

Abstract
Under physiological conditions, perivascular adipose tissue (PVAT) attenuates agonist‐induced vasoconstriction by releasing vasoactive molecules including hydrogen peroxide, angiotensin 1–7, adiponectin, methyl palmitate, hydrogen sulfide, NO and leptin. This anticontractile effect of PVAT is lost under conditions of obesity. The central mechanism underlying this PVAT dysfunction in obesity is likely to be an ‘obesity triad’ (consisting of PVAT hypoxia, inflammation and oxidative stress) that leads to the impairment of PVAT‐derived vasoregulators. The production of hydrogen sulfide, NO and adiponectin by PVAT is reduced in obesity, whereas the vasodilator response to leptin is impaired (vascular leptin resistance). Strikingly, the vasodilator response to acetylcholine is reduced only in PVAT‐containing, but not in PVAT‐free thoracic aorta isolated from diet‐induced obese mice, indicating a unique role for PVAT in obesity‐induced vascular dysfunction. Furthermore, PVAT dysfunction has also been observed in small arteries isolated from the gluteal/visceral fat biopsy samples of obese individuals. Therefore, PVAT may represent a new therapeutic target for vascular complications in obesity. A number of approaches are currently being tested under experimental conditions. Potential therapeutic strategies improving PVAT function include body weight reduction, enhancing PVAT hydrogen sulfide release (e.g. rosiglitazone, atorvastatin and cannabinoid CB1 receptor agonists) and NO production (e.g. arginase inhibitors), inhibition of the renin–angiotensin–aldosterone system, inhibition of inflammation with melatonin or cytokine antagonists, activators of AMP‐activated kinase (e.g. metformin, resveratrol and diosgenin) and adiponectin releasers or expression enhancers.
Linked Articles
This article is part of a themed section on Molecular Mechanisms Regulating Perivascular Adipose Tissue – Potential Pharmacological Targets? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.20/issuetoc
Abbreviations
- ADRF
adipocyte‐derived relaxing factor
- Ang
angiotensin
- BAT
brown adipose tissue
- BKCa
large‐conductance Ca2 +‐activated K+ channels
- CBS
cystathionine‐β‐synthase
- CSE
cystathionine‐γ‐lyase
- EDHF
endothelium‐derived hyperpolarizing factor
- eNOS
endothelial NOS
- ET‐1
endothelin‐1
- H2S
hydrogen sulfide
- HFD
high‐fat diet
- KATP
ATP‐dependent K+ channels
- Kv
voltage‐gated K+ channels
- L‐NAME
NG‐nitro‐L‐arginine methyl ester
- MCP‐1
monocyte chemoattractant protein‐1
- PAME
palmitic acid methyl ester
- PVAT
perivascular adipose tissue
- PVRF
perivascular‐derived relaxing factors
- RAAS
renin–angiotensin–aldosterone system
- RAS
renin–angiotensin system
- sGC
soluble guanylyl cyclase
- VSMC
vascular smooth muscle cells
- WAT
white adipose tissue
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Nuclear hormone receptors d |
| TNF‐α | PPARγ |
| Adipo1 receptor | Catalytic receptors e |
| GPCRs b | Leptin receptor |
| β3 adrenoceptor | Enzymes f |
| Ang 1‐7 receptor (MAS1) | ACE |
| AT1 receptor | AMPK |
| Voltage‐gated ion channels c | Akt (PKB) |
| BKCa (KCa1.1) channel | CBS |
| IKCa (KCa3.1) channel | eNOS |
| KATP (Kir6.1) channel | PKG |
| KCNQ (Kv7.1) channel | sGC |
| KV channels | |
| SKCa (KCa2.1) channel |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,d,e,fAlexander et al., 2015a,b,c,d,e,f).
Introduction
The central role of the cardiovascular system is the transportation of oxygen, nutrients, biomolecules and signalling molecules to organs, tissues and cells. In addition, the circulation system is important in host defence by the immune system and in blood haemostasis/coagulation (Daiber et al., 2017). A dysregulation of vascular function may result in increased peripheral vascular resistance and blood pressure. Furthermore, vascular dysfunction promotes atherogenesis, and exacerbates insulin resistance by limiting the nutritive flow downstream in the microcirculation (Yudkin et al., 2005; Greenstein et al., 2009; Fuster et al., 2016).
For decades, the endothelium has been the focus of vascular research. In recent years, however, the importance of other vascular cells has been increasingly recognized, including vascular smooth muscle cells (VSMCs) (Lacolley et al., 2012), adventitia cells (Stenmark et al., 2013; Wu et al., 2015) and cells in the perivascular adipose tissue (PVAT). Moreover, communications exist between the different vascular cells and between the different layers of the vascular wall (Campbell et al., 2012). Therefore, the vascular wall should be regarded as a whole rather than separate layers. The present article focusses on the role of PVAT in regulating vascular function.
PVAT: general aspects
PVAT surrounds large arteries and veins, small and resistance vessels, and skeletal muscle microvessels. Other microvasculatures and the cerebral vasculature are free of PVAT (Brown et al., 2014; Gil‐Ortega et al., 2015). In contrast to humans and other large experimental animals (including rabbits and pigs), the murine coronary artery is without PVAT (Brown et al., 2014). Large vessels (such as the saphenous vein) are separated from PVAT by an anatomical barrier that is rich in collagen bundles, elastin networks, VSMCs, fibroblasts, autonomic nerve endings and vasa vasorum (Ahmed et al., 2004). In small vessels and microvessels, however, PVAT is an integral part of the vascular wall without laminar structures or any organized barrier separating PVAT from the adventitia (Szasz and Webb, 2012; Gil‐Ortega et al., 2015).
It is proposed that factors released by the PVAT reach the medial and endothelial layer of blood vessels either by direct diffusion or via the vasa vasorum (Gil‐Ortega et al., 2015). Another possible way for PVAT‐derived factors to reach the inner layers of the vascular wall is via the small media conduits, a dense reticular network of collagenous conduits connecting the medial layer with the underlying adventitia (Grabner et al., 2009; Campbell et al., 2012). These conduits enable soluble molecules to traffic between the PVAT/adventitia and media/intima.
In a murine model with VSMC‐specific PPAR‐γ deletion, the animals are completely devoid of PVAT in the aortic and mesenteric regions. In contrast, interscapular brown adipose tissue (BAT) and gonadal/inguinal/subcutaneous white adipose tissue (WAT) in these animals remain intact. These results indicate that the origin of the adipocytes in the PVAT is different from those in WAT and BAT (Chang et al., 2012; Brown et al., 2014). This notion is consistent with a previous observation that PVAT is a functionally specialized type of adipose tissue and PVAT adipocytes differ inherently in developmental and secretory properties from adipocytes in other fat depots (Chatterjee et al., 2009). Indeed, PVAT adipocytes are likely to arise from VSMC progenitors (Brown et al., 2014; Omar et al., 2014; Gil‐Ortega et al., 2015).
Importantly, regional phenotypic and functional differences exist among PVAT depots (Brown et al., 2014; Gil‐Ortega et al., 2015). Depending on the vascular bed, PVAT can be WAT‐like (e.g. murine mesenteric PVAT), BAT‐like (e.g. PVAT of the murine thoracic aorta) or mixed adipose tissue (e.g. PVAT of the murine abdominal aorta) and may have different vascularization, innervation and adipokine profiles (Szasz and Webb, 2012; Padilla et al., 2013; Brown et al., 2014; Gil‐Ortega et al., 2015; Drosos et al., 2016; Victorio et al., 2016b).
The morphological properties of PVAT in other species are less well‐defined than murine PVAT. Human coronary PVAT exhibits a histological appearance and gene expression pattern more consistent with WAT rather than BAT (Chatterjee et al., 2009; 2013).
Anticontractile function of PVAT
PVAT plays a role in intravascular thermoregulation (Chang et al., 2012), vascular inflammation (Chatterjee et al., 2013), VSMC proliferation (Miao and Li, 2012; Ozen et al., 2015), the development of atherosclerosis (Greif et al., 2009; Verhagen and Visseren, 2011; Chang et al., 2012; Kawahito et al., 2013; van Hinsbergh et al., 2015; Gomez‐Hernandez et al., 2016) and hypertension (Galvez et al., 2006; Lee et al., 2011; Chang et al., 2013). The present article focuses on the role of PVAT in regulating vascular function under physiological conditions as well as in obesity.
A role for PVAT in vascular function was first indicated by the observation that PVAT decreased the contractile responses to noradrenaline in rat aorta (Soltis and Cassis, 1991). Now, it is known that PVAT attenuates the vascular responsiveness to several (hormonal) agonists, including phenylephrine, angiotensin II (Ang II), 5‐HT and endothelin‐1 (ET‐1) (Gollasch, 2012; Szasz and Webb, 2012).
PVAT may regulate vascular tone by releasing bioactive molecules, which can be regarded as an endocrine‐related effect. In addition, PVAT also exerts a more direct, local effect on the vascular wall via paracrine mechanisms. The PVAT is composed mainly of adipocytes and releases a wide range of biologically active molecules that modulate vascular function (Szasz and Webb, 2012). Factors that mediate the anticontractile effects of PVAT are referred to as adipocyte‐derived relaxing factor (ADRF) (Lohn et al., 2002; Gollasch, 2012), or perivascular‐derived relaxing factors (PVRF) (Lee et al., 2011).
The mechanism of action of some ADRF has been shown to rely on the opening of smooth muscle K+ channels, especially the KCNQ‐type of the voltage‐gated (Kv) K+ channels (Lohn et al., 2002; Gollasch, 2012), whereas ATP‐dependent K+ (KATP) channels have only minor effects (Schleifenbaum et al., 2010; Gil‐Ortega et al., 2015). Indeed, pharmacological openers of KCNQ channels mimic the effects of ADRF in spontaneously hypertensive rats as well as in a mouse model of metabolic syndrome (the New Zealand Obese mouse) (Zavaritskaya et al., 2013; Tano et al., 2014).
The identity of ADRF/PVRF is a matter of ongoing debate and it is likely to be a combination of several different molecules, depending on the stimulus applied, the vascular bed examined, and the phenotypic state of the PVAT (Figure 1).
Figure 1.
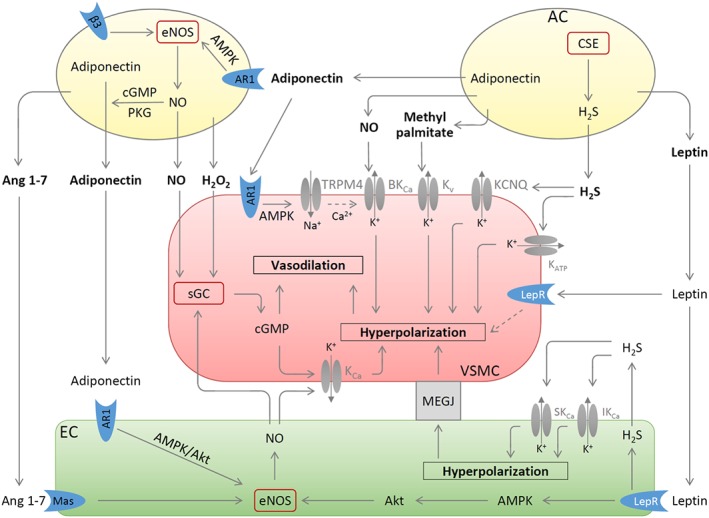
PVAT‐derived vasoactive molecules. Methyl palmitate produced by PVAT adipocytes (AC) causes vasodilatation by opening the Kv channels on VSMC. H2S is synthesized in PVAT by CSE and induces VSMC hyperpolarization by stimulating KCNQ‐type Kv or KATP channels. Leptin induces endothelium‐dependent vasodilatation by stimulating leptin receptor (LepR), which leads to activation of eNOS via a pathway involving AMPK and Akt and to H2S production. This H2S functions as an EDHF and activates endothelial small (SKCa) and intermediate (IKCa) conductance calcium‐dependent K+ channels via autocrine mechanisms. The resulting hyperpolarization of endothelial cells can be transmitted to VSMC by electrical coupling through myoendothelial gap junction (MEGJ). Leptin also causes endothelium‐independent vasodilatation by inducing VSMC hyperpolarization through unknown mechanisms. NO and H2O2 released from PVAT can elicit vasodilatation by activating sGC leading to the synthesis of cGMP. Adiponectin released from PVAT AC can be enhanced by stimulation of β3 adrenoceptors (β3) and by the NO‐cGMP‐PKG pathway. Adiponectin exerts multiple vascular effects: it stimulates NO production from PVAT and from endothelial cells and induces VSMC hyperpolarization by activating TRPM4 channels followed by opening BKCa; Ang 1–7 produced by PVAT acts on endothelial Ang 1–7 receptor (Mas; MAS1 receptor) thereby stimulating endothelial NO production. Besides stimulating sGC activity, NO from PVAT and endothelial cells can also induce/potentiate VSMC hyperpolarization through KCa or BKCa. Partly adopted from (Beltowski, 2013; Weston et al., 2013; Withers et al., 2014a).
