

Abstract
The endothelium is an established modulator of vascular tone; however, the recent discovery of the anti‐contractile nature of perivascular adipose tissue (PVAT) suggests that the fat, which surrounds many blood vessels, can also modulate vascular tone. Both the endothelium and PVAT secrete vasoactive substances, which regulate vascular function. Many of these factors are common to both the endothelium and PVAT; therefore, this review will highlight the potential shared mechanisms in the modulation of vascular tone. Endothelial dysfunction is a hallmark of many vascular diseases, including hypertension and obesity. Moreover, PVAT dysfunction is now being reported in several cardio‐metabolic disorders. Thus, this review will also discuss the mechanistic insights into endothelial and PVAT dysfunction in order to evaluate whether PVAT modulation of vascular contractility is similar to that of the endothelium in health and disease.
Linked Articles
This article is part of a themed section on Molecular Mechanisms Regulating Perivascular Adipose Tissue – Potential Pharmacological Targets? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.20/issuetoc
Abbreviations
- BAT
brown adipose tissue
- CSE
cystathionine γ‐lyase
- CVD
cardiovascular disease
- H2O2
Hydrogen peroxide
- H2S
hydrogen sulphide
- MLC
myosin light chain
- O2−
superoxide
- PVAT
perivascular adipose tissue
- PVRFs
PVAT‐derived relaxant factors
- VSM
vascular smooth muscle
- WAT
white adipose tissue
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Enzymes e |
| TNF‐α | Adenylate cyclase |
| GPCRs b | AMPK |
| IP receptor | COX‐1 |
| Voltage‐gated ion channels c | COX‐2 |
| KV channels | CSE |
| Ligand‐gated ion channels d | eNOS |
| BKCa channel | Guanylyl cyclase |
| IKCa channel | NOS |
| KCa channel | PKA |
| KATP (Kir6.1) channel | PKG |
| SKCa channel |
| LIGANDS | |
|---|---|
| 5‐HT | L‐arginine |
| Adiponectin | MCP‐1 (CCL2) |
| ATP | NO |
| cAMP | Noradrenaline |
| cGMP | PGD2 |
| GTP | PGE2 |
| H2O2 | PGF2α |
| IL‐6 | PGI2 |
| Indomethacin | TXA2 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,d,eAlexander et al., 2015a,b,c,d,e).
Introduction
Blood vessels are composed of three layers: tunica adventitia, tunica media and tunica intima, and many blood vessels are surrounded by perivascular adipose tissue (PVAT). The adventitia mainly contains connective tissue, while the tunica media (especially in arteries) consists mainly of vascular smooth muscle (VSM). The endothelium, the inner layer of all blood vessels, consists of a thin layer of squamous endothelial cells. Vascular contractility is primarily regulated by the level of contraction of the VSM that can be influenced by the endothelium. The recent discovery of the anti‐contractile nature of PVAT suggests that the fat around blood vessels can also modulate vascular contractility rather than merely providing a structural support. The principal role of VSM is regulation of blood vessel tone/diameter and blood pressure (Owens et al., 2004). VSM contraction may be modulated by vasoconstrictive hormones, vasodilating agents as well as electrical and mechanical stimuli. Some vasoconstricting agonists can activate the RhoA/Rho‐kinase pathway, which sensitizes the VSM contractile machinery to Ca2 + inducing greater contraction at lower intracellular Ca2 + levels; mechanistically this is achieved via inhibition of dephosphorylation of the MLC by myosin light chain phosphatase (Fukata et al., 2001). Increasing [Ca2 +]i also activates potassium (K+) channels, which results in VSM hyperpolarisation and subsequent vasodilatation (Eichhorn and Dobrev, 2007).
The endothelium functions as a semi‐selective barrier between the circulating blood in the lumen and the tissues of the vessel wall and extracellular compartment. Fluid and solutes easily pass across the endothelial layer, and white blood cells may also pass into and out of the blood stream. The endothelium is an important endocrine and paracrine organ that plays a key role in the regulation of vascular tone by releasing factors that modulate VSM contractility (Edwards et al., 2010). Furchgott and Zawadzki (1980) were the first to observe a role for the endothelium in the control of vascular contractility when they demonstrated the release of a diffusible factor, an endothelium‐derived relaxing factor, which promoted vasorelaxation (Furchgott and Zawadzki, 1980). Numerous research studies have shown that the endothelium releases both vasodilating (such as endothelium‐derived hyperpolarising factors, the prostaglandin PGI2 and NO) and vasoconstricting agents (such as endothelin‐1, TXA2 and angiotensin II) and, in the healthy individual, controls vascular tone by balancing their release (see Feletou and Vanhoutte, 2006). The endothelium can modulate VSM contraction via myo‐endothelial gap junctions, which permit ions and polar molecules to pass from cell to cell via an aqueous central pore (Fleming, 2000). It is well known that the endothelium plays a key role in the control of vascular function as its dysfunction is a major factor in the pathogenesis of many cardiovascular diseases (CVD) including hypertension and atherosclerosis (Shaul, 2002; Gollasch and Dubrovska, 2004; Nagata et al., 2004; Boily et al., 2008; Horman et al., 2008; Malinowski et al., 2008; Maenhaut and Van de Voorde, 2011). Endothelial cell dysfunction is also associated with diabetes and metabolic syndrome (Kim et al., 2006; Avogaro et al., 2011).
Although the importance of the endothelium in the regulation of vascular tone is undisputed, it is now clear that it is not the only significant regulator of arterial tone. PVAT surrounds many blood vessels in the body; it is very abundant around the aorta and mesenteric arteries, but it is absent in the cerebral vasculature and microvasculature (Horman et al., 2008; Miao and Li, 2012). At first it was thought to provide mechanical support. However, since the original publication by Soltis and Cassis in 1991 (Soltis and Cassis, 1991), a number of groups have demonstrated that PVAT can significantly influence vascular contractility; these include Lohn's observations on the existence of a adipocyte derived relaxing factor (ADRF) in 2002 (Lohn et al., 2002). PVAT is composed of both white (WAT) and brown adipose tissue (BAT). WAT serves mainly as an energy store (Miao and Li, 2012), whereas BAT is mainly associated with thermogenesis, is more vascularized and is highly active metabolically (Smith and Roberts, 1964). The ratio of WAT/BAT is not constant across vascular beds; for example, PVAT in rats and mice surrounding the abdominal aorta and mesenteric vessels is mainly composed of WAT, while PVAT associated with the thoracic aorta is predominantly BAT (Frontini and Cinti, 2010; Cinti, 2011; Miao and Li, 2012). Interestingly, recently it has been proposed that adipocytes can undergo ‘browning’ (Young et al., 1984), a process whereby WAT transforms into BAT via activation of the PI3K/Akt and AMPK signalling pathways (Than et al., 2015).
