

Abstract
Perivascular adipose tissue (PVAT) plays a critical role in the pathogenesis of cardiovascular disease. In vascular pathologies, perivascular adipose tissue increases in volume and becomes dysfunctional, with altered cellular composition and molecular characteristics. PVAT dysfunction is characterized by its inflammatory character, oxidative stress, diminished production of vaso‐protective adipocyte‐derived relaxing factors and increased production of paracrine factors such as resistin, leptin, cytokines (IL‐6 and TNF‐α) and chemokines [RANTES (CCL5) and MCP‐1 (CCL2)]. These adipocyte‐derived factors initiate and orchestrate inflammatory cell infiltration including primarily T cells, macrophages, dendritic cells, B cells and NK cells. Protective factors such as adiponectin can reduce NADPH oxidase superoxide production and increase NO bioavailability in the vessel wall, while inflammation (e.g. IFN‐γ or IL‐17) induces vascular oxidases and eNOS dysfunction in the endothelium, vascular smooth muscle cells and adventitial fibroblasts. All of these events link the dysfunctional perivascular fat to vascular dysfunction. These mechanisms are important in the context of a number of cardiovascular disorders including atherosclerosis, hypertension, diabetes and obesity. Inflammatory changes in PVAT's molecular and cellular responses are uniquely different from classical visceral or subcutaneous adipose tissue or from adventitia, emphasizing the unique structural and functional features of this adipose tissue compartment. Therefore, it is essential to develop techniques for monitoring the characteristics of PVAT and assessing its inflammation. This will lead to a better understanding of the early stages of vascular pathologies and the development of new therapeutic strategies focusing on perivascular adipose tissue.
Linked Articles
This article is part of a themed section on Molecular Mechanisms Regulating Perivascular Adipose Tissue – Potential Pharmacological Targets? To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.20/issuetoc
Abbreviations
- AAA
aortic abdominal aneurysm
- ADRF
adipocyte‐derived relaxing factor
- Apoe
apolipoprotein E
- ATLO
adventitial tertiary lymphoid organ
- BAT
brown adipose tissue
- CD
cluster of differentiation
- EDRFs
endothelium‐derived relaxing factors
- PVAT
perivascular adipose tissue
- STAT
signal transducer and activator transcription
- TH17
IL‐17‐producing T cells
- TLO
tertiary lymphoid organs
- Treg
T regulatory lymphocytes
- TRM
tissue‐resident memory T cell
- VSMCs
vascular smooth muscle cells
- WAT
white adipose tissue
Tables of Links
| TARGETS | |
|---|---|
| Other protein targets a | Nuclear hormone receptors c |
| TNF‐α | PPAR‐γ |
| CD3 | Catalytic receptors d |
| GPCRs b | TLR4 |
| Angiotensin 1‐7 (MAS1) receptor | Enzymes e |
| AT1 receptor | APMK |
| CCR2 | eNOS |
| CCR3 | |
| CCR4 | |
| CXCR3 | |
| IL‐17 receptor | |
| S1P1 receptor |
| LIGANDS | |
|---|---|
| Adiponectin | GM‐CSF |
| Ang II | ICAM‐1 |
| CCL12 | IFN‐γ |
| CCL2 (MCP‐1) | IL‐10 |
| CCL20 | IL‐17A |
| CCL5 (RANTES) | IL‐17C |
| CCL7 | IL‐1β |
| CCL8 | IL‐23 |
| CXCL1 | IL‐6 |
| CXCL10 | Leptin |
| CXCL3 | NO |
| CXCL5 | PGI2 |
| G‐CSF |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,c,d,eAlexander et al., 2015a,b,c,d,e).
Introduction
Most blood vessels, apart from the vasculature in the brain, are surrounded or embedded in perivascular adipose tissue (PVAT) (Gao, 2007). It represents around 3% of the total body adipose tissue mass (Siegel‐Axel and Haring, 2016). While initially considered to provide primarily mechanistic support for the vasculature, in recent years it has become clear that PVAT is critical for the regulation of vascular/endothelial function in both physiology and pathology. In normal conditions, PVAT releases substances key for maintaining vasomotor tone and modulating vessel function (Gollasch and Dubrovska, 2004; Galvez et al., 2006; Gao, 2007). This includes beneficial adipocyte‐derived relaxing factor (ADRF), which has been shown to affect vasomotor tone and regulate important homeostatic blood vessel functions. In spite of vast research, the nature of this ADRF remains unidentified with adiponectin, hydrogen peroxide, H2S (hydrogen sulfide), prostacyclin, angiotensin 1–7 or EDHF (endothelium‐derived hyperpolarizing factor) being primary candidates (Szasz et al., 2013; Brown et al., 2014). Studies leading to the discovery of this novel vasorelaxant molecule have been initiated by a finding of Soltis and Cassis (1991) that the presence of PVAT may decrease contractile responses to vasoconstrictive agents. At that time, however, endothelial NO and its role in the regulation of vascular function were at the centre stage of vascular biology; thus, this report was not sufficiently appreciated, until further studies showed the release of classical vascular relaxing factor from the PVAT (Lohn et al., 2002). Several interesting investigations such as those of Gollasch (Lohn et al., 2002; Gollasch and Dubrovska, 2004; Galvez et al., 2006; Fesus et al., 2007) and Gao (Gao et al., 2006, 2007; Gao, 2007) provided further insights into the pharmacology and physiology of these mediators in the PVAT.
The physiological importance of PVAT is emphasized further by studies showing that loss of adipose tissue in lipoatrophic mice (A‐ZIP/F1) enhances the contractile responses of blood vessels, resulting ultimately in hypertension partially linked to an up‐regulation of vascular angiotensin II type 1 (AT1) receptors (Takemori et al., 2007). Moreover, the deletion of PPARγ in vascular smooth muscle cells (VSMCs) causes the loss of PVAT in the aorta (Chang et al., 2012). The interactions between PVAT and vascular function are tightly regulated by a number of metabolic factors including AMPK (5′ AMP‐activated protein kinase) (Almabrouk et al., 2014, 2017). In pathologies associated with vascular dysfunction, release of ADRF is diminished, while PVAT releases a number of paracrine factors such as adipokines (resistin, leptin and visfatin), cytokines (IL‐6 and TNF‐α), chemokines [regulated upon activation, normal T cell expressed and secreted (RANTES, CCL5) and monocyte chemoattractant protein 1 (MCP‐1, CCL2)] – all of which can directly affect VSMCs and endothelial cells and which initiate and orchestrate vascular inflammation. This imbalance between the production and release of protective factors and pro‐inflammatory molecules has been termed, similarly to endothelial dysfunction, the PVAT dysfunction (Guzik et al., 2006, 2007b). Such dysfunctional PVAT has been reported in a range of vascular pathologies including atherosclerosis, hypertension, diabetes and obesity (Guzik et al., 2006; Ignacak et al., 2012). While specific mechanisms and characteristics of PVAT dysfunction may differ, its inflammatory characteristics constitute an important common denominator in a number of vascular pathologies (Figure 1), which will be the primary focus of the current review. Characteristics of inflammation in the PVAT are unique and different to mechanisms observed in typical visceral fat in obesity (Mikolajczyk et al., 2016). This difference results not only from its direct location next to vascular wall but also most likely from the differential release of adipokines and chemokines/cytokines.
Figure 1.
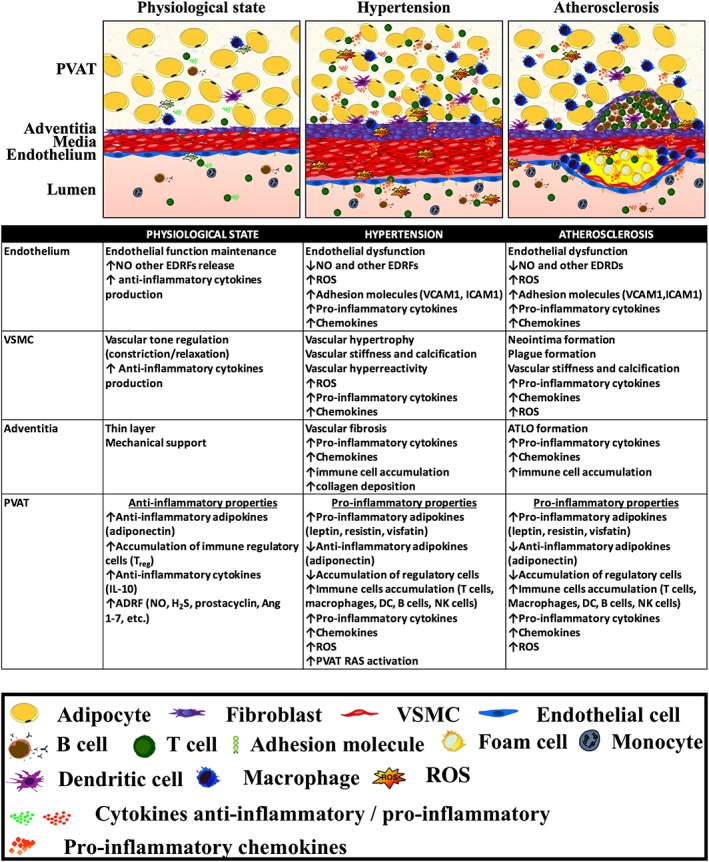
Central role of PVAT inflammation in the regulation of vascular disease. Differential role of PVAT and vascular compartments in the normal physiological state and in the development of vascular pathology in hypertension and atherosclerosis.
PVAT expands in a number of pathologies in humans. This can be systemic, for example, in obesity (Greif et al., 2009; Mahabadi et al., 2010). Local expansion of PVAT has been reported to be associated with atherosclerotic plaque development and vascular calcifications (Lehman et al., 2010), hypertension or aortic abdominal aneurysm (AAA). While the presence of PVAT inflammation is a common feature of vascular disease states, its characteristic varies between different pathologies (Figure 1).
PVAT – brown adipose tissue or white adipose tissue?
Adipose tissue is typically classified as white (WAT), brown (BAT) or beige according to the characteristic colour, but mostly in relation to mitochondrial properties and uncoupling protein 1 (UCP‐1) content. BAT is associated with thermogenesis, while WAT serves as a lipid storage. WAT is less vascular and less metabolically active in comparison with BAT. Both BAT and WAT are under the control of the sympathetic nervous system, but the nerve supply is denser in BAT than in WAT (Harms and Seale, 2013). These differences are reviewed elsewhere (Harms and Seale, 2013) and have been summarized in Table 1.
Table 1.
Key differences between WAT, BAT and PVAT
| WAT | BAT | PVAT | References | |
|---|---|---|---|---|
| Location | Subcutaneous and visceral | Suprarenal, interscapular, neck region in human infants | Surrounds blood vessels | Brown et al. (2014) |
| Morphology | Large adipocytes | Small adipocytes | Small adipocytes | Cedikova et al. (2016); Chatterjee et al. (2009) |
| Lipid droplet | Single, large | Multiple, small | Multiple, small | Brown et al. (2014); Cedikova et al. (2016); Chang et al. (2012) |
| Origin/development | Pdgfr‐α progenitors | Myf5+ progenitors | SM22α + progenitors | Brown et al. (2014); Harms and Seale (2013) |
| Major function |
Energy storage |
Heat production | Vascular regulation, heat production | Chang et al. (2012); Harms and Seale (2013) |
| Mitochondria/UCP1 | +/+ (nearly undetectable) | +++/+++ | ++(+)/++(+) | Cedikova et al. (2016) |
| Adipocyte‐specific genes | PPAR‐γ, PLIN1, HOXC8, TCF21, TLE3, C/EBPα, Rb, RIP140, APOL7C, DAPL1, NANT, SNCG, STAP1, GRAP2, MEST |
ZIC1, LHX8, EVA1, PDK4, EPSTI1, PRDM16, CIDEA, ELOVL3, SCL27A2, COX7A1, CPT1B, KNG2m ACOT11, DIO2, BMP7 |
Similar to BAT | Cedikova et al. (2016); Fitzgibbons et al. (2011); Harms and Seale (2013) |
Differences in both their histological and metabolic profile are also linked to differential immuno‐inflammatory properties of these different types of adipose tissue (Galvez‐Prieto et al., 2008).