PVAT‐derived vasoregulators
A number of PVAT‐derived biologically active molecules with vascular effects have been identified so far; these include adipokines (e.g. leptin, adiponectin, omentin, visfatin, resistin and apelin), cytokines/chemokines [e.g. IL‐6; TNF‐α; monocyte chemoattractant protein‐1 (MCP‐1) and plasminogen activator inhibitor−1], some gaseous molecules (e.g. NO and H2S), prostacyclin, Ang 1–7, Ang II, methyl palmitate, ET‐1 and ROS (e.g. superoxide, H2O2) (Gollasch, 2012; Szasz and Webb, 2012; Almabrouk et al., 2014; Tano et al., 2014). Some of these are potential candidates for ADRF/PVRF.
H2O2
Bioassay experiments indicate that PVAT can exert its anticontractile effects through two distinct mechanisms: by releasing transferable relaxing factors and by an endothelium‐independent mechanism involving H2O2 and subsequent activation of soluble guanlylyl cyclase (sGC) (Gao et al., 2007).
PVAT‐derived H2O2 has been shown to be one of the factors mediating the endothelium‐independent anticontractile property of PVAT. Because H2O2 is membrane‐permeable, PVAT‐derived H2O2 can easily diffuse to underlying smooth muscle cells and induce vasodilatation by acting as a non‐NO sGC activator (Gao et al., 2007; Fernandez‐Alfonso et al., 2013). Moreover, H2O2 can directly activate cGMP‐dependent PKGIα independently of cGMP (Burgoyne et al., 2007).
The PVAT‐derived transferable relaxing factors have been suggested by later studies to include Ang 1–7 (Lee et al., 2009; Gil‐Ortega et al., 2015), adiponectin (Greenstein et al., 2009; Lynch et al., 2013; Weston et al., 2013) and methyl palmitate (Lee et al., 2011).
Ang 1–7 and Ang II
All components of the renin–angiotensin system (RAS), except renin, are found in aortic and mesenteric PVAT, including angiotensinogen, ACE, ACE2, chymase, Ang I, Ang II, AT1 and AT2 receptors (Galvez‐Prieto et al., 2008), as well as Ang 1–7 (Lee et al., 2009).
Ang 1–7 produced by PVAT is one of the diffusible factors that induces vasodilatation indirectly through an endothelium‐dependent mechanism. It binds to the endothelial Ang 1–7 receptor (Mas) and thereby stimulates endothelial NO production. The latter acts as a hyperpolarizing factor mediated through calcium‐dependent K+ (KCa) channels to cause relaxation of the blood vessels (Lee et al., 2009; Gil‐Ortega et al., 2015).
PVAT also produces the vasoconstrictor Ang II (Huang et al., 2010; Lu et al., 2010). The release of Ang II from PVAT is enhanced in spontaneously hypertensive rats compared with normotensive rats (Lee et al., 2011). PVAT‐specific RAS activation has been recently implicated in the development of atherosclerosis associated with chronic kidney disease (Kawahito et al., 2013).
Adiponectin
It is controversial whether adiponectin is an ADRF/PVRF. Aortae and mesenteric arteries from adiponectin‐knockout mice have been shown to maintain their anticontractile properties (Fesus et al., 2007). However, recent studies demonstrate that the anticontractile activity of PVAT is significantly reduced in adiponectin‐deficient mice (Lynch et al., 2013; Withers et al., 2014b), and the anticontractile function of PVAT can be largely abolished by blocking adiponectin receptors (Greenstein et al., 2009; Lynch et al., 2013). Undisputedly, adiponectin is produced in adipose tissues (also in PVAT) and is a potent vasodilator. Circulating adiponectin has been shown to be directly related to endothelial function in the general population (Saarikoski et al., 2010), although some controversy exists (Ran et al., 2010). Adiponectin is an independent predictor of endothelial function in a well‐phenotyped cohort of patients with coronary artery disease (Margaritis et al., 2013), and genome‐wide association studies have revealed that genetically‐determined lower circulating adiponectin levels are related to worse endothelial function (Margaritis et al., 2013).
Adiponectin induces vasodilatation through multiple mechanisms. (i) Adiponectin can act directly on VSMC, causing myocyte hyperpolarization by activating TRPM4 channels followed by the opening of large‐conductance Ca2 +‐activated K+ channels (BKCa) (Lynch et al., 2013; Weston et al., 2013). (ii) Adiponectin stimulates NO release from adjacent adipocytes through paracrine mechanisms. This adipocyte‐derived NO potentiates BKCa opening in VSMC (Weston et al., 2013). (iii) Adiponectin can enhance endothelial NO production by stimulating the binding of HSP90 to endothelial NOS (eNOS) (Xi et al., 2005), by enhancing eNOS phosphorylation at serine 1177, which is catalysed by either PI3K/Akt (Xi et al., 2005; Cerqueira et al., 2012; Margaritis et al., 2013) or AMP‐activated kinase (AMPK) (Withers et al., 2014a), and by increasing the biosynthesis of tetrahydrobiopterin, an essential cofactor of eNOS (Margaritis et al., 2013). Consistently, adiponectin‐knockout mice exhibit reduced eNOS phosphorylation at serine 1177 and impaired endothelial function (Cao et al., 2009).
Moreover, adiponectin produced in PVAT has also been shown to exert a paracrine effect on the underlying arterial wall by suppressing NADPH oxidase activity via a PI3K/Akt‐mediated inhibition of Rac1 and down‐regulation of p22phox gene expression (Antonopoulos et al., 2015b).
Importantly, adiponectin plays a central role in mediating crosstalk between different vascular cells (Figure 1). Through paracrine mechanisms, adiponectin from PVAT adipocytes regulates NO production in adjacent adipocytes (Withers et al., 2014a,b). As mentioned above, PVAT adiponectin induces vasodilatation by stimulating endothelial cells and VSMC hyperpolarization, and reduces vascular oxidative stress by inhibiting NADPH oxidase activity. As a feedback mechanism, oxidative stress in the vascular wall up‐regulates PVAT adiponectin expression by activating PPARγ (Antonopoulos et al., 2015b).
Methyl palmitate
A study using the superfusion bioassay cascade technique has revealed that PVAT releases a lipophilic, heat‐stable factor that causes vasodilatation by opening the Kv channels on VSMC (Lee et al., 2011). This factor has been identified as palmitic acid methyl ester (PAME). PAME derives from PVAT adipocytes and is released from PVAT spontaneously in Ca2 +‐containing solutions. PAME release is inhibited in Ca2 +‐free conditions and enhanced by the calcium ionophore A23187 (Lee et al., 2011). The anticontractile function of PVAT was found to be reduced in spontaneously hypertensive rats as well as the release of PAME; both mechanisms contributing to the pathogenesis of hypertension (Lee et al., 2011).
H2S
H2S is synthesized from L‐cysteine by cystathionine‐β‐synthase (CBS) or cystathionine‐γ‐lyase (CSE), and is enzymatically metabolized in mitochondria by sulfide:quinone oxidoreductase (Papapetropoulos et al., 2015). In endothelial cells, H2S is produced by CSE and is considered the most likely candidate for endothelium‐derived hyperpolarizing factor (EDHF) (Yang et al., 2008; Wang et al., 2015). In an autocrine mode, endothelial H2S activates small and intermediate conductance calcium‐dependent K+ channels on endothelial cells (Mustafa et al., 2011). The resulting hyperpolarization of endothelial cells can be transmitted to VSMC by electrical coupling through myoendothelial gap junction or by increased K+ efflux, which then activates VSMC inwardly‐rectifying K+ channels and/or Na+/K+‐ATPase (Feletou and Feletou and Vanhoutte, 2009; Jamroz‐Wisniewska et al., 2014). In a paracrine mode, endothelium‐generated H2S can directly induce VSMC hyperpolarization by opening KATP channels in these cells (Wang et al., 2015).
Recent studies indicate that PVAT also produces H2S via CSE activity, and that PVAT‐derived H2S is involved in the anticontractile effect of PVAT on adjacent vessels (Fang et al., 2009; Schleifenbaum et al., 2010; Kohn et al., 2012). H2S donors produce a strong vasorelaxation of VSMC by opening the KCNQ‐type Kv channels (Kohn et al., 2012), perhaps also partly by opening KATP channels (Jamroz‐Wisniewska et al., 2014). The anticontractile effect of PVAT can be reduced by inhibition of CSE (but not CBS) in aortas from rats but not from mice, indicating that endogenous H2S is an ADRF from rat (but not mouse) aortic PVAT (Kohn et al., 2012).
NO
It has been demonstrated that the anti‐contractile effects of PVAT cannot be blocked by inhibiting NOS (Lohn et al., 2002). It was, therefore, concluded that that the PVAT‐derived relaxing factor was not NO. Now, it is clear that ADRF represents more than one single molecule, and blockade of NO synthesis alone may not be sufficient to prevent the effect of PVAT. Recent studies demonstrate that NO is produced within PVAT and this PVAT‐derived NO contributes to the anticontractile effect of PVAT (Dashwood et al., 2007; Gil‐Ortega et al., 2010; Withers et al., 2014b; Aghamohammadzadeh et al., 2015; Bussey et al., 2016; Xia et al., 2016; Victorio et al., 2016b).
Immunohistochemistry analysis has shown eNOS staining in adipocytes as well as endothelial cells of the capillaries and vasa vasorum within PVAT (Dashwood et al., 2007). The NO produced in PVAT can be directly visualized in situ with fluorescence imaging (Gil‐Ortega et al., 2010; Xia et al., 2016; Victorio et al., 2016b). Adipocyte‐derived NO is supposed to be released into the interstitial fluid and to diffuse into the capillaries and adjacent arterioles causing vasodilatation (Mastronardi et al., 2002).
In myograph experiments, NG‐nitro‐L‐arginine methyl ester (L‐NAME) induces vasoconstriction, which reflects the amount of basal NO released. The L‐NAME‐induced vasoconstriction in small arteries isolated from visceral fat of healthy individuals is reduced by the removal of PVAT (Virdis et al., 2015), indicating that PVAT contributes to vascular NO production. In PVAT‐intact, endothelium‐denuded rat mesenteric arteries, eNOS inhibitors significantly enhance noradrenaline‐induced contractions, indicating that PVAT‐derived NO contributes to the anticontractile effect of PVAT independently of the endothelium (Aghamohammadzadeh et al., 2015; Bussey et al., 2016).
Multiple mechanisms may mediate the vasorelaxant action of PVAT‐derived NO (Figure 1): (i) PVAT NO may diffuse into adjacent smooth muscle cells and induce vasodilatation by stimulating cGMP synthesis; (ii) NO derived from eNOS in adipocytes positively regulates adiponectin release (Withers et al., 2014b); and (iii) adipocyte‐derived NO modulates BKCa in smooth muscle cells and potentiates hyperpolarization (Weston et al., 2013).
Leptin
Leptin is mainly produced by adipocytes and plays important roles in regulating appetite, energy expenditure, inflammation and immune responses (Molica et al., 2015). At the clinical level, its role in coronary heart disease still remains controversial (Antonopoulos et al., 2015a). Leptin is also produced by PVAT (Dashwood et al., 2011; Galvez‐Prieto et al., 2012), although it is not considered an ADRF. The anticontractile effect of PVAT is maintained in the Zucker fa/fa rats that lack functional leptin receptors (Lohn et al., 2002).
Under physiological conditions, leptin has no acute effect on blood pressure because it activates both pressor (sympathetic nervous system) and depressor (vasodilatation and natriuresis) mechanisms in a balanced manner (Jamroz‐Wisniewska et al., 2014). Through central mechanisms, leptin increases sympathetic nervous activity to the kidney, the adrenal gland, the hindlimbs and to BAT (Haynes et al., 1997; Mark, 2013). However, leptin also directly induces relaxation of blood vessels. Acute infusion of leptin has no significant effect on blood pressure in rats (Haynes et al., 1997) or in humans (Brook et al., 2007). Interestingly, leptin's dual effect on blood pressure can be revealed by various procedures (Fruhbeck, 1999). When NO synthesis is inhibited, leptin administration to Wistar rats produces a statistically significant increase in blood pressure, whereas a hypotensive response is induced by leptin in the presence of ganglionic blockade (Fruhbeck, 1999).
