

Abstract
Caspases are proteases of clan CD and were described for the first time more than two decades ago. They play critical roles in the control of regulated cell death pathways including apoptosis and inflammation. Due to their involvement in the development of various diseases like cancer, neurodegenerative diseases or autoimmune disorders, caspases have been intensively investigated as potential drug targets, both in academic and industrial laboratories. This review presents a thorough, deep, and systematic assessment of all technologies developed over the years for the investigation of caspase activity and specificity using substrates and inhibitors, as well as activity based probes, which in recent years have attracted considerable interest due to their usefulness in the investigation of biological functions of this family of enzymes.
Graphical Abstract
1. INTRODUCTION
Caspases (Cysteine Asp-specific proteases) are conserved throughout metazoans and play a central role in many biological events including apoptosis, cell survival, inflammation and differentiation.1–4 Since their discovery over two decades ago they have been extensively studied in academia and industry. Caspases are excellent therapeutic targets since their dysregulation is linked to a plethora of diseases, e.g. cancer and other proliferative diseases, heart disease, neurodegenerative diseases, osteoarthritis, rheumatoid arthritis, and many more.5–10 To date several biological tools including antibodies, endogenous protein inhibitors and substrates have been discovered or developed for studying caspases biology. Although biologics are very useful, they have also some limitations and often are problematic to use. The second “family” of tools for investigating caspases encompasses small molecule active-site directed substrates, inhibitors and activity based-probes.11–13 Hundreds of peptides and peptidomimetics have been developed for analyzing caspases and their use has provided massive amounts of information regarding specificity, activation, regulation and networking. New more and tailored specific caspase probes are under development to allow tracking of individual caspase activity in vitro and in vivo. Several review papers have described aspects of caspase synthetic substrates or inhibitors, however a comprehensive compilation of this topic has not been described.11,12,14–17 Moreover, the rapidly growing area of small molecule activity based probes design and synthesis encourages us to describe in details the most important aspects of this approach.13,18–21 Accordingly, this review describes and highlights all known classes of caspase tools of synthetic origin, which together have made an enormous impact in understanding caspase activity.
2. CASPASE SUBSTRATES
2.1. Peptide based substrates
As with other protease families, strategies to identify substrate specificity of caspases encompass: 1) individual substrates equipped with reporter group (Figure 1), 2) Positional Scanning-Substrate Combinatorial Libraries (PS-SCL), 3) microarray techniques, 4) phage display libraries, and 5) proteomic-based techniques. All of these methods constitute powerful and reliable tools in determining detailed substrate specificity of an enzyme, although no single one can describe the complete in vivo specificity that leads to a biological outcome.
Figure 1.

Conventional measurement of protease activity. Examples of reporter groups.25
The first extensive studies on caspase substrate selectivity, seeding the foundational knowledge of individual caspase substrate specificity, were conducted in 1997.22–24 Rano and colleagues22 employed PS-SCL methods to study caspases, initially focusing on interleukin-1β converting enzyme (ICE, caspase-1),22 and subsequently the inherent subsite preferences of almost all members of the human caspase family.24 PS-SCL is based on libraries of peptidic substrates with conjugated reporter groups, such as fluorophores, luminophores or chromophores. Fluorophores are probably the most commonly used, as they are quite easy to synthesize, have relatively small size and posses high sensitivity (luminophores posses the highest sensitivity, while chromophores - the lowest).25 In such fluorogenic substrate libraries the fluorophore is fixed at the P1′ position (nomenclature of Schechter and Berger26 – see Figure 1) where it is quenched, and as soon as protease cleavage takes place the fluorophore is released and emits fluorescence after excitation by an appropriate wavelength (Figure 1).
The fluorescence signal can be quantitatively measured, providing data on reaction kinetics and enabling selection of the best and the worst recognized substrates. PS-SCL permits the capture of reliable substrate specificity profiles of an enzyme in a short time. This technique constitutes a powerful tool in determining non-prime residues of a peptide substrate (the residues N-terminal of the scissile bond). For a wider exploration of the enzyme catalytic cleft (residues C-terminal of the scissile bond) other approaches must be applied (as described later). In their pioneering description of caspase-1 substrate specificity Rano and colleagues designed and synthesized three sublibraries of tetrapeptidic substrates.22 Each sublibrary was anchored by Asp acid at P1, one position fixed with a proteinogenic amino acids and the remaining positions contained equimolar mixture of natural amino acids as indicated by Ostresh et al.27 This library architecture was consistent with previous studies revealing a strong requirement for Asp in P1 position.28–30 As a reporter group 7-amino-4-methyl-coumarin (AMC) was employed. The general construction of this library is illustrated in Figure 2.
Figure 2.

Structure of the combinatorial library used by Rano et al. 22 The library is composed of 3 sublibraries. Position P1′ is occupied by a fluorogenic reporter (AMC), position P1 is fixed with aspartic acid, the outlined position represents a spatially addressed natural amino acid while the other positions represent isokinetic mixtures.
The studies conducted by Rano and colleagues highlighted the important principle that the optimal substrate recognition sequence does not necessarily match the sequence of natural substrates. This concept was championed initially by Madison and colleagues who explored preferred substrate sequences of plasminogen activators identified by phage display in comparison with the natural substrate plasminogen.31,32 The general conclusion was that secondary interactions with natural substrates influence specificity in vivo, and this result is conformed when the “best” peptide sequence for caspase-1 (WEHD) does not match the natural cleavage site in pro-IL-1β (YVHD).22 Kinetic parameters kcat/KM measured on individual substrates were evidence that WEHD-AMC 1 tetrapeptide was better than the initially championed YVAD-AMC 2 and YVHD-AMC 3 sequences with kcat/KM value around 50-fold and 12-fold higher, respectively. Additionally, inhibitors that were designed based on substrates, Ac-YVAD-CHO 4 and Ac-WEHD-CHO 5 with Ki values 760 nM and 56 nM respectively, provided evidence that inhibitors can be selected based on substrate sequences.22 Thornberry and coworkers employed the same library to profile nine human caspases.24 This research constituted milestone in studies of caspases inherent subsite preferences and resulted in grouping the enzymes based on specificity profiles. The division proposed by Thornberry et al. is shown in Table 1.
Table 1.
Division of caspases and optimal sequences cleaved by caspases (X represents that several amino acids are tolerated in this position) proposed by Thornberry et al..24
Caspase-10 and caspase-14 substrate specificities were established by other groups employing PS-SCL. The optimal recognition sequence for caspase-10 was found to be LEXD (X symbolize that several amino acids may occupy this position)33,35 and for that reason it was classified in group III. Caspase-14 with WEHD as the most preferred tetrapeptide recognition motif34 was placed in group I.
One may notice all caspases show selectivity for glutamic acid at P3. The positions P4 and P2 played an important role in assigning caspases to groups. It is worth noting that the library constructed by Rano et al. may be used only for proteases with strong preferences for aspartic acid in the S1 subsite, since this residue is fixed. Later there appeared some attempts to overcome this limitation,36,37 however it is still not easy to examine proteases with unknown P1 specificity using PS-SCL technology.
Talanian et al. examined substrate specificities of several caspases by using of sets of individual peptide substrates38 with either fluorogenic (AMC) or chromogenic (pNA) reporter groups revealing that caspase-2 cleaves more efficiently when substrates are extended to the P5 position, with a preference for hydrophobic residues (Figure 1). The kcat/KM value for the pentapeptidic substrate Ac-VDVAD-pNA 6 was 84000 M−1s−1 while tetrapeptidic substrates, such as Ac-DEVD-pNA 7, Ac-YVAD-pNA 8, Ac-VEID-pNA 9, Ac-VQVD-pNA 10 were not cleaved. Tang et al. confirmed that indeed caspase-2 has a strong requirement for S5 pocket to be filled.39 Two pentapeptides VDVAD 11 and ADVAD 12 were compared with the tetrapeptide DVAD 13 (all three substrates were equipped with the AFC fluorophore) and the kcat/KM values were 24000 M−1s−1, 5500 M−1s−1, 1300 M−1s−1 respectively. Later structural investigations revealed that residues Thr-380 and Tyr-420 of caspase-2 are crucial for P5 residue binding,39 and suggested that caspase-2 may be an outlier within the caspase family as it has strong requirement for S5 pocket to be filled and its activity on tetrapeptides is very low.
A breakthrough in synthesis of fluorogenic libraries came with application of a bifunctional fluorogenic group 7-amino-4-carbamoylmethylcoumarin (ACC) in 2000,40 which allowed for solid phase synthesis of PS-SCLs.41 The strategy gave the advantage of incorporating any amino acid in any position, enabling complete diversification of fluorogenic libraries. Furthermore, incorporation of ACC significantly improved assay sensitivity compared to libraries equipped with AMC since ACC has a higher fluorescent yield. Consequently, smaller amount of substrate and enzyme can be used for each assay. Caspase-3 substrate specificity was examined by this approach42 demonstrating that the specificity profile was in line with Thornberry’s analysis.24 Detailed protocols for determination of caspases substrate specificity profiles using PS-SCL approach have been described by Poreba et al..43 More recently, ACC-based libraries have formed the basis of a new approach christened Hybrid Combinatorial Substrate Library (HyCoSuL) introduced by Kasperkiewicz et al.44 which is an extension of traditional PS-SCL method. In this technique, in addition to natural amino acids, a diverse series of commercially-available unnatural amino acids are used. Poreba and colleagues conducted a broad study of six caspases applying HyCoSuL containing 110 unnatural amino.45 This methodology resulted in a solution to the problem of overlapping specificities of the caspases and for the first time allowed a high degree of discrimination between individual caspases (as described later).
Interestingly, in contrast to methods based on measurements of increasing fluorescence after enzyme hydrolysis, a method that utilizes decreasing fluorescence measurements was reported.46 Lozanov et al. applied 2-aminoacridone (AMAC) as a reporter group for caspase-3 substrate (Ac-DEVD-AMAC 14) and measured decrease of fluorescence intensity after enzyme cleavage.46 The catalytic parameters of caspase-3 hydrolysis point that the AMAC group was well tolerated in the P1′ position. The substrate, obtained using standard Fmoc-based solid phase peptide synthesis provides hydrolysis products that are not fluorescent therefore a background correction is not necessary. Hence, this technique gives some advantages over the commonly used gain in fluorescence ones.
Importantly, all the approaches described above constitute powerful tools to determine non-prime residues of a peptide substrate (the residues N-terminal of the scissile bond). Therefore, in order to examine prime side substrate specificity other methods has to be employed. Stennicke et al. used internally quenched fluorescence peptide substrates that overcome this limitation, to examine five caspases (−1, −3, −6, −7, −8).47 This approach is based on earlier work on fluorescence quenched substrates for collagenase48 where a fluorogenic group and a quenching group are placed on the opposite sides of the scissile bond. Hence, until protease cleavage occurs the quencher absorbs energy emitted by the donor and there is no fluoresce signal (or a very weak one). After peptide hydrolysis by the protease the quencher and fluorophores are disconnected and increased fluorescence emission is observed (Figure 3).
Figure 3.

Principle of Fluorescence-Quenched (FQ) substrates.
Stennicke et al. used anthranilic acid (ABz) and 3-nitro-tyrosine [Tyr(NO2)] as a quencher-fluorophore pair (Figure 4). Position P1′, P1 and P4 were examined for caspases −1,−3,−6,−7 and −8. The results revealed that small residues, like Gly, Ser and Ala are preferred by all caspases at P1′. Surprisingly, peptides with large aromatic residues such as Phe and Tyr were also hydrolyzed efficiently (Table 2). On the other hand, substrates with charged groups, branched aliphatic residues and proline were not well tolerated.47 The analysis of P1 and P4 position of a substrate essentially confirmed previous findings.24 This study was the first to evaluate P1 preference for a number of caspases, and S1 for all examined caspases showed preference for aspartic acid. Moreover, the selectivity toward aspartic acid in the position P1 was emphasized as substrates with glutamic acid, the next best tolerated residue, had substantially lower catalytic efficiency.47 This preference is rare among proteases, and is accounted for by strict conservation of residues Arg-179, Arg-341, and Gln-283 (caspase-1 based numbering convention) in all caspases.49
Figure 4.

Structures of FQ pairs used by Stennicke et al.47 (Abz-Tyr(NO2) and Petrassi et al.50 (MCA-DNP).
Table 2.
Table showing kcat/KM (in M−1s−1) values for the most preferred residues in position P4 in a substrate Abz-GXEVD-GVY(NO2)D and position P1′ in substrate Abz-GDEVD-XVY(NO2)D.47
| Enzyme | P4 | kcat/KM | P1′ | kcat/KM |
|---|---|---|---|---|
| Caspase-1 | Tyrosine | 75000 | Glycine | 2700 |
| Phenylalanine | 44500 | Serine | 1100 | |
|
| ||||
| Caspase-3 | Aspartic acid | 200000 | Glycine | 193000 |
| Serine | 200000 | |||
| Alanine | 157000 | |||
| Tyrosine | 63500 | |||
| Phenylalanine | 63000 | |||
|
| ||||
| Caspase-6 | Valine | 12000 | Glycine | 1100 |
| Threonine | 11000 | |||
|
| ||||
| Caspase-7 | Aspartic acid | 33000 | Glycine | 55100 |
| Serine | 33000 | |||
| Alanine | 25900 | |||
| Tyrosine | 14500 | |||
| Phenylalanine | 16600 | |||
|
| ||||
| Caspase-8 | Leucine | 83000 | Glycine | 38900 |
| Serine | 21400 | |||
Positional scanning using fluorescence quenched (FQ)-based substrate libraries represents a powerful technique to simultaneously profile prime and non-prime substrate specificity of proteases, however it has some limitations. First, it is not easy to determine location of the cleavage site. Second, a large number of substrates in such a library constitutes a problem, due to a very low concentration of each substrate – a fluorescence signal may not be observed for poor substrates. Accordingly, Petrassi et al. used a combination of two techniques, positional scanning of ACC-based substrate libraries and FRET-based substrate libraries, to determine the substrate specificity of caspase-3.50 The two-stage method consisted of a first step where the optimal non-prime sequence of a substrate was determined with a use of an ACC-based PS-SCL. In the second step, the preferred unprimed site sequence was inserted into an FQ-based substrate library to investigate the prime optimal sequence. In this study, 7-methoxycoumarin-4-acetic acid (MCA) was used as a fluorophore and N-(2,4-dinitrophenyl) (Dnp) was used as a quencher (Figure 4), revealing an optimal substrate sequence for caspase-3 of DEVD↓GGFV (P4-P1↓P1′-P4′; where ↓ indicates the site of cleavage26). Additionally, in the P1′ position, both alanine and serine were well tolerated,50 confirming earlier work.47 This method allows for fast and reliable determination of prime and non-prime substrate specificity of proteases.
Microarray techniques have been developed as another method to profile substrate specificity of proteases, allowing the use of much smaller amounts of substrate and enzyme. To investigate caspase-3 substrate preferences para-nitroanilide(PNA)-encoded libraries of rhodamine-based fluorogenic substrates were used.51 In general, fluorogenic substrates are linked through a polyethylene (PEG) spacer to PNA, which in turn is hybridized to a DNA microarray after protease treatment. Such a hybridization allows for spatial deconvolution of the substrate library and detection of optimal substrates. After cleavage of substrates by protease rhodamine yields a fluorescent signal and the most preferred sequences can be identified by the location of fluorescent spots. Wissinger et al. used a mix and split technique to synthesize a library with 192 substrates, which was evaluated with caspase-3. The results were in agreement with already determined enzyme specificity, revealing that this method constitutes another reliable techniquefor profiling substrate specificity of proteases.
In addition to these chemical diversity based methods there are also biological based diversity methods for determining protease substrate specificity. Phage display originally developed to address metalloprotease specificity,52 has been adapted to investigate caspase-3 and −8 substrate specificity.53 In this approach nucleotide sequences that encode a high diversity of amino acids are inserted into a phage genome and bacteria are infected with the phage library. Phage are immobilized by virtue of an encoded ligand binding sequence and cleaved by a particular protease. Substrate phage that are the most specific are released upon hydrolysis and several rounds of amplification and selection are employed to increase stringency of the analysis, and thus the sequences of the best substrates are defined. In the study conducted by Lien et al. a monovalent hGH-phagemid display system was used, each substrate was flanked by a variant of human growth hormone (hGH; ligand) and a truncated form of the gen III protein of M13.53 Phage was captured on a hGHbp-coated support and released by proteolysis. Results obtained from the phage display utilizing fully randomized libraries of four and six residues were generally in line with previously determined substrate specificity analyses for caspase-3 and −8 using the chemical diversity based approaches described above. However some variations in canonical motifs were discovered, it was reported that peptide DLVD was cleaved by caspase-3 up to 170% faster than peptide DEVD. Although this method may generate longer diverse peptide sequences than combinatorial chemistry approaches, making it suitable for examination of proteases that recognize long substrate sequences, the disadvantage is that non-natural amino acids cannot be placed in the peptide substrates. Moreover, to obtain kinetic values of substrate cleavage there is a need to prepare synthetic substrates to enable quantitative measurements.
Cellular libraries of peptide substrates (CLiPS) is another biology based approach used to investigate caspase substrate specificity.54 This method is analogous to the one described above, but instead of providing substrates on they are displayed on a mutated outer membrane protein X (OmpX) on the surface of bacteria. Fluorescence-activated cell sorting (FACS) is applied to find optimal substrates for a particular enzyme, enabling quantitative measurements of whole-cell fluorescence and establishment of kinetic parameters. The protease cleavage removes a fluorescent-probe peptide ligand and as a consequence reduction of cellular fluorescence is observed. A study of caspase-3 substrate specificity with CLiPS revealed DXVD↓G sequence as an optimal one, in line with previous reports.24,47 The main advantage of this technique over phage display library is a quantitative measurement of whole-cell fluorescence allowing determination of kinetic parameters without synthesis of additional substrates.
2.2. Overlapping substrate specificity – problem solved
For many years there has been a problem of substantial cross-reactivity among caspases, as even the best defined substrates do not constitute the selective ones that enable distinction between individual caspases. Commercially available peptide “specific” substrates containing natural amino acids, developed to examine the activity of caspases, lacked required specificity.55 Three studies conducted by McStay et al.,55 Pereira et al.56 and Benkowa et al.57 drew attention to this problem. It was revealed that the consensus sequences of caspases are overlapping, therefore one caspase is able to hydrolyze efficiently a substrate intended for other caspase. It is especially confusing while studying complex mixtures, such as cell lysates. Therefore such substrates should be applied only to study individual purified caspases.55,58 To illustrate this problem we summarize the results of three studies that utilized conventional or commercial caspase tetrapeptide substrates coupled to fluorophores or chromophores, demonstrating the large degree of overlap between individual caspases.55–57 These results originated from three independent research groups where various caspases and substrate concentrations were used, thus to unify this data we adopt the following symbols: ◆ indicates that the substrate is cleaved, × stands for no cleavage under the experimental conditions and - not determined. Moreover we define “substrate is hydrolyzed by caspase” when the activity is higher than 5% comparing to the best substrate from the series. For example, the preferred cleavage motif for caspase-2 is VDVAD-reporter (100%), however this enzyme also displays some activity toward LEHD-reporter (7–8%). All the data regarding caspases overlapping substrate specificity we collected in Table 3.
Table 3.
Cumulative results of three studies that examined substrate specificities of caspases (◆ indicates that the substrate is cleaved, × stands for not cleaved substrates and - not determined).
| Substrate cleavage Ac-X-X-X-Asp-reporter: AFC55, pNA56, AMAC57 |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ref. | YVAD | WEHD | VDVAD | DEVD | LEVD | VEID | IETD | LEHD | AEVD | |
| Caspase-1 | 55 | - | - | - | - | - | - | - | - | - |
| 56 | ◆ | - | × | ◆ | - | ◆ | ◆ | ◆ | - | |
| 57 | × | ◆ | × | × | ◆ | × | × | ◆ | × | |
| Caspase-2 | 55 | - | - | ◆ | × | - | × | × | ◆ | - |
| 56 | × | - | ◆ | × | × | × | × | ◆ | - | |
| 57 | × | × | ◆ | × | × | × | × | × | × | |
| Caspase-3 | 55 | - | - | ◆ | ◆ | - | ◆ | ◆ | ◆ | - |
| 56 | × | - | ◆ | ◆ | - | ◆ | × | × | - | |
| 57 | × | × | ◆ | ◆ | ◆ | ◆ | × | × | ◆ | |
| Caspase-4 | 55 | - | - | - | - | - | - | - | - | - |
| 56 | - | - | - | - | - | - | - | - | - | |
| 57 | × | ◆ | × | × | ◆ | × | × | ◆ | × | |
| Caspase-5 | 55 | - | - | - | - | - | - | - | - | - |
| 56 | - | - | - | - | - | - | - | - | - | |
| 57 | × | ◆ | × | × | ◆ | × | × | ◆ | × | |
| Caspase-6 | 55 | - | - | × | ◆ | - | ◆ | ◆ | ◆ | - |
| 56 | × | - | × | ◆ | - | ◆ | ◆ | ◆ | - | |
| 57 | × | × | × | ◆ | ◆ | ◆ | ◆ | ◆ | ◆ | |
| Caspase-7 | 55 | - | - | ◆ | ◆ | - | × | × | × | - |
| 56 | × | - | ◆ | ◆ | - | ◆ | × | × | - | |
| 57 | × | × | ◆ | ◆ | ◆ | × | × | × | ◆ | |
| Caspase-8 | 55 | - | - | × | ◆ | - | ◆ | ◆ | ◆ | - |
| 56 | × | - | × | ◆ | - | ◆ | ◆ | ◆ | - | |
| 57 | × | × | × | ◆ | ◆ | ◆ | ◆ | ◆ | ◆ | |
| Caspase-9 | 55 | - | - | × | × | - | × | ◆ | ◆ | - |
| 56 | × | - | × | ◆ | - | ◆ | ◆ | ◆ | - | |
| 57 | × | × | ◆ | ◆ | ◆ | ◆ | ◆ | ◆ | ◆ | |
| Caspase-10 | 55 | - | - | ◆ | ◆ | - | ◆ | ◆ | ◆ | - |
| 56 | - | - | - | - | - | - | - | - | - | |
| 57 | × | × | ◆ | ◆ | ◆ | ◆ | ◆ | ◆ | × | |
All three studies revealed that the examined substrates lacked useful selectivity toward individual caspase, clearly describing the problem of overlapping substrate specificity among caspases and drawing the attention to the fact that commercially available substrates containing natural amino acids are useful in studying individual purified caspases, but are not appropriate in dissecting individual caspase activity in complex mixtures, such as cell lysates. This problem was solved in 2014 by Poreba and coworkers by application of new approach called Hybrid Combinatorial Substrate Library (HyCoSuL), an extension of traditional PS-SCL method.45 The heart of this technique incorporates both natural (proteinogenic) and unnatural amino acids allowing a large amplification of chemical space and substantially increasing diversity. This allows more precise exploration of enzyme catalytic clefts and can lead to identification of more active substrates for an enzyme and, importantly, more selective substrates allowing for discrimination between closely related enzymes.
Poreba and colleagues determined specificity profiles of six recombinant human apoptotic caspases (−3,−6,−7,−8,−9,−10) by HyCoSuL screening.45 Individual caspase substrates allowing caspase discrimination were designed and kinetic parameters were determined. Subsequently, the utility of hybrid substrates was confirmed in a cell-free model of apoptosis where several caspases can be activated. The general formula of HyCoSuL employed in the study was: Ac-P4-P3-P2-Asp-ACC. In order to determine caspases preferences at S4-S2 subsites based on this scaffold three sub-libraries (P4, P3, P2) of tetrapeptidic substrates were synthesized. Each sublibrary was built from aspartic acid in P1, one position fixed with one of 129 amino acids (19 natural - cysteine was omitted and 110 unnatural amino acids) and the remaining positions contained equimolar mixture of 19 amino acids. A scheme illustrating HyCoSuL architecture and its utility in finding the most active and most selective substrates is shown in Figure 5.
Figure 5.

Scheme illustrating HyCoSuL architecture and its utility in finding the most active and most selective substrates.45
Based on the results obtained from HyCoSuL screening selective substrates that allowed for discrimination between caspases were designed and synthesized. To quantify the degree of substrate specificity we introduce here “discrimination factors” which are calculated by dividing the substrate kcat/KM for a targeted caspase by the kcat/KM for other caspases. Substrates that discriminate between caspases by factors of at least 40-fold (2.5% of activity) were identified, with the exception of caspases-3 and −7 where discriminatory substrates could not been found. In Table 4 and Figure 6 we present the structures, kinetic data and “discrimination factors” of caspase “unnatural” substrates as well as kinetic data of commercially available substrates.
Table 4.
Selectivity factors calculated for 5 fluorogenic substrates containing unnatural amino acids designed based on HyCoSuL profiling. Substrates that discriminate between caspases by factors lower than 40-fold are bolded.
| Substrate | Target | Selectivity factor (kcat/KM casp-of interest : kcat/KM casp-X)
|
|||||
|---|---|---|---|---|---|---|---|
| → Casp-3 | → Casp-6 | → Casp-7 | → Casp-8 | → Casp-9 | → Casp-10 | ||
| MPP39 15 | Casp-3 | - | 5000 | 4.5 | 120 | > 10000 | 600 |
| Casp-7 | 0.22 | 1125 | - | 27 | 2500 | 135 | |
|
| |||||||
| MPP36 16 | Casp-6 | 19.8 | - | 47.5 | 45.1 | > 10000 | 4.6 |
|
| |||||||
| MPP30 17 | Casp-8 | 65.5 | 980 | 150 | - | 73 | 53 |
|
| |||||||
| MPP8 18 | Casp-9 | > 10000 | 1070 | > 10000 | 12.4 | - | 33.5 |
|
| |||||||
| MPP52 19 | Casp-10 | 147 | 515 | 286 | 79 | 32 | - |
Figure 6.


Structures and kinetic data of specific HyCoSuL-derived caspases substrates with unnatural amino acids.45 X axis represents kcat/KM expressed in M−1s−1 × 105. Black bars - substrate activity toward caspase of interest. Grey bars - non-specific cleavage.
The usefulness of the designed hybrid specific substrates was confirmed in a consensus cell-free model of cytochrome-c programmed apoptosis, where several caspases are sequentially activated.59–61 Intact cells were avoided due to uncertainty of cellular penetration. For the first time it was possible to observe activity of individual caspases during cytochrome-c triggered apoptosis in a complex system. The results demonstrate the utility of HyCoSuL in designing selective substrates that allow for discrimination between closely related enzymes.
In addition to discovering selective substrates, HyCoSuL is also useful in discovering highly sensitive substrates, as defined by a higher activity (turnover rates) compared to previous substrates. For most of six examined caspases analyzed, substrates were found with higher activity than substrates containing natural amino acids. Caspase-10 was the exception, as the best substrate increased activity only slightly, around 20%. This substrate (MPP43 25) contained serine benzyl ester (Ser(Bzl) at P2 instead of histidine (Leu-Glu-Ser(Bzl)-Asp). For the remaining five caspases the activity was increased by 2 to 6-fold compared to substrates containing the best natural amino acids. The kcat/KM values for a new substrate designed for caspase-3 and −7 (threonine benzyl ester introduced at P2 - MPP41 26) were 2-fold higher than for the reference Ac-DEVD-ACC 20 substrate. Interestingly, glutamic acid at P3 could be replaced by many others amino acids, such as 2-thienyl Ala, with a little decrease in overall activity (compound 26a). New substrates for caspase-6 (MPP48 27, MPP49 28) were 4–5 fold better than the reference one, Ac-VEID-ACC 21. It was revealed that Val and Glu are required in positions P4, P3 respectively, but position P2 tolerates several amino acids, especially bulky ones, such as threonine benzyl ester Thr(Bzl) or homophenylalanine (hPhe). The best substrate for caspase-8 (MPP46 29) contained three unnatural amino acids, tert-leucine (Tle), homoglutamic acid (Aad) and threonine benzyl ester (Thr(Bzl)) at P4, P3 and P2, respectively, resulting in a 5.5-fold increase in kcat/KM compared to the reference Ac-IETD-ACC 22 substrate. Replacement of Glu at P3 by tert-leucine in a substrate for caspase-9 (MPP47 30) made it 3-fold better than the reference Ac-LEHD-ACC 23. The structures of sensitive substrates for six caspases with their kinetic parameters, guided by HyCoSuL, are shown below in Figure 7.
Figure 7.


The structures of new highly sensitive substrates containing unnatural amino acids for all six caspases with their kinetic parameters.45
In a search to provide acceptable discrimination between caspases-3 and −7 Vickers and coworkers presented a substrate that is selectively recognized by active caspase-3 over other apoptotic caspases and applied it to image caspase-3 activity in live cells after apoptosis induction.62 The substrate 31 employed pentapeptide recognition sequence 3Pal-D-βhLeu-F-D (termed DW3, that had been reported before63), an N-terminal cell-penetrating peptide sequence (KKKRKV) and a fluorophore/quencher pair (Cy5 and QSY21). Additionally, an alanine residue was placed at the P1′ position, resulting in higher catalytic efficiency (Figure 8). This FQ substrate 31, termed DW3-FQ, was first subjected to in vitro selectivity studies, where recombinant apoptotic caspases (−3, −6, −7, −8, −9) were used. The results demonstrated that none of caspase-6, −7, −8, −9 recognized DW3-FQ 31 substrate, only caspase-3 hydrolyzed it, while an analogous DEVD-FQ 32 substrate was cleaved by all of examined caspases. Subsequently these two substrates were used to monitor caspase activity in live cells by fluorescence microscopy. The studies on live cells confirmed that DW3-FQ 31 is selectively cleaved by caspase-3 and therefore can be used to examine the distinct role of caspase-3 in biological processes.
Figure 8.

Structure of DW3-FQ caspase substrate.62
2.3. Substrates for cell and in vivo imaging
Caspases, as central engines of apoptosis, constitute potentially useful direct markers of this process. Since many cancer chemotherapeutics are thought to act by inducing apoptosis in tumor cells, real-time in vivo imaging of the process would be invaluable in early assessment of treatment response in patients with cancer. Moreover, it would be very useful in drug development, enabling imaging of drug effect and evaluation of its efficacy. Therefore, development of sensitive, selectively acting probes for caspases is important for preclinical and clinical applications.
In this section we will discuss imaging probes targeting caspases, focusing on imaging in cell lines and in vivo. As optical imaging is less expensive and more convenient than other imaging techniques like magnetic resonance imaging or positron emission tomography, methods based on fluorescence and bioluminescence signals are the most widely utilized for protease activity imaging. Therefore, these two main imaging strategies are demonstrated. As nanotechnology is gaining more and more popularity and increased application, diverse types of nanoparticle-based activatable probes are also presented.
Fluorescence imaging activatable probes consist of three main components: a protease cleavage sequence, a fluorophore (with or without a quencher) that generates fluorescence signal upon enzyme hydrolysis, and a carrier that optimizes pharmacokinetics or enhances cell penetration of the probe. Standard activatable peptide-based probes are optically silent until protease hydrolysis occurs revealing strong fluorescence signals only after cleavage. Different types of fluorescence probes such as auto-quenched probes where a fluorophore is fixed in a position P1′ or fluorophore-quencher pair labeled probes that are internally quenched based on the resonance energy transfer. Another type represent nanoparticle-based fluorescent probes, which are finding increased applications. Nanoparticles are employed in order to improve pharmacokinetics of probes. They can incorporate good carriers that accumulate at tumor sites by the enhanced permeability and retention (EPR) effect.64–68
The bioluminescence imaging probes have become a powerful visualization tool for in vitro as well as for in vivo studies. This method presents exceptionally high signal-to-noise levels. It is based on the expression of luciferase, an enzyme that oxidizes its substrate luciferin in the presence of oxygen and ATP thereby emitting light. There are two main strategies in such imaging, luciferase reporter gene substrates can be modified to indicate caspase activity, or a luciferase reporter gene can be modified: either the substrate structure or the enzyme structure is changed. This means that when the substrate is modified it cannot be oxidized by luciferase before caspase hydrolysis. In the second situation luciferase is inactive and restores its activity after caspase cleavage. There are also probes that utilize bioluminescence resonance energy transfer (BRET).64,65
Importantly, bioluminescence imaging is more sensitive than fluorescence imaging as an external excitation light source is not required.
2.3.1. Fluorescence imaging
Fluorescence imaging is a very popular and frequently used method for imaging various processes in cell lines as well as in vivo. Weissleder et al. were the first to demonstrated in vivo optical imaging of protease activity69 and subsequently diverse imaging probes have been developed, including auto-quenched probes or fluorophore-quencher pair labeled probes. The first do not efficiently reduce a background signal, so the second - based on fluorescence resonance energy transfer - are considered to be more promising.64 It was revealed that in such probes the distance between the donor and acceptor and their relative orientation strongly influence the energy transfer efficiency. The standard distance between fluorophore and quencher in most assays is 1 to 10 nm.70 Over the past years a wide range of diverse activatable fluorescence imaging probes have been developed.
In 1999 an attempt was made to find appropriate compounds for biological studies on caspases.71 The substrates containing AMC and AFC as a fluorophore have short wavelengths, low extinction coefficients and high fluorescent backgrounds, limiting their biological application. A substrate based on a rhodamine dye, (Z-DEVD)2-R 110 33, improves these parameters71 with a longer excitation and emission wavelength and lower background, with 10-fold more sensitivity than the same substrate with AFC - Z-DEVD-AFC 34 - and a reportedly higher turnover rate. However, the general architecture of a bis-peptide resulted in two-step hydrolysis leading to a limitation inlinear dynamic range of (Z-DEVD)2-R 110 substrate. The second disadvantage of this substrate was its poor cell permeability, hence it could not be used for in vivo studies,71 but the principle of rhodamine-based substrates was sufficiently productive to entice other groups to investigate. Compounds with one caspase-cleavable amide bond, such as N-Ac-DEVD-N′-octyloxycarbonyl-R 110 35,72 N-Ac-DEVD-N′-(poly-fluorobenzoyl)-R 110 36,73 and N-Ac-DEVD-N′-morpholinecarbonyl-R 110 3774 are shown in Figure 9. All of them were cell-permeable, however only the latter exhibited higher sensitivity than the bis-peptide substrate (Z-DEVD)2-R 110 33. This substrate with a morpholine derived fluorescent dye also showed superior caspase turnover rate in solution as well as in apoptotic Jurkat cells, and thus appears to be a promising compound for biological studies on caspase activities in living cells.74 Later, Kushida et al. developed another fluorescent dye based on rhodamine, 2 Me SiR600.75 The oxygen atom of the xanthene moiety was replaced by a Si atom. This substitution resulted in red fluorescence of the compound instead of green. A substrate 38 for caspase-3 based on the core DEVD recognition sequence, equipped with novel fluorescent dye was synthesized and examined (Figure 9). It was reported that caspase-3 cleaved the substrate efficiently and it is expected that such probes will be valuable in multi-color imaging, because they provide additional a color window. Moreover, because tissues are more transparent to red than to green light, the probes have superior characteristics for in vivo imaging and appear to be more suitable for such applications.75
Figure 9.


Structures of caspase substrates equipped with rhodamine dyes.71–73,75,76
Cen et al. developed a method utilizing NucView488 as a excellent reporter group for biological studies on cells.77 It was revealed that substrate DEVD-NucView488 39 is highly cell-permeable, efficiently cleaved by caspase-3 in vitro and is very sensitive, allowing detection of caspase-3 activity in real time. These features make NucView488 suitable for examination of cell systems.77
It is worth noting that FQ substrates were also applied for cell system studies and for in vivo imaging. In one of the studies a far-red quencher QSY 21, a fluorophore Alexa Fluor 647 and a cell penetrating Tat peptide were joined with DEVD and the resulting probe (TcapQ (647) 40) was used to image apoptosis in cells.78 The Tat peptide sequence (Ac-RKKRRORRR) was incorporated into the structure in order to enhance cell penetration. TcapQ647 40 was reported to have high quenching efficiencies and a low fluorescent background. The main advantage of this probe is its far-red fluorescence, so there is a little interference from autofluorescence of cells. Later, the same probe was employed for in vivo studies.79,80 Afterwards, the same group developed a second-generation apoptosis imaging probe, KcapQ 41, with a modified cell-penetrating peptide sequence (Ac-KKKRKV).81 The TcapQ penetrating sequence contains numerous arginine residues while KcapQ penetrating sequence is lysine-rich. This change resulted in higher quenching efficiency and lower toxicity to cells.81
Another example of using FQ-based methods for measuring caspase activity is the application of a substrate containing TAMRA (5′-tetramethylrhodamine-5(6)-carboxyamide (donor) and Cy5 (acceptor) molecules (TAMRA-SSELSGDEVDSGK(SC)Cy542).82 The probe contains the caspase DEVD/S cleavage motif, which was flanked by both fluorophores. Because TAMRA was attached to the N-terminal, cell-penetrating peptides or another agents for intracellular probe uptake can be coupled to the free Cys residue that was also incorporated into peptide sequence. Such fluorogenic substrates displays several advantages: strong fluorescence signal, large FRET effect (the alteration of TAMRA/Cy5 emission ratio in response to caspase-3 cleavage was much larger than the one measured in the previous assays 83,84) and it was reported to be an efficient and selective substrate towards caspase-3 with very low KM = 1.60 ± 0.23 μM. The authors postulated that the strategy utilized in the design of this probe can be applied in real-time measurement of caspases in living cells.
Karvinen and coworkers developed homogenous time-resolved fluorescence quenching assay for caspase detection that they termed (TR-FQAs).85 The authors utilized fluorescent LANCE™ europium chelate and dabcyl as a donor-quencher pair between which a hexapeptide with the DEVD motif was inserted, revealing that TR-FQAs was far more sensitive than commercially available AFC-based assays. Additionally, this method was applied to identify caspase-3 inhibitors using the Micro Arrayed Compound Screening (μARCS) technology.86 In the μARCS format, 8,640 compounds were spotted onto a polystyrene sheet. Next, the enzyme and the substrate were cast into agarose gels separately. The assay is initiated by placing the enzyme gel on a polystyrene sheet with test compounds, followed by incubation for 10 minutes. Next, the substrate gel was placed on top of the enzyme geland after 15 min incubation the fluorescence signal was detected using a ViewLux charge-coupled device imaging system. The potential caspase-3 inhibitors appeared as a dark spots on a bright fluorescence background. The authors described this method as simple, robust and reproducible, and allowing for the screening of more than 80,000 potential caspase-3 inhibitors per 8h.
Mizukami et al. designed FQ probes utilizing dichlorofluorescein (CDCF) and luciffer yellow (LY) as donors and tetramethylrhodamine (CTMR), X-rhodamine (CXR) as acceptors, with an internal GDEVDGVK peptide sequence.87 Three different probes were synthesized, LY-GDEVDGVK-CTMR 43, CDCF-GDEVDGVK-CTMR 44, CDCF-GDEVDGVK-CXR 45, and examined on recombinant caspases (−3, −7, −6, −1). Caspase-3 and −7 cleaved them efficiently giving strong fluorescent signals, while caspase-1 and −6 displayed little activity. Subsequently the probes were used to study cell lysates and the CDCF-GDEVDGVK-CTMR 44 probe was applied in HeLa-S3 cells stimulated with etoposide, with the authors reporting imaging of enzyme activity within the cells.
Leriche and colleagues developed an interesting FQ-based probe with a chemically deactivatable quencher.88 This concept enables turn-on of fluorescence of the FQ probe by an enzymatic cleavage or by a chemical reagent (sodium dithionite), allowing the analysis of unreacted probes in cell-based experiments according to the following principle. First, enzymatic cleavage turns on the fluorescence in FQ probe that allows detection of protease activity. Next, treatment with appropriate chemical reagent deactivates the quencher in intact probes thus allowing detection of inactivated probes (Figure 10). In this study a CDQ-DEVD-G-DEAC 46 probe [where CDQ ((4-hydroxy-2-metoxy-phenylazo) benzoic acid) is the chemically deactivable quencher and DEAC (7-diethylaminocoumarin-3-carboxylic acid) is the fluorescent dye] was developed and applied to in vitro studies with recombinant caspase-3 and in cells.
Figure 10.