PVAT consists of a variety of cells, including adipocytes, macrophages, lymphocytes, endothelial cells, fibroblasts and adipocyte stem/progenitor cells. PVAT is an endocrine organ that releases free fatty acids/non‐esterified fatty acids by lipolysis and also secretes a number of bioactive proteins, called adipokines. These adipokines have autocrine, paracrine and endocrine functions, and they can modulate vascular function under normal physiological conditions. Some adipokines including adiponectin, nesfatin, vaspin, chemerin and omentin may be involved in the regulation of cardiovascular function; however, the mechanisms are still unclear and, for some, their expression in PVAT has not been conclusively demonstrated (Yamawaki, 2011).
PVAT has also been shown to modulate vascular contractility through the release of PVAT‐derived relaxant factors (PVRFs) (Chang et al., 2013). Although it is still unclear how PVRFs exert their anti‐contractile effects, a number of potential PVRFs have been suggested including hydrogen sulphide H2S (Wojcicka et al., 2011), NO (Gao et al., 2007) and angiotensin 1–7 (Lee et al., 2009). Data from a number of groups suggest that this PVAT‐induced vasodilatation is not species or vessel subtype specific (Soltis and Cassis, 1991; Dubrovska et al., 2004; Gollasch and Dubrovska, 2004; Guzik et al., 2007; Greenstein et al., 2009). In addition to endothelial dysfunction, it has also been demonstrated that PVAT dysfunction is evident in many cardio‐metabolic diseases, including hypertension and obesity (Greenstein et al., 2009; Zou et al., 2016).
The endothelium is a well‐established modulator of vascular tone; however, more recently, PVAT has also been shown to control arterial contractility. Both the endothelium and PVAT play a part in the pathogenesis of many vascular diseases. Therefore, this review will focus on the control of vascular tone by PVAT and its similarities in the control of contractility by the endothelium.
Anti‐contractile effects
Both the endothelium and PVAT release vasodilators and vasoconstrictors with the net effect in a healthy individual tending towards vasodilatation. The landmark discovery of NO (Palmer et al., 1987), which has subsequently been demonstrated to be the main vasodilator released by the endothelium, established its key role in the endothelium‐dependent modulation of vascular contractility. More recently, PVAT has been similarly shown to have the ability to reduce vascular contractility through the release of PVRFs (Lohn et al., 2002). The mechanisms by which PVAT exerts this anti‐contractile effect are not as well established as the control of vascular contractility by the endothelium; however, a number of studies have noted that the endothelium and PVAT release many common vasoactive factors that modulate vascular tone, and these are discussed below.
Nitric oxide
NO is a free‐radical, endogenous gas that is involved in many physiological and pathophysiological processes of the cardiovascular system. In the endothelium, NO is synthesized from endothelial NOS (eNOS), which is sensitive to changes in [Ca2 + i] concentration. eNOS catalyses the oxidation of L‐arginine into NO and L‐citrulline, a reaction requiring the presence of cofactors such as calmodulin, flavin mononucleotide, flavin adenine dinucleotide and nicotinamide adenine dinucleotide phosphate. eNOS is regulated by phosphorylation at two major sites with opposing effects; Ser1177 phosphorylation leads to its activation while Thr495 phosphorylation decreases its activity (Dudzinski and Michel, 2007).
Palmer et al. showed that endothelial cells are able to generate NO that is responsible for the vasodilating properties of the endothelium‐derived relaxing factor (Palmer et al., 1987) described 7 years earlier (Furchgott and Zawadzki, 1980). NO regulates basal vascular tone and causes relaxation of VSM by stimulation of soluble GC to produce cGMP (Murad, 1986). Increased cGMP levels activate calcium sensitive potassium channels (KCa channels) and KATP channels leading to a decreased [Ca2 +]i concentration and subsequent relaxation of VSM (Warner et al., 1994). NO can also directly activate KCa channels (Bolotina et al., 1994).
eNOS is expressed in adipocytes (Ribiere et al., 1996) and has now also been demonstrated in PVAT (Gao et al., 2007). The anti‐contractile effect of PVAT in various vascular beds – including rat aorta (Gao et al., 2007), mouse mesenteric arteries (Lynch et al., 2013), rat mesenteric artery (Bussey et al., 2016; Zaborska et al., 2016) and human subcutaneous arteries – has been shown to be abolished by NOS inhibition (Greenstein et al., 2009; Aghamohammadzadeh et al., 2015). NO leads to increased cGMP levels and subsequent protein kinase G activation (Rapoport and Murad, 1983). The anti‐contractile effect of PVAT is also lost in mesenteric arteries from protein kinase G knockout mice (Withers et al., 2014).
To summarize, inhibition of NOS has been shown to modulate the effects of both the endothelium and PVAT on vascular tone suggesting that NO is an important vasodilator, which is synthesized and released from both the endothelium and PVAT.
Prostacyclin
Prostaglandins were one of the first vasoactive substances produced by the vasculature to be identified (Moncada et al., 1977). They are synthesized from arachidonic acid by two COX enzymes (COX1 and COX2). The five primary prostaglandins include PGE2, prostacyclin (PGI2), PGD2, PGF2α and TXA2. These species modulate vascular contractility by activation of specific PG receptors either inducing vasoconstriction (PGF2α; TXA2) or vasodilatation (PGD2; PGI2) or both (PGE2). PGI2 is the most abundant prostaglandin produced by the endothelium (Moncada et al., 1977) and is an important endogenous vasodilator that binds to IP receptors on the VSM. Via stimulation of the GS subunit, activation of adenylate cyclase promotes cAMP production and subsequent VSMC relaxation (Alfranca et al., 2006). This prostacyclin‐mediated vasorelaxation is associated with the opening/activation of potassium channels in the VSM cells, including ATP‐sensitive potassium channels (KATP) and delayed‐rectifier potassium channels (Kdr); both subtypes are sensitive to the increasing intracellular cAMP levels (Li et al., 1997).