Leptin may cause vasodilatation in different vessels through different mechanisms. In large arteries (e.g. the aorta), leptin induces an endothelium‐dependent vasodilatation that is the result of the release of NO (Lembo et al., 2000). Leptin‐stimulated endothelial NO production is mediated by the sequential activation of an AMPKα1‐Akt‐eNOS pathway leading to eNOS phosphorylation at serine 1177 (Vecchione et al., 2002; Procopio et al., 2009). In small arteries (e.g. mesenteric arteries), both NO and EDHF play a role in leptin‐induced relaxation (Lembo et al., 2000; Jamroz‐Wisniewska et al., 2014). The leptin‐induced vasorelaxation mediated by EDHF, is at least in part, a result of CSE‐dependent H2S production in endothelial cells (Jamroz‐Wisniewska et al., 2014). In human saphenous vein and internal mammary artery (Momin et al., 2006), and to a small extent in rat mesenteric artery (Jamroz‐Wisniewska et al., 2014), leptin induces an endothelium‐independent vasodilatation that is mediated by hyperpolarization of the VSMCs.
In coronary artery bypass surgeries, saphenous vein grafts with PVAT exhibit superior patency rate and better preserved intimal, medial and adventitial architecture compared with those without PVAT (Verma et al., 2014; Kopjar and Dashwood, 2016), demonstrating the beneficial effects of PVAT in vivo. Leptin concentrations in the PVAT of saphenous vein grafts are within the concentration range causing relaxation of bypass conduits (Dashwood et al., 2011). It has therefore been proposed that PVAT‐derived leptin may play a role in preserving a ‘healthy graft’ by reducing vasospasm at graft harvesting as well as after its implantation into the coronary circulation (Dashwood et al., 2011; Dashwood and Tsui, 2013). However, there is no evidence for a causal link between PVAT‐derived leptin and the superior performance of PVAT‐containing grafts. The beneficial effects of PVAT in this regard can also be attributed to other vasoactive molecules released by PVAT.
PVAT dysfunction in obesity
Obesity has numerous adverse effects on the circulation and cardiovascular structure and function (Lavie et al., 2009). Obese patients are more likely to develop hypertension and cardiomyopathy, and have a higher risk of stroke (Lavie et al., 2009). Obesity and atherosclerosis have long been linked in observational studies. The two conditions share similar pathophysiological pathways, including dyslipidaemia and chronic inflammatory processes (Rocha and Libby, 2009).
In humans, the thoracic peri‐aortic fat mass correlates with hypertension, diabetes and aortic/coronary calcification, when corrected for body‐mass index (but not if corrected for visceral adipose tissue) (Lehman et al., 2010). Clinical studies have shown that abdominal adiposity is associated with vascular dysfunction, as measured by flow‐mediated dilation of the brachial artery (Brook et al., 2001; Hamburg et al., 2008). In human small arteries, the anticontractile effect of PVAT is completely lost in obese patients with metabolic syndrome (Greenstein et al., 2009).
In animal models of obesity, the PVAT mass and adipocyte size are increased (Marchesi et al., 2009; Ketonen et al., 2010), which is accompanied by other structural modifications in the PVAT (Szasz and Webb, 2012). The anticontractile effect of PVAT is completely lost in the mouse model of diet‐induced obesity (Ketonen et al., 2010) and genetic model of metabolic syndrome (the New Zealand obese mouse) (Marchesi et al., 2009), or significantly reduced in the ob/ob mice (Agabiti‐Rosei et al., 2014). In addition, the PVAT of obese animals inhibits the vasodilator responses to acetylcholine (Ketonen et al., 2010; Ma et al., 2010; Xia et al., 2016). Obesity‐induced dysfunctions of the PVAT correlate with a rise in blood pressure in rodent models of diet‐induced obesity (Aghamohammadzadeh et al., 2015), demonstrating the importance of PVAT function in vascular pathology in vivo.
Based on current research results, an ‘obesity triad’ consisting of PVAT hypoxia, inflammation and oxidative stress can be proposed as the central mechanism in obesity‐induced PVAT dysfunction (Figure 2). Cellular hypoxia in PVAT is thought to be due to adipocyte hypertrophy twinned with reductions in capillary density and angiogenesis (Pasarica et al., 2009; Fuster et al., 2016). Hypoxia, in turn, stimulates the production of inflammatory cytokines and chemokines from PVAT adipocytes and infiltrating macrophages (Greenstein et al., 2009). Importantly, PVAT inflammation precedes macrophage infiltration. A short‐term high‐fat diet (HFD) fed to mice for 2 weeks results in a marked up‐regulation of pro‐inflammatory leptin and MIP1α (also known as CCL3), and down‐regulation of anti‐inflammatory PPARγ (Chatterjee et al., 2009; Cheang et al., 2015) and adiponectin (Chatterjee et al., 2009). At this early stage, there are no signs of macrophage infiltration in PVAT, although the infiltration of T cells in PVAT is likely (Chatterjee et al., 2009). Thus, the infiltration of macrophages may be a response to PVAT inflammation, probably triggered by chemokines such as MCP‐1. PVAT adipocytes produce 50‐fold more MCP‐1 than adipocytes from other fat regional depots (Chatterjee et al., 2009), and a HFD further enhances MCP‐1 expression in aortic PVAT (Ketonen et al., 2010; Xia et al., 2016). Macrophages, in turn, potentiate the PVAT inflammatory response and enhance the activity of NADPH oxidase, a major source of superoxide anion in the vasculature that is also expressed in PVAT (Gao et al., 2006). Indeed, the expression of p67phox (Ketonen et al., 2010) and Nox2 (Xia et al., 2016), subunits in the NADPH oxidase complex, is increased in the PVAT of obese mice. Moreover, HFD‐fed mice also show a reduced SOD3 expression and glutathione levels in their mesenteric PVAT (Gil‐Ortega et al., 2014). The resulting oxidative stress further enhances PVAT inflammation, leading to a vicious circle (Figure 2).
Figure 2.
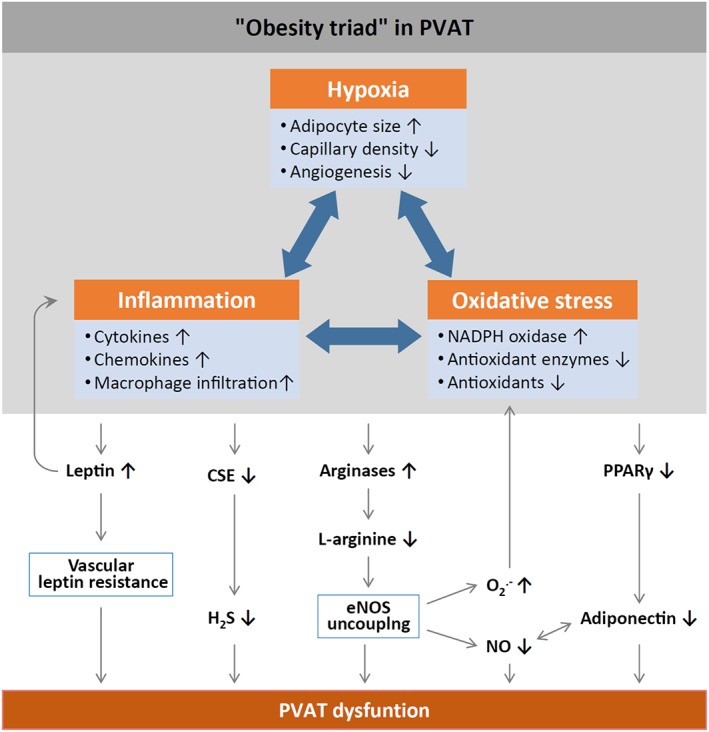
Mechanisms of PVAT dysfunction in diet‐induced obesity. HFD‐induced adipocyte hypertrophy leads to hypoxia and the production of pro‐inflammatory cytokines and chemokines, activation of NADPH oxidase and down‐regulation of antioxidant enzymes (e.g. superoxide dismutase and peroxiredoxin‐1) and non‐enzymatic antioxidants (e.g. glutathione). Infiltrating immune cells potentiate PVAT inflammation and oxidative stress. Chronic hyperleptinaemia leads to vascular leptin resistance (loss of leptin‐induced vasodilatation) and potentiation of PVAT inflammation. Long‐term obesity decreases PVAT H2S production by down‐regulating CSE expression. The up‐regulation of arginases leads to L‐arginine deficiency and eNOS uncoupling (enhanced superoxide production and reduced NO production by eNOS). PVAT adiponectin expression is reduced in obesity, very likely due to a down‐regulation of PPARγ. Normally, NO stimulates adiponectin secretion and adiponectin increases PVAT NO production. This positive feedback mechanism is impaired in obesity.
In support of this concept, PVAT dysfunction induced by in vitro incubation with aldosterone (Withers et al., 2011) or cytokines (Greenstein et al., 2009) can be fully restored using a combination of catalase and superoxide dismutase. Ex vivo incubation with superoxide dismutase and catalase also restores the anticontractile function of PVAT from obese individuals (Aghamohammadzadeh et al., 2013, 2015). Similarly, hypoxia‐induced PVAT dysfunction can be normalized by in vitro incubation with an anti‐TNF‐α antibody or an anti‐IL‐6 antibody (Greenstein et al., 2009), confirming the role of inflammation and oxidative stress in PVAT dysfunction. In agreement with this, ex vivo incubation with the anti‐TNF‐α antibody infliximab improves NO production in small arteries isolated from obese patients. Moreover, this effect is more pronounced in PVAT‐containing vessels than in PVAT‐free arteries (Virdis et al., 2015). The removal of macrophages prevents hypoxia‐ and aldosterone‐induced PVAT dysfunction (Withers et al., 2011). Also, preventing macrophage infiltration ameliorates PVAT dysfunction in mice with diet‐induced obesity (Wang et al., 2012).
Local inflammation in PVAT is potentiated by systemic inflammation and inflammation of other adipose tissues in the obesity state, where adipose tissue mass can range from 30 to 50% of total body mass (Fuster et al., 2016). Adipose tissue is the major source of IL‐6, contributing as much as one third of total circulating IL‐6 (Mohamed‐Ali et al., 1997). Similarly, leptin is an adipose tissue‐specific adipokine. It is highly expressed in adipocytes, and circulating leptin levels increase in parallel with adipose tissue mass (Fuster et al., 2016). Thus, IL‐6, leptin and other pro‐inflammatory factors released from other adipose tissues can reach PVAT via the circulation and contribute to PVAT inflammation.
The obesity triad of hypoxia, inflammation and oxidative stress in PVAT ultimately leads to a dysregulated production/secretion of adipokines and cytokines. In PVAT of HFD‐fed mice, pro‐inflammatory adipokines/cytokines (e.g. leptin, IL‐6, TNF‐α and interferon‐γ) are up‐regulated, whereas anti‐inflammatory adipokines/cytokines (e.g. adiponectin and IL‐10) are down‐regulated (Chatterjee et al., 2009; Ketonen et al., 2010; Xia et al., 2016). The resulting exacerbation of inflammation and oxidative stress promotes a dysregulation of biomolecule production in the PVAT and vascular dysfunction (Almabrouk et al., 2014; Gil‐Ortega et al., 2014; Omar et al., 2014; Molica et al., 2015).
Dysregulation of PVAT‐derived vasoregulators in obesity
Under conditions of obesity, the synthesis, secretion or action of PVAT‐derived vasoactive molecules are impaired (Figure 2).
H2S production by PVAT in obesity
Experimental obesity induced by a high‐calorie (but not high‐fat) ‘cafeteria’ diet is associated with a time‐dependent effect on PVAT‐produced H2S in rats. Feeding for 3 months results in obesity with signs of metabolic syndrome, whereas 1 month feeding leads to adiposity without insulin resistance. Interestingly, short‐term obesity (induced by 1 month of cafeteria diet) in rats increases production of H2S by PVAT and this is associated with an enhanced anticontractile effect of PVAT, whereas long‐term obesity (3 months on cafeteria diet) reduces H2S production and the anticontractile effects of PVAT (Beltowski, 2013). The enhanced H2S production in the early phase of obesity may represent a compensatory mechanism and result from reduced H2S oxidation due to hypoxia. In contrast, H2S production is decreased in long‐lasting obesity and metabolic syndrome because of a down‐regulation of the CSE enzyme in PVAT (Beltowski, 2013).
Plasma H2S levels are reduced in overweight people and patients with type 2 diabetes, independently of diabetes. Waist circumference has been shown to be an independent predictor of plasma H2S and remains an independent predictor of plasma H2S after adjustment for systolic blood pressure, microvascular function, insulin sensitivity, glycaemic control and lipid profile (Whiteman et al., 2010).
Adiponectin expression in PVAT and its secretion in obesity
In humans, circulating adiponectin levels are decreased with increasing obesity (Weiss et al., 2004) and type 2 diabetes (Antonopoulos et al., 2015b). Hypoadiponectinemia is closely associated with endothelial dysfunction (Shimabukuro et al., 2003).
Reduced adiponectin levels are uniformly found in PVAT of obese subjects (Aghamohammadzadeh et al., 2013) and in PVAT samples from different animal models of obesity, including the ob/ob mice (Agabiti‐Rosei et al., 2014), the db/db mice (Meijer et al., 2013) and diet‐induced obese mice (Chatterjee et al., 2009).