Schematic representation of FQ-based probe with chemically deactivatable quencher. First, enzymatic hydrolysis of the substrate turns on the fluorescence in FQ probes enabling protease activity to be detected. Next, treatment with an appropriate chemical reagent deactivates the quencher thus allowing detection of inactivated probes.88
Genetically engineered probes based on fluorescence resonance energy transfer have also developed, the most popular being CFP-YFP fusion proteins (CFP-cyan fluorescent protein, YFP-yellow fluorescent protein) with caspase cleavage sequence in between. Kawai et al. reported measuring changes in the initiator or effector caspase activity in single living cells employing two kinds of CFP-YFP probes varying in inserted caspase cleavage sequences.89 The first sequence was derived from procaspase-3, and was expected to be hydrolyzed by the initiator caspases-8, −9, and −10, while the second was from PARP, and was expected to be hydrolyzed by the effector caspases-3, −6, and −7. The authors claimed the ability to monitor activities of the initiator caspases and effector caspases, although the caveats mentioned earlier regarding the non-specificity of such sequences were not taken into account. Rehm et al. used a CFP-DEVD-YFP 47 probe to monitor the rate of caspase activation after apoptotic induction.90 Luo and coworkers conducted similar studies employing a FRET-based probe with CFP-YFP pair seeking to examine caspase-3 activation during UV-induced apoptosis91 and caspase-8 activation during TNF-α-induced apoptosis.92 Tyas et al. used CFP-DEVD-YFP 48 probe and similarly studied the timescale of caspase-3 activation.84 In another example, He and colleagues applied a CFP-LEVD-YFP 49 probe to monitor caspase activity in living cells using flow cytometry,93 while Onuki et al. developed a YFP-Bid-CFP 50 probe designed for cleavage by for caspase-8 and studied the relationship between caspase-8 activation and β-amyloid toxicity (Bid is a pro-apoptotic member of the Bcl-2 protein family and an endogenous substrate of caspase-8).94 CFP-YFP based probes were also used in studies correlating cell cycle with apoptosis in Jurkat cells95 and for drug screening.96 Mahajan and colleagues employed probes with a CFP-YFP donor-acceptor pair with caspase-1 and caspase-3 cleavage sequences to determine activity in COS-7 cells, while for in vitro studies probes with blue fluorescent protein (BFP) and green fluorescent protein (GFP) pair were utilized.97 Xu et al. used enhanced blue fluorescent protein (EBFP) and enhanced green fluorescent protein (EGFP) as a donor-acceptor pair to monitor cleavage of a caspase-3 sequencein cells.98 Brophy et al. employed a ECFP-DEVD-EYFP 51 probe (ECFP-enhanced cyan fluorescent protein, EYFP-enhanced yellow fluorescent protein) to investigate the role of the proteasome in apoptosis in COS-7 cells.99 In order to improve the FRET imaging methods, to deal with disadvantage of low brightness and autofluorescence of blue (BFP) or cyan (CFP) fluorescent proteins, Harpur et al. presented a fluorescence lifetime imaging microscopy (FLIM) approach.100 In this method the fluorescence lifetime of the combined donor/acceptor emission is determined, abrogating the need for spectral separation of the employed GFPs. Similar and bright yellow and green fluorescent proteins (EYFP/EGFP) (a pair unsuitable for FRET applications before) can be successfully employed. In the Harpur et al. study, caspase activity in individual cells during apoptosis was monitored providing evidence for the benefits of this technique in the analysis of single-cell signaling. It was revealed that this approach provides a sensitive, reproducible, and intrinsically calibrated FRET measurement. Interestingly, in 2005 a study was published where various color versions of the FRET-based probes for caspase activity were designed to expand the choice of fluorescent range.101 Six color versions were developed by a combination of cyan fluorescent protein (CFP), GFP, yellow fluorescent protein (YFP), and DsRed. It was reported that all probes could detect caspase activation following apoptotic induction. The pairs GFP-DsRed and YFP-DsRed presented similar sensitivity to CFP-YFP, and CFP-DsRed pair also demonstrated a high signal increase. Two of them, CFP-DsRed and YFP-DsRed, were chosen and used in simultaneous multi-FRET studies seeking to dissect initiator- and effector-caspase activation. Such a method, employing FRET probes in combination, would be valuable in analyzing multi-events simultaneously in single cells.
Takemoto and colleagues tried to improve probes that were based on CFP-YFP donor-acceptor pair.102 It was revealed that such probes (mainly because of acceptor properties) are highly sensitive to proton (H+) and chloride ion (Cl−) concentration, which can change during apoptosis in vivo. Thus, precise caspase activation monitoring may be hindered. Therefore, new H+ and Cl− insensitive indicators of caspase activation, called SCAT 52 (a sensor for activated caspases based on FRET), was designed. Enhanced cyan fluorescent protein (ECFP) was used as a donor and Venus as an acceptor. This is a variant of EYFP that exhibits fast and efficient maturation and is significantly less sensitive to H+ and Cl− changes. In order to monitor caspase activity a DEVD peptide sequences was inserted between donor and acceptor, forming SCAT3 53. SCAT9 54 contained LEHD sequence, cleaved mainly by caspase-9 in a purified system. With a use of this probes it was possible to monitor enzymes activity in living cells. It was reported that SCAT is highly resistant to changes in H+ and Cl− levels in vitro and insensitive to environmental effects in living cells. Subsequently, Nagai et al. examined the length of linker regions in SCAT3 53 between ECFP and Venus by use of a PCR technique in order to enhance FRET efficacy.103 Improved SCAT3.1 55 (ECFP-ΔC7-DEVD-GT-Venus) provided a 10-fold higher signal during apoptosis in examined cells than SCAT 52, what allowed visualization of caspase-3 activation with better spatial resolution than previous SCATs. Wang et al. applied SCAT3 53 to measure dynamics of caspase-3 activation in living cells during apoptosis induced by high fluence low-power laser irradiation (LPLI).76 It was revealed that with a use of this probe it was possible to observe that high fluence LPLI can induce apoptosis in human lung adenocarcinoma cells (ASTC-a-1) (according to RP Photonics Encyclopedia104,105: “In the general physics, the fluence is defined as the time-integrated flux of some radiation or particle stream”). Joseph et al. also employed SCAT3 53 as well as SCAT9 54 and SCAT8 56 to detect live cell caspase activation.106 In this study a novel high throughput platform for multiparameter apoptosis detection and high content drug screening was developed. Karaswa et al. were another group that put an effort in developing H+ concentration insensitive probe.107 Two fluorescent proteins employed as donor-acceptor pair were derived from stony coral animals, cyan fluorescent protein from Acropara sp. and orange fluorescent protein from Fungia concinna. They were called MiCy and KO, respectively. The probe containing this donor (MiCy) – acceptor (monomeric KO) pair enabled for caspase activity imaging during apoptosis in HeLa cells. Moreover, it was revealed that donor as well as acceptor mKO (monomeric KO) are completely insensitive to pH changes, constituting a new generation useful donor-acceptor pair.
Suzuki and coworkers developed several probes for caspase-3 imaging consisting of chemically engineered intramolecular fluorescence resonance energy transfer mutants of green fluorescent protein.108 The green fluorescent protein was modified in order to introduce a chemical fluorophore (near to the C-terminus) that could act as a quenching moiety. The DEVD sequence was inserted at the mobile C-terminal region of GFP, near to Cys that was used for the chemical derivatization of such construct. Several different fluorophores (eosin-5-maleimide, BODIPY 530/550, Alexa Fluor 532 and tetramethylrhodamine) were coupled to Cys to obtain a caspase sensitive FRET-based substrate. After measuring the fluorescence spectra of the probes it was revealed that the ones with eosin-5-maleimide 57 and Alexa Fluor 532 58 were the best. Therefore this two probes were applied to study cell lysates prepared from HeLa cells undergoing apoptosis to monitor caspase-3 activity. Caspase activity was demonstrated for both probes in a cell lysate system, however the protein construct with Alexa Fluor 532 58 was reported to be superior. Thus it was used to study apoptosis in HeLa cells revealing that such intramolecular-FRET-based bioprobes can be applied as sensitive indicators of caspase activity in living cells.
Wu et al. developed an expanded FRET-based probe for observing activity of two distinct caspases at the same time in living cells.109 The general architecture of a probe was CFP-YFP-mRFP (CFP-cyan fluorescent protein, YFP-yellow fluorescent protein, mRFP-monomeric red fluorescent protein). The caspase-3 preferred substrate, DEVD, was inserted between CFP and YFP, while a caspase-6 preferred substrate, VEID, between YFP and mRFP (CFP-C3-YFP-C6-mRFP). After protease hydrolysis flow cytometry was employed to reportedly distinguish activities of the two enzymes (caspase-3 and −6), allowing for separate detection of two FRET signals.109
Nicholls and coworkers constructed also interesting genetically encoded dark-to-bright activatable GFP reporter for caspase activity monitoring.110 The dark-to-bright change in direct response to enzyme hydrolysis forms another class of fluorescent probes. A version of GFP with a quenching peptide that tetramerizes GFP preventing maturation was developed with the concept that GFP fluorescence can be entirely restored by enzyme cleavage of the short quenching peptide. Caspase hydrolysis of the caspase-activatable GFP (CA-GFP 59) released GFP, which can then undergo required conformational rearrangements enabling GFP maturation and fluorescence signal emission. Caspase-activatable GFP (CA-GFP 59) can be applied for in vitro as well as for in vivo studies, and was successfully applied to monitor real-time apoptosis in live cells.
In 2012 Vuojola et al. presented a new approach for the in vitro screening of caspase-3 inhibitors, based on upconversion fluorescence energy transfer (UC-FRET).111 This study utilized lanthanide-containing inorganic nanocrystals, which have a unique ability to emit light in the visible spectrum when excitated by near-infrared wavelengths, thus eliminating autofluorescence of biological samples. The assay technique is similar to the one described below112 and relies on dual-step energy transfer, however here Black Hole Quencher 3 (BHQ-3) was used as a quencher. Its main advantage (over the BlackBerry Quencher 650) is a good stability in the presence of reducing agents (DTT). One of the benefits of the use of near-infrared excitable UCPs for caspase activity detection was also the elimination of autofluorescence thus a higher signal-to-noise ratio was obtained. The feasibility of this method was confirmed by using a Z-DEVD-CH2F (or Z-DEVD-FMK) 60 control inhibitor at varying concentrations.
Another example of using lanthanide-based reporters for caspase detection was described shortly after by the same group.111 Vuojola and coworkers designed a genetically encoded substrate 61 that contains terbium-ion-containing lanthanide-binding peptide (Tb3+-LBP) (luminescent donor complex) and green fluorescent protein (GFP) (acceptor) that flanked a peptide containing caspase cleavage site. The recombinant protein construct when excited at 280 nm was successfully used for caspase-3 activity detection. In the intact substrate the energy is transferred by FRET to the GFP acceptor, which emits light at 520 nm. Once the probe is cleaved Tb3+-LBP (donor) is released and emits luminescence at 545 nm. The use of LBP donor has the following advantages compared to conventional fluorophores: long emission lifetime enabling time-gated detection, ease of incorporation into recombinant proteins, and photobleching resistance or site-specificity of the label. Furthermore, by inserting a specific peptide linker, the method can be used to detect a wide variety of proteases.
2.3.2. Nanoparticle-based fluorescent probes
Another subgroup of imaging agents constitutes nanoparticle-based fluorescent probes, which are gaining more attention and are becoming more and more popular. Nanoparticles are excellent carriers that can substantially improve the pharmacokinetics of probes.113 This results from their small size and large surface area. Standard probes are difficult to modify because of lack of functional groups whereas nanoparticles have numerous functional sites that can be derivatized efficiently with diverse molecules. Nanoparticles-based fluorescent probes also present higher quenching efficiency and lower background signals in comparison to standard ones. In standard probes the donor-acceptor pairs appear in one-to-one combination64 whereas nanoprobes form a platform where diverse quencher-fluorophore combinations can be attached, such as multiple-to-one or multiple-to-multiple donor-acceptor pairs, which can lead to signal amplification. Their high sensitivity and accuracy make nanoparticles an excellent platform for the screening potential caspase inhibitors, because very low enzyme concentration is needed in these assays.
Lee and colleagues developed a nanoprobe based on a polymer nanoparticle platform.114 The probe is composed of dual-quenched (dye-dark quencher and dye-dye quenching mechanisms) caspase cleavable peptides, which are located on the surface of hyaluronic acid-based, self assembled polymeric nanoparticles (HA-NPs). The caspase activatable peptides consisted of Cy5.5 a NIR dye, sequence recognized by the enzyme and BHQ-3 a NIR dark-quencher specified for Cy5.5, Cy5.5-Gly-DEVDAPKGC-BHQ-3 62 (Figure 11). The probe is delivered efficiently into cells as the nanoparticles serve as good carriers. Such a probe provides fluorescence signal amplification enabling high resolution imaging of apoptotic processes in cells and in vivo as well. The nanoplatform employed in this study is flexible and can be extended to develop various specific probes.
Figure 11.

Schematic illustration of a caspase nanoprobe constructed on the basis of a polymer nanoparticle platform.114
In another example, Liu and coworkers described the development of an efficient strategy for imaging caspase activity in the central nervous system.115 To achieved this goal, the authors designed and synthesized a brain-targeted nano-device, in which a biodegradable synthetic polymer, dendrigraft poly-L-lysines (DGLs) was used as a scaffold. The FQ pair (Cy5 and QSY-21) and a nine residue caspase-3 preferred sequence were attached to the DGLs. The key challenge was to develop an efficient device to achieve brain-targeted apoptosis detection. To provide specific uptake by neurons, the brain-targeted peptide RVG29 was conjugated to DGLs (RVG29 is a peptide derived from the rabies virus glycoprotein, which is able to pass the blood-brain barrier through receptor-mediated transcytosis). Next, the DGLs-RVG29-FRET 63 nano-device was used to detect caspase activity in vivo (as a biological model male Sprague-Dawley rats were selected). Once DGLs-RVG29-FRET enters the central nervous system caspases recognizes the DEVD motif and shed the Cy5 fluorophore from the nano-device surface resulting in the rapid liberation of fluorescence. The authors conclude that before the nano-device could be applied in clinical diagnostic further studies are necessary, however this kind of tool open doors for the early diagnosis of neurodegenerative diseases.
Gold nanoparticles (AuNPs) were also employed in the design of various caspase-activatable probes.116–118 Sun and coworkers designed a simple probe 64 in which a DEVD sequence was attached to the AuNP surface and Cy5.5, a near-infrared fluorescence dye, was attached to the DEVD sequence.116 To enable the adhesion of the peptide sequence to the AuNP surface it was combined with mussel-inspired adherent peptides, 3,4-dihydroxy phenylalanines (DOPA) and lysines. The fluorescence of such a probe is quenched by gold NPs. It was reported that substrate cleavage and fluorescence signal appear very fast, thus there is no need to fix the cells for imaging. Early detection of apoptosis is possible with a use of this system. Gold nanoparticles were also involved in construction of crown nanoparticle plasmon rulers.117 Such a plasmon ruler comprises peptide-linked gold nanoparticle satellites around a core particle. The peptides crosslink the core and satellite nanoparticles via avidin-biotin interactions (Figure 12). In the Jun et al. study, plasmon rulers were employed for in vivo assays to monitor long trajectories of caspase activity at the single-molecule level. This technique appears to be a powerful tool for single-molecule imaging in live cells, as conventional single-molecule imaging techniques are limited by shorter continuous observation windows. Application of different types of plasmon rulers should allow for multicolor imaging of different signaling molecules in live cells.
Figure 12.

Schematic representation of crown nanoparticle probes.117
Lin and coworkers developed photoluminescent gold quantum dots 65 (GQDs) functionalized with NES-linker-DEVD-linker-NLS peptides [where NES-nuclear export signal and NLS-nuclear localization signal sequences, used to mimic actions of nuclear shuttle proteins].118 The probe 65 was used to monitor cellular apoptosis via cleavage of the recognition site within GQD that changes the subcellular distribution of GQD fragments. By the ratios of GQDs photoluminescence in the nucleus to that in the cytoplasm it is possible to quantify the distribution changes. With a use of this approach it was possible to monitor activation and transportation through the nuclear pore complex. Interestingly, a label-free colorimetric assay employing unmodified gold nanoparticles (AuNPs) and an unlabelled DEVD-containing peptide for the detection of caspase activity was also developed.119 Because of their unique optical properties related to surface plasmon resonance AuNPs can be successfully employed in simple colorimetric assays that do not require complicated instruments as the results can be observed by the naked eye. In the Pan et al. study an octapeptide sequence, GDEVDCCR (GR-8), was used as caspase recognition site and was attached to citrate-capped AuNPs through -SH cysteine groups, forming Au-S bonds 66. The total charge of GR-8 at pH 7.4 is negative and binding of the peptide does not induce AuNPs aggregation (because of negative charge density on the AuNPs surface). After caspase cleavage of the peptides GR-8, positively charged peptides CCR (CR-3) are formed. Their binding decreases the negative charge density on the AuNPs surface and breaks NPs electrostatic stability. As a result AuNPs aggregation occurs and the color changes from red to violet or blue and this color change can reportedly be used to detect apoptosis. This technique was validated using recombinant human caspase-3 and subsequently applied to detect apoptosis in Jurkat cells, revealing that the method was successful in discriminating apoptotic cells from normal cells. Bifunctional combined Au-Fe2O3 nanoparticles for induction of cancer cell-specific apoptosis and real-time imaging were also reported.120 On the Au surface αVβ3 integrin-targeting peptide (RGD) and fluorescein isothiocyanate (FITC)-labeled DEVD caspase recognition sequence were attached to obtain 67. These nanoparticles bind preferentially to integrin αVβ3 –rich in human liver cancer cells, and initiate formation of hydroxyl radicals that induce apoptosis, enabling monitoring of apoptosis in targeted cancer cells. Because of the Au-Fe2O3 interface polarization effect, catalytic activity of such a probe is reportedly much higher than that of individual γ-Fe2O3 NPs. As it presents simultaneous targeting, therapeutic and imaging functions this approach has great potential in future therapeutic applications in cancer.
Wang and colleagues reported graphene oxide–peptide conjugate as a sensor 68 for caspase activation imaging in live cells.121 The unique ability of graphene oxide (GO) in adsorbing biomolecules with high fluorescence quenching efficiency, forms a robust platform for biosensor development. A new intracellular sensor 68 based on the nanoconjugate of GO and peptide substrates contained a DEVD caspase recognition sequence and fluorescein amidite (FAM)-labeled lysine at the C terminus. Cell penetrating peptides TAT were fixed to the GO surface in order to improve the efficiency of intracellular delivery and endosomal escape of the GO–peptide conjugate. As GO with TAT peptides serves as a good carrier, the peptides conjugated with fluorophores are delivered inside live cells and are cleaved by active caspases to generate enhanced fluorescence due to release of fluorophores from the GO surface, enabling live cell apoptosis imaging.
Chen et al. also utilized graphene oxide to detect caspase activity however their novel approach differs from traditional fluorogenic assays. The authors described an efficient, label-free and highly sensitive electrochemical method for simple apoptosis assays.122 In this approach an acetylated peptide (Ac-GGHDEVD-HGGGC) was immobilized onto the gold electrode surface via a covalent Au-Cys bond to obtain 69. In the presence of caspase-3, the substrate 69 was hydrolyzed, which resulted in formation of two peptides: the NH2-HGGGC fragment with a free N-terminal amino group was then covalently conjugated with GO. Next, the electrochemical active molecule - methylene blue (MB) was attached to an electrode through the π-π stacking and electrostatic interaction between GO and MB. Finally the electrochemical signal, obtained due to methylene blue redox reaction, was measured. The method was successfully applied to detect apoptosis in human the pulmonary carcinoma A549 cell line. One of the key merits of this approach is that only 0.06 pg/ml (low detection limit) of caspase-3 is needed to obtain an accurate electrochemical signal, making it very sensitive.
Another example of using immobilized caspase-3 peptide substrates on the gold surface was described by Hung et al..123 The authors designed an electrochemical impedance spectroscopy (ESI)-based biosensor 70 to measure caspase activity in biological samples. In this approach a caspase-3 recognition peptide (GDGDEVDGC) was covalently attached to the surface of screen-printed gold electrodes (SPGE) using N-hydroxysuccinimide-activated lipoic acid esters. Once the GDGDEVD peptide was shed from the surface, the changes in the immobilized peptide film was measured using apparent charge transfer resistance (RAPP) thus revealing proteolytic activity. The feasibility of this approach was confirmed on apoptotic human SH-SY5Y neuroblastoma cell lysates (only 2 μL sample was used). This method appears as a useful tool for rapid and cost-effective screening of caspase activity and can be adapted in various apoptotic cell lysates.
Kihara and coworkers developed another interesting system to evaluate caspase activity using a nanoneedle and a fluorescent probe.124 Importantly, using nanoneedles smaller than 400 nm in diameter penetration do not induce lethal damage to the plasma membrane and such cantilevers can be kept inside the cell for more than 1h due to their low invasiveness.125 A new FRET probe (NHGcas546 71) was constructed and fixed to a nanoneedle. The probe was composed of an engineered GFP (donor) with an N-terminal (His)6 tag, DEVD peptide sequence, and a cysteine for site-directed modification with Alexa Fluor 546 dye (acceptor) at its C-terminus. It was attached to the nanoneedle by chelate bonding of the (His)6 tag, and the derivatized nanoneedle was inserted into apoptotic cells, with the expectation that the probe would react with caspase inside the cell. Successful detection of caspase activity in a single cell using NHGcas546 71 probe immobilized on the nanoneedle was presented.124 This new practical approach is called MOlecular MEter with Nanoneedle Technology (MOMENT), and has food potential to be expanded and monitor various intracellular phenomena.
Boeneman et al. presented the use of a hybrid fluorescent protein semiconductor quantum dot (QD) sensor 72 to monitor caspase activity.126 Monomeric red fluorescent mCherry protein was modified to express a caspase cleavage site and a polyhistidine sequence His6. The sequence was self-assembled to the surface of CdSe-ZnS dihydrolipoic acid (DHLA)-funtionalized QDs via metal-affinity coordination, resulting in a sensitive FRET-based protease sensor, where quenching of the QD and sensitized emission from mCherry acceptor was observed. Caspase cleavage reduces the FRET efficiency allowing monitoring enzyme activity. The QD serves in such bioconjugates as both a central nanoscaffold and an excitation donor.
Valanne and coworkers developed a novel dual-step fluorescence resonance energy transfer-based assay method for screening potential inhibitors of caspase-3.112 In this assay europium(III)-chelated-doped nanoparticles (coated with streptavidin) were conjugated with a biotinylated consensus caspase-3 substrate equipped with Alexa Fluor 680 (fluorescent acceptor) on the N-termini and BlackBerry Quencher 650 on the C-termini. In this assay europium(III)-chelated-doped nanoparticle 73 is excited at 340 nm and the energy is transferred onto Alexa Fluor 680 acceptor. In inhibitory conditions (substrate is intact) the close inherence of BlackBerry Quencher 650 attenuates the energy so no fluorescence signal is observed. However, once caspase-3 cleaves the peptide, quencher is released and the Alexa Fluor 680 produces the fluorescence (730 nm). This method provides a sensitized fluorescence signal proportional to the enzyme activity with very low background of complete enzyme inhibition. Furthermore, the europium-chelate-doped nanoparticle improve the generality and cost-efficiency of the method. The results obtained using this assay was in line with those obtained by other methods described in the literature.71,127 The authors also mentioned same weakness of their dual-step FRET techniques such as moderate stability of the labels in the presence of reducing agents or the large size of nanoparticles (smaller particles produce weaker fluorescence background).
2.3.3. Bioluminescence imaging probes
Bioluminescence imaging appears to be more sensitive than fluorescence imaging as external excitation light source is not involved, yielding exceptionally high signal-to-noise levels making it a powerful visualization tool for in vitro as well as for in vivo studies. Based on expression of luciferase, enzyme that oxidizes its substrate luciferin in the presence of oxygen and ATP to emit light,64 bioluminescence imaging employs one of two strategies. The substrate is modified and it cannot be oxidized by luciferase until a protease hydrolyses it; or the enzyme is modified, it is inactive and restores its activity only after protease cleavage. Probes have also been developed to utilize bioluminescence resonance energy transfer (BRET). In such probes luminescence is quenched by a fluorescent acceptor protein and between these two nonradiative transfer of energy occurs.128 As long as they are connected there is no change in BRET signal. The first successfully demonstrated BRET system used Renilla luciferase (Rluc) as the donor and an enhanced yellow fluorescent protein (EYFP) as the acceptor.129 Later, new pairs were developed, the commercial BRET system (BRET2) employed Rluc as a donor, a highly blue-shifted phenylcoelenterazine as its substrate and green fluorescent protein (GFP) as an acceptor. It is worth noting that BRET technique may be applied to study protein-protein interactions in living cells.130–133 Bioluminescence imaging is employed in different kinds of assays where diverse strategies are applied.
Angers and coworkers presented a BRET technique in cells using Renilla luciferase (Rluc) as a donor and yellow fluorescent protein (YFP) as an acceptor linked through a peptide sequence recognized by caspase-3 with the general architecture Rluc-DEVD-YFP 74.134 After caspase activity stimulation in cells by staurosporine, a change in BRET ratio was observed indicating that the designed protein was cleaved and the donor-acceptor pair was disconnected. The specificity of the effect was confirmed by using a caspase inhibitor, revealing that BRET is appropriate to monitor dynamic processes in living cells.
In 2009 Gammon and colleagues examined a new BRET systems employing caspases studies.135 A series of novel BRET pairs based on luciferases that oxidize D-luciferin (instead of the coelenterazine derivative) were reported. The choice of luciferase that utilizes another substrate resulted in favorable biochemical attributes red-shifted photonic outputs, and increased efficacy. Such red-shifted BRET pairs are especially useful for in vivo imaging as tissues are more transparent to red than to green light. BRET systems measuring blue to green color ratios limit in vivo imaging to superficial structures as the tissues absorb and scatter light at such wavelengths. In this study click beetle green luciferase (CBG) was employed as a donor (due to the low cost and favorable pharmacokinetics of the substrate) and tdTomato was chosen as the optimal red fluorophore acceptor, and a DEVD caspase recognition sequence was inserted between CBG and tdTomato. The fusion protein 75 was examined in vitro on recombinant caspases as well as in cellulo. High signal-to-noise ratios and Z′ factors were reported, rendering it useful tool for imaging of caspase activity on both short (minutes) and long (days) time scale. It is also worth noting that an issue concerning peptide-linker length was raised in this study. It was revealed that in order to achieve a maximal change in BRET signal after protease hydrolysis the peptide linker should be as short as possible because the fluorophore acceptor has to be as close as possible to the active site of luciferase. Nevertheless, one has to be aware that such a construction may influence the structures of donor and acceptor and reduce protease activation.
One of the bioluminescence strategies forimaging of caspases uses a caspase-recognition peptide sequence with aminoluciferin attached in position P1′ and firefly luciferase. Protease cleavage releases aminoluciferin, which is oxidized by luciferase and light is emitted. Aminoluciferin linked with a peptide does not constitute a substrate for luciferase, only after proteolysis is an appropriate luciferase substrate produced (Figure 13).
Figure 13.

Schematic representation of the luminescent caspase assay.
The Promega Corporation developed and commercialized a caspase substrate that contains aminoluciferin in P1′, Z-DEVD-aminoluciferin 76 (VivoGlo™ Caspase-3/7 Substrate). This approach was applied in several studies for examining cell systems136,137 and in vivo studies.138–141 O’Brien explored this bioluminescent imaging technique on caspase-3 to define sensitivity, speed and stability of the signal136 revealing that this method has lower background levels and higher sensitivity than available fluorescent assays. Additionally, it is notably faster. In vivo studies138–141 reported that it is possible to follow apoptosis induction by caspase activation with a use of Z-DEVD-aminoluciferin 76 and demonstrate that this approach could perhaps be used to validate drug efficacy during chemotherapy and aid in drug discovery drug discovery and development.
In 2010 another bioluminescent substrate was developed and employed for biological studies on cell lysates, in order to monitor caspase-1 activity.142 In this study, an activity-based probe (ABP) for the enzyme was designed by the Reverse Design concept. The caspase-1 inhibitor, Pralnacasan (or VX-74077), was converted into selective substrate ABP, CM-269 78. The inhibitor warhead was replaced by a peptide bond and amino-luciferin. It was reported that CM-269 78 revealed very high selectivity and sensitivity for caspase-1. Thus, this method constitutes a powerful tool in studies of complex proteomic samples, such as cell lysates.
Bittner et al. developed a strategy for dual-analyte bioluminescence detection in vivo.143 This method enables visualization of two different biochemical processes by employing a pair of caged complementary luciferin precursors, which are unmasked by processes that leads to in situ luciferin formation. Luciferin is formed and bioluminescence signals may be observed only if both biochemical processes occur. In this study simultaneous presence of H2O2 and caspase activity was examined. A peroxy Caged Luciferin-2 (PCL-2) probe 79 that releases 6-hydroxy-2-cyanobenzothiazole (HCBT) upon reacting with H2O2 and a peptide based probe, Z-IETD-D-Cys 80, that releases D-cysteine after caspase cleavage were developed. Only if the two events take place (HCBT and D-cysteine are released) does in situ luciferin formation occur and bioluminescence may be observed (Figure 14). Therefore the two probes form an AND-type molecular logic gate. Employing this new chemical tool it is possible to study simultaneous oxidative stress and inflammation processes in vivo. Moreover, this approach appears to be versatile for concurrent monitoring of various biochemical processes (as diverse probes can be utilized). An in situ luciferin formation method was also used in a study where caspase-3/7 activity was reportedly monitored.144
Figure 14.

Strategy of dual-analyte detection that employs in situ Luciferin formation from two caged precursors.143
The second bioluminescence strategy utilizes the modification of a luciferase reporter gene to respond to caspases. Laxman et al. constructed a reporter gene containing firefly luciferase gene, a DEVD linker (protease cleavage site) and on the opposite site estrogen receptor (281–599 of the modified mouse estrogen receptor sequence).145 Fusion with the estrogen receptor regulatory domain silenced firefly luciferase activity. After caspase cleavage luciferase is separated from the silencing domain and its activity is restored and can be detected with bioluminescence imaging in cell lines and in vivo.145 Kanno and colleagues engineered a genetically encoded cyclic luciferase with an inserted DEVD substrate sequence for caspase-3.146 In such a recombinant circularized molecule 81 luciferase loses its bioluminescence activity, and activity is restored after caspase hydrolysis. This cyclic luciferase was employed for quantitative detection of DEVD-ase activity in cells and in vivo.146–148 Another bioluminescent system 82 was developed by Coppola and coworkers149 who split luciferase into NLuc and CLuc fragments with each linked to one of two high affinity peptides: peptide A and peptide B. Between these two fragments, pepANLuc and pepBCLuc, a DEVD sequence was incorporated. Caspase cleavage results in restoration of luciferase activity because interaction of the two peptides leads to NLuc and CLuc complementation.
Ray and colleagues created a more complicated system for bioluminescence imaging of caspase activity.150 A fusion protein that combined three different reporter proteins was developed. Red fluorescent protein (mRFP1), firefly luciferase (FL), and HSV1-sr39 truncated thymidine kinase (TK), linked through a sequence recognized by caspase-3 were combined. Activity of all three proteins was silenced in the fused form and after caspase hydrolysis significant signal increase of mRFP1, FL, and TK activity was reported. With this approach caspase activity can be monitored effectively and noninvasively ranging from single live cells to a multicellular tumor environment. In an additional example, Shah et al. employed dual substrate/reporter bioluminescence imaging (Fluc: firefly luciferase– luciferin and Rluc: Renilla luciferase – coelenterazine) that enabled monitoring in real time both gene delivery and efficacy of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in tumors in vivo.151
In summary, over the past years a wide range of diverse activable probes for caspase activity imaging in cellulo and in vivo was developed, most of them based either on fluorescence or bioluminescence measurements. The probes based on nanoparticles are becoming more and more popular and are applied increasingly. Although there appeared some other methods to image caspase activity such as electrochemical detection using ferrocene-labeled peptide152 or methods based on chemiluminescence-resonance-energy transfer (CRET)153 the methods presented above based on fluorescence and on bioluminescence measurements constitute the most popular ones. A substantial problem is that most of existing probes have poor specificity and sensitivity limiting their imaging ability and applications. Furthermore, some probes have low cell permeability thus decreasing the possibility of reaching cytosolic target molecules. Although huge progress has been made in imaging of caspase activity, these issues that have to be addressed to enhance specificity and delivery to appropriate sites of caspase activation
2.4. Future perspectives in substrates design
A good probe for imaging intracellular (cytosolic) enzymes should have four main features, it should interact specifically with the examined target, have low background signals, high output signals (high signal-to-noise ratio) and should be cell permeable (for in cellulo, in vivo studies). As one may notice, a huge number of probes for caspase activity imaging in cellulo and in vivo have been developed, but most of them do not satisfy the main requirements. Almost all the probes described above lack selectivity, some have problems with cell permeability, and some of them need an improved signal-to-background ratio. The broad study conducted by Poreba and colleagues resulted in substantial advance in the problem of overlapping specificities of the caspases and enabled to distinguish between them. Therefore probes for in vivo studies designed on the basis of these hybrid natural/unnatural peptide sequences should act far more selectively and should give more reliable results for individual caspases. There is need to work on a carrier that improves the cellular uptake of the probe. Solutions presented in this review could be employed in creating new specific probes, for example cell penetrating Tat peptide Ac-RKKRRORRR or the modified lysine-rich Ac-KKKRKV should enhance cell penetration of the probe. Probes based on nanoparticles constitute a promising set of versatile imaging tools and are becoming more and more popular as nanpoparticles are demonstrated to serve as good carriers. In standard probes the donor-acceptor pairs appear in a one-to-one combination, so the versatility of NPs may enable delivery of probes to the target site with a by employing different cellular uptake mechanisms and may enhance signal-to background ratios thus improving detection sensitivity.
All things considered, the combination of specific caspases peptide sequences with systems that improve cellular uptake and enhance signal-to background ratio may lead to development of important probes enabling the monitoring activity of one chosen caspase. Such probes will allow for even more precise determination of the roles of individual enzymes in diverse biological processes and should constitute invaluable tools in clinical applications. Real-time in vivo imaging of the process of apoptosis would be invaluable in early assessment of the treatment response in patients undergoing chemotherapy and would help to establish the utility of therapeutic treatments tailored for individual patents. Furthermore, it would be very useful in drug development, enabling imaging of a drug effect and evaluation of its efficacy.
3. CASPASE INHIBITORS
3.1. Introduction
Caspases are cysteine proteases that have an preference for aspartic acid in P1 position of their substrates. Attempts have been made to differentiate caspases within the family by inhibitors containing an appropriate sequence on the left to the scissile bond (non prime site) and a specific electrophilic warhead that can bind to the prime region of caspases active site. The P1′ region, known also as cysteine trap, has been extensively studied with numerous of different functional groups. The main goal of this area of research is to design and synthesize caspase-specific inhibitors with good ADMET parameters while maintaining high potency toward targeted caspase. Since the discovery of caspase-129,154 and caspase-3155–157 various types of inhibitors have been described in the literature including peptides, peptidomimetics and small molecule, nonpeptidic compounds. Most caspases inhibitors use their electrophilic warhead to covalently modify the catalytic cysteine residue that leads to enzyme inactivation.158 The entire collection of chemical caspases inhibitors can be assigned as reversible (if the enzyme is inactivated through formation of reversible thiohemiketal) or irreversible (if the enzyme is permanently inactivated via formation of thioether complex) (Figure 15).23,159 Some compounds display a bimodal pattern of inhibition, with the thiohemiketal slowly transformed into a thioether adduct.160,161 In this review we do not consider naturally-occurring protein inhibitors of caspases, which often have entirely different inhibitory mechanisms (see4 for a review).
Figure 15.

