibitor (such as SAHA and SNDX‐275) to erlotinib or SAHA combined with gefitinib has become a novel clinical strategy to treat EGFR‐TKI resistance in patients suffering from NSCLC (NCT00503971, NCT00750698 and NCT02151721; https://www.clinicaltrials.gov/; accessed: April 11, 2017). The combination of romidepsin and erlotinib also exhibited inhibitory effects on relevant molecular targets in an unselected advanced NSCLC phase 1 clinical trial (Gerber et al., 2015). These findngs have led to the notion that HDAC inhibitors are promising drugs to potentiate the therapeutic efficacy of EGFR‐TKIs.
YF454A, a novel HDAC inhibitor synthesized by our group, exhibited potent inhibitory activities against breast cancer through the up‐regulation of acetyl‐histone H3 and H4, down‐regulation of paxillin expression and inhibition of tumour epithelial‐mesenchymal transition (Yang et al., 2014). In this study, we explored the novel ability of YF454A to potentiate the effects of erlotinib in both EGFR‐TKI‐resistant cells in vitro and in vivo. YF454A exhibited more potent anti‐proliferative activity than SAHA in a panel of EGFR‐TKI‐resistant NSCLC cell lines (Supporting Information Table S2). Furthermore, YF454A and erlotinib exhibited a synergistic action to suppress cell growth and colony formation by triggering cell cycle arrest and apoptosis in resistant cells. Consistently, the suppressive activity of the combined therapy was also observed in two different mouse models in vivo. Importantly, the YF454A alone or when given as a combined treatment was well‐tolerated in tumour‐bearing mice at the tested dosage. Although combining a traditional HDAC inhibitor SAHA with standard anti‐NSCLC therapy showed moderate effects (Azad et al., 2013), the excellent tolerability, limited toxicity and potent anti‐cancer activity of YF45A may potentially make it an effective therapeutic candidate in clinical trials to treat EGFR‐TKI‐resistant NSCLC.
The cell cycle, like other important cellular functions such as cell proliferation and apoptosis, are regulated by ‘epigenetic modifiers’ known as HDACs (Bose et al., 2014). HDAC inhibitors have been reported to cause G1/S or G2/M cell cycle arrest, suppress cyclin D, cyclin A and acetylation of pericentromeric chromatin and impair kinetochore assembly that activates mitotic check points (Gui et al., 2004; Robbins et al., 2005; Sandor et al., 2000). In our study, we characterized an important mechanism of action of YF454A in combination with erlotinib in EGFR‐TKI‐resistant NSCLC. Cell cycle analysis in PC9/ER cells showed that addition of YF454A to erlotinib blocked cell cycle progression in G1/S phase and consistently suppressed the cyclin D1/Rb/E2F signalling pathway. These phenomena were related to a decrease in the mRNA level of CHK1, CDC25A and CCND1 (Figure 6D). Our microarray analysis consistently revealed that the cell cycle pathway was strongly implicated in the synergy of YF454A and erlotinib. Transcription of MCM7, MSH6, TIPIN, E2F3, SUV39H2, KAT2B and other genes involved in the cell cycle pathways were also affected by the combined treatment (Figure 7). All these results indicate that YF454A augments the therapeutic efficacy of erlotinib through primarily targeting the cell‐cycle regulation in EGFR‐TKI‐resistant NSCLC cells.
Additionally, ‘bypass’ receptor tyrosine kinases, including Her2, IGF1R, c‐Met and AXL, play crucial roles in acquired EGFR‐TKI resistance. Previous studies showed that HDAC inhibitors can down‐regulate receptor tyrosine kinases to overcome EGFR‐TKI resistance in NSCLC (Rho et al., 2014; Wang et al., 2013). However, the limited knowledge of the effects of HDAC inhibitors on receptor tyrosine kinases restricts their clinical application. Our data showed that YF454A either as a single agent or in combination with erlotinib simultaneously blocked the activity of receptor tyrosine kinases by inhibiting their protein or phosphorylation levels (Figure 5A, B). Meanwhile, the enhanced activity of EGFR downstream molecules such as Akt and ERK was also remarkably diminished by the combined regimen. These results additionally suggested that YF454A might prevent the emergence of resistance to EGFR‐TKIs through interfering with multiple ‘bypass’ receptor tyrosine kinase pathways.