A recent study has identified PPARγ as a positive regulator of adiponectin expression in PVAT (Antonopoulos et al., 2015b). Interestingly, PPARγ is down‐regulated in the PVAT of diet‐induced obese mice (Chatterjee et al., 2009); this may represent a molecular mechanism for the reduced adiponectin expression in PVAT found during obesity.
Furthermore, adiponectin secretion is also reduced during obesity. Normally, adiponectin secretion from PVAT adipocytes is fine‐tuned; the mechanisms include β3‐adrenoceptor stimulation (Weston et al., 2013), NO production and PKG activation (Withers et al., 2014b). Under conditions of obesity, adiponectin secretion is decreased, which may be attributable to β3 adrenoceptor desensitization, reduced NO production and PKG down‐regulation in PVAT adipocytes (Withers et al., 2014a).
PVAT NO production in obesity
Although an adaptive overproduction of NO from mesenteric PVAT has been observed at the early phase of diet‐induced obesity in C57BL/6J mice (Gil‐Ortega et al., 2010), a longer time on a HFD leads to a reduction in PVAT NO production (Aghamohammadzadeh et al., 2015; Xia et al., 2016).
Inhibition of NO synthesis enhances the agonist‐induced contraction of PVAT‐intact, endothelium‐denuded mesenteric arteries from healthy rats. However, this effect of NO synthesis inhibition is lost in PVAT‐intact vessel segments from diet‐induced obese animals (Aghamohammadzadeh et al., 2015; Bussey et al., 2016). These results indicate that the anticontractile effect of healthy PVAT is partially mediated by PVAT‐derived NO, whereas obesity reduces PVAT NO production to a level that is too low to be functionally relevant.
Consistent with these data, basal NO release has been shown to be reduced in small arteries from obese patients compared with non‐obese controls. Interestingly, this obesity‐induced reduction in basal NO production is only evident in PVAT‐containing but not in PVAT‐removed arteries (Virdis et al., 2015), supporting the concept that a reduced production of NO by PVAT is involved in the obesity‐induced loss of the anticontractile effect of PVAT.
Several molecular mechanisms may be responsible for the reduced PVAT NO production in obesity: (i) A reduced expression of eNOS has been reported in the mesenteric PVAT of diet‐induced obese rats (Bussey et al., 2016) and mice (Gil‐Ortega et al., 2014). (ii) In the PVAT of thoracic aorta from diet‐induced obese mice, however, we did not observe any changes in eNOS expression but have obtained evidence that the function of eNOS is impaired (eNOS uncoupling as well as reduced eNOS phosphorylation at serine 1177 residue) (Xia et al., 2016). (iii) The reduction in PVAT NO can also be partially attributed to a deficiency in PVAT adiponectin that normally stimulates eNOS activity in PVAT adipocytes (Weston et al., 2013).
In addition to PVAT NO, NO production from the endothelium is also reduced in experimental obesity (Korda et al., 2008; Marchesi et al., 2009; Yu et al., 2014).
Vascular leptin resistance in obesity
Hyperleptinaemia in diet‐induced obesity leads to selective leptin resistance with preservation of leptin‐induced increases in renal sympathetic nerve activity and blood pressure despite resistance to the anorexic and weight‐reducing actions of leptin (Rahmouni et al., 2005; Coppari and Bjorbaek, 2012; Mark, 2013). Cerebroventricular administration of leptin antagonists significantly decreases mean arterial pressure and renal sympathetic nerve activity in diet‐induced obese rabbits, indicating that the central actions of leptin play a role in the elevation of blood pressure and renal sympathetic nerve activity induced by a HFD in rabbits (Lim et al., 2013). Similarly, the elevation in blood pressure induced by chronic leptin infusion can be prevented with combined α‐ and β‐adrenoceptor blockade (Shek et al., 1998; Carlyle et al., 2002).
Hyperleptinaemia shows a biphasic effect on vascular function. In the early stage of diet‐induced obesity (4 weeks), the NO‐mediated component of leptin‐induced relaxation of mesenteric artery is impaired, which is compensated for by an up‐regulation of EDHF‐mediated vasodilatation (Beltowski et al., 2010; Jamroz‐Wisniewska et al., 2014). Interestingly, the impaired NO component of leptin‐induced vasodilatation in obese rats can be restored by antagonizing the leptin receptors, indicating that hyperleptinaemia induces vascular leptin resistance in diet‐induced obesity. Consistently, the impairment of leptin‐induced, NO‐mediated vasodilatation can also be observed in rats treated for 8 days with exogenous leptin (Jamroz‐Wisniewska et al., 2014). In the later phase of obesity (3 months), both the NO‐ and EDHF‐mediated effects of leptin are reduced leading to an increase in blood pressure (Beltowski et al., 2009; Jamroz‐Wisniewska et al., 2014). Chronic hyperleptinaemia‐induced vascular leptin resistance (loss of leptin‐induced vasodilatation) has been shown both in vivo in anaesthetised dogs and ex vitro in coronary rings isolated from diet‐induced obese dogs (Knudson et al., 2005). Mechanistically, leptin‐induced endothelial dysfunction is associated with inflammation, oxidative stress and eNOS uncoupling (Korda et al., 2008).
Leptin levels are uniformly increased in diet‐induced obesity, not only in the blood but also in PVAT (Ketonen et al., 2010; Schroeter et al., 2013; Gil‐Ortega et al., 2014; Xia et al., 2016). In a mouse model of diet‐induced obesity, the up‐regulation of leptin in aortic PVAT is paralleled by a reduced anticontractile effect of PVAT and correlates with a loss of PVAT‐derived NO and PVAT eNOS (Gil‐Ortega et al., 2014).
Metabolic syndrome in Ossabaw miniature swine increases epicardial PVAT leptin protein and coronary leptin receptor expression. Coronary arteries from swine with metabolic syndrome display significant endothelial dysfunction that is markedly exacerbated by PVAT. A leptin antagonist reverses the metabolic syndrome effect of PVAT on endothelium‐dependent vasodilatation, indicating that epicardial PVAT‐derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome (Payne et al., 2010).
The role of PVAT in obesity‐induced vascular dysfunction
In a recent study, we analysed the vascular function of thoracic aorta isolated from diet‐induced obese C57BL/6J mice (Xia et al., 2016). Unexpectedly, the endothelium‐dependent, NO‐mediated vasodilator response to acetylcholine remained unchanged in aortas from mice fed a HFD for 20 weeks (Figure 3A). Even after varying the experimental conditions (60 or 45 kcal% fat; 20 or 12 weeks on a HFD), we did not observe any endothelial dysfunction in the PVAT‐free thoracic aorta of obese mice.
Figure 3.
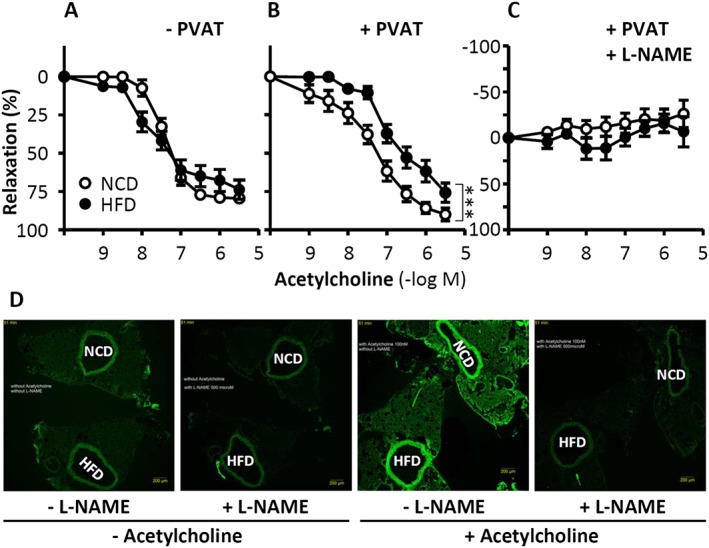
Role of PVAT in obesity‐induced vascular dysfunction. C57BL/6J mice were fed a HFD or normal control diet (NCD) for 20 weeks starting at the age of 8 weeks. The vasodilator response to acetylcholine (A–C) was performed in noradrenaline‐precontracted aorta with or without PVAT in the absence or presence of the NO synthase inhibitor L‐NAME. ***P < 0.001, n = 8. To detect PVAT NO production, NCD and HFD aorta samples were mounted back‐to‐back on the same slide to guarantee identical staining conditions for the two samples (D). NO production in PVAT‐containing aorta was determined by 4,5‐diaminofluorescein diacetate (DAF‐2 DA) staining. From Xia et al. (2016) with permission of Wolters Kluwer Health, Inc. Copyright © 2016, Wolters Kluwer Health.
Strikingly, when the aortic PVAT was left in place, a clear reduction in the vasodilator response to acetylcholine was observed in the aorta of obese animals as compared with lean controls (Figure 3B). Acetylcholine‐induced vasodilatation in the mouse aorta (either with or without PVAT) can be completely blocked by inhibition of NO synthesis (Figure 3C), indicating that this response is NO‐dependent. Thus, the reduced vasomotor function in the aorta of HFD‐fed mice (Figure 3B) results from eNOS dysfunction in the PVAT, but not in the endothelium. Indeed, we found evidence for PVAT eNOS dysfunction in diet‐induced obese mice (Xia et al., 2016).
Notably, all the pathological changes (arginase induction, L‐arginine deficiency, Akt inhibition and reduced eNOS phosphorylation) in diet‐induced obese mice were observed only in the PVAT but not in the aorta itself (Xia et al., 2016). These data provide a compelling mechanistic explanation for our initial observation that vascular dysfunction in obese mice is only evident in PVAT‐containing aorta but not in PVAT‐free aorta. These findings further substantiate our hypothesis that the role of PVAT may be even more important than that of the endothelium in obesity‐induced vascular dysfunction under certain experimental settings.
Similar evidence also exists in studies with human samples. In small arteries isolated from obese patients, an ET‐1/NO imbalance is evident, which is associated with vascular dysfunction. Interestingly, the reduced NO availability and enhanced ET‐1 signalling in arteries from obese patients can be reversed by PVAT removal (Virdis et al., 2015), demonstrating the crucial role of PVAT in obesity‐induced vascular dysfunction.
Human studies
The role of PVAT in regulating vascular function in humans has been studied by using either small arteries from gluteal/visceral fat biopsy samples or coronary bypass graft materials such as the internal mammary artery or the saphenous vein.
The anticontractile effect of human PVAT has been shown for the internal mammary artery (Gao et al., 2005; Malinowski et al., 2008; Foudi et al., 2011; Malinowski et al., 2013; Ozen et al., 2013), the saphenous vein (Ford et al., 2006; Dashwood et al., 2007, 2011; Foudi et al., 2011; Ozen et al., 2013) and for small arteries (Aghamohammadzadeh et al., 2013; Greenstein et al., 2009; Virdis et al., 2015).
Currently, PVAT‐removed graft materials are mostly used in coronary artery bypass operations. Intimal hyperplasia and vasospasm with a reduction in patency rate are common problems that occur after bypass surgery (Ozen et al., 2015). In this regard, saphenous vein grafts harvested by the ‘no‐touch’ technique with intact adventitia and PVAT have been shown to be superior to those obtained by open vein harvesting or endoscopic vein harvesting with adventitia and PVAT removed (Souza et al., 2001; Dashwood and Tsui, 2013; Dreifaldt et al., 2013; Verma et al., 2014; Kopjar and Dashwood, 2016). Unfortunately, there are yet no such clinical studies addressing the role of internal mammary artery PVAT in coronary artery bypass operations.
Obesity in humans is associated with endothelial dysfunction in vivo (Steinberg et al., 1996; Al Suwaidi et al., 2001; Grassi et al., 2010). In human small arteries taken from subcutaneous gluteal fat biopsies, the anticontractile effect of PVAT is shown to be completely lost in obese patients (Greenstein et al., 2009; Aghamohammadzadeh et al., 2013).
In obese patients, PVAT undergoes hypoxia, oxidative stress and inflammation, leading to the up‐regulation of pro‐inflammatory cytokines (e.g. TNF‐α) (Greenstein et al., 2009), enhanced ROS production, a down‐regulation of antioxidant enzymes (e.g. SOD1, peroxiredoxin‐1) (Aghamohammadzadeh et al., 2015) and reduced release of vasodilating adipokines (e.g. adiponectin) (Greenstein et al., 2009) as well as a decreased NO production from PVAT (Aghamohammadzadeh et al., 2015). Ex vivo incubation of arteries from obese individuals with superoxide dismutase and catalase restores the anticontractile effects of PVAT (Aghamohammadzadeh et al., 2013; 2015).