Two possible ways of caspases inactivation – reversible and irreversible. For kobs calculation a Kitz-Wilson equation was used.162 LG is a leaving group.
3.2. Structural determinants of caspases inhibition
A detailed knowledge of caspase specificity is invaluable for the design and development of potent and selective inhibitors. Caspases substrate/inhibitor profiles can be acquired form substrate combinatorial library screening, inhibitory SAR studies or caspase crystal structure analysis. Thornberry and coworkers divided human caspases into three distinct groups: group I exhibiting preferences for substrates containing WEHD motif (caspases -1, -4, -5), group II with a preference for DExD (caspases -2, -3, and -7) and group III with a preference for (V/L)ExD (caspases -6, -8, -9, and -10)24, and as discussed above this analysis has stood the test of time. All these caspases display a strong preference for aspartic acid at P1 position of their substrates or inhibitors and all recognize at least four amino acids on the left to the cleavage site.29,163 These specificities profiles are important to the apoptotic cascade and inflammatory cell death, where certain groups of proteins are cleaved by ordered proteolysis rather than general protein degradation.164 To date numerous caspase crystal structures in complex with peptide and peptidomimetic inhibitors have been solved and deposited in RCSB protein data bank (Table 5–11). Many of the commonly used inhibitors contain peptide sequences derived from natural substrates. For instance caspase-1 have been co-crystallized with Ac-YVAD-CHO 4165 (sequence derived from cleavage motif of Il-1β), while caspase-3 have been bound with Ac-DEVD-CHO 83166 (sequence derived from cleavage motif of poly(ADP-ribose)polymerase) and Ac-DVAD-CH2F 84167. Sometimes, to investigate similarities and differences in inhibitor recognition processes and to study possible cross-reactivity within the caspases family, these enzymes have been co-crystallized with non-naturally occurring or non-optimal sequences. Below we present an analysis based on the current knowledge of structural determinants for caspases inhibition, highlighting the most conservative patterns common for all caspases as well as differences discriminating caspases within a family.
Table 5.
Multiple examples of x-ray crystal structures of caspase-1 adapted from PDB database.
| CASPASE-1 | |||||
|---|---|---|---|---|---|
|
| |||||
| PDB entry | Mutation | Res. | Enzyme info | Ligand | Ref. |
| inhibitor free | |||||
| 3E4C | C285A | 2.05 | procaspase 1 zymogen domain(UNP residues 104-404) | ligand free (only Mg2+ ion) | 165 |
| 1SC1 | C285A | 2.60 | p10 and p20 subunits | ligand free (only Cl− ion) | 181 |
| 1SC4 | C285A | 2.10 | p10 and p20 subunits | ligand free (after removal of malonate ion – see 1SC3) | 181 |
|
| |||||
| with inhibitors | |||||
| 1SC3 | C285A | 1.80 | p10 and p20 subunits |
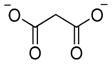 malonate ion (2M sodium salt) (reversible bounding in the caspase-1 S1 pocket) |
181 |
| 1IBC | D381A | 2.73 | chain A – length 194 chain B – length 88 |
Ac-WEHD-CHO (covalent, reversible, peptide inhibitor) |
22 |
| 1ICE | wild type | 2.60 | chain A – length 167 chain B – length 88 |
Ac-YVAD-CHO (covalent, reversible, peptide inhibitor) |
182 |
| 3NS7 | wild type | 2.60 | chain A (res.136-297) chain B (res. 317-404) |
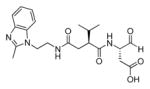 (covalent, reversible, peptidomimetic aldehyde inhibitor) |
183 |
| 1BMQ | wild type | 2.50 | chain A – length 167 chain B – length 88 |
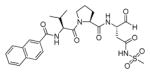 (covalent, reversible, peptidomimetic aldehyde inhibitor) |
184 |
| 1RWM | wild type | 2.70 | p10 and p20 subunits(N120-D297 and A317-H404) |
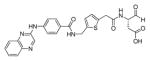 (covalent, reversible, peptidomimetic aldehyde inhibitor) |
185 |
| 1RWN | wild type | 2.00 | p10 and p20 subunits(N120-D297 and A317-H404) |
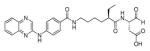 (covalent, reversible, peptidomimetic aldehyde inhibitor) |
185 |
| 1RWO | wild type | 2.10 | p10 and p20 subunits(N120-D297 and A317-H404) |
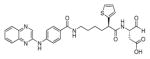 (covalent, reversible, peptidomimetic aldehyde inhibitor) |
185 |
| 1RWP | wild type | 2.20 | p10 and p20 subunits(N120-D297 and A317-H404) |
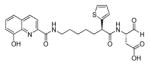 (covalent, reversible, peptidomimetic aldehyde inhibitor) |
185 |
| 1RWV | wild type | 2.10 | p10 and p20 subunits |
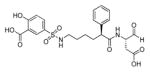 (covalent, reversible, peptidomimetic aldehydeinhibitor) |
186 |
| 1RWW | wild type | 2.80 | p10 and p20 subunits |
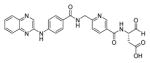 (covalent, reversible, peptidomimeticaldehydeinhibitor) |
187 |
| 1RWX | wild type | 1.85 | p10 and p20 subunits |
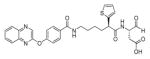 (covalent, reversible, peptidomimetic aldehyde inhibitor) |
187 |
| 2HBQ | wild type | 1.80 | p20 subunit (120-297), and p10 subunit(317-404) |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
188 |
| 2HBR | R286A | 2.20 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
188 |
| 2HBY | E390A | 2.10 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
188 |
| 2HBZ | R286A, E390A | 1.90 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
188 |
| 2H48 | C362A, C364A, C397A | 2.20 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
188 |
| 2H4W | E390D | 2.00 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
189 |
| 2H4Y | R286K | 1.90 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
189 |
| 2H51 | E390D, R286K | 2.10 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
189 |
| 2H54 | T388A | 1.80 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
189 |
| 3D6F | R240Q | 1.90 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
190 |
| 3D6H | N263S | 2.00 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
190 |
| 3D6M | K319R | 1.80 | p10 and p20 subunits |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
190 |
| 1RWK | wild type | 2.30 | p10 and p20 subunits(N120-D297 and A317-H404) |
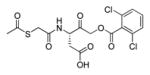 (covalent, irreversible, peptidomimetic inhibitor; AOMK) |
185 |
| 2FQQ | C285A, C362A, C364A, C397A | 3.30 | p10 and p20 subunits |
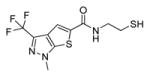 (covalent, allosteric inhibitor – Cys331) |
188 |
Table 11.
Multiple examples of x-ray crystal structures of caspase-3 adapted from PDB database.
| CASPASE-3 | |||||
|---|---|---|---|---|---|
|
| |||||
| PDB entry | Mutation | Res. | Enzyme info | Ligand | Ref. |
| inhibitors free | |||||
|
| |||||
| 1QX3 | wild type | 1.90 | casp-3 p17/p12 subunits (length 257) | ligand-free | 229 |
| 4JQY | C163A | 2.49 | procasp-3 (UNP residues 34–277) | ligand-free | 227 |
| 4JQZ | C163A | 2.89 | procasp-3 (UNP residues 34–277) | ligand-free | 227 |
| with inhibitors | |||||
|
| |||||
| 1I3O | C285A | 2.70 | casp-3 p17 subunit and casp-3 p12 subunit |
second baculoviral IAP repeat of XIAP (XIAP-BIR2, protein inhibitor) |
230 |
| 2XZD | - | 2.10 | casp-3 p17 subunit and casp-3 p12 subunit |
DARPIN-3.4 (competitive protein inhibitor, residues 1–136) |
231 |
| 2Y0B | - | 2.10 | casp-3 p17 subunit and casp-3 p12 subunit |
DARPIN-3.4_S76R (competitive protein inhibitor, residues 1–136) |
231 |
| 1PAU | wild type | 2.50 | casp-3: chain A (length 147) and chain B (length 102) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
166 |
| 2H5I | wild type | 1.69 | p17 subunit (34–174) and p12 subunit (186–277) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
232 |
| 2H5J | wild type | 2.00 | p17 subunit (34–174) and p12 subunit (186–277) |
Ac-DMQD-CHO (covalent, reversible, peptide inhibitor) |
232 |
| 2H65 | wild type | 2.30 | p17 subunit (34–174) and p12 subunit (186–277) |
Ac-VDVAD-CHO (covalent, reversible, peptide inhibitor) |
232 |
| 3EDQ | wild type | 1.61 | chains A,C (UNP residues 29–175), chains B, D (UNP residues 176–283) |
Ac-LDESD-CHO (covalent, reversible, peptide inhibitor) |
179 |
| 3GJQ | wild type | 2.60 | p17 subunit (length 147) and p12 subunit (length 108) |
Ac-WEHD-CHO (covalent, reversible, peptide inhibitor) |
233 |
| 3GJS | wild type | 1.90 | p17 subunit (length 147) and p12 subunit (length 108) |
Ac-YVAD-CHO (covalent, reversible, peptide inhibitor) |
233 |
| 3GJT | wild type | 2.20 | p17 subunit (length 147) and p12 subunit (length 108) |
Ac-IEPD-CHO (covalent, reversible, peptide inhibitor) |
233 |
| 4DCJ | L168D | 1.70 | p17 subunit (length 147) and p12 subunit (length 108) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
234 |
| 4DCO | L168Y | 1.70 | p17 subunit (length 147) and p12 subunit (length 108) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
234 |
| 4DCP | L168F | 1.70 | p17 subunit (length 147) and p12 subunit (length 108) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
234 |
| 1RE1 | wild type | 2.50 | p17 and p12 subunits |
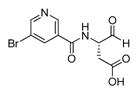 (reversible nicotinic acid aldehyde inhibitor) |
235 |
| 1RHM | wild type | 2.50 | p17 and p12 subunits |
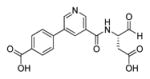 (reversible nicotinic acid aldehyde inhibitor) |
235 |
| 1CP3 | wild type | 2.30 | p17 subunit (35–173) and p12 subunit (185–277) |
Ac-DVAD-CH2F (covalent, irreversible, peptide inhibitor) |
167 |
| 3GJR | wild type | 2.20 | p17 subunit (length 147) and p12 subunit (length 108) |
Ac-D(Me)-CH2F (covalent, irreversible, peptide inhibitor) |
233 |
| 2DKO | D175A | 1.06 | p17 subunit (29–174) and p12 subunit (175–277) |
Z-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
177 |
| 2CJX | D175A | 1.70 | p17 subunit (29–175) and p12 subunit (176–277) |
Z-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
177 |
| 2CJY | wild type | 1.67 | p17 subunit (29–175) and p12 subunit (176–277) |
Z-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
177 |
| 2J30 | wild type | 1.40 | casp-3 residues 29–277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
236 |
| 2J31 | E167A | 1.50 | casp-3 residues 29–277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
236 |
| 2J32 | E173A | 1.30 | casp-3 residues 29–277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
236 |
| 2J33 | Y203F | 2.00 | casp-3 residues 29–277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
236 |
| 3ITN | V266E | 1.63 | casp-3 length 250 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
42 |
| 3PCX | E246AK242A | 1.50 | casp-3 (UNP residues 29–277) |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
237 |
| 3PD1 | K242A | 1.62 | casp-3 (UNP residues 29–277) |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
237 |
| 3PD0 | E246A | 2.00 | casp-3 (UNP residues 29–277) |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
237 |
| 4EHA | V266H | 1.70 | casp-3 length 277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
238 |
| 4EHD | Y193C | 1.58 | casp-3 length 277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
238 |
| 4EHF | Y193CV266H | 1.65 | casp-3 length 277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
238 |
| 4EHH | E124A | 1.78 | casp-3 length 277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
238 |
| 4EHK | E124AY197C | 1.67 | casp-3 length 277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
238 |
| 4EHL | E124AV266H | 1.80 | casp-3 length 277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
238 |
| 4EHN | E124AY197CV266H | 1.69 | casp-3 length 277 |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
238 |
| 4JR0 | D175A | 1.80 | procasp-3 (UNP residues 34–277) |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
227 |
| 2XYG | wild type | 1.54 | p17 subunit (res 29–174) and p12 subunit (res 185–277) |
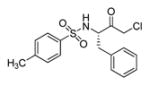 (active site inhibitor without P1 Asp moiety; -CH2Cl) |
239 |
| 2XYH | wild type | 1.89 | p17 subunit (res 29–174) and p12 subunit (res 185–277) |
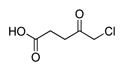 (active site inhibitor; -CH2Cl) |
239 |
| 2XYP | wild type | 1.86 | p17 subunit (res 29–174) and p12 subunit (res 185–277) |
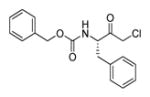 (active site inhibitor without P1 Asp moiety; -CH2Cl) |
239 |
| 1NMS | wild type | 1.70 | large subunit (p17) |
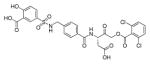 (irreversible, peptidomimetic inhibitor; AOMK) |
240 |
| 1NMQ | wild type | 1.60 | large subunit (p17) |
(irreversible, peptidomimetic inhibitor; AOMK) |
240 |
| 1NME | wild type | 2.40 | large subunit (p17) and small subunit (p12) |
(irreversible, peptidomimetic inhibitor; AOMK) |
240 |
| 3KJF | wild type | 2.00 | chain A (length 147) and casp-3 chain B (length 109) |
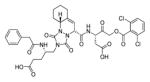 (irreversible, peptidomimetic inhibitor; AOMK) |
211 |
| 4JJE | wild type | 1.48 | casp-3 (length 257) |
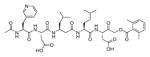 (irreversible unnatural inhibitor; AOMK) |
20 |
| 2C1E | wild type | 1.77 | p17 subunit (29–175) and p12 subunit (176–277) |
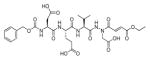 (irreversible, aza-peptide inhibitor – Michael acceptor; trans) |
212 |
| 2C2K | wild type | 1.87 | p17 subunit (29–175) and p12 subunit (176–277) |
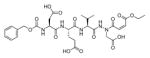 (irreversible, aza-peptide inhibitor – Michael acceptor; cis) |
212 |
| 2C2M | wild type | 1.94 | p17 subunit (29–175) and p12 subunit (176–277) |
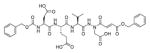 (irreversible, aza-peptide inhibitor – Michael acceptor) |
212 |
| 2C2O | wild type | 2.45 | p17 subunit (29–175) and p12 subunit (176–277) |
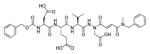 (irreversible, aza-peptide inhibitor – Michael acceptor) |
212 |
| 2CNK | wild type | 1.75 | p17 subunit (29–175) and p12 subunit (176–277) |
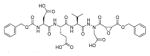 (irreversible, aza-peptide epoxide inhibitor – Michael acceptor) |
168 |
| 2CNL | wild type | 1.67 | p17 subunit (29–175) and p12 subunit (176–277) |
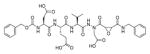 (irreversible, aza-peptide epoxide inhibitor – Michael acceptor) |
168 |
| 2CNN | wild type | 1.70 | p17 subunit (29–175) and p12 subunit (176–277) |
(irreversible, aza-peptide epoxide inhibitor – Michael acceptor) |
168 |
| 2CNO | wild type | 1.95 | p17 subunit (29–175) and p12 subunit (176–277) |
(irreversible, aza-peptide epoxide inhibitor – Michael acceptor) |
168 |
| 2CDR | wild type | 1.70 | p17 subunit (29–175) and p12 subunit (176–277) |
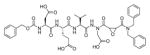 (irreversible, aza-peptide epoxide inhibitor – Michael acceptor) |
168 |
| 1RHJ | wild type | 2.20 | p17 and p12 subunits |
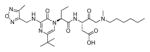 (reversible inhibitor) |
235 |
| 1RHK | wild type | 2.50 | p17 and p12 subunits |
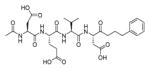 (reversible peptide inhibitor) |
235 |
| 1RHQ | wild type | 3.00 | p17 and p12 subunits |
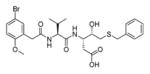 (reversible peptide inhibitor) |
235 |
| 1RHR | wild type | 3.00 | p17 and p12 subunits |
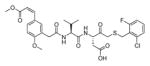 (reversible peptide inhibitor) |
235 |
| 1RHU | wild type | 2.51 | p17 and p12 subunits |
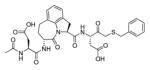 (covalent, irreversible peptide inhibitor) |
235 |
| 3DEH | wild type | 2.50 | casp-3 (length 249) |
 (irreversible, non-peptide inhibitor) |
241 |
| 3DEI | wild type | 2.80 | casp-3 (length 249) |
 (irreversible, non-peptide inhibitor) |
241 |
| 3DEJ | wild type | 2.60 | casp-3 (length 249) |
 (irreversible, non-peptide inhibitor) |
241 |
| 3DEK | wild type | 2.40 | casp-3 (length 249) |
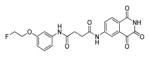 (irreversible, non-peptide inhibitor) |
241 |
| 3H0E | wild type | 2.00 | casp-3 p12 and p17 subunits; total length 255 |
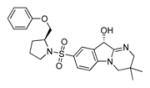 (covalent, reversible, non-peptidic inhibitor) |
242 |
| 1GFW | wild type | 2.80 | p20 subunit (29–175) and casp-3p10 subunit (181–277) |
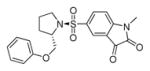 (reversible, non-peptide inhibitor) |
243 |
We focus on the structural recognition pattern of caspases -1, -3 (and -7) and -8 spanning three different groups of caspases and preferring different cleavage motifs (YVAD, DEVD and LETD respectively).24 The crystal structures of these caspases with caaspase-3 preferred Ac-DEVD-CHO 83 inhibitor show that although different side chains are involved in inhibitor binding, the overall organization of inhibitor binding is highly conserved.
At the P1′ position (referred to also as the cysteine trap) several different warheads have been tested among which aldehydes and activated ketones are of special interest. In the case of aldehyde inhibitors the classic oxyanion hole is not observed while ketones (as fluoromethylketones) display this mode of binding. Powers and Grutter have explored the S1′ caspase-3 pocket using various aza-peptide epoxide inhibitors.168 The five crystal structures of caspase-3 in complex with aza-peptide epoxides revealed the basis for inhibition specificity on the right to the scissile bond. It was found that S1′ pocket is large and can accommodate bulky, hydrophobic warheads. This observation is at odds with the preference of S1′ for small side-chains in substrates described above, leading us to speculate that substrate binding and aza-peptide epoxide binding at the P1′ position are not congruent. The favorable inhibitor interactions in this pocket are formed by the catalytic histidine residue, an oxyanion hole, and water molecules, which are present in all five described caspase-3-inhibitor complexes.
It is well documented in the literature that aspartate residue at P1 position is highly desired by caspases for efficient proteolysis and interactions in this pocket are conserved among all human caspases.12,169 The aspartic acid side chain is buried into a narrow, positively charged S1 pocket displaying high electrostatic potential.39,170 The Asp carboxyl group is stabilized by two highly conserved arginine side residues as well as amide nitrogen glutamine. In addition there is a hydrogen bond formed between P1 amide nitrogen and carbonyl group of highly conserved serine backbone (in caspase-2, this serine is replaced by alanine, but the bond is also present 171).
The S2 pocket is not very selective and liberal substituents are tolerated. Several lines of evidence indicate that this pocket is considered to be of minor importance for discrimination between caspases.24 Generally, the S2 pocket likes hydrophobic residues that are bounded by hydrophobic interactions. In the case of caspase-1 this pocket is constituted by two hydrophobic residues Val338 and Trp340, and crystal structure analysis confirmed that various amino acids side chains from branched valine to bulky and basic histidine can be accommodated in this pocket.22,165 Caspase-3 (and caspase-7) possesses an additional phenylalanine residue, thus alanine, valine or larger leucine are tolerated.170,172,173 Caspase-8 also possesses 3 hydrophobic residues in S2 (Val and two Tyr).174,175 Interestingly, none of these caspases involve the P2 amide nitrogen in inhibitor-enzyme binding, which opens the door for utility of peptidomimetics as caspase inhibitors, where the P3-P2 region in replaced by mono- or bicylic platforms. Furthermore, Weber and coworkers discovered by crystal structure analysis that the S2 pocket of caspase-7 displays some plasticity and can accommodate methionine, leucine, glutamine, histidine or even proline.173
Glutamic acid at the P3 position is considered to be required for efficient catalysis, but a few others amino acids are also tolerated in S3 pocket. Glu makes a charge-charge interaction with a conserved arginine side chain, which is also important for binding P1 Asp residue.170 The second polar interaction is created by a water molecule in caspases -3, -7, and -8, although caspase-1 lacks this interaction. There is evidence in the literature that other amino acids can be introduced at P3 with only slight decrease in activity.167,176 For instance, replacement of glutamic acid with a hydrophobic residue does not result in significant decrease of activity, since this hydrophobic side chain forms favorable van der Waals interactions with hydrophobic proline residue.
The most striking differences among caspases -1, -3 and -8 can be observed in the S4 pocket. Caspase-1 prefers bulky, hydrophobic residues (tyrosine and tryptophan),22,24 caspase-3 has a near absolute requirement for aspartic acid,168,177 while caspase-8 can accommodate a number of residues, but branched leucine and valine are most preferable.170,174 These different P4 requirements are determined by the size and shape of S4 pocket of particular caspase. The S4 pocket of caspase-1 is large and hydrophobic thus can bind bulky Tyr, Trp or bicyclic substituents.178 Caspase-3 binds its substrates or inhibitors in S4 using the Phe250 backbone nitrogen, Asn208 side chain and one water molecule. Additionally, the carbonyl oxygen from the acetyl group of inhibitors makes an interaction with both hydroxyl group and amide nitrogen of Ser209.170 The caspase-8 S4 pocket prefers hydrophobic residues as leucine, but also small, polar amino acids are tolerated. Asp is bound in the S4 subsite directly by Trp420 and Asn414, while the methyl group of the N-terminal acetyl moiety interacts with hydrophobic Phe415 and the acetyl carbonyl makes interaction with Asn414.170
Caspase-2 seems unusual among the family members since it strongly prefers a pentapeptide sequence (VDVAD or ADVAD) over a tetrapeptide, which is a common requirement for efficient catalysis by other caspases. AFC-labeled substrates with ADVAD 12 and VDVAD 11 sequences are 10–40 fold more active toward caspase-2 than a truncated tetrapeptide (DVAD) 13, and VDVAD is an optimal sequence. The crystal structure of caspase-2 in complex with pentapeptide aldehydes revealed that two residues, Thr380 and Tyr420, are crucial for the P5 recognition. Thr380 interacts with both P5 carbonyl oxygen and amide nitrogen, while Tyr420 is involved in accommodating P5 Val or Ala.39 Schweizer and coworkers described the crystal structure of caspase-2 in complex with the pentapeptide Ac-LDESD-CHO 85.171 The structural analysis confirmed a previously mentioned feature that P5 amino acid (here leucine) is positioned in a productive orientation via two hydrogen bonds between amide the NH and carbonyl CO and Thr343. Recently Weber and coworkers investigated the influence of P5 position on the substrate and inhibitor recognition by three executioner caspases (-3, -6 and -7).179,180 It was found that caspase-3 slightly prefers a pentapeptide LDEVD over tetrapeptide DEVD (1.46 fold more active), caspase-6 cleaves QDEVD and LDEVD more efficiently than DEVD, while additional P5 position does not increase caspase-7 activity. These kinetic data can be explained by crystal structures of these caspases in complex with Ac-LDESD-CHO 85 where two hydrophobic residues, Phe250 and Phe252 bind leucine at P5 and thus increase the LDESD activity toward the enzyme. The same pocket in caspase-7 is formed by three hydrophilic residues, Gln276, Ser277 and Asp278 that are not able to stabilize hydrophobic leucine. Finally caspase-6 possesses S5 pocket of assorted, hydrophilic and hydrophobic character that can accommodate both glutamine and leucine. In Figure 16 we present the schematic representation of chemical interactions between caspase and their inhibitors.
Figure 16.

Schematic representation of hydrogen bonds and ionic interactions between caspases and their peptide inhibitors: caspase-1-Ac-YVAD-CHO,165 caspase-3-Ac-DEVD-CHO,166,170 caspase-8-Ac-IETD-CHO,175 caspase-2-Ac-LDESD-CHO.171
The structural data are overall in excellent agreement with substrate/inhibitor kinetic analysis making structure-based inhibitor design potentially useful for caspases. Data from crystal structure analysis has helped to answer some classic questions: how do caspases recognize their short substrates? Are longer substrates preferred for particular caspases, and why? And finally, are there any structural attributes dedicated to a particular caspase that can be useful for development of specific caspase inhibitors?
We have listed crystal structures of caspases (Tables 5–11) that have been deposited in the RCSB protein data bank so far. These structures contain some caspase zymogens as well as active enzymes in complex with active site directed ligands, allosteric inhibitors, protein-like inhibitors or ligand free.
3.3. Peptide and peptidomimetic caspase inhibitors
Human caspases are mainly involved either in inflammation processes (caspases -1, -4, and -5) and apoptosis cascades (initiator caspases -8, -9, -10 and executioners -3, -6, -7).164 Caspase-14 is involved in keratinocyte maturation244 and the function of caspase-2 is still highly controversial.245,246 The main catalytic feature for efficient proteolysis is that they all required aspartate residue at P1 and recognize at least four amino acid residues to the left of cleavage site.29,163 Truncation of tetrapeptide to tri- or dipeptide leads to significant decrease of activity.247,248 As mentioned earlier, caspases are divided into subgroups depending on their preferred substrate recognition of tetrapeptides. Inhibitors based on these preferences with appropriate peptide or peptidomimetic backbone can be useful evidence for their role in particular biological processes. For instance, the finding that the caspase-1 directed inhibitor, Ac-YVAD-CHO 4, prevents the release of mature interleukin 1-β from monocytes was evidence that caspase-1 is responsible for interleukin 1-β maturation, upon specific proteolysis29. Most peptide-based inhibitors cannot be used therapeutically due to their toxic nature or metabolites. A good example are Cbz-VAD-CH2F 86 and Boc-D-CH2F 87, a broad spectrum caspase inhibitors. Cbz-VAD-CH2F 86 and Boc-D-CH2F 87 are excellent probes for enzyme kinetic analysis, but are unsuitable for use in vivo due to production of highly toxic fluoroacetate.249,250 Another example of broad spectrum caspase inhibitor is Q-VD-OPh (OPH-001, 88) equipped with 2,6-difluorophenoxymethylketone warhead. It represents a new generation of caspase inhibitor which is non-toxic, more stable is aqueous media and displays higher activity than CH2F-based inhibitors. This compound was used to block caspases activity in both, in vivo and in vitro models.251,252 All these three compounds (86, 87, and 88) are by far the most widely used apoptosis inhibitors, however they do not inhibit caspases equally. A detailed study regarding their affinity toward particular caspases in in vitro and in cellula models have been described by Chauvier et al..253 The prototype caspase inhibitors contained simple warheads attached to the peptide backbone, inhibiting caspases via covalent modification of cysteine residue. The most commonly used warheads are aldehydes and ketones with their derivatives (halomethylketones, aryloxymethylketones or acyloxymethylketones), which tend to be highly reactive with cysteine proteases.158 The specificity for particular family (i.e. caspases) is achieved by optimization the non-prime backbone (usually peptide or peptidomimetic). Based on Thornberry’s specificity profile of 9 human caspases,24 Garcia-Calvo et al. used peptide aldehydes and potent, broad spectrum Cbz-VAD-CH2F 86 for caspases inhibitory profile investigation (Figure 17).254 Ac-WEHD-CHO 5 was designed for inflammatory caspases -1, -4 and -5 (group I), Ac-DEVD-CHO 83 for caspases -2, -3, and -7 (group II), and Ac-IETD-CHO 89 for caspases -6, -8, -9, and -10. Quantitative analysis revealed that these inhibitors are potent caspase inhibitors, but absolute specificity had not been achieved. Ac-WEHD-CHO 5 is specific for caspases from group I, but it can also inhibit caspase-8 efficiently. The caspase -3 and -7 specific Ac-DEVD-CHO 83 does not inhibit caspase -2, which also belongs to the group II, but it displays high potency toward caspase -8. The third aldehyde, Ac-IETD-CHO 89 is specific only for caspase -6, -8 (group III) and caspase -1 (group I). Finally, Cbz-VAD-CH2F 86 displays broad spectrum of activity, with the exception of caspase -2. In Table 12 we collected kinetic parameters of the above described inhibitors. These results strongly indicate that knowledge about caspases specificity is not enough for discriminating these enzymes within the family and further efforts are desirable.
Figure 17.

Structures of four potent caspases inhibitors designed based on substrate specificity profile.254
Table 12.
Kinetic parameters of potent peptide caspase inhibitors. A high degree of specificity has not been obtained for any caspase. 254
| Enzyme | Ki, nM | k, M−1s−1 | |||
|---|---|---|---|---|---|
|
| |||||
| Ac-WEHD-CHO 5 | Ac-DEVD-CHO 83 | Ac-IETD-CHO 89 | Cbz-VAD-CH2F 86 | ||
| Group I | Casp-1 | 0.056 | 18 | <6 | 280000 |
| Casp-4 | 97 | 132 | 400 | 5500 | |
| Casp-5 | 43 | 205 | 223 | 130000 | |
|
| |||||
| Group II | Casp-2 | > 10000 | 1710 | 9400 | 290 |
| Casp-3 | 1960 | 0.23 | 195 | 16000 | |
| Casp-7 | > 10000 | 1.6 | 3 280 | 18000 | |
|
| |||||
| Group III | Casp-6 | 3090 | 31 | 5.6 | 7100 |
| Casp-8 | 21.1 | 0.92 | 1.05 | 280000 | |
| Casp-9 | 508 | 60 | 108 | 180000 | |
| Casp-10 | 330 | 12 | 27 | not determined | |
To address this problem researchers from academia and industry have developed more tailored peptide and peptidomimetic backbones for caspases. Analyzing the literature of caspase inhibitory profiles we suggest dividing caspase inhibitor structures into three regions: N-terminal P4 position, dipeptide P3-P2 region (P1 is fixed as Asp), and P1′ cysteine trap (warhead) (Figure 18). In a typical approach, a series of inhibitors are synthesized where a particular region is varied while the rest of the scaffold is kept constant. This strategy provides an excellent structure-activity relationship that can be used for specific caspase inhibitor discovery.
Figure 18.

Caspase inhibitor architecture presented using Ac-WEHD-CHO 5 – a potent caspase-1 reversible inhibitor – as an example.
3.3.1. Inflammatory caspases
The group of inflammatory caspases is constituted by caspases -1, -4, -5, but to date only caspase-1 has been extensively studied in the context of design of specific and potent inhibitors. Caspase-1 is a highly selective cysteine protease that is responsible for maturation of inactive IL-1β pro-form to the active cytokine via proteolysis.29,255,256 It has been reported that this enzyme requires a tetrapeptide substrate with appropriate amino acid sequence for efficient cleavage.22 To date numerous reversible and irreversible caspase-1 inhibitors have been discovered. Many of them contain common tetrapeptide motif Ac-Tyr-Val-Ala-Asp based on IL-1β sequence, a natural caspase-1 substrate (native His at P2 position was replaced with hydrophobic Ala). However the YVAD sequence is not optimal for caspase-1, and Thornberry et al reported that an aldehyde inhibitor with Ac-Trp-Glu-His-Asp sequence is 13-fold more potent.22 These two prototypic caspase-1 peptide inhibitors shed light on caspase-1 structural requirements for inhibition making this protease an early topical target of research. To date various types of caspase-1 inhibitors have been described in details. Structural analysis of potent and selective caspase-1 inhibitors led to conclusion that this enzyme requires a few specific structural attributes from its inhibitors. In many caspase-1 inhibitors the P3-P2 region is constructed with Val-Ala residues. This dipeptide scaffold maintains important interactions of the inhibitor backbone with the caspase-1 substrate cleft, crucial for proper orientation of the P1 aspartate residue and the P4 hydrophobic moiety in the enzyme pocket.257 Appropriate orientation of the Val-Ala dipeptide within an inhibitor can induce a number of hydrogen bonds between amido hydrogens (P1, P3) and the carbonyl group (P1) and caspase-1 active site residues, which are necessary for efficient enzyme inhibition.257,258 Caspase-1 prefers bulky, hydrophobic substituents in P4 position, such as Tyr (IL-1β sequence), Trp (AMC-tagged substrate combinatorial library assay) and Cbz or Naphthyl groups (from inhibitor library screening). The P4-P1 scaffold architecture has a significant impact on inhibitor potency, however optimization of the P1′ region is as important as the non-prime side. So far numerous warheads of different types have been synthesized. Generally, caspase-1 prefers hydrophobic substituents with phenyl/benzyl derivatives. Most of caspase-1 inhibitors display a β-sheet pattern of hydrogen bond formation between peptide inhibitor and substrate binding cleft backbone, as revealed by X-ray structural analysis.
3.3.1.1. P1 position
Caspase-1 prefers aspartic acid residue at P1 position and chemical modification of Asp results in dramatic loss of inhibitory activity. For example, O-capped aspartic acid is not able to form hydrogen bonds with Arg179, Arg341 and Gln283 residues from the caspase-1 active site. Moreover methylation of the P1 amide carbon causes the same effect, because the P1 NH group works as a hydrogen bond donor and is crucial for maintain inhibitor-enzyme binding potency.259
3.3.1.2. P1′ position (tri- and tetrapeptides/peptidomimetics)
Do date many different cysteine traps have been described in the literature. All these inhibitors can be divided based on their mode of inhibition: covalent or non covalent, reversible or irreversible and more. In early 1990s many efforts were focused on the design specific caspase-1 inhibitors. One of the first and best most known caspase-1 inhibitors was Ac-YVAD-CHO 4 containing aldehyde warhead.165,260 This compound was used in X-ray structural analysis and numerous cell based assays, providing invaluable information about caspase-1 specificity and biological function. The aldehyde warhead is highly reactive and can display non specific cross-reactivity with bionucleophiles. However, the Asp-CHO moiety can be easily transformed into a hemiacetal prodrug form. Additionally, the hydroxyl can be capped by an ethyl group making Asp-aldehydes more bioavailable. Another, early caspase-1 peptide based inhibitors contained fluoromethylketones as reactive warhead.249 This electrophilic moiety also limited the utility of caspase-1 inhibitors in biological assays, so efforts to design therapeutically useful caspase-1 probes shifted to the use of activated ketones as leaving groups. Early work in this area was made by Merck laboratories, where Mjalli et al. described the preparation of peptide phenylalkyl ketones displaying a reversible mode of inhibition. The most potent inhibitor 92 had a -C(O)(CH2)5Ph warhead attached to the Ac-Tyr-Val-Ala-Asp scaffold.261 This electrophilic moiety was further optimized by Harter et al., who introduced a sulfonamide group into the alkyl chain resulting in discovery of more potent caspase-1 inhibitors.262 Mjalli et al. also described phenoxymethyl ketones and heterosubstitued ketones.261 In other project, Dolle et al. proposed several different electrophilic warheads for caspase-1 inactivation. They described the synthesis and kinetic evaluation of a Cbz-Val-Ala-Asp sequence conjugated to α-((2,6-Dichlorobenzoyl)oxy)methyl,263 α-((1-Phenyl-3-(trifluoromethyl)-pyrazol-5-yl)oxy)methyl,264 and α-((Diphenylphosphinyl)oxy)methyl ketones265 as time-dependent, irreversible inhibitors of this enzyme. Shortly thereafter, Thornberry et al. proposed (acyloxy)methyl ketones as new set of caspase-1 inhibitors with high potency, yet chemical inert properties.266 Boxer and coworkers examined the potential of nitriles as an electrophile for reversible, covalent inhibition of caspase-1.267 These authors synthesized cyanopropanoate derivatives based on the peptidic scaffold of the prodrug VX-765 93 (please see the “P2–P3 region” section). Compound NCG00183434 94 (Figure 19) was found as a potent and reportedly selective caspase-1 inhibitor. Furthermore, ester and tetrazole analogues of this compound also exhibited good inhibitory potency. The hydrolytic stability studies and profiles of selected ADME properties also demonstrated that this novel agent can be utilized in in vivo studies. To date many (Q)SAR studies have been performed for design of optimal P1’ warheads to satisfy caspase-1 requirements. In Table 13 and Table 14 we have collected multiple examples of reported leaving groups.
Figure 19.

Structures of peptidomimetic caspase-1 inhibitors.
Table 13.
Several examples of early ICE (caspase-1) reversible and irreversible inhibitors with various leaving groups attached to Cbz-Val-Ala-Asp-scaffold
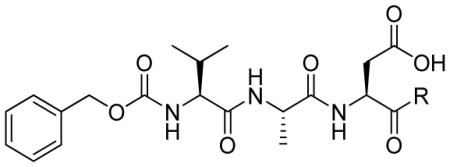
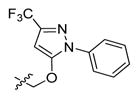
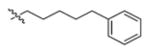
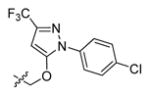
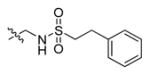
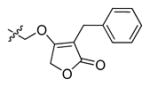
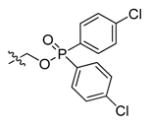
Table 14.
Examples of early caspase-1 irreversible inhibitors with various leaving groups attached to the PhCH2C(O)-Val-Ala-Asp-scaffold.266
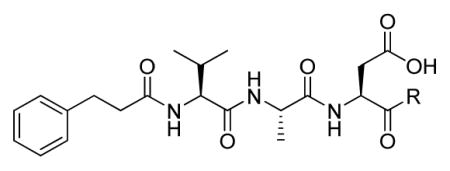 105–110 | |||||
|---|---|---|---|---|---|
|
| |||||
| R = | kobs/[I], M−1s−1 | R = | kobs/[I], M−1s−1 | R = | kobs/[I], M−1s−1 |
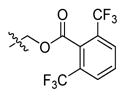 105 |
900000 |
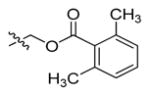 107 |
1200000 |
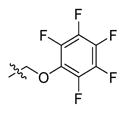 109 |
1100000 |
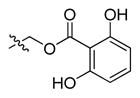 106 |
710000 |
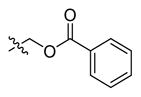 108 |
280000 |
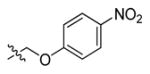 110 |
1300000 |
Löser and coworkers developed potent noncovalent caspase-1 inhibitors by replacing the conventional electrophile (e.g., aldehyde) by secondary amine isosteres.270 The peptidic moiety 2-Nap-Val-Ala-Asp was used as a scaffold for the synthesis of ten benzyl- and cyclohexylamine inhibitors for which Ki values were determined toward caspase-1 (Table 15). One compound with a 2-hydroxybenzylamine group in P1′ position 111 appeared as potent (Ki = 47 ± 7 nM) and stable noncovalent caspase-1 inhibitor. Compound 113 was also evaluated for caspase selectivity by testing toward caspases-3 and -8. There was no inhibition of caspase-3 up to micromolar concentration and some inhibition of caspase-8 at high micromolar concentration was detected. This approach of incorporating benzylamine in place of the electrophile could be applied in the design of inhibitors targeting other cysteine proteases.
Table 15.
Examples of benzyl- and cyclohexylamine caspase-1 inhibitors.270
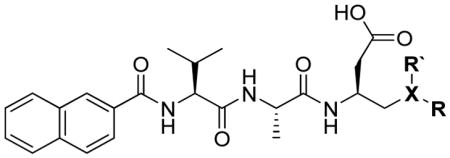 111-121 | |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| R | R′ | X | Ki, nM | R | R′ | X | Ki, nM |
 111 |
H | N | 600 |
 117 |
H | N | 209 |
 112 |
H | N | 47 |
 118 |
H | N | 945 |
 113 |
H | N | 1200 |
119 |
CH3 | N | 1890 |
 114 |
H | N | 965 |
120 |
CH3 | N | 128 |
 115 |
H | N | 1370 |
121 control |
H | CH | >50000 |
 116 |
H | N | 1820 | ||||
3.3.1.3. P1′ position (truncated peptides/peptidomimetics)
One of the biggest challenge in the design of inhibitors targeting caspase-1 is to select a rational compromise between inhibitor potency and desirable pharmacokinetic profile. Tetra- and tripeptides with appropriate amino acids sequences labeled with an electrophilic warhead are very potent caspase-1 inhibitors. Unfortunately their peptide character and presence of negatively charged Asp residue substantially limits their utility as therapeutic agents. On the other hand non peptide or peptidomimetic inhibitors that posses biologically desirable attributes are not always potent/selective enough for targeting caspase-1. Thus many efforts focused on design and synthesis of truncated caspase-1 inhibitors. Dipeptide-based derivatives has significantly reduced peptide character and the use of appropriate substituents at both N- and C-terminal ends maintains high potency and selectivity. To date various dipeptide and single amino acid based inhibitors have been proposed in the literature.
Mjalli et al. reported that truncation of the potent caspase-1 inhibitor Ac-Tyr-Val-Ala-Asp-CO(CH2)4Ph 122 to its single amino acid analogue alloc-Asp-OC(O)(CH2)4Ph 123 resulted in complete loss of activity.271 To address this problem they synthesized a library of single amino acid based caspase 1inhibitors with various P1′ leaving groups. An inhibitor with CH2-CO-(CH2)2-cyclohexyl leaving group 124 displayed good potency toward caspase-1 and was one of the first examples of single amino acids based compound labeled with appropriate warhead that can be used for targeting caspase-1. Shortly thereafter, Mjalli et al. described another group of N-acyl-aspartylaryloxymethyl ketones labeled with 2-naphthyl derivatives at the C terminus.272 Two best single amino acid based caspase-1 inhibitors from this series were 125 (Ki of 0.09 μM and kon of 12000 M−1s−1) and 126 (Ki of 0.32 μM and kon of 5600 M−1s−1). In other work, Shahripour et al. used Cbz-Asp-CO-(CH2)5-Ph 127, a very weak caspase-1 inhibitor (Ki = 119 μM), as a starting point structure for molecular modeling and SAR studies. As a result they synthesized a few N-capped single amino acids aldehydes that displayed good potency against caspase-1.273 Harter et al. discovered Cbz-Asp-sulfonamides as caspase-1 inhibitors with moderate potency.262 Recently Walker’s group also demonstrated that P1′ leaving group can be key determinant in design of caspase-1 inhibitors.269 They demonstrated that Cbz-Asp-based compounds can display nanomolar potency toward caspase-1. Elongation of the peptide chain by adding additional amino acid at P2 significantly enhanced inhibitory potency. Ullman et al. described a library of dipeptide based caspase-1 inhibitors with variable P1′ positions.274 Furthermore Linton and coworkers demonstrated that dipeptide aldehydes with appropriate N-terminal substituents can display broad spectrum activity labeling not only caspase-1, but also caspases -3, -6, -7, and -8.275,276 All these examples demonstrate that truncated peptides equipped with an appropriate warhead and N-capped with suitable substituents can also be potent caspase inhibitors while maintaining good pharmacokinetic profile. In Table 16 we have collected structures and inhibition parameters of single amino acid caspase-1 inhibitors.
Table 16.
Structures of single amino acid ICE (caspase-1) inhibitors with various leaving groups.
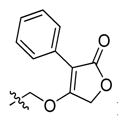
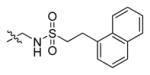
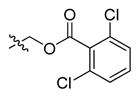
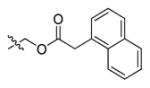
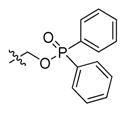
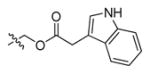
3.3.1.4. P2-P3 region
The most common P3-P2 motif of caspase-1 inhibitors is Val-Ala based on results from positional scanning (Figure 16, Figure 20), but it was also reported that replacing P2 Ala by other amino acids including secondary piperidine or proline do not affect inhibitory potency.264 One such examples is VX-043198 135, a potent and selective caspase-1 inhibitor with unnatural tert-leucine in P3 position and secondary proline in P2 (Figure 19). This inhibitor is an active metabolite of VX-765 93, an orally absorbed prodrug (Figure 20). VX-765 93 is a rare example of peptide-based caspase-1inhibitor possessing good pharmacokinetic profile and displaying the utility for treatment of inflammatory diseases.277 Most peptide-based inhibitors with appropriate sequence are good caspase-1 inhibitors, but despite their high potency and selectivity they are not promising for drug development efforts (poor cell penetration, moderate stability under biological conditions). To overcome this problem a number of peptidomimetic caspase-1 inhibitors have been proposed. It was previously shown that P4 and P1 positions in substrate or inhibitor are crucial for caspase-1 inhibitory activity. Residues at P2 and P3 positions were found to be less significant for efficient proteolysis, but the proper orientation of P3 NH and CO backbone groups is still required since these atoms participate in formation of hydrogen bonds.165 Therefore, the challenge is to mimic a peptide P3 carbonyl and amide group while maintaining the high potency and selectivity of the inhibitor of interest. Dolle et al. have discovered that P3-P2 (Val-Ala) region can be successfully replaced by pyrimidone moiety retaining crucial hydrogen bonding functionality in the first example of a caspase-1 peptidomimetic inhibitor.258 There are additional reports describing the utility of pyrimidone (or pyridone) moiety for mimicking the P3-P2 region.278–280 Pyrimidone and pyridone groups were substituted with different ligands to explore the S2 active site pocket of caspase-1. This new scaffold significantly reduced peptidic character, but also decreased the inhibitory potency toward caspase-1. This may be explained by the stiffness of the pyrimidone (or pyridone) moiety, which led to the suboptimal presentation of both the P3 amide group and the backbone to the caspase-1 active site. To address this problem a new, more flexible scaffold for caspase-1 inhibitors have been proposed. Pyridazinodiazepines-based caspase-1 inhibitors have been described by Dolle et al.257 This moiety, which occupies P3-P2 region significantly reduced peptide character of inhibitors while maintaining the high affinity against caspase-1. Moreover the utility of this scaffold was demonstrated for both reversible and irreversible inhibitors. The use of a pyridazinodiazepine group for mimicking Val-Ala sequence resulted in the discovery of Pralnacasan (VX-740) 77 (Figure 20), which was tested in advanced clinical trials as a drug candidate for rheumatoid arthritis (failed due to toxicological issues).16,257,281 This drug possesses a 2-naphthyl hydrophobic moiety at P4 position, a pyridazinodiazepine-based bicyclic core mimicking the Val-Ala region (P3-P2 residues) and a lactone moiety at the C-terminus, which masks an aspartate residue at P1 position (Figure 20). Since Pralnacasan was discovered many different inhibitors with geometrically constrained mono- or bicyclic backbone mimicking P3-P2 have been developed. To date Pralnacasan 77 is the best-known peptidomimetic caspase-1 inhibitor displaying therapeutic utility. There are several more approaches concerning the chemical modification of Pralnacasan bicyclic core to better explore the P3-P2 region. Lauffer and Mullican described the practical synthesis of caspase-1 inhibitors based on a 5-benzodiazepinone scaffold which allows for the broad exploration of P3 position.282 Others have evaluated numbers of different mono-, bi- or tricyclic scaffolds mimicking the P3-P2 region. Monocyclic 8-membered lactams have been designed to specifically target caspase-1.283 This new scaffold allows introduction of numerous substituents at nitrogen-5 of the lactam moiety to efficiently explore new binding interactions with the caspase-1 S3 pocket. This group of inhibitors can selectively target caspase-1 over caspase-3 and -8, but the most potent inhibitor displays lower affinity for caspase-1 than Pralnacasan 77 (IC50 of 15 nM and 2 nM respectively). Shortly afterwards the same group reported the synthesis and biological evaluation of unsaturated caprolactams.284 This new group of compounds displayed high affinity and selectivity towards caspase-1- IC50 for best characterized caprolactam-based derivatives was of 1 nM.
Figure 20.

