

Abstract
Background and Purpose
We have recently shown that a reduced function of endothelial nitric oxide synthase (eNOS) in the perivascular adipose tissue (PVAT) contributes crucially to obesity‐induced vascular dysfunction in mice. The current study was conducted to test the hypothesis that vascular dysfunction in obesity can be reversed by in vivo improvement of PVAT eNOS activity.
Experimental Approach
Male C57BL/6J mice were fed a high‐fat diet (HFD) for 22 weeks to induce obesity. During the last 4 weeks of HFD feeding, the obese mice were treated p.o. with the standardized Crataegus extract WS® 1442, which has been shown previously to improve eNOS activity.
Key Results
Diet‐induced obesity in mice markedly reduced the vasodilator response of thoracic aorta to acetylcholine in wire myograph experiments. Strikingly, this vascular dysfunction was only evident in PVAT‐containing aorta but not in PVAT‐free aorta. In vivo treatment of obese mice with WS® 1442 had no effect on body weight or epididymal fat mass, but completely restored the vascular function of PVAT‐containing aorta. Feeding a HFD led to a reduced phosphorylation and an enhanced acetylation of PVAT eNOS, both effects were reversed by WS® 1442 treatment.
Conclusion and Implications
PVAT plays a key role in vascular dysfunction in diet‐induced obese mice. Not obesity itself, but a PVAT dysfunction is responsible for obesity‐induced vascular disorders. Improving PVAT function by pharmacological means (e.g. with WS® 1442) can ameliorate vascular function even without reducing body weight or fat mass.
Linked Articles
This article is part of a themed section on Molecular Mechanisms Regulating Perivascular Adipose Tissue – Potential Pharmacological Targets? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.20/issuetoc
Abbreviations
- AMPK
AMP‐activated protein kinase
- DAF‐2 DA
4,5‐diaminofluorescein diacetate
- eNOS
endothelial NOS
- HFD
high‐fat diet
- iNOS
inducible NOS
- LVEF
left ventricular ejection fraction
- NAM
nicotinamide
- NAMPT
nicotinamide phosphoribosyltransferase
- NFD
normal‐fat diet
- nNOS
neuronal NOS
- PVAT
perivascular adipose tissue
- SIRT1
sirtuin 1
- TBP
TATA‐binding protein
- WS
Crataegus extract WS® 1442
Tables of Links
| TARGETS | |
|---|---|
| Akt (PKB) | eNOS |
| AMPK | iNOS |
| Arginase 1 | nNOS |
| Arginase 2 | SIRT1 |
| LIGANDS |
|---|
| Acetylcholine |
| L‐NAME |
| NAD |
| NO |
| Noradrenaline |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
With the economic growth, industrialization, mechanized transport, urbanization, an increasingly sedentary lifestyle and a nutritional transition to processed foods and high‐calorie diets over the last 30 years, the epidemic of overweight and obesity has become a major challenge to health around the world (Hruby and Hu, 2015). Obesity is an established risk factor for type 2 diabetes mellitus, cardiovascular diseases and many cancer types (Flegal et al., 2013). Obese patients have higher risk to develop hypertension, cardiomyopathy and stroke (Lavie et al., 2009).
Despite these well‐established facts, endothelial dysfunction is not always evident in in vitro studies. Recently, we examined the vascular function of thoracic aorta isolated from diet‐induced obese mice. To our surprise, the vasodilator response to acetylcholine remained normal in the aorta of mice fed a high‐fat diet (HFD) for 20 weeks (Xia et al., 2016). In traditional vascular physiology studies, the surrounding adipose tissue is usually cleaned from the blood vessel. Interestingly, by leaving the perivascular adipose tissue (PVAT) intact, a clear reduction in the vasodilator response to acetylcholine was observed in the aorta of obese animals as compared with lean controls (Xia et al., 2016). These results indicate that PVAT plays a key role in obesity‐induced vascular dysfunction.
In the aforementioned study, we have found out that the endothelial NOS (eNOS) in PVAT is in a dysfunctional state based on at least two mechanisms: (i) a deficiency of L‐arginine because of arginase induction; and (ii) a reduction in eNOS phosphorylation at serine 1177 residue due to Akt inhibition (an upstream kinase for eNOS serine 1177 phosphorylation) (Xia et al., 2016). The vascular dysfunction of PVAT‐containing aorta from obese mice could be normalized by an improvement of L‐arginine availability (a combination of L‐arginine supplementation and arginase inhibition) (Xia et al., 2016). These results suggest that the obesity‐induced vascular dysfunction is reversible and can be corrected by improving PVAT eNOS functionality.
In the current study, we have addressed the second mechanisms of PVAT eNOS dysfunction in obesity. We aimed to find out whether obesity‐induced vascular dysfunction can be reversed by normalization of eNOS phosphorylation status. For this purpose, we treated obese mice with the standardized Crataegus extract WS® 1442 (WS), which has been shown in a previous study to enhance eNOS phosphorylation at serine 1177 residue by stimulating Akt activity (Anselm et al., 2009).
Methods
Animals
C57BL/6J mice are a well‐established mouse model of diet‐induced obesity (Wang and Liao, 2012). A total of 75 male C57BL/6J mice from Janvier Labs (Le Genest‐Saint‐Isle, France) were used, and the animals were randomized into three groups. The control group received a normal‐fat diet (NFD, 13% energy from fat; ssniff® E15748‐04, Soest, Germany) for 22 weeks starting at the age of 8 weeks whereas the HFD and HFD + WS groups received a HFD (45% energy from fat; ssniff® E15744‐34) for 22 weeks starting at the age of 8 weeks. Animals in the HFD + WS group were treated with WS® 1442 (150 mg·kg−1 day−1, suspended in 0.2% agar) via gavage during the last 4 weeks of HFD feeding (week 19–22) while the mice in the control and HFD groups were treated with the same volume (10 mL·kg−1) of the vehicle. The daily dose of WS® 1442 for mice was extrapolated from the standard human dose (900 mg·day−1) (Holubarsch et al., 2008) using the body surface area normalization method of dose conversion (Reagan‐Shaw et al., 2008).