Both COX1 and COX2 are also expressed in adipocytes (Bolduc et al., 2004), endothelial cells (Hla and Neilson, 1992) and macrophages (Fels et al., 1986); all of which are present in PVAT. Adipocytes also serve as a major source of PGs induced by catecholamine stimulation (Shaw and Ramwell, 1968) suggesting PVAT has the ability to produce and release prostaglandins and that these PGs may be involved in the mediation of the anti‐contractile effect of PVAT on the vascular tone. Experimentally, the data are inconsistent; the early work by Lohn et al. (2002) suggested that COX is not an ADRF, as PVAT still exerted an anti‐contractile effect in response to 5‐HT stimulation, in the presence of a non‐specific COX inhibitor, indomethacin, in the rat aorta (Lohn et al., 2002), while others have determined that COX does contribute to the noradrenaline‐induced anti‐contractile effect of PVAT in mouse mesenteric artery (Lynch et al., 2013) and human saphenous vein (Ozen et al., 2013). The latter data suggests that prostaglandins (specifically PGE2 and PGI2) only mediate noradrenaline‐induced vasodilatation by PVAT (Ozen et al., 2013) (i.e. the involvement of products of COX enzymic activity is agonist‐dependent). PGI2 has also been shown to be responsible for the PVAT‐mediated endothelium‐dependent vasodilatation in mouse carotid artery (Chang et al., 2012). TXA2 release from mouse aortic PVAT is increased in obesity in response to 5‐HT stimulation (Meyer et al., 2013).
To summarize, both the endothelium and PVAT modulate vascular contractility by releasing a variety of prostaglandins.
Hydrogen peroxide
Hydrogen peroxide (H2O2) is a ROS produced in the endothelium (Breton‐Romero and Lamas, 2014) either by spontaneous dismutation of superoxide (O2 −) or dismutation of O2 − by superoxide dismutases. It is clear from the literature that H2O2 significantly alters vascular tone; however, the data are inconsistent as some studies report vasodilatation (Matoba et al., 2000; Matoba et al., 2002; Shimokawa and Matoba, 2004; Liu et al., 2011) while others vasoconstriction (Gao and Lee, 2005; Mills et al., 2009; Suvorava and Kojda, 2009). H2O2 has been shown to vasodilate mouse and human mesenteric arteries (Matoba et al., 2000; Matoba et al., 2002); these vasodilator effects of H2O2 may be mediated by activation of large conductance calcium‐activated potassium channels (BKCa) on the VSM cells (Liu et al., 2011) and activation of endothelial NO production (Drummond et al., 2000).
H2O2 has also been shown to be produced in adipocytes (Luoma et al., 1998) and has been proposed as a PVRF. PVAT‐intact rat aortae incubated with catalase (a H2O2 scavenger) show enhanced vessel contraction (Gao et al., 2007) suggesting that H2O2 may play a role in the anti‐contractile effect of PVAT. This PVAT‐derived H2O2 exerts vasodilatation through activation of soluble guanylyl cyclase within VSMC (Gao et al., 2007).
To summarize, H2O2 modulates vascular contractility. Endothelium‐derived H2O2 can induce either vasoconstriction or vasodilatation depending on the concentration, species and vessel type. However, PVAT‐derived H2O2 has only been shown to contribute to the anti‐contractile effect through activation of soluble GC in VSM.
Hydrogen sulphide
H2S is a gaseous transmitter synthesized from L‐cysteine by the enzyme cystathionine γ‐lyase (CSE) (Kimura, 2011). Endothelium‐dependent relaxation has been shown to be impaired in CSE−/− knockout mice (Mustafa et al., 2011), suggesting that H2S is a vasorelaxant produced/released by the endothelium. Additionally, CSE expression has been demonstrated in mouse endothelial cells, where it is activated by Ca2 +/calmodulin (Yang et al., 2008). H2S induces relaxation by activating endothelial SKCa and IKCa channels; it has also been shown to activate KATP channels in VSM cells (Zhao et al., 2001; Mustafa et al., 2011). CSE expression has also been demonstrated in rat aortic PVAT where it has been proposed to be responsible for the anti‐contractile effects of PVAT (Kohn et al., 2012). It has also been suggested that H2S mediates the anti‐contractile effect of PVAT via opening of KV channels (Schleifenbaum et al., 2010). However, the minutiae of how this gas acts within the cell to promote vasodilatation are yet to be fully elucidated.
To summarize, H2S is a gaseous relaxant that activates K+ channels when released from the endothelium. It may contribute to the anti‐contractile effect of PVAT, although the mechanism is currently unknown.
Potassium channels
VSM membrane potential is the major determinant of vascular tone due to its influence on voltage‐gated Ca2 + channels, which permit Ca2 + entry into the cell and subsequent constriction. K+ channels are key regulators of the membrane potential, their activation leads to K+ efflux and closure of voltage‐dependent Ca2 + channels, which results in vasodilatation. However, K+ channel inhibition results in depolarisation and subsequently contraction. SKCa and IKCa channels are expressed in endothelial cells (Marchenko and Sage, 1996; Burnham et al., 2002) and were proposed to modulate VSM contraction via endothelial hyperpolarization that can be transmitted to VSM through myo‐endothelial gap junctions (Marrelli et al., 2003; Gluais et al., 2005). Activation of these channels also results in an increase in extracellular K+, which promotes hyperpolarization of VSM through activation of Na+/K+‐ATPase or inwardly rectifying K+ channels (Kir) (Weston et al., 2002).
Experimentally, an increase in extracellular K+ concentration (60 mM KCl) abolishes the anti‐contractile effect of PVAT, suggesting that K+ channels are involved in the modulation of vascular tone by PVAT [since a high extracellular K+ concentration attenuates the effects of K+ channels by reducing the K+ gradient across the cell membrane (Lohn et al., 2002)]. In addition to this, PVAT also hyperpolarises the membrane potential in rat mesenteric (Verlohren et al., 2004; Weston et al., 2013) and gracillis muscle arteries (Zavaritskaya et al., 2013) further supporting the role of K+ in the anti‐contractile capacity of PVAT. Moreover, BKCa and KV channels have also been proposed to modulate the effect of PVAT on vascular contractility (Verlohren et al., 2004; Galvez et al., 2006; Schleifenbaum et al., 2010; Lynch et al., 2013) in numerous vascular beds.