Structures of prototypic peptide-based (Ac-YVAD-CHO 4) and two peptidomimetic caspase-1 inhibitors.
Thiazepines are another group of monocyclic peptidomimetic caspase-1 inhibitors.285 A sulfur atom (or sulfonyl group when sulfur is oxidized) is responsible for induction of positive interactions with the caspase-1 S3 pocket. This group also displays a high affinity and selectivity toward caspase-1, but still there is no better inhibitor than Pralnacasan 77 among thiazepines peptidomimetics. Another of caspase-1 inhibitors based on the monocyclic scaffold are 1-(2-acylhydrazinocarbonyl)cycloalkylcarboxamides,178 with their potency depending on the number of atoms building the monocyclic moiety. Cyclohexyloderivatives are preferred over the shorter homologues cyclopentyl, cyclobutyl or cyclopropyl. However the replacement of a cyclohexyl group with the 2-Indanyl moiety as P3-P2 scaffold results in increased inhibitor potency. Peptidomimetics based on a monocyclic moiety occupying the Val-Ala region are good caspase-1 inhibitors, but none of them was more potent than Pralnacasan 77. To enhance potency new classes of 8,5- and 8,6-fused bicyclic peptidomimetics have been developed.286,287 The design of these compounds followed a concept that 8,5- and 8,6-fused bicyclic scaffolds are able to occupy S2–S3 caspase-1 pockets more tightly than their monocyclic counterparts. Indeed some of these peptidomimetics displayed a higher affinity toward caspase-1 than Pralnacasan. In summary, most of these peptidomimetics display desirable pharmacokinetic profiles while maintaining high potency and selectivity toward caspase-1. Compounds from these series possess good activity in whole cell assays. These observations confirm that this group of caspase-1 inhibitors is very promising as therapeutic agents. In Table 17 there are collected some examples of mono- and bi-cyclic peptidomimetics acting as potent and selective caspase-1 inhibitors.
Table 17.
Multiple examples of caspase-1 peptidomimetic inhibitors based on various P3-P2 scaffolds.
| Structure | R = | No. | Casp-1 IC50, nM | Casp-3 IC50, nM | Casp-8 IC50, nM | THP-1 IC50, nM | Ref |
|---|---|---|---|---|---|---|---|
| Reference - PRALNACASAN | |||||||
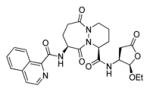
|
- | 77 | 3.6 | 1 300 | 40 | 190 | 286 |
|
| |||||||
| UNSATURATED CAPROLACTAMS | |||||||
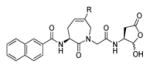
|
- H | 137 | 14 | > 10000 | 2160 | 100 | 284 |
| - Me | 138 | 19 | > 10000 | 2688 | 153 | ||
| - CH2OH | 139 | 22 | > 10000 | 2110 | 543 | ||
| - CH2NHCOPh | 140 | 2 | 1698 | 1085 | 466 | ||
| - CH2NHCO-3-OMePh | 141 | 1 | 1079 | 448 | 243 | ||
| - CH2NHCO-4-OMePh | 142 | 1 | 1128 | 471 | 95 | ||
|
|
|||||||
| MONOCYCLIC 8-MEMBERED LACTAMS | |||||||
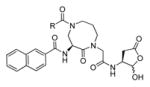
|
- Ph(2-OMe) | 143 | 38 | > 10000 | > 10000 | 246 | 283 |
| - Ph(3-OMe) | 144 | 15 | > 10000 | > 10000 | 229 | ||
| - Ph(3-F) | 145 | 22 | > 10000 | > 10000 | 397 | ||
| - Piperonyl | 146 | 24 | > 10000 | > 10000 | 285 | ||
| - N-morpholine | 147 | 44 | > 10000 | > 10000 | 224 | ||
|
|
|||||||
| 8,5-FUSED BICYCLIC PEPTIDOMIMETICS | |||||||
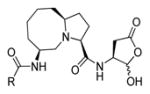
|
- Phenyl | 148 | 3.6 | 1300 | 40 | 190 | 286 |
| - 3-Chlorophenyl | 149 | 2.3 | > 10000 | 2900 | 200 | ||
| - 1-Naphthyl | 150 | 1 | > 10000 | 66 | 3 | ||
| - 2-Naphthyl | 151 | < 1 | > 10000 | 390 | 32 | ||
| - 1-Isoquinolyl | 152 | < 1 | > 10000 | 160 | 54 | ||
|
|
|||||||
| 8,6-FUSED BICYCLIC PEPTIDOMIMETICS | |||||||
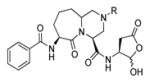
|
- H | 153 | 10 | > 10000 | 384 | 245 | 287 |
| - Me | 154 | 5 | > 10000 | 247 | 1820 | ||
| - COPh | 155 | 7 | 8070 | 38 | 1420 | ||
| - SO2Me | 156 | 6 | > 10000 | 384 | 174 | ||
| - SO2Ph | 157 | 2 | > 10000 | 106 | 159 | ||
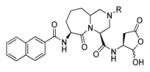
|
-H | 158 | 1 | > 10000 | 506 | 75 | |
| -SO2Me | 159 | 1 | > 10000 | 283 | 46 | ||
| -SO2Ph | 160 | 1 | > 10000 | 50 | 153 | ||
|
|
|||||||
| THIAZEPINES | |||||||
|
- Phenyl | 161 | 2 300 | > 10000 | > 10000 | 3200 | 285 |
| - 3-CF3Phenyl | 162 | - | - | - | 3400 | ||
| - 2-Benzo[b]thiphene | 163 | 320 | > 10000 | 8500 | 1700 | ||
| - 1-Isoquinolyl | 164 | 130 | > 10000 | 5000 | 1400 | ||
| - 2-Naphthyl | 165 | 150 | > 10000 | > 10000 | 1100 | ||
|
|
|||||||
| 1-(2-ACYLHYDRAZINOCARBONYL)-CYCLOALKYL CARBOXAMIDES | |||||||
|
- m-Methylphenyl | 166 | 430 | - | 3120 | ≥2500 | 178 |
| - 1-Naphthyl | 167 | 40 | - | 420 | 690 | ||
| - 2-Naphthyl | 168 | 30 | - | 2100 | 500 | ||
|
|
|||||||
3.3.1.5. P4 position
The first inhibitors designed for caspase-1 contained a Tyr residue at P4, based on the IL-1β cleavage motif, and next Thornberry et al. found that optimal tetrapeptide sequence for caspase-1 is WEHD with Trp at the N-terminus24, clearly demonstrating that caspase-1 prefers hydrophobic residues at P4. Moreover, commonly used caspase-1 tripeptide inhibitors are usually N-capped with benzyloxycarbonyl (Cbz or Z) group that mimics a hydrophobic P4 residue.269 This requirement for bulky substituents at P4 has important impact on discriminating caspase-1 from caspase-3 since it was found that caspase-3 displays a high preference for aspartic acid at P4. Over the past two decades many different functional groups have been proposed to mimic the Ac-Tyr moiety. The most commonly used chemical groups for capping N-termini of caspase-1 inhibitors are 2-naphtyl, 1-naphthyl, 1-isoquinolyl and their derivatives.283,284,286 These classes of substituents fit well to the caspase-1 S4 pocket, thus are very useful in the design of small molecule probes, and have been coupled with both peptidomimetics and traditional peptide based inhibitors. Perhaps the best known caspase-1 inhibitor capped by a 1-isoquinolyl is Pralnacasan.
More bulky, hydrophobic P4 substituents have been described in the literature. For instance Dolle et al. designed and synthesized a 6-membered collection of inhibitors with benzyl derivatives at the N-terminus.257 Some inhibitors containing these ligands displayed higher potency against caspase-1 than ones with traditional Cbz groups, and some of these substituents increased solubility of the inhibitors. In another approaches Dolle et al. used a pyrimidone-scaffold258 and Golec et al. used a pyridone-scaffold for P4 optimization.278 Recently Galatias et al. reported caspase-1 inhibitors containing succinic acid amides as P3-P2 replacements.183 Based on this scaffold they synthesized a set of compounds with variable P4 substituents. The best hit 169 displayed Ki of 0.58 nM and IC50 of 5.7 nM, making this compound more potent toward caspase-1 than well-known full tetrapeptide Ac-YVAD-CHO 4. In Table 18 and Table 19 we collected selected examples of P4 SAR studies. The current strategy for the discovery of new, potent P4 substituents include SAR studies assisted by parallel synthesis,258 tethering approaches185,187 or “click chemistry”.288
Table 18.
Examples of irreversible, peptidomimetic caspase-1 inhibitors with diverse P4 position.
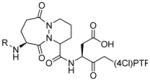
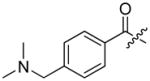
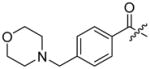
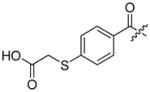
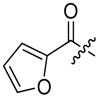
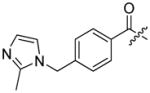
Table 19.
Examples of reversible and irreversible caspase-1 inhibitors with diverse P4 position
|
R = | Ki, nM | Ref |
|
R = | Ki, nM | Ref |
|---|---|---|---|---|---|---|---|
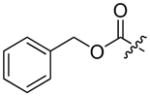 93 |
Cbz | 55 | 268 |
183 |
Cbz | 590 | 279 |
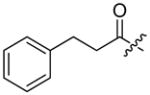 182 |
DCA | 54 | 278 |
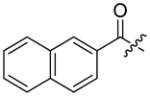 184 |
2-Naphthyl | 160 | |
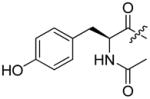 4 |
Ac-Tyr | 9 | 278 |
 185 |
1-Naphthyl | 95 |
3.3.2. Apoptotic caspases
Apoptotic caspases can be divided based on their role in programmed cell death for initiators (caspases -8, -9, -10) and executioners (caspases -3, -6, -7) as well as based on their substrate specificity toward small peptides - group II preferring DE(V/H)D motifs (caspases -2, -3, -7) and group III recognizing (V/L)EHD sequences (caspases -6, -8, -9).163,169 Over the last decade the design and development of apoptotic caspases inhibitors has been a very active area of research in both academia and industry.12,23,289 Work in this area has resulted in the identification of numerous apoptotic caspase inhibitors that can be assigned as reversible (aldehydes, ketones, nitriles) or irreversible (activated ketones like acyloxymethylketones, halomethylketones or diazomethylketones).158,169 Initially, small molecule inhibitors of apoptotic caspases were short peptides equipped with an appropriate electrophilic warhead. The peptide sequence usually is often derived from natural caspases substrates (like PARP for caspase-3) or preferred sequences defined from combinatorial substrate library analyses that has accelerated the development of new, potent small molecule tools for caspase investigations. More recently the development of new caspases inhibitors shifted more to peptidomimetics such as azapeptides or conformationally constrained derivatives rather than simple peptides with natural amino acids.
3.3.2.1. P1 position
Small molecule inhibitors designed for apoptotic caspase inactivation took advantage of the preference of theses enzymes for aspartic acid in P1 position (one of the known exceptions is Drosophila caspase Dronc that cleaves substrates with glutamic acid in P1 position almost as good as these with Asp290). Thus, all known potent caspases inhibitors share the same motif in P1 (Asp residue).
3.3.2.2. P1′ position (cysteine trap)
Prototypic apoptotic caspase inhibitors encompass aldehydes, fluoromethylketones or chloromethylketones. These small leaving groups bind tightly to the S1′ pocket, in the good agreement with the observation that caspase recognize small residues in P1′ position of their substrates.47 Modern warheads for caspase inactivation go beyond S1′ pocket and bind to the S2′, S3′ and so on. Efforts to identify good leaving groups are usually assisted by SAR studies, molecular modeling as well as iterative synthesis, combinatorial approaches and “click” chemistry. It is well documented in the literature that commonly used caspases inhibitors equipped with simple reactive groups like aldehydes (−CHO), fluoromethylketones (−CH2F or FMK), chloromethylketones (−CH2Cl or CMK) or acyloxymethylketones (AOMK) lack specificity.254,291 Peptides with these warheads are broad spectrum inhibitors, that inactivate caspases by indiscriminate processes and they are not tailored for targeting particular caspases specifically. To address this problem many groups from both academia and industry have utilized SAR studies to explore P1′ substituents. Grimm et al used a solid phase strategy to synthesize caspase-3 inhibitors.176 As an optimal peptide backbone Ac-DEVD-sequence was chosen, based on previously reported results. Next, several warheads were anchored to the Ac-DEVD platform, which resulted in the discovery of new caspase-3 inhibitors. All these inhibitors have been tested on caspases -1, -3, -7, and -8 and additionally in NT2 cells. The most promising group for all of these caspases is a-(CH2)3-naphthyl-1-yl warhead, however caspase-3 was inhibited most efficiently (12-fold better than caspase-7 and 19-fold better than caspase-8). In addition, these activated ketones display higher potency comparing to aldehyde counterparts (Table 20).
Table 20.
Some examples of Ac-DEVD-based caspases inhibitors with various P1′ substituents.
|
enzyme assays IC50, nM | cellular assays IC50, μM | Ref | |||
|---|---|---|---|---|---|---|
| rh-Casp-1 | rh-Casp-3 | rh-Casp-7 | rh-Casp-8 | NT2 | ||
| - H 83 | 292 | 44 | 716 | 286 | > 100 | 176 |
 186 |
3775 | 1.3 | 12 | 185 | n.d. | |
|
187 |
4575 | 1.9 | 14 | 140 | n.d. | |
 188 |
1395 | 46 | 363 | 656 | n.d. | |
 189 |
307 | 0.8 | 7.7 | 16 | 30 | |
 190 |
24 | 0.8 | 7.2 | 9.6 | n.d. | |
 191 |
14 | 0.1 | 1.2 | 1.9 | 26 | |
 192 |
451.5 (Ki) | 0.261 (Ki) | - | 159.3 (Ki) | - | 292 |
Another group also investigated S1′ pocket using several Ac-DEVD-ketone inhibitors.293 Fifteen tetrapeptide aspartic acid ketone derivatives were designed and synthesized. The potency of all inhibitors was tested to determine the influence of substitution in the P1′ position on caspase-3 and -7 inhibition. It was demonstrated that, in the case of straight-chain aliphatic series 187 and 193–195 (Table 21), chain elongation correlates with the increase of inhibition potency. This observation was in line with previous studies.235 On the other hand, truncating the linker enhances potency of unsubstituted aromatic P1′ inhibitors. Furthermore, incorporating cyclic group at P1′ dramatically decreases potency (except for cyclobutane). Molecular modeling studies showed that compound 202 (the best of the series) with a cyclobutyl substituent reduces steric interaction between ring system and the cysteine/histidine active site pair, making substantial caspase inhibition possible. It is worth to notice that simple modification of the benzyl moiety (incorporating the second methyl group in orto-position) increases selectivity by 20-fold toward caspase-3. Results presented in that study clearly demonstrate that modification of group in P1′ position in combination with optimizing P4-P2 region is a good approach for obtaining potent and selective caspases inhibitors.
Table 21.
Examples of Ac-DEVD-ketone inhibitors with various P1′ substituents.293
|
|
enzyme assays Ki, nM | |
|---|---|---|
| Casp-3 | Casp-7 | |
| - H 83 | 5.8 | 19.7 |
|
187 |
5.3 | 41.7 |
|
193 |
3.3 | 19.7 |
|
194 |
1.1 | 18.2 |
|
195 |
1.3 | 12.2 |
 196 |
94.2 | 653 |
 197 |
1.5 | 14.5 |
 198 |
3.5 | 16.4 |
 189 |
8.3 | 97.7 |
 199 |
0.7 | 3.0 |
 200 |
0.2 | 4.0 |
 201 |
172 | 420 |
 202 |
2.1 | 3.5 |
 203 |
272 | 573 |
 204 |
2255 | 15842 |
 205 |
905 | 5993 |
The Powers lab has aza-peptide epoxides and Michael acceptors as a new class of cysteine proteases inhibitors.294,295 These inhibitors were designed to closely resemble of optimal peptide based substrates with the conversion of an α-carbon of the Asp residue into nitrogen and the introduction an epoxide or Michael acceptor moiety at P1′. Several warheads were developed and tested against a diverse group of caspases. Initial work from 2002 was the proof of concept that these peptide-based compounds can act as highly potent caspase inhibitors while displaying no cross reactivity with other cysteine proteases. Aza-epoxide and aza-Michael platforms are suitable for being extended in P1′ direction, so subsequent work focused on P1′ optimization to discover potent and selective caspases inhibitors. To achieve this goal a set of inhibitors with different warheads at the prime site and appropriate peptide sequence at the non-prime site for particular caspases was synthesized. For aza-epoxides it was found that S,S stereoisomers are preferred over R,R > trans > cis respectively. Importantly, these inhibitors are more resistant to proteolysis than traditional peptide based inhibitors, which is a desirable attribute required for candidate therapeutic agents. At P1′ caspase-3 and -6 prefer a -COOCH2Ph bulky, hydrophobic warhead, while caspase-8 preferred the small and aliphatic -COOEt group. Four caspases (including pro-inflammatory caspase-1) were discriminated by a combination strategy: appropriate peptide backbone on the left of scissile bond and optimal warhead anchored to an aza-Asp-epoxide moiety (Table 22).247,294 The next two papers from the Powers group provided an excellent study on aza-peptide Michael acceptors targeting seven apoptotic caspases (−2, −3, −6, −7, −8, −9, and −10).212,295 This α,β-unsaturated scaffold, tailored to explore the P′ region, revealed that most caspases prefer trans stereochemistry in their inhibitors. The only exception is caspase-2 that prefers cis stereochemistry. Various warheads of different type were synthesized resulting to develop potent and selective caspase inhibitors. Caspase-2 prefers CH=CH-COOEt (cis) over CH=CH-COOEt (trans) stereoisomer. Caspase-3 and −7 favor bulky, hydrophobic warheads (like CH=CON(CH2-1-Naphth)2. Caspase-6 is sensitive to CH=CH-CONHPh, caspases −8 and −10 prefer trans isomer of -CH=CH-COOEt and caspase-9 is inhibited by -CON(CH3)CH2-1-Naphth (Table 23). This study provided great insight into architecture of the caspase active site on the right to the scissile bond and opened a window for further optimization efforts. The authors mentioned that there is no relevant reason that aza-expoxides or aza-Michaels acceptors could not be useful as therapeutic agents.
Table 22.
Multiple examples of Aza-peptide epoxides with various leaving groups as caspases inhibitors.247
| Aza-peptide Epoxides |
k2, M−1s−1 | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| EP | Casp-1 | Casp-3 | Casp-6 | Casp-8 | ||
| Cbz-Val-Ala-Asp-CH2F | 86 | - | 290 | 16000 | 7100 | 18000 |
| Cbz-Asp-Glu-Val-azaAsp-EP-R - caspase 3/7 specific sequence | ||||||
| - COOEt | 206 | S,S | 11800 | 1070000 | 5440 | 95500 |
| - CO-Phe-NH2 | 207 | S,S | 9250 | 722000 | 6140 | 29600 |
| - CONHCH2Ph | 208 | S,S | 25400 | 1090000 | 6000 | 84400 |
| - COOCH2Ph | 209 | S,S | 54700 | 1910000 | 12700 | 188000 |
| Cbz-Ile-Glu-Thr-azaAsp-EP-R - caspase 6/8 specific sequence | ||||||
| - COOEt | 210 | S,S | 12400 | 4080 | 45800 | 52800 |
| - COOCH2Ph | 211 | S,S | 45800 | 9500 | 86200 | 58500 |
| - CONHCH2Ph | 212 | R,R | 18100 | 6500 | 60100 | 6500 |
| - CO-Ala-NHCH2Ph | 213 | S,S | ND | 3050 | 38200 | 56000 |
Table 23.
Multiple examples of caspase inhibitors based on Aza-peptide Michael acceptors with various leaving groups.212
| Aza-peptide Michael Acceptors |
k2, M−1s−1 | |||||||
|---|---|---|---|---|---|---|---|---|
| Casp-2 | Casp-3 | Casp-6 | Casp-7 | Casp-8 | Casp-9 | Casp-10 | ||
| Cbz-Val-Ala-Asp-CH2F | 86 | 290 | 16000 | 7100 | 18000 | 280000 | 180000 | - |
| Cbz-Asp-Glu-Val-azaAsp-CH=CH-R- caspase 3/7 specific sequence | ||||||||
| -COOEt (cis) | 214 | 26 200 | 1060000 | 11000 | 139000 | 181000 | - | 13500 |
| -COOEt (trans) | 215 | 2 640 | 2130000 | 35575 | 239000 | 272960 | - | 49900 |
| -CON(CH3)CH2Ph | 216 | 660 | 2640000 | 9500 | 275000 | 90300 | 820 | 29400 |
| -CON(CH2Ph)2 | 217 | 110 | 3000000 | 5100 | 359000 | 8600 | 450 | 6300 |
| -CON(CH2-1-Naphth)2 | 218 | 410 | 5620000 | 29700 | 875000 | 9460 | 150 | 32500 |
| Cbz-Ile-Glu-Thr-azaAsp-CH=CH-R - caspase 6/8 specific sequence | ||||||||
| -COOEt | 219 | 300 | 6740 | 88700 | 530 | 56500 | - | 6900 |
| -COOCH2Ph | 220 | 110 | 2300 | 23350 | 660 | 148400 | - | 15200 |
| -CONHPh | 221 | 365 | 7030 | 99200 | 920 | 245000 | 940 | 9210 |
| -CON(CH3)CH2Ph | 222 | 95 | 6000 | 45900 | 390 | 59700 | 845 | 8500 |
| -CON(CH2Ph)2 | 223 | 76 | 9570 | 83900 | 1 140 | 39500 | 1930 | 7930 |
| Cbz-Leu-Glu-Thr-azaAsp-CH=CH-R- caspase 8 specific sequence | ||||||||
| -COOEt | 224 | NI | 5560 | 18700 | NI | 237000 | 37 | NI |
| -COOCH2Ph | 225 | 480 | 4600 | 47600 | 1 570 | 98400 | - | 18900 |
| -CONHPh | 226 | 290 | 4700 | 11400 | 730 | 176000 | 1190 | 6050 |
| -CON(CH3)CH2Ph | 227 | 140 | 6000 | 10800 | 520 | 169000 | 4320 | 8430 |
| -CON(CH2Ph)2 | 228 | 110 | 8630 | 14100 | 760 | 129000 | 1105 | 8080 |
| -CON(CH3)CH21naphth | 229 | 290 | 11200 | 21700 | 1 100 | 179000 | 5030 | 13600 |
In another study of P1′ position, Merck researchers described an extensive SAR study on pyrazinonemonoamides supported by iterative chemical synthesis296 (Table 24). The pyrazinone moiety introduced at P3 led to the discovery of a novel series of inhibitors. The most active and caspase-3 selective inhibitors posses long, aliphatic warheads at P1′ that satisfy caspase requirements on the prime region in the active site. Among these peptidomimetics, two of them (M826 230 and M867 231) displayed high inhibitory activity against recombinant caspases −3, −7 and −8, and good cellular potency in a NT2 cell system.
Table 24.
Examples of peptidomimetics with various leaving groups as caspases inhibitors.296
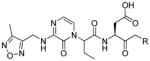
|
enzyme assays IC50, μM | cellular assays IC50, μM | ||||
|---|---|---|---|---|---|---|
| rh-Casp-1 | rh-Casp-3 | rh-Casp-6 | rh-Casp-8 | NT2 | ||
|
232 | 0.15 | 0.078 | 1.00 | 0.99 | 3.56 |
|
233 | 0.22 | 0.0079 | 0.73 | 2.46 | 1.39 |
|
234 | 0.0087 | 0.014 | 0.13 | 4.29 | 1.52 |
|
235 | 0.013 | 0.013 | 0.083 | 7.38 | 1.25 |
|
236 | 1.75 | 0.10 | 1.01 | > 10.0 | 3.99 |
|
|
237 | 0.028 | 0.011 | 0.14 | > 10.0 | 0.13 |
|
|
238 | 0.025 | 0.009 | 0.10 | > 10.0 | 0.15 |
|
| ||||||
|
enzyme assays IC50, μM | cellular assays IC50, μM | ||||
| rh-Casp-1 | rh-Casp-3 | rh-Casp-6 | rh-Casp-8 | NT2 | ||
|
| ||||||
|
239 | 1.23 | 0.064 | 0.48 | 10.0 | 5.12 |
|
240 | 5.13 | 0.014 | 0.14 | 5.28 | 1.38 |
|
241 | 0.62 | 0.021 | 0.13 | > 10.0 | 0.29 |
|
242 | 0.73 | 0.11 | 0.54 | > 10.0 | 8.62 |
|
|
230 | 0.067 | 0.006 | 0.042 | 6.48 | 0.021 |
|
|
231 | 0.11 | 0.0001 | 0.036 | 5.95 | 0.0027 |
SAR on the P1′ position of truncated peptides has been described in context of apoptotic caspases. Ullman and coworkers have studied the effect of leaving group modifications on inhibitory potency toward caspases.274 They synthesized a set of dipeptide 1-Naphthyl-CO-Val-Asp-R analogues, determining that the best leaving group for caspase-3 is (O-1-naphthyl), while caspase-6 and caspase-8 prefer O(2,6-Cl2-Ph) and O(2,3,5,6-F4-Ph) respectively. Moreover, this library of inhibitors have been used in cellular assay with Concanavalin A stimulated monocytes and Jurkat cells. Interestingly, several inhibitors from the series displayed a broad spectrum of activity. A conclusion that can be extracted from this data is that the incorporation of fluorine atom(s) onto the phenyl ring results in increasing cellular potency making these warheads promising candidates for further optimization. In other work the group of Wu used the previously reported 1-Naphthyl-CO-Val-Asp platform for further leaving group optimization,297 synthesizing a set of inhibitors with heterocyclic warheads that were based on a PTP structure (one of the first leaving groups dedicated to caspase family members, especially to caspase-1). These inhibitors were tested against murine caspase-1, human apoptotic caspases −3, −6, −8, and in Jurkat cellular system. Among the warheads were isoxazoles, thioethers, pyrimides or benzofused analogues. Unfortunately, these inhibitors displayed only moderate potency toward caspases and, compared to the previously mentioned phenyl derivatives, appear to be significant weaker in Jurkat cell assays, which confirms that the PTP scaffold and its derivatives are not tailored caspase warheads. In Table 25 we collected some examples of 1-Naphthyl-CO-Val-Asp-based inhibitors. The same group have also used oxamyl dipeptides to investigate P1′298 (Table 26). SAR studies led to the discovery of new group of caspase inhibitors with improved cellular potency compared to the previously reported 1-Naphthyl-CO-Val-Asp derivatives. Moreover oxamyl dipeptides displayed a broad spectrum of action, targeting caspase-1, −3, −6, and −8 efficiently. The inhibitor with -O(2,3,5,6-F4-Ph) was used for further SAR investigation. Soon after the researchers from Idun Pharmaceuticals developed “first-in-class” potent pan-caspase inhibitor with liver disease as a therapeutic indication.299 As a result of extensive SAR studies IDN-6556 243 was discovered (Table 27). This compound was the first irreversible pan-caspase inhibitor to enter Phase II clinical trials targeting liver disease. The optimization of P1′ resulted in the discovery of several additional potent PCIs (pan-caspase inhibitors), with enzyme assays (murine caspase-1, human caspases −3, −6, and −8), cellular assays (JFas, TMP) and a-Fas liver studies clearly demonstrating the utility of these novel inhibitors. All reports concerning P1′ optimization clearly demonstrate that this position is of special interest for caspases investigation since the leaving group can be used for discriminate between individual caspases or can act as a broad spectrum inactivator targeting inflammatory, initiator and apoptotic caspases at the same time.
Table 25.
Examples of dipeptide caspases inhibitors based on 1-Naphthyl-CO-Val-Asp-scaffold with various leaving groups.
|
enzyme assays k3/Ki, M−1s−1 | cellular assays IC50, μM | Ref | ||||||
|---|---|---|---|---|---|---|---|---|---|
| mCasp-1 | Casp-3 | Casp-6 | Casp-8 | Con A | Monocyte | Jurkat | |||
| -F | 246 | 129000 | 207000 | 36842 | 71667 | 3.05 | 0.95 | 0.24 | 274 |
| -OPh | 247 | 3630 | 19152 | 2399 | 4258 | 38.00 | 11.17 | 13.95 | |
| -OCO(2,6-Cl2-Ph) | 248 | 52109 | 725222 | 56883 | 17010 | 2.15 | 11.50 | 10.61 | |
| -O(1-Naphthylyl) | 249 | 354241 | 1958905 | 0 | 0 | 2.65 | 19 | > 100 | |
| -O(2-Naphthylyl) | 250 | 263 | 7521 | 0 | 0 | > 50 | 15.90 | > 100 | |
| -O(2,6-F2-Ph) | 251 | 53875 | 200052 | 9979 | 9075 | 2.40 | > 100 | 2.04 | |
| -O(2,3,6-F3-Ph) | 252 | 47081 | 187700 | 81749 | 16972 | 2.433 | - | 1.195 | |
| -O(2,3,5,6-F4-Ph) | 253 | 77528 | 267953 | 78887 | 30173 | 2.17 | 6.25 | 4.50 | |
| -O(2,6-Cl2-Ph) | 254 | 25259 | 178629 | 87715 | 2713 | 2.75 | 15.32 | 3.00 | |
|
255 | 7572 | 71138 | 23909 | 2266 | n.d. | n.d. | 28 | 297 |
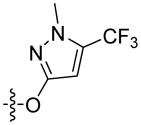
|
256 | 9396 | 35478 | 509 | 295 | n.d. | n.d. | 46 | |
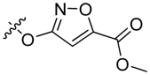
|
257 | 42463 | 143850 | 5804 | 6344 | n.d. | n.d. | 71 | |
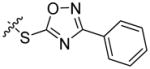
|
258 | 1191 | 38119 | 975 | 3059 | n.d. | n.d. | 102 | |
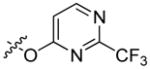
|
259 | 24834 | 113766 | 5147 | 3199 | n.d. | n.d. | 37 | |
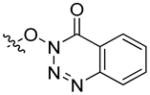
|
260 | 45379 | 168736 | 44007 | 18230 | n.d. | n.d. | 137 | |
Table 26.
Examples of oxamyl dipeptide caspases inhibitors with various leaving groups.298
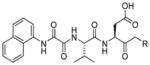
|
Enzyme assays k3/Ki, M−1s−1 | Cellular assays IC50, μM | ||||
|---|---|---|---|---|---|---|
| mCasp-1 | Casp-3 | Casp-6 | Casp-8 | THP-1 | Con A | |
| -O-CO(2,6-Cl2-Ph) 261 | 1052775 | 740466 | 234318 | 119197 | 0.52 | 0.35 |
| -O-(2,3,5,6-F4-Ph) 262 | 892596 | 288748 | 142903 | 447761 | 0.770 | 0.55 |
| -O-POPh2 263 | 2950000 | 876478 | 222420 | 2447090 | 0.65 | 0.65 |
| -O-PO(CH3)Ph 264 | 1404853 | 529376 | 165406 | 847197 | 0.09 | 2.00 |
| -O-(2,6-F2-Ph) 265 | 1295045 | 492401 | 49970 | 311465 | 0.48 | 0.45 |
| -O-(2-CF3-4pyrimidyl) 266 | 1069270 | 293818 | 13349 | 60347 | 0.25 | 2.00 |
| -O-SOPh 267 | 2969 | 342 | 459 | 1 | 8.30 | > 50 |
| -O-(2-naphthyl) 268 | 155894 | 9455 | 31 | 20 | 1.93 | 7.30 |
| -O-(1-naphthyl) 268 | 378200 | 11653 | 268 | 12 | 3.06 | 2.70 |
| -O-Ph(4-Ph) 269 | 58032 | 1659 | 0 | 0 | 1.12 | > 50 |
Table 27.
Examples of oxamyl dipeptide caspases inhibitors with various leaving groups.299
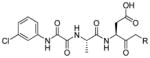
|
enzyme assays k3/Ki, M−1s−1 | cellular assays IC50, μM | - | ||||
|---|---|---|---|---|---|---|---|
| mCasp-1 | Casp-3 | Casp-6 | Casp-8 | JFas | THP-1 | a-Fas liver, IP, ED50 mg.kg | |
| -O(2,3,5,6-F4-Ph) 243 IDN-6556 |
689000 | 75700 | 58700 | 2940000 | 0.025 | 0.27 | 0.08 |
| -OPOPh2 271 | 2158000 | 99100 | 52100 | 2010000 | 0.081 | 9.09 | 0.14 |
| -OCO(2,6-Cl2-Ph) 272 | 8330000 | 236000 | 105000 | 4300 | 0.143 | 11.8 | 45.8 |
Guo et al reported on the utility of N-nitrosoanilines as caspase-3 inhibitors.300 These compounds posses a nitric oxide group (NO), which had previously been demonstrated to inhibit cysteine proteases (protease inactivation was due to S-nitrosylation of catalytic cysteine thiol group). Eight low molecular weight N-nitrosoaniline derivatives were initially screened as candidates for caspase inhibitors, and two molecules were chosen as a platform for synthesis of caspase-3 inhibitors Ac-DVAD-NNO 244 and Ac-DV-AMO 245. These NO donating inhibitors were reported to block caspase-3 activity with Ki values of 2 μM and 22 μM respectively.
Newton and co-workers reported on the synthesis and evaluation of vinyl sulfones as caspase-3 inhibitors. Kinetic analysis of tetrapeptides as well as truncated derivatives showed that these Michael acceptor warheads are not suitable for caspase inhibition. The best compounds from the series displayed only moderate activity.301
3.3.2.3. P2-P3 region
It had been shown by Thornberry and co-workers that various residues are tolerated in substrates at the P3-P2 region without major loss of potency. To date, two main chemical strategies for optimizing the P3-P2 backbone have been developed. The first, traditional, one replaced one or both P2 and P3 substituents in a tetrapeptide platform (peptide inhibitors). The second, more tricky approach is to introduce internally to the peptide backbone mono-, bi-, or tricyclic nonpeptidic moiety mimicking typical P3-P2 amino acids functionality (peptidomimetic inhibitors). Such conformationally constrained inhibitors posses reduced peptide character thus are more chemically stable toward proteolysis and have enhanced bioavailability while retaining high potency and selectivity. Peptidomimetics are highly desired since traditional peptide based inhibitors lack biological utility. For instance Ac-DEVD-phenylpropyl ketone 273, a potent caspase-3 inhibitor (IC50 = 0.8 nM), is not efficient in NT2 cells studies due to its polar nature and poor membrane permeability (IC50 shifted dramatically to 30 μM).176
They are several reports described in the literature concerning optimization of the P3-P2 region without destroying the peptide character of inhibitors. The group of Tomaselli from Idun Pharmaceuticals reported on the utility of acyl dipeptides as caspase inhibitors.275,276 This group of compounds is less potent than tetrapeptide inhibitors, but the appropriate choice of a P3 surrogate can retain nanomolar and broad spectrum activity against caspases. Selected examples of acyl dipeptides and their SAR analysis are presented in Table 28. Wang and others have also truncated Ac-DEVD, a caspase-3 sequence, to the dipeptide Val-Asp dipeptide. Based on this, they discovered that Cbz-Val-Asp-CH2F (MX1013) 274 is a potent, irreversible dipeptide caspase inhibitor that displays good properties both in vitro and in vivo.302 274 have been revealed as an effective cytoprotective agent in several animal models. In further work 274 was modified to explore P2. Several structurally different amino acids were used, for example Ile (aliphatic), Phe (hydrophobic), Lys (basic) and non-proteinogenic homoalanine, phenylglycine, cyclohexylglycine and (2-thienyl)alanine, but no better substitution for valine at P2 was found (Table 29).303 Next, using 274 structure as a starting point, Cai and coworkers identified a new group of potent and broad spectrum caspase inhibitors.304 By SAR of the N-protecting group they found that 2,4-Cl2-PhCH2O-(MX1122) 275 is the most tailored against caspase-3 in enzyme kinetic studies and effective in cell apoptosis protection assays (Table 30).304 Shortly thereafter, starting with the 274 structure Wang et al. synthesized new derivatives with an optimized P3 moiety.305 In this study P2 nitrogen atom was replaced by oxygen to form α-carbamoyl-alkyl-carbonyl-Asp-CH2F inhibitor. Replacement of P3 NH with O was possible since the X-ray crystal structure of caspase-3 with inhibitor showed that this nitrogen has little to no contribution to inhibitory binding. Some newly synthesized inhibitors displayed better activity than 274 in both enzyme and cellular assays. Moreover, MX1153 276 displayed a broad spectrum activity against caspases −1, −3, −6, −7, −8, and −9 and good properties in preventing induction of apoptosis by anti-Fas antibody. A truncated Val-Asp sequence has been also used by the group of Zamboni,306 who synthesized a series of tripeptide inhibitors with the general formula R-Val-Asp-CH2-S-Ph (R-valine-aspartic acid-thiobenzylmethylketone). These iterative SAR studies led to the discovery of series potent caspases inhibitors with improved cellular potency compared to tetrapeptide aldehydes (Table 28). The same group studied the hydrogen-bonding effect in P3 using the previously reported scaffold (Val-Asp-CH2-S-Ph).307 The synthesis and biological evaluation of a new series of truncated caspases inhibitors led to the discovery that two binding mode exist in P3 region.
Table 28.
Examples of dipeptide caspases inhibitors with various N-capping substituents occupying P3 position.
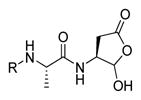
|
enzyme assays IC50, μM | ||||||
|---|---|---|---|---|---|---|---|
| mCasp-1 | Casp-3 | Casp-6 | Casp-7 | Casp-8 | Ref | ||
| Ac-DEVD-CHO | 83 | 0.05 | 0.0035 | 0.01 | 0.01 | 0.08 | 275 |
| 1-Naphthyloxy acetyl | 277 | 0.570 | 0.135 | 0.940 | 1.81 | 0.770 | |
| 2- Naphthyloxy acetyl | 278 | 10 | 0.944 | 18.56 | 8.87 | > 10 | |
| 2-(1-Naphthyloxy)propionyl | 279 | 3.99 | 0.376 | 1.28 | 1.32 | 2.43 | |
| 1-Naphthylmercapto acetyl | 280 | 2.75 | 0.195 | 1.43 | 1.74 | 7.42 | |
| 3-(1-Naphthyloxy)propionyl | 281 | 0.686 | 0.059 | 0.305 | 1.37 | 9.81 | |
| 4-Methoxy-1-naphthyloxy acetyl | 282 | 0.831 | 0.263 | 22.6 | 4.08 | 1.45 | |
| 4-Chloro-1-naphthyloxy acetyl | 283 | 0.429 | 0.231 | 12.0 | 3.38 | 1.69 | |
| 2,4-Dichloro-1-naphthyloxy acetyl | 284 | 0.141 | 0.357 | 21.4 | 3.61 | 3.04 | |
| 2-Biphenoxy acetyl | 285 | 0.636 | 0.095 | 0.717 | 2.02 | 1.71 | |
| (2-Cyclopentyl)phenoxy acetyl | 286 | 0.538 | 0.197 | 3.37 | 1.49 | 1.86 | |
|
| |||||||
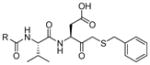
|
enzyme assays IC50, μM | Cell assay IC50, μM | |||||
| Casp-1 | Casp-3 | Casp-7 | Casp-8 | NT2 cells | Ref | ||
|
| |||||||
| Ac-DEVD-CHO | 83 | 0.19 | 0.027 | 0.087 | 0.13 | > 100 | 306 |
| 2,5-di-(OCH3)-Ph-CH2- | 287 | 6.0 | 0.048 | 3.2 | 6.6 | 10 | |
| 2,4-di-Br-Ph-CH2- | 288 | 0.86 | 0.14 | 1.5 | 8.7 | 63.0 | |
| 5-fluoro-1H-indole- | 289 | 2.6 | 0.083 | 1.4 | 3.3 | 55.0 | |
| 2-CH3O-Ph-CH2- | 290 | 28 | 0.31 | 2.7 | 15.5 | - | |
| 3-CH3O-Ph-CH2- | 291 | 14 | 0.21 | 3.1 | 14 | 60.0 | |
| Ph-CH2- | 292 | 22 | 0.75 | 8.4 | 22.5 | - | |
| 2-CH3O-4-Br-Ph-CH2- | 293 | 3.1 | 0.06 | 3.1 | 2.3 | 9.0 | |
| 2-CH3O-4-acetyl-Ph-CH2- | 294 | 5.9 | 0.01 | 0.13 | 2.9 | 2.0 | |
Table 29.
Examples of dipeptide caspase-3 inhibitors with various P2 substituents.303
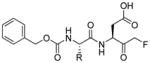
|
enzyme assays IC50, μM Casp-3 |
||
|---|---|---|---|
| Valine, Val MX1013 | 274 |
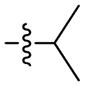
|
0.030 |
| Isoleucine, Ile | 295 |
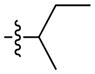
|
0.070 |
| Leucine, Leu | 296 |
|
0.20 |
| Phenylalanine, Phe | 297 |
|
0.40 |
| Alanine, Ala | 298 |
|
0.60 |
| Glycine, Gly | 299 |
|
1.9 |
| Lysine, Lys | 300 |
|
1.6 |
| Glutamicacid, Glu | 301 |
|
14.0 |
| 2-aminobutyric acid, Abu | 302 |
|
0.10 |
| Phenylglycine, Phg | 303 |
|
0.10 |
| Cyclohexylglycine, Chg | 304 |
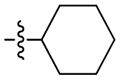
|
0.10 |
| 2-thienylalanine, 2-Tha | 305 |
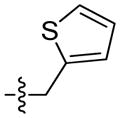
|
0.15 |
Table 30.
Examples of dipeptide caspase -3 inhibitors with various P3 substituents.304
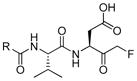
|
enzyme assays IC50, μM Casp-3 |
50% cell protection, nM | |
|---|---|---|---|
| PhCH2O- (MX1013) | 274 | 30 | 250 |
| CH3- | 306 | 250 | - |
| CH3CH2- | 307 | 81 | - |
| PhCH2- | 308 | 61 | - |
| CH3O- | 309 | 37 | 500 |
| PhCH2CH2- | 310 | 98 | - |
| Isopropyl-O- | 311 | 35 | - |
| CyclopentylCH2O- | 312 | 30 | 300 |
| PhCH2CH2O- | 313 | 110 | - |
| PhCH2CH2CH2O- | 314 | 46 | - |
| 2-Cl-PhCH2O- | 315 | 36 | - |
| 3-Cl-PhCH2O- | 316 | 36 | - |
| 4-Cl-PhCH2O- | 317 | 34 | - |
| 2-F-PhCH2O- | 318 | 38 | - |
| 3-F-PhCH2O- | 319 | 29 | - |
| 4-F-PhCH2O- | 320 | 28 | 150 |
| 2,4-di-Cl-PhCH2O- (MX1122) | 275 | 25 | 100 |
| 3,4-di-Cl-PhCH2O- | 321 | 21 | 150 |
| 2,5-di-Cl-PhCH2O- | 322 | 15 | 100 |
| 2,4-di-F-PhCH2O- | 323 | 35 | - |
The discovery of MX1013 274 and MX1122 275 strongly demonstrates that design of caspase inhibitors with potential therapeutic utility is not always about potency and selectivity. For example Ac-DEVD-CHO 83 is very potent caspase-3 inhibitor with IC50 of 0.8 nM, but this peptide aldehyde is useless in cellular system assays (IC50 for NT2 cells is greater than 100 μM). This feature is probably due the high polarity of the DEVD sequence, resulting in poor cell penetration, and also high affinity of aldehydes for non-specific reaction with bionucleophiles. To address this problem Grimm and coworkers synthesized three peptide inhibitors with various P3 alterations: reference glutamic acid, double oxidized methionine and alanine.176 Replacement of glutamic acid with other amino acids resulted in 2-fold decrease in activity on recombinant caspase-3, but it also led to the improvement of biological properties (lower IC50 values in NT2 cellular assay). This approach demonstrates that inhibitors with Glu in P3 are not suitable for cellular assays likely due to its polar characteristic and poor cell permeability.176
Natural amino acids have been extensively explored for the synthesis of apoptotic caspases inhibitors, however modern inhibitors (not only for caspases) go beyond the utility of only proteinogenic amino acids. The group of Luisi reported that prototypic peptide caspase-3 inhibitor Ac-DEVD-CHO 83 can be truncated to the Cbz-tert-Leu-Asp-CHO 324 dipeptide still maintaining the high inhibitory potency.308 Non-pretoinogenic tert-leucine possess a bulky and lipophilic side chain, which can induce many productive van der Waals interaction with the hydrophobic S2 pocket of caspase-3. The utility of this inhibitor was demonstrated using a biological assay in DLD-1 cells, where Cbz-tert-Leu-Asp-CHO 324 showed anti-apoptotic activity. Ferrucci and coworkers demonstrate that enzyme activity can be inhibited by peptidyl aldehydes containing only two amino acids.309 To overcome weak stability and poor cell permeability of standard caspase-3 inhibitors the authors synthesized di- and tri-peptidyl aldehydes. These new compounds inhibit caspase-3 activity in vitro with Ki∘ values ranging between 6.5 nM and 110 nM (Ki∘ = Ki at pH 7.5, and 25.0°C). The capacity of Ac-tert-Leu-Asp-CHO 325 to inhibit apoptosis implied good cell permeability of this inhibitor giving significant opportunities for in vivo applications. A much more extensive studies engaging non-proteinogenic amino acid for the synthesis of caspases inhibitors has been made by the group of Bogyo.18 This work explored more than 40 unnatural amino acids for the synthesis of libraries encompassing P2, P3, and P4, equipped with acyloxymethylketone warheads (AOMKs). After combinatorial library positional screening, individual inhibitors were tested against caspases −3, −7, −8 and −9. This work demonstrated the utility of inhibitor screening incorporating peptide sequences containing unnatural (non-proteinogenic) amino acids.
Caspases specific inhibitors with optimized P3-P2 region have been also obtained via a combined substrate-inhibitor strategy. Leyva and coworkers synthesized and screened a substrate library with an aminocoumarin as a reporting group.310 The best hits from the library contain 1,2,3-triazole scaffold occupying P3-P2 region. Next, these were transformed into inhibitors by the replacement of this fluorophore with the 2,3,5,6-tetrafluorophenoxymethyl ketone leaving group. Three inhibitors from this series appeared as pan-caspases inhibitors and inhibit caspase-3 and −6 activity and preventing huntingtin (HTT) proteolysis at the embedded caspases cleavage site (Table 31).310 This feature is important since caspase-dependent huntingtin cleavage is associated with development of Huntington’s disease.
Table 31.
Kinetic data for three best 2,3,5,6-tetrafluorophenoxymethyl caspases inhibitors.311
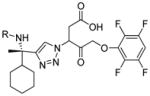
|
enzyme assays kinact/KI, M−1s−1 | ||||||
|---|---|---|---|---|---|---|---|
| mCasp-1 | Casp-2 | Casp-3 | Casp-6 | Casp-7 | Casp-8 | Casp-9 | |
| Ph-CH2-SO2-326 | 55200 | 33.0 | 47000 | 23100 | 27700 | 82850 | 18250 |
| Ph-CH2-CO-327 | 243250 | 2.0 | 81700 | 32950 | 57250 | 30650 | 7550 |
| HOOC-(CH2)3-CO-328 | 55200 | 6.90 | 29850 | 45750 | 34200 | 60000 | 28100 |
In another approach, Allen and coworkers from Sunesis Pharmaceuticals used a Tethering strategy to investigate the caspase P2 pocket.312 Their initial work resulted in the discovery of a caspase inhibitor series whose structures bypasses the S2 and S3 active site pockets. In further work they synthesized several inhibitors with various substituents at P2. The best hits from the series contained phenyl, 3-chloro-phenyl and 2-thienyl ligands. Incorporation of these groups at P2 significantly enhanced potency toward caspase-3 compared to a non-substituted prototype inhibitor.
Starting from the thesis that tetrapeptide caspase-3 inhibitors exhibit poor cell penetration, Isabel et al. attempted to identify novel cell-penetrating inhibitors.313 In the first step the authors synthesized libraries of capped aspartic acid aldehydes with the best hit from the library containing 5-bromonicotinic acid at P2. This scaffold was then used to optimize P1′ in which thioethers displayed the highest potency. The last step was to optimize the P3 position by introducing various groups in position 5 of the pyridine ring. Sulfonamides displayed the highest biological activity from the series. This champion compound 332 was finally transformed into an acyloxymethylketone making it a potent and irreversible caspase-3 inhibitor (IC50 = 1.3 μM in a NT2 cell assay) (Figure 21).
Figure 21.