PVAT is different from other fat depots in the body through its possible dynamic interplay between white and brown adipocytes, which results in differential functional properties (Table 1). In rodents, PVAT surrounding the thoracic aorta is mainly brown, although a narrow strip immediately adjacent to the vascular adventitia is WAT. The abdominal aorta is surrounded by adipose tissue that is a mixture of brown and white adipocytes, whereas mesenteric arteries are surrounded by mesenteric fat composed mainly of white adipocytes in which these vessels are embedded (Wang et al., 2009; Brown et al., 2014). In humans, PVAT has been attributed to more histological properties of WAT. However, its comparison with typical subcutaneous WAT shows clear differences. In larger vessels, which are of interest in relation to their propensity for atherosclerosis, PVAT commonly displays distinct morphology with adipocytes of a smaller size and of a much less differentiated phenotype than in typical WAT (Galvez‐Prieto et al., 2008; Chatterjee et al., 2009; Chang et al., 2012), as indicated by a less efficient lipid storing capacity, and lower expression levels of WAT adipocyte‐specific genes, with some clear similarities with those in BAT. Thus, PVAT gene and protein profile is clearly different from that of WAT (Chang et al., 2012). PVAT is characterized by a less differentiated phenotype than classical visceral fat, closer to pre‐adipocytes with a particular propensity for the release of pro‐inflammatory factors and growth factors. PVAT, to a greater extent than other adipose tissue compartments, is a conglomerate of various cell types, including adipocytes, pre‐adipocytes and mesenchymal stem cells. Pathological conditions such as Ang II or pro‐atherosclerotic factors increase de‐differentiation in PVAT adipocytes (Tomono et al., 2008; Iwai et al., 2009). This coincides with NFκB‐mediated increases in pro‐inflammatory cytokines such as IL‐6, IL‐8 or chemokines such as MCP‐1 or RANTES (Skurk et al., 2004, 2005; Mikolajczyk et al., 2016).
While clear evidence of the origin of perivascular adipocytes is still lacking, their origin is most likely distinct from other adipocytes. The fact that VSMC PPARγ is essential for the generation of PVAT indicates that plasticity of VSMCs is essential. This is particularly important in the light of the fact that vascular macrophages in atherosclerosis may be largely derived from VSMCs or common precursors. A common embryological origin for perivascular adipocytes and VSMC is therefore very likely and would explain the difference between BAT, WAT and PVAT (Chang et al., 2012; Omar et al., 2014).
PVAT inflammation in vascular pathologies
Morphological, structural and functional alterations of PVAT have been observed in the major vascular pathologies and in association with cardiovascular risk factors including atherosclerosis, hypertension, AAA and diabetic vasculopathies (Figure 1).
Atherosclerosis
A role for the immune system and inflammation in atherosclerosis has been known for several decades now (Hansson and Hermansson, 2011). Studies of immune mechanisms of atherosclerosis have initially focused on neo‐intima and atherosclerotic plaques. Recent evidence suggests a key role for perivascular inflammation at various stages of atherosclerosis (Skiba et al., 2016). Importantly, perivascular inflammation precedes atherosclerotic plaque formation and even the development of endothelial dysfunction and oxidative stress in apolipoprotein E−/− (Apoe−/−) mice (Skiba et al., 2016). While the majority of data in atherosclerosis are focused on adventitial inflammation, clear links to PVAT are evident from these studies. Atherosclerotic mice (Apoe−/− or LDL receptor knockout mice) are characterized by increased production of pro‐inflammatory cytokines such as IL‐6 and IL‐1 in PVAT (Lohmann et al., 2009). Furthermore, perivascular inflammation is associated with a marked increase in chemokines such as MCP‐1 (CCL2) (Manka et al., 2014), MIP‐1α (macrophage inflammatory protein 1‐α, CCL3) (Moos et al., 2005) and RANTES (Sakamoto et al., 2014), which attract immune cells into injury sites.
During the progression of atherosclerosis in Apoe−/− mice, macrophages, T cells and DCs are recruited into perivascular adventitia and adipose tissue (Moos et al., 2005; Galkina et al., 2006) and are correlated with age and lesion size (Moos et al., 2005). An increased number of T cells and macrophages in adventitial layer of abdominal aorta of Apoe−/− has been reported (Sakamoto et al., 2014). Adventitial T and beta cells are present at early stages of atherosclerosis as loose aggregates (Moos et al., 2005; Galkina et al., 2006), while in later stages, they can form adventitial tertiary lymphoid organs (ATLOs) (Akhavanpoor et al., 2014; Hu et al., 2015). Recently, the Galkina group has shown that smooth muscle cell‐derived IL‐17C plays a pro‐atherogenic role by supporting the perivascular recruitment of TH17 cells. IL‐17c−/−Apoe−/− displayed a reduced accumulation of aortic leukocytes (Butcher et al., 2016). Pro‐inflammatory IL‐17A‐producing T cells are present in the adventitia, and blockade of IL‐17A leads to reduction in the accumulation of macrophages and atherosclerosis (Smith et al., 2010).
While many of the above studies focus on adventitial inflammation, which is best characterized in animal models of atherosclerosis, a close interrelationship and lack of clear anatomical border with PVAT makes PVAT essential for this process. Indeed, transplantation of PVAT on the carotid artery increased vascular remodelling after a wire‐induced injury in LDL receptor knockout animals, through adventitial inflammation and angiogenesis (Manka et al., 2014). Endovascular injury significantly up‐regulates pro‐inflammatory MCP‐1, TNF‐α, IL‐6 and plasminogen activator inhibitor‐1 and down‐regulates anti‐inflammatory adipokines, such as adiponectin, within PVAT (Takaoka et al., 2010). Immunohistochemical analyses of periadventitial fat revealed increased macrophages and T cells in Apoe−/− animals compared with WT mice fed a cholesterol diet (Lohmann et al., 2009). There are more macrophages (CD68+ cells) in the PVAT and adventitia in the LDL receptor−/− animals than in the media and intima, in both atherosclerotic and non‐atherosclerotic areas of the vessels (Ding et al., 2013). Consistently, Yamashita et al. (2008) showed that macrophages in the media and adventitia, but not in the intima, are critically involved in expansive atherosclerotic remodelling via matrix degradation and smooth muscle cell reduction. In human atherosclerosis, perivascular macrophages near atherosclerotic lesions are polarized towards the M2 phenotype (Stoger et al., 2012), but their role in atherosclerosis is still controversial.
Molecular mechanisms of PVAT inflammation in atherosclerosis indicate several key targets. Signal transducer and activator transcription 4 (STAT4) is expressed in adipocytes and immune cells and may participate in PVAT inflammation. A deficiency in STAT4 reduces the development of atherosclerosis and PVAT inflammation in Apoe−/− mouse (Dobrian et al., 2015) and in insulin‐resistant obese Zucker rats (Pei et al., 2006). Apoe−/− animals show higher numbers of CD45+ cells in PVAT, but not in visceral fat, compared with Apoe−/−STAT4−/− mouse. In particular, the number of CD8+ T cells is dramatically increased in PVAT of Apoe−/−mice. A reduction of PVAT inflammation was also associated with a diminished expression of CCL5, CXCL10, CX3CL1 and TNF‐α in STAT4‐and Apoe‐deficient mice. Furthermore, a deficiency in STAT4 induces a bias towards anti‐inflammatory macrophages producing IL‐10 and IL‐4 in PVAT of Apoe−/− mouse without affecting their total number (Dobrian et al., 2015). Also, tetrahydrobiopterin treatment markedly reduces leukocyte infiltration into atherosclerotic lesions and the vascular adventitia via endothelial cell signalling (Schmidt et al., 2010). These studies are further supported by findings that vasoprotective compounds such as Mas receptor agonists prevent atherosclerosis through a reduction of chemokine expression and accumulation on immune cells in PVAT (Skiba et al., 2016).
While macrophages and T cells regulate PVAT inflammation in atherosclerosis, an important role for perivascular mast cells has recently been identified (Kennedy et al., 2013). During plaque progression, activated mast cells accumulate in the arterial adventitia and promote macrophage apoptosis and microvascular leakage (Wu et al., 2015). Furthermore, perivascular mast cell activation promotes monocyte adhesion in a CXCR2‐ and vascular cell adhesion molecule 1 (VCAM1)‐dependent manner (Bot et al., 2007).
Hypertension
Hypertension is associated with the activation of the renin–angiotensin–aldosterone system (RAS) and increased vascular oxidative stress. Both Ang II and ROS play a crucial role in the initiation and maintenance of vascular inflammation. The primary site of the initial inflammation in hypertension is within the PVAT and PVAT/adventitial border (Harrison et al., 2011; Kirabo et al., 2014; Mikolajczyk et al., 2016). Almost all components of the RAS, except renin, are expressed in the PVAT (Galvez‐Prieto et al., 2008; Nguyen Dinh Cat and Touyz, 2011), which may play a key role in modulating perivascular inflammation in hypertension. Additionally, PVAT expresses a complex ROS machinery‐containing NADPH, endothelial NOS (eNOS) and antioxidative enzymes (Guzik et al., 2005; Szasz et al., 2013). PVAT‐derived ROS can promote endothelial dysfunction, which could be induced either by endothelial NO scavenging by PVAT‐derived ROS or through the modulation of perivascular inflammation that then affects endothelial function (Ketonen et al., 2010; Even et al., 2014). During the progression of hypertension, immune cells accumulate mainly in perivascular fat tissue surrounding both large and resistance vessels such as the aorta and mesenteric arteries. It is interesting to note that while inflammation is particularly pronounced in PVAT, non‐perivascular visceral fat immune cell infiltration is much less pronounced in non‐obesity‐induced hypertension (Guzik et al., 2007a; Mikolajczyk et al., 2016).
Mice lacking T cells or monocytes exhibit a reduced inflammatory reaction in response to various hypertensive stimuli (Guzik et al., 2007a; Wenzel et al., 2011), whereas loss of lymphocyte adaptor protein (Lnk) gene, encoding a negative regulator of T cell activation, markedly enhances perivascular inflammation (Saleh et al., 2015). Moreover, pro‐hypertensive stimuli increase tissue‐homing markers on leukocytes as well as pro‐inflammatory chemokines, both of which further promote chemotaxis toward adipose tissue (Guzik et al., 2007a; Hoch et al., 2009; Mikolajczyk et al., 2016). The accumulation of leukocytes is markedly reduced in IL‐17−/− and IL‐6−/− Ang II‐infused animals (Madhur et al., 2011). Chronic oxidative stress promotes vascular inflammation in hypertension. Mice lacking NADPH oxidase components such as p47phox, NOX1 and NOX4 are protected against hypertension (Landmesser et al., 2002; Matsuno et al., 2005), while mice with smooth muscle‐targeted overexpression of p22phox (NADPH catalytic subunit) exhibit increased vascular superoxide production, which is associated with an elevation in the total number of leukocytes in PVAT (Wu et al., 2016) and increased susceptibility to vascular dysfunction.
Aneurysms
Abdominal aortic aneurysm is an inflammatory disease associated with marked changes in the cellular composition of the aortic wall and PVAT. Aneurysm formation often coexists with atherosclerosis. Numerous inflammatory cells are involved in AAA formation such as neutrophils, macrophages, T and B cells and mast cells (Sagan et al., 2012; Spear et al., 2015). These immune cells are observed both within PVAT and within luminal thrombi and are partially linked to advanced atherosclerotic plaques (Clement et al., 2015), but they clearly increase susceptibility to AAA formation (Police et al., 2009). A deficiency in TLR4 or myeloid differentiation factor 88 (MyD88) reduced perivascular inflammation and AAA formation (Owens et al., 2011). Apart from contributing to general inflammation, leukocytes in the PVAT may produce proteases such as cathepsins that promote the degradation of aortic wall cells (Folkesson et al., 2016).