The animal experiment was approved by the regional competent authority (Regierungspräsidium Karlsruhe, A 40/09) and was conducted in accordance with the German animal protection law, the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals, and with the principles of the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Assessment of vascular function
Thoracic aortas were isolated and dissected into rings of 2–3 mm in length, with perivascular fat and connective tissues either removed or left intact. Isometric tension was recorded using a wire myograph system (Danish Myo Technology, Aarhus, Denmark). The rings were equilibrated for 60 min and contracted two times with 120 mM KCl. For assessment of vascular function, the rings were pre‐contracted with noradrenaline to reach the submaximal tension (80% of that obtained with 120 mM KCl), before vasodilatation was induced with acetylcholine (Xia et al., 2016).
Direct assessment of NO release
NO in PVAT was detected with 4,5‐diaminofluorescein diacetate (DAF‐2 DA), a cell‐permeable derivative of the fluorescent NO probe DAF‐2 that is hydrolyzed to DAF‐2 by intracellular esterases. DAF 2‐DA can be used in fluorescence microscopy to measure real‐time changes in NO levels (Hu et al., 2009; Cortese‐Krott et al., 2012; Cheang et al., 2014). After an incubation with or without the NOS inhibitor NG‐nitro‐L‐arginine methyl ester (L‐NAME, 500 μM), aorta cryostat sections were loaded with DAF‐2 DA (20 μM) in the presence of acetylcholine (100 nM). Then, fluorescence imaging was immediately performed by real‐time time‐lapse imaging in a heated (37°C) incubator chamber with a Zeiss 710 confocal laser scanning microscope (Zeiss, Germany). DAF‐2 DA was excited at 488 nm and fluorescence emitted between 495 and 550 nm was collected (Xia et al., 2016). Fluorescence intensity in PVAT was quantified after subtraction of that in L‐NAME‐treated samples.
Gene expression analyses
RNA was isolated using peqGOLD TriFast™ (PEQLAB), and cDNA was generated with the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative real time RT‐PCR (qPCR) reactions were performed on a StepOnePlus™ Real‐Time PCR System (Applied Biosystems) using SYBR® Green JumpStart™ Taq ReadyMix™ (Sigma‐Aldrich) and 20 ng cDNA. Relative mRNA levels of target genes were quantified using comparative threshold CT normalized to housekeeping gene TATA‐binding protein (TBP) (Xia et al., 2010; Xia et al., 2013). mRNA expression in control animals with NFD were set at 100%. The following target genes were studied: neuronal NOS (nNOS), inducible NOS (iNOS), eNOS, arginase 1 (Arg1), arginase 2 (Arg2), the macrophage marker CD68, the phagocyte NADPH oxidase NOX2, sirtuin 1 (SIRT1) and nicotinamide phosphoribosyltransferase (NAMPT). The qPCR primer sequences were as follows: nNOS_forward: TCC ACC TGC CTC GAA ACC, nNOS_reverse: TTG TCG CTG TTG CCA AAA AC; iNOS_forward: CAG CTG GGC TGT ACA AAC CTT, iNOS_reverse: CATTGGAAGTGAAGCGTTTCG; eNOS_forward: CCT TCC GCT ACC AGC CAG A, eNOS_reverse: CAG AGA TCT TCA CTG CAT TGG CTA; arginase 1_forward: GGA ACC CAG AGA GAG CAT GA, arginase 1_reverse: TTT TTC CAG CAG ACC AGC TT; arginase 2_forward: ACC AGG AAC TGG CTG AAG TG, arginase 2_reverse: TGA GCA TCA ACC CAG ATG AC; CD68_forward: CTT CCC ACA GGC AGC ACA G, CD68_reverse: AAT GAT GAG AGG CAG CAA GAG G; NOX2_forward: CCA ACT GGG ATA ACG AGT TCA, NOX2_reverse: GAG AGT TTC AGC CAA GGC TTC; SIRT1_forward: GCC AAA CTT TGT TGT AAC CCT GTA, SIRT1_reverse: TGG TGG CAA CTC TGA TAA ATG AA; NAMPT_forward: TTC CCG AGG GCT CTG TCA, NAMPT_reverse: GTA GCA CTC TGG GTC TGT GTT TTC; TBP_forward: CTT CGT GCA AGA AAT GCT GAA T, TBP_reverse: CAG TTG TCC GTG GCT CTC TTA TT (Xia et al., 2010; Xia et al., 2013).
Western blot analyses
Western blot analyses were performed with total protein samples (30 μg each) from aorta or PVAT. The following primary antibodies were used: rabbit monoclonal antibody against β‐tubulin I (catalogue number T7816, Sigma‐Aldrich; 1:200 000), mouse monoclonal antibody against eNOS (catalogue number 610297, BD Transduction Laboratories; 1:2000), phospho‐eNOS (Ser1177) (catalogue number 9571, Cell Signaling Technology, Boston, MA, USA; 1:1000), Akt (catalogue number 4691, Cell Signaling Technology; 1:5000), phospho‐Akt (Thr308) (catalogue number 2965, Cell Signaling Technology; 1:1000), phospho‐Akt (Ser473) (catalogue number 4060, Cell Signaling Technology; 1:2000), AMP‐activated protein kinase (AMPKα) (catalogue number 2532, Cell Signaling Technology; 1:1000), phospho‐AMPKα1 (Ser485)/AMPKα2 (Ser491) (catalogue number 4185, Cell Signaling Technology; 1:1000), phospho‐AMPKα (Thr172) (catalogue number 2535, Cell Signaling Technology; 1:1000), SIRT1 (catalogue number LS‐B1564, from LifeSpan BioSciences, Seattle, WA, USA; 1:500) and NAMPT (catalogue number ARP42255‐T100, Aviva Systems Biology, San Diego, CA, USA; 1:500).
Western blot was carried out as previously described (Li et al., 2006; Wu et al., 2015). Protein samples were separated on a Bis‐Tris gel and transferred to a nitrocellulose membrane. Blots were blocked in 5% milk powder in TBST (10 mM Tris–HCl, pH 7.4, 150 mM NaCl with 0.1% Tween 20) for 1 h at room temperature. The primary antibodies were diluted in the same solution used for blocking at 4°C overnight. Blots were then washed in TBST and incubated with a horseradish peroxidase‐conjugated secondary antibody diluted in 5% milk in TBST for 1 h. After being washed in TBST and then in TBS, the immunocomplexes were visualized using an enhanced horseradish peroxidase/luminol chemiluminescence reagent (PerkinElmer Life and Analytical Sciences, Boston, MA, USA) according to the manufacturer's instructions. Densitometric analysis of scanned blots was performed using the Quantity One software (Bio‐Rad, Munich, Germany).