Thus, in this study, we have provided the first evidence that YF454A and erlotinib synergistically inhibit the growth of EGFR‐TKI‐resistant NSCLC by blocking the cell cycle pathways and the receptor tyrosine kinase pathways. These preclinical data further indicate that HDAC inhibitors could be used to sensitize EGFR‐TKIs in the treatment of NSCLC. Therefore, the addition of YF454A to erlotinib may be warranted as part of a clinical therapy to overcome EGFR‐TKI‐resistance in patients with advanced NSCLC.
Author contributions
W.Y., W.L. and X.P. wrote the manuscript. X.P. and M.L. contributed to the experimental design. W.Y., W.L., G.C., F.C., Y.C. and H.S. performed experiments and analysed the data. All authors edited the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Chemical structures of YF454A, YF513 and YF441B.
Figure S2 YF454A and erlotinib are not synergistic in non‐EGFR‐TKI‐resistant NSCLC cells.
Figure S3 The combined treatment of YF454A and erlotinib has little effect on body weight in mice.
Table S1 Sequence of primers for PCR amplification.
Table S2 Cytotoxicity of novel HDAC inhibitors in EGFR‐TKI‐resistant NSCLC cells.
Table S3 Genes that were significantly affected by the combined treatment of YF454A and erlotinib.
Table S4 Differential expression analysis for five selected cell cycle‐related genes.
Acknowledgements
This research was supported by a grant 16ZR1410400 from the Science and Technology Commission of Shanghai Municipality (grant to X.P.), a grant 81672758 from the National Natural Science Foundation of China (grant to X.P.). The authors thank Dr Bin‐bing S. Zhou (Department of Hematology and Oncology, Shanghai Children's Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China) by providing HCC827/ER cells. The authors also thank Dr George Leiman (Center for Cancer Research, National Cancer Institute, the National Institute of Health) for revising the manuscript.
Yu, W. , Lu, W. , Chen, G. , Cheng, F. , Su, H. , Chen, Y. , Liu, M. , and Pang, X. (2017) Inhibition of histone deacetylases sensitizes EGF receptor‐TK inhibitor‐resistant non‐small‐cell lung cancer cells to erlotinib in vitro and in vivo . British Journal of Pharmacology, 174: 3608–3622. doi: 10.1111/bph.13961.
References
- Abend A, Kehat I (2015). Histone deacetylases as therapeutic targets – from cancer to cardiac disease. Pharmacol Ther 147: 55–62. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad N, Zahnow CA, Rudin CM, Baylin SB (2013). The future of epigenetic therapy in solid tumours – lessons from the past. Nat Rev Clin Oncol 10: 256–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bivona TG, Hieronymus H, Parker J, Chang K, Taron M, Rosell R et al (2011). FAS and NF‐kappaB signalling modulate dependence of lung cancers on mutant EGFR. Nature 471: 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose P, Dai Y, Grant S (2014). Histone deacetylase inhibitor (HDACI) mechanisms of action: emerging insights. Pharmacol Ther 143: 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M et al (2013). An epithelial‐mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 19: 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research N (2014). Comprehensive molecular profiling of lung adenocarcinoma. Nature 511: 543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MC, Chen CH, Wang JC, Tsai AC, Liou JP, Pan SL et al (2013). The HDAC inhibitor, MPT0E028, enhances erlotinib‐induced cell death in EGFR‐TKI‐resistant NSCLC cells. Cell Death Dis 4: e810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong CR, Janne PA (2013). The quest to overcome resistance to EGFR‐targeted therapies in cancer. Nat Med 19: 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC (2006). Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58: 621–681. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P (1984). Quantitative analysis of dose‐effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai F, Chen Y, Song Y, Huang L, Zhai D, Dong Y et al (2012). A natural small molecule harmine inhibits angiogenesis and suppresses tumour growth through activation of p53 in endothelial cells. PLoS One 7: e52162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL et al (2014). Cancer treatment and survivorship statistics, 2014. CA Cancer J Clin 64: 252–271. [DOI] [PubMed] [Google Scholar]