Three step optimization of 3-(5-bromonicotinamido)-4-oxobutanoic acid to obtain caspase-3 potent inhibitor 332.
Conformationally constrained peptides with appropriate warheads are also potent caspase inhibitors. In their structure the common P3-P2 dipeptide motif is replaced by a mono-, bi- or tricyclic core, which maintains key hydrogen bonds with the caspase active site and explores the S2 and S3 subsites. Karanewsky et al. synthesized several inhibitors based on the Cbz-Val-Asp-CHO scaffold in which the P2 amide nitrogen was “tied back” to the either or both the P3 and P2 side chain.314 This strategy opened the door for others to pursue new caspases inhibitors with constrained peptide backbone. For example, Karanewsky and Linton described the synthesis and evaluated oxoazepinoindoles (OAIs) as a new scaffold for caspases inhibitor discovery.315 Some of these inhibitors were employed in biological assays, for instance IDN-5370 333 and IDN-7866 334 were tested in a mouse stroke model.316 In Figure 22 we present structures 333–337 of conformationally constrained caspase inhibitors.
Figure 22.

Structures of conformationally constrained apoptotic caspases inhibitors. Above – examples where the P2 amide nitrogen was “tied back” to the either or both P3 and P2 side chain. Below – two examples of oxoazepinoindoles (OAIs).
Analogously to what was observed by Karanewsky, another group of researchers synthesized a monocyclic conformationally constrained analogue of Ac-DEVD-CHO 83 in which the dipeptide Glu-Val was replaced by a 1,4-benzodiazepine moiety.317 This inhibitor exhibited lower inhibitory activity (Ki = 36 ± 9 nM) than the commonly used tetrapeptide 83 (Ki = 3.2 ± 6 nM), however its selectivity toward caspase-3 and the capability to impair apoptosis in live cells makes it an attractive target for further studies.
Vazquez and coworkers proposed a new scaffold for the inhibition of caspase-3 activity focused on incorporation of a hydantoin ring at P3, which induces conformational restriction in the backbone of inhibitors.318 Preliminary studies disclosed that the hydantoin moiety can occupy the S3 pocket of the enzyme, thus the next step was to optimize the side chain by incorporating selected amino acids in the hydantoin ring. Aldehyde or phenylpropyl ketone warheads were employed and aminobutyric acids was used as a spacer between the aspartyl and hydantoin moiety. Compound 338 with Pbf-protected guanidine group showed good inhibitory activity (IC50 = 3.9 μM), thus was selected for further optimization. Substitution of the guanidine group in the Arg side chain revealed that aromatic sulfonyl groups or nitrated and compounds with tetra-alkylated guanidines showed almost the same inhibitory activity as inhibitor 338. Finally, the authors decided to replace the aldehyde group with a less active, but more caspase specific P1′ trap. Several nucleophiles including thiol, alcohols and secondary amines were tested, however only compound 339 with an S-methylphenyl chain exhibited similar biological activity to that of 338. In Figure 23 we present the two step optimization of hydantoin-containing caspase-3 inhibitors.
Figure 23.

Optimization of a hydantoin-based lead compound to obtain novel scaffold for a caspase-3 inhibitor.
3.3.2.4. P4 position
Because peptide-based inhibitors lack specificity among caspases further efforts for their optimization in the context of selectivity are highly desired. We previously mentioned that typical truncation of tetrapeptide inhibitors to their mono- or dipeptide derivatives results in decreasing potency against targeted caspases. But on the other hand these truncated inhibitors have better pharmacokinetic profile (better cell permeability, higher metabolic stability), thus are more useful in cellular and in vivo conditions. The compromise between potency in vitro and in vivo can be achieved by rational optimization of P4, a main determinant of selectivity within the caspase family. Group from Sunesis Pharmaceuticals used a Tethering strategy to explore S4 of caspase-3,319,312 They synthesized several aldehyde inhibitors based on a pyridine scaffold with various P4 substituents. The best inhibitor from the series 340 displayed a Ki value of 0.05 μM and was used for further, iterative optimization (P3 position and leaving group). This “step by step” strategy led to the discovery of a caspase-3 inhibitors 340–349 with moderate potency that have a structural potential for further optimization (Table 32). Extensive SAR studies on P4 of caspases inhibitors have been also performed by Han et al..296 The tripeptide scaffold based on a pyridine core was N-capped by various heterocyclic substituents. It was found that the best group at P4 is a 1,2,5-oxadiazole moiety 357 (Table 33). The lead inhibitor N-capped with this group was optimized in other positions (P3 and P1′), finally leading to the discovery of the previously reported M826 230 and M867 231, potent and selective caspase-3 inhibitors with therapeutic utility. Shortly thereafter Linton et al. used oxamyl dipeptides for SAR studies directed toward P1′, P2 and P4 regions.298 Almost 50 compounds with diverse P4 were synthesized and tested toward murine caspase-1, human caspases −3, −6, and −8 and also used in four cellular assays (Jurkat, THP-1, Con A and SKW 6.4). This study demonstrated that many different substituents are tolerated in P4. Several interesting examples 358–369 are presented in Table 34. Recently oxamyl dipeptides have also been investigated by Ueno and coworkers.320 A new collection of aryloxymethylketones with various P4 moieties was screened with caspases-3, −8 and −9 and tested in a Fas cellular assay. Selected compounds from the series displayed high in vivo activity, making the oxamyl derivatives an interesting scaffold for further development.
Table 32.
Examples of caspase-3 inhibitors with various P4 substituents designed using tethering technology.
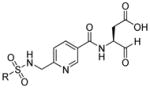
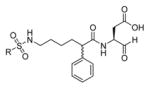
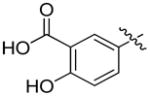
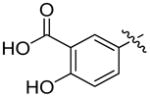
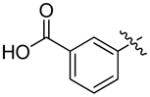
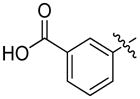
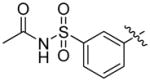
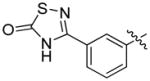
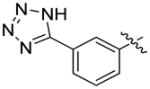
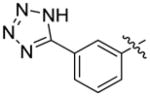
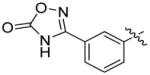
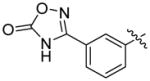
Table 33.
Examples of caspases inhibitors with various P4 substituents designed using tethering technology.296
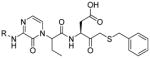
|
enzyme assays IC50, μM | cellular assays IC50, μM | ||||
|---|---|---|---|---|---|---|
| rh-Casp-1 | rh-Casp-3 | rh-Casp-7 | rh-Casp-8 | NT2 | ||
|
350 | 0.90 | 0.080 | > 10.0 | 9.10 | 15.30 |
|
351 | 0.087 | 0.14 | 3.28 | 8.22 | 30.0 |
|
352 | 0.065 | 0.036 | 0.81 | 2.09 | 4.56 |
|
|
353 | 0.92 | 0.071 | 9.73 | 7.65 | > 10.0 |
|
354 | 1.93 | 0.018 | 9.71 | > 10.0 | 5.74 |
|
355 | 0.13 | 0.026 | 1.13 | 1.75 | 3.08 |
|
356 | 0.19 | 0.012 | 0.49 | 6.59 | 3.42 |
|
357 | 0.22 | 0.0079 | 0.73 | 2.46 | 1.39 |
Table 34.
Examples of oxamyl dipeptides with various P4 substituents as caspases inhibitors.298
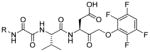
|
enzyme assays K3/Ki, M−1s−1 | cellular assays IC50, μM | |||||||
|---|---|---|---|---|---|---|---|---|---|
| mCasp-1 | Casp-3 | Casp-6 | Casp-8 | Jurkat | THP-1 | Con A | SKW 6.4 | ||
| Me- | 358 | 16846 | 149284 | 126292 | 21165 | 0.11 | 1.23 | 7 | 1.04 |
| Ph- | 359 | 158722 | 327155 | 274616 | 281943 | 0.43 | 0.07 | 1.5 | 0.94 |
| PhCH2- | 360 | 259386 | 388350 | 218579 | 12416 | 0.0771 | 0.57 | 1.5 | 0.91 |
| 1-Naphthyl- | 361 | 892596 | 288748 | 142903 | 447761 | 0.0061 | 0.77 | 0.55 | 1.61 |
| 2-Naphthyl- | 362 | 625087 | 395393 | 483945 | 588771 | 0.0007 | 0.7 | 3.28 | 0.38 |
| 1-Anthryl- | 363 | 17 606236 | 260083 | 263666 | 1917858 | 0.01 | 0.22 | 0.85 | 1.96 |
| 2-tert-Bu-Ph- | 364 | 955812 | 268006 | 424103 | 2302210 | 0.0022 | 1.85 | 5.68 | 0.34 |
| 4-F-Ph- | 365 | 106344 | 8161648 | 247449 | 179386 | 0.019 | 1.21 | 2 | 0.92 |
| 2-Cl-Ph- | 366 | 2 019148 | 1173738 | 654890 | 2137136 | 0.016 | 0.16 | 0.3 | 0.11 |
| 2-Br-Ph- | 367 | 7 157535 | 1274281 | 504107 | 2346190 | 0.356 | 0.54 | 0.4 | 1.27 |
| 2-I-Ph- | 368 | 1 980934 | 563535 | 373103 | 2328063 | 0.081 | 1.5 | 1 | 1.54 |
| 2-F;4-I-Ph | 369 | >10000000 | 206962 | 132506 | 83952 | 0.01 | 0.41 | 0.7 | 0.15 |
3.4. Focus on inhibitors containing unnatural amino acids
As discussed earlier, the finite pool of natural amino acids does not allow for the design of highly selective caspase inhibitors – ones that are able to distinguish between members of the family. The solution to this problem was proposed by the Bogyo group who modified the essence of PS-SCL to generate a new chemical tool for caspase investigations – PSCL-AOMK (the Positional Scanning Combinatorial Libraries based on AOMKs inhibitors).18 These nitrophenyl acetate (NP) capped tetrapeptide libraries contained natural and an additional 41 unnatural amino acids allowing exploration of enhanced chemical space (Figure 24). In first step authors demonstrated that inhibitors with only natural amino acids lack the specificity and then they tested unnatural libraries on four recombinant apoptotic caspases: executioner −3 and −7, and apical (initiator) −8 and −9. The screening results were presented as “affinity fingerprints” providing detailed information regarding caspases preferences. NP-DEVD-AOMK (AB09 370) is a very potent caspase-3 and −7 inhibitor, however it also inhibited caspase-8. By introducing unnatural amino acid (NN3 or NN34) in the P3 position the authors retained highly potent inhibitors (AB06 371 and AB13 372) but with improved selectivity against caspase-8. Another natural inhibitor 373 (NP-LETD-AOMK, AB08) binds not only to its intended enzyme caspase-8 but also cross-reacts with caspase-3. To achieve new, caspase-8 specific sequence the authors synthesized two new AOMK inhibitors: AB20 374 (with unnatural amino acids at P4, NN29) and AB19 375 (with NN31 at P4 and NN23 at P2). AB20 374 is more potent than the natural amino acid-containing AB08 373 but still is recognized by caspase-3 and additionally by caspase-9. On the other hand AB19 375 is not recognized by caspase-9 but still inhibits caspase-3. Most difficult was to design new probes selective for caspase-9. The natural NP-LEHD-AOMK 376 was a good inhibitor for caspase-9 and −7 and much better for caspase-3 and −8. Through PSCL analysis it was possible to design only two inhibitors (AB38 377 and AB42 378) that displayed a slightly enhancement of selectivity, however both of them contain only natural amino acids. The kinetic parameters of both natural and new unnatural inhibitors were gathered in Table 35.
Figure 24.

Nitrophenylacetate (NP) capped tetrapeptide AOMK library containing natural and unnatural amino acids for caspases investigation.18
Table 35.
Kinetic parameters of caspases AOMK inhibitors containing natural and unnatural amino acids. The chemical structures of unnatural amino acids are also presented.18
| NP-peptide-AOMK | kobs/[I], M−1s−1 | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Target | Inhibitor | Sequence | Casp-3 | Casp-7 | Casp-8 | Casp-9 |
| Casp-3/7 | AB09 370 | D-E-V-D | 10922261 | 1529040 | 1077839 | < 5000 |
| AB06 371 | D-3-V-D | 7456511 | 968070 | 32909 | NI | |
| AB13 372 | D-34-V-D | 3416050 | 279519 | < 5000 | NI | |
| Casp-8 | AB08 373 | L-E-T-D | 127835 | 19424 | 599788 | < 5000 |
| AB20 374 | 29-E-T-D | 570900 | 181332 | 1071401 | 41300 | |
| AB19 375 | 31-E-23-D | 179086 | 42994 | 396225 | NI | |
| Casp-9 | AB07 376 | L-E-H-D | 75295 | 10447 | 506912 | 20141 |
| AB38 377 | P-L-A-D | 46108 | 27814 | 19676 | 18004 | |
| AB42 378 | I-F-P-D | 892045 | 42594 | 22544 | 44709 | |
|
| ||||||
|
| ||||||
The wide range of commercially available unnatural amino acids suitable for solid phase synthesis offers an excellent tool to discover new, more specific caspase probes.45,321 However, there are several questions regarding this new opportunity and the most important of them seems to be: which amino acids should be used in libraries synthesis. There is no simple answer for this question. The Bogyo group employed 41 unnatural amino acids to find new, more specific inhibitors for caspases −3, −7, −8, and −9, however the absolute specificity has not been achieved for none of these caspases. The best hit for caspase-3 372 (AB13) displayed 9-fold selectivity over caspase-7 and >680-fold selectivity over caspase-8. Another inhibitor 375 (AB19) engineered for caspase-8 was 2.2-fold and 9.2-fold more selective over caspases-3 and −7 respectively. None of these inhibitors (372 and 375) inhibits caspase-9. Recently the Wolan group returned to the idea of using unnatural amino acids in inhibitor libraries for studying caspases. They initially focused on developing a caspase-3 specific inhibitor and activity based probe.20 The commonly used active site directed DEVD-based inhibitors are not caspase-3 specific labeling also caspases −6, −8, −9 and especially caspase-7 which shares 77% active site identity with caspase-3. To solve this problem the authors synthesized 120 aldehyde-trapped peptides possessing the DEVD core. This pool of inhibitors was equally divided into four groups: Ac-P5DEVD-CHO, Ac-P4EVD-CHO, Ac-DP3VD-CHO, and Ac-DEP2-CHO where 30 unnatural amino acids were applied. Next these inhibitors were tested against caspase-3 and −7 and then P5-P2 “affinity matrixes” were made. The authors found that the tetrapeptide Ac-D-βhLeu-hLeu-D-CHO 379 displays 60-fold selectivity for caspase-3 over caspase-7, however its affinity to caspase-3 comparing with Ac-DEVD-CHO 83 decreased over 50-fold. To maintain both high affinity and selectivity to caspase-3 the authors added to this tetrapeptide 3-pyridinyl-Ala occupying the P5 position and exchanged the aldehyde moiety to an AOMK warhead (Ac-CV3-AOMK 381). The new inhibitor indeed had a nanomolar range IC50 value and high index of selectivity. These inhibitors 379–381 were also tested against other apoptotic caspases (−6, −8, and −9) showing only little to moderate inhibition of these enzymes. In further studies Vickers et al. sought new warheads binding specifically to S1, to improve the potency of their champion inhibitor toward caspase-3 and reduce off target interaction with other caspases.63 To reach this goal the authors synthesized 24 Ac-DEVD-based acyloxymethylketones and tested them against caspases. From this pool the 5-methoxy-2-thiophene (KE) AOMK-warhead appeared to be most specific toward caspase-3, so in next step this moiety was attached to the previously published Ac-CV3 sequence to obtain (Ac-CV3-KE) inhibitor 382. This two step approach (first finding the optimal recognition element and combining it with the optimal warhead) resulted in discovery of a new very specific caspase-3 inhibitor containing unnatural amino acids. All the kinetic data as well as the structures of these unnatural inhibitors are presented in Table 36. Encouraged by these results, the Wolan group used the same approach to design a new, specific inhibitor for apical caspase-8.322 The chemical library consisted on 184 LETD-based aldehydes peptides divided equally into four 46-elements groups: Ac-P5LETD-CHO, Ac-P4ETD-CHO, Ac-LP3TD-CHO, and Ac-LEP2-CHO where 16 natural and 30 unnatural amino acids were used. This library was then screened against caspase-3 and -8. Analysis of the P2, P3 and P4 libraries indicated respectively hydroxyproline (Hyp), glutamine (Gln) and leucine (Leu) to be most specific for caspase-8 in these positions. Analysis of a P5 library did not provide any “specific” amino acids however to improve water solubility the authors introduced NH2-β-Ala into P5 of the inhibitor. The C-terminal aldehyde library screening provided a new caspase-8 recognition element with the sequence NH2-β-Ala-Leu-Gln-Hyp-Asp (CV8/9) which was further attached to two warheads AOMK and KE generating CV8/9-AOMK 384 and CV8/9-KE 385. Detailed kinetic analysis of these two caspase-8 inhibitors demonstrated that indeed these compounds displayed high selectivity over the executioner caspases (-3,−6 and −7), however they also both inhibit caspase-9 (Table 37). In addition, these new inhibitors have significantly reduced activity toward caspase-8 than the reference LETD-based probes. Based on the sequences of the inhibitors the group also synthesized specific Activity Based Probes (described in the next section). It is noticing that the application of unnatural amino acids in peptide inhibitors libraries is still an under-explored area and the use of wider ranges of these amino acids might result in the discovery of even more potent and selective inhibitors.
Table 36.
Kinetic data of novel peptide based caspase inhibitors containing unnatural amino acids. Ac-CV3-AOMK and Ac-CV3-KE are caspase-3 specific inhibitors.63
| INHIBITOR | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| Casp-3 | Casp-6 | Casp-7 | Casp-8 | Casp-9 | C-7/C-3 ratio | |
| Ac-DEVD-CHO 83 | 0.021 | 0.048 | 0.040 | 0.038 | 2.00 | 1.90 |
| Ac-D-βhL-hL-D-CHO 379 | 1.1 | > 200 | 66 | > 100 | > 100 | 60 |
| Ac-D-βhL-hL-D-AOMK 380 | 0.080 | 4.2 | 0.81 | 1.1 | 10 | 10 |
| Ac-3Pal-D-βhL-hL-D-AOMK(Ac-CV3-AOMK) 381 | 0.023 | 3.4 | 0.73 | 0.40 | 4.6 | 29 |
| Ac-3Pal-D-βhL-hL-D-KE(Ac-CV3-KE) 382 | 0.0068 | > 10 | 0.730 | 0.190 | n.d. | 107 |
|
| ||||||
|
| ||||||
Table 37.
Kinetic data of novel peptide based caspase inhibitors containing unnatural amino acids. CV8/9-AOMK and CV8/9-KE are caspases 8/9 specific inhibitors.322
| INHIBITOR | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| Casp-3 | Casp-6 | Casp-7 | Casp-8 | Casp-9 | C-3/C-8 ratio | |
| Ac-LETD-AOMK 383 | 0.020 | 0.068 | 0.46 | 0.028 | 0.089 | 0.71 |
| NH2-βA-L-Q-Hyp-D-AOMK(CV8/9-AOMK) 384 | 43 | 220 | 250 | 0.30 | 0.32 | 143 |
| NH2-βA-L-Q-Hyp-D-KE(CV8/9-KE) 385 | 150 | 320 | 400 | 0.30 | 0.64 | 433 |
|
| ||||||
| kinact/Ki (mM−1s−1) | ||||||
| Ac-LETD-AOMK 383 | 490 | n.d. | n.d. | 880 | 1300 | 0.56 |
| NH2-βA-L-Q-Hyp-D-AOMK(CV8/9-AOMK) 384 | 0.14 | n.d. | n.d. | 150 | 190 | 0.0009 |
|
| ||||||
| ||||||
3.5. Nonpeptidic caspase inhibitors
So far numerous of peptide based inhibitors have been developed for targeting caspases. Despite their high potency and selectivity caspase inhibitors usually fail clinical trials due to their poor bioavailability and metabolic stability, or toxicity (usually hepatic toxicity). To address this problem many low molecular weight, non peptide-based caspase inhibitors have been evaluated. Most of them display the winning combination of high affinity for caspases and desirable pharmacokinetics properties. The family of non-peptide caspase inhibitors contain several different scaffolds in their structure 386–393: isatins, indolones, isoquinoline-1,3,4-triones, quinolines, quinazolines, pyridazines, quinones and more (Figure 25).323–327 Most of them act as reversible inhibitors and share a common mechanism of caspases inactivation. A few of them are also referred to as Michael acceptors or epoxide derivatives, and because of this they can display other inhibition modes, depending on their nature. Here we present multiple examples of non peptide small molecule caspase inhibitors.323–327
Figure 25.

Highly functionalized core structures of non-peptidic caspase inhibitors.243,324,327,328
3.5.1. Isatin- and indolone-based inhibitors
Isatin and indolone derivatives are small, non-peptidic compounds that act as alkylating agents of the thiol group in the active site of caspases.323 The mechanism of inhibition is based on a tetrahedral intermediate, which is formed between the carbonyl group of the isatin moiety and the catalytic cysteine residue. Inhibitors containing an isatin or indolone core are a very attractive starting point for activity based probe and drug development since they possess a few “anchor” points that can be easily extended or chemically modified to obtain very powerful and selective inhibitors.
Lee et al. were first to notice that isatin derivatives can inhibit caspases activity.323 They demonstrated that 5-nitroisatin 394 inhibited caspase-3 with a Ki(app) of 500 nM and caspase-7 with a Ki(app) of 290 nM. They modified the isatin structure by replacing the 5-nitro group with a (S)-1-methyl-5-(2-(phenoxymethyl)pyrrolidin-1-yl)sulfonyl moiety. The best compound they found 395 inhibits caspase-3 with a Ki(app) of 15 nM and caspase-7 with a Ki(app) of 47 nM (Figure 14). It was the first isatin based caspase-3/7 specific inhibitor, since its ability to inhibit other caspases is reported to be very poor. X-ray co-crystal structure of isatin sulfonamide based inhibitor with caspase-3 revealed that the pyrrolidine ring from isatin binds to the S2 hydrophobic pocket, which is constituted by three hydrophobic residues (Tyr204, Trp206 and Phe256). This feature, which is unique to caspase-3 and -7 is a very good discriminating agent between caspases in the context of specific inhibitors and activity based probes design. Isatin based inhibitors do not bind to S1, S3 and S4 pockets. This is of interest because inhibitor/substrate interaction with S1 was previously believed to be critical for caspase recognition and activity. Moreover, caspase active site determinants S3 and S4 were believed to be critical for selectivity within the caspase family.24 Next, these compounds were tested as apoptosis inhibitors in chondrocytes.323 It was found that isatin sulfonamide analogues can block chondrocyte apoptosis, which strongly confirms that this group of compounds plays a role as efficient caspase-3 and -7 inhibitors. The next effort to further investigate isatin inhibitory activity was made also by Lee et al., who designed and synthesized a new set of isatin sulfonamides analogues by (1) substitution of the pyrrolidine ring and (2) alkylation of the isatin nitrogen.329 The most potent inhibitor they found 396 inhibits caspase-3 with a Ki(app) of 1.2 nM and caspase-7 with a Ki(app) of 6.0 nM. Ki values for other caspases were in micromolar range (except caspase-9, Ki(app) = 120 nM). These results confirmed previous observations that selectivity toward caspase-3 and -7 can be achieved through interaction of inhibitor (pyrrolidine ring) with the S2 pocket. None of synthesized inhibitors possesses acidic functionality, to positively interact with S1 pocket, nevertheless they are very potent caspase-3 and -7 inhibitors. The next effort to further investigate isatin inhibitory activity was made also by Lee et al., who designed and synthesized a new set of isatin sulfonamides analogues by (1) substitution of the pyrrolidine ring and (2) alkylation of the isatin nitrogen.329 The most potent inhibitor they found 396 inhibits caspase-3 with a Ki(app) of 1.2 nM and caspase-7 with a Ki(app) of 6.0 nM. Ki values for other caspases were in micromolar range (except caspase-9, Ki(app) = 120 nM). These results confirmed previous observations that selectivity toward caspase-3 and -7 can be achieved through interaction of inhibitor (pyrrolidine ring) with the S2 pocket. None of synthesized inhibitors possesses acidic functionality, to positively interact with S1 pocket, nevertheless they are very potent caspase-3 and -7 inhibitors. The key structural feature that is related to high potency for inhibiting effector caspases -3 and -7 is a benzyl moiety attached to the isatin nitrogen atom and (S)-2-phenoxymethy group in the 2-position of the pyrrolidine ring. It has been also demonstrated that these inhibitors can interact with cytosolic constituents, which results in a shift in inhibitory activity of isatin sulfonamides between in vitro isolated caspase-3 and cell based assays. In additional work Chu et al. extended an isatin sulfonamide library by using a simple SAR strategy.330 They modified an isatin based inhibitor by (1) replacing the pyrrolidine ring with an azetidine ring, (2) replacing the benzene ring with a pyridine ring and (3) substitution the para position of the N-benzyl moiety or replacing it with a pyridine ring. This strategy produced several new N-benzylisatin sulfonamides whose analogues are more potent toward caspase-3 and -7 than the previously reported compounds.323 Inhibitor 397 appeared to be the most potent of the series. The authors found that pyrrolidine and azetidine analogues have similar affinity for caspase-3 and -7 and pyridine analogues have higher potency than their phenyl congeners. This work provided insight into SAR between isatin sulfonamide inhibitors and caspase-3 and -7. The group of Kopka developed a 5-pyrrolidinylsulfonyl isatin structure to obtain a new, potent caspase-3 inhibitor 398 with the Ki of 0.2 nM.331 In this work they synthesized a new set of inhibitors by attaching different groups to the isatin core nitrogen atom. Some of these compounds were tested via Western blot analysis to confirm their affinity to caspase-3. The same approach was used by Krause-Heuer et al. for the synthesis of a new set of fluorinated 5-pyrrolidinylsulfonyl isatin derivatives.332 Some of these new compounds were more potent against caspase-3 than the previously described 398. The main goal of this work was to find new compounds applicable in PET imaging of apoptosis, which is described in the next section. A new class of isatin based caspase inhibitors was discovered by Chu et al.333 who synthesized and evaluated a series of isatin derivatives containing a Michael acceptor (IMA). These inhibitors are potent and selective of caspase-3 and -7 and exhibit low potency towards caspase-1, -6 and -8. Compound 399 displayed the highest affinity against caspase-3 within the library, and it appeared that a new inhibition mechanism had been found. One inhibitor possessing a Michael acceptor group was treated with benzylmercaptane mimicking the thiol active site of caspases. Two products formed that confirmed that this new class of isatin based inhibitors can be useful for targeting caspases. The same group designed and synthesized a new series of isatin derivatives containing a Michael acceptor as selective caspase-6 inhibitors.334 They replaced the L-phenoxymethylpyrolidine ring in isatin sulfonamide compounds with another nitrogen heterocycle and demonstrated that the thiomorpholine analogue 400 exhibited a high nanomolar potency for caspase-6 with enhanced selectivity towards caspase-6 versus caspase-3. This compound could serve as a leading structure for development of new, highly potent caspase-6 inhibitors.
One of the main problem with the isatin sulfonamide inhibitors is their only moderate biological stability. To overcome this difficulty Smith et al. synthesized a novel series of isatin inhibitors containing a 1,2,3-triazole moiety in their structure.335 This chemical modification resulted in an improved metabolic profile and biodistribution, without altering the high affinity for caspases, compared to inhibitors containing other substituents on the nitrogen atom. The best inhibitor was 401. Jiang and Hansen also reported the synthesis of isatin derivatives with a 1,2,3-triazole moiety,336 demonstrating that introduction of a 1,2,3-triazole group to the nitrogen atom of isatin 402 can result in enhanced inhibitory activity compared to isatin non-substituted analogues, 401.
The next problem that needed to be solved was a very similar potency of isatin sulfonamide analogues for caspase-3 and caspase-7. Almost all tested inhibitors bind to these enzymes with similar affinity. Podichetty et al. synthesized and tested a new series of N-substitued (S)-5-[1-(2-methoxymethylpyrrolidinyl)2sulfonyl]isatin derivatives.337 Interestingly, N-propyl 403 and N-butyl 404 isatins derivatives were shown to be potent inhibitors having different affinities for caspase-3 and -7. Moreover, both inhibitors failed to bind caspases -1, -6 and -8. In another paper Podichetty et al. designed and synthesized two new classes of isatin analogues (the bromofluorides, 405 and fluorohydrins, 406).338 This research confirms an assumption that there are many chemically distant groups that can be introduced at the isatin nitrogen atom with minimal consequences in affinity for caspase-3 and -7. Recently the Haufe group used a previously published (S)-5-[1-(2-methoxymethylpyrrolidinyl)2sulfonyl]isatin 407 structure as a lead compound for discovery of new caspase-3 inhibitors. Initially they synthesized a set of 407 derivatives where two position were changed: position 2 on the pyrrolidinyl ring (to explore caspase-3 S3 pocket) and the nitrogen atom on isatin core (to explore caspase-3 S1 pocekt). The best moiety in position 2 on the pyrrolidinyl ring is still a methoxy group (previously published), however by introducing the n-Butyl group on the isatin nitrogen 404 enhanced potency against caspase-3 was achieved.339 In the second paper the same group synthesized a library of 4- and 5-substitued pyrrolidine derivatives of (S)-5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatin to investigate the preferences of the caspase-3 in S2 pocket. In position 4 –OMe, -OPEG4, -CF3, -F, and –F,F moieties were used. All these new compounds were less active against caspase-3 than the lead structure, confirming that caspase-3 prefers hydrophobic residues in the S2 pocket.340 In Figure 26 we present the strategy for the discovery of very potent isatin sulfonamide-like caspase-3 inhibitors and in Figure 27 we collected several examples of isatin-based caspase-3 and -7 inhibitors.
Figure 26.

A rational strategy for the optimization of isatin sulfonamides structures to develop new, specific caspases inhibitors. Adapted from 339,340
Figure 27.

Multiple examples of isatin-based caspase-3, -6 and -7 inhibitors.243,329,332,336,339,342–344 Kinetic constants are presented for caspase-3.
Search for new potent and selective isatin sulfonamides analogues has been also supported by computer-aided drug design. Wang et al. designed a series of 59 compounds and docked them to the X-ray structure of caspase-3341 and analyzed the structures by 3D-QSAR. The results confirmed previous data concerning the essentials of the binding mode for isatin sulfonamide analogues and also provided insight on their structure-activity relationship. These data also indicated that hydrophobicity of isatin analogues and their ability to form numbers of hydrogen bonds with targeted enzymes are key factors for understanding their high affinity towards caspase-3 and -7.
It was previously shown that the carbonyl atom in the isatin core is key for the inhibitory activity. The same motif can be found in pyrimidoindolones, which makes these compounds potential caspase inhibitors. Havran et al. screened a library of compounds against caspase-3 and detected one lead structure 408, which was further optimized.345 Several structurally different ligands were attached to 3,4-dihydropyrimido(1,2-α)indol-10(2H)-on scaffold, leading to the discovery of a set of potent caspase inhibitors. Compound 409 displayed the highest potency from the series but only moderate chemical stability. To solve this problem spiropentacyclic derivatives were synthesized. Two analogues 410 and 411 displayed high caspase-3 potency and high stability under biological conditions. The structures of pyrimidoindolone-based caspase inhibitors are presented in Figure 28.
Figure 28.

3.5.2. 1,2-benzisothiazol-3-one derivatives
Another scaffold for developing nonpeptide caspases inhibitors was described by Liu et al.347 who tested a 4000-membered diverse small-molecule library on human caspase-3, resulting in the discovery of 1,2-benzthiazol-3-one 387, a non-peptide compound displaying IC50 of 45.74 μM against this enzyme. The authors demonstrated that the sulfur atom is necessary for the inhibitory potency, because after sulfur oxidation (S to S(O)2) the new compound 412 had a two-fold higher IC50 value (110.5 μM). Next the urea group at the N position of the lead structure was investigated. The authors synthesized a set of 1,2-benzthiazol-3-one derivatives from which the compound 413 displayed the highest caspase-3 potency (IC50 of 31 nM). Such high affinity was explained by docking studies such that the carbonyl group located on 1,2-benzthiazol-3-one 387 binds to the caspase-3 S1 pocket forming two hydrogens bond with H121 and Gly122, the second carbonyl (on the urea group) interacts with the S2 pocket via hydrogen bonding and finally the hydrophobic phenyl moiety forms hydrophobic interactions within the S3 pocket, which also stabilizes the 413-caspase-3 complex. 413 lacks a P4 moiety, so in further work the authors synthesized a new set of derivatives with the ability to explore the chemical space in the caspase-3 S4 pocket.348 The best hit 414 possesses a morpholine ring attached to the phenyl moiety, which resulted in a significant increase of potency against caspase-3 (IC50 value shifted from 31 nM to 1.15 nM). The analysis of these results clearly demonstrates how important the caspase-3 S4 pocket is in terms of developing new, potent inhibitors. Although 1,2-benzisothiazol-3-one derivatives display good inhibitory potency against caspases, their poor solubility limits their application, especially in cell experiments. In order to overcome this problem, Li and coworkers synthesized a novel series of N-acyl-substituted 1,2-benzisothiazol-3-one derivatives by replacing the urea moiety with the amide group.349 The most potent compound against caspase-3 and -7 was m-methoxyl-substituted derivative 415 (IC50 = 34.9 ± 20 nM, IC50 = 54 ± 8.6 nM, respectively). Biological studies performed on human Jurkat T cells demonstrated that the N-acyl-substituted 1,2-benzisothiazol-3-one derivatives exhibited better cell permeability than urea-containing derivatives. Shortly after, the same group discovered that incorporation of 1,2,3-triazole ring into 1,2-benzisothiazol-3-one drivatives improves their inhibitory potency against caspase-3.350 The best hit, compound 416, (IC50 = 11.0 ± 1.2 nM) was 3-fold more potent against caspase-3 than its 1,2,3-triazol-less precursor, 413. In the Figure 29 we present the two step optimization of 1,2-benzisothiazol-3-one structure to obtain 414 caspase-3 potent inhibitor.
Figure 29.

Optimization of 1,2-benzisothiazol-3-one lead compound to obtain a caspase-3 potent nonpeptide inhibitor.347–350
3.5.3. Quinoline- and quinazoline-based inhibitors
Quinoline derivatives constitute another group possessing the ability for caspase inhibition. Kim et al. synthesized and evaluated a library of quinoline-based inhibitors toward caspases.325 Compounds 417, 418 and 419 displayed the highest potency of the series with IC50 values below 14 μM and around 91–92 % of inhibition in concentration of 20 μM.
1,3-Dioxo-2,3-dihydro-1H-pyrrolo[3,4]quinolines, another group of quinone derivatives, display a noncompetitive and reversible model of caspases inhibition.351 The mechanism of caspases inactivation is due to a nuclophilic attack of the catalytic cysteine thiol on the phtalimide-like carbon atoms. A few quinoline based inhibitors have been developed and their biological activity toward caspase-3 determined. The main 1,3-dioxo-2,3-dihydro-1H-pyrrolo[3,4]quinoline scaffold contains three regions (R1, R2 and R3) that can be easily modified to obtain potent and selective caspase-3 inhibitors. A strategy applied by Kravchenko and coworkers was to synthesize sets of quinoline based inhibitors where two of three R-positions were fixed with a specific group and the remaining position was changed.352,353 It was demonstrated that the best substituent in the R1 position is morpholin-4-ylsulfonyl group, which binds to the S3 pocket of caspase-3. A methyl group was the best in the R2 position, probably due to its small size. As a result of this part of the optimization, two compounds 420 and 421 were found as promising candidates for further optimization. Finally, it was demonstrated that the most active substituents in the R3 region are 1,3,5-trimethyl-1H-pyrazol and pyridine.354 This last step of optimization resulted in finding two caspase-3 inhibitors 422 and 423 with nanomolar potency. This group of quinoline based inhibitors seems to be very promising not only for enzyme assays but also for in vivo studies (Figure 30).
Figure 30.