Analysis of eNOS acetylation
PVAT samples were ground into a fine powder in liquid nitrogen and homogenized in lysis buffer (catalogue number 87788 from Thermo Fisher Scientific). Immunoprecipitation of eNOS was carried out by incubating 3 μg of antibody (catalogue number LS‐C288673 from LifeSpan BioSciences) with 1 mg of PVAT lysate for 1 h at 4°C, followed by 50 μL of Pierce™ Protein A/G Magnetic Beads (catalogue number 88802 from Thermo Fisher Scientific) for 1 h at room temperature. After washing, immunoprecipitates were boiled at 95°C for 5 min in loading buffer, subjected to SDS/PAGE, transferred to a nitrocellulose membrane, and probed with anti‐eNOS (catalogue number 610297, BD Transduction Laboratories) and anti‐acetylated lysine (catalogue number 9441, Cell Signaling Technology) for immunoblotting respectively.
Materials
WS®1442 is the active ingredient of Crataegutt® (Dr. Willmar Schwabe GmbH & Co. KG, Karlsruhe, Germany). It is a dry extract from hawthorn leaves with flowers obtained by extraction with 45% (w.w‐1) aqueous ethanol (drug/extract ratio 4–6.6 : 1). The extract is adjusted to a content of 17.3–20.1% oligomeric procyanidins (Bubik et al., 2012; Idris‐Khodja et al., 2012) and was provided by Dr. Willmar Schwabe GmbH & Co. KG, Karlsruhe, Germany. Chemicals (acetylcholine, KCl, L‐NAME and noradrenaline) were obtained from Sigma‐Aldrich, Taufkirchen, Germany. DAF‐2 DA was from Thermo Fisher Scientific, Darmstadt, Germany. PCR primers were synthesized by Eurofins Genomics, Ebersberg, Germany.
Statistics
Results are expressed as mean ± SEM. Student's t‐test was used for comparison of HFD group with NFD group. Two‐way ANOVA was used to compare the curves. P values <0.05 were considered significantly different. For statistical analysis GraphPad Prism (GraphPad Software, La Jolla, CA, USA) was used. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Results
WS® 1442 restores vascular function without changing body weight or fat mass
Feeding of male C57BL/6J mice with HFD led to a significant increase in body weight, epididymal fat mass (Figure 1) and concentration of serum lipids (Table 1). Treatment with WS® 1442 for 4 weeks starting after 18 weeks of HFD feeding had no effect on body weight, epididymal fat mass (Figure 1), serum HDL or triglyceride content but slightly elevated serum LDL concentration (Table 1). Obese mice had also increased PVAT mass, which was not changed by WS® 1442 treatment (PVAT cross section area: NFD 2.13 ± 0.31 mm2; HFD 3.47 ± 0.12 mm2, P < 0.05 compared to NFD; HFD + WS 3.73 ± 0.28 mm2; P < 0.05 compared to NFD; n = 6).
Figure 1.
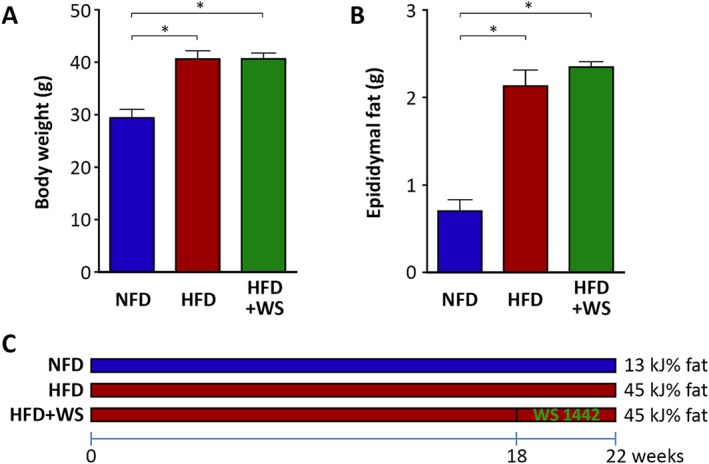
WS® 1442 had no effect on body weight or fat mass. Male C57BL/6J mice were put on a NFD or HFD for 22 weeks starting at the age of 8 weeks. A subgroup of HFD animals were treated with WS® 1442, p.o., during the last 4 weeks (from week 19 to week 22) of HFD feeding. Body weight and epididymal fat were measured at the end of the experiment (week 22). Columns represent mean ± SEM. *P < 0.05, n = 10.
Table 1.
Serum lipid profile at the age of 30 weeks
| NFD | HFD | HFD+WS | |
|---|---|---|---|
| Cholesterol (mmol·L−1) | 3.34 ± 0.11 | 5.12 ± 0.19 * | 5.65 ± 0.16 * |
| LDL (mmol·L−1) | 1.83 ± 0.09 | 2.69 ± 0.09 * | 3.34 ± 0.19 * , # |
| HDL (mmol·L−1) | 1.60 ± 0.09 | 2.43 ± 0.16 * | 2.43 ± 0.11 * |
| Triglyceride (mmol·L−1) | 1.08 ± 0.09 | 1.02 ± 0.08 | 0.99 ± 0.04 |
Serum lipids were measured with the scil Reflovet® Plus (Viernheim, Germany).
P<0.05, compared to NFD;
P<0.05, compared to HFD; n=6‐11.
In the thoracic aortas without PVAT, no difference in acetylcholine‐induced endothelium‐dependent vasodilatation was observed between NFD and HFD mice (Figure 2). In contrast, a marked reduction in the acetylcholine response was evident in HFD‐fed mice if PVAT was left in place (Figure 2). These results indicate that obesity‐induced vascular dysfunction is PVAT‐dependent. Oral treatment with WS® 1442 (150 mg·kg−1·day−1) for 4 weeks completely restored vascular function in HFD mice (Figure 2).
Figure 2.