In summary, PVAT inflammation is a characteristic feature of vascular pathologies. While there is a number of similarities between perivascular inflammation in hypertension and atherosclerosis, there are also key differences (Figure 1). While in atherosclerosis, perivascular immune infiltrates relatively quickly form organized structures, forming eventually adventitial tertiary organs (ATLOs), in hypertension T cell and B cell infiltration is more scattered. Macrophage infiltration of PVAT is more prominent in atherosclerosis than in hypertension. Aneurysms are so far the only pathology in humans, in which clear PVAT/adventitial ATLO structures have been identified. This either may be related to specific aneurysm pathology or may be aligned to advanced atherosclerosis, which typically accompanies AAA.
How is PVAT inflammation initiated?
Endothelial dysfunction is a key early mechanism of vascular disease. It is characterized by the loss of NO bioavailability accompanied by reduced production of vasoprotective substances, such as prostacyclin (PGI2) and increased production of vascular damaging and pathologically activating molecules such as ROS, endothelin and thromboxane (Channon and Guzik, 2002). Importantly, the vasoprotective substances such as NO have potent anti‐inflammatory properties, which are conveyed through inhibitory effects on adhesion molecule and chemokine expression. Thus, dysfunctional endothelial cells release chemokines such as RANTES, CCL2 and CXCL10 (Mateo et al., 2006; Ide et al., 2008), which can induce leukocyte migration or activation.
Increased ICAM‐1 (intracellular adhesion molecule) and VCAM‐1 expression, on the vascular endothelium, is one of the hallmarks of endothelial dysfunction, linking it to inflammation. When this dysfunction occurs in microvessels and vasa vasorum of PVAT, it will lead to the development of perivascular infiltration, indicating a bidirectional relationship between the vascular endothelium and PVAT.
Oxidative stress, characterized by the overproduction of superoxide anion and hydrogen peroxide, is a key feature of endothelial dysfunction. It results in rapid scavenging of NO in the blood vessel wall – a key mechanism of endothelial dysfunction in a number of vascular pathologies, but it also leads to activation of redox‐sensitive genes within the endothelium, VSMCs and adventitia. Numerous pro‐inflammatory genes including cytokines and chemokines as well as adhesion molecules are redox sensitive, linking vascular oxidative stress to inflammatory processes (Shah et al., 2011).
VSMCs are a significant source of chemokines and cytokines, such as CCL2, CCL7, CCL20, CXCL1, CX3CL1, CXCL5 and IL‐6, IL‐23a and IL‐1β (Butcher et al., 2016). All of these can be essential for the induction of perivascular inflammation. Increased expression of key chemokines in the vascular wall is observed at the early stages of atherosclerosis or hypertension. Chemokine receptors, such as CCR2, CCR5 and CXCR4, are also up‐regulated by oxygen radicals (Zhang et al., 2005; Chan et al., 2012). Thus, endothelial dysfunction and vascular oxidative stress may initiate and exacerbate PVAT inflammation evoked by key risk factors for atherosclerosis, and chemokines are key mediators of this process.
Chemokines in PVAT inflammation
The role of chemokines in initiating and orchestrating inflammation and specific immune responses is widely recognized (Henrichot et al., 2005). These small molecular weight molecules (7–12 kDa) can be divided into four subclasses, C, CC, CXC and CX3C chemokines, based on the position of the N‐terminal cysteine (van der Vorst et al., 2015). Chemokines and their receptors are widely expressed on vascular cells and on leukocytes and play a key role in the recruitment of immune cells to the sites of inflammation or injury in response to a chemokine gradient in many cardiovascular diseases. Conditioned media from PVAT induces a chemotaxis of monocytes and T cells (Miao and Li, 2012; Chatterjee et al., 2013; Mikolajczyk et al., 2016). The role of CCL2, CCL5 and CX3CL1 in the recruitment of circulating monocytes and T cells in atherosclerosis is well established (Charo and Taubman, 2004; van der Vorst et al., 2015). CCL2 produced by adipocytes has been identified as a potential factor contributing to macrophage infiltration into adipose tissue (Kanda et al., 2006; Chan et al., 2012). RANTES (chemokine also known as CCL5), in turn, can be produced by T cells, macrophages, VSMCs and endothelial cells as well as PVAT adipocytes (Mateo et al., 2006; Krensky and Ahn, 2007; Surmi and Hasty, 2010) and is a key factor in the recruitment of leukocytes into inflammatory or infection sites (Marques et al., 2013). RANTES is increased in PVAT in hypertension (Guzik et al., 2007a) and is a characteristic of early stages of atherosclerosis (Veillard et al., 2004; Podolec et al., 2016). RANTES receptors (CCR1, CCR3 and CCR5) are elevated in vascular diseases and are clearly associated with PVAT inflammation (Guzik et al., 2007a; de Jager et al., 2012; Marques et al., 2013; Mikolajczyk et al., 2016). Recently, we demonstrated that RANTES−/− reduces Ang II‐induced accumulation of T cells, macrophages and DCs in the PVAT (Mikolajczyk et al., 2016). Genetic deletion or blockade of RANTES, using the peptide antagonist Met‐RANTES, inhibits leukocyte infiltration to the site of inflammation (Marques et al., 2013) and is effective in modulating perivascular and plaque inflammation in hypertension (Mikolajczyk et al., 2016) and atherosclerosis (Veillard et al., 2004).
CXCL10 (IP‐10) is an IFN‐γ‐inducible protein produced by T cells, NK and NKT cells, monocytes and DCs but also by fibroblasts and endothelial cells (Bondar et al., 2014). It is particularly important in chronic inflammation, including atherosclerosis and hypertension (Ide et al., 2008). Circulating levels of CXCL10 are increased in hypertension (Antonelli et al., 2008) and coronary heart disease (Safa et al., 2016). CXCL10 exerts its biological effects mainly by binding to CXCR3. The CXCL10/CXCR3 axis is important in regulating T cell responses in atherosclerosis. A deficiency of CXCR3 or using CXCR3 antagonist reduces lesion formation in Apoe−/− animals, reduces T cell migration and up‐regulates the expression of anti‐inflammatory molecules (Veillard et al., 2005; van Wanrooij et al., 2008). The expression of CXCL10 is reduced in the PVAT of STAT4−/−Apoe−/− mice, which are protected from PVAT inflammation (Dobrian et al., 2015). The expression of CXCL10 correlates with STAT1 phosphorylation in vascular cells in plaques from human carotid arteries (Chmielewski et al., 2014), and STAT1 and NFκB both regulate CXCL10 (Veillard et al., 2005). CXCL10 has direct effects on vascular wall cells as it induces the migration and proliferation of endothelial cells and VSMCs. Ide et al. demonstrated that CXCL10 (IP‐10) increases the expression of RAS components in endothelial cells (Ide et al., 2008), making it almost a prototypical ‘bidirectional’ cytokine in vascular biology, through which the vessel wall can regulate inflammation and inflammatory cells that can produce CXCL10 affect vascular wall biology.
Immune cells in PVAT inflammation
PVAT inflammation in vascular pathologies appears to differ from typical visceral adipose tissue inflammation in obesity.
In diseases such as hypertension, hypercholesterolaemia and diabetes, PVAT inflammation may occur in the absence of obesity or metabolic syndrome. This may be related to the vicinity of the blood vessel wall, which affects the development of vascular inflammation, and to the presence of vasa vasorum, enabling greater metabolic activity and a clear route for immune cells to migrate into PVAT. There are numerous differences in cellular and humoral characteristics of PVAT inflammation when compared with well‐described inflammation within classical visceral adipose tissue depots. This is manifested by a unique cellular composition and inflammatory cytokine signature (Skiba et al., 2016).
T cells
PVAT T cell infiltration may precede and exceed macrophage infiltration in animal models and in humans with hypertension and hypercholesterolaemia. This is in contrast to typical visceral fat where macrophage‐dependent inflammation predominates from the earliest stages of the disease (Wu et al., 2007). Perivascular T cells represent a morphologically and functionally heterogeneous cellular compartment. Both T helper cells (CD4+) and CD8+ cytotoxic cells are present in the PVAT with a high proportion of CD3+CD4‐CD8‐ T cells, which are predominantly γδ T cells (Guzik et al., 2007a; Mikolajczyk et al., 2016). Recent studies of PVAT T cells indicate their effector and memory functions (Itani et al., 2016). These include primarily TH1 and TH17 cells (producing IFN‐γ and TNF‐α or IL‐17, respectively) or in some stages of pathology TH2 cells. CD8+ lymphocyte‐infiltrated PVAT may also functionally differ depending on their content of granzyme B/perforin or IFN‐γ/TNF‐α (Broere et al., 2011). Ang II and hypertension increase the percentage of circulating T cells with an effector phenotype, which next accumulate in PVAT to trigger inflammation and promote vascular dysfunction (Guzik et al., 2007a; Mikolajczyk et al., 2016). PVAT T cells express CD69, CD25 and CD44 markers, which may confer activation as well as tissue phenotype, and they commonly express high levels of receptors for inflammatory chemokines (CCR1, CCR5 and CCR3) (Vinh et al., 2010) and adhesion molecules (CD44), which are key to their recruitment to PVAT (Guzik et al., 2007a; Mikolajczyk et al., 2016). A substantial proportion of PVAT CD4+ and CD8+ T cells express the activation marker CD25 and produce IFN‐γ and TNF‐α. Ang II induces a shift of T cells towards TH1 that produces IFN‐γ, which is dependent on T cell AT1 receptors (Shao et al., 2003). T regulatory cells (Treg) represent a small but functionally significant population of T cells in the PVAT. They are characterized by high CD25 levels and the presence of the forkhead transcription factor (FOXP3) and through the release of suppressive anti‐inflammatory cytokines (IL‐10 and TGF‐β) play a critical role in immune homeostasis and prevent excessive immune responses (Sakaguchi et al., 2010). Interestingly, adoptive transfer of Tregs ameliorates vascular dysfunction, reduces blood pressure and the infiltration of immune cells in blood vessels and perivascular tissue in Ang II‐treated mice (Matrougui et al., 2011). Treg also prevents monocyte/macrophage and T lymphocyte PVAT infiltration associated with various vascular insults such as wire injury, atherosclerosis and Ang II or aldosterone (Kasal et al., 2012). Finally, a subset of CD8+ regulatory cells, which are also found in the PVAT, may mediate cell death through perforin/granzyme‐dependent pathways (Grossman et al., 2004), controlling immune responses but also affecting apoptosis and the function of adjacent vascular cells. While other subpopulations of T cells such as invariant NK T cells have been reported in PVAT, their functional importance is not clear.
B cells
In atherosclerosis B cells are primarily localized within the plaque and ATLOs (Sage and Mallat, 2014). Little is known about the characteristics of B cells and their function in the PVAT. This is interesting because recent studies show that B cells constitute up to 20% of PVAT leukocytes where they interact with T cells (Parker, 1993; Wei et al., 2014) but are also scattered independently of other immune cells. Chan et al. found that Ang II‐induced hypertension was associated with an increased activation of B cells in the PVAT. Moreover, this was associated with an elevation of serum and aortic antibody deposition of IgG2b and IgG3. Depletion of B cells protected against hypertension (Chan et al., 2015). B regulatory cells have also been described in atherogenesis (Strom et al., 2015); thus, a better understanding of the links between pro‐ and anti‐inflammatory B cells in PVAT is needed. The links between a well‐characterized role of adventitial and ATLO B cells in atherosclerosis to their PVAT infiltration need to be better understood.
Macrophages
Macrophages typically represent about 10–15% of stromal‐vascular fraction, while their number increases to 45–50% during obesity (Wynn et al., 2013). Macrophage infiltration in adipose tissue was first described in a form of crown structures in obesity; it has been linked to the expression of chemokines and adhesion molecules in the fat (Cancello et al., 2005; Kolak et al., 2007). Macrophages accumulate in PVAT and the adventitia during hypercholesterolaemia and hypertension, and also in the absence of obesity (Chan et al., 2012; Moore et al., 2015), and release free radicals via NOX2 NADPH oxidase (Kotsias et al., 2013). Infiltrating macrophages produce cytokines such as IL‐6, IFN‐γ and TNF‐α that change the vascular and PVAT cell biology. While M1 macrophages were classically defined to be associated with obesity and atherosclerosis, recent studies point to a significant infiltration of M2 macrophages in PVAT, which may regulate PVAT adipokine release, as well as perivascular fibrosis. Classically, M1 macrophages produce IL‐12 and IL‐23 and promote TH1 and TH17 cells (Wynn et al., 2013), while M2 produce IL‐10 and participate in TH2‐type and pro‐fibrotic responses (Murray and Wynn, 2011). PVAT macrophages are also important in the regulation of T cell activation through antigen presentation, the expression of co‐stimulatory ligands and release of mediators that modulate their function and/or chemotaxis (Shirai et al., 2015). T cell‐dependent responses may reciprocally regulate PVAT macrophage infiltration. For example, loss of the Lnk gene, which increases T cell activation, enhances macrophage (F4/80+ cells) infiltration into PVAT, and Ang II infusion enhances this effect (Saleh et al., 2015).