To summarize, activation of K+ channels may be involved in the anti‐contractile effects of both PVAT and the endothelium on vascular contractility.
Endothelial/PVAT dysfunction
Resistance artery dysfunction observed in obesity can arise from, and contribute to, hypertension (de Jongh et al., 2004; Frisbee, 2005) leading to a ‘vicious cycle’ in which the resistance vasculature maintains or even augments the initial rise in blood pressure (Levy et al., 2001). Obesity has detrimental effects on health; it can lead to insulin resistance, the metabolic syndrome, Type 2 diabetes mellitus and increased blood pressure, all being major risk factors for CVD.
Endothelial dysfunction refers to the inability of the endothelium to maintain vascular homeostasis resulting from an imbalance in the production of vasodilating and vasoconstricting factors from the endothelium. This dysfunction results in a shift towards the production of vasoconstricting factors, thus increasing peripheral resistance and consequently blood pressure (Drexler, 1998). This impairment in endothelial function is observed early in the progression of CVD; hence, endothelial dysfunction is an early predictor of future CVD, hypertension and atherosclerosis (Schachinger et al., 2000). A blunted endothelium‐dependent response to vasodilating agents (such as acetylcholine) has been reported in studies on hypertension and obesity (Aalkjaer et al., 1987; Izzard et al., 1996; Jonk et al., 2007).
PVAT dysfunction is also now becoming evident in disease states. Age, environmental conditions and nutritional status also change the percentage of the cellular subtypes within PVAT. Normal adipose tissue tends to be in a non‐inflammatory state, while obesity results in abnormal adipokine production, oxidative stress, hypoxia and inflammation of the adipose tissue (Achike et al., 2011). PVAT dysfunction is also associated with an up‐regulation of vasoconstricting factors (Almabrouk et al., 2014) and has been implicated in obesity (Greenstein et al., 2009; Meijer et al., 2015) and hypertension (Aghamohammadzadeh et al., 2015). PVAT dysfunction has been shown to contribute to the obesity‐related endothelial dysfunction (Ketonen et al., 2010) and smooth muscle cell dysfunction (Greenstein et al., 2009).
Obesity‐related hypertension is associated with endothelial (de Jongh et al., 2004) and PVAT dysfunction (Aghamohammadzadeh et al., 2015), and several factors have been proposed to play a role and are discussed in more detail below.
Endothelial/PVAT dysfunction – nitric oxide
The endothelial dysfunction observed in obesity is associated with reduced NO bioavailability due to increased production of ROS (Bakker et al., 2000). eNOS uncoupling leads to the production of O2 − (Kalinowski and Malinski, 2004) instead of NO. O2 − can cause vasoconstriction directly due to the production of peryoxynitrate (from O2 − + NO), which in turn increases the production of vasoconstricting prostaglandins by interacting with prostaglandin endoperoxide synthase (Goodwin et al., 1999); O2 − can also cause vasoconstriction indirectly by reducing NO bioavailability. In obesity, endothelial dysfunction is associated with the decreased NO bioavailability due to eNOS uncoupling (Lobato et al., 2011).
Similarly, obesity‐linked PVAT dysfunction is also associated with reduced NO bioavailability from PVAT (Bussey et al., 2016), which is responsible for the reduced anti‐contractile effect observed in obesity. Uncoupling of eNOS and increased O2 − production were also proposed to contribute to the PVAT‐induced endothelial dysfunction in obese mice (Xia et al., 2016).
Endothelial/PVAT dysfunction – prostanoids
Another mechanism involved in endothelial and PVAT dysfunction in obesity is the impairment of prostaglandin release. Both COX1 and COX2 activities are increased in the endothelium from obese rats; however, in lean rats, COX2 does not influence the endothelial function, suggesting that the shift in COX2 activity observed in obesity contributes to the endothelial dysfunction (Sanchez et al., 2010). Indeed the increased production of the TXA2 and reduced PGI2 release observed in obesity were attributed to the increased prostaglandin production from arachidonic acid by COX2, induced by inflammation (Lobato et al., 2011).
Obesity is also associated with increased activity of both COX1 and COX2 in adipose tissue (Hsieh et al., 2010; Farb et al., 2014). Recently, Meyer et al. (2013) reported that obesity is associated with increased COX activity that leads to the release of a pro‐contractile factor (TXA2) from PVAT, suggesting that COX activity may play a part in obesity‐induced PVAT dysfunction (Meyer et al., 2013).
Endothelial/PVAT dysfunction – K+ channels
Various potassium channels (such as KCa, Kir and KV) have been implicated in the endothelial dysfunction observed in obesity and hypertension (reviewed by (Climent et al., 2014). The diminished anti‐contractile effect of PVAT observed in hypertension can be restored using KCNQ channel openers in rat Gracilis muscle arteries from spontaneously hypertensive rats, suggesting that a dysfunction in K+ channels activation mediates the altered PVAT regulation of vascular tone (Zavaritskaya et al., 2013).
Endothelial/PVAT dysfunction – inflammation
Obesity also leads to an increased production of the pro‐inflammatory cytokines, such as monocyte chemoattractant protein‐1 (also known as CCL2), TNF‐α and IL‐6 by endothelial cells and pre‐adipocytes (Wellen and Hotamisligil, 2003). Obesity‐linked inflammation contributes to endothelial dysfunction (Ziccardi et al., 2002) by stimulating ROS generation and reducing NO bioavailablity (Virdis et al., 2011). Local inflammation and hypoxia of PVAT observed in obesity were also proposed to attenuate its anti‐contractile properties (Greenstein et al., 2009).
In summary, endothelial dysfunction is well recognized as an important predictor of CVD, but more recently, PVAT has also been proposed to play a role. Obesity‐linked PVAT and endothelial dysfunction are associated with altered NO and prostaglandin production, as well as inflammation and impaired K+ channel activation.