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO et al (2007). MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043. [DOI] [PubMed] [Google Scholar]
- Falkenberg KJ, Johnstone RW (2014). Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov 13: 673–691. [DOI] [PubMed] [Google Scholar]
- Gandara DR, Li T, Lara PN, Kelly K, Riess JW, Redman MW et al (2014). Acquired resistance to targeted therapies against oncogene‐driven non‐small‐cell lung cancer: approach to subtyping progressive disease and clinical implications. Clin Lung Cancer 15: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber DE, Boothman DA, Fattah FJ, Dong Y, Zhu H, Skelton RA et al (2015). Phase 1 study of romidepsin plus erlotinib in advanced non‐small cell lung cancer. Lung Cancer 90: 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui CY, Ngo L, Xu WS, Richon VM, Marks PA (2004). Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter‐associated proteins, including HDAC1. Proc Natl Acad Sci U S A 101: 1241–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B, Surowiak P, Budczies J, Lanczky A (2013). Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non‐small‐cell lung cancer. PLoS One 8: e82241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, Lane HA (2005). ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5: 341–354. [DOI] [PubMed] [Google Scholar]
- Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA et al (2010). Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non‐small‐cell lung cancer. J Clin Oncol 28: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SE, Chester JD (2015). Personalised cancer medicine. Int J Cancer 137: 262–266. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo Y, Gemma A, Noro R, Seike M, Kataoka K, Matsuda K et al (2005). Reduction of PTEN protein and loss of epidermal growth factor receptor gene mutation in lung cancer with natural resistance to gefitinib (IRESSA). Br J Cancer 92: 1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmaiah KC, Jacob LA, Aparna S, Lokanatha D, Saldanha SC (2014). Epigenetic therapy of cancer with histone deacetylase inhibitors. J Cancer Res Ther 10: 469–478. [DOI] [PubMed] [Google Scholar]
- Lee JK, Shin JY, Kim S, Lee S, Park C, Kim JY et al (2013). Primary resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) in patients with non‐small‐cell lung cancer harboring TKI‐sensitive EGFR mutations: an exploratory study. Ann Oncol 24: 2080–2087. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG (2006). Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 6: 38–51. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Takeuchi S, Yamada T, Ebi H, Sano T, Nanjo S et al (2013). EGFR‐TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res 73: 2428–2434. [DOI] [PubMed] [Google Scholar]
- Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko TK, Teo AS et al (2012). A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med 18: 521–528. [DOI] [PubMed] [Google Scholar]
- Pang X, Yi Z, Zhang X, Sung B, Qu W, Lian X et al (2009). Acetyl‐11‐keto‐beta‐boswellic acid inhibits prostate tumor growth by suppressing vascular endothelial growth factor receptor 2‐mediated angiogenesis. Cancer Res 69: 5893–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M et al (2005). KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2: e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA et al (2005). Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A 102: 3697–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho JK, Choi YJ, Kim SY, Kim TW, Choi EK, Yoon SJ et al (2014). MET and AXL inhibitor NPS‐1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res 74: 253–262. [DOI] [PubMed] [Google Scholar]