Okun et al. used a “focused diversity” approach to select around 15,000 small molecules as potential protease inhibitors from their 650,000 compounds collection.355 Next, this pool of compounds was tested against caspase-3. One of the selection criteria was based on the warhead concept, which from prior art should possess an electrophilic moiety. The caspase-3 recognition binding motif (core structure) consisted of a variety of heterocyclic structures such as quinoxalinones, pyridines, pyrimidinones, pyrimidinediones, qiunolones, etc. As the result of the screening 11 different inhibitory scaffold were detected. Further kinetic analysis revealed a new, small molecule caspase-3 inhibitor scaffold - 8-sulfono-pyrrolo[3,4-c]quinoline-1,3-dione (SPQ), which was used for the synthesis of potent inhibitors (the general architecture of these compounds is presented in Figure 30; chemotype 1 and chemotype 2). Next, both chemotypes were subsequently changed by introducing various ligands in R1 and R2 positions, revealing that bulky lipophilic groups (cycloheptyl, cyclohexyl) in the R2 position significantly reduced the inhibitory potency of the compounds, and more polar and hydrophilic groups enhanced the potency. Next, the R1 position was optimized, however no better ligand than 4-methyl-piperidine was found. The best compound for the SPQ series (CD-001-0011, 424) exhibited fairly good inhibition potency (IC50 = 130 ± 23 nM) and effectively prevents apoptosis in various cell lines (Jurkat, NIH-3T3, SH-SY5Y) as well as in zebrafish model.
Quinazoline derivatives are known for their potency toward caspases. Scott et al. synthesized a library of caspases inhibitors based on aminoquinazoline scaffold.326 Compounds 425, 426 and 427 display high potency for caspase-3 and high selectivity (> 40-fold in terms of Ki) against caspases -1, -2, -7, and -8 (Figure 31). Inhibitor 425 was additionally selective against caspase-6 and efficient in inhibition of caspase-3 activity in staurosporine-treated SH-SY5Y cells (IC50 = 14.9 μM). Interestingly, none of these compounds were reported to inhibit caspase-7, whose specificity and inhibition profile is very similar to caspase-3. Moreover compound 427 displays high potency toward caspase-6 and at the same time does not inhibit caspase-8, which exhibits analogous substrate specificity profile. This study showed that aminoquinazolines can be used for targeting caspases and distinguishing even closely related isoforms.326
Figure 31.

Caspase-3 inhibitors based on quinazoline scaffold.328
3.5.4. Isoquinoline-1,3,4-trione based inhibitors
Isoquinoline-1,3,4-trione derivatives have been reported as slow-binding caspase inhibitors, displaying the same mechanism of inhibition of cysteine proteases as quinoline derivatives. The ability of isoquinoline-1,3,4-trione analogues for blocking caspase-3 activity was first identified by Chen et al.324 who screened a diverse, small-molecule library of 8000 compounds against caspase-3, resulting in discovery of an isoquinoline lead structure 428. Next this hit structure was modified to obtain a few compounds 429, 430 and 431 that displayed nanomolar potency toward caspase-3. For further optimization numerous hydrophobic groups were introduced to the structures to enhance cell penetration (see 432 and 433). It was reported that 430 exhibited a good protection against apoptosis induced by β-amyloid in primary neuronal cells and PC12 cells.324 Further work on isoquinoline-1,3,4-trione derivatives showed that these inhibitors can irreversibly block caspase-3 activity also in a presence of DTT through redox cycling.324 Some representative examples of isoquinoline-1,3,4-trione derivatives are presented in Figure 32.
Figure 32.

Isoquinoline-1,3,4-trione derivatives as caspase-3 inhibitors.324
Small molecule, non peptidic caspase-3 inhibitors can be also synthesized via multicomponent reactions (MCRs). Many dihydropyrrole derivatives exhibit low potency toward caspase-3 due to their inappropriate structure, but these compounds posses highly functionalizable core structure that can be easily modified. Zhu et al. synthesized a series of tetra- and penta-substitued dihydropyrroles.356 Their biological evaluation led to few hit structures with only moderate potency toward caspases. However, the polyfunctional nature of these compounds is promising for future SAR studies.
3.5.5. Non-peptide, natural inhibitors containing Michael acceptor or epoxide moieties
In the previous section we mentioned that Michael acceptors or epoxides are good inhibitors of cysteine proteases. The mechanism of inhibition is based on irreversible thioalkylation of the catalytic cysteine group via nucleophilic addition.158 This feature was used to design and synthesize potent and selective peptide-based caspases inhibitors. However some non peptide inhibitors also posses Michael acceptor or epoxide group in their structure, making them very interesting targets for drug discovery efforts.357–359
It has been also reported that some bacterial and fungal metabolites can selectively inhibit caspase-1. EI-1507-1 434 and EI-1507-2 435 were discovered as Streptomyces sp. metabolites by Tsukadaet al..357 These two epoxide-based natural products inhibit human caspase-1 with IC50 values of 230 and 420 nM respectively and at the same time they do not inhibit the unrelated proteases cathepsin B and elastase. Moreover it has been reported the EI-1507 close derivative 436, lacking the epoxide moiety, displayed significantly lower affinity toward caspase-1 (IC50 = 10000 nM) confirming that the epoxide group is crucial for inhibitory potency. The next class of Streptomyces sp. epoxide-based metabolites with ability to block caspase-1 was described by Tanaka et al.358,360,361 EI-1511-3 437, EI-1551-5 438 and EI-1625-2 439 inhibited caspase-1 with IC50 values of 90, 380 and 200 nM, respectively. Moreover these compounds inhibited also the secretion of mature interleukin-1β from THP cells with IC50 in micromolar range (5.4, 3.6 and 2.2 μM respectively). Another caspase-1 inhibitor, EI-2128-1 440 is a metabolite produced by Penicilliumsp.359 This epoxide-containing compound inhibits caspase-1 with an IC50 value of 590 nM and also inhibits mature interleukin-1β secretion from THP-1 cells with an IC50 value of 280 nM. Koizumi et al. described the isolation of two novel caspase-1 inhibitors from culture broths of Farrowiasp..362,363 EI-1941-1 441 and EI-1941-2 442 block the caspase-1 activity with IC50 values of 86 and 6 nM respectively and do not inhibit cathepsin B and elastase.362 Natural bacterial and fungal metabolites appeared as good caspase-1 inhibitors for enzyme and cell culture assays. It has been also proved that epoxide ring is crucial for their activity and these compounds are very promising for further synthetic development (Figure 33).
Figure 33.

Examples of caspase-3 epoxide-containing inhibitors.358,360,361,364,365.
Compounds containing a Michael acceptor can be produced not only by chemical synthesis, but also via metabolic pathways in humans, microorganisms or plants. It was reported that hydroquinone 443, a toxic metabolite of benzene can bind to caspase-1 and inhibit maturation of interleukin-1β, which leads to several disorders like aplastic anemia or acute leukemia.366 Moreover hydroquinone can also deregulate apoptosis processes.327 1,4-benzoquinone 444, a hydroquinone derivative with a Michael acceptor group, is formed via hydroquinone oxidation, and displays even higher affinity toward caspase-1 than hydroquinone and can inhibit apoptosis.367 These results show that quinone-based Michael acceptors can inhibit cysteine proteases via cysteine active site thioalkylation. High-throughput screening utilizing several diverse groups of natural compounds with Michael acceptor has reported hits as caspase-1 inhibitors. L-741,494 445 is a metabolite produced by Xylaria (genus of ascomycetous fungi).368 It was found that this water-soluble xalaric acid can inhibit caspase-1 with IC50 and Ki values of 33 and 8 μM respectively. Moreover 445 doesnot inhibit papain and trypsin. L-741,494 is one of the first quinone-based natural caspase-1 inhibitors. The group of Omura described that Pentenocin B 446 obtained from cultured broth of Trichodermahamatum FO-6903 displayed only weak potency toward caspase-1 (IC50 = 250 μM)369. Another quinone-based caspase-1 inhibitor is EI-2346 447. This compound was isolated from culture broths of Streptomyces sp. and biological evaluation have provided that it binds weakly to caspase-1 (IC50 = 3.9 μM) and displays only average selectivity toward elastase and cathepsin B (IC50 > 23 μM).365 These natural compounds display only moderate inhibitory potency for caspase-1 and due to possessing a reactive α,β-unsaturated moiety are not selective for targeted enzymes. Structures of above described α,β-unsaturated caspase-1 inhibitors are presented in Figure 34.
Figure 34.

3.5.6. Other non peptide caspase inhibitors
The last group of caspases inhibitors, which cannot be assigned to any of previously described group, are steroid and non-steroid-based compounds containing inorganic nitric oxide (NO). Nitric oxide is a highly reactive molecule, that is involved in diverse physiological processes including, but not limited to, programmed cell death regulation.370,371 The mechanism of caspase inhibition by NO-donors is due to S-nitrosylation of catalytic cysteine residue. The binding mode is suggested to be reversible since addition of dithiothreitol (DTT) significantly enhance caspases activity.372 So far only a few NO-donating compounds have been characterized as caspases inhibitors. NCX-4016 448, a NO-derivative of acetyl salicylic acid, is a potent caspase-1 inhibitor (Figure 35), consistent with the report that 448 inhibits release of proinflammatory cytokines from endotoxin-challenged monocytes.373 NCX-1000 449, another NO-donating caspases inhibitor, represents a steroid-based, nitric oxide-containing family of ursodeoxycholic acid (UDCA) derivatives (Figure 35). NCX-1000 possesses an ability to reversibly inhibit caspase-3, -8 and -9 and to protection against N-acetyl-para-aminophenol acetaminophen-induced hepatotoxicity.374 449 was tested in phase II clinical trials as a drug candidate for liver diseases, including portal hypertension. This confirms that NO-releasing caspases inhibitors are promising compounds for further studies.
Figure 35.

Nonsteroid and steroid based NO-donors for targeting caspase-1.373,374
Another example of non-peptidyl small molecular inhibitors are N-(substituted-phenyl)-oxalamic acid derivatives described by Sengupta et al..375 Aspartyl fluoromethyl ketone, 2,3,5,6-tetrafluorophenoxy and 2,6-difluorophenoxy oxalamides were synthesized and evaluated as caspase-3 inhibitors. Compounds 451, 452 and 460 (Table 38) exhibited low micromolecular inhibitory activity towards caspase-3.
Table 38.
Examples of novel oxalamido derivatives as caspase-3 inhibitors.375
| No. |
|
Enzyme assays IC50, μM | |
|---|---|---|---|
|
| |||
| R | R′ | Casp-3 | |
| 450 | 4-OCH3 | fluoro | 20.14 |
| 451 | 4-CH3 | fluoro | 9.67 |
| 452 | 4-F | fluoro | 3.47 |
| 453 | 4-OCH3 | 2,3,5,6-tetrafluorophenoxy | >50 |
| 454 | 4-CH3 | 2,3,5,6-tetrafluorophenoxy | 12.26 |
| 455 | 4-F | 2,3,5,6-tetrafluorophenoxy | >50 |
| 456 | 4-Br | 2,3,5,6-tetrafluorophenoxy | >50 |
| 457 | 4-Cl | 2,3,5,6-tetrafluorophenoxy | >50 |
| 458 | 4-OCH3 | 2,6-difluorophenoxy | >50 |
| 459 | 3-OCH3 | 2,6-difluorophenoxy | >50 |
| 460 | 2-OCH3 | 2,6-difluorophenoxy | 8.4 |
| 461 | 4-CH3 | 2,6-difluorophenoxy | >50 |
| 462 | 3-CH3 | 2,6-difluorophenoxy | 25.2 |
| 463 | 2-CH3 | 2,6-difluorophenoxy | >50 |
| 464 | 4-F | 2,6-difluorophenoxy | 14.2 |
| 465 | 2-F | 2,6-difluorophenoxy | >50 |
Finally, there are few more non peptidic caspase inhibitor groups including metal ions,376 arsene-based compounds,377 dithiocarbamates378 or disulfiram.379 Unfortunately, due to their broad range of enzymes inhibition and lack of selectivity toward caspases they cannot be used as therapeutic agents.
4. ACTIVITY BASED PROBES
4.1. Introduction
Activity Based Probe Profilling (ABPP) is relatively new discipline that employs chemical molecules to detect and track active enzymes within a complex proteome.380–382 For this purpose, small molecules, usually possessing a substrate recognition moiety, termed Activity Based Probes (ABPs) are used. These probes are molecules that bind directly to an enzyme via an electrophilic group to form a covalent bond with catalytic amino acid side chain nucleophile (Cys, Ser, Thr).158 In this elegant method, zymogens or inactive forms of proteases are not labeled and therefore ABPP facilitates analysis of alteration in enzyme activity during multiple processes rather than simply protein abundance.
The first protease ABPs employed biotinylated isocoumarins for detection of serine proteases.383 This method has been progressively improved and today it is commonly used in protease research. The number of proteases targeted by this method has increased dramatically in the last few years, and the technique is being continuously developed by obtaining more selective, cell permeable and sensitive probes. Of note, ABPP is currently confined to proteases that operate via a covalent intermediate and thus not available for metalloproteases and aspartic proteases.
Apoptosis is a multistep process initiated and executed by caspases. Unfortunately these enzymes display overlapping substrate specificity, thus it is very challenging to design very specific and selective chemical probes to label and track only one caspase at the time. The activity based profilling of caspases has progressively evolved from simple nonspecific fluorescent labeled pan-caspase inhibitors to more and more selective probes with tailored peptide sequence to detect only one particular caspase in a complex system. However, although two decades have seen significant progress, specific tools for some caspases have yet to be developed.
4.2. Design principles
Activity based probes are small molecules that posses specified structural features required for proteases labeling.384 Design principles of caspases specific ABPs are based on a few main criteria, however their overlapping substrate specificity and cross reactivity with other chemical probes (legumain, cathepsins) significantly impede reaching this goal. Moreover poor cell permeability of many peptide-based probes prevent their in vivo application,331,385 which needs be taken into account during probe design.
In general, the majority of caspases ABP consist of (1) the reactive functional group “warhead” (at the C-terminus) that binds to the active site cysteine residue, (2) a tag that enables the enzyme detection (mostly on the N-terminus) and (3) a peptide based sequence (or linker) that connects the warhead with a tag and provides the selectivity toward a caspase of interest.386,387 So far for each of these groups a broad range of chemical elements has been tested and evaluated to design more potent, selective and cell permeable caspase probes. In Figure 36 we present some of the most commonly used chemical building blocks.
Figure 36.

The general structure of an Activity Based Probe with some examples of most commonly used tags, linkers, and warheads.
4.2.1. Warheads
There are several types of warheads used in caspases ABP design, however all of them work in similar a manner. Their mechanism of action relies on nucleophilic attack of the active site cysteine on the ABP electrophilic centre, thus forming a transitional (reversible) or covalent (irreversible) caspase-inhibitor complex.388
The most significant feature of a particular warhead is its ability to target only active site cysteine residue, omitting other free nucleophiles in the proteome. Thus the catalytic mechanism of the targeted proteolytic enzyme plays an important role in the selection of an appropriate warhead. It is important that the thiol group of the catalytic cysteine residue in cysteine proteases is more polarizable than the hydroxyl group on the catalytic serine or threonine, therefore electrophiles used as a warhead for cysteine proteases can be softer then ones for serine or threonine proteases.158 Thus caspases are most extensively studied with ABP containing warheads such as diazomethylketons, epoxides and halo- and acyloxy-methylketons (Figure 37). The major advantages of these warheads are their ease of synthesis, good bioavailability and active site Cys-selective reactivity.21,158
Figure 37.

Mechanism-based warheads used for caspase covalent labeling.384
The most commonly used and commercially available caspases ABPs are fluoro- (FMK or -CH2F), chloro- (CMK or -CH2Cl) or acyloxy-methylketones (AOMK) (the AOMKs are the weakest electrophile among this group, thus they are most suitable for investigating caspases because of little cross reactivity with other bionucleophiles). FMK-based inhibitors were the first and so far they have dominated research in this field. FMK-based ABPs are usually labeled with different fluorophores to make them suitable for flow cytometry. Fluorophore-labeled FMK inhibitors are called FLICAs (Fluorescent Labeled Inhibitors of Caspases). These probes will be discussed in details in the next section. FLICAs are often used to estimate the kinetic of cell death in response for different stimuli using flow cytometry and microscopy.389 Moreover, FLICA-based probes bind covalently to caspases active site via fluoromethylketone residue with 1:1 stoichiometry, and therefore label only active enzyme. One of the advantages of FMKs is that ketone reagents penetrate the plasma membrane and is relatively non-toxic to the cell.390 Use of FMK inhibitors has also some disadvantages: cross-reactivity of such probes with other cysteine proteases such as legumains, cathepsin B and cathepsin H,391,392 and production of high labeling background from non-specific binding of reactive the FMK group.393,394
Many of protease ABPs derived from E-64 (pepidyl epoxysuccinates), however E-64 is not equipped with a suitable P1 residue for reaction with caspases, thus in 2002 Asgian et al. reported the synthesis of azaepoxide inhibitors that posses a fragment mimicking P1 Asp294 (Figure 38). In 2005, Kato et al. described a general solid phase method of diverse azapeptide probes synthesis, and the Bogyo group developed an efficient and convenient method for the preparation of azapeptide epoxide and aza-AOMK ABPs dedicated to cysteine protease from CD clan to which caspases belong (Figure 39).395
Figure 38.

General structure of ABP derived from E-64.395
Figure 39.

General scheme of aza-ABP synthesis. This method allows synthesis of caspase ABPs with different warheads.395
Aza-Asp ABPs contain an epoxide or Michael acceptor as a warhead and thus can label caspases in apoptotic cells extracts, however they also effectively label legumain (belonging to clan CD) and, unexpectedly, cathepsin B (CA clan). Aza-peptides with AOMK were weaker, but more selective toward caspases, thus no crossreactivity toward other cysteine proteases was observed.395 In further studies, Kato et al. also demonstrated that commercially available biotinylated fluoromethylketones (bVAD-CH2F 466) produce a high background signal in contrast to alternatively acyloxymethylketone derivatives (bEVD-AOMK 467), which facilitated caspase-9 visualization (Figure 40).
Figure 40.

Structures of three broad spectrum caspase Activity Based Probes.291,395
These results demonstrated that AOMK probes produce lower background signal than their FMK counterparts, and since AOMKs have weaker electrophilicity their react with caspases more specific and display reduced cross-reactivity with other Cys-dependent proteases. An Asp-AOMK probe efficiently label caspase-3, -6, -7 and -8 and does not bind to caspase-9. Biotin-EVD-AOMK 467 probe with an extended peptide binding motif allowed detection of caspases -3, -7, -8 and -9 but not caspase-6. Additionally, Kato et al. using AOMK probes with six different amino acids (Gly, Arg, Leu, Lys, Asp, Asn) at P1, demonstrated that caspase-3 tolerates only Asp at the S1 pocket (Figure 41).396
Figure 41.

AOMK probes with six different amino acids (Gly, Arg, Leu, Lys, Asp, Asn) in P1 position. Caspase-3 was detected only with the probe containing Asp at P1.396
In 2007 the Bogyo group evaluated a series of aza-aspartate ABPs containing epoxide, Michael acceptor, dimethyl- and dichloro-benzoic acid leaving groups to examine the reactivity of these electrophiles toward various caspases. These ABPs contained aza-Asp or EVD-azaAsp motifs linked with biotin tag via hexanoic acid spacer. In this work the authors demonstrated that b-azaD-epoxide 470 and b-aD-Michael acceptor 472 selectively labeled executioner caspase-3 and -7. Likewise aza-aspartate ABP with an elongated peptide motif (b-EVazaD-epoxide 471) effectively labeled caspase-3/7 and additionally showed labeling of caspase-9, in a good agreement with previously characterized b-EVD-AOMKMe probe 476.397
Deployment of aza-epoxide or aza-Michael acceptor probes in micromolar concentrations resulted in non-specific reactivity and therefore produced high background in caspase labeling assays. As predicted based on kinetic parameters (Table 39), b-azaD-AOMKMe 474 and b-azaD-AOMKCl 473 showed no labeling, and the extension of binding sequences by adding additional amino acid residues (from D to EVD) resulted in labeling caspase-3 and -7. Moreover, Sexton et al. pointed out differences between kinetic measurements using recombinant enzymes and experiments on cell lysates.397
Table 39.
Structures and second order rate constants for the inhibition of caspase-3 by pointed ABP.397
| Compound | K2, M−1s−1 |
|---|---|
| biotin-azaD-epoxide 470 | 4241±345 |
| biotin-EVazaD-epoxide 471 | 358233±14669 |
| biotin-azaD-Michael acceptor 472 | 324±85 |
| biotin-azaD-AOMKCl 473 | <100 |
| biotin-azaD-AOMKMe 474 | NI |
| biotin-EVazaD-AOMKMe 475 | 1832±137 |
| biotin-EVD-AOMKMe 476 | 20156±1507 |
Berger et al. developed highly selective AOMK active site directed probes containing both natural and unnatural amino acids, which were used to dissect caspase activation cascade. Similarly to experiments described earlier, the authors demonstrated that FMK (-CH2F) probes displayed higher background and off-target effect compared to AOMK probes. Additionally, the use of unnatural amino acids in the specificity region of the probe reduced its cross-reactivity (for more details regarding probes selectivity please see the “Recognition peptide sequence” section).13,18
In 2012 Puri et al. used an AOMK probe to label caspase-1,394 comparing the commercially available FLICA probe (6-FAM-YVA-Asp(OMe)-CH2F 477) with AWP28 AOMK-probe 478 that contains one unnatural amino acid (FMK-Y-Tle-P-D-AOMK). The probe containing an FMK electrophile produced a strong non-specific background signal in contrast to the AOMK probe. Additionally the new caspase-1 AOMK probe 478 was over 10-fold more potent against caspase-1 than to other caspases (Table 40). Active caspase-1 is able to co-localize with the adapter protein ASC, which oligemerizes into large macromolecular inflammasome foci, building up an activating platform for procaspase-1. Using fluorescence microcopythe authors found that 478 labeled ASC foci whereas the FLICA probe failed to completely label all ASC foci at probe concentrations from 0.01–5 μM. Moreover, similarly to the gel experiments, FLICA probes also produced higher background.394
Table 40.
Ki(app) (or kobs/[I]) value calculated for AWP28 toward caspases and granzyme B.394
| AWP28 476 | ||
|---|---|---|
|
| ||
| Enzyme | Ki(app), M−1sec−1 | Fraction Caspase-1 activity |
| Caspase 1 | 16 764 000 ± 882 000 | 1.000 |
| Caspase 3 | 1 505 000 ± 445 000 | 0.09 |
| Caspase 4 | 7000 ± 1000 | 3.89 × 104 |
| Caspase 6 | 2000 ± 1000 | 1.11 × 104 |
| Caspase 7 | 1 412 000 ± 321 000 | 0.08 |
| Caspase 8 | 594 000 ± 184 000 | 0.04 |
| Granzyme B | <1000 | - |
Recently, novel AOMK and KE (ketoesters) ABPs with unnatural amino acids incorporated into the peptide recognition motifs were reported by Wolan and colleagues.63 To extract the most potent and selective warhead, a library of 24 inhibitors with the general formula of Ac-DEVD-R was tested. That 5-methyl-2-thiophene carboxylate-derived ketoester group (termed “KE”) binds to caspase-3 and is selective over caspase-7 and -8. Next, this inhibitor was converted into an ABP - Rho-Ahx2-DW3-KE 479 (Figure 42), which displayed 140 and 37 fold caspase-3 selectivity over caspases-7 and -8 respectively.63 It was also demonstrated that FAM-Ahx2-CV3-AOMK 480 interacted more rapidly with caspase-3 than caspase-7, while Rho-Ahx2-DW3-KE combined robust and rapid binding to caspase-3 with slow binding to caspase-7 with no caspase-7 labeling reported even in large excess of the probe.63
Figure 42.

The structure of Rho-Ahx2-DW3-KE probe, which specifically labels caspase-3.63
In conclusion, AOMK and KE as warheads are excellent electrophiles for caspases profiling in complex proteomes, and both are more selective than FMK-probes. However AOMK inhibitors still display some cross-reactive with other cysteine proteases including cathepsins and legumain, and therefore additional modification of thepeptide recognition sequence is required to make these probes more specific.
Activity based probes with peptide sequences are not the only tools for caspases visualization. Another large group of compounds suitable for protease imaging are non peptidic inhibitors equipped with an appropriate tag. Isatins are small molecule covalent inhibitors of various caspases and therefore provide attractive probes for imaging apoptosis. In 2006 5-pyrrolidinylsulfonyl isatins were proposed as nonpeptide active site directed caspases markers.331 The Levkau group synthesized a series of (S)-5-[1-(2-methoxymethylpyrrolidinyl)sulfonyl]isatins to dissect caspases in vitro and in vivo. These compounds are termed Caspases Binding Radioligands (CBRs) and a series of these compounds was examined as caspases inhibitors with some of them demonstrating high selectivity for the effector caspase-3 and -7 in vitro. Next, a library of eight 5-pyrrolidinylsulfonyl analogs as potential ABPs were selected for isotopic modifications.331 One year later the group demonstrated that 18F-fluorination of a prototype PET-compatible CbR [18F] can be successfully employed as a molecular imagining probes for studying apoptosis.398 Radiolabeled isatins can accumulate in liver, as demonstrated in mice models.334,399 This kind of probes was reported to display a subnanomolar affinity for caspase-3 and -7, high stability in vivo, rapid uptake, reduced lipophilicity, elimination in healthy tissues (and tumor) and high metabolic stability and signal intensity.334,335,338,399,400 As an example, one of these compounds - [18F]WC-II-89 481 has good biodistribution and reactivity with caspases -1,-6 and -8.401 Isotopically-labeled isatin sulfonamide [18F]ICMT-11 482 was selected by QuIC-ConCePT as a candidate for cell death imaging in humans 402 (for more details please see Radioisotopes section).
4.2.2. Labeling groups
4.2.2.1. Radioisotopes labeled ABPs
The primary advantage of using radioisotopes is that the produce very small modification of the inhibitor with minimal structural change, compared to fluorescent or biotinylated probes, and hence have cell permeability essentially identical to the parent inhibitor. 125I has often been used as an ABP tag gamma ray emitter. In addition this kind of ABP provides a highly sensitive signal easy to detect by radiography with minimal low background. Facile detection, chemical stability and in vivo activity are other benefits of radioisotopes. Radioisotopes are also commonly used in detection of proteases following electrophoresis.
Radiolabeled probes such as 125I labeled fluoromethyl ketone probes like 125I-M808 483 have been used in biological samples such as cells, lysates or even whole organisms,396,403 providing the first example of caspase activity based probes used in animal research, in this case to detect caspases in septic mice. However, use of 483 probe had some limitations regarding specificity of the inhibitor - M808. Radioiodinated probes may give low background, but do not provide for a method to isolate and identifying labeled targets. The availability of other, safer and more sensitive tags decreased usage of 125I as a tag.331
Another example of ABP with radioisotope is [18F]WC-II-89 481, a non-peptidic based isatin sulfonamide analog reacting with caspase-3. The IC50 value for isatin-derivative inhibitor WC-II-89 482 toward a few caspases demonstrated that 482 binds to caspase-3 and -7 with high affinity and specificity versus caspases -1, -6, -8 (Table 41). Based on this data a potential radiotracer [18F]WC-II-89 481 was synthesized and used in in vitro and in vivo. For the first time it was demonstrated that apoptosis can be measured and imaged in an animal model with PET –using a 18F-labeled isatin ABP.401
Table 41.
IC50 values for WC-II-89 toward caspases -1, -3, -6, -7, and -8.401
[18F]WC-II-89 481
| |
|---|---|
|
| |
| Enzyme | IC50 (nM) |
| Casp-1 | > 50000 |
| Casp-3 | 9.7 ± 1.3 |
| Casp-6 | 3700 ± 390 |
| Casp-7 | 23.5 ± 3.5 |
| Casp-8 | > 50000 |
In next report the same group has synthesized a new potent caspase-3 inhibitor isotopically labeled with 11C 483 (Figure 43) and used it to examine a Fas liver injury apoptosis model in mice.399 The experiment demonstrated that 11C-labeled caspase-3 and -7 inhibitor ([11C]WC-98, 480) displays high uptake in liver of Fas-treated animals, compared to the control. Increased caspase-3 activation was verified by a fluorometric caspase-3 kinetic assay with Ac-DEVD-AMC 484 substrate. This makes this probe a promising agent for imaging caspases activation during apoptosis.
Figure 43.

Two isatin-based radiolabeled probes for caspases 3/7 imaging.399
There are more reports of the use of isotopically labeled isatins as a lead compound with modification of its N-position for studying caspases activity. In 2006, Kopka et al developed a few isatin derivatives as a potential tools for the imagining caspases in apoptosis. They suggested that isotopically labeled the most promiscuous inhibitors will resulted in development of an exclusive apoptosis-imagining agents through visualizating active caspases in vivo.331 Faust and coworkers used 18FCbR (caspases binding radioligand) to investigate its biodistribution profile in nude mice.398
Independently Kopka and Mach demonstrated uptake of [18F]-fluoroethyl phenyl ether 485 as a putative tracer for PET.330,331 Instead of an iodinated derivative replaced the fluorinated one however this compound revealed poor biological stability.331 In 2008 the Aboagye group designed, synthesized and biologically characterized a series of a caspase-3 and -7 selective isatin inhibitors labeled with 2-[18F]fluoroethylazide with an improved metabolic profile, reduced lipophilicity and subnanomolar affinity for caspase-3.335 Inhibition of recombinant human caspases by novel fluorinated probes was assayed with fluorogenic substrate Ac-DEVD-AMC 484 (kinetic data are collected in Table 41).
Based on kinetic results, 498 appeared to be the most potent ABP toward caspase-3 (Table 42). The authors suggested that introduction of fluorine into aromatic group should result in the development of a more stable isatin in vivo. They tested 498 and [18F]498 in cell based-assays and analyzed their stability in mice overtime. A substantial increase in uptake of [18F]498 overtime was observed in treated cells (Table 43), and a rapid clearance of [18F]498 in the untreated tumor and the heart makes this probe useful for imaging caspase-3 and -7 in these tissues in contrast to high localization of [18F]498 in liver, and therefore excluded this probe from in vivo assay of apoptosis in this tissue.335
Table 42.
Kinetic data of isatin-based fluorinated inhibitors - precursors of radioisotope-labeled probes.330,335,342.
| structure | No. | EC50,nM | eLog P | ||
|---|---|---|---|---|---|
|
| |||||
| Casp-3 | Casp-7 | Casp-1/6/8 | |||
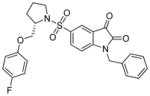
|
486 | 41.8 | 29.4 | >5000 | 3.65 |
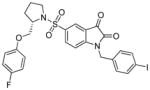
|
487 | 59.9 | 25.3 | >5000 | 4.68 |
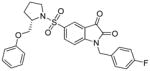
|
488 | 50.5 | 19.8 | >5000 | 3.7 |
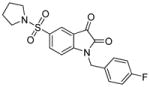
|
489 | 199.5 | 78.6 | >5000 | 1.93 |
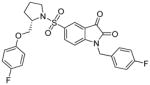
|
490 | 26.1 | 8.0 | >5000 | 3.83 |
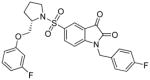
|
491 | 17.0 | 13.5 | >5000 | 3.8 |
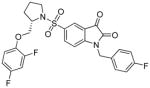
|
492 | 12.4 | 13.0 | >5000 | 3.87 |
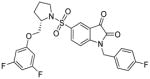
|
493 | 10.4 | 16.8 | >5000 | 3.94 |
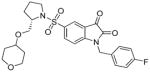
|
494 | 10.7 | 14.4 | >5000 | 1.87 |
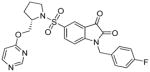
|
495 | 5.5 | 2.3 | >5000 | 2.06 |
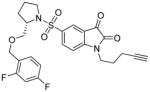
|
496 | 50.1 | 60.4 | >5000 | 2.42 |
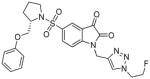
|
497 | 16.7 | 28.2 | >5000 | 1.38 |
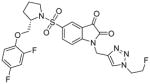
|
498 | 0.5 | 2.2 | >5000 | 1.55 |
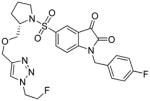
|
499 | 12.6 | 18.3 | >5000 | 1.55 |
Table 43.
The metabolism of [18F]498 measured by the accumulation of this compound in liver and plasma at selected time points.335
| Time (min) | plasma | liver | ||
|---|---|---|---|---|
|
| ||||
| parent | recovery | parent | recovery | |
| 2 | 86.1±3.7 | 92.1±3.4 | 35.7/43.4 | 79.7/86.9 |
| 15 | 61.3±5.9 | 76.8±5.2 | 9.7/10.2 | 67.1/68.9 |
| 60 | 64.8±7.0 | 50.9±3.7 | 27.0/29.7 | 46.4/65.2 |
In 2008 new N-substituted 5-pyrrolidinylsulfonyl isatins as a potential ABP for caspases imagining were synthesized, however only kinetic assays were conducted to demonstrate the inhibitory potency improvement. From many of 5-[1-(2methoxtmethylpyrrolidinyl)sulfonyl]isatin analogues, compound 500 was selected as binding to caspase-3 and selective over caspase-1, -6 and -7. This compound could be used in the future as a radiotracer for molecular imagining of activated caspase-3, and therefore imaging apoptosis (Figure 44).338
Figure 44.

Two examples of isatin-5-sulfonamide radiolabeled caspase probes.404,405
In 2009 Nguyen and coworkers used isatin-5-sulfonamide ICMT-11 as a leading sequence, and radiolabeled it by ‘click radiochemistry’, demonstrating that 18F-ICMT-11 501 displays high metabolic stability and therefore could be used as a PET imagining tracer. The authors reported high affinity for target (active caspase-3), high metabolic stability, reduced lipophilicity and convenient synthesis. 501 was tested on four different cell lines and with three different anticancer agents to observe radiotracer uptake. This group demonstrated that 501 is able to trace apoptosis in several drug-induced apoptotic cancer cells and additionally in 38C13 murine lymphoma xenografts.400 Moreover, 501 binds to caspase-3 and -7 and is reported to demonstrate around 10,000-fold selectivity over other caspases (Figure 44).
Radioisotopes were also applied in the development of peptide-based irreversible caspase probes. In 2006 Kato et al. described a series of AOMK-equipped activity based probes for cysteine proteases. The broad spectrum caspase probe contained Asp at P1 position, linker, 125I-Tyr and a biotin tag, and this probe is able to detect caspase-3 and legumain.
A recent report used [19F]FB-VAD-CH2F 502, which although unspecific, was able to detect activated caspases -3, -6, -7, -8 in an apoptotic process with no signal demonstrated in necrosis cells. 502 was used to evaluate prodrug AZD-1152 503 treatment response with CRC cell line models (SW620 and DLD-1). With this probe it was demonstrated that combination therapy led to an increase in apoptosis compared to vehicle-treated or single-agent-treated CRC xenograft bearing mice. The authors hypothesized that this probe could be used in non-invasive imaging to monitor the response to complicated therapeutic multidrug regimens. Moreover, the probe could be used to estimate the clinical response to therapeutics, however the relatively modest uptake and low selectivity are drawbacks of this probe.406
Lee at al. obtained probes LS498 504 and 64Cu-LS498 505 consisting of DOTA for chelating the PET-friendly radionuclide 64Cu, a near-infrared fluorescent dye separated with a quencher dye via a cleavable peptide substrate where fluorescence is extinguished until caspase-3 cleavage, which occurs with a kcat/KM value 4.91×105 s−1M−1. During experiments in an activated caspase-3 mice model, NIR fluorescence was 5-times increased whereas radioactivity was the same in comparison to caspase-3 positive and negative control. This probe offers the opportunity to define both localization and distribution of active caspase-3 in mice.407 The structures of 504 and 505 are presented in Figure 45.
Figure 45.

The structure of LS-498, caspase PET probe containing DOTA - a 64Cu chelating agent.
In 2007 a new methodology for in vivo caspase-3 imaging, based on MRI signal quenching from the intramolecular paramagnetic effect of Gd3+ was described.408 19F MRI probe 506 consists of three main parts: (1) caspase-3 cleavable linker sequence DEVD, (2) Gd3+ complex and (3) 19F-radiolabeled moiety (Gd-DOTA-DEVD-Tfb presented in Figure 46). The Gd3+ complex has strong paramagnetic relaxation enhancement (PRE) because of the seven unpaired electrons in the 4f orbital of Gd3+.408,409 Proximity of 19F to Gd3+ in the probe weakened the MRI signal which significantly increases after hydrolysis with decrease in the PRE signal.409,410 It was demonstrated that T1 and T2 values for Gd3+ were higher than for the Gd3+ free compound. Peptide bond hydrolysis by caspase-3 lead to the enhancement of 19F-MRI signal in time dependent manner (from 3 to 70 minutes).408
Figure 46.

The structure of Gd3+ labeled caspase specific activity based probe 506.408
The main advantage of MRI probes is the amount of information obtained with high spatial resolution, even in deep areas of animal bodies.410 Disadvantages include the relatively short half-life of appropriate radionuclides and needs for handling and use with special care. Hence, these probes must be used immediately after radionuclide chelation.
4.2.2.2. Biotin labeled ABPs
Biotin (also known as Vitamin H or coenzyme R) is the most common tag used in activity-based proteomics. The use of biotin for the investigation of caspase activation and apoptosis was pioneered by Thornberry and Nicholson,411,412 however biotin as an ABP tag may have been used for the first time in 1980 for the visualization of acetylcholine receptors.413 Work on simple caspase probes opened the door for others to pursue more specific and potent biotin-containing affinity labels to track caspase activation. In this approach caspase inhibitors are converted into activity based probes by simple attachment of a biotin moiety to the free amine group (usually at the N-terminal end, but also inside inhibitors to available lysine residues) (Figure 47). The use of biotin for protease investigation is optimal since this tag displays diffusion rate-limited binding to streptavidin.414 This phenomenon makes both the detection of labeled enzyme and affinity purification of the active protease using avidin possible. Importantly, these probes label only active caspases at the substrate binding site. However, the use of biotin for affinity labeling has some limitations. Because of poor cell permeability biotin-probes are not often useful in in vivo assays.414 Secondly, this method is not completely specific since some endogenous biotinylated polypeptides and proteins can be also detected on blots and histochemistry.414 Nonetheless, as mentioned previously, biotin tagged caspase inhibitors have been widely used, mostly in proof of concept experiments as well as in many biological assays dedicated to apoptosis investigation.
Figure 47.