Conclusions
The endothelium and PVAT produce both vasodilating and vasoconstricting factors that have been clearly demonstrated to modulate vascular contractility. There are many common pathways by which the endothelium and PVAT control vascular tone; these include NO, prostaglandins, K+ channels, H2O2 and H2S (Figure 1). An imbalance in the production of vasodilating and vasoconstricting factors that promotes an increased release of the latter leads to endothelial and PVAT dysfunction. These cellular dysfunctions are observed in both human obesity and animal models of disease, and the mechanisms involved are similar in both the endothelium and PVAT. The role of the endothelium in the control of vascular contractility is well established, more so than for PVAT; however, recent studies have shown that PVAT releases similar vasoactive substances to those released from the endothelium. The ability of PVAT to alter vascular reactivity by the production and release of vasoactive factors would ensure that the perfusion of local blood vessels could be attuned by PVAT to more accurately meet the needs of the tissues. However, more research is needed with regards to the modulation of vascular tone mediated by PVAT, as the mechanisms have yet to be fully elucidated.
Figure 1.
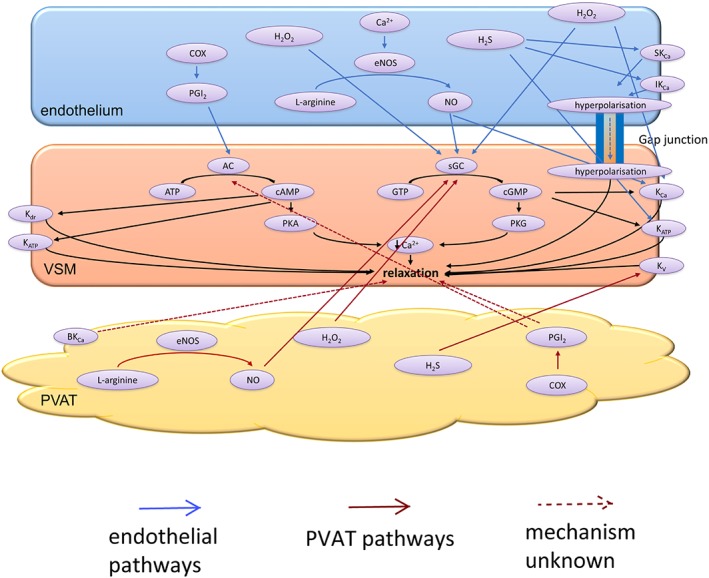
The vasodilating factors released by the endothelium and PVAT. The conversion of L‐arginine into NO by endothelial (e) NOS in both the endothelium and PVAT stimulate soluble guanylyl cycles (sGC) in the VSM, which in turn converts GTP into cGMP. PKG activation leads to vasorelaxation. The enzymatic activity of COX leads to PGI2 production within the endothelium and PVAT. Endothelial PGI2 stimulates AC, which converts ATP into cAMP. PKA is then activated, which leads to relaxation of the VSM. Increasing levels of cAMP can also activate delayed rectifier potassium channels (Kdr) ATP‐sensitive potassium channels (KATP). PVAT‐derived PGI2 causes relaxation of VSM via an unknown mechanism; however, it is most likely to stimulate AC. H2O2 is released by both the endothelium and PVAT and exerts its anti‐contractile effects via sGC activation within the VSM. Endothelial H2O2 can also directly activate KCa on the VSM. H2S mediates the anti‐contractile effects of PVAT via voltage‐gated potassium channels (KV) on VSM. Within the endothelium, H2S activates small‐conductance (SKCa) and intermediate‐conductance (IKCa) calcium‐activated potassium as well as KATP channels on VSM. Activation of SKCa and IKCa on the endothelium leads to hyperpolarisation, which can spread to VSM via myo‐endothelial gap junctions. Large‐conductance calcium‐activated potassium channels (BKCa) are also thought to mediate the anti‐contractile effect of PVAT.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
K.E.Z. is a PhD student supported by the British Heart Foundation (FS/12/68/30006) and The Maternal & Fetal Health Research Centre receives support from Tommy's; let's talk baby.
Zaborska, K. E. , Wareing, M. , and Austin, C. (2017) Comparisons between perivascular adipose tissue and the endothelium in their modulation of vascular tone. British Journal of Pharmacology, 174: 3388–3397. doi: 10.1111/bph.13648.
References
- Aalkjaer C, Heagerty AM, Petersen KK, Swales JD, Mulvany MJ (1987). Evidence for increased media thickness, increased neuronal amine uptake, and depressed excitation–contraction coupling in isolated resistance vessels from essential hypertensives. Circ Res 61: 181–186. [DOI] [PubMed] [Google Scholar]
- Achike FI, To NH, Wang H, Kwan CY (2011). Obesity, metabolic syndrome, adipocytes and vascular function: a holistic viewpoint. Clin Exp Pharmacol Physiol 38: 1–10. [DOI] [PubMed] [Google Scholar]
- Aghamohammadzadeh R, Unwin RD, Greenstein AS, Heagerty AM (2015). Effects of obesity on perivascular adipose tissue vasorelaxant function: nitric oxide, inflammation and elevated systemic blood pressure. J Vasc Res 52: 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015e). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfranca A, Iniguez MA, Fresno M, Redondo JM (2006). Prostanoid signal transduction and gene expression in the endothelium: role in cardiovascular diseases. Cardiovasc Res 70: 446–456. [DOI] [PubMed] [Google Scholar]
- Almabrouk TA, Ewart MA, Salt IP, Kennedy S (2014). Perivascular fat, AMP‐activated protein kinase and vascular diseases. Br J Pharmacol 171: 595–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avogaro A, Albiero M, Menegazzo L, de KS , Fadini GP (2011). Endothelial dysfunction in diabetes: the role of reparatory mechanisms. Diabetes Care 34 (Suppl 2): S285–S290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker SJ, IJzerman RG, Teerlink T, Westerhoff HV, Gans RO, Heine RJ (2000). Cytosolic triglycerides and oxidative stress in central obesity: the missing link between excessive atherosclerosis, endothelial dysfunction, and beta‐cell failure? Atherosclerosis 148: 17–21. [DOI] [PubMed] [Google Scholar]
- Boily G, Seifert EL, Bevilacqua L, He XH, Sabourin G, Estey C et al. (2008). SirT1 regulates energy metabolism and response to caloric restriction in mice. PLoS One 3: e1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc C, Larose M, Lafond N, Yoshioka M, Rodrigue MA, Morissette J et al. (2004). Adipose tissue transcriptome by serial analysis of gene expression. Obes Res 12: 750–757. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA (1994). Nitric oxide directly activates calcium‐dependent potassium channels in vascular smooth muscle. Nature 368: 850–853. [DOI] [PubMed] [Google Scholar]
- Breton‐Romero R, Lamas S (2014). Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol 2: 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham MP, Bychkov R, Feletou M, Richards GR, Vanhoutte PM, Weston AH et al. (2002). Characterization of an apamin‐sensitive small‐conductance Ca(2+)‐activated K(+) channel in porcine coronary artery endothelium: relevance to EDHF. Br J Pharmacol 135: 1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussey CE, Withers SB, Aldous RG, Edwards G, Heagerty AM (2016). Obesity‐related perivascular adipose tissue damage is reversed by sustained weight loss in the rat. Arterioscler Thromb Vasc Biol 36: 1377–1385. [DOI] [PubMed] [Google Scholar]