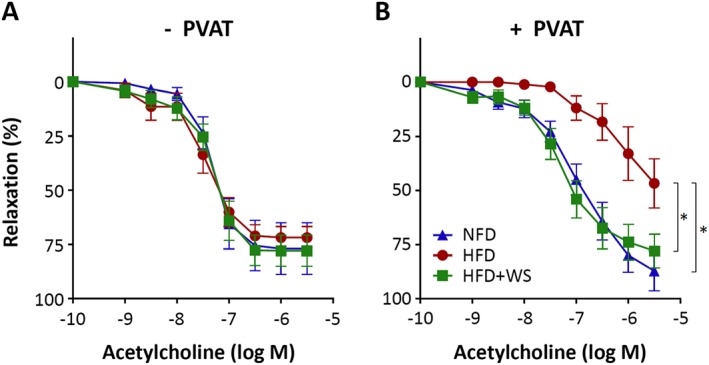
WS® 1442 restores vascular function in HFD mice. Male C57BL/6J mice were put on a NFD or HFD for 22 weeks starting at the age of 8 weeks. A subgroup of HFD animals were treated with WS® 1442, p.o., during the last 4 weeks of HFD feeding. The vasodilator response to acetylcholine was performed in noradrenaline‐precontracted aorta with or without PVAT. Symbols represent mean ± SEM. *P < 0.05, n = 6.
WS® 1442 improves NO production in PVAT
Immunohistochemistry staining indicates that eNOS is expressed in the endothelium as well as in the PVAT (Figure 3A), which is consistent with previous studies (Dashwood et al., 2007; Xia et al., 2016). To directly access NO production from PVAT, fluorescence imaging was performed with aorta sections stained with the fluorescent NO probe DAF‐2 DA. As shown in Figure 3B,C, HFD‐induced obesity significantly reduced the PVAT NO production, which was improved by WS® 1442 treatment (Figure 3).
Figure 3.
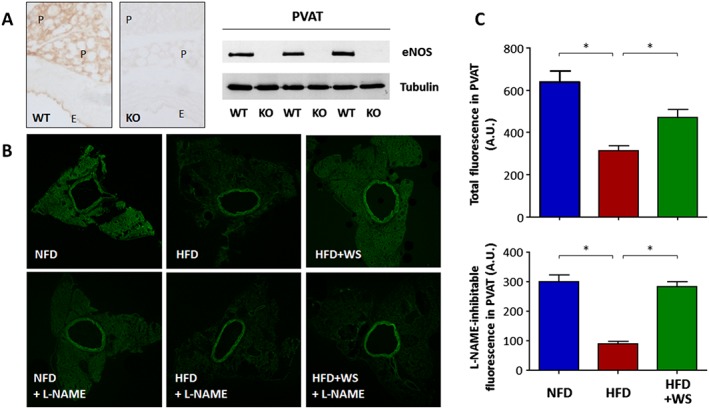
WS® 1442 improves PVAT NO production in HFD mice. (A) eNOS immunohistochemistry staining and western blot analyses were performed using PVAT‐containing aorta samples from C57BL/6J wild‐type mice (WT) or global eNOS knockout mice (KO). E and P indicate endothelium and PVAT respectively. (B) Male C57BL/6J mice were put on a NFD or HFD for 22 weeks starting at the age of 8 weeks. A subgroup of HFD animals were treated with WS® 1442, p.o., during the last 4 weeks of HFD feeding. NO production in PVAT was determined by DAF‐2 DA staining in the absence or presence of the NOS inhibitor L‐NAME. The confocal images shown are representative for five independent experiments with similar results. (C) Shows the quantification fluorescence intensity in PVAT. *P < 0.05, n = 5.
WS® 1442 had no effect on the expression of NO synthases or arginases
To study the molecular mechanisms underlying the observed PVAT NO production changes, we analysed the expression of NO synthase isoforms. As shown in Figure 4A,B, HFD feeding had no effect on the expression of nNOS, iNOS or eNOS, neither in aorta nor in PVAT, which is consistent with our previous findings (Xia et al., 2016). Also in accordance with our previous study, we have observed a HFD‐induced up‐regulation of Arg1 in the PVAT but not in the aorta (Figure 4C,D). The up‐regulation of Arg1 in PVAT was accompanied by an enhanced expression of the macrophage marker CD68 and phagocyte NADPH oxidase NOX2 (Figure 4E,F). Treatment with WS® 1442 had no effect on the expression of NO synthases, arginases, CD68 or NOX2 (Figure 4).
Figure 4.
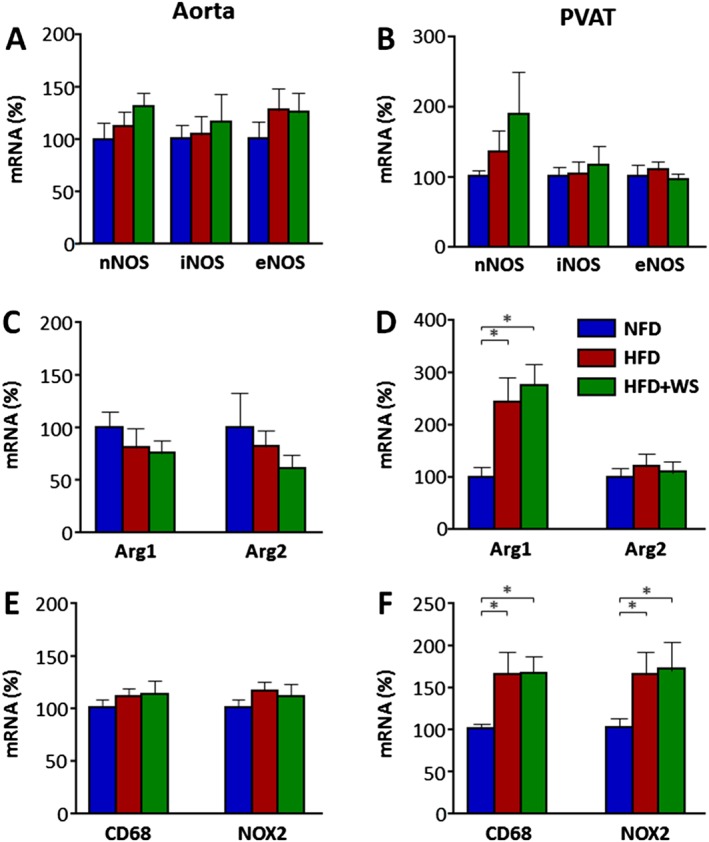
WS® 1442 had no effect on NO synthases or arginases. Male C57BL/6J mice were put on a NFD or HFD or for 22 weeks starting at the age of 8 weeks. A subgroup of HFD animals were treated with WS® 1442, p.o., during the last 4 weeks of HFD feeding. The mRNA expression of NO synthases (nNOS, iNOS and eNOS), arginases (Arg1 and Arg2), the macrophage marker CD68 and phagocyte NADPH oxidase NOX2 was studied with qPCR in aorta (A, C and E) and PVAT (B, D and F). Columns represent mean ± SEM. *P < 0.05, n = 9.