Dendritic cells
DCs are key in regulating adaptive immune responses in cardiovascular diseases. They are located primarily on the adventitia–PVAT border but have been reported in PVAT (Wei et al., 2014; Mikolajczyk et al., 2016). This has been identified in hypertension and is enhanced by chronic oxidative stress leading to the formation of immunogenic isoketal–protein adducts, which can accumulate in DCs and promote T cell activation (Kirabo et al., 2014; Wu et al., 2016). Dendritic cells release mediators such as IL‐1β, IL‐6 and IL‐23 that polarize T cells to produce IL‐17A as well as TNF‐α and IFN‐γ, which has been implicated in hypertension and PVAT inflammation (Guzik et al., 2007a; Marko et al., 2012) (Figure 2). Moreover, blocking the CD28/CD80/CD86 co‐stimulation axis between DC and T cells prevents PVAT inflammation (Vinh et al., 2010). However, the role of DCs either in PVAT or the adventitia still raises more questions and answers especially in relation to their migratory capacity into secondary lymphoid organs and in relation to understanding the possible antigens/neo‐antigens they would be presenting to activate T cells (Kirabo et al., 2014) (Figure 2).
Figure 2.
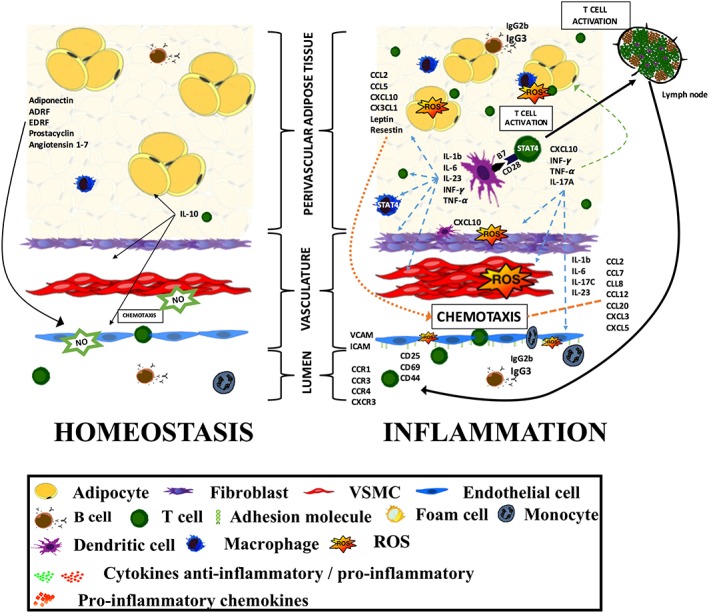
Cellular and humoral components of PVAT inflammation and their interactions in the regulation of vascular homeostasis and vascular dysfunction. A detailed description is provided within the text.
Natural killer cells
NK cells have been identified in PVAT although their role is much less clearly defined than in visceral adipose tissue, where they link obesity‐induced adipose stress to inflammation and insulin resistance in part through IFN‐γ release (Wensveen et al., 2015).
Adventitial tertiary lymphoid organs
Antigen‐presenting cell–T cell interactions occur primarily in secondary lymphoid organs such as lymph nodes and the spleen (Junt et al., 2008). Such interactions have however been demonstrated in vascular adventitia (Koltsova et al., 2012) and possibly PVAT (A. Vinh, personal communication) in the context of chronic vascular inflammation, in atherosclerosis or in hypertension (Figure 1). Such interactions could trigger the development of and be sustained by tertiary lymphoid organs (TLOs) (Hansson and Hermansson, 2011). TLOs are organized aggregates of immune cells formed in post‐embryonic life (GeurtsvanKessel et al., 2009). They can be found around blood vessels in chronic allograft rejection, atherosclerosis and pulmonary hypertension and in patients with chronic obstructive pulmonary disease (Neyt et al., 2012; Perros et al., 2012; Yadava et al., 2016). Interestingly, TLO formation is reversible when inflammation is resolved or after therapeutic intervention (Drayton et al., 2006).
The development of TLOs is orchestrated by various chemokines and cytokines such as CXCL12, CXCL13, CCL19, CCL20, CCL21, lymphotoxin‐α and lymphotoxin‐β (Rangel‐Moreno et al., 2011; Akhavanpoor et al., 2014). Interestingly, IL‐17 also contributes to the formation of TLOs (Rangel‐Moreno et al., 2011). Immune cells can be organized in follicle‐like structures called ATLOs. They can be found in murine models of atherosclerosis and AAA (Hu et al., 2015; Spear et al., 2015). Recently, Hu et al. in a very elegant study showed that an aging immune system employs ATLOs to control atherosclerosis‐related T cell immunity. VSMC‐lymphotoxin β receptors (LTβRs) maintain the ATLO structure and attenuate atherosclerosis (Hu et al., 2015). These structures are evident in human aorta in the context of aortic abdominal aneurysms (Clement et al., 2015).
Origins of PVAT immune cells
While a substantial number of immune cells are recruited by chemotaxis during perivascular inflammation (Henrichot et al., 2005), some immune cells in the vascular wall are chronically resident within the vessel wall. This includes primarily resident macrophages (Robbins et al., 2013; Ensan et al., 2016), which can proliferate in atherosclerotic plaques and potentially in PVAT, as well as resident memory T cells (Schenkel et al., 2014). Resident macrophages are important as they drive the influx of subsequent inflammatory leukocytes, such as monocytes, neutrophils and T cells (Asano et al., 2015). The propensity for this recruitment, based on peripheral blood subpopulations of either monocytes or T cells, remains controversial (Weber et al., 2016). Using multiple fate mapping approaches, it has recently been shown that arterial macrophages arise embryonically from CX3CR1(+) precursors and postnatally from bone marrow‐derived monocytes that colonize the tissue immediately after birth (Ensan et al., 2016). The survival of resident arterial macrophages depends on chemokines, in particular on the fractalkine (CX3CL1) axis, the expression of which is critical in human atherosclerosis and vascular disease (Lucas et al., 2003).
Similar to the myelomonocytic cell lineage, PVAT T cells are either acutely recruited during the development of pathology or may have tissue‐resident memory T cell (TRM cells) characteristics, identified on the basis of phenotypic markers CD69 and CD103 (Mackay et al., 2013; Clark, 2015). TRM cells express low level of receptors such as CCR7 (Bromley et al., 2005; Clark, 2015) and sphingosine‐1‐phosphate receptor 1 (S1P1 receptor; Resop et al., 2016), which promote exit cells from the tissues. TRM cells express high levels of CD44 and low levels of CD62L and release a number of effector cytokines such as IFN‐γ or TNF‐α (Slifka and Whitton, 2000). A subset of TRM cells mediates the protective immunity; however, dysregulation of TRM can contribute to autoimmune and inflammatory diseases. While the potential role of TRM in vascular pathologies is of great interest, other lymphocytes, including classical effector T cells, NK T cells, NK cells and Treg cells, have been described in PVAT. Most of these are likely to be acutely recruited into PVAT.
T cell recruitment to PVAT may be controlled by the sympathetic nervous system in PVAT and the adventitia (Marvar et al., 2010; Guzik and Mikolajczyk, 2014; Itani et al., 2016). Recent evidence suggests a central role for T cells of splenic origin in the initiation of inflammation in hypertension (Carnevale et al., 2014, 2016). These studies from Lembo and Carnevale's group show elegantly that hypertensive challenges activate splenic sympathetic nerve discharge to prime immune response and stimulate immune cell egression from the spleen into target organs, including PVAT (Carnevale et al., 2014, 2016).
The characteristics of PVAT dendritic cells may be divergent. This is particularly important in the light of recent discoveries that plasmacytoid DCs play a key role in atherosclerosis and infiltrate atherosclerotic plaques (Sage et al., 2014). Their role in the PVAT remains unclear.
Mechanisms linking PVAT inflammation to vascular dysfunction
Conditioned media from dysfunctional PVAT in models of vascular disease induce VSMC proliferation and endothelial dysfunction (Miao and Li, 2012; Chatterjee et al., 2013; Mikolajczyk et al., 2016). This is in part mediated by adipokines, which has been reviewed elsewhere (Tilg and Moschen, 2006; Mattu and Randeva, 2013), but may also be dependent on cytokines released by activated inflammatory cells in the PVAT. Most evidence points to the key role of IFN‐γ, IL‐17, IL‐6 and TNF‐α in regulating this process (Matusik et al., 2012) (Figure 2).
Pro‐inflammatory cytokines and endothelial function
IFN‐γ is one of the key cytokines produced by T cells, NK cells as well as some vascular cells. The classical function of IFN‐γ is in the activation of monocytes/macrophages along with polarization of immune cells into a pro‐inflammatory phenotype (Knorr et al., 2014). Importantly, acting on endothelial cells, IFN‐γ impairs endothelium‐dependent relaxation, as demonstrated in ex vivo studies (Mikolajczyk et al., 2016) as well as in vivo using IFN‐γ knockout mice (Kossmann et al., 2013). Furthermore, a reduced recruitment of IFN‐γ‐producing cells into PVAT in RANTES−/− hypertensive animals protects them from impaired endothelium‐dependent relaxation, while having no effect on endothelium‐independent relaxation (Mikolajczyk et al., 2016).
IL‐6, which is produced by macrophages, T cells, DCs and PVAT adipocytes, can directly affect endothelial cells (Pietrowski et al., 2011). It mediates the increase in superoxide production and endothelial dysfunction by affecting the NO‐cGMP signalling pathway (Orshal and Khalil, 2004; Schramm et al., 2012). IL‐6 deficiency prevents vascular dysfunction in spite of various damaging stimuli (Schrader et al., 2007). Treatment of C57BL/6J animals in vivo, or ex vivo by incubating with blood vessels, with IL‐6 impairs endothelium‐dependent relaxation (Wassmann et al., 2004). IL‐6 is also necessary for TH17 cell differentiation (Bettelli et al., 2006), another T cell subpopulation with a strong pro‐inflammatory effect on endothelial cells and VSMCs. IL‐17 is a potent activator of endothelial cells promoting the expression of adhesion molecules (Roussel et al., 2010). IL‐17A activates RhoA/Rho‐kinase and increases inhibitory eNOS Thr495 phosphorylation in endothelial cells leading to decreased NO production (Nguyen et al., 2013). IL‐17A, IFN‐γ and IL‐6 have a synergistic effect with TNF‐α to modulate inflammatory responses (Ruddy et al., 2004). TNF‐α is produced by a wide range of cell types including immune cells, vascular cells and adipocytes (Mendizabal et al., 2013). Stimulation of endothelial cells with this pro‐inflammatory cytokine decreases eNOS expression (Hot et al., 2012) by destabilization of eNOS mRNA (Neumann et al., 2004). TNF‐α, through NFκB, enhances ROS production by endothelial NADPH oxidases. In hypertension, Ang II infusion stimulates T cells to produce TNF‐α and etanercept (TNF‐α antagonist) blunts vascular superoxide production (Guzik et al., 2007a). Moreover, TNF‐α increases the expression of endothelial adhesion molecules and production of pro‐inflammatory chemokines such as CCL5, CCL7, CCL8 or CXCL9 (Hot et al., 2012). Combined treatment with TNF‐α and IL‐17 promotes the synergistic activation of endothelial cells to express adhesion molecules and chemokines that enhance immune cell migration (Griffin et al., 2012). An opposing action is performed by IL‐10, produced by T regulatory cells, selected macrophages and DCs (Saraiva and O'Garra, 2010; Krause et al., 2015). This anti‐inflammatory cytokine reduces NADPH‐dependent oxidative stress and increases the production of NO by enhancing the phosphorylation and activation of eNOS (Kassan et al., 2011). IL‐10 inhibits the activation of p38 MAPK, which contributes to the stimulation of pro‐inflammatory cytokines but can also regulate NADPH oxidases (Kontoyiannis et al., 2001; Konior et al., 2014).