Several examples of caspase activity based probes tagged with biotin.
Caspase biotinylated ABPs were first used to investigate interleukin-1β-converting enzyme (ICE, Caspase-1) almost two decades ago. This first generation of probes had a very simple structure composed of warhead (AOMK - acyloxymethylketone), enzyme recognition sequence (peptide sequence) and biotin as a labeling indicator. In an initial probe application, active site binding L-742,395 507, was used to monitor the activation of p45 caspase-1 by stepwise proteolytic processing. This provided information on the activation pathway where the full length precursor contained little activity, but was enhanced dramatically following processing to p22 and p20 proteolytic derivatives .415
The group of Lazebnik reported on the use of biotin-labeled caspase inhibitors for studying apoptosis.416 They used three affinity labels: biotin-VAD-CH2Cl 508, biotin-YVAD-AOMK 509 and biotin-DEVD-CH2Cl 510 to monitor caspase activation in response to various apoptosis-inducing triggers (etoposide, staurosporine, α-Fas). The results demonstrated that the major active caspases in apoptotic Jurkat cells were executioner caspase-3 and -6. Moreover these caspases are present as several catalytically active bands in gels, revealing that the activation process produces several proteolytically-processed intermediates. This work confirmed the utility of simple biotinylated inhibitors for biological purposes, but a few questions remain unanswered. Firstly, it is not known whether only caspase-3 and -6 were activated during the apoptosis process, or whether more active caspases were present, but at much lower levels. Additionally, the authors used only probes directed to caspases -1, -3, and -6 so they could miss labeling and detection of some others enzymes. In other work, the same group studied oncogene-depended apoptosis in drug resistant cell lines.417 Since apoptosis can be induced by expression of various oncogenes, the question how the caspase become activated during such expression remained unanswered at that time. To dissect this problem the affinity labeling method was applied (biotin-YVAD-AOMK 509, caspase-1 directed probe) in combination with caspases fluorogenic substrates (Ac-DEVD-AFC 510), with results demonstrating that the HEK293 cell line, upon treatment with an anticancer drug, does not activate caspase-1-like caspases.
Concomitantly (1997) Martins et al. used a novel activity based probe Cbz-EK(biotin)D-AOMK 511 examine apoptosis induction in HL-60 cells.418 They demonstrated that untreated HL-60 cells contain transcripts for nine caspases, so the question was, which caspases become activated during apoptosis. The study with 511 probe demonstrated that at least two distinct enzymes are activated during apoptosis (one of them hydrolyzes Ac-DEVD-AFC 510 substrate, and the second hydrolyzes Ac-VEID-AMC 512). Moreover two-dimensional gel electrophoresis revealed that several others active species are present in the cytosolic fraction of apoptotic cells. The possibility to detect distinct caspases was limited by the peptide structure of the biotin-containing probe authors used 511, whichcan label only caspases that recognize Cbz-EKD- tripeptide motif. Accordingly the authors supported their work with other chemical tools (for example the fact that procaspase-1 did not become activated during this type of apoptosis was demonstrated by the lack of hydrolysis of Ac-YVAD-AFC 513, a caspase-1 specific substrate).418 The same tripeptide Cbz-capped probe, but equipped with another electrophilic warhead - Cbz-EK(biotin)D-CH2F 514 was used by Saunders et al. to detect caspases activity upon apoptosis initiation. The authors mentioned that penetration of this ABP into cells is problematic due to its poor cell permeability, but the biotin-containing affinity tag could be directly used in cell extracts.419 More recently, Shelton and coworkers used a biotin-VAD-FMK 466 probe to demonstrate that activation of the initiator caspase-9 is required for heat-induced apoptosis in Jurkats and that two other presumptive initiator caspase-2 and -8 are not essential for this type of programmed cell-death.420 These examples strongly indicate that caspases inhibitors equipped with a biotin moiety are useful for affinity labeling of apoptotic cell extracts. But it is worth noting that these activity based probes contained biotin linked to the inhibitory moiety by a short arm, which could cause problem in affinity caspase purification and identification. To overcome this problem Henzing and coworkers synthesized several caspase activity based probes where biotin was coupled with the inhibitor via extended linkers.421 Kinetic analysis showed that one of the new probes that was labeled on N-ε-lysine by -CO(CH2)5NH-biotin displays almost 2-fold higher affinity toward caspase-3 than the previously reported 511. Moreover, using this probe the authors were able to purify nondenaturated EGFP-caspase 6 from apoptotic cell extracts, demonstrating the importance of utilizing linkers to separate biotin from the specificity region of the probe – presumably decreasing steric constraints for caspase binding. The structures of these probes is based on prototypic caspases substrates and inhibitors. Unfortunately the probes lack specificity and produce high background labeling during crude proteome assays. Because of this, more specific caspase activity based probes are highly desired. Some of biotinylated caspase probes are presented in Figure 47.
A more complicated strategy for protease identification (including caspase-3) was proposed by Lathia et al..422,423 This approach was designed to investigate protease activity based on an inductively coupled plasma - mass spectrometry (ICP-MS) phenomenon. To test the utility of this method the authors synthesized several orthogonal substrates targeting calpain-1, caspase-3, MMP-9 ADAM10, α-chymotrypsin. Substrate 515 was dedicated to caspase-3 and contained an appropriate tetrapeptide DEVD sequence, lanthanide chelator, DTPA tagged by holium ion (Ho3+) at the N-terminus and Gly-Lys(biotin)-amide at the C-terminus. Upon hydrolysis the C-terminal biotin-containing moiety was capped by streptavidin and N-terminal DTPA(Ho3+)-(βAla)4-DEVD-OH was detected ICP-MS. The experiment performed in simple buffers containing proteases and in HeLa cell lysates demonstrated that this method displays high sensitivity and linearity. Moreover this approach is an example how systems containing distinct proteases can be fingerprinted.
4.2.2.3. Fluorophore labeled ABPs
Probes with fluorescent tags, similarly to biotin labeled inhibitors, can be visualized by the use of SDS-PAGE gel approaches, thus these probes enable direct read-out from SDS-PAGE gels using laser scanner, omitting the additional step of transfer to membrane and subsequent avidin probing. Moreover, these probes can be also visualized directly in cells using fluorescence microscopy. This feature opens a window for studying protease biology in living cells 424–427. Moreover, the large diversity of commercially available fluorophores makes it possible to perform many tailored experiments of caspase biology. In 2006 Sadaghiani et al. described the chemical and physical properties of commonly used fluorophores in activity based proteomics highlighting their advantages and disadvantages (Table 44).414
Table 44.
Collected tags which were used in caspases labeling. Adapted from 414
| Tag | Advantage | Disadvantage | Application + Ref. |
|---|---|---|---|
| Biotin | Fast and specific, pH stable, high sensitivity, low price | Time-consuming detection | Aza-peptide ABP 20,395,397 |
| Rhodamineand derivatives | Readily excited | Lack of cell permeability,low quantum yield | Aza-peptide ABP, FLICA with CaspaTag |
| Cyanine | Stable in SPPS, stable in broad pH range, hydrophilic, DMSO stable, photo stable | High price | Peptide-bases ABP 13,19,428 |
| Radiolabeling | Small size, easy detection, high activity, high sensitivity, easy synthesis, chemical stability, use in biological samples in vivo | Short half-life, harmful, gel-based analysis | Mainly isatin-based probes 334,335,338,398–401,403 |
| Fluorescein derivatives | High absorptivity, hydrophilic, inexpensive, stable in SPPS | Light-sensitive, unstable in pH>7, lack cell permeability, more sensitive at lower pH | Using in FLICA, FAM-probes 13,20 |
The idea of fluorescently labeled probes for caspases was initiated in 2000 by the group of Darzynkiewicz.17,390,429,430 They used fluorophores labeled inhibitors of caspases (FLICA) to detect these enzymes in cells undergoing apoptosis. The probe FAM-VAD-CH2F 516 was a broad spectrum caspases inhibitor that penetrates cell membrane, so it is suitable for in situ experiments. The authors showed that HL-60 cells undergoing apoptosis were able to bind FLICA with a frequency strongly correlated with DNA strand breaks detected using Terminal deoxynucleotidyl transferase-mediated d-UTP Nick End Labeling (TUNEL) – a conventional apoptosis readout. However, the authors also noticed that caspase-3 null MCF-7 cells were labeled with the “caspase -3 specific” FAM-DEVD-CH2F 517 probe, indicating that other caspases must recognize the DEVD tetrapeptide sequence. These observations together indicate that FLICA offers a promising assay for studying caspases biology, but lack of specificity limits its utility and suggests that more specific peptide or peptidomimetic caspases sequences are desired. At the same time Amstad and coworkers reported on use of the same FLICA probe for caspase detection in staurosporine-treated Jurkat and HeLa cells.83 Likewise, they demonstrated that caspases, when activated upon apoptosis, can be visualized by fluorescence spectroscopy using 96-well plates, which confirmed that 516 can be used for high throughput apoptotic drug screening. This early work in the area of fluorescent probes tailored for caspases shows that FAM-labeled peptide based inhibitors can be successfully used to detect caspase activation in various apoptotic cell lines (for examples of FLICA probes please see Warhesds section). FLICAs were used in many biological assays that strongly contributed to the understanding of apoptosis phenomenon.389,431–435
In 2000 the Darzynkiewicz group used two FLICA probes FAM-VEID-CH2F 518 (for caspase-6) and FAM-VAD-CH2F 516 (pan-caspase probe) in Laser scanning cytometry to detect caspasesin HL-60 cells induced by CPT (camptothecin) or TNF-α + cycloheximide. An increase of frequency of cells bound to the ABPs was observed after 30–90’ of apoptosis stimuli by TNF-α + cycloheximide and between 2–4h after the administration of CPT.429 In cells treated with CPT, 518 was bound 30% less than 516, what led to conclusion that activation of caspase-6 was delayed or not induced in some cells. Likewise, human breast carcinoma MCF-7 cells treated with CPT and examined by microscopy revealing that most cells labeled by FLICA had characteristic changes for apoptosis like shrinkage. This suggested that FLICA probes can penetrate into the cells and bind to activated caspases.429 However, what was not demonstrated was whether FLICA could penetrate cells before or after the membrane changes associated with apoptosis. In Table 45 we collected some examples of FLICA-labeled FMK (-CH2F) probes for caspases.
Table 45.
Some examples of FAM labeled caspases FMK inhibitors
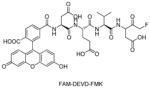
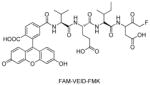
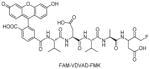
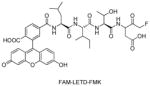
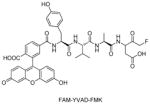
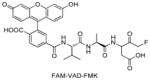
Smolewski et al. suggested that FLICA penetration through a plasma membrane may be relatively slow in comparison to apoptosis progression. Moreover, additional difficulties with FMK-inhibitors as an apoptotic labeling ABP was considered. Firstly, this kind of probe with ketone as an electrophile has an influence in apoptosis progression by covalently binding to caspases. Another problem when using FLICA in apoptotic-cell detection is the removal of unbounded FLICA from nonapoptotic cells, and subsequent loss of fragile apoptotic cells, mainly during centrifugation.390 In the same year Amstad group used the 516 probe to detect caspases in living cells using flow cytometry, fluorescence microscopy, and a fluorescence plate reader. They demonstrated that this probe is able to bind to apoptotic and nonapoptotic cells, however for apoptotic cells fluorescence increase 30-fold in comparison to nonapoptotic ones (autofluorescence from non-apoptotic cells is around two times higher in comparison to unstained cells).83
In other studies the Grabarek group tested few FLICA ABPs with different peptide sequences on Jurkat leukemic cells treated with STS (staurosporine). 516 targeted nonspecifically protein fragments of approximately 20kDa, the large catalytic subunits of caspase-3 (p19 and p20), caspase-6 (p18) and/or caspase-7 (p22). Additional unknown protein labeling was observed between 30 and 40 kDa. After cell treatment with STS or death ligand receptor Fas/CD95 three bands in a range from 17 to 22 kDa with FAM-LETD-CH2F 520 and FAM-AEVD-CH2F 522 and three bands between 18 and 20 kDa with FAM-LEHD-CH2F 523 were detected. FAM-YVAD-CH2F 521 (a supposedly caspase-1 dedicated probe) labeled three proteins corresponding to large subunit of caspase-3 p20 and p17, caspase-6 p18 and caspase-7 p22. The FAM-DEVD-CH2F 517 probe labeled not only caspase-3 (p20 subunit, main signal), but also proteins with molecular weight between 17 and 22 kDa. FAM-VEID-CH2F 518 targeted two proteins with molecular masses close to 20 kDa (one of them is the p18 large subunit of caspase-6) in STS treated cells. Another probe, FAM-LETD-CH2F 520 labeled a protein from Jurkat cells treated with anti-Fas monoclonal antibody, which may have corresponded to large subunit of caspase-8. These results demonstrate that FAM labeled FMK (−CH2F) inhibitors are useful in measurement of caspases activation in situ with laser scanning cytometry and flow cytometry,431,432 however their lack of selectivity causes current FLICA tools to be suitable only for overall apoptosis detection, rather than tracking of individual caspases.
In another study 517 was used in head-to-head comparison with caspase antibodies in gentamicin-treated chick chochlea and leukemic JURL-MK1 cells,389,437 demonstrating that caspases activation and caspase processing were similar in both cases.
A common fluorescent dye for caspases inhibitor labeling is Cy5 – a near infrared fluorescent tag (NIRF) that allows for the non-invasive detection of caspases, an approach in studying caspases pioneered by the Bogyo lab.13 In early studies, the AB50-Cy5 524 probe was synthesized to detect and image caspase-3 activity in dexamethasone-treated thymocytes. Next, to enhance the cell permeability of the probe, aderivative was synthesized by attaching a Tat peptide at the N-terminus. This approach led to the discovery of a fluorescent probe with good caspase-3 potency and excellent cell penetration ability. In 2012 the same group demonstrated that another executioner caspase, caspase-6 can be activated in the absence of caspases -3 or -7.19 To confirm this phenomenon a caspase-6 targeted probe was developed (LE22, 525) with a Cy5 fluorophore at the N-terminus, AOMK as a warhead, and an optimal caspase-6 VEID sequence. Using this probe the authors demonstrated that caspase-6 undergoes a conformational changes upon activation and can bind substrates prior to proenzyme cleavage. These initial studies with the use of Cy5 demonstrated that caspase activity can be detected in many biological assays, from cell free apoptotic lysates, to tissues, to whole organisms. Importantly, potent and cell permeable caspase activity based probes possess diagnostic and therapeutic potential. Some FLICAs structures as well as fluorescent dyes are presented in Figure 48.
Figure 48.

Some examples of FLICAs for caspase activity detection.13,19,83,430,438
Another near infrared probe IR780-Val-Ala-Glu(OMe)-CH2F 530, engineered as a caspase-9 monitoring agent, was used in biochemical experiments on DU145 cells undergoing apoptosis. This probe was reported to provide high quantum yield and cell permeability. Surprisingly, the P1 site of the probe contains a glutamic acid ester instead of aspartic acid, distinguishing it from all other caspases probes (Figure 49). The authors postulated that this probe could be used for in vitro imagining to monitor tumor cell progression and apoptotic cells.439
Figure 49.
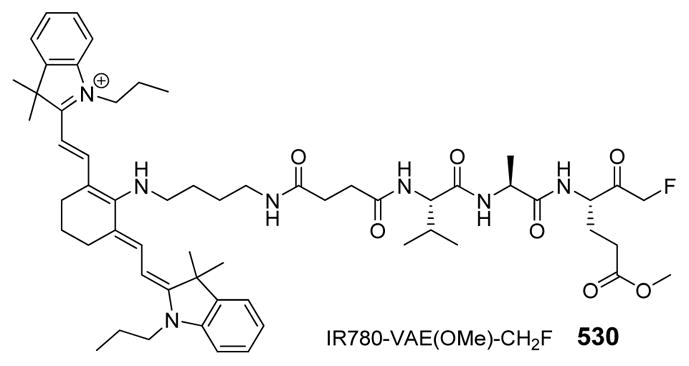
The structure of P1-Glu near infrared broad spectrum caspase probe
Recent work on activity based probe research has shown that many efforts are directed towards selectivity of these markers rather than their potency against targeted enzymes. As we have described earlier, one of the key problem of caspases probes is their lack of specificity. Commercially available caspase activity based probes label various caspases within the family as well as other cysteine proteases such as legumain and cathepsin B. To date many efforts have been made to overcome this problem. Edgington and coworkers synthesized a library of AOMKs inhibitors containing natural and unnatural amino acids.13 Following screening of caspase-3, legumain and cathepsin B the authors extracted three caspase-3 specific sequences and converted them into fluorophores-labeled activity based probes. The most specific inhibitor/probe for caspase-3 (AB53-Cy5 531, Figure 50) contained proline (Pro) in P2 (not recognized by cathepsin B) and nonproteinogenic biphenylalanine (Bip) in P3 (not recognized by legumain). However, this probe displayed relatively poor cell penetration. In 2012 Puri and coworkers using 532 (Figure 50), a caspase-1 directed activity based probe, showed that this protease activity is critical for pyroptosis (a form of inflammatory cell death) during bacterial infection.394 532, labeled with FAM carboxyfluorescein at N-terminal and equipped with an AOMK warhead at the C terminus, contained a tert-leucine residue in P3 position making it highly selective for caspase-1 over apoptotic caspases. These studies suggest that the specificity of activity based probes for particular caspases detection and imaging can be improved by the use of non-proteinogenic amino acids. Moreover, the wide (almost endless) group of unnatural amino acids offers numerous chemically distinct structures, from small and hydrophilic to large and hydrophobic.
Figure 50.

Potent and specific caspases activity based probes containing non-proteinogenic amino acids.13,63,394,440
4.2.3. Linkers
4.2.3.1. Recognition sequence design
A linker can be defined as the part of an ABP that connects the electrophilic reactive group on C-termini with a tag thatis usually located on N-termini. In principle the linker encompasses the peptide specificity region (see Figure 36). In addition to specificity, the length of the linker has an influence on effectiveness of an ABP. For instance a single amino acid probe biotin-Asp-AOMK 533 labels caspases -3, -6, -7, -8 but not -9. Peptide chain elongation to tripeptide biotin-EVD-AOMK 534 provides labeling of caspase-9, but on the other hand does not detect caspase-6, thus the EVD fragment is not able to bind in caspase-6 pockets. Commercially available, unspecific biotin-VAD-CH2F 535 is able to detect caspases -3, -6, -7, 8 and -9.396
In 2006, the Bogyo group used a positional scanning combinatorial library (PSCL) approach to screen caspases using a library of nitrophenyl acetate capped tetrapeptide acyloxymethyl ketones containing both natural and unnatural amino acids, aiming to obtain specific activity based probes. By testing a library of AOMK peptide inhibitors the authors selected the best hits transformed into activity based probes using biotin as a tag (the detailed analysis of these inhibitors is described here18). Novel ABPs, constructed from a pool of natural and unnatural amino acids, enabled identification of single caspase in the mixture of purified recombinant proteins. To demonstrate the utility of those probes, a broad range of probes concentrations were used. bAB06 536 and bAB13 537 selectively labeled caspase-3, bAB19 538 selectively labeled caspase-8 and bAB38 539 labeled both caspase -8 and -9 (Figure 51).
Figure 51.

Structures of ABPs developed by Bogyo group.18
Recently, the Wolan group demonstrated an improved potency and selectivity of probes with unique peptide sequences containing unnatural amino acids.20,63,322 Similarly to above described approach, authors obtain caspases selective activity based probes by attaching tags (biotin, carboxyfluorescein, rhodamine) to the N-terminal of selected inhibitors (Table 47 and Figure 52).
Table 47.
| ABP | IC50, nM | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Casp-3 | Casp-6 | Casp-7 | Casp-8 | Casp-9 | ||
| FAM-DEVD-AOMK | 540 | 18 | 26 | 24 | 29 | 1100 |
| FAM-CV3-AOMK | 541 | 27 | 3000 | 1300 | 100 | 3100 |
| Biotin-DEVD-AOMK | 542 | 39 | 75 | 38 | 50 | 1100 |
| Biotin-CV3-AOMK | 543 | 13 | 1200 | 420 | 53 | 2500 |
| Rho-Ahx2-DW3-KE | 544 | 25 | 4000 | 3500 | 930 | ND |
| Rho-DEVD-AOMK | 545 | 10 | 65 | 17 | 12 | ND |
| Biotin-CV8/9-KE | 546 | 4500 | ND | ND | 170 | ND |
Figure 52.

Recognition elements (containing unnatural amino acids) of ABPs for caspases-3, -8, and -9 developed by Wolan group.20,63,322
In addition to specific sequences and separation linkers, another important modification in ABPs is the attachment of Tat peptide to improve its cell permeability. Edgington et al. used the caspase probe AB50-Cy5 524 for the basis of obtaining a cell permeable tAB50-Cy5 547. Both probes – 524 and 547 label caspase-3, however the 547 exhibits increased cell permeability. These probes were used in non-invasive optical imagining of caspases, resulting in discrimination between apoptotic and non-apoptotic tumors.13 That this Tat modification strategy was earlier used also to increase cell update in caspases substrates.78,441
After two decades of caspases activity based probes research, the holy grail of exquisitely selective reagents has yet to be found. The studies described above clearly demonstrate that caspase ABPs discovered to date have the potential to be used in both in vitro and in vivo assays. From the very beginning small, unspecific probes were used to monitor caspases activation in basic biochemical assays. Probes have been used in animal research and some of them accumulated in tissues like liver or kidneys leading to high background fluorescence. Over time the structure and therefore selectivity and sensitivity have been improved to allow more sophisticated and challenging research including noninvasive imaging of apoptosis in both preclinical and clinical settings.
5. CONCLUSION
Because caspases participate in a wide range of cell signaling processes implicated in health and disease there has been a great need to develop sensitive and selective chemical tools that can detect and track the active form of these enzymes within complex proteomes. Substantial strides have been made in the diversity of substrates and inhibitors for demonstrating and ablating caspase activity in vitro as well as in whole animals. These advancements include numerous fluorescent and radiolabeled probes for whole body imaging, and a wide diversity of electrophilic warheads that help to selectively visualize caspases over other proteolytic enzymes. But the “holy grail” – the goal of achieving highly selective reagents to distinguish between individual enzymes of the closely related caspase family – has yet to be achieved.
The main goal of this review has been to provide a comprehensive assessment of technologies and approaches designed to pursue the development of selective caspase specific motifs. Peptidic recognition elements based on proteinogenic (natural) amino acids, which were demonstrated as early as 1997 to be recognized by several caspases, have been confirmed as largely non-specific. Therefore, in light of these studies, our current knowledge strongly suggests that probes based on proteinogenic amino acids lack selectivity and therefore the potential to target one caspase. Their application is useful in observing total caspase activity in a sample, and they are very useful from this perspective. There have been many reports describing substrates, inhibitors and activity based probes as selective for a particular caspase, but it is only in the last two years that these claims can be seriously justified.
The specificity sites of caspases may have been “built” to recognize natural amino acids, but the regions around the active site contain surfaces available to non-natural chemotypes, different for each caspases. The recent development and application of non-proteinogenic (unnatural) amino acid peptidic substrates and inhibitors has demonstrated that this particular path to specificity and selectivity shows outstanding promise, because it allows for the exploitation of radically enhanced chemical space. This review demonstrates that, although there remains a huge gap in understanding the biological function of individual caspases determined using active site-directed probes, pan-specific probes can be very useful, and the future of individual selectivity is likely going to follow on from the use of non-proteinogenic peptidic elements (Figure 53).
Figure 53.

Notable achievements in the development of active site-targeted chemical tools for studying human caspases.
Table 6.
Multiple examples of x-ray crystal structures of caspase-2 adapted from PDB database.
| CASPASE-2 | |||||
|---|---|---|---|---|---|
|
| |||||
| PDB entry | Mutation | Res. | Enzyme info | Ligand | Ref. |
| inhibitors free | |||||
| 3R7S | wild type | 2.25 | p18 subunit (length 160) and p12 subunit (length 112) | ligand free | 39 |
|
| |||||
| with inhibitors | |||||
| 2P2C | wild type | 3.24 | p18 subunit (167-333) andp12 subunit (348-452) |
AR_F8 (designed ankyrin repeat protein) |
191 |
| 1PYO | wild type | 1.65 | p18 subunit (151-316) andp12 subunit (331-435) |
Ac-LDESD-CHO (covalent, reversible, peptide inhibitor) |
171 |
| 3R5J | wild type | 1.77 | p18 subunit (length 160) and p12 subunit (length 112) |
Ac-ADVAD-CHO (covalent, reversible, peptide inhibitor) |
39 |
| 3R6G | wild type | 2.07 | p18 subunit (length 160) and p12 subunit (length 112) |
Ac-VDVAD-CHO (covalent, reversible, peptide inhibitor) |
39 |
| 3R7B | wild type | 1.80 | p18 subunit (length 160) and p12 subunit (length 112) |
Ac-DVAD-CHO (covalent, reversible, peptide inhibitor) |
39 |
| 3R7N | wild type | 2.33 | p18 subunit (length 160) and p12 subunit (length 112) |
Ac-DVAD-CHO two copies (covalent, reversible, peptide inhibitor) |
39 |
| 3R6L | T380A | 1.90 | p18 subunit (length 160) and p12 subunit (length 112) |
Ac-VDVAD-CHO (covalent, reversible, peptide inhibitor) |
39 |
| 3RJM | wild type | 2.55 | p18 subunit (167-333) and p12 subunit (348-452) |
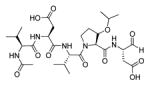 (covalent, reversible, peptide aldehyde inhibitor) |
192 |
Table 7.
Multiple examples of x-ray crystal structures of caspase-6 adapted from PDB database.
| CASPASE-6 | |||||
|---|---|---|---|---|---|
|
| |||||
| PDB entry | Mutation | Res. | Enzyme info | Ligand | Ref. |
| inhibitors free | |||||
| 2WDP | wild type | 1.95 | dimer – two large and two small subunits | ligand free (only phosphate ion) | 193 |
| 3NR2 | C163A | 2.90 | delta prodomain; subunit p18 and subunit p11 | ligand free | 194 |
| 3K7E | D179 CT | 3.00 | delta prodomain | ligand free | 195 |
| 3NKF | D179A | 2.90 | delta prodomain | ligand free | 196 |
| 3P45 | wild type | 2.53 | mature apo-enzyme: subunit p20 and subunit p10 | ligand free | 197 |
| 4IYR | H121A | 2.70 | zymogen – length 304 | ligand free | 198 |
| 4EJF | wild type | 2.65 | casp-6 (length 279 aa) and phage-derived peptide 419 (length 18 aa) | ligand free (only phosphate ion) | 199 |
|
| |||||
| with inhibitors | |||||
| 4FXO | wild type | 2.85 | Length 299 aa |
Zinc ion (Zn2+) (exosite inhibitor: Lys36, Glu244, His287) |
200 |
| 3OD5 | wild type | 1.60 | delta prodomain; subunit p18 and subunit p11 |
Ac-VEID-CHO (covalent, reversible, peptide inhibitor) |
194 |
| 3S70 | wild type | 1.63 | length 278 aa |
Ac-VEID-CHO (covalent, reversible, peptide inhibitor) |
201 |
| 3P4U | wild type | 1.90 | UNP residues 24-179 and 193-293 | Ac-VEID-CHO(covalent, reversible, peptide inhibitor) | 202 |
| 3QNW | wild type | 2.65 | p20 subunit (24-179) and p10 subunit (193-293) |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
203 |
| 4HVA | wild type | 2.07 | Caspase 6 (length 265 aa) bound to VEID-aldehyde |
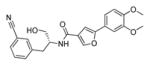 (uncompetitive inhibitor) |
204 |
| 4N6G | C163A | 2.14 | procasp-6; UNP residues 24-293 |
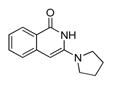 (allosteric inhibitor) |
205 |
| 4NBK | C163A | 1.94 | procasp-6; UNP residues 24-293 |
 (allosteric inhibitor) |
205 |
| 4NBL | C163A | 1.76 | procasp-6; UNP residues 24-293 |
 (allosteric inhibitor) |
205 |
| 4N5D | C163A | 2.06 | procasp-6; UNP residues 24-293 |
 (allosteric inhibitor) |
205 |
| 4N7J | C163A | 1.67 | procasp-6; UNP residues 24-293 |
(allosteric inhibitor) |
205 |
| 4NBN | C163A | 1.75 | procasp-6; UNP residues 24-293 |
 (allosteric inhibitor) |
205 |
Table 8.
Multiple examples of x-ray crystal structures of caspase-8 adapted from PDB database.
| CASPASE-8 | |||||
|---|---|---|---|---|---|
|
| |||||
| PDB entry | Mutation | Res. | Enzyme info | Ligand | Ref. |
| inhibitors free | |||||
| 2K7Z | C360A | NMR | monomeric unprocessed catalytic domain (procaspase-8) | ligand free | 206 |
|
| |||||
| with inhibitors | |||||
| 1I4E | wild type | 3.00 | casp-8 p18 subunit and p11 subunit |
baculoviral p35 protein (covalent, protein inhibitor) |
207 |
| 2FUN | wild type | 3.00 | casp-8 p18 subunit and p11 subunit |
baculoviral p35 protein (covalent, protein inhibitor) |
208 |
| 3H11 | D359A D369A | 1.90 | Casp-8 zymogen bound to Ac-IETD-aldehyde inhibitor | protease like domain of FLIP | 209 |
| 1QTN | wild type | 1.20 | casp-8 p18 subunit (length 164) and p11 subunit (length 95) |
Ac-IETD-CHO (covalent, reversible, peptide inhibitor) |
175 |
| 1F9E | wild type | 2.90 | casp-8 p18 subunit (length 153) and p11 subunit (length 89) |
Z-DEVD-CHO (covalent, reversible, peptide inhibitor) |
210 |
| 1QDU | wild type | 2.80 | casp-8 p18 subunit (length 153) and p11 subunit (length 88) |
Z-EVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
174 |
| 3KJQ | wild type | 1.80 | casp-8 p18 subunit (211-374) and casp-8 p10 subunit (385-479) |
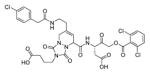 (covalent, irreversible, peptidomimetic inhibitor; AOMK) |
211 |
| 3KJN | wild type | 1.80 | casp-8 p18 subunit (211-374) and casp-8 p10 subunit (385-479) |
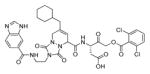 (covalent, irreversible, peptidomimetic inhibitor; AOMK) |
211 |
| 4JJ7 | wild type | 1.18 | casp-8 (length 275) |
(covalent, irreversible inhibitor; AOMK) |
20 |
| 2C2Z | wild type | 1.95 | casp-8 p18 subunit (218-374) and casp-8 p10 subunit (376-479) |
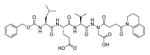 (covalent, irreversible, aza-peptide inhibitor) |
212 |
Table 9.
Multiple examples of x-ray crystal structures of caspase-9 adapted from PDB database.
| CASPASE-9 | |||||
|---|---|---|---|---|---|
|
| |||||
| PDB entry | Mutation | Res. | Enzyme info | Ligand | Ref. |
| inhibitors free | |||||
|
| |||||
| 3YGS | wild type | 2.50 | APAF1 (CARD, length 95) and procaspase 9 (prodomain, length 97) | ligand free | 213 |
| 2AR9 | C287S, G402C, C403I, F404V, N405S, F406M | 2.80 | dimeric casp-9 catalytic domain (residues 140-416) | D-malate | 214 |
|
| |||||
| with inhibitors | |||||
| 1NW9 | wild type | 2.40 | casp-9 catalytic domain (residues 139-416) |
third baculoviral IAP repeat of XIAP (XIAP-BIR3, protein inhibitor) |
215 |
| 1JXQ | wild type | 2.80 | delta CARD caspase-9 |
Z-VAD-CH2F (covalent, irreversible, peptide inhibitor) |
216 |
Table 10.
Multiple examples of x-ray crystal structures of caspase-7 adapted from PDB database.
| CASPASE-7 | |||||
|---|---|---|---|---|---|
|
| |||||
| PDB entry | Mutation | Res. | Enzyme info | Ligand | Ref. |
| inhibitors free | |||||
|
| |||||
| 1GQF | C285A | 2.90 | procasp-7 (length 265) | ligand free | 217 |
| 1K86 | C186A | 2.60 | casp-7 (length 253) | ligand free | 218 |
| 1K88 | C186A | 2.70 | procasp-7 (length 253) | ligand free | 218 |
| 3IBF | wild type | 2.50 | casp-7 p20 subunit (length 173) and casp-7 p10 subunit (length 97) | ligand free | 219 |
| 3R5K | R210CC246S | 2.86 | redox controlled version of caspase-7 | ligand free | 220 |
| 4FDL | wild type | 2.80 | casp-7 (length 305) | ligand free | 221 |
| 4HQ0 | D198A | 3.00 | procasp-7 (UNP residues 47-303) | ligand free | 222 |
|
| |||||
| with inhibitors | |||||
| 1I51 | D169A | 2.45 | casp-7 p20 subunit(length 148), casp-7 p11 subunit (length 105) |
second baculoviral IAP repeat of XIAP (XIAP-BIR2, protein inhibitor) |
223 |
| 1KMC | C285A | 2.90 | casp-7 (length 303) |
second baculoviral IAP repeat of XIAP (XIAP-BIR2, protein inhibitor) |
224 |
| 1I4O | wild type | 2.40 | casp-7 (length 280) |
second baculoviral IAP repeat of XIAP( XIAP-BIR2, protein inhibitor) |
225 |
| 1F1J | wild type | 2.35 | casp-7 p20/p10 catalytic domain (length 305) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
170 |
| 2QL5 | wild type | 2.34 | casp-7 p20 subunit (length 173), casp-7 p10 subunit (length 97 |
Ac-DMQD- CHO (covalent, reversible, peptide inhibitor) |
173 |
| 2QL7 | wild type | 2.40 | casp-7 p20 subunit (length 173), casp-7 p10 subunit (length 97 |
Ac-IEPD-CHO (covalent, reversible, peptide inhibitor) |
173 |
| 2QL9 | wild type | 2.14 | casp-7 p20 subunit (length 173), casp-7 p10 subunit (length 97 |
Ac-DQMD-CHO (covalent, reversible, peptide inhibitor) |
173 |
| 2QLB | wild type | 2.25 | casp-7 p20 subunit (length 173), casp-7 p10 subunit (length 97 |
Ac-ESMD-CHO (covalent, reversible, peptide inhibitor) |
173 |
| 2QLF | wild type | 2.80 | casp-7 p20 subunit (length 173), casp-7 p10 subunit (length 97 |
Ac-DNLD-CHO (covalent, reversible, peptide inhibitor) |
173 |
| 2QLJ | wild type | 2.60 | casp-7 p20 subunit (length 173), casp-7 p10 subunit (length 97 |
Ac-WEHD-CHO (covalent, reversible, peptide inhibitor) |
173 |
| 3H1P | I213A | 2.61 | caspase-7 (length 260) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
226 |
| 3IBC | wild type | 2.75 | casp-7 p20 subunit (length 173) and casp-7 p10 subunit (length 97) |
Ac-YVAD-CHO (covalent, reversible, peptide inhibitor) |
219 |
| 4HQR | D198A | 3.00 | procasp-7 (UNP residues 47-303) |
Ac-DEVD-CHO (covalent, reversible, peptide inhibitor) |
222 |
| 4JR1 | D198A | 2.15 | procasp-7 (UNP residues 57-303) |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
227 |
| 4JR2 | D198A | 1.65 | procasp-7/ caspase-7 heterodimer |
Ac-DEVD-CH2Cl (covalent, irreversible, peptide inhibitor) |
227 |
| 4JJ8 | wild type | 2.94 | casp-7 length 255 |
(irreversible unnatural aa inhibitor; AOMK) |
20 |
| 1SHJ | D169A | 2.80 | casp-7 (length 262) |
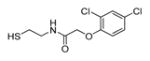 (DICA, allosteric inhibitor) |
228 |
| 1SHL | D192A | 3.00 | casp-7 (length 245) |
 (FICA, allosteric inhibitor) |
228 |
| 4FEA | wild type | 3.79 | casp-7 p20/p10 catalytic domain (UNP residues 57-303) |
 (allosteric inhibitor) |
221 |
Table 46.
Kinetic parameters of ABPs developed by Bogyo group.18
| Code | Specificity region | Ki(app), M−1s−1 | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| Casp Target | Casp-3 | Casp-7 | Casp-8 | Casp-9 | ||
| bAB06 536 | D-30-V-D | 3/7 | 2,528,900 | 412,413 | 29,124 | NI |
| bAB13 537 | D-34-V-D | 3/7 | 6,829,900 | 456,884 | <1,000 | NI |
| bAB19 538 | 31-E-23-D | 8 | 192,225 | 40,011 | 152,956 | NI |
| bAB38 539 | P-L-A-D | 9 | 28,809 | 18,685 | 24,000 | NI |
Acknowledgments
This work was supported by National Science Center in Poland (grant 2011/03/B/ST5/01048) to MD, a statutory activity subsidy for the Faculty of Chemistry at Wroclaw University of Technology to MD and MP, and NIH grant 1R01GM099040 to GSS and a Corporate Sponsored Research Agreement from Genentech, Inc. to GSS. MP is a beneficiary of the Marie Curie Global Fellowship (Horizon2020, PROVIST project). MP and PK are beneficiaries of START scholarship from Foundation for Polish Science.
Biographies

Marcin Poreba obtained his Master degree in Chemical Technology (2010) with specialization in Medicinal Chemistry from Wroclaw University of Technology (Poland) and PhD degree (2014) in Chemistry from the Faculty of Chemistry at the same university under supervision of Marcin Drag. In 2014 he obtained Research Assistant position at the Faculty of Chemistry, Wroclaw University of Technology. During his Ph.D. program he did two research internships: Prof. Boris Turk lab. (Jozef Stefan Institute, Slovenia, 2011) and Prof. Guy Salvesen lab. (Sanford Burnham Prebys Medical Discovery Institute, USA, 2013). He specializes in Organic Synthesis, Peptide Chemistry, and Biochemistry. In 2015 he obtained Marie Curie Global Fellowship for a three-year post-doc in Guy Salvesen lab. and Marcin Drag lab. His research interest is focused on the development of new chemical tools for selective visualization of caspases and cysteine cathepsins in complex biological systems.

Aleksandra Szalek (Strozyk) obtained her Master degree (2011) with specialization in Medicinal Chemistry from Wroclaw University of Technology in Poland. In 2012 she has started Ph.D. study in the Department of Bioorganic Chemistry, Wroclaw University of Technology under the supervision of Marcin Darg. During her studying Aleksandra did two research trainings: Pharmaceutical Production Company HASCO-LEK, Poland (2007) and Pierre and Marie Curie University, France (2011). In her research she focuses on the development of new internally-quenched substrates for the investigation of medicinally important metalloproteases.

Paulina Kasperkiewicz obtained her M.Sc. in 2010, in Pharmaceutical Biotechnology and Bioinformatics at Wroclaw University of Technology, Wroclaw, Poland. That same year she started her Ph.D. program in Chemistry at Wroclaw University of Technology, Wroclaw, Poland and she defend her PhD thesis in 2014 under Dr. Marcin Drag’s supervision. She gained broad experience in Biological Chemistry, Peptide Chemistry, and Biochemistry. Following graduation in 2015 she has started a postdoctoral fellow in Sanford Burnham Prebys Medical Discovery Institute under Professor Guy S. Salvesen supervision, La Jolla, USA. Currently she is an Research Assistant at Faculty of Chemistry, Wroclaw University of Technology in Poland. In her research Dr. Kasperkiewicz focuses in particular on specific activity based probes investigation for neutrophil serine proteases and their application in the visualization of neutrophil traps formation.

Wioletta Rut received her Bachelor’s degree in Biotechnology from Rzeszow University of Technology. In 2013 she received her Master’s degree in Biotechnology at Wroclaw University of Technology, Wroclaw, Poland. Since 2013 Wioletta has been Ph.D. student in chemistry at Wroclaw University of Technology under the supervision of Marcin Drag. Her research interest is focused on combinatorial peptide chemistry and determination of threonine (proteasome) and cysteine (deubiquitinating enzymes) proteases substrate specificity.

Guy Salvesen earned his Ph.D. in biochemistry from Cambridge University in 1980. He conducted postdoctoral research at the University of Georgia, Strangeways Laboratory and MRC Laboratory of Molecular Biology in Cambridge. He obtained his first independent position as Assistant Professor of Pathology at Duke University. In 1996, he moved his laboratory to Sanford Burnham Prebys Medical Discovery Institute in San Diego to work with world leaders in cell death research. Dr. Salvesen is Dean of the Graduate Program in Biomedical Sciences and Director of Scientific Training at Sanford Burnham Prebys, and holds an Adjunct Professorship in Molecular Pathology at the University of California, San Diego. He is on the editorial board of several journals, Reviews Editor of the Biochemical Journal, and Co-founder of the International Proteolysis Society. More information can be found at http://www.salvesenlab.org.