WS® 1442 restores eNOS phosphorylation in PVAT
We have shown in our previous study that HFD feeding led to a reduced eNOS phosphorylation at serine 1177 residue selectively in the PVAT, with no effect in the aorta (Xia et al., 2016). Therefore, in the present study we analysed the phosphorylation status of eNOS and two of its upstream kinases, Akt and AMP‐activated protein kinase (AMPK), in PVAT.
As shown in Figure 5, diet‐induced obesity in HFD mice resulted in a reduction in PVAT eNOS phosphorylation at serine 1177. This was associated with a reduction in the phosphorylation of Akt at serine 473 and AMPK at threonine 172 respectively (Figure 5). Treatment with WS® 1442 restored the phosphorylation level of eNOS and Akt, but had no significant effect on AMPK (Figure 5). Interestingly, HFD feeding also reduced eNOS serine 1177 phosphorylation in epididymal visceral adipose tissue (VAT), with no significant effects on Akt or AMPK (Figure 5), indicating that different protein kinases are responsible for eNOS phosphorylation in PVAT and VAT. Treatment with WS® 1442 had no effect on eNOS, Akt or AMPK in epididymal VAT (Figure 5), indicating that the effect of WS® 1442 is relatively specific for PVAT.
Figure 5.
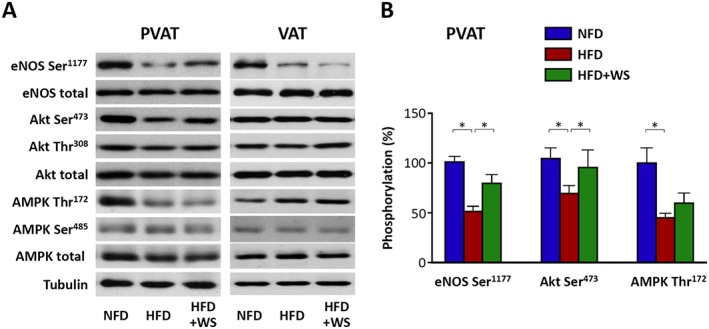
WS® 1442 restores eNOS phosphorylation in PVAT. Male C57BL/6J mice were put on a NFD or HFD for 22 weeks starting at the age of 8 weeks. A subgroup of HFD animals were treated with WS® 1442, p.o., during the last 4 weeks of HFD feeding. The expression and phosphorylation of eNOS at serine 1177 and of upstream kinases for eNOS serine 1177 phosphorylation, Akt and AMPK were analysed by western blotting using samples from aortic PVAT or epididymal visceral adipose tissue (VAT). The blots shown are representative of six experiments with similar results (A). (B) Shows the results of densitometric analyses. Columns represent mean ± SEM. *P < 0.05, n = 6.
WS® 1442 normalizes eNOS acetylation level in PVAT
In addition to phosphorylation, eNOS activity is also regulated by acetylation as a post‐translational modification. The NAD+‐dependent histone/protein deacetylase SIRT1 has been shown to enhance eNOS activity by deacetylating eNOS (Mattagajasingh et al., 2007). We therefore analysed eNOS acetylation in PVAT.
As shown in Figure 6A,B, eNOS acetylation was enhanced in the PVAT from HFD‐fed mice, which was normalized by WS 1422 treatment. The expression level of SIRT1 was not changed, neither by HFD nor by WS® 1442. However, HFD feeding reduced the expression level of NAMPT, a rate‐limiting enzyme in NAD+ biosynthesis. Treatment with WS® 1442 reversed the NAMPT down‐regulation by HFD (Figure 6C–E).
Figure 6.
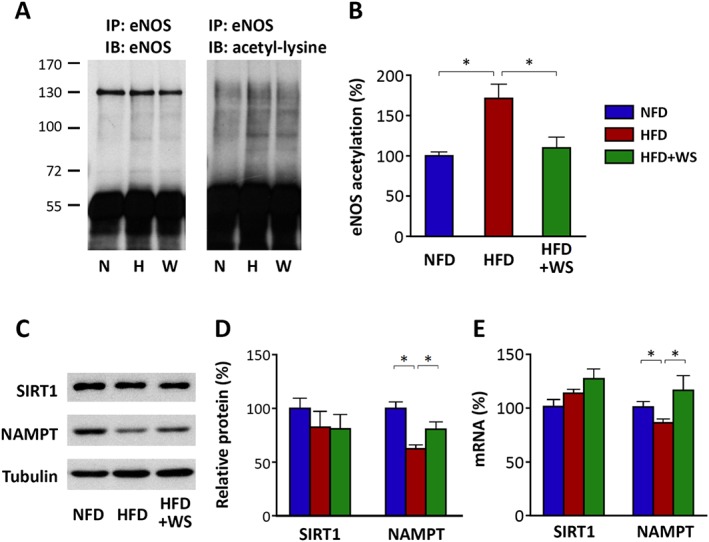
WS® 1442 normalizes eNOS acetylation level in PVAT. Male C57BL/6J mice were put on a NFD or HFD for 22 weeks starting at the age of 8 weeks. A subgroup of HFD animals were treated with WS® 1442, p.o., during the last 4 weeks of HFD feeding. Immunoprecipitation (IP) was performed with an anti‐eNOS antibody using PVAT samples followed by immunoblotting (IB) with antibodies against eNOS or acetyl‐lysine respectively. The band detected with the antibody against acetyl‐lysine at the level of eNOS was considered acetyl‐eNOS. In (A) the blots shown are representative of six independent experiments. (B) Results of densitometric analyses for acetyl‐eNOS normalized to total eNOS; n = 6. (C,D and E) Expression of SIRT1 and NAMPT at the protein (C and D) or mRNA (E) levels was analysed by western blotting and qPCR respectively. In (C) the blots shown are representative of six independent experiments. (D and E) Results of densitometric analyses are shown; n = 6. Columns represent mean ± SEM. *P < 0.05.