In small arteries isolated from visceral fat of obese patients, the expression of TNF‐α is increased both in the vascular wall and in PVAT (Virdis et al., 2015). This increase in TNF‐α reduces the production of adiponectin, triggers eNOS uncoupling by activating NADPH oxidase activity and stimulates ET‐1 generation. The resulting ET‐1/NO imbalance is implicated in obesity‐induced vascular dysfunction (Virdis et al., 2015). Moreover, the reduced NO availability and enhanced ET‐1 signalling in arteries from obese patients can be reversed by PVAT removal (Virdis et al., 2015), indicating that the vasorelaxant effect of PVAT under physiological conditions is transformed into an inflammatory pro‐contractile phenotype in obesity (Virdis et al., 2015).
Therapeutic approaches to improve PVAT function
Weight loss
Calorie restriction‐induced sustained weight loss in obese rats leads to an improvement in PVAT function associated with a reversal in obesity‐induced hypertension, the restoration of adipocyte size, PVAT eNOS function, PVAT TNF‐α expression and normalization of plasma adipokine levels, including leptin and insulin (Bussey et al., 2016).
In humans, bariatric surgery restores the anticontractile activity of PVAT in severely obese individuals (Aghamohammadzadeh et al., 2013). The normalization of PVAT function after surgery is accompanied by improvements in insulin sensitivity, serum glycaemic indexes, inflammatory cytokines, adipokine profile and systolic blood pressure together with increased PVAT adiponectin and NO bioavailability (Aghamohammadzadeh et al., 2013). Strikingly, these changes are evident despite the patients remaining obese (BMI declines from 51 before surgery to 38 after surgery) (Aghamohammadzadeh et al., 2013).
Exercise training
In familial hypercholesterolaemic pigs, exercise training significantly reduces the contractile response of left circumflex coronary artery to ET‐1. This effect has been shown to be independent of PAVT (Bunker and Laughlin, 2010).
Exercise training of Wistar rats for 8 weeks decreases plasma triglyceride levels with no effect on adipokine profiles. Exercise training up‐regulates the expression of eNOS in PVAT but not in the aorta. Exercise reduces the amount of thoracic PVAT without changing the effects of PVAT on relaxation or constriction (Araujo et al., 2015). However, this study was performed in healthy rats on normal chow. Future studies should investigate the effect of exercise training on PVAT function in obesity models.
Improving the function of PVAT eNOS
A major mechanism for the PVAT eNOS dysfunction in experimental obesity is eNOS uncoupling due to a deficiency in L‐arginine, which is attributable to an up‐regulation of arginase found in PVAT of diet‐induced obese mice (Xia et al., 2016). Arginases are major L‐arginine‐consuming enzymes that metabolize L‐arginine to urea and L‐ornithine. The up‐regulation of arginase limits L‐arginine bioavailability for NO production and leads to eNOS uncoupling (Yang and Ming, 2013). An uncoupled eNOS produces superoxide at the expense of NO (Li and Forstermann, 2013; Li et al., 2013) and thus contributes to oxidative stress in PVAT (Xia et al., 2016).
The molecular mechanisms responsible for the increase in arginase and the subsequent L‐arginine deficiency observed in diet‐induced obesity remain to be determined. We propose that PVAT inflammation plays an important role in this effect. The expression/activity of vascular arginases can be enhanced by a variety of stimuli (Pernow and Jung, 2013), including Ang II (Shatanawi et al., 2011), high glucose (Romero et al., 2008), thrombin (Ming et al., 2004) and oxidized low‐density lipoprotein (Ryoo et al., 2006), conditions that inevitably lead to vascular inflammation.
To ascertain that eNOS uncoupling is causally involved in obesity‐induced vascular dysfunction, we incubated PVAT‐containing aorta with a combination of L‐arginine and an arginase inhibitor for 30 min ex vivo in organ chambers. This treatment had no effect on acetylcholine‐induced vasodilatation in aorta from control mice, but restored the vasodilator effects of PVAT‐containing aortas from HFD‐fed mice, indicating that L‐arginine deficiency is indeed a reason for PVAT eNOS dysfunction (Xia et al., 2016).
Another mechanism for PVAT eNOS dysfunction is the reduced phosphorylation of eNOS at the serine 1177 residue associated with Akt inhibition (one of the upstream kinases for serine 1177 phosphorylation) (Xia et al., 2016). In a recent study, we treated diet‐induced obese mice with the standardized Crataegus extract WS® 1442, which is known to enhance eNOS phosphorylation at the serine 1177 residue by stimulating Akt activity. The in vivo treatment with WS® 1442 improved the phosphorylation levels of Akt and eNOS, and completely normalized the vasodilator response of PVAT‐containing aorta from diet‐induced obese mice, confirming the functional importance of the Akt‐eNOS axis in obesity‐induced PVAT dysfunction (Xia N et al. unpublished data 2016).
In addition, obesity has been shown to reduce eNOS expression in mesenteric PVAT of diet‐induced obese rats (Bussey et al., 2016) and mice (Gil‐Ortega et al., 2014). Sustained weight loss in rats restores eNOS expression and improves PVAT NO production (Bussey et al., 2016). In severely obese individuals, bariatric surgery improves NO bioavailability in PVAT of small subcutaneous arteries (Aghamohammadzadeh et al., 2013).
Enhancing PVAT H2S production
In a rat model of obesity and metabolic syndrome (cafeteria diet, fed for 3 months), treatment with the PPARγ agonist rosiglitazone increases insulin sensitivity, reduces fasting insulin levels and triglyceride concentration, increases CSE expression and activity as well as PVAT H2S production, and improves the anticontractile effect of PVAT on aortic rings (Beltowski, 2013).
After treatment of rats for 3 weeks with lipophilic atorvastatin (20 mg·kg–1·day−1), but not hydrophilic pravastatin (40 mg·kg−1·day−1), the PVAT H2S levels were increased as its mitochondrial oxidation was inhibited, and the anticontractile effect of PVAT was augmented (Wojcicka et al., 2011). Inhibition of H2S metabolism results from the atorvastatin‐induced decrease in coenzyme Q, which is a cofactor of H2S oxidation by sulfide:quinone oxidoreductase. In contrast to H2S, statins do not impair mitochondrial oxidation of organic substrates (Beltowski and Jamroz‐Wisniewska, 2012). The effect of atorvastatin on PVAT H2S levels is independent of the lipid‐lowing action of the drug because both statins at the doses used had comparable effects on the plasma lipid profile. Hydrophilic statins act mainly in the liver whereas lipophilic statins are equally active in hepatocytes and extrahepatic tissues (Shitara and Sugiyama, 2006) and are supposed to accumulate in triglyceride‐rich PVAT (Beltowski, 2013). The effect of statins on PVAT function has not yet been verified in obesity models. Atorvastatin has been shown to improve PVAT function in spontaneously hypertensive rats (Zeng et al., 2009). It is not yet clear whether these effects are mediated by H2S.
In addition, cannabinoid CB1 receptor agonists elevate PVAT H2S by inhibiting its mitochondrial oxidation (Beltowski, 2013). The imidazoline I1 receptor agonist moxonidine has been shown to increase myocardial CSE expression and H2S production in streptozotocin‐induce diabetic rats (El‐Sayed et al., 2016).
Sulfhydrylated ACE inhibitors may be of particular interest because of their dual properties: inhibition of ACE and potentiation of the H2S pathway in vivo, as shown for S‐zofenopril in spontaneous hypertensive rats (Bucci et al., 2014).
Inhibition of the renin–angiotensin–aldosterone system
In wire myograph experiments with rat mesenteric small arteries, the in vitro creation of a hypoxic environment causes the loss of PVAT's anticontractile function. This hypoxia‐induced loss of PVAT's anticontractile effect can be prevented by incubation with the ACE inhibitor captopril or the AT1 receptor antagonist termisartan (Rosei et al., 2015), or with the aldosterone antagonist eplerenone (Withers et al., 2011), whereas the β‐blocker atenolol was without effect (Rosei et al., 2015). Mechanistically, the renin–angiotensin–aldosterone system (RAAS) is thought to play a key role in the exacerbation of the hypoxia‐induced inflammation and oxidative stress. Hypoxia stimulates ROS production and may activate RAAS by up‐regulating ACE expression and inhibiting ACE2 activity (Zhang et al., 2009). Ang II and aldosterone enhance oxidative stress, macrophage infiltration and inflammation (Withers et al., 2011; Rosei et al., 2015). Both Ang II and mitoROS are potent activators of HIF‐1α (Patten et al., 2010). In vivo treatment with the AT1 receptor antagonist losartan improves PVAT function in fructose‐induced hypertensive rats (Huang et al., 2010). The aldosterone antagonist spironolactone has also been shown to restore PVAT function in a rat model of β‐adrenoceptor overstimulation (Victorio et al., 2016a). Nevertheless, the effect of RAAS inhibitors on PVAT function has not been tested in obesity models in vivo so far.
Anti‐inflammatory strategies
Chronic administration of melatonin has been shown to reduce body weight, circulating insulin, glucose and triglyceride serum levels in HFD‐fed rats (Prunet‐Marcassus et al., 2003; Rios‐Lugo et al., 2010). In the leptin‐deficient ob/ob mice, the anticontractile function of mesenteric PVAT is reduced (Agabiti‐Rosei et al., 2014). Treatment with melatonin in the drinking water for 8 weeks improves PVAT function. Importantly, melatonin improves vascular function only in the presence of PVAT, indicating the importance of PVAT in the vascular dysfunction observed in ob/ob mice (Agabiti‐Rosei et al., 2014). The improvement of PVAT function by melatonin is likely to be attributable to its antioxidant/anti‐inflammatory properties. Melatonin reduces the expression of ET‐1, IL‐6 and metalloproteases 2 and 9 in the aorta, decreases TNF‐α and CD68 levels in visceral fat and increases the expression of adiponectin and adiponectin receptor 1 in PVAT of ob/ob mice (Agabiti‐Rosei et al., 2014).
Cytokine antagonists also represent attractive anti‐inflammatory approaches. Ex vivo incubation with the anti‐TNF‐α antibody infliximab has been shown to improve PVAT function and PVAT NO production in small arteries isolated from obese patients (Virdis et al., 2015). The effect of in vivo anti‐TNF‐α therapy on PVAT function has not been reported, so far.
AMPK activators
In vitro treatment of PVAT from rat thoracic aorta with AMPK activators AICAR, salicylate, metformin, resveratrol or diosgenin down‐regulates the expression of pro‐inflammatory factors (TNF‐α, IL‐6 and MCP‐1) and increases anti‐inflammatory molecules (adiponectin and PPARγ) in PVAT (Sun et al., 2014; Chen et al., 2016). This is associated with an improvement in eNOS phosphorylation and PVAT function (Sun et al., 2014; Chen et al., 2016).
Fructose‐feeding in rats leads to a dysregulation of adipokine/cytokine expression in plasma and PVAT, accompanied by a vascular dysfunction. Oral administration of fructose‐fed rats with resveratrol and metformin improves adipokine/cytokine profiles, enhances AMPK activity and SIRT1 expression in PVAT, and restores PVAT eNOS phosphorylation and acetylcholine‐induced vasodilatation (Sun et al., 2014). Importantly, in vitro treatment of aortic rings from normal rats with conditioned media of PVAT from fructose‐fed rats reduces acetylcholine‐induced vasodilatation. This inhibition of vascular function is reversed by conditioned media of PVAT from resveratrol‐ or metformin‐treated rats (Sun et al., 2014), indicating the key role of PVAT in the improvement of vascular function by the two compounds.
In HFD‐fed rats, oral administration of diosgenin or resveratrol normalizes PVAT size, restores PVAT expression of TNF‐α, IL‐6, MCP‐1, adiponectin, PPARγ and PVAT eNOS phosphorylation. Diosgenin and resveratrol also restores the effect of PVAT on vascular function (Chen et al., 2016). In contrast to these stimulating effects of diosgenin or resveratrol, another natural compound, genistein, showed no beneficial effects on PVAT despite preventing weight gain in obese ob/ob mice (Simperova et al., 2016).
Adiponectin releasers/expression enhancers
A recent meta‐analysis demonstrates a significant increase in plasma adiponectin levels following statin therapy (Chrusciel et al., 2016). Adiponectin release from PVAT can be enhanced by stimulating the eNOS‐NO‐cGMP‐PKG pathway (Weston et al., 2013; Withers et al., 2014b). Thus, the mechanisms of statin‐induced adiponectin secretion may involve vascular NO production (Forstermann and Li, 2011; Li and Forstermann, 2014). Theoretically, PVAT adiponectin release may also be stimulated by activators of eNOS, sGC or PKG. As PPARγ plays a role in governing adiponectin expression (Antonopoulos et al., 2015b), PPARγ agonists (e.g. glitazones) and AMPK activators may enhance the expression of adiponectin in vivo, in addition to their effects on PVAT H2S production (see above). However, these concepts remain speculations at this stage. The effect of such approaches on adiponectin expression/release and PVAT function need to be investigated in future studies.