- Chang L, Milton H, Eitzman DT, Chen YE (2013). Paradoxical roles of perivascular adipose tissue in atherosclerosis and hypertension. Circ J 77: 11–18. [DOI] [PubMed] [Google Scholar]
- Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C et al. (2012). Loss of perivascular adipose tissue on peroxisome proliferator‐activated receptor‐gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 126: 1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinti S (2011). Between brown and white: novel aspects of adipocyte differentiation. Ann Med 43: 104–115. [DOI] [PubMed] [Google Scholar]
- Climent B, Simonsen U, Rivera L (2014). Effects of obesity on vascular potassium channels. Curr Vasc Pharmacol 12: 438–452. [DOI] [PubMed] [Google Scholar]
- de Jongh RT, Serne EH, IJzerman RG, de VG , Stehouwer CD (2004). Impaired microvascular function in obesity: implications for obesity‐associated microangiopathy, hypertension, and insulin resistance. Circulation 109: 2529–2535. [DOI] [PubMed] [Google Scholar]
- Drexler H (1998). Endothelium as a therapeutic target in heart failure. Circulation 98: 2652–2655. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG (2000). Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res 86: 347–354. [DOI] [PubMed] [Google Scholar]
- Dubrovska G, Verlohren S, Luft FC, Gollasch M (2004). Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol Heart Circ Physiol 286: H1107–H1113. [DOI] [PubMed] [Google Scholar]
- Dudzinski DM, Michel T (2007). Life history of eNOS: partners and pathways. Cardiovasc Res 75: 247–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Feletou M, Weston AH (2010). Endothelium‐derived hyperpolarising factors and associated pathways: a synopsis. Pflugers Arch 459: 863–879. [DOI] [PubMed] [Google Scholar]
- Eichhorn B, Dobrev D (2007). Vascular large conductance calcium‐activated potassium channels: functional role and therapeutic potential. Naunyn Schmiedebergs Arch Pharmacol 376: 145–155. [DOI] [PubMed] [Google Scholar]
- Farb MG, Tiwari S, Karki S, Ngo DT, Carmine B, Hess DT et al. (2014). Cyclooxygenase inhibition improves endothelial vasomotor dysfunction of visceral adipose arterioles in human obesity. Obesity (Silver Spring) 22: 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM (2006). Endothelium‐derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol 26: 1215–1225. [DOI] [PubMed] [Google Scholar]
- Fels AO, Pawlowski NA, Abraham EL, Cohn ZA (1986). Compartmentalized regulation of macrophage arachidonic acid metabolism. J Exp Med 163: 752–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I (2000). Myoendothelial gap junctions: the gap is there, but does EDHF go through it? Circ Res 86: 249–250. [DOI] [PubMed] [Google Scholar]
- Frisbee JC (2005). Hypertension‐independent microvascular rarefaction in the obese Zucker rat model of the metabolic syndrome. Microcirculation 12: 383–392. [DOI] [PubMed] [Google Scholar]
- Frontini A, Cinti S (2010). Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab 11: 253–256. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Amano M, Kaibuchi K (2001). Rho‐Rho‐kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non‐muscle cells. Trends Pharmacol Sci 22: 32–39. [DOI] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373–376. [DOI] [PubMed] [Google Scholar]
- Galvez B, de CJ , Herold D, Dubrovska G, Arribas S, Gonzalez MC et al. (2006). Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 26: 1297–1302. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Lee RM (2005). Hydrogen peroxide is an endothelium‐dependent contracting factor in rat renal artery. Br J Pharmacol 146: 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Lu C, Su LY, Sharma AM, Lee RM (2007). Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol 151: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluais P, Edwards G, Weston AH, Falck JR, Vanhoutte PM, Feletou M (2005). Role of SK(Ca) and IK(Ca) in endothelium‐dependent hyperpolarizations of the guinea‐pig isolated carotid artery. Br J Pharmacol 144: 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollasch M, Dubrovska G (2004). Paracrine role for periadventitial adipose tissue in the regulation of arterial tone. Trends Pharmacol Sci 25: 647–653. [DOI] [PubMed] [Google Scholar]
- Goodwin DC, Landino LM, Marnett LJ (1999). Effects of nitric oxide and nitric oxide‐derived species on prostaglandin endoperoxide synthase and prostaglandin biosynthesis. FASEB J 13: 1121–1136. [DOI] [PubMed] [Google Scholar]
- Greenstein AS, Khavandi K, Withers SB, Sonoyama K, Clancy O, Jeziorska M et al. (2009). Local inflammation and hypoxia abolish the protective anticontractile properties of perivascular fat in obese patients. Circulation 119: 1661–1670. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Marvar PJ, Czesnikiewicz‐Guzik M, Korbut R (2007). Perivascular adipose tissue as a messenger of the brain‐vessel axis: role in vascular inflammation and dysfunction. J Physiol Pharmacol 58: 591–610. [PubMed] [Google Scholar]
- Hla T, Neilson K (1992). Human cyclooxygenase‐2 cDNA. Proc Natl Acad Sci U S A 89: 7384–7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horman S, Morel N, Vertommen D, Hussain N, Neumann D, Beauloye C et al. (2008). AMP‐activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J Biol Chem 283: 18505–18512. [DOI] [PubMed] [Google Scholar]
- Hsieh PS, Lu KC, Chiang CF, Chen CH (2010). Suppressive effect of COX2 inhibitor on the progression of adipose inflammation in high‐fat‐induced obese rats. Eur J Clin Invest 40: 164–171. [DOI] [PubMed] [Google Scholar]
- Izzard AS, Bund SJ, Heagerty AM (1996). Myogenic tone in mesenteric arteries from spontaneously hypertensive rats. Am J Physiol 270: H1–H6. [DOI] [PubMed] [Google Scholar]