Discussion
In the present study, we demonstrated that (i) vascular dysfunction in thoracic aorta of diet‐induced obese mice is PVAT‐dependent; the acetylcholine‐induced vasodilator response is reduced only in PVAT‐containing aorta but remains completely normal in PVAT‐free aorta. (ii) In vivo treatment of obese mice with WS® 1442 normalizes vascular function without changing body weight or fat mass. (iii) The normalization of vascular function in obesity by WS® 1442 is associated with a restoration of eNOS phosphorylation and acetylation levels in PVAT.
Although it is well known that obesity increases cardiovascular risk, to date there are no efficient pharmacological options available for the treatment of obesity‐induced vascular complications. For the development of novel therapy, it is essential to understand the pathophysiology. The present study, along with our previous publication (Xia et al., 2016), provides evidence for a novel mechanism underlying vascular dysfunction in diet‐induced obesity. The experimental settings in the two studies are slightly different, with 60 cal% HFD in the previous and 45 cal% HFD in the current study. Despite this difference, the principle findings of the two studies are highly consistent. Both studies show that the PVAT plays a crucial role in obesity‐induced vascular dysfunction.
PVAT has been recognized as an important player in vascular biology. The role of PVAT in vascular function was first discovered by the observation that PVAT decreases agonists‐induced contractile responses in isolated blood vessels (Soltis and Cassis, 1991). Now, it is known that PVAT can produce many biologically active molecules regulating vascular function via endocrine or paracrine mechanisms, including adipokines, cytokines/chemokines and reactive oxygen species (Gollasch, 2012; Szasz and Webb, 2012; Xia and Li, 2017).
Recent studies have shown that eNOS is expressed in the PVAT and PVAT eNOS plays an important role in regulating vascular function (Dashwood et al., 2007; Aghamohammadzadeh et al., 2015; Virdis et al., 2015; Bussey et al., 2016; Xia and Li, 2017; Xia et al., 2016). Consistently, we show in the present study that eNOS is expressed in PVAT and is functionally active (Figure 3). Stimulation with acetylcholine enhances NO production in PVAT, which can be prevented by NOS inhibitor L‐NAME. In diet‐induced obesity, NO production in PVAT is reduced. At least three mechanisms contribute to the reduced eNOS functionality in PVAT: arginine deficiency, reduction in eNOS serine 1177 phosphorylation and enhanced eNOS acetylation. That PVAT eNOS dysfunction is causally linked to vascular dysfunction in diet‐induced obesity is supported by findings that vascular function can be normalized by improving PVAT eNOS function, either by improving L‐arginine availability (as in our previous study) or by restoring eNOS phosphorylation and acetylation (as in the present study).
For the restoration of eNOS phosphorylation and acetylation levels, we used WS® 1442 in the present study. The Crataegus special extracts WS® 1442 is an herbal drug registered as a traditional herbal medicinal product to support cardiovascular function. In clinical trials, WS® 1442 improved maximal workload, left ventricular ejection fraction (LVEF) and typical symptoms in early heart failure patients (Eggeling et al., 2011). In a randomized, double‐blind, placebo‐controlled multicentre study involving 2681 patients, WS® 1442 (900 mg·day−1) was used as an add‐on therapy to standard pharmacological therapy for heart failure (β‐blockers, angiotensin converting enzyme inhibitors, etc.) (Holubarsch et al., 2008). After a treatment period for 24 months, WS® 1442 showed no significant difference in the composite endpoint including cardiac death, non‐fatal myocardial infarction and hospitalization due to progressive heart failure. It trended to reduce the cardiac mortality (9.7% at month 24). This effect, however, was not statistically significant. Nevertheless in patients with less compromised left ventricular function (LVEF between 25% and 35%), WS® 1442 (900 mg·day−1) reduced sudden cardiac death by 39.7% (Holubarsch et al., 2008). The excellent safety profile of WS® 1442 makes it an attractive herbal remedy (Holubarsch et al., 2008).
In cell culture experiments, WS® 1442 has been shown to increase both Akt phosphorylation at serine 473 and eNOS phosphorylation at serine 1177 (Anselm et al., 2009). The PI3K/Akt pathway is likely to mediate eNOS phosphorylation and activation because the WS® 1442‐stimulated eNOS phosphorylation can be abolished by the PI3K inhibitor wortmannin (Anselm et al., 2009). The Akt phosphorylation, in turn, is preventable by intracellular scavengers of reactive oxygen species or by Src‐kinase inhibitor PP2, indicating that the Src‐kinase, a redox‐sensitive protein kinase, acts upstream of the PI3K/Akt pathway (Anselm et al., 2009).
Serine 1177 is the best studied eNOS regulatory site (Heiss and Dirsch, 2014). Phosphorylation of serine 1177 increases the sensitivity of the eNOS enzyme to calcium (Fleming, 2010), inhibits calmodulin dissociation from eNOS and enhances the internal rate of electron transfer and thus leads to activation of the eNOS enzyme (Dudzinski and Michel, 2007). Several kinases have been reported to phosphorylate eNOS serine 1177, with Akt and AMPK being the most prominent ones (Fleming, 2010). In the present study, we showed that the reduction of serine 1177 phosphorylation of PVAT eNOS is associated with inhibition of Akt and AMPK. Interestingly, treatment with WS® 1442 normalized the phosphorylation status of Akt, but not that of AMPK (Figure 5). Nevertheless, serine 1177 phosphorylation of PVAT eNOS was largely restored by WS® 1442, indicating that Akt may play a greater role than AMPK under this experimental conditions.