Conclusion
PVAT plays a crucial role in obesity‐induced vascular dysfunction. Hypoxia, inflammation and oxidative stress in PVAT lead to an impairment in the release of vasoactive factors from PVAT, and the normal anticontractile function of PVAT is lost in obesity. PVAT may, therefore, represent a new therapeutic target for vascular complications in obesity. Various approaches have been shown to improve PVAT function; however, their therapeutic potential needs to be further verified in obesity models.
Author contributions
N.X. and H.L. wrote the manuscript. Both authors agreed to its publication.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
This study was supported by the university intramural grant Stufe I of Johannes Gutenberg University Medical Center, Mainz, Germany.
Xia, N. , and Li, H. (2017) The role of perivascular adipose tissue in obesity‐induced vascular dysfunction. British Journal of Pharmacology, 174: 3425–3442. doi: 10.1111/bph.13650.
References
- Agabiti‐Rosei C, De Ciuceis C, Rossini C, Porteri E, Rodella LF, Withers SB et al. (2014). Anticontractile activity of perivascular fat in obese mice and the effect of long‐term treatment with melatonin. J Hypertens 32: 1264–1274. [DOI] [PubMed] [Google Scholar]
- Aghamohammadzadeh R, Greenstein AS, Yadav R, Jeziorska M, Hama S, Soltani F et al. (2013). Effects of bariatric surgery on human small artery function: evidence for reduction in perivascular adipocyte inflammation, and the restoration of normal anticontractile activity despite persistent obesity. J Am Coll Cardiol 62: 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM (2015). Effects of obesity on perivascular adipose tissue vasorelaxant function: nitric oxide, inflammation and elevated systemic blood pressure. J Vasc Res 52: 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed SR, Johansson BL, Karlsson MG, Souza DS, Dashwood MR, Loesch A (2004). Human saphenous vein and coronary bypass surgery: ultrastructural aspects of conventional and “no‐touch” vein graft preparations. Histol Histopathol 19: 421–433. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015e). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015f). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almabrouk TA, Ewart MA, Salt IP, Kennedy S (2014). Perivascular fat, AMP‐activated protein kinase and vascular diseases. Br J Pharmacol 171: 595–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Suwaidi J, Higano ST, Holmes DR Jr, Lennon R, Lerman A (2001). Obesity is independently associated with coronary endothelial dysfunction in patients with normal or mildly diseased coronary arteries. J Am Coll Cardiol 37: 1523–1528. [DOI] [PubMed] [Google Scholar]
- Antonopoulos AS, Antoniades C, Tousoulis D (2015a). Unravelling the “adipokine paradox”: when the classic proatherogenic adipokine leptin is deemed the beneficial one. Int J Cardiol 197: 125–127. [DOI] [PubMed] [Google Scholar]
- Antonopoulos AS, Margaritis M, Coutinho P, Shirodaria C, Psarros C, Herdman L et al. (2015b). Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: the regulatory role of perivascular adipose tissue. Diabetes 64: 2207–2219. [DOI] [PubMed] [Google Scholar]
- Araujo HN, Valgas da Silva CP, Sponton AC, Clerici SP, Davel AP, Antunes E et al. (2015). Perivascular adipose tissue and vascular responses in healthy trained rats. Life Sci 125: 79–87. [DOI] [PubMed] [Google Scholar]
- Beltowski J (2013). Endogenous hydrogen sulfide in perivascular adipose tissue: role in the regulation of vascular tone in physiology and pathology. Can J Physiol Pharmacol 91: 889–898. [DOI] [PubMed] [Google Scholar]
- Beltowski J, Jamroz‐Wisniewska A (2012). Modulation of h(2)s metabolism by statins: a new aspect of cardiovascular pharmacology. Antioxid Redox Signal 17: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltowski J, Wojcicka G, Jamroz‐Wisniewska A, Marciniak A (2009). Resistance to acute NO‐mimetic and EDHF‐mimetic effects of leptin in the metabolic syndrome. Life Sci 85: 557–567. [DOI] [PubMed] [Google Scholar]
- Beltowski J, Wojcicka G, Jamroz‐Wisniewska A, Wojtak A (2010). Chronic hyperleptinemia induces resistance to acute natriuretic and NO‐mimetic effects of leptin. Peptides 31: 155–163. [DOI] [PubMed] [Google Scholar]
- Brook RD, Bard RL, Bodary PF, Eitzman DT, Rajagopalan S, Sun Y et al. (2007). Blood pressure and vascular effects of leptin in humans. Metab Syndr Relat Disord 5: 270–274. [DOI] [PubMed] [Google Scholar]
- Brook RD, Bard RL, Rubenfire M, Ridker PM, Rajagopalan S (2001). Usefulness of visceral obesity (waist/hip ratio) in predicting vascular endothelial function in healthy overweight adults. Am J Cardiol 88: 1264–1269. [DOI] [PubMed] [Google Scholar]
- Brown NK, Zhou Z, Zhang J, Zeng R, Wu J, Eitzman DT et al. (2014). Perivascular adipose tissue in vascular function and disease: a review of current research and animal models. Arterioscler Thromb Vasc Biol 34: 1621–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci M, Vellecco V, Cantalupo A, Brancaleone V, Zhou Z, Evangelista S et al. (2014). Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ACE inhibition. Cardiovasc Res 102: 138–147. [DOI] [PubMed] [Google Scholar]
- Bunker AK, Laughlin MH (2010). Influence of exercise and perivascular adipose tissue on coronary artery vasomotor function in a familial hypercholesterolemic porcine atherosclerosis model. J Appl Physiol (1985) 108: 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne JR, Madhani M, Cuello F, Charles RL, Brennan JP, Schroder E et al. (2007). Cysteine redox sensor in PKGIa enables oxidant‐induced activation. Science 317: 1393–1397. [DOI] [PubMed] [Google Scholar]
- Bussey CE, Withers SB, Aldous RG, Edwards G, Heagerty AM (2016). Obesity‐related perivascular adipose tissue damage is reversed by sustained weight loss in the rat. Arterioscler Thromb Vasc Biol 36: 1377–1385. [DOI] [PubMed] [Google Scholar]
- Campbell KA, Lipinski MJ, Doran AC, Skaflen MD, Fuster V, McNamara CA (2012). Lymphocytes and the adventitial immune response in atherosclerosis. Circ Res 110: 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Tao L, Yuan Y, Jiao X, Lau WB, Wang Y et al. (2009). Endothelial dysfunction in adiponectin deficiency and its mechanisms involved. J Mol Cell Cardiol 46: 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyle M, Jones OB, Kuo JJ, Hall JE (2002). Chronic cardiovascular and renal actions of leptin: role of adrenergic activity. Hypertension 39: 496–501. [DOI] [PubMed] [Google Scholar]
- Cerqueira FM, Brandizzi LI, Cunha FM, Laurindo FR, Kowaltowski AJ (2012). Serum from calorie‐restricted rats activates vascular cell eNOS through enhanced insulin signaling mediated by adiponectin. PLoS One 7: e31155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Milton H, Eitzman DT, Chen YE (2013). Paradoxical roles of perivascular adipose tissue in atherosclerosis and hypertension. Circ J 77: 11–18. [DOI] [PubMed] [Google Scholar]
- Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C et al. (2012). Loss of perivascular adipose tissue on peroxisome proliferator‐activated receptor‐gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 126: 1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee TK, Aronow BJ, Tong WS, Manka D, Tang Y, Bogdanov VY et al. (2013). Human coronary artery perivascular adipocytes overexpress genes responsible for regulating vascular morphology, inflammation, and hemostasis. Physiol Genomics 45: 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G et al. (2009). Proinflammatory phenotype of perivascular adipocytes: influence of high‐fat feeding. Circ Res 104: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang WS, Tian XY, Wong WT, Huang Y (2015). The peroxisome proliferator‐activated receptors in cardiovascular diseases: experimental benefits and clinical challenges. Br J Pharmacol 172: 5512–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Xu X, Zhang Y, Liu K, Huang F, Liu B et al. (2016). Diosgenin regulates adipokine expression in perivascular adipose tissue and ameliorates endothelial dysfunction via regulation of AMPK. J Steroid Biochem Mol Biol 155: 155–165. [DOI] [PubMed] [Google Scholar]
- Chrusciel P, Sahebkar A, Rembek‐Wieliczko M, Serban MC, Ursoniu S, Mikhailidis DP et al. (2016). Impact of statin therapy on plasma adiponectin concentrations: A systematic review and meta‐analysis of 43 randomized controlled trial arms. Atherosclerosis 253: 194–208. [DOI] [PubMed] [Google Scholar]
- Coppari R, Bjorbaek C (2012). Leptin revisited: its mechanism of action and potential for treating diabetes. Nat Rev Drug Discov 11: 692–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daiber A, Steven S, Weber A, Shuvaev VV, Muzykantov VR, Laher I et al. (2017). Targeting vascular (endothelial) dysfunction. Br J Pharmacol 174: 1591–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashwood MR, Dooley A, Shi‐Wen X, Abraham DJ, Dreifaldt M, Souza DS (2011). Perivascular fat‐derived leptin: a potential role in improved vein graft performance in coronary artery bypass surgery. Interact Cardiovasc Thorac Surg 12: 170–173. [DOI] [PubMed] [Google Scholar]
- Dashwood MR, Dooley A, Shi‐Wen X, Abraham DJ, Souza DS (2007). Does periadventitial fat‐derived nitric oxide play a role in improved saphenous vein graft patency in patients undergoing coronary artery bypass surgery? J Vasc Res 44: 175–181. [DOI] [PubMed] [Google Scholar]
- Dashwood MR, Tsui JC (2013). ‘No‐touch’ saphenous vein harvesting improves graft performance in patients undergoing coronary artery bypass surgery: a journey from bedside to bench. Vascul Pharmacol 58: 240–250. [DOI] [PubMed] [Google Scholar]
- Dreifaldt M, Souza D, Bodin L, Shi‐Wen X, Dooley A, Muddle J et al. (2013). The vasa vasorum and associated endothelial nitric oxide synthase is more important for saphenous vein than arterial bypass grafts. Angiology 64: 293–299. [DOI] [PubMed] [Google Scholar]
- Drosos I, Chalikias G, Pavlaki M, Kareli D, Epitropou G, Bougioukas G et al. (2016). Differences between perivascular adipose tissue surrounding the heart and the internal mammary artery: possible role for the leptin‐inflammation‐fibrosis‐hypoxia axis. Clin Res Cardiol 105: 887–900. [DOI] [PubMed] [Google Scholar]
- El‐Sayed SS, Zakaria MN, Abdel‐Ghany RH, Abdel‐Rahman AA (2016). Cystathionine‐gamma lyase‐derived hydrogen sulfide mediates the cardiovascular protective effects of moxonidine in diabetic rats. Eur J Pharmacol 783: 73–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Zhao J, Chen Y, Ma T, Xu G, Tang C et al. (2009). Hydrogen sulfide derived from periadventitial adipose tissue is a vasodilator. J Hypertens 27: 2174–2185. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM (2009). EDHF: an update. Clin Sci (Lond) 117: 139–155. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Alfonso MS, Gil‐Ortega M, Garcia‐Prieto CF, Aranguez I, Ruiz‐Gayo M, Somoza B (2013). Mechanisms of perivascular adipose tissue dysfunction in obesity. Int J Endocrinol 2013: 402053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC et al. (2007). Adiponectin is a novel humoral vasodilator. Cardiovasc Res 75: 719–727. [DOI] [PubMed] [Google Scholar]
- Ford CA, Mong K, Tabrizchi R (2006). Influence of tangential stress on mechanical responses to vasoactive agents in human saphenous vein with and without perivascular adipose tissue. Can J Cardiol 22: 1209–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstermann U, Li H (2011). Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br J Pharmacol 164: 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi N, Kotelevets L, Gomez I, Louedec L, Longrois D, Chastre E et al. (2011). Differential reactivity of human mammary artery and saphenous vein to prostaglandin E(2): implication for cardiovascular grafts. Br J Pharmacol 163: 826–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruhbeck G (1999). Pivotal role of nitric oxide in the control of blood pressure after leptin administration. Diabetes 48: 903–908. [DOI] [PubMed] [Google Scholar]
- Fuster JJ, Ouchi N, Gokce N, Walsh K (2016). Obesity‐induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res 118: 1786–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez‐Prieto B, Bolbrinker J, Stucchi P, de Las Heras AI, Merino B, Arribas S et al. (2008). Comparative expression analysis of the renin‐angiotensin system components between white and brown perivascular adipose tissue. J Endocrinol 197: 55–64. [DOI] [PubMed] [Google Scholar]