Effects of cytokines produced by immune cells on VSMCs
Inflammatory cytokines released in PVAT modulate smooth muscle cell constriction, proliferation and migration (McMaster et al., 2015). Similar to its effects in endothelial cells, IL‐6 significantly increases Ang II‐mediated ROS production in VSMCs (Wassmann et al., 2004). In vivo treatment of C57BL6 animals with IL‐6 increases the expression of vascular AT1 receptors and mediates medial hypertrophy (Schrader et al., 2007). It also enhances the constriction of the blood vessels (Orshal and Khalil, 2004). Furthermore, IL‐6 has been reported to play role in VSMC migration and proliferation (Chava et al., 2009). IL‐17 receptors are also present on VSMCs (Jin and Dong, 2013). IL‐17A induces the expression of mRNA for collagens I, III and V in a p38 MAPK‐dependent fashion leading to collagen deposition and loss of aortic compliance (Wu et al., 2014). Blood vessels from Ang II‐treated IL‐17A−/− mice are protected from vascular dysfunction with dramatically blunted superoxide production and fibrosis (Madhur et al., 2010). This is because IL‐17A induces NADPH oxidases to produce superoxide anion and hydrogen peroxide and therefore can regulate redox‐sensitive pro‐inflammatory cytokines [IL‐6, MCP‐1, granulocyte‐colony stimulating factor (G‐CSF), granulocyte macrophage colony‐stimulating factor (GM‐CSF)] (Pietrowski et al., 2011). Synergistically with TNF‐α, IL‐17A increases the expression of CCL8, CSF3, CXCL2 and CCL7 in human aortic smooth muscle cells (Madhur et al., 2010).
IFN‐γ can also act directly on VSMCs to induce proliferation (Wang et al., 2007) or apoptosis (Rosner et al., 2006). Neutralization of IFN‐γ prevents outward vascular remodelling of human coronary arteries induced by allogenic T cells in SCID/beige mice (Wang et al., 2004). IFN‐γ induces ICAM‐1 mRNA expression in smooth muscle cells (Chung et al., 2002). IFN‐γ also has a strong impact on superoxide production by up‐regulation of the expression and activity of NOXs in human aortic smooth muscle cells (Manea et al., 2014).
Effects of cytokines produced by immune cells on perivascular adipocytes
As discussed above, part of the effects, through which inflammation mediates vascular function, are dependent on the regulation of classical adipokine expression and release. Adiponectin has a wide range of anti‐inflammatory effects, whereas leptin has pro‐inflammatory effects (Tilg and Moschen, 2006). Both are also critical in regulating vascular function making them prototypical bidirectional adipokines in vascular biology (Antonopoulos et al., 2015, 2016; Woodward et al., 2016) abd also have potent NO‐releasing vasorelaxant properties (Cheng et al., 2007). The production of adiponectin can be inhibited by pro‐inflammatory cytokines such as TNF‐α, IL‐6 and IL‐17A (Maeda et al., 2002; Fasshauer et al., 2003; Noh, 2012). Leptin is produced mainly by adipocytes and is structurally similar to IL‐6, IL‐12 and IL‐15. IL‐17A and TNF‐α increase leptin production (La Cava and Matarese, 2004; Noh, 2012). Leptin apart from direct effects on endothelial NO production and VSMCs can affect leukocyte chemotaxis, the release of oxygen radicals, VSMC proliferation and expression of adhesion molecules on endothelial cells and VSMCs (La Cava and Matarese, 2004). While adiponectin and leptin have been well investigated, PVAT shows particularly high expression of resistin, which also exerts pro‐inflammatory effects. Resistin up‐regulates the expression of VCAM‐1 and ICAM and/or the induction of CCL2 as well as endothelin‐1 from endothelial cells (Bokarewa et al., 2005) and can induce endothelial dysfunction. The gene expression of resistin is induced by pro‐inflammatory cytokines including IL‐1, IL‐6 and TNF‐α (Kaser et al., 2003). Finally, dysfunctional adipocytes in PVAT can produce high levels of classical chemokines MCP‐1, IL‐8 and IL‐6, further contributing to PVAT inflammation.
Conclusions
A dual role of PVAT in the regulation of vascular function is closely linked with PVAT as a site of the development of vascular inflammation. A protective role of PVAT in physiological conditions linked to ADRF release has been demonstrated by numerous studies including seminal studies showing increased vascular dysfunction and hypertension in lipoatrophic mice. This led to the conclusion that ‘fat is not always bad’. Before long, however, in parallel with endothelial dysfunction, the concept of a dysfunctional PVAT was developed, characterized by the loss of PVAT's protective properties. This was initially linked to changes in the adipokine profile, but it soon became apparent that PVAT dysfunction is orchestrated by inflammatory responses. In such conditions, perivascular adipocytes de‐differentiate and are no longer primarily lipid‐storing cells but become a metabolically active synthetic tissue that produces pro‐inflammatory cytokines and chemokines and precipitates the key role of inflammation in cardiovascular disease (Figure 3). This occurs in a number of pathologies including hypertension, early atherosclerosis, hypercholesterolaemia and diabetes. Importantly, the loss of perilipin, which directly induces this change in PVAT phenotype, results in the development of spontaneous hypertension and vascular dysfunction with striking PVAT adipocyte de‐differentiation and inflammatory cell infiltration (Zou et al., 2016). These studies show that PVAT plays a mechanistic role in the development of vascular dysfunction, closing a vicious circle of vascular disease pathogenesis. It still remains unclear how dysfunctional, inflamed PVAT affects vascular dysfunction, remodelling and disease. Is it just an entry point for adventitial inflammation, or is it itself a source of cytokines and chemokines which affect intimal and medial layers of the vessel as well? Whatever the exact mechanism – PVAT inflammation appears to be a tightly regulated process, which occurs early on in the pathogenesis vascular disease, and can constitute a valuable target for future therapies.
Figure 3.
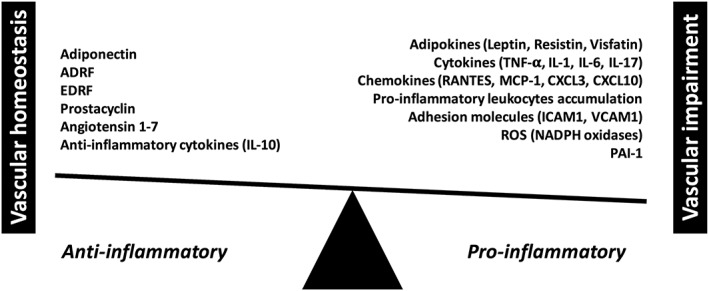
Balancing anti‐ versus pro‐inflammatory properties and functions of perivascular adipose tissue (PVAT).
Author contributions
R.N. drafted the manuscript and prepared the figures; T.J.G. drafted the manuscript and approved the final version to be published.
Conflict of interest
The authors declare no conflicts of interest.
Nosalski, R. , and Guzik, T. J. (2017) Perivascular adipose tissue inflammation in vascular disease. British Journal of Pharmacology, 174: 3496–3513. doi: 10.1111/bph.13705.
References
- Akhavanpoor M, Wangler S, Gleissner CA, Korosoglou G, Katus HA, Erbel C (2014). Adventitial inflammation and its interaction with intimal atherosclerotic lesions. Front Physiol 5: 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015d). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015e). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almabrouk TA, Ewart MA, Salt IP, Kennedy S (2014). Perivascular fat, AMP‐activated protein kinase and vascular diseases. Br J Pharmacol 171: 595–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almabrouk TA, Ugusman AB, Katwan OJ, Salt IP, Kennedy S (2017). Deletion of AMPKa1 attenuates the anticontractile effect of perivascular adipose tissue (PVAT) and reduces adiponectin release. Br J Pharmacol 174: 3398–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli A, Fallahi P, Rotondi M, Ferrari SM, Romagnani P, Ghiadoni L et al. (2008). High serum levels of CXC chemokine ligand 10 in untreated essential hypertension. J Hum Hypertens 22: 579–581. [DOI] [PubMed] [Google Scholar]
- Antonopoulos AS, Margaritis M, Coutinho P, Shirodaria C, Psarros C, Herdman L et al. (2015). Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: the regulatory role of perivascular adipose tissue. Diabetes 64: 2207–2219. [DOI] [PubMed] [Google Scholar]
- Antonopoulos AS, Margaritis M, Verheule S, Recalde A, Sanna F, Herdman L et al. (2016). mutual regulation of epicardial adipose tissue and myocardial redox state by PPAR‐gamma/adiponectin signalling. Circ Res 118: 842–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano K, Takahashi N, Ushiki M, Monya M, Aihara F, Kuboki E et al. (2015). Intestinal CD169(+) macrophages initiate mucosal inflammation by secreting CCL8 that recruits inflammatory monocytes. Nat Commun 6: 7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M et al. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441: 235–238. [DOI] [PubMed] [Google Scholar]
- Bokarewa M, Nagaev I, Dahlberg L, Smith U, Tarkowski A (2005). Resistin, an adipokine with potent proinflammatory properties. J Immunol 174: 5789–5795. [DOI] [PubMed] [Google Scholar]
- Bondar C, Araya RE, Guzman L, Rua EC, Chopita N, Chirdo FG (2014). Role of CXCR3/CXCL10 axis in immune cell recruitment into the small intestine in celiac disease. PLoS One 9: e89068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C et al. (2007). Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E‐deficient mice. Circulation 115: 2516–2525. [DOI] [PubMed] [Google Scholar]
- Broere F, Apasov SG, Sitkovsky MV, van Eden W (2011). A2 T cell subsets and T cell‐mediated immunity. 15–27.