Another important post‐translational modification of eNOS is acetylation. Although SIRT1 is known as a class III histone deacetylase, its targets also include some non‐histone proteins and cytosolic molecules such as eNOS. SIRT1 increases eNOS activity by deacetylating eNOS at lysine residues 494 and 504 in the calmodulin‐binding domain of eNOS and thus enhancing calmodulin binding (Mattagajasingh et al., 2007).
SIRT1 is a NAD+‐dependent deacetylase. The SIRT1‐mediated deacetylation reaction consumes NAD+ as co‐substrate and generates nicotinamide (NAM) as by‐product (Zhang and Kraus, 2010). Therefore, SIRT1 activity is stimulated by NAD+ and inhibited by NAM (product inhibition).
NAMPT is the rate‐limiting enzyme for NAD+ biosynthesis in the salvage pathway and in vivo studies demonstrate that NAMPT expression levels closely correlate with cellular NAD+ production (Houtkooper et al., 2010; Zhang and Kraus, 2010). In addition, NAMPT also directly reduces cellular NAM levels because NAM is utilized as a precursor for NAD+ biosynthesis. Both actions of NAMPT (increasing NAD+ and reducing NAM levels) have the potential to stimulate SIRT1 activity (Houtkooper et al., 2010; Zhang and Kraus, 2010).
In the present study, we report for the first time that acetylation of PVAT eNOS is enhanced in diet‐induced obesity and WS® 1442 treatment normalized eNOS acetylation status (Figure 6). Interestingly, these effects were not caused by SIRT1 expression changes but are very likely to be mediated by SIRT1 activity alterations secondary to NAMPT (Figure 6). The molecular mechanisms on how HFD and WS® 1442 regulate NAMPT expression need to be explored in future studies.
A very interesting observation in the present study is that WS® 1442 is capable of normalizing vascular function in diet‐induced obese mice without any effect on body weight and fat mass (Figures 1 and 2). This finding suggests that the reason for vascular dysfunction in obesity is not body weight itself but a PVAT dysfunction. Therefore, analysing the molecular mechanisms underlying PVAT dysfunction represents a key step for the development of novel therapy to treatment obesity‐induced vascular complications.
We started WS® 1442 treatment 18 weeks after HFD feeding, which was after the establishment of obesity and vascular dysfunction (Xia et al., 2016). The intension was to test WS® 1442 as a therapy and not as a prevention. The impressing efficacy of WS® 1442 in normalizing vascular function indicate that vascular dysfunction in obesity is reversible, at least under our experimental condition.
Our study has several limitations. Firstly, we have no definitive evidence for a causal relationship between the effect of WS® 1442 on eNOS phosphorylation/acetylation and the effect of WS® 1442 on vascular function. Our data only show that the normalization of vascular function in diet‐induced obese mice by WS® 1442 is associated with an improvement of eNOS phosphorylation/acetylation. Secondly, although we provide compelling evidence that a reduced eNOS activity plays an important role in obesity‐induced PVAT dysfunction, we cannot rule out the contribution of other PVAT‐derived factors. WS® 1442 may also improve PVAT function through eNOS‐independent mechanisms. The fact that vascular function is fully normalized by WS® 1442 whereas the restoration of eNOS phosphorylation is only partial indicates that additional, still unknown mechanisms may contribute to the effect of WS® 1442. Thirdly, we cannot absolutely exclude the contribution of endothelial eNOS to obesity‐induced vascular dysfunction. The relative roles of PVAT eNOS and endothelial eNOS should be addressed in future studies by using endothelium‐denuded vessels and by using cell type‐specific eNOS‐knockout mice. Fourthly, we do not have sufficient data on the specificity of WS® 1442. The observation that WS® 1442 improves eNOS phosphorylation in PVAT but not in visceral VAT (Figure 5) indicates that the effect of WS® 1442 is likely to be relatively tissue‐specific. This is not surprising, because PVAT and VAT have different developmental origins (Xia and Li, 2017). However, WS® 1442 has more effects (Bubik et al., 2012) than stimulating Akt activity, strengthening the assumption that more mechanisms may underlie the effect of WS® 1442 on vascular function than the mechanisms identified in the present study.
In conclusion, diet‐induced obesity leads to PVAT dysfunction, which is associated with dysfunction of eNOS in the PVAT (e.g. reduced eNOS serine 1177 phosphorylation and enhanced eNOS acetylation). Pharmacological treatments (e.g. WS® 1442) that improve PVAT eNOS function can restore vascular function even without reducing body weight or fat mass.
Author contributions
N.X. and H.L. designed the study. N.X., S.K. and G.R. performed the experiments and analysed the data. N.X. and H.L. wrote the initial draft of the manuscript. E.K., M.B. and U.F. critically reviewed and revised the manuscript. All authors contributed to manuscript preparation and agreed to its publication.
Conflict of interest
The study was supported by a grant from Dr. Willmar Schwabe GmbH & Co. KG, Karlsruhe, Germany. S.K., E.K. and M.B. are employees of Dr. Willmar Schwabe GmbH & Co. KG.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by a grant from Dr. Willmar Schwabe GmbH & Co. KG (Karlsruhe, Germany), and by the university intramural grant Stufe I of Johannes Gutenberg University Medical Center, Mainz, Germany.
Xia, N. , Weisenburger, S. , Koch, E. , Burkart, M. , Reifenberg, G. , Förstermann, U. , and Li, H. (2017) Restoration of perivascular adipose tissue function in diet‐induced obese mice without changing bodyweight. British Journal of Pharmacology, 174: 3443–3453. doi: 10.1111/bph.13703.