- Robbins AR, Jablonski SA, Yen TJ, Yoda K, Robey R, Bates SE et al (2005). Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle 4: 717–726. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandor V, Senderowicz A, Mertins S, Sackett D, Sausville E, Blagosklonny MV et al (2000). P21‐dependent g(1)arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer 83: 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartore‐Bianchi A, Di Nicolantonio F, Nichelatti M, Molinari F, De Dosso S, Saletti P et al (2009). Multi‐determinants analysis of molecular alterations for predicting clinical benefit to EGFR‐targeted monoclonal antibodies in colorectal cancer. PLoS One 4: e7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T et al (2013). Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 31: 3327–3334. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A (2016). Cancer statistics, 2016. CA Cancer J Clin 66: 7–30. [DOI] [PubMed] [Google Scholar]
- Soucheray M, Capelletti M, Pulido I, Kuang Y, Paweletz CP, Becker JH et al (2015). Intratumoral heterogeneity in EGFR‐mutant NSCLC results in divergent resistance mechanisms in response to EGFR tyrosine kinase inhibition. Cancer Res 75: 4372–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CS, Gilligan D, Pacey S (2015). Treatment approaches for EGFR‐inhibitor‐resistant patients with non‐small‐cell lung cancer. Lancet Oncol 16: e447–e459. [DOI] [PubMed] [Google Scholar]
- Vaziri C, Stice L, Faller DV (1998). Butyrate‐induced G1 arrest results from p21‐independent disruption of retinoblastoma protein‐mediated signals. Cell Growth Differ 9: 465–474. [PubMed] [Google Scholar]
- Wang B, Yu W, Guo J, Jiang X, Lu W, Liu M et al (2015). The antiparasitic drug, potassium antimony tartrate, inhibits tumor angiogenesis and tumor growth in nonsmall‐cell lung cancer. J Pharmacol Exp Ther 352: 129–138. [DOI] [PubMed] [Google Scholar]
- Wang J, Pursell NW, Samson ME, Atoyan R, Ma AW, Selmi A et al (2013). Potential advantages of CUDC‐101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol Cancer Ther 12: 925–936. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhang L, Chen G, Zhang J, Li Z, Lu W et al (2014). Small molecule 1′‐acetoxychavicol acetate suppresses breast tumor metastasis by regulating the SHP‐1/STAT3/MMPs signaling pathway. Breast Cancer Res Treat 148: 279–289. [DOI] [PubMed] [Google Scholar]
- Webster RM (2016). Combination therapies in oncology. Nat Rev Drug Discov 15: 81–82. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Dunn EF, Harari PM (2010). Understanding resistance to EGFR inhibitors‐impact on future treatment strategies. Nat Rev Clin Oncol 7: 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, De Bruin A, Wang H, Simmons T, Cleghorn W, Goldenberg LE et al (2015). Selective roles of E2Fs for ErbB2‐ and Myc‐mediated mammary tumorigenesis. Oncogene 34: 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Zhang T, Wu H, Yang Y, Liu N, Chen A et al (2014). Design and optimization of novel hydroxamate‐based histone deacetylase inhibitors of Bis‐substituted aromatic amides bearing potent activities against tumor growth and metastasis. J Med Chem 57: 9357–9369. [DOI] [PubMed] [Google Scholar]
- Yeh HH, Ogawa K, Balatoni J, Mukhapadhyay U, Pal A, Gonzalez‐Lepera C et al (2011). Molecular imaging of active mutant L858R EGF receptor (EGFR) kinase‐expressing nonsmall cell lung carcinomas using PET/CT. Proc Natl Acad Sci U S A 108: 1603–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Geng Y, Sicinski P (2001). Specific protection against breast cancers by cyclin D1 ablation. Nature 411: 1017–1021. [DOI] [PubMed] [Google Scholar]
- Zhang T, Chen Y, Li J, Yang F, Wu H, Dai F et al (2014). Antitumor action of a novel histone deacetylase inhibitor, YF479, in breast cancer. Neoplasia 16: 665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Chemical structures of YF454A, YF513 and YF441B.
Figure S2 YF454A and erlotinib are not synergistic in non‐EGFR‐TKI‐resistant NSCLC cells.
Figure S3 The combined treatment of YF454A and erlotinib has little effect on body weight in mice.
Table S1 Sequence of primers for PCR amplification.
Table S2 Cytotoxicity of novel HDAC inhibitors in EGFR‐TKI‐resistant NSCLC cells.
Table S3 Genes that were significantly affected by the combined treatment of YF454A and erlotinib.
Table S4 Differential expression analysis for five selected cell cycle‐related genes.