- Bromley SK, Thomas SY, Luster AD (2005). Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat Immunol 6: 895–901. [DOI] [PubMed] [Google Scholar]
- Brown NK, Zhou Z, Zhang J, Zeng R, Wu J, Eitzman DT et al. (2014). Perivascular adipose tissue in vascular function and disease: a review of current research and animal models. Arterioscler Thromb Vasc Biol 34: 1621–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher MJ, Waseem TC, Galkina EV (2016). Smooth muscle cell‐derived interleukin‐17C plays an atherogenic role via the recruitment of proinflammatory interleukin‐17A+ T cells to the aorta. Arterioscler Thromb Vasc Biol 36: 1496–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancello R, Henegar C, Viguerie N, Taleb S, Poitou C, Rouault C et al. (2005). Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery‐induced weight loss. Diabetes 54: 2277–2286. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Pallante F, Fardella V, Fardella S, Iacobucci R, Federici M et al. (2014). The angiogenic factor PlGF mediates a neuroimmune interaction in the spleen to allow the onset of hypertension. Immunity 41: 737–752. [DOI] [PubMed] [Google Scholar]
- Carnevale D, Perrotta M, Pallante F, Fardella V, Iacobucci R, Fardella S et al. (2016). A cholinergic–sympathetic pathway primes immunity in hypertension and mediates brain‐to‐spleen communication. Nat Commun 7: 13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedikova M, Kripnerová M, Dvorakova J, Pitule P, Grundmanova M, Babuska V et al. (2016). Mitochondria in white, brown, and beige adipocytes. Stem Cells Int 2016: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CT, Moore JP, Budzyn K, Guida E, Diep H, Vinh A et al. (2012). Reversal of vascular macrophage accumulation and hypertension by a CCR2 antagonist in deoxycorticosterone/salt‐treated mice. Hypertension 60: 1207–1212. [DOI] [PubMed] [Google Scholar]
- Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H et al. (2015). Obligatory role for B cells in the development of angiotensin II‐dependent hypertension. Hypertension 66: 1023–1033. [DOI] [PubMed] [Google Scholar]
- Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C et al. (2012). Loss of perivascular adipose tissue on peroxisome proliferator‐activated receptor‐gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation 126: 1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channon KM, Guzik TJ (2002). Mechanisms of superoxide production in human blood vessels: relationship to endothelial dysfunction, clinical and genetic risk factors. J Physiol Pharmacol 53: 515–524. [PubMed] [Google Scholar]
- Charo IF, Taubman MB (2004). Chemokines in the pathogenesis of vascular disease. Circ Res 95: 858–866. [DOI] [PubMed] [Google Scholar]
- Chatterjee TK, Aronow BJ, Tong WS, Manka D, Tang Y, Bogdanov VY et al. (2013). Human coronary artery perivascular adipocytes overexpress genes responsible for regulating vascular morphology, inflammation, and hemostasis. Physiol Genomics 45: 697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee TK, Stoll LL, Denning GM, Harrelson A, Blomkalns AL, Idelman G et al. (2009). Proinflammatory phenotype of perivascular adipocytes: influence of high‐fat feeding. Circ Res 104: 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chava KR, Karpurapu M, Wang D, Bhanoori M, Kundumani‐Sridharan V, Zhang Q et al. (2009). CREB‐mediated IL‐6 expression is required for 15(S)‐hydroxyeicosatetraenoic acid‐induced vascular smooth muscle cell migration. Arterioscler Thromb Vasc Biol 29: 809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KK, Lam KS, Wang Y, Huang Y, Carling D, Wu D et al. (2007). Adiponectin‐induced endothelial nitric oxide synthase activation and nitric oxide production are mediated by APPL1 in endothelial cells. Diabetes 56: 1387–1394. [DOI] [PubMed] [Google Scholar]
- Chmielewski S, Olejnik A, Sikorski K, Pelisek J, Blaszczyk K, Aoqui C et al. (2014). STAT1‐dependent signal integration between IFNgamma and TLR4 in vascular cells reflect pro‐atherogenic responses in human atherosclerosis. PLoS One 9: e113318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HK, Lee IK, Kang H, Suh JM, Kim H, Park KC et al. (2002). Statin inhibits interferon‐gamma‐induced expression of intercellular adhesion molecule‐1 (ICAM‐1) in vascular endothelial and smooth muscle cells. Exp Mol Med 34: 451–461. [DOI] [PubMed] [Google Scholar]
- Clark RA (2015). Resident memory T cells in human health and disease. Sci Transl Med 7: 269rv261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement M, Guedj K, Andreata F, Morvan M, Bey L, Khallou‐Laschet J et al. (2015). Control of the T follicular helper‐germinal center B‐cell axis by CD8(+) regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 131: 560–570. [DOI] [PubMed] [Google Scholar]
- de Jager SC, Bongaerts BW, Weber M, Kraaijeveld AO, Rousch M, Dimmeler S et al. (2012). Chemokines CCL3/MIP1alpha, CCL5/RANTES and CCL18/PARC are independent risk predictors of short‐term mortality in patients with acute coronary syndromes. PLoS One 7: e45804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Mizeracki AM, Hu C, Mehta JL (2013). LOX‐1 deletion and macrophage trafficking in atherosclerosis. Biochem Biophys Res Commun 440: 210–214. [DOI] [PubMed] [Google Scholar]
- Dobrian AD, Hatcher MA, Brotman JJ, Galkina EV, Taghavie‐Moghadam P, Pei H et al. (2015). STAT4 contributes to adipose tissue inflammation and atherosclerosis. J Endocrinol 227: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drayton DL, Liao S, Mounzer RH, Ruddle NH (2006). Lymphoid organ development: from ontogeny to neogenesis. Nat Immunol 7: 344–353. [DOI] [PubMed] [Google Scholar]
- Ensan S, Li A, Besla R, Degousee N, Cosme J, Roufaiel M et al. (2016). Self‐renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat Immunol 17: 159–168. [DOI] [PubMed] [Google Scholar]
- Even SE, Dulak‐Lis MG, Touyz RM, Dinh Cat AN (2014). Crosstalk between adipose tissue and blood vessels in cardiometabolic syndrome: implication of steroid hormone receptors (MR/GR). Horm Mol Biol Clin Investig 19: 89–101. [DOI] [PubMed] [Google Scholar]
- Fasshauer M, Kralisch S, Klier M, Lossner U, Bluher M, Klein J et al. (2003). Adiponectin gene expression and secretion is inhibited by interleukin‐6 in 3T3‐L1 adipocytes. Biochem Biophys Res Commun 301: 1045–1050. [DOI] [PubMed] [Google Scholar]
- Fesus G, Dubrovska G, Gorzelniak K, Kluge R, Huang Y, Luft FC et al. (2007). Adiponectin is a novel humoral vasodilator. Cardiovasc Res 75: 719–727. [DOI] [PubMed] [Google Scholar]
- Fitzgibbons TP, Kogan S, Aouadi M, Hendricks GM, Straubhaar J, Czech MP (2011). Similarity of mouse perivascular and brown adipose tissues and their resistance to diet‐induced inflammation. Am J Physiol Heart Circ Physiol 301: H1425–H1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkesson M, Vorkapic E, Gulbins E, Japtok L, Kleuser B, Welander M et al. (2016). Inflammatory cells, ceramides, and expression of proteases in perivascular adipose tissue adjacent to human abdominal aortic aneurysms. J Vasc Surg. doi:10.1016/j.jvs.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K (2006). Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L‐selectin dependent. J Exp Med 203: 1273–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez B, de Castro J, Herold D, Dubrovska G, Arribas S, Gonzalez MC et al. (2006). Perivascular adipose tissue and mesenteric vascular function in spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol 26: 1297–1302. [DOI] [PubMed] [Google Scholar]
- Galvez‐Prieto B, Bolbrinker J, Stucchi P, de Las Heras AI, Merino B, Arribas S et al. (2008). Comparative expression analysis of the renin‐angiotensin system components between white and brown perivascular adipose tissue. J Endocrinol 197: 55–64. [DOI] [PubMed] [Google Scholar]
- Gao YJ (2007). Dual modulation of vascular function by perivascular adipose tissue and its potential correlation with adiposity/lipoatrophy‐related vascular dysfunction. Curr Pharm Des 13: 2185–2192. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Lu C, Su LY, Sharma AM, Lee RM (2007). Modulation of vascular function by perivascular adipose tissue: the role of endothelium and hydrogen peroxide. Br J Pharmacol 151: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Takemori K, Su LY, An WS, Lu C, Sharma AM et al. (2006). Perivascular adipose tissue promotes vasoconstriction: the role of superoxide anion. Cardiovasc Res 71: 363–373. [DOI] [PubMed] [Google Scholar]
- GeurtsvanKessel CH, Willart MA, Bergen IM, van Rijt LS, Muskens F, Elewaut D et al. (2009). Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus‐infected mice. J Exp Med 206: 2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollasch M, Dubrovska G (2004). Paracrine role for periadventitial adipose tissue in the regulation of arterial tone. Trends Pharmacol Sci 25: 647–653. [DOI] [PubMed] [Google Scholar]
- Greif M, Becker A, von Ziegler F, Lebherz C, Lehrke M, Broedl UC et al. (2009). Pericardial adipose tissue determined by dual source CT is a risk factor for coronary atherosclerosis. Arterioscler Thromb Vasc Biol 29: 781–786. [DOI] [PubMed] [Google Scholar]
- Griffin GK, Newton G, Tarrio ML, Bu DX, Maganto‐Garcia E, Azcutia V et al. (2012). IL‐17 and TNF‐alpha sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J Immunol 188: 6287–6299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ (2004). Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity 21: 589–601. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S et al. (2007a). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, Mangalat D, Korbut R (2006). Adipocytokines – novel link between inflammation and vascular function? J Physiol Pharmacol 57: 505–528. [PubMed] [Google Scholar]
- Guzik TJ, Marvar PJ, Czesnikiewicz‐Guzik M, Korbut R (2007b). Perivascular adipose tissue as a messenger of the brain‐vessel axis: role in vascular inflammation and dysfunction. J Physiol Pharmacol 58: 591–610. [PubMed] [Google Scholar]
- Guzik TJ, Mikolajczyk T (2014). In search of the T cell involved in hypertension and target organ damage. Hypertension 64: 224–226. [DOI] [PubMed] [Google Scholar]
- Guzik TJ, Olszanecki R, Sadowski J, Kapelak B, Rudzinski P, Jopek A et al. (2005). Superoxide dismutase activity and expression in human venous and arterial bypass graft vessels. J Physiol Pharmacol 56: 313–323. [PubMed] [Google Scholar]
- Hansson GK, Hermansson A (2011). The immune system in atherosclerosis. Nat Immunol 12: 204–212. [DOI] [PubMed] [Google Scholar]
- Harms M, Seale P (2013). Brown and beige fat: development, function and therapeutic potential. Nat Med 19: 1252–1263. [DOI] [PubMed] [Google Scholar]
- Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR et al. (2011). Inflammation, immunity, and hypertension. Hypertension 57: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrichot E, Juge‐Aubry CE, Pernin A, Pache JC, Velebit V, Dayer JM et al. (2005). Production of chemokines by perivascular adipose tissue: a role in the pathogenesis of atherosclerosis? Arterioscler Thromb Vasc Biol 25: 2594–2599. [DOI] [PubMed] [Google Scholar]
- Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P et al. (2009). Regulation of T‐cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 296: R208–R216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hot A, Lenief V, Miossec P (2012). Combination of IL‐17 and TNFalpha induces a pro‐inflammatory, pro‐coagulant and pro‐thrombotic phenotype in human endothelial cells. Ann Rheum Dis 71: 768–776. [DOI] [PubMed] [Google Scholar]
- Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P et al. (2015). Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin beta receptors. Immunity 42: 1100–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide N, Hirase T, Nishimoto‐Hazuku A, Ikeda Y, Node K (2008). Angiotensin II increases expression of IP‐10 and the renin–angiotensin system in endothelial cells. Hypertens Res 31: 1257–1267. [DOI] [PubMed] [Google Scholar]
- Ignacak A, Kasztelnik M, Sliwa T, Korbut RA, Rajda K, Guzik TJ (2012). Prolactin–not only lactotrophin. A “new” view of the “old” hormone. J Physiol Pharmacol 63: 435–443. [PubMed] [Google Scholar]
- Itani HA, McMaster WG Jr, Saleh MA, Nazarewicz RR, Mikolajczyk TP, Kaszuba AM et al. (2016). Activation of human T cells in hypertension: studies of humanized mice and hypertensive humans. Hypertension 68: 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai M, Tomono Y, Inaba S, Kanno H, Senba I, Mogi M et al. (2009). AT2 receptor deficiency attenuates adipocyte differentiation and decreases adipocyte number in atherosclerotic mice. Am J Hypertens 22: 784–791. [DOI] [PubMed] [Google Scholar]