- Jonk AM, Houben AJ, de Jongh RT, Serne EH, Schaper NC, Stehouwer CD (2007). Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity‐associated insulin resistance and hypertension. Physiology (Bethesda) 22: 252–260. [DOI] [PubMed] [Google Scholar]
- Kalinowski L, Malinski T (2004). Endothelial NADH/NADPH‐dependent enzymatic sources of superoxide production: relationship to endothelial dysfunction. Acta Biochim Pol 51: 459–469. [PubMed] [Google Scholar]
- Ketonen J, Shi J, Martonen E, Mervaala E (2010). Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet‐induced obese C57Bl/6 mice. Circ J 74: 1479–1487. [DOI] [PubMed] [Google Scholar]
- Kim JA, Montagnani M, Koh KK, Quon MJ (2006). Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113: 1888–1904. [DOI] [PubMed] [Google Scholar]
- Kimura H (2011). Hydrogen sulfide: its production, release and functions. Amino Acids 41: 113–121. [DOI] [PubMed] [Google Scholar]
- Kohn C, Schleifenbaum J, Szijarto IA, Marko L, Dubrovska G, Huang Y et al. (2012). Differential effects of cystathionine‐gamma‐lyase‐dependent vasodilatory H2S in periadventitial vasoregulation of rat and mouse aortas. PLoS One 7: e41951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RM, Lu C, Su LY, Gao YJ (2009). Endothelium‐dependent relaxation factor released by perivascular adipose tissue. J Hypertens 27: 782–790. [DOI] [PubMed] [Google Scholar]
- Levy BI, Ambrosio G, Pries AR, Struijker‐Boudier HA (2001). Microcirculation in hypertension: a new target for treatment? Circulation 104: 735–740. [DOI] [PubMed] [Google Scholar]
- Li PL, Zou AP, Campbell WB (1997). Regulation of potassium channels in coronary arterial smooth muscle by endothelium‐derived vasodilators. Hypertension 29: 262–267. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD (2011). H2O2 is the transferrable factor mediating flow‐induced dilation in human coronary arterioles. Circ Res 108: 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobato NS, Filgueira FP, Akamine EH, Davel AP, Rossoni LV, Tostes RC et al. (2011). Obesity induced by neonatal treatment with monosodium glutamate impairs microvascular reactivity in adult rats: role of NO and prostanoids. Nutr Metab Cardiovasc Dis 21: 808–816. [DOI] [PubMed] [Google Scholar]
- Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM (2002). Periadventitial fat releases a vascular relaxing factor. FASEB J 16: 1057–1063. [DOI] [PubMed] [Google Scholar]
- Luoma JS, Stralin P, Marklund SL, Hiltunen TP, Sarkioja T, Yla‐Herttuala S (1998). Expression of extracellular SOD and iNOS in macrophages and smooth muscle cells in human and rabbit atherosclerotic lesions: colocalization with epitopes characteristic of oxidized LDL and peroxynitrite‐modified proteins. Arterioscler Thromb Vasc Biol 18: 157–167. [DOI] [PubMed] [Google Scholar]
- Lynch FM, Withers SB, Yao Z, Werner ME, Edwards G, Weston AH et al. (2013). Perivascular adipose tissue‐derived adiponectin activates BK(Ca) channels to induce anticontractile responses. Am J Physiol Heart Circ Physiol 304: H786–H795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maenhaut N, Van de Voorde J (2011). Regulation of vascular tone by adipocytes. BMC Med 9: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinowski M, Deja MA, Golba KS, Roleder T, Biernat J, Wos S (2008). Perivascular tissue of internal thoracic artery releases potent nitric oxide and prostacyclin‐independent anticontractile factor. Eur J Cardiothorac Surg 33: 225–231. [DOI] [PubMed] [Google Scholar]
- Marchenko SM, Sage SO (1996). Calcium‐activated potassium channels in the endothelium of intact rat aorta. J Physiol 492 (Pt 1): 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrelli SP, Eckmann MS, Hunte MS (2003). Role of endothelial intermediate conductance KCa channels in cerebral EDHF‐mediated dilations. Am J Physiol Heart Circ Physiol 285: H1590–H1599. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Kubota H, Morikawa K, Fujiki T, Kunihiro I et al. (2002). Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun 290: 909–913. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K et al. (2000). Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest 106: 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer RI, Serne EH, Korkmaz HI, van der Peet DL, de Boer MP, Niessen HW et al. (2015). Insulin‐induced changes in skeletal muscle microvascular perfusion are dependent upon perivascular adipose tissue in women. Diabetologia 58: 1907–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Fredette NC, Barton M, Prossnitz ER (2013). Regulation of vascular smooth muscle tone by adipose‐derived contracting factor. PLoS One 8: e79245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao CY, Li ZY (2012). The role of perivascular adipose tissue in vascular smooth muscle cell growth. Br J Pharmacol 165: 643–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills TA, Wareing M, Shennan AH, Poston L, Baker PN, Greenwood SL (2009). Acute and chronic modulation of placental chorionic plate artery reactivity by reactive oxygen species. Free Radic Biol Med 47: 159–166. [DOI] [PubMed] [Google Scholar]
- Moncada S, Higgs EA, Vane JR (1977). Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregation. Lancet 1: 18–20. [DOI] [PubMed] [Google Scholar]
- Murad F (1986). Cyclic guanosine monophosphate as a mediator of vasodilation. J Clin Invest 78: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK et al. (2011). Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata D, Takeda R, Sata M, Satonaka H, Suzuki E, Nagano T et al. (2004). AMP‐activated protein kinase inhibits angiotensin II‐stimulated vascular smooth muscle cell proliferation. Circulation 110: 444–451. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84: 767–801. [DOI] [PubMed] [Google Scholar]
- Ozen G, Topal G, Gomez I, Ghorreshi A, Boukais K, Benyahia C et al. (2013). Control of human vascular tone by prostanoids derived from perivascular adipose tissue. Prostaglandins Other Lipid Mediat 107: 13–17. [DOI] [PubMed] [Google Scholar]