References
- Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM (2015). Effects of obesity on perivascular adipose tissue vasorelaxant function: nitric oxide, inflammation and elevated systemic blood pressure. J Vasc Res 52: 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anselm E, Socorro VF, Dal‐Ros S, Schott C, Bronner C, Schini‐Kerth VB (2009). Crataegus special extract WS 1442 causes endothelium‐dependent relaxation via a redox‐sensitive Src‐ and Akt‐dependent activation of endothelial NO synthase but not via activation of estrogen receptors. J Cardiovasc Pharmacol 53: 253–260. [DOI] [PubMed] [Google Scholar]
- Bubik MF, Willer EA, Bihari P, Jurgenliemk G, Ammer H, Krombach F et al. (2012). A novel approach to prevent endothelial hyperpermeability: the Crataegus extract WS(R) 1442 targets the cAMP/Rap1 pathway. J Mol Cell Cardiol 52: 196–205. [DOI] [PubMed] [Google Scholar]
- Bussey CE, Withers SB, Aldous RG, Edwards G, Heagerty AM (2016). Obesity‐related perivascular adipose tissue damage is reversed by sustained weight loss in the rat. Arterioscler Thromb Vasc Biol 36: 1377–1385. [DOI] [PubMed] [Google Scholar]
- Cheang WS, Tian XY, Wong WT, Lau CW, Lee SS, Chen ZY et al. (2014). Metformin protects endothelial function in diet‐induced obese mice by inhibition of endoplasmic reticulum stress through 5′ adenosine monophosphate‐activated protein kinase‐peroxisome proliferator‐activated receptor delta pathway. Arterioscler Thromb Vasc Biol 34: 830–836. [DOI] [PubMed] [Google Scholar]
- Cortese‐Krott MM, Rodriguez‐Mateos A, Kuhnle GG, Brown G, Feelisch M, Kelm M (2012). A multilevel analytical approach for detection and visualization of intracellular NO production and nitrosation events using diaminofluoresceins. Free Radic Biol Med 53: 2146–2158. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashwood MR, Dooley A, Shi‐Wen X, Abraham DJ, Souza DS (2007). Does periadventitial fat‐derived nitric oxide play a role in improved saphenous vein graft patency in patients undergoing coronary artery bypass surgery? J Vasc Res 44: 175–181. [DOI] [PubMed] [Google Scholar]
- Dudzinski DM, Michel T (2007). Life history of eNOS: partners and pathways. Cardiovasc Res 75: 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling T, Regitz‐Zagrosek V, Zimmermann A, Burkart M (2011). Baseline severity but not gender modulates quantified Crataegus extract effects in early heart failure‐‐a pooled analysis of clinical trials. Phytomedicine 18: 1214–1219. [DOI] [PubMed] [Google Scholar]
- Flegal KM, Kit BK, Orpana H, Graubard BI (2013). Association of all‐cause mortality with overweight and obesity using standard body mass index categories: a systematic review and meta‐analysis. JAMA 309: 71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I (2010). Molecular mechanisms underlying the activation of eNOS. Pflugers Arch 459: 793–806. [DOI] [PubMed] [Google Scholar]
- Gollasch M (2012). Vasodilator signals from perivascular adipose tissue. Br J Pharmacol 165: 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiss EH, Dirsch VM (2014). Regulation of eNOS enzyme activity by posttranslational modification. Curr Pharm Des 20: 3503–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holubarsch CJ, Colucci WS, Meinertz T, Gaus W, Tendera M (2008). The efficacy and safety of Crataegus extract WS 1442 in patients with heart failure: the SPICE trial. Eur J Heart Fail 10: 1255–1263. [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Canto C, Wanders RJ, Auwerx J (2010). The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 31: 194–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruby A, Hu FB (2015). The epidemiology of obesity: a big picture. Pharmacoeconomics 33: 673–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Xu X, Zhu G, Atzler D, Kimoto M, Chen J et al. (2009). Vascular endothelial‐specific dimethylarginine dimethylaminohydrolase‐1‐deficient mice reveal that vascular endothelium plays an important role in removing asymmetric dimethylarginine. Circulation 120: 2222–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idris‐Khodja N, Auger C, Koch E, Schini‐Kerth VB (2012). Crataegus special extract WS® 1442 prevents aging‐related endothelial dysfunction. Phytomedicine 19: 699–706. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavie CJ, Milani RV, Ventura HO (2009). Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J Am Coll Cardiol 53: 1925–1932. [DOI] [PubMed] [Google Scholar]
- Li H, Witte K, August M, Brausch I, Godtel‐Armbrust U, Habermeier A et al. (2006). Reversal of endothelial nitric oxide synthase uncoupling and up‐regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J Am Coll Cardiol 47: 2536–2544. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB et al. (2007). SIRT1 promotes endothelium‐dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 104: 14855–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan‐Shaw S, Nihal M, Ahmad N (2008). Dose translation from animal to human studies revisited. FASEB J 22: 659–661. [DOI] [PubMed] [Google Scholar]
- Soltis EE, Cassis LA (1991). Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A 13: 277–296. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szasz T, Webb RC (2012). Perivascular adipose tissue: more than just structural support. Clin Sci (Lond) 122: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdis A, Duranti E, Rossi C, Dell'Agnello U, Santini E, Anselmino M et al. (2015). Tumour necrosis factor‐alpha participates on the endothelin‐1/nitric oxide imbalance in small arteries from obese patients: role of perivascular adipose tissue. Eur Heart J 36: 784–794. [DOI] [PubMed] [Google Scholar]
- Wang CY, Liao JK (2012). A mouse model of diet‐induced obesity and insulin resistance. Methods Mol Biol 821: 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Siuda D, Xia N, Reifenberg G, Daiber A, Munzel T et al. (2015). Maternal treatment of spontaneously hypertensive rats with pentaerythritol tetranitrate reduces blood pressure in female offspring. Hypertension 65: 232–237. [DOI] [PubMed] [Google Scholar]
- Xia N, Daiber A, Habermeier A, Closs EI, Thum T, Spanier G et al. (2010). Resveratrol reverses endothelial nitric‐oxide synthase uncoupling in apolipoprotein E knockout mice. J Pharmacol Exp Ther 335: 149–154. [DOI] [PubMed] [Google Scholar]
- Xia N, Horke S, Habermeier A, Closs EI, Reifenberg G, Gericke A et al. (2016). Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet‐induced obese mice. Arterioscler Thromb Vasc Biol 36: 78–85. [DOI] [PubMed] [Google Scholar]
- Xia N, Li H (2017). The role of perivascular adipose tissue in obesity‐induced vascular dysfunction. Br J Pharmacol 174: 3425–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia N, Strand S, Schlufter F, Siuda D, Reifenberg G, Kleinert H et al. (2013). Role of SIRT1 and FOXO factors in eNOS transcriptional activation by resveratrol. Nitric Oxide 32: 29–35. [DOI] [PubMed] [Google Scholar]
- Zhang T, Kraus WL (2010). SIRT1‐dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta 1804: 1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]