- Jin W, Dong C (2013). IL‐17 cytokines in immunity and inflammation. Emerg Microbes Infect 2: e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junt T, Scandella E, Ludewig B (2008). Form follows function: lymphoid tissue microarchitecture in antimicrobial immune defence. Nat Rev Immunol 8: 764–775. [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R et al. (2006). MCP‐1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116: 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A et al. (2012). T regulatory lymphocytes prevent aldosterone‐induced vascular injury. Hypertension 59: 324–330. [DOI] [PubMed] [Google Scholar]
- Kaser S, Kaser A, Sandhofer A, Ebenbichler CF, Tilg H, Patsch JR (2003). Resistin messenger‐RNA expression is increased by proinflammatory cytokines in vitro. Biochem Biophys Res Commun 309: 286–290. [DOI] [PubMed] [Google Scholar]
- Kassan M, Galan M, Partyka M, Trebak M, Matrougui K (2011). Interleukin‐10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol 31: 2534–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy S, Wu J, Wadsworth RM, Lawrence CE, Maffia P (2013). Mast cells and vascular diseases. Pharmacol Ther 138: 53–65. [DOI] [PubMed] [Google Scholar]
- Ketonen J, Shi J, Martonen E, Mervaala E (2010). Periadventitial adipose tissue promotes endothelial dysfunction via oxidative stress in diet‐induced obese C57Bl/6 mice. Circ J 74: 1479–1487. [DOI] [PubMed] [Google Scholar]
- Kirabo A, Fontana V, de Faria AP, Loperena R, Galindo CL, Wu J et al. (2014). DC isoketal‐modified proteins activate T cells and promote hypertension. J Clin Invest 124: 4642–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr M, Munzel T, Wenzel P (2014). Interplay of NK cells and monocytes in vascular inflammation and myocardial infarction. Front Physiol 5: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolak M, Westerbacka J, Velagapudi VR, Wagsater D, Yetukuri L, Makkonen J et al. (2007). Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity. Diabetes 56: 1960–1968. [DOI] [PubMed] [Google Scholar]
- Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR et al. (2012). Dynamic T cell–APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest 122: 3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konior A, Schramm A, Czesnikiewicz‐Guzik M, Guzik TJ (2014). NADPH oxidases in vascular pathology. Antioxid Redox Signal 20: 2794–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontoyiannis D, Kotlyarov A, Carballo E, Alexopoulou L, Blackshear PJ, Gaestel M et al. (2001). Interleukin‐10 targets p38 MAPK to modulate ARE‐dependent TNF mRNA translation and limit intestinal pathology. EMBO J 20: 3760–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M et al. (2013). Angiotensin II‐induced vascular dysfunction depends on interferon‐gamma‐driven immune cell recruitment and mutual activation of monocytes and NK‐cells. Arterioscler Thromb Vasc Biol 33: 1313–1319. [DOI] [PubMed] [Google Scholar]
- Kotsias F, Hoffmann E, Amigorena S, Savina A (2013). Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid Redox Signal 18: 714–729. [DOI] [PubMed] [Google Scholar]
- Krause P, Morris V, Greenbaum JA, Park Y, Bjoerheden U, Mikulski Z et al. (2015). IL‐10‐producing intestinal macrophages prevent excessive antibacterial innate immunity by limiting IL‐23 synthesis. Nat Commun 6: 7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krensky AM, Ahn YT (2007). Mechanisms of disease: regulation of RANTES (CCL5) in renal disease. Nat Clin Pract Nephrol 3: 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Cava A, Matarese G (2004). The weight of leptin in immunity. Nat Rev Immunol 4: 371–379. [DOI] [PubMed] [Google Scholar]
- Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H et al. (2002). Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension 40: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman SJ, Massaro JM, Schlett CL, O'Donnell CJ, Hoffmann U, Fox CS (2010). Peri‐aortic fat, cardiovascular disease risk factors, and aortic calcification: the Framingham Heart Study. Atherosclerosis 210: 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmann C, Schafer N, von Lukowicz T, Sokrates Stein MA, Boren J, Rutti S et al. (2009). Atherosclerotic mice exhibit systemic inflammation in periadventitial and visceral adipose tissue, liver, and pancreatic islets. Atherosclerosis 207: 360–367. [DOI] [PubMed] [Google Scholar]
- Lohn M, Dubrovska G, Lauterbach B, Luft FC, Gollasch M, Sharma AM (2002). Periadventitial fat releases a vascular relaxing factor. FASEB J 16: 1057–1063. [DOI] [PubMed] [Google Scholar]
- Lucas AD, Bursill C, Guzik TJ, Sadowski J, Channon KM, Greaves DR (2003). Smooth muscle cells in human atherosclerotic plaques express the fractalkine receptor CX3CR1 and undergo chemotaxis to the CX3C chemokine fractalkine (CX3CL1). Circulation 108: 2498–2504. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML et al. (2013). The developmental pathway for CD103(+)CD8+ tissue‐resident memory T cells of skin. Nat Immunol 14: 1294–1301. [DOI] [PubMed] [Google Scholar]
- Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE et al. (2011). Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e‐deficient mice. Arterioscler Thromb Vasc Biol 31: 1565–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ et al. (2010). Interleukin 17 promotes angiotensin II‐induced hypertension and vascular dysfunction. Hypertension 55: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H et al. (2002). Diet‐induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 8: 731–737. [DOI] [PubMed] [Google Scholar]
- Mahabadi AA, Reinsch N, Lehmann N, Altenbernd J, Kalsch H, Seibel RM et al. (2010). Association of pericoronary fat volume with atherosclerotic plaque burden in the underlying coronary artery: a segment analysis. Atherosclerosis 211: 195–199. [DOI] [PubMed] [Google Scholar]
- Manea SA, Todirita A, Raicu M, Manea A (2014). C/EBP transcription factors regulate NADPH oxidase in human aortic smooth muscle cells. J Cell Mol Med 18: 1467–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manka D, Chatterjee TK, Stoll LL, Basford JE, Konaniah ES, Srinivasan R et al. (2014). Transplanted perivascular adipose tissue accelerates injury‐induced neointimal hyperplasia: role of monocyte chemoattractant protein‐1. Arterioscler Thromb Vasc Biol 34: 1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ et al. (2012). Interferon‐gamma signaling inhibition ameliorates angiotensin II‐induced cardiac damage. Hypertension 60: 1430–1436. [DOI] [PubMed] [Google Scholar]
- Marques RE, Guabiraba R, Russo RC, Teixeira MM (2013). Targeting CCL5 in inflammation. Expert Opin Ther Targets 17: 1439–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C et al. (2010). Central and peripheral mechanisms of T‐lymphocyte activation and vascular inflammation produced by angiotensin II‐induced hypertension. Circ Res 107: 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo T, Naim Abu Nabah Y, Abu Taha M, Mata M, Cerda‐Nicolas M, Proudfoot AEI et al. (2006). Angiotensin II‐induced mononuclear leukocyte interactions with arteriolar and venular endothelium are mediated by the release of different CC chemokines. J Immunol 176: 5577–5586. [DOI] [PubMed] [Google Scholar]
- Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez‐Villalobos RA et al. (2011). Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol 178: 434–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M et al. (2005). Nox1 is involved in angiotensin II‐mediated hypertension: a study in Nox1‐deficient mice. Circulation 112: 2677–2685. [DOI] [PubMed] [Google Scholar]
- Mattu HS, Randeva HS (2013). Role of adipokines in cardiovascular disease. J Endocrinol 216: T17–T36. [DOI] [PubMed] [Google Scholar]
- Matusik P, Guzik B, Weber C, Guzik TJ (2012). Do we know enough about the immune pathogenesis of acute coronary syndromes to improve clinical practice? Thromb Haemost 108: 443–456. [DOI] [PubMed] [Google Scholar]
- McMaster WG, Kirabo A, Madhur MS, Harrison DG (2015). Inflammation, immunity, and hypertensive end‐organ damage. Circ Res 116: 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal Y, Llorens S, Nava E (2013). Hypertension in metabolic syndrome: vascular pathophysiology. Int J Hypertens 2013: 230868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao CY, Li ZY (2012). The role of perivascular adipose tissue in vascular smooth muscle cell growth. Br J Pharmacol 165: 643–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikolajczyk TP, Nosalski R, Szczepaniak P, Budzyn K, Osmenda G, Skiba D et al. (2016). Role of chemokine RANTES in the regulation of perivascular inflammation, T‐cell accumulation, and vascular dysfunction in hypertension. FASEB J 30: 1987–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JP, Vinh A, Tuck KL, Sakkal S, Krishnan SM, Chan CT et al. (2015). M2 macrophage accumulation in the aortic wall during angiotensin II infusion in mice is associated with fibrosis, elastin loss, and elevated blood pressure. Am J Physiol Heart Circ Physiol 309: H906–H917. [DOI] [PubMed] [Google Scholar]
- Moos MP, John N, Grabner R, Nossmann S, Gunther B, Vollandt R et al. (2005). The lamina adventitia is the major site of immune cell accumulation in standard chow‐fed apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol 25: 2386–2391. [DOI] [PubMed] [Google Scholar]
- Murray PJ, Wynn TA (2011). Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 11: 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann P, Gertzberg N, Johnson A (2004). TNF‐alpha induces a decrease in eNOS promoter activity. Am J Physiol Lung Cell Mol Physiol 286: L452–L459. [DOI] [PubMed] [Google Scholar]
- Neyt K, Perros F, GeurtsvanKessel CH, Hammad H, Lambrecht BN (2012). Tertiary lymphoid organs in infection and autoimmunity. Trends Immunol 33: 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Dinh Cat A, Touyz RM (2011). A new look at the renin‐angiotensin system–focusing on the vascular system. Peptides 32: 2141–2150. [DOI] [PubMed] [Google Scholar]
- Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM (2013). Interleukin‐17 causes Rho‐kinase‐mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh M (2012). Interleukin‐17A increases leptin production in human bone marrow mesenchymal stem cells. Biochem Pharmacol 83: 661–670. [DOI] [PubMed] [Google Scholar]
- Omar A, Chatterjee TK, Tang Y, Hui DY, Weintraub NL (2014). Proinflammatory phenotype of perivascular adipocytes. Arterioscler Thromb Vasc Biol 34: 1631–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orshal JM, Khalil RA (2004). Interleukin‐6 impairs endothelium‐dependent NO‐cGMP‐mediated relaxation and enhances contraction in systemic vessels of pregnant rats. Am J Physiol Regul Integr Comp Physiol 286: R1013–R1023. [DOI] [PubMed] [Google Scholar]
- Owens AP 3rd, Rateri DL, Howatt DA, Moore KJ, Tobias PS, Curtiss LK et al. (2011). MyD88 deficiency attenuates angiotensin II‐induced abdominal aortic aneurysm formation independent of signaling through toll‐like receptors 2 and 4. Arterioscler Thromb Vasc Biol 31: 2813–2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker DC (1993). T cell‐dependent B cell activation. Annu Rev Immunol 11: 331–360. [DOI] [PubMed] [Google Scholar]
- Pei H, Gu J, Thimmalapura PR, Mison A, Nadler JL (2006). Activation of the 12‐lipoxygenase and signal transducer and activator of transcription pathway during neointima formation in a model of the metabolic syndrome. Am J Physiol Endocrinol Metab 290: E92–E102. [DOI] [PubMed] [Google Scholar]
- Perros F, Dorfmuller P, Montani D, Hammad H, Waelput W, Girerd B et al. (2012). Pulmonary lymphoid neogenesis in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 185: 311–321. [DOI] [PubMed] [Google Scholar]
- Pietrowski E, Bender B, Huppert J, White R, Luhmann HJ, Kuhlmann CR (2011). Pro‐inflammatory effects of interleukin‐17A on vascular smooth muscle cells involve NAD(P)H‐ oxidase derived reactive oxygen species. J Vasc Res 48: 52–58. [DOI] [PubMed] [Google Scholar]
- Podolec J, Kopec G, Niewiara L, Komar M, Guzik B, Bartus K et al. (2016). Chemokine RANTES is increased at early stages of coronary artery disease. J Physiol Pharmacol 67: 321–328. [PMC free article] [PubMed] [Google Scholar]