- Galvez‐Prieto B, Somoza B, Gil‐Ortega M, Garcia‐Prieto CF, de Las Heras AI, Gonzalez MC et al. (2012). Anticontractile effect of perivascular adipose tissue and leptin are reduced in hypertension. Front Pharmacol 3: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez B, de Castro J, Herold D, Dubrovska G, Arribas S, Gonzalez MC et al. (2006). Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 26: 1297–1302. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Lu C, Su LY, Sharma AM, Lee RM (2007). Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol 151: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM et al. (2006). Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res 71: 363–373. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Zeng ZH, Teoh K, Sharma AM, Abouzahr L, Cybulsky I et al. (2005). Perivascular adipose tissue modulates vascular function in the human internal thoracic artery. J Thorac Cardiovasc Surg 130: 1130–1136. [DOI] [PubMed] [Google Scholar]
- Gil‐Ortega M, Condezo‐Hoyos L, Garcia‐Prieto CF, Arribas SM, Gonzalez MC, Aranguez I et al. (2014). Imbalance between pro and anti‐oxidant mechanisms in perivascular adipose tissue aggravates long‐term high‐fat diet‐derived endothelial dysfunction. PLoS One 9: e95312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil‐Ortega M, Somoza B, Huang Y, Gollasch M, Fernandez‐Alfonso MS (2015). Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol Metab 26: 367–375. [DOI] [PubMed] [Google Scholar]
- Gil‐Ortega M, Stucchi P, Guzman‐Ruiz R, Cano V, Arribas S, Gonzalez MC et al. (2010). Adaptative nitric oxide overproduction in perivascular adipose tissue during early diet‐induced obesity. Endocrinology 151: 3299–3306. [DOI] [PubMed] [Google Scholar]
- Gollasch M (2012). Vasodilator signals from perivascular adipose tissue. Br J Pharmacol 165: 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Hernandez A, Beneit N, Escribano O, Diaz‐Castroverde S, Garcia‐Gomez G, Fernandez S et al. (2016). Severe brown fat lipoatrophy aggravates atherosclerotic process in male mice. Endocrinology 157: 3517–3528. [DOI] [PubMed] [Google Scholar]
- Grabner R, Lotzer K, Dopping S, Hildner M, Radke D, Beer M et al. (2009). Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE−/− mice. J Exp Med 206: 233–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassi G, Seravalle G, Scopelliti F, Dell'Oro R, Fattori L, Quarti‐Trevano F et al. (2010). Structural and functional alterations of subcutaneous small resistance arteries in severe human obesity. Obesity (Silver Spring) 18: 92–98. [DOI] [PubMed] [Google Scholar]
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M et al. (2009). Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 119: 1661–1670. [DOI] [PubMed] [Google Scholar]
- Greif M, Becker A, von Ziegler F, Lebherz C, Lehrke M, Broedl UC et al. (2009). Pericardial adipose tissue determined by dual source CT is a risk factor for coronary atherosclerosis. Arterioscler Thromb Vasc Biol 29: 781–786. [DOI] [PubMed] [Google Scholar]
- Hamburg NM, Larson MG, Vita JA, Vasan RS, Keyes MJ, Widlansky ME et al. (2008). Metabolic syndrome, insulin resistance, and brachial artery vasodilator function in Framingham Offspring participants without clinical evidence of cardiovascular disease. Am J Cardiol 101: 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI (1997). Receptor‐mediated regional sympathetic nerve activation by leptin. J Clin Invest 100: 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Lezama MA, Ontiveros JA, Bravo G, Villafana S, del ‐Rio‐Navarro BE et al. (2010). Effect of losartan on vascular function in fructose‐fed rats: the role of perivascular adipose tissue. Clin Exp Hypertens 32: 98–104. [DOI] [PubMed] [Google Scholar]
- Jamroz‐Wisniewska A, Gertler A, Solomon G, Wood ME, Whiteman M, Beltowski J (2014). Leptin‐induced endothelium‐dependent vasorelaxation of peripheral arteries in lean and obese rats: role of nitric oxide and hydrogen sulfide. PLoS One 9: e86744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahito H, Yamada H, Irie D, Kato T, Akakabe Y, Kishida S et al. (2013). Periaortic adipose tissue‐specific activation of the renin‐angiotensin system contributes to atherosclerosis development in uninephrectomized apoE−/− mice. Am J Physiol Heart Circ Physiol 305: H667–H675. [DOI] [PubMed] [Google Scholar]
- Ketonen J, Shi J, Martonen E, Mervaala E (2010). Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet‐induced obese C57Bl/6 mice. Circ J 74: 1479–1487. [DOI] [PubMed] [Google Scholar]
- Knudson JD, Dincer UD, Dick GM, Shibata H, Akahane R, Saito M et al. (2005). Leptin resistance extends to the coronary vasculature in prediabetic dogs and provides a protective adaptation against endothelial dysfunction. Am J Physiol Heart Circ Physiol 289: H1038–H1046. [DOI] [PubMed] [Google Scholar]
- Kohn C, Schleifenbaum J, Szijarto IA, Marko L, Dubrovska G, Huang Y et al. (2012). Differential effects of cystathionine‐gamma‐lyase‐dependent vasodilatory H2S in periadventitial vasoregulation of rat and mouse aortas. PLoS One 7: e41951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopjar T, Dashwood MR (2016). Endoscopic versus “No‐Touch” saphenous vein harvesting for coronary artery bypass grafting: a trade‐off between wound healing and graft patency. Angiology 67: 121–132. [DOI] [PubMed] [Google Scholar]
- Korda M, Kubant R, Patton S, Malinski T (2008). Leptin‐induced endothelial dysfunction in obesity. Am J Physiol Heart Circ Physiol 295: H1514–H1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB (2012). The vascular smooth muscle cell in arterial pathology: a cell that can take on multiple roles. Cardiovasc Res 95: 194–204. [DOI] [PubMed] [Google Scholar]
- Lavie CJ, Milani RV, Ventura HO (2009). Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J Am Coll Cardiol 53: 1925–1932. [DOI] [PubMed] [Google Scholar]
- Lee RM, Lu C, Su LY, Gao YJ (2009). Endothelium‐dependent relaxation factor released by perivascular adipose tissue. J Hypertens 27: 782–790. [DOI] [PubMed] [Google Scholar]
- Lee YC, Chang HH, Chiang CL, Liu CH, Yeh JI, Chen MF et al. (2011). Role of perivascular adipose tissue‐derived methyl palmitate in vascular tone regulation and pathogenesis of hypertension. Circulation 124: 1160–1171. [DOI] [PubMed] [Google Scholar]
- Lehman SJ, Massaro JM, Schlett CL, O'Donnell CJ, Hoffmann U, Fox CS (2010). Peri‐aortic fat, cardiovascular disease risk factors, and aortic calcification: the Framingham Heart Study. Atherosclerosis 210: 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo G, Vecchione C, Fratta L, Marino G, Trimarco V, d'Amati G et al. (2000). Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes 49: 293–297. [DOI] [PubMed] [Google Scholar]
- Li H, Forstermann U (2013). Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr Opin Pharmacol 13: 161–167. [DOI] [PubMed] [Google Scholar]
- Li H, Forstermann U (2014). Pharmacological prevention of eNOS uncoupling. Curr Pharm Des 20: 3595–3606. [DOI] [PubMed] [Google Scholar]
- Li H, Horke S, Forstermann U (2013). Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol Sci 34: 313–319. [DOI] [PubMed] [Google Scholar]
- Lim K, Burke SL, Head GA (2013). Obesity‐related hypertension and the role of insulin and leptin in high‐fat‐fed rabbits. Hypertension 61: 628–634. [DOI] [PubMed] [Google Scholar]
- Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM (2002). Periadventitial fat releases a vascular relaxing factor. FASEB J 16: 1057–1063. [DOI] [PubMed] [Google Scholar]
- Lu C, Su LY, Lee RM, Gao YJ (2010). Mechanisms for perivascular adipose tissue‐mediated potentiation of vascular contraction to perivascular neuronal stimulation: the role of adipocyte‐derived angiotensin II. Eur J Pharmacol 634: 107–112. [DOI] [PubMed] [Google Scholar]
- Lynch FM, Withers SB, Yao Z, Werner ME, Edwards G, Weston AH et al. (2013). Perivascular adipose tissue‐derived adiponectin activates BK(Ca) channels to induce anticontractile responses. Am J Physiol Heart Circ Physiol 304: H786–H795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Ma S, He H, Yang D, Chen X, Luo Z et al. (2010). Perivascular fat‐mediated vascular dysfunction and remodeling through the AMPK/mTOR pathway in high‐fat diet‐induced obese rats. Hypertens Res 33: 446–453. [DOI] [PubMed] [Google Scholar]
- Malinowski M, Deja MA, Golba KS, Roleder T, Biernat J, Wos S (2008). Perivascular tissue of internal thoracic artery releases potent nitric oxide and prostacyclin‐independent anticontractile factor. Eur J Cardiothorac Surg 33: 225–231. [DOI] [PubMed] [Google Scholar]
- Malinowski M, Deja MA, Janusiewicz P, Golba KS, Roleder T, Wos S (2013). Mechanisms of vasodilatatory effect of perivascular tissue of human internal thoracic artery. J Physiol Pharmacol 64: 309–316. [PubMed] [Google Scholar]
- Marchesi C, Ebrahimian T, Angulo O, Paradis P, Schiffrin EL (2009). Endothelial nitric oxide synthase uncoupling and perivascular adipose oxidative stress and inflammation contribute to vascular dysfunction in a rodent model of metabolic syndrome. Hypertension 54: 1384–1392. [DOI] [PubMed] [Google Scholar]
- Margaritis M, Antonopoulos AS, Digby J, Lee R, Reilly S, Coutinho P et al. (2013). Interactions between vascular wall and perivascular adipose tissue reveal novel roles for adiponectin in the regulation of endothelial nitric oxide synthase function in human vessels. Circulation 127: 2209–2221. [DOI] [PubMed] [Google Scholar]
- Mark AL (2013). Selective leptin resistance revisited. Am J Physiol Regul Integr Comp Physiol 305: R566–R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronardi CA, Yu WH, McCann SM (2002). Resting and circadian release of nitric oxide is controlled by leptin in male rats. Proc Natl Acad Sci U S A 99: 5721–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer RI, Bakker W, Alta CL, Sipkema P, Yudkin JS, Viollet B et al. (2013). Perivascular adipose tissue control of insulin‐induced vasoreactivity in muscle is impaired in db/db mice. Diabetes 62: 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao CY, Li ZY (2012). The role of perivascular adipose tissue in vascular smooth muscle cell growth. Br J Pharmacol 165: 643–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming XF, Barandier C, Viswambharan H, Kwak BR, Mach F, Mazzolai L et al. (2004). Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation 110: 3708–3714. [DOI] [PubMed] [Google Scholar]
- Mohamed‐Ali V, Goodrick S, Rawesh A, Katz DR, Miles JM, Yudkin JS et al. (1997). Subcutaneous adipose tissue releases interleukin‐6, but not tumor necrosis factor‐alpha, in vivo. J Clin Endocrinol Metab 82: 4196–4200. [DOI] [PubMed] [Google Scholar]
- Molica F, Morel S, Kwak BR, Rohner‐Jeanrenaud F, Steffens S (2015). Adipokines at the crossroad between obesity and cardiovascular disease. Thromb Haemost 113: 553–566. [DOI] [PubMed] [Google Scholar]
- Momin AU, Melikian N, Shah AM, Grieve DJ, Wheatcroft SB, John L et al. (2006). Leptin is an endothelial‐independent vasodilator in humans with coronary artery disease: Evidence for tissue specificity of leptin resistance. Eur Heart J 27: 2294–2299. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK et al. (2011). Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar A, Chatterjee TK, Tang Y, Hui DY, Weintraub NL (2014). Proinflammatory phenotype of perivascular adipocytes. Arterioscler Thromb Vasc Biol 34: 1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozen G, Daci A, Norel X, Topal G (2015). Human perivascular adipose tissue dysfunction as a cause of vascular disease: Focus on vascular tone and wall remodeling. Eur J Pharmacol 766: 16–24. [DOI] [PubMed] [Google Scholar]
- Ozen G, Topal G, Gomez I, Ghorreshi A, Boukais K, Benyahia C et al. (2013). Control of human vascular tone by prostanoids derived from perivascular adipose tissue. Prostaglandins Other Lipid Mediat 107: 13–17. [DOI] [PubMed] [Google Scholar]