- Palmer RM, Ferrige AG, Moncada S (1987). Nitric oxide release accounts for the biological activity of endothelium‐derived relaxing factor. Nature 327: 524–526. [DOI] [PubMed] [Google Scholar]
- Rapoport RM, Murad F (1983). Endothelium‐dependent and nitrovasodilator‐induced relaxation of vascular smooth muscle: role of cyclic GMP. J Cyclic Nucleotide Protein Phosphor Res 9: 281–296. [PubMed] [Google Scholar]
- Ribiere C, Jaubert AM, Gaudiot N, Sabourault D, Marcus ML, Boucher JL et al. (1996). White adipose tissue nitric oxide synthase: a potential source for NO production. Biochem Biophys Res Commun 222: 706–712. [DOI] [PubMed] [Google Scholar]
- Sanchez A, Contreras C, Martinez P, Villalba N, Benedito S, Garcia‐Sacristan A et al. (2010). Enhanced cyclooxygenase 2‐mediated vasorelaxation in coronary arteries from insulin‐resistant obese Zucker rats. Atherosclerosis 213: 392–399. [DOI] [PubMed] [Google Scholar]
- Schachinger V, Britten MB, Zeiher AM (2000). Prognostic impact of coronary vasodilator dysfunction on adverse long‐term outcome of coronary heart disease. Circulation 101: 1899–1906. [DOI] [PubMed] [Google Scholar]
- Schleifenbaum J, Kohn C, Voblova N, Dubrovska G, Zavarirskaya O, Gloe T et al. (2010). Systemic peripheral artery relaxation by KCNQ channel openers and hydrogen sulfide. J Hypertens 28: 1875–1882. [DOI] [PubMed] [Google Scholar]
- Shaul PW (2002). Regulation of endothelial nitric oxide synthase: location, location, location. Annu Rev Physiol 64: 749–774. [DOI] [PubMed] [Google Scholar]
- Shaw JE, Ramwell PW (1968). Release of prostaglandin from rat epididymal fat pad on nervous and hormonal stimulation. J Biol Chem 243: 1498–1503. [PubMed] [Google Scholar]
- Shimokawa H, Matoba T (2004). Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pharmacol Res 49: 543–549. [DOI] [PubMed] [Google Scholar]
- Smith RE, Roberts JC (1964). Thermogenesis of brown adipose tissue in cold‐acclimated rats. Am J Physiol 206: 143–148. [DOI] [PubMed] [Google Scholar]
- Soltis EE, Cassis LA (1991). Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A 13: 277–296. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvorava T, Kojda G (2009). Reactive oxygen species as cardiovascular mediators: lessons from endothelial‐specific protein overexpression mouse models. Biochim Biophys Acta 1787: 802–810. [DOI] [PubMed] [Google Scholar]
- Than A, He HL, Chua SH, Xu D, Sun L, Leow MK et al. (2015). Apelin enhances brown adipogenesis and browning of white adipocytes. J Biol Chem 290: 14679–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verlohren S, Dubrovska G, Tsang SY, Essin K, Luft FC, Huang Y et al. (2004). Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension 44: 271–276. [DOI] [PubMed] [Google Scholar]
- Virdis A, Santini F, Colucci R, Duranti E, Salvetti G, Rugani I et al. (2011). Vascular generation of tumor necrosis factor‐alpha reduces nitric oxide availability in small arteries from visceral fat of obese patients. J Am Coll Cardiol 58: 238–247. [DOI] [PubMed] [Google Scholar]
- Warner TD, Mitchell JA, Sheng H, Murad F (1994). Effects of cyclic GMP on smooth muscle relaxation. Adv Pharmacol 26: 171–194. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS (2003). Obesity‐induced inflammatory changes in adipose tissue. J Clin Invest 112: 1785–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AH, Egner I, Dong Y, Porter EL, Heagerty AM, Edwards G (2013). Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: involvement of myocyte BKCa channels and adiponectin. Br J Pharmacol 169: 1500–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston AH, Richards GR, Burnham MP, Feletou M, Vanhoutte PM, Edwards G (2002). K + −induced hyperpolarization in rat mesenteric artery: identification, localization and role of Na+/K + −ATPases. Br J Pharmacol 136: 918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers SB, Simpson L, Fattah S, Werner ME, Heagerty AM (2014). cGMP‐dependent protein kinase (PKG) mediates the anticontractile capacity of perivascular adipose tissue. Cardiovasc Res 101: 130–137. [DOI] [PubMed] [Google Scholar]
- Wojcicka G, Jamroz‐Wisniewska A, Atanasova P, Chaldakov GN, Chylinska‐Kula B, Beltowski J (2011). Differential effects of statins on endogenous H2S formation in perivascular adipose tissue. Pharmacol Res 63: 68–76. [DOI] [PubMed] [Google Scholar]
- Xia N, Horke S, Habermeier A, Closs EI, Reifenberg G, Gericke A et al. (2016). Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet‐induced obese mice. Arterioscler Thromb Vasc Biol 36: 78–85. [DOI] [PubMed] [Google Scholar]
- Yamawaki H (2011). Vascular effects of novel adipocytokines: focus on vascular contractility and inflammatory responses. Biol Pharm Bull 34: 307–310. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma‐lyase. Science 322: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young P, Arch JR, Ashwell M (1984). Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett 167: 10–14. [DOI] [PubMed] [Google Scholar]
- Zaborska KE, Wareing M, Edwards G, Austin C (2016). Loss of anti‐contractile effect of perivascular adipose tissue in offspring of obese rats. Int J Obes (Lond) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavaritskaya O, Zhuravleva N, Schleifenbaum J, Gloe T, Devermann L, Kluge R et al. (2013). Role of KCNQ channels in skeletal muscle arteries and periadventitial vascular dysfunction. Hypertension 61: 151–159. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J 20: 6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziccardi P, Nappo F, Giugliano G, Esposito K, Marfella R, Cioffi M et al. (2002). Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation 105: 804–809. [DOI] [PubMed] [Google Scholar]
- Zou L, Wang W, Liu S, Zhao X, Lyv Y, Du C et al. (2016). Spontaneous hypertension occurs with adipose tissue dysfunction in perilipin‐1 null mice. Biochim Biophys Acta 1862: 182–191. [DOI] [PubMed] [Google Scholar]