- Police SB, Thatcher SE, Charnigo R, Daugherty A, Cassis LA (2009). Obesity promotes inflammation in periaortic adipose tissue and angiotensin II‐induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol 29: 1458–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel‐Moreno J, Carragher DM, de la Luz Garcia‐Hernandez M, Hwang JY, Kusser K, Hartson L et al. (2011). The development of inducible bronchus‐associated lymphoid tissue depends on IL‐17. Nat Immunol 12: 639–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resop RS, Douaisi M, Craft J, Jachimowski LC, Blom B, Uittenbogaart CH (2016). Sphingosine‐1‐phosphate/sphingosine‐1‐phosphate receptor 1 signaling is required for migration of naive human T cells from the thymus to the periphery. J Allergy Clin Immunol 138: 551–557 e558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL et al. (2013). Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 19: 1166–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner D, Stoneman V, Littlewood T, McCarthy N, Figg N, Wang Y et al. (2006). Interferon‐γ induces Fas trafficking and sensitization to apoptosis in vascular smooth muscle cells via a PI3K‐ and Akt‐dependent mechanism. Am J Pathol 168: 2054–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel L, Houle F, Chan C, Yao Y, Berube J, Olivenstein R et al. (2010). IL‐17 promotes p38 MAPK‐dependent endothelial activation enhancing neutrophil recruitment to sites of inflammation. J Immunol 184: 4531–4537. [DOI] [PubMed] [Google Scholar]
- Ruddy MJ, Wong GC, Liu XK, Yamamoto H, Kasayama S, Kirkwood KL et al. (2004). Functional cooperation between interleukin‐17 and tumor necrosis factor‐alpha is mediated by CCAAT/enhancer‐binding protein family members. J Biol Chem 279: 2559–2567. [DOI] [PubMed] [Google Scholar]
- Safa A, Rashidinejad HR, Khalili M, Dabiri S, Nemati M, Mohammadi MM et al. (2016). Higher circulating levels of chemokines CXCL10, CCL20 and CCL22 in patients with ischemic heart disease. Cytokine 83: 147–157. [DOI] [PubMed] [Google Scholar]
- Sagan A, Mrowiecki W, Mikolajczyk TP, Urbanski K, Siedlinski M, Nosalski R et al. (2012). Local inflammation is associated with aortic thrombus formation in abdominal aortic aneurysms. Relationship to clinical risk factors. Thromb Haemost 108: 812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage AP, Mallat Z (2014). Multiple potential roles for B cells in atherosclerosis. Ann Med 46: 297–303. [DOI] [PubMed] [Google Scholar]
- Sage AP, Murphy D, Maffia P, Masters LM, Sabir SR, Baker LL et al. (2014). MHC class II‐restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity. Circulation 130: 1363–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010). FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10: 490–500. [DOI] [PubMed] [Google Scholar]
- Sakamoto S, Tsuruda T, Hatakeyama K, Imamura T, Asada Y, Kitamura K (2014). Impact of age‐dependent adventitia inflammation on structural alteration of abdominal aorta in hyperlipidemic mice. PLoS One 9: e105739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR et al. (2015). Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end‐organ inflammation. J Clin Invest 125: 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva M, O'Garra A (2010). The regulation of IL‐10 production by immune cells. Nat Rev Immunol 10: 170–181. [DOI] [PubMed] [Google Scholar]
- Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D (2014). T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346: 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TS, McNeill E, Douglas G, Crabtree MJ, Hale AB, Khoo J et al. (2010). Tetrahydrobiopterin supplementation reduces atherosclerosis and vascular inflammation in apolipoprotein E‐knockout mice. Clin Sci (Lond) 119: 131–142. [DOI] [PubMed] [Google Scholar]
- Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP (2007). IL‐6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol 27: 2576–2581. [DOI] [PubMed] [Google Scholar]
- Schramm A, Matusik P, Osmenda G, Guzik TJ (2012). Targeting NADPH oxidases in vascular pharmacology. Vascul Pharmacol 56: 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D, Wanchu A, Bhatnagar A (2011). Interaction between oxidative stress and chemokines: possible pathogenic role in systemic lupus erythematosus and rheumatoid arthritis. Immunobiology 216: 1010–1017. [DOI] [PubMed] [Google Scholar]
- Shao J, Nangaku M, Miyata T, Inagi R, Yamada K, Kurokawa K et al. (2003). Imbalance of T‐cell subsets in angiotensin II‐infused hypertensive rats with kidney injury. Hypertension 42: 31–38. [DOI] [PubMed] [Google Scholar]
- Shirai T, Hilhorst M, Harrison DG, Goronzy JJ, Weyand CM (2015). Macrophages in vascular inflammation–from atherosclerosis to vasculitis. Autoimmunity 48: 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel‐Axel DI, Haring HU (2016). Perivascular adipose tissue: an unique fat compartment relevant for the cardiometabolic syndrome. Rev Endocr Metab Disord 17: 51–60. [DOI] [PubMed] [Google Scholar]
- Skiba DS, Nosalski R, Mikolajczyk TP, Siedlinski M, Rios FJ, Montezano AC et al. (2016). Antiatherosclerotic effect of Ang‐ (1–7) non‐peptide mimetic (AVE 0991) is mediated by inhibition of perivascular and plaque inflammation in early atherosclerosis. Br J Pharmacol. doi:10.1111/bph.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurk T, Herder C, Kraft I, Muller‐Scholze S, Hauner H, Kolb H (2005). Production and release of macrophage migration inhibitory factor from human adipocytes. Endocrinology 146: 1006–1011. [DOI] [PubMed] [Google Scholar]
- Skurk T, van Harmelen V, Hauner H (2004). Angiotensin II stimulates the release of interleukin‐6 and interleukin‐8 from cultured human adipocytes by activation of NF‐kappaB. Arterioscler Thromb Vasc Biol 24: 1199–1203. [DOI] [PubMed] [Google Scholar]
- Slifka MK, Whitton JL (2000). Activated and memory CD8+ T cells can be distinguished by their cytokine profiles and phenotypic markers. J Immunol 164: 208–216. [DOI] [PubMed] [Google Scholar]
- Smith E, Prasad KM, Butcher M, Dobrian A, Kolls JK, Ley K et al. (2010). Blockade of interleukin‐17A results in reduced atherosclerosis in apolipoprotein E‐deficient mice. Circulation 121: 1746–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis EE, Cassis LA (1991). Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin Exp Hypertens A 13: 277–296. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP, et al. (2016) The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44:D1054‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear R, Boytard L, Blervaque R, Chwastyniak M, Hot D, Vanhoutte J et al. (2015). Adventitial tertiary lymphoid organs as potential source of MicroRNA biomarkers for abdominal aortic aneurysm. Int J Mol Sci 16: 11276–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoger JL, Gijbels MJ, van der Velden S, Manca M, van der Loos CM, Biessen EA et al. (2012). Distribution of macrophage polarization markers in human atherosclerosis. Atherosclerosis 225: 461–468. [DOI] [PubMed] [Google Scholar]
- Strom AC, Cross AJ, Cole JE, Blair PA, Leib C, Goddard ME et al. (2015). B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL‐10. Thromb Haemost 114: 835–847. [DOI] [PubMed] [Google Scholar]
- Surmi BK, Hasty AH (2010). The role of chemokines in recruitment of immune cells to the artery wall and adipose tissue. Vascul Pharmacol 52: 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szasz T, Bomfim GF, Webb RC (2013). The influence of perivascular adipose tissue on vascular homeostasis. Vasc Health Risk Manag 9: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka M, Suzuki H, Shioda S, Sekikawa K, Saito Y, Nagai R et al. (2010). Endovascular injury induces rapid phenotypic changes in perivascular adipose tissue. Arterioscler Thromb Vasc Biol 30: 1576–1582. [DOI] [PubMed] [Google Scholar]
- Takemori K, Gao YJ, Ding L, Lu C, Su LY, An WS et al. (2007). Elevated blood pressure in transgenic lipoatrophic mice and altered vascular function. Hypertension 49: 365–372. [DOI] [PubMed] [Google Scholar]
- Tilg H, Moschen AR (2006). Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 6: 772–783. [DOI] [PubMed] [Google Scholar]
- Tomono Y, Iwai M, Inaba S, Mogi M, Horiuchi M (2008). Blockade of AT1 receptor improves adipocyte differentiation in atherosclerotic and diabetic models. Am J Hypertens 21: 206–212. [DOI] [PubMed] [Google Scholar]
- van der Vorst EP, Doring Y, Weber C (2015). Chemokines. Arterioscler Thromb Vasc Biol 35: e52–e56. [DOI] [PubMed] [Google Scholar]
- van Wanrooij EJ, de Jager SC, van Es T, de Vos P, Birch HL, Owen DA et al. (2008). CXCR3 antagonist NBI‐74330 attenuates atherosclerotic plaque formation in LDL receptor‐deficient mice. Arterioscler Thromb Vasc Biol 28: 251–257. [DOI] [PubMed] [Google Scholar]
- Veillard NR, Kwak B, Pelli G, Mulhaupt F, James RW, Proudfoot AE et al. (2004). Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res 94: 253–261. [DOI] [PubMed] [Google Scholar]
- Veillard NR, Steffens S, Pelli G, Lu B, Kwak BR, Gerard C et al. (2005). Differential influence of chemokine receptors CCR2 and CXCR3 in development of atherosclerosis in vivo. Circulation 112: 870–878. [DOI] [PubMed] [Google Scholar]
- Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ et al. (2010). Inhibition and genetic ablation of the B7/CD28 T‐cell costimulation axis prevents experimental hypertension. Circulation 122: 2529–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY (2009). Perivascular adipose tissue‐derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res 81: 370–380. [DOI] [PubMed] [Google Scholar]
- Wang Y, Bai Y, Qin L, Zhang P, Yi T, Teesdale SA et al. (2007). Interferon‐gamma induces human vascular smooth muscle cell proliferation and intimal expansion by phosphatidylinositol 3‐kinase dependent mammalian target of rapamycin raptor complex 1 activation. Circ Res 101: 560–569. [DOI] [PubMed] [Google Scholar]
- Wang Y, Burns WR, Tang PC, Yi T, Schechner JS, Zerwes HG et al. (2004). Interferon‐gamma plays a nonredundant role in mediating T cell‐dependent outward vascular remodeling of allogeneic human coronary arteries. FASEB J 18: 606–608. [DOI] [PubMed] [Google Scholar]
- Wassmann S, Stumpf M, Strehlow K, Schmid A, Schieffer B, Bohm M et al. (2004). Interleukin‐6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ Res 94: 534–541. [DOI] [PubMed] [Google Scholar]
- Weber C, Shantsila E, Hristov M, Caligiuri G, Guzik T, Heine GH et al. (2016). Role and analysis of monocyte subsets in cardiovascular disease. Joint consensus document of the European Society of Cardiology (ESC) Working Groups “Atherosclerosis & Vascular Biology” and “Thrombosis”. Thromb Haemost 116: 626–637. [DOI] [PubMed] [Google Scholar]
- Wei Z, Spizzo I, Diep H, Drummond GR, Widdop RE, Vinh A (2014). Differential phenotypes of tissue‐infiltrating T cells during angiotensin II‐induced hypertension in mice. PLoS One 9: e114895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S et al. (2015). NK cells link obesity‐induced adipose stress to inflammation and insulin resistance. Nat Immunol 16: 376–385. [DOI] [PubMed] [Google Scholar]
- Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S et al. (2011). Lysozyme M‐positive monocytes mediate angiotensin II‐induced arterial hypertension and vascular dysfunction. Circulation 124: 1370–1381. [DOI] [PubMed] [Google Scholar]
- Woodward L, Akoumianakis I, Antoniades C (2016). Unravelling the adiponectin paradox: novel roles of adiponectin in the regulation of cardiovascular disease. Br J Pharmacol. doi:10.1111/bph.13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL et al. (2007). T‐cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation 115: 1029–1038. [DOI] [PubMed] [Google Scholar]
- Wu J, Grassia G, Cambrook H, Ialenti A, MacRitchie N, Carberry J et al. (2015). Perivascular mast cells regulate vein graft neointimal formation and remodeling. PeerJ 3: e1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Saleh MA, Kirabo A, Itani HA, Montaniel KR, Xiao L et al. (2016). Immune activation caused by vascular oxidation promotes fibrosis and hypertension. J Clin Invest 126: 50–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L et al. (2014). Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen‐activated protein kinase. Circ Res 114: 616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496: 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadava K, Bollyky P, Lawson MA (2016). The formation and function of tertiary lymphoid follicles in chronic pulmonary inflammation. Immunology 149: 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Shoji K, Tsuruda T, Furukoji E, Takahashi M, Nishihira K et al. (2008). Medial and adventitial macrophages are associated with expansive atherosclerotic remodeling in rabbit femoral artery. Histol Histopathol 23: 127–136. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen X, Song H, Chen HZ, Rovin BH (2005). Activation of the Nrf2/antioxidant response pathway increases IL‐8 expression. Eur J Immunol 35: 3258–3267. [DOI] [PubMed] [Google Scholar]
- Zou L, Wang W, Liu S, Zhao X, Lyv Y, Du C et al. (2016). Spontaneous hypertension occurs with adipose tissue dysfunction in perilipin‐1 null mice. Biochim Biophys Acta 1862: 182–191. [DOI] [PubMed] [Google Scholar]