- Padilla J, Jenkins NT, Vieira‐Potter VJ, Laughlin MH (2013). Divergent phenotype of rat thoracic and abdominal perivascular adipose tissues. Am J Physiol Regul Integr Comp Physiol 304: R543–R552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Whiteman M, Cirino G (2015). Pharmacological tools for hydrogen sulphide research: a brief, introductory guide for beginners. Br J Pharmacol 172: 1633–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE et al. (2009). Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58: 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patten DA, Lafleur VN, Robitaille GA, Chan DA, Giaccia AJ, Richard DE (2010). Hypoxia‐inducible factor‐1 activation in nonhypoxic conditions: the essential role of mitochondrial‐derived reactive oxygen species. Mol Biol Cell 21: 3247–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne GA, Borbouse L, Kumar S, Neeb Z, Alloosh M, Sturek M et al. (2010). Epicardial perivascular adipose‐derived leptin exacerbates coronary endothelial dysfunction in metabolic syndrome via a protein kinase C‐beta pathway. Arterioscler Thromb Vasc Biol 30: 1711–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernow J, Jung C (2013). Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res 98: 334–343. [DOI] [PubMed] [Google Scholar]
- Procopio C, Andreozzi F, Laratta E, Cassese A, Beguinot F, Arturi F et al. (2009). Leptin‐stimulated endothelial nitric‐oxide synthase via an adenosine 5′‐monophosphate‐activated protein kinase/Akt signaling pathway is attenuated by interaction with C‐reactive protein. Endocrinology 150: 3584–3593. [DOI] [PubMed] [Google Scholar]
- Prunet‐Marcassus B, Desbazeille M, Bros A, Louche K, Delagrange P, Renard P et al. (2003). Melatonin reduces body weight gain in Sprague Dawley rats with diet‐induced obesity. Endocrinology 144: 5347–5352. [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Morgan DA, Morgan GM, Mark AL, Haynes WG (2005). Role of selective leptin resistance in diet‐induced obesity hypertension. Diabetes 54: 2012–2018. [DOI] [PubMed] [Google Scholar]
- Ran J, Xiong X, Liu W, Guo S, Li Q, Zhang R et al. (2010). Increased plasma adiponectin closely associates with vascular endothelial dysfunction in type 2 diabetic patients with diabetic nephropathy. Diabetes Res Clin Pract 88: 177–183. [DOI] [PubMed] [Google Scholar]
- Rios‐Lugo MJ, Cano P, Jimenez‐Ortega V, Fernandez‐Mateos MP, Scacchi PA, Cardinali DP et al. (2010). Melatonin effect on plasma adiponectin, leptin, insulin, glucose, triglycerides and cholesterol in normal and high fat‐fed rats. J Pineal Res 49: 342–348. [DOI] [PubMed] [Google Scholar]
- Rocha VZ, Libby P (2009). Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol 6: 399–409. [DOI] [PubMed] [Google Scholar]
- Romero MJ, Platt DH, Tawfik HE, Labazi M, El‐Remessy AB, Bartoli M et al. (2008). Diabetes‐induced coronary vascular dysfunction involves increased arginase activity. Circ Res 102: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosei CA, Withers SB, Belcaid L, De Ciuceis C, Rizzoni D, Heagerty AM (2015). Blockade of the renin‐angiotensin system in small arteries and anticontractile function of perivascular adipose tissue. J Hypertens 33: 1039–1045. [DOI] [PubMed] [Google Scholar]
- Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D et al. (2006). Oxidized low‐density lipoprotein‐dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res 99: 951–960. [DOI] [PubMed] [Google Scholar]
- Saarikoski LA, Huupponen RK, Viikari JS, Marniemi J, Juonala M, Kahonen M et al. (2010). Adiponectin is related with carotid artery intima‐media thickness and brachial flow‐mediated dilatation in young adults‐‐the Cardiovascular Risk in Young Finns Study. Ann Med 42: 603–611. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T et al. (2010). Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens 28: 1875–1882. [DOI] [PubMed] [Google Scholar]
- Schroeter MR, Eschholz N, Herzberg S, Jerchel I, Leifheit‐Nestler M, Czepluch FS et al. (2013). Leptin‐dependent and leptin‐independent paracrine effects of perivascular adipose tissue on neointima formation. Arterioscler Thromb Vasc Biol 33: 980–987. [DOI] [PubMed] [Google Scholar]
- Shatanawi A, Romero MJ, Iddings JA, Chandra S, Umapathy NS, Verin AD et al. (2011). Angiotensin II‐induced vascular endothelial dysfunction through RhoA/Rho kinase/p38 mitogen‐activated protein kinase/arginase pathway. Am J Physiol Cell Physiol 300: C1181–C1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shek EW, Brands MW, Hall JE (1998). Chronic leptin infusion increases arterial pressure. Hypertension 31: 409–414. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Higa N, Asahi T, Oshiro Y, Takasu N, Tagawa T et al. (2003). Hypoadiponectinemia is closely linked to endothelial dysfunction in man. J Clin Endocrinol Metab 88: 3236–3240. [DOI] [PubMed] [Google Scholar]
- Shitara Y, Sugiyama Y (2006). Pharmacokinetic and pharmacodynamic alterations of 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitors: drug–drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther 112: 71–105. [DOI] [PubMed] [Google Scholar]
- Simperova A, Al‐Nakkash L, Faust JJ, Sweazea KL (2016). Genistein supplementation prevents weight gain but promotes oxidative stress and inflammation in the vasculature of female obese ob/ob mice. Nutr Res 36: 789–797. [DOI] [PubMed] [Google Scholar]
- Soltis EE, Cassis LA (1991). Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A 13: 277–296. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza DS, Bomfim V, Skoglund H, Dashwood MR, Borowiec JW, Bodin L et al. (2001). High early patency of saphenous vein graft for coronary artery bypass harvested with surrounding tissue. Ann Thorac Surg 71: 797–800. [DOI] [PubMed] [Google Scholar]
- Steinberg HO, Chaker H, Leaming R, Johnson A, Brechtel G, Baron AD (1996). Obesity/insulin resistance is associated with endothelial dysfunction. Implications for the syndrome of insulin resistance. J Clin Invest 97: 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark KR, Yeager ME, El Kasmi KC, Nozik‐Grayck E, Gerasimovskaya EV, Li M et al. (2013). The adventitia: essential regulator of vascular wall structure and function. Annu Rev Physiol 75: 23–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Li J, Xiao N, Wang M, Kou J, Qi L et al. (2014). Pharmacological activation of AMPK ameliorates perivascular adipose/endothelial dysfunction in a manner interdependent on AMPK and SIRT1. Pharmacol Res 89: 19–28. [DOI] [PubMed] [Google Scholar]
- Szasz T, Webb RC (2012). Perivascular adipose tissue: more than just structural support. Clin Sci (Lond) 122: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tano JY, Schleifenbaum J, Gollasch M (2014). Perivascular adipose tissue, potassium channels, and vascular dysfunction. Arterioscler Thromb Vasc Biol 34: 1827–1830. [DOI] [PubMed] [Google Scholar]
- van Hinsbergh VW, Eringa EC, Daemen MJ (2015). Neovascularization of the atherosclerotic plaque: interplay between atherosclerotic lesion, adventitia‐derived microvessels and perivascular fat. Curr Opin Lipidol 26: 405–411. [DOI] [PubMed] [Google Scholar]
- Vecchione C, Maffei A, Colella S, Aretini A, Poulet R, Frati G et al. (2002). Leptin effect on endothelial nitric oxide is mediated through Akt‐endothelial nitric oxide synthase phosphorylation pathway. Diabetes 51: 168–173. [DOI] [PubMed] [Google Scholar]
- Verhagen SN, Visseren FL (2011). Perivascular adipose tissue as a cause of atherosclerosis. Atherosclerosis 214: 3–10. [DOI] [PubMed] [Google Scholar]
- Verma S, Lovren F, Pan Y, Yanagawa B, Deb S, Karkhanis R et al. (2014). Pedicled no‐touch saphenous vein graft harvest limits vascular smooth muscle cell activation: the PATENT saphenous vein graft study. Eur J Cardiothorac Surg 45: 717–725. [DOI] [PubMed] [Google Scholar]
- Victorio JA, Clerici SP, Palacios R, Alonso MJ, Vassallo DV, Jaffe IZ et al. (2016a). Spironolactone prevents endothelial nitric oxide synthase uncoupling and vascular dysfunction induced by beta‐adrenergic overstimulation: role of perivascular adipose tissue. Hypertension 68: 726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victorio JA, Fontes MT, Rossoni LV, Davel AP (2016b). Different anti‐contractile function and nitric oxide production of thoracic and abdominal perivascular adipose tissues. Front Physiol 7: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdis A, Duranti E, Rossi C, Dell'Agnello U, Santini E, Anselmino M et al. (2015). Tumour necrosis factor‐alpha participates on the endothelin‐1/nitric oxide imbalance in small arteries from obese patients: role of perivascular adipose tissue. Eur Heart J 36: 784–794. [DOI] [PubMed] [Google Scholar]
- Wang H, Luo W, Wang J, Guo C, Wang X, Wolffe SL et al. (2012). Obesity‐induced endothelial dysfunction is prevented by deficiency of P‐selectin glycoprotein ligand‐1. Diabetes 61: 3219–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Szabo C, Ichinose F, Ahmed A, Whiteman M, Papapetropoulos A (2015). The role of H2S bioavailability in endothelial dysfunction. Trends Pharmacol Sci 36: 568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss R, Dziura J, Burgert TS, Tamborlane WV, Taksali SE, Yeckel CW et al. (2004). Obesity and the metabolic syndrome in children and adolescents. N Engl J Med 350: 2362–2374. [DOI] [PubMed] [Google Scholar]
- Weston AH, Egner I, Dong Y, Porter EL, Heagerty AM, Edwards G (2013). Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: involvement of myocyte BKCa channels and adiponectin. Br J Pharmacol 169: 1500–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Gooding KM, Whatmore JL, Ball CI, Mawson D, Skinner K et al. (2010). Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia 53: 1722–1726. [DOI] [PubMed] [Google Scholar]
- Withers SB, Agabiti‐Rosei C, Livingstone DM, Little MC, Aslam R, Malik RA et al. (2011). Macrophage activation is responsible for loss of anticontractile function in inflamed perivascular fat. Arterioscler Thromb Vasc Biol 31: 908–913. [DOI] [PubMed] [Google Scholar]
- Withers SB, Bussey CE, Saxton SN, Melrose HM, Watkins AE, Heagerty AM (2014a). Mechanisms of adiponectin‐associated perivascular function in vascular disease. Arterioscler Thromb Vasc Biol 34: 1637–1642. [DOI] [PubMed] [Google Scholar]
- Withers SB, Simpson L, Fattah S, Werner ME, Heagerty AM (2014b). cGMP‐dependent protein kinase (PKG) mediates the anticontractile capacity of perivascular adipose tissue. Cardiovasc Res 101: 130–137. [DOI] [PubMed] [Google Scholar]
- Wojcicka G, Jamroz‐Wisniewska A, Atanasova P, Chaldakov GN, Chylinska‐Kula B, Beltowski J (2011). Differential effects of statins on endogenous H2S formation in perivascular adipose tissue. Pharmacol Res 63: 68–76. [DOI] [PubMed] [Google Scholar]
- Wu J, Grassia G, Cambrook H, Ialenti A, MacRitchie N, Carberry J et al. (2015). Perivascular mast cells regulate vein graft neointimal formation and remodeling. Peer J 3: e1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi W, Satoh H, Kase H, Suzuki K, Hattori Y (2005). Stimulated HSP90 binding to eNOS and activation of the PI3‐Akt pathway contribute to globular adiponectin‐induced NO production: vasorelaxation in response to globular adiponectin. Biochem Biophys Res Commun 332: 200–205. [DOI] [PubMed] [Google Scholar]
- Xia N, Horke S, Habermeier A, Closs EI, Reifenberg G, Gericke A et al. (2016). Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet‐induced obese mice. Arterioscler Thromb Vasc Biol 36: 78–85. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma‐lyase. Science 322: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Ming XF (2013). Arginase: the emerging therapeutic target for vascular oxidative stress and inflammation. Front Immunol 4: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Rajapakse AG, Montani JP, Yang Z, Ming XF (2014). p38 mitogen‐activated protein kinase is involved in arginase‐II‐mediated eNOS‐uncoupling in obesity. Cardiovasc Diabetol 13: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudkin JS, Eringa E, Stehouwer CD (2005). “Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet 365: 1817–1820. [DOI] [PubMed] [Google Scholar]
- Zavaritskaya O, Zhuravleva N, Schleifenbaum J, Gloe T, Devermann L, Kluge R et al. (2013). Role of KCNQ channels in skeletal muscle arteries and periadventitial vascular dysfunction. Hypertension 61: 151–159. [DOI] [PubMed] [Google Scholar]
- Zeng ZH, Zhang ZH, Luo BH, He WK, Liang LY, He CC et al. (2009). The functional changes of the perivascular adipose tissue in spontaneously hypertensive rats and the effects of atorvastatin therapy. Clin Exp Hypertens 31: 355–363. [DOI] [PubMed] [Google Scholar]
- Zhang R, Wu Y, Zhao M, Liu C, Zhou L, Shen S et al. (2009). Role of HIF‐1alpha in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 297: L631–L640. [DOI] [PubMed] [Google Scholar]