Marcin Drag obtained his Master degree (1999) with specialization in Environmental Chemistry from University of Wroclaw in Poland; PhD degree (2003) in Chemistry from the Institute of Organic Chemistry, Biochemistry and Biotechnology at Wroclaw University of Technology (Poland) under supervision of prof. Pawel Kafarski. After PhD, with interest in Chemical Biology, he gained expertise in medicinal chemistry, chemical biology, proteomics, biochemistry and molecular biology working for 3 years as postdoc under supervision of prof. Guy. S. Salvesen at Sanford Burnham Prebys Medical Discovery Institute (formerly Sanford-Burnham Medical Research Institute), La Jolla, California, USA. Currently he is an Associate Professor at Faculty of Chemistry, Wroclaw University of Technology in Poland. His research group is interested in developing and applying combinatorial libraries, substrates, inhibitors and activity-based probes to decipher the mechanism of action and the function of proteases in health and disease.
Footnotes
Notes
The authors declare no conflict of interest
References
- 1.Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- 2.Cohen GM. Biochem J. 1997;326(Pt 1):1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salvesen GS, Dixit VM. Cell. 1997;91:443–446. doi: 10.1016/s0092-8674(00)80430-4. [DOI] [PubMed] [Google Scholar]
- 4.Fuentes-Prior P, Salvesen GS. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Philchenkov A, Zavelevich M, Kroczak TJ, Los M. Exp Oncol. 2004;26:82–97. [PubMed] [Google Scholar]
- 6.Narula J, Chandrasekhar Y, Dec GW. Apoptosis. 1998;3:309–315. doi: 10.1023/a:1009673501809. [DOI] [PubMed] [Google Scholar]
- 7.Friedlander RM. N Engl J Med. 2003;348:1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 8.Schulz JB, Weller M, Moskowitz MA. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 9.D’Lima D, Hermida J, Hashimoto S, Colwell C, Lotz M. Arthritis Rheum. 2006;54:1814–1821. doi: 10.1002/art.21874. [DOI] [PubMed] [Google Scholar]
- 10.Kay J, Calabrese L. Rheumatology (Oxford) 2004;43(Suppl 3):iii2. doi: 10.1093/rheumatology/keh201. [DOI] [PubMed] [Google Scholar]
- 11.Poreba M, Strozyk A, Salvesen GS, Drag M. Cold Spring Harb Perspect Biol. 2013;5:a008680. doi: 10.1101/cshperspect.a008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Callus BA, Vaux DL. Cell Death Differ. 2007;14:73–78. doi: 10.1038/sj.cdd.4402034. [DOI] [PubMed] [Google Scholar]
- 13.Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, Bogyo M. Nat Med. 2009;15:967–973. doi: 10.1038/nm.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crawford ED, Wells JA. Annu Rev Biochem. 2011;80:1055–1087. doi: 10.1146/annurev-biochem-061809-121639. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien T, Lee D. Mini Rev Med Chem. 2004;4:153–165. doi: 10.2174/1389557043487448. [DOI] [PubMed] [Google Scholar]
- 16.Linton SD. Curr Top Med Chem. 2005;5:1697–1717. doi: 10.2174/156802605775009720. [DOI] [PubMed] [Google Scholar]
- 17.Grabarek J, Amstad P, Darzynkiewicz Z. Hum Cell. 2002;15:1–12. doi: 10.1111/j.1749-0774.2002.tb00094.x. [DOI] [PubMed] [Google Scholar]
- 18.Berger AB, Witte MD, Denault JB, Sadaghiani AM, Sexton KM, Salvesen GS, Bogyo M. Mol Cell. 2006;23:509–521. doi: 10.1016/j.molcel.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Edgington LE, van Raam BJ, Verdoes M, Wierschem C, Salvesen GS, Bogyo M. Chem Biol. 2012;19:340–352. doi: 10.1016/j.chembiol.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vickers CJ, Gonzalez-Paez GE, Wolan DW. ACS Chem Biol. 2013;8:1558–1566. doi: 10.1021/cb400209w. [DOI] [PubMed] [Google Scholar]
- 21.Sanman LE, Bogyo M. Annu Rev Biochem. 2014;83:249–273. doi: 10.1146/annurev-biochem-060713-035352. [DOI] [PubMed] [Google Scholar]
- 22.Rano TA, Timkey T, Peterson EP, Rotonda J, Nicholson DW, Becker JW, Chapman KT, Thornberry NA. Chem Biol. 1997;4:149–155. doi: 10.1016/s1074-5521(97)90258-1. [DOI] [PubMed] [Google Scholar]
- 23.Talanian RV, Brady KD, Cryns VL. J Med Chem. 2000;43:3351–3371. doi: 10.1021/jm000060f. [DOI] [PubMed] [Google Scholar]
- 24.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, et al. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 25.Poreba M, Drag M. Curr Med Chem. 2010;17:3968–3995. doi: 10.2174/092986710793205381. [DOI] [PubMed] [Google Scholar]
- 26.Schechter I, Berger A. Biochem Biophys Res Commun. 1967;27:157–162. doi: 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- 27.Ostresh JM, Winkle JH, Hamashin VT, Houghten RA. Biopolymers. 1994;34:1681–1689. doi: 10.1002/bip.360341212. [DOI] [PubMed] [Google Scholar]
- 28.Sleath PR, Hendrickson RC, Kronheim SR, March CJ, Black RA. J Biol Chem. 1990;265:14526–14528. [PubMed] [Google Scholar]
- 29.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, et al. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 30.Howard AD, Kostura MJ, Thornberry N, Ding GJ, Limjuco G, Weidner J, Salley JP, Hogquist KA, Chaplin DD, Mumford RA, et al. J Immunol. 1991;147:2964–2969. [PubMed] [Google Scholar]
- 31.Ding L, Coombs GS, Strandberg L, Navre M, Corey DR, Madison EL. Proc Natl Acad Sci USA. 1995;92:7627–7631. doi: 10.1073/pnas.92.17.7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ke SH, Coombs GS, Tachias K, Corey DR, Madison EL. J Biol Chem. 1997;272:20456–20462. doi: 10.1074/jbc.272.33.20456. [DOI] [PubMed] [Google Scholar]
- 33.Wachmann K, Pop C, van Raam BJ, Drag M, Mace PD, Snipas SJ, Zmasek C, Schwarzenbacher R, Salvesen GS, Riedl SJ. Biochemistry. 2010;49:8307–8315. doi: 10.1021/bi100968m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikolajczyk J, Scott FL, Krajewski S, Sutherlin DP, Salvesen GS. Biochemistry. 2004;43:10560–10569. doi: 10.1021/bi0498048. [DOI] [PubMed] [Google Scholar]
- 35.Garcia-Calvo M, Peterson EP, Rasper DM, Vaillancourt JP, Zamboni R, Nicholson DW, Thornberry NA. Cell Death Differ. 1999;6:362–369. doi: 10.1038/sj.cdd.4400497. [DOI] [PubMed] [Google Scholar]
- 36.Edwards PD, Mauger RC, Cottrell KM, Morris FX, Pine KK, Sylvester MA, Scott CW, Furlong ST. Bioorg Med Chem Lett. 2000;10:2291–2294. doi: 10.1016/s0960-894x(00)00460-1. [DOI] [PubMed] [Google Scholar]
- 37.Backes BJ, Harris JL, Leonetti F, Craik CS, Ellman JA. Nat Biotechnol. 2000;18:187–193. doi: 10.1038/72642. [DOI] [PubMed] [Google Scholar]
- 38.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 39.Tang Y, Wells JA, Arkin MR. J Biol Chem. 2011;286:34147–34154. doi: 10.1074/jbc.M111.247627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris JL, Backes BJ, Leonetti F, Mahrus S, Ellman JA, Craik CS. Proc Natl Acad Sci USA. 2000;97:7754–7759. doi: 10.1073/pnas.140132697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maly DJ, Leonetti F, Backes BJ, Dauber DS, Harris JL, Craik CS, Ellman JA. J Org Chem. 2002;67:910–915. doi: 10.1021/jo016140o. [DOI] [PubMed] [Google Scholar]
- 42.Walters J, Pop C, Scott FL, Drag M, Swartz P, Mattos C, Salvesen GS, Clark AC. Biochem J. 2009;424:335–345. doi: 10.1042/BJ20090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poreba M, Szalek A, Kasperkiewicz P, Drag M. Methods Mol Biol. 2014;1133:41–59. doi: 10.1007/978-1-4939-0357-3_2. [DOI] [PubMed] [Google Scholar]
- 44.Kasperkiewicz P, Poreba M, Snipas SJ, Parker H, Winterbourn CC, Salvesen GS, Drag M. Proc Natl Acad Sci USA. 2014;111:2518–2523. doi: 10.1073/pnas.1318548111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poreba M, Kasperkiewicz P, Snipas SJ, Fasci D, Salvesen GS, Drag M. Cell Death Differ. 2014;21:1482–1492. doi: 10.1038/cdd.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lozanov V, Ivanov IP, Benkova B, Mitev V. Amino Acids. 2009;36:581–586. doi: 10.1007/s00726-008-0136-4. [DOI] [PubMed] [Google Scholar]
- 47.Stennicke HR, Renatus M, Meldal M, Salvesen GS. Biochem J. 2000;350(Pt 2):563–568. [PMC free article] [PubMed] [Google Scholar]
- 48.Stack MS, Gray RD. J Biol Chem. 1989;264:4277–4281. [PubMed] [Google Scholar]
- 49.Chowdhury I, Tharakan B, Bhat GK. Comp Biochem Physiol B, Biochem Mol Biol. 2008;151:10–27. doi: 10.1016/j.cbpb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 50.Petrassi HM, Williams JA, Li J, Tumanut C, Ek J, Nakai T, Masick B, Backes BJ, Harris JL. Bioorg Med Chem Lett. 2005;15:3162–3166. doi: 10.1016/j.bmcl.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 51.Winssinger N, Damoiseaux R, Tully DC, Geierstanger BH, Burdick K, Harris JL. Chem Biol. 2004;11:1351–1360. doi: 10.1016/j.chembiol.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 52.Smith MM, Shi L, Navre M. J Biol Chem. 1995;270:6440–6449. doi: 10.1074/jbc.270.12.6440. [DOI] [PubMed] [Google Scholar]
- 53.Lien S, Pastor R, Sutherlin D, Lowman HB. Protein J. 2004;23:413–425. doi: 10.1023/b:jopc.0000039555.92058.51. [DOI] [PubMed] [Google Scholar]
- 54.Boulware KT, Daugherty PS. Proc Natl Acad Sci USA. 2006;103:7583–7588. doi: 10.1073/pnas.0511108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McStay GP, Salvesen GS, Green DR. Cell Death Differ. 2008;15:322–331. doi: 10.1038/sj.cdd.4402260. [DOI] [PubMed] [Google Scholar]
- 56.Pereira NA, Song Z. Biochem Biophys Res Commun. 2008;377:873–877. doi: 10.1016/j.bbrc.2008.10.101. [DOI] [PubMed] [Google Scholar]
- 57.Benkova B, Lozanov V, Ivanov IP, Mitev V. Anal Biochem. 2009;394:68–74. doi: 10.1016/j.ab.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 58.Timmer JC, Salvesen GS. Cell Death Differ. 2007;14:66–72. doi: 10.1038/sj.cdd.4402059. [DOI] [PubMed] [Google Scholar]
- 59.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 60.Jiang X, Wang X. J Biol Chem. 2000;275:31199–31203. doi: 10.1074/jbc.C000405200. [DOI] [PubMed] [Google Scholar]
- 61.Denault JB, Eckelman BP, Shin H, Pop C, Salvesen GS. Biochem J. 2007;405:11–19. doi: 10.1042/BJ20070288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vickers CJ, Gonzalez-Paez GE, Wolan DW. ACS Chem Biol. 2014:2199–2203. doi: 10.1021/cb500586p. [DOI] [PubMed] [Google Scholar]
- 63.Vickers CJ, Gonzalez-Paez GE, Wolan DW. J Am Chem Soc. 2013;135:12869–12876. doi: 10.1021/ja406399r. [DOI] [PubMed] [Google Scholar]
- 64.Huang X, Lee S, Chen X. Am J Nucl Med Mol Imaging. 2011;1:3–17. [PMC free article] [PubMed] [Google Scholar]
- 65.Niu G, Chen X. J Nucl Med. 2010;51:1659–1662. doi: 10.2967/jnumed.110.078584. [DOI] [PubMed] [Google Scholar]
- 66.Kobayashi H, Choyke PL. Acc Chem Res. 2011;44:83–90. doi: 10.1021/ar1000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee S, Park K, Kim K, Choi K, Kwon IC. Chem Commun (Camb) 2008:4250–4260. doi: 10.1039/b806854m. [DOI] [PubMed] [Google Scholar]
- 68.Lee S, Xie J, Chen X. Curr Top Med Chem. 2010;10:1135–1144. doi: 10.2174/156802610791384270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weissleder R, Tung CH, Mahmood U, Bogdanov A., Jr Nat Biotechnol. 1999;17:375–378. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 70.Li C, Lee CJ, Simeone DM. Methods Mol Biol. 2009;568:161–173. doi: 10.1007/978-1-59745-280-9_10. [DOI] [PubMed] [Google Scholar]
- 71.Liu J, Bhalgat M, Zhang C, Diwu Z, Hoyland B, Klaubert DH. Bioorg Med Chem Lett. 1999;9:3231–3236. doi: 10.1016/s0960-894x(99)00566-1. [DOI] [PubMed] [Google Scholar]
- 72.Cai SX, Zhang HZ, Guastella J, Drewe J, Yang W, Weber E. Bioorg Med Chem Lett. 2001;11:39–42. doi: 10.1016/s0960-894x(00)00590-4. [DOI] [PubMed] [Google Scholar]
- 73.Zhang HZ, Kasibhatla S, Guastella J, Tseng B, Drewe J, Cai SX. Bioconjug Chem. 2003;14:458–463. doi: 10.1021/bc0256188. [DOI] [PubMed] [Google Scholar]
- 74.Wang ZQ, Liao J, Diwu Z. Bioorg Med Chem Lett. 2005;15:2335–2338. doi: 10.1016/j.bmcl.2005.02.081. [DOI] [PubMed] [Google Scholar]
- 75.Kushida Y, Hanaoka K, Komatsu T, Terai T, Ueno T, Yoshida K, Uchiyama M, Nagano T. Bioorg Med Chem Lett. 2012;22:3908–3911. doi: 10.1016/j.bmcl.2012.04.114. [DOI] [PubMed] [Google Scholar]
- 76.Wang F, Chen TS, Xing D, Wang JJ, Wu YX. Lasers Surg Med. 2005;36:2–7. doi: 10.1002/lsm.20130. [DOI] [PubMed] [Google Scholar]
- 77.Cen H, Mao F, Aronchik I, Fuentes RJ, Firestone GL. FASEB J. 2008;22:2243–2252. doi: 10.1096/fj.07-099234. [DOI] [PubMed] [Google Scholar]
- 78.Bullok K, Piwnica-Worms D. J Med Chem. 2005;48:5404–5407. doi: 10.1021/jm050008p. [DOI] [PubMed] [Google Scholar]
- 79.Bullok KE, Maxwell D, Kesarwala AH, Gammon S, Prior JL, Snow M, Stanley S, Piwnica-Worms D. Biochemistry. 2007;46:4055–4065. doi: 10.1021/bi061959n. [DOI] [PubMed] [Google Scholar]
- 80.Barnett EM, Zhang X, Maxwell D, Chang Q, Piwnica-Worms D. Proc Natl Acad Sci USA. 2009;106:9391–9396. doi: 10.1073/pnas.0812884106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maxwell D, Chang Q, Zhang X, Barnett EM, Piwnica-Worms D. Bioconjug Chem. 2009;20:702–709. doi: 10.1021/bc800516n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee GH, Lee EJ, Hah SS. Anal Biochem. 2014;446:22–24. doi: 10.1016/j.ab.2013.10.013. [DOI] [PubMed] [Google Scholar]
- 83.Amstad PA, Yu G, Johnson GL, Lee BW, Dhawan S, Phelps DJ. Biotechniques. 2001;31:608–610. doi: 10.2144/01313pf01. [DOI] [PubMed] [Google Scholar]
- 84.Tyas L, Brophy VA, Pope A, Rivett AJ, Tavare JM. EMBO Rep. 2000;1:266–270. doi: 10.1093/embo-reports/kvd050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Karvinen J, Hurskainen P, Gopalakrishnan S, Burns D, Warrior U, Hemmila I. J Biomol Screen. 2002;7:223–231. doi: 10.1177/108705710200700306. [DOI] [PubMed] [Google Scholar]
- 86.Gopalakrishnan SM, Karvinen J, Kofron JL, Burns DJ, Warrior U. J Biomol Screen. 2002;7:317–323. doi: 10.1177/108705710200700403. [DOI] [PubMed] [Google Scholar]
- 87.Mizukami S, Kikuchi K, Higuchi T, Urano Y, Mashima T, Tsuruo T, Nagano T. FEBS Lett. 1999;453:356–360. doi: 10.1016/s0014-5793(99)00755-3. [DOI] [PubMed] [Google Scholar]
- 88.Leriche G, Budin G, Darwich Z, Weltin D, Mely Y, Klymchenko AS, Wagner A. Chem Commun (Camb) 2012;48:3224–3226. doi: 10.1039/c2cc17542h. [DOI] [PubMed] [Google Scholar]
- 89.Kawai H, Suzuki T, Kobayashi T, Mizuguchi H, Hayakawa T, Kawanishi T. Biochim Biophys Acta. 2004;1693:101–110. doi: 10.1016/j.bbamcr.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 90.Rehm M, Dussmann H, Janicke RU, Tavare JM, Kogel D, Prehn JH. J Biol Chem. 2002;277:24506–24514. doi: 10.1074/jbc.M110789200. [DOI] [PubMed] [Google Scholar]
- 91.Luo KQ, Yu VC, Pu Y, Chang DC. Biochem Biophys Res Commun. 2001;283:1054–1060. doi: 10.1006/bbrc.2001.4896. [DOI] [PubMed] [Google Scholar]
- 92.Luo KQ, Yu VC, Pu Y, Chang DC. Biochem Biophys Res Commun. 2003;304:217–222. doi: 10.1016/s0006-291x(03)00559-x. [DOI] [PubMed] [Google Scholar]
- 93.He L, Wu X, Meylan F, Olson DP, Simone J, Hewgill D, Siegel R, Lipsky PE. Am J Pathol. 2004;164:1901–1913. doi: 10.1016/S0002-9440(10)63751-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Onuki R, Nagasaki A, Kawasaki H, Baba T, Uyeda TQ, Taira K. Proc Natl Acad Sci USA. 2002;99:14716–14721. doi: 10.1073/pnas.232177599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Donahue CJ, Santoro M, Hupe D, Jones JM, Pollok B, Heim R, Giegel D. Cytometry. 2001;45:225–234. doi: 10.1002/1097-0320(20011101)45:3<225::aid-cyto1166>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 96.Jones J, Heim R, Hare E, Stack J, Pollok BA. J Biomol Screen. 2000;5:307–318. doi: 10.1177/108705710000500502. [DOI] [PubMed] [Google Scholar]
- 97.Mahajan NP, Harrison-Shostak DC, Michaux J, Herman B. Chem Biol. 1999;6:401–409. doi: 10.1016/s1074-5521(99)80051-9. [DOI] [PubMed] [Google Scholar]
- 98.Xu X, Gerard AL, Huang BC, Anderson DC, Payan DG, Luo Y. Nucleic Acids Res. 1998;26:2034–2035. doi: 10.1093/nar/26.8.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Brophy VA, Tavare JM, Rivett AJ. Arch Biochem Biophys. 2002;397:199–205. doi: 10.1006/abbi.2001.2679. [DOI] [PubMed] [Google Scholar]
- 100.Harpur AG, Wouters FS, Bastiaens PI. Nat Biotechnol. 2001;19:167–169. doi: 10.1038/84443. [DOI] [PubMed] [Google Scholar]
- 101.Kawai H, Suzuki T, Kobayashi T, Sakurai H, Ohata H, Honda K, Momose K, Namekata I, Tanaka H, Shigenobu K, Nakamura R, Hayakawa T, Kawanishi T. J Pharmacol Sci. 2005;97:361–368. doi: 10.1254/jphs.fp0040592. [DOI] [PubMed] [Google Scholar]
- 102.Takemoto K, Nagai T, Miyawaki A, Miura M. J Cell Biol. 2003;160:235–243. doi: 10.1083/jcb.200207111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nagai T, Miyawaki A. Biochem Biophys Res Commun. 2004;319:72–77. doi: 10.1016/j.bbrc.2004.04.147. [DOI] [PubMed] [Google Scholar]
- 104.Paschotta Rd. RP Photonics Consulting. RP Photonics Consulting; Zürich, Switzerland: [Google Scholar]
- 105.Paschotta Rd. Encyclopedia of laser physics and technology. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 106.Joseph J, Seervi M, Sobhan PK, Retnabai ST. PLoS One. 2011;6:e20114. doi: 10.1371/journal.pone.0020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Karasawa S, Araki T, Nagai T, Mizuno H, Miyawaki A. Biochem J. 2004;381:307–312. doi: 10.1042/BJ20040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Suzuki M, Ito Y, Sakata I, Sakai T, Husimi Y, Douglas KT. Biochem Biophys Res Commun. 2005;330:454–460. doi: 10.1016/j.bbrc.2005.02.178. [DOI] [PubMed] [Google Scholar]
- 109.Wu X, Simone J, Hewgill D, Siegel R, Lipsky PE, He L. Cytometry A. 2006;69:477–486. doi: 10.1002/cyto.a.20300. [DOI] [PubMed] [Google Scholar]
- 110.Nicholls SB, Chu J, Abbruzzese G, Tremblay KD, Hardy JA. J Biol Chem. 2011;286:24977–24986. doi: 10.1074/jbc.M111.221648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vuojola J, Syrjanpaa M, Lamminmaki U, Soukka T. Anal Chem. 2012;85:1367–1373. doi: 10.1021/ac302030q. [DOI] [PubMed] [Google Scholar]
- 112.Valanne A, Malmi P, Appelblom H, Niemela P, Soukka T. Anal Biochem. 2008;375:71–81. doi: 10.1016/j.ab.2007.12.032. [DOI] [PubMed] [Google Scholar]
- 113.Olson ES, Jiang T, Aguilera TA, Nguyen QT, Ellies LG, Scadeng M, Tsien RY. Proc Natl Acad Sci USA. 2010;107:4311–4316. doi: 10.1073/pnas.0910283107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lee S, Choi KY, Chung H, Ryu JH, Lee A, Koo H, Youn IC, Park JH, Kim IS, Kim SY, Chen X, Jeong SY, Kwon IC, Kim K, Choi K. Bioconjug Chem. 2011;22:125–131. doi: 10.1021/bc1004119. [DOI] [PubMed] [Google Scholar]
- 115.Liu Y, Hu Y, Guo Y, Ma H, Li J, Jiang C. J Control Release. 2012;163:203–210. doi: 10.1016/j.jconrel.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 116.Sun IC, Lee S, Koo H, Kwon IC, Choi K, Ahn CH, Kim K. Bioconjug Chem. 2010;21:1939–1942. doi: 10.1021/bc1003026. [DOI] [PubMed] [Google Scholar]
- 117.Jun YW, Sheikholeslami S, Hostetter DR, Tajon C, Craik CS, Alivisatos AP. Proc Natl Acad Sci USA. 2009;106:17735–17740. doi: 10.1073/pnas.0907367106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lin SY, Chen NT, Sun SP, Chang JC, Wang YC, Yang CS, Lo LW. J Am Chem Soc. 2010;132:8309–8315. doi: 10.1021/ja100561k. [DOI] [PubMed] [Google Scholar]
- 119.Pan Y, Guo M, Nie Z, Huang Y, Peng Y, Liu A, Qing M, Yao S. Chem Commun (Camb) 2012;48:997–99. doi: 10.1039/c1cc15407a. [DOI] [PubMed] [Google Scholar]
- 120.Gao W, Ji L, Li L, Cui G, Xu K, Li P, Tang B. Biomaterials. 2012;33:3710–3718. doi: 10.1016/j.biomaterials.2012.01.047. [DOI] [PubMed] [Google Scholar]
- 121.Wang H, Zhang Q, Chu X, Chen T, Ge J, Yu R. Angew Chem. 2011;50:7065–7069. doi: 10.1002/anie.201101351. [DOI] [PubMed] [Google Scholar]
- 122.Chen HX, Zhang JJ, Gao YM, Liu SY, Koh K, Zhu XL, Yin YM. Biosens Bioelectron. 2015;68:777–782. doi: 10.1016/j.bios.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 123.Hung VWS, Veloso AJ, Chow AM, Ganesh HVS, Seo K, Kenduzler E, Brown IR, Kerman K. Electrochim Acta. 2015;162:79–85. [Google Scholar]
- 124.Kihara T, Nakamura C, Suzuki M, Han SW, Fukazawa K, Ishihara K, Miyake J. Biosens Bioelectron. 2009;25:22–27. doi: 10.1016/j.bios.2009.05.036. [DOI] [PubMed] [Google Scholar]
- 125.Han S, Nakamura C, Obataya I, Nakamura N, Miyake J. Biochem Biophys Res Commun. 2005;332:633–639. doi: 10.1016/j.bbrc.2005.04.059. [DOI] [PubMed] [Google Scholar]
- 126.Boeneman K, Mei BC, Dennis AM, Bao G, Deschamps JR, Mattoussi H, Medintz IL. J Am Chem Soc. 2009;131:3828–3829. doi: 10.1021/ja809721j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hug H, Los M, Hirt W, Debatin KM. Biochemistry. 1999;38:13906–13911. doi: 10.1021/bi9913395. [DOI] [PubMed] [Google Scholar]
- 128.Xia Z, Rao J. Curr Opin Biotechnol. 2009;20:37–44. doi: 10.1016/j.copbio.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu Y, Piston DW, Johnson CH. Proc Natl Acad Sci USA. 1999;96:151–156. doi: 10.1073/pnas.96.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lechardeur D, Dougaparsad S, Nemes C, Lukacs GL. J Biol Chem. 2005;280:40216–40225. doi: 10.1074/jbc.M502220200. [DOI] [PubMed] [Google Scholar]
- 131.De A, Gambhir SS. FASEB J. 2005;19:2017–2019. doi: 10.1096/fj.05-4628fje. [DOI] [PubMed] [Google Scholar]
- 132.Arai R, Nakagawa H, Kitayama A, Ueda H, Nagamune T. J Biosci Bioeng. 2002;94:362–364. doi: 10.1263/jbb.94.362. [DOI] [PubMed] [Google Scholar]
- 133.Otsuji T, Okuda-Ashitaka E, Kojima S, Akiyama H, Ito S, Ohmiya Y. Anal Biochem. 2004;329:230–237. doi: 10.1016/j.ab.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 134.Angers S, Salahpour A, Joly E, Hilairet S, Chelsky D, Dennis M, Bouvier M. Proc Natl Acad Sci USA. 2000;97:3684–3689. doi: 10.1073/pnas.060590697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gammon ST, Villalobos VM, Roshal M, Samrakandi M, Piwnica-Worms D. Biotechnol Prog. 2009;25:559–569. doi: 10.1002/btpr.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.O’Brien MA, Daily WJ, Hesselberth PE, Moravec RA, Scurria MA, Klaubert DH, Bulleit RF, Wood KV. J Biomol Screen. 2005;10:137–148. doi: 10.1177/1087057104271865. [DOI] [PubMed] [Google Scholar]
- 137.Liu JJ, Wang W, Dicker DT, El-Deiry WS. Cancer Biol Ther. 2005;4:885–892. doi: 10.4161/cbt.4.8.2133. [DOI] [PubMed] [Google Scholar]
- 138.Scabini M, Stellari F, Cappella P, Rizzitano S, Texido G, Pesenti E. Apoptosis. 2011;16:198–207. doi: 10.1007/s10495-010-0553-1. [DOI] [PubMed] [Google Scholar]
- 139.Hickson J, Ackler S, Klaubert D, Bouska J, Ellis P, Foster K, Oleksijew A, Rodriguez L, Schlessinger S, Wang B, et al. Cell Death Differ. 2010;17:1003–1010. doi: 10.1038/cdd.2009.205. [DOI] [PubMed] [Google Scholar]
- 140.Shah K, Tung CH, Breakefield XO, Weissleder R. Mol Ther. 2005;11:926–931. doi: 10.1016/j.ymthe.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 141.Kizaka-Kondoh S, Itasaka S, Zeng L, Tanaka S, Zhao T, Takahashi Y, Shibuya K, Hirota K, Semenza GL, Hiraoka M. Clin Cancer Res. 2009;15:3433–3441. doi: 10.1158/1078-0432.CCR-08-2267. [DOI] [PubMed] [Google Scholar]
- 142.Kindermann M, Roschitzki-Voser H, Caglic D, Repnik U, Miniejew C, Mittl PR, Kosec G, Grutter MG, Turk B, Wendt KU. Chem Biol. 2010;17:999–1007. doi: 10.1016/j.chembiol.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 143.Van de Bittner GC, Bertozzi CR, Chang CJ. J Am Chem Soc. 2013;135:1783–1795. doi: 10.1021/ja309078t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Godinat A, Park HM, Miller SC, Cheng K, Hanahan D, Sanman LE, Bogyo M, Yu A, Nikitin GF, Stahl A, et al. ACS Chem Biol. 2013;8:987–999. doi: 10.1021/cb3007314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Laxman B, Hall DE, Bhojani MS, Hamstra DA, Chenevert TL, Ross BD, Rehemtulla A. Proc Natl Acad Sci USA. 2002;99:16551–16555. doi: 10.1073/pnas.252644499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kanno A, Yamanaka Y, Hirano H, Umezawa Y, Ozawa T. Angew Chem. 2007;46:7595–7599. doi: 10.1002/anie.200700538. [DOI] [PubMed] [Google Scholar]
- 147.Ozaki M, Haga S, Ozawa T. Theranostics. 2012;2:207–214. doi: 10.7150/thno.3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang F, Zhu L, Liu G, Hida N, Lu G, Eden HS, Niu G, Chen X. Theranostics. 2011;1:302–309. doi: 10.7150/thno/v01p0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Coppola JM, Ross BD, Rehemtulla A. Clin Cancer Res. 2008;14:2492–2501. doi: 10.1158/1078-0432.CCR-07-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ray P, De A, Patel M, Gambhir SS. Clin Cancer Res. 2008;14:5801–5809. doi: 10.1158/1078-0432.CCR-07-5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shah K, Tang Y, Breakefield X, Weissleder R. Oncogene. 2003;22:6865–6872. doi: 10.1038/sj.onc.1206748. [DOI] [PubMed] [Google Scholar]
- 152.Xiao H, Liu L, Meng F, Huang J, Li G. Anal Chem. 2008;80:5272–5275. doi: 10.1021/ac8005268. [DOI] [PubMed] [Google Scholar]
- 153.Waud JP, Bermudez Fajardo A, Sudhaharan T, Trimby AR, Jeffery J, Jones A, Campbell AK. Biochem J. 2001;357:687–697. doi: 10.1042/0264-6021:3570687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Black RA, Kronheim SR, Sleath PR. FEBS Lett. 1989;247:386–390. doi: 10.1016/0014-5793(89)81376-6. [DOI] [PubMed] [Google Scholar]
- 155.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 156.Fernandes-Alnemri T, Litwack G, Alnemri ES. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 157.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 158.Powers JC, Asgian JL, Ekici OD, James KE. Chem Rev. 2002;102:4639–4750. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 159.Graczyk PP. Prog Med Chem. 2002;39:1–72. doi: 10.1016/s0079-6468(08)70068-7. [DOI] [PubMed] [Google Scholar]
- 160.Brady KD, Giegel DA, Grinnell C, Lunney E, Talanian RV, Wong W, Walker N. Bioorg Med Chem. 1999;7:621–631. doi: 10.1016/s0968-0896(99)00009-7. [DOI] [PubMed] [Google Scholar]
- 161.Brady KD. Biochemistry. 1998;37:8508–8515. doi: 10.1021/bi9803325. [DOI] [PubMed] [Google Scholar]
- 162.Kitz R, Wilson IB. J Biol Chem. 1962;237:3245–3249. [PubMed] [Google Scholar]
- 163.Pop C, Salvesen GS. J Biol Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Denault JB, Salvesen GS. Chem Rev. 2002;102:4489–4500. doi: 10.1021/cr010183n. [DOI] [PubMed] [Google Scholar]
- 165.Elliott JM, Rouge L, Wiesmann C, Scheer JM. The J Biol Chem. 2009;284:6546–6553. doi: 10.1074/jbc.M806121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M, Peterson EP, Rasper DM, Ruel R, Vaillancourt JP, et al. Nat Struct Biol. 1996;3:619–625. doi: 10.1038/nsb0796-619. [DOI] [PubMed] [Google Scholar]
- 167.Mittl PR, Di Marco S, Krebs JF, Bai X, Karanewsky DS, Priestle JP, Tomaselli KJ, Grutter MG. J Biol Chem. 1997;272:6539–6547. doi: 10.1074/jbc.272.10.6539. [DOI] [PubMed] [Google Scholar]
- 168.Ganesan R, Jelakovic S, Campbell AJ, Li ZZ, Asgian JL, Powers JC, Grutter MG. Biochemistry. 2006;45:9059–9067. doi: 10.1021/bi060364p. [DOI] [PubMed] [Google Scholar]
- 169.Thornberry NA. Chem Biol. 1998;5:R97–103. doi: 10.1016/s1074-5521(98)90615-9. [DOI] [PubMed] [Google Scholar]
- 170.Wei Y, Fox T, Chambers SP, Sintchak J, Coll JT, Golec JM, Swenson L, Wilson KP, Charifson PS. Chem Biol. 2000;7:423–432. doi: 10.1016/s1074-5521(00)00123-x. [DOI] [PubMed] [Google Scholar]
- 171.Schweizer A, Briand C, Grutter MG. J Biol Chem. 2003;278:42441–42447. doi: 10.1074/jbc.M304895200. [DOI] [PubMed] [Google Scholar]
- 172.Yoshimori A, Sakai J, Sunaga S, Kobayashi T, Takahashi S, Okita N, Takasawa R, Tanuma S. BMC Pharmacol. 2007;7:8. doi: 10.1186/1471-2210-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Agniswamy J, Fang B, Weber IT. FEBS J. 2007;274:4752–65. doi: 10.1111/j.1742-4658.2007.05994.x. [DOI] [PubMed] [Google Scholar]
- 174.Blanchard H, Kodandapani L, Mittl PR, Marco SD, Krebs JF, Wu JC, Tomaselli KJ, Grutter MG. Structure. 1999;7:1125–1133. doi: 10.1016/s0969-2126(99)80179-8. [DOI] [PubMed] [Google Scholar]
- 175.Watt W, Koeplinger KA, Mildner AM, Heinrikson RL, Tomasselli AG, Watenpaugh KD. Structure. 1999;7:1135–1143. doi: 10.1016/s0969-2126(99)80180-4. [DOI] [PubMed] [Google Scholar]
- 176.Grimm EL, Roy B, Aspiotis R, Bayly CI, Nicholson DW, Rasper DM, Renaud J, Roy S, Tam J, Tawa P, et al. Bioorg Med Chem. 2004;12:845–851. doi: 10.1016/j.bmc.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 177.Ganesan R, Mittl PR, Jelakovic S, Grutter MG. J Mol Biol. 2006;359:1378–1388. doi: 10.1016/j.jmb.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 178.Soper DL, Sheville J, O’Neil SV, Wang Y, Laufersweiler MC, Oppong KA, Wos JA, Ellis CD, Fancher AN, Lu W, et al. Bioorg Med Chem Lett. 2006;16:4233–4236. doi: 10.1016/j.bmcl.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 179.Fu G, Chumanevich AA, Agniswamy J, Fang B, Harrison RW, Weber IT. Apoptosis. 2008;13:1291–1302. doi: 10.1007/s10495-008-0259-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Fang B, Boross PI, Tozser J, Weber IT. J Mol Biol. 2006;360:654–666. doi: 10.1016/j.jmb.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 181.Romanowski MJ, Scheer JM, O’Brien T, McDowell RS. Structure. 2004;12:1361–1371. doi: 10.1016/j.str.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 182.Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, et al. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 183.Galatsis P, Caprathe B, Gilmore J, Thomas A, Linn K, Sheehan S, Harter W, Kostlan C, Lunney E, Stankovic C, et al. Bioorg Med Chem Lett. 2010;20:5184–5190. doi: 10.1016/j.bmcl.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 184.Okamoto Y, Anan H, Nakai E, Morihira K, Yonetoku Y, Kurihara H, Sakashita H, Terai Y, Takeuchi M, Shibanuma T, et al. Chem Pharm Bull (Tokyo) 1999;47:11–21. doi: 10.1248/cpb.47.11. [DOI] [PubMed] [Google Scholar]
- 185.O’Brien T, Fahr BT, Sopko MM, Lam JW, Waal ND, Raimundo BC, Purkey HE, Pham P, Romanowski MJ. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:451–458. doi: 10.1107/S1744309105010109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.O’Brien T, Fahr BT, Sopko MM, Lam JW, Waal ND, Raimundo BC, Purkey HE, Pham P, Romanowski MJ. Protein Data Bank. 2004:1RWV. [Google Scholar]
- 187.Fahr BT, O’Brien T, Pham P, Waal ND, Baskaran S, Raimundo BC, Lam JW, Sopko MM, Purkey HE, Romanowski MJ. Bioorg Med Chem Lett. 2006;16:559–562. doi: 10.1016/j.bmcl.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 188.Scheer JM, Romanowski MJ, Wells JA. Proc Natl Acad Sci USA. 2006;103:7595–7600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Datta D, Scheer JM, Romanowski MJ, Wells JA. J Mol Biol. 2008;381:1157–1167. doi: 10.1016/j.jmb.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Rosen-Wolff A, Romanowski MJ, Ritter L, Flecks S, Quoos N, Gramatt J, Petzold C, Nguyen HD, Gahr M, Roesler J. Ann Rheum Dis. 2008;67(Suppl II):106. [Google Scholar]
- 191.Schweizer A, Roschitzki-Voser H, Amstutz P, Briand C, Gulotti-Georgieva M, Prenosil E, Binz HK, Capitani G, Baici A, Pluckthun A, et al. Structure. 2007;15:625–636. doi: 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 192.Maillard MC, Brookfield FA, Courtney SM, Eustache FM, Gemkow MJ, Handel RK, Johnson LC, Johnson PD, Kerry MA, Krieger FC, et al. Bioorg Med Chem. 2011;19:5833–5851. doi: 10.1016/j.bmc.2011.08.020. [DOI] [PubMed] [Google Scholar]
- 193.Baumgartner R, Meder G, Briand C, Decock A, D’Arcy A, Hassiepen U, Morse R, Renatus M. Biochem J. 2009;423:429. doi: 10.1042/BJ20090540. [DOI] [PubMed] [Google Scholar]
- 194.Wang XJ, Cao Q, Liu X, Wang KT, Mi W, Zhang Y, Li LF, LeBlanc AC, Su XD. EMBO Rep. 2010;11:841. doi: 10.1038/embor.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Vaidya S, Abbruzzese G, Velazquez E, Witkowski W, Hardy J. Protein Data Bank. 2010:3K7E. [Google Scholar]
- 196.Vaidya S, Velazquez-Delgado EM, Abbruzzese G, Hardy JA. J Mol Biol. 2011;406:75–91. doi: 10.1016/j.jmb.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Muller I, Lamers MB, Ritchie AJ, Park H, Dominguez C, Munoz-Sanjuan I, Maillard M, Kiselyov A. J Mol Biol. 2011;410:307–315. doi: 10.1016/j.jmb.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 198.Cao Q, Wang XJ, Li LF, Su XD. Acta Crystallogr D Biol Crystallogr. 2014;70:58–67. doi: 10.1107/S1399004713024218. [DOI] [PubMed] [Google Scholar]
- 199.Stanger K, Steffek M, Zhou L, Pozniak CD, Quan C, Franke Y, Tom J, Tam C, Krylova I, Elliott JM, et al. Nat Chem Biol. 2012;8:655–660. doi: 10.1038/nchembio.967. [DOI] [PubMed] [Google Scholar]
- 200.Velazquez-Delgado EM, Hardy JA. J Biol Chem. 2012;287:36000–36011. doi: 10.1074/jbc.M112.397752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Liu X, Zhang H, Wang XJ, Li LF, Su XD. PLoS One. 2011;6:e24227. doi: 10.1371/journal.pone.0024227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mueller I, Lamers MBAC, Ritchie AJ, Dominguez C, Munoz I, Maillard M, Kiselyov A. Protein Data Bank. 2011:3P4U. [Google Scholar]
- 203.Muller I, Lamers MB, Ritchie AJ, Dominguez C, Munoz-Sanjuan I, Kiselyov A. Bioorg Med Chem Lett. 2011;21:5244–5247. doi: 10.1016/j.bmcl.2011.07.041. [DOI] [PubMed] [Google Scholar]
- 204.Heise CE, Murray J, Augustyn KE, Bravo B, Chugha P, Cohen F, Giannetti AM, Gibbons P, Hannoush RN, Hearn BR, et al. PloS One. 2012;7:e50864. doi: 10.1371/journal.pone.0050864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Murray J, Giannetti AM, Steffek M, Gibbons P, Hearn BR, Cohen F, Tam C, Pozniak C, Bravo B, Lewcock J, et al. ChemMedChem. 2014;9:73–77. doi: 10.1002/cmdc.201300424. [DOI] [PubMed] [Google Scholar]
- 206.Keller N, Mares J, Zerbe O, Grutter MG. Structure. 2009;17:438–448. doi: 10.1016/j.str.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 207.Xu G, Cirilli M, Huang Y, Rich RL, Myszka DG, Wu H. Nature. 2001;410:494–497. doi: 10.1038/35068604. [DOI] [PubMed] [Google Scholar]
- 208.Lu M, Min T, Eliezer D, Wu H. Chem Biol. 2006;13:117–122. doi: 10.1016/j.chembiol.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 209.Yu JW, Jeffrey PD, Shi Y. Proc Natl Acad Sci USA. 2009;106:8169–8174. doi: 10.1073/pnas.0812453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Blanchard H, Donepudi M, Tschopp M, Kodandapani L, Wu JC, Grutter MG. J Mol Biol. 2000;302:9–16. doi: 10.1006/jmbi.2000.4041. [DOI] [PubMed] [Google Scholar]
- 211.Wang Z, Watt W, Brooks NA, Harris MS, Urban J, Boatman D, McMillan M, Kahn M, Heinrikson RL, Finzel BC, et al. Biochim Biophys Acta. 2010;1804:1817–1831. doi: 10.1016/j.bbapap.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 212.Ekici OD, Li ZZ, Campbell AJ, James KE, Asgian JL, Mikolajczyk J, Salvesen GS, Ganesan R, Jelakovic S, Grutter MG, et al. J Med Chem. 2006;49:5728–5749. doi: 10.1021/jm0601405. [DOI] [PubMed] [Google Scholar]
- 213.Qin H, Srinivasula SM, Wu G, Fernandes-Alnemri T, Alnemri ES, Shi Y. Nature. 1999;399:549–557. doi: 10.1038/21124. [DOI] [PubMed] [Google Scholar]
- 214.Chao Y, Shiozaki EN, Srinivasula SM, Rigotti DJ, Fairman R, Shi Y. PLoS Biol. 2005;3:e183. doi: 10.1371/journal.pbio.0030183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shiozaki EN, Chai J, Rigotti DJ, Riedl SJ, Li P, Srinivasula SM, Alnemri ES, Fairman R, Shi Y. Mol Cell. 2003;11:519–527. doi: 10.1016/s1097-2765(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 216.Renatus M, Stennicke HR, Scott FL, Liddington RC, Salvesen GS. Proc Natl Acad Sci USA. 2001;98:14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Riedl SJ, Fuentes-Prior P, Renatus M, Kairies N, Krapp S, Huber R, Salvesen GS, Bode W. Proc Natl Acad Sci USA. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chai J, Wu Q, Shiozaki E, Srinivasula SM, Alnemri ES, Shi Y. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]
- 219.Agniswamy J, Fang B, Weber IT. Apoptosis. 2009;14:1135–1144. doi: 10.1007/s10495-009-0388-9. [DOI] [PubMed] [Google Scholar]
- 220.Witkowski WA, Hardy JA. Protein Sci. 2011;20:1421–1431. doi: 10.1002/pro.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Feldman T, Kabaleeswaran V, Jang SB, Antczak C, Djaballah H, Wu H, Jiang X. Mol Cell. 2012;47:585–595. doi: 10.1016/j.molcel.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kang HJ, Lee YM, Bae KH, Kim SJ, Chung SJ. Acta Crystallogr D Biol Crystallogr. 2013;69:1514–1521. doi: 10.1107/S0907444913010196. [DOI] [PubMed] [Google Scholar]
- 223.Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y. Cell. 2001;104:769–780. doi: 10.1016/s0092-8674(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 224.Riedl SJ, Salvesen GS, Bode W. Protein Data Bank. 2002:1KMC. [Google Scholar]
- 225.Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Cell. 2001;104:781–790. [PubMed] [Google Scholar]
- 226.Witkowski WA, Hardy JA. Protein Sci. 2009;18:1459–1468. doi: 10.1002/pro.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Thomsen ND, Koerber JT, Wells JA. Proc Natl Acad Sci USA. 2013;110:8477–8482. doi: 10.1073/pnas.1306759110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Hardy JA, Lam J, Nguyen JT, O’Brien T, Wells JA. Proc Natl Acad Sci USA. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ni CZ, Li C, Wu JC, Spada AP, Ely KR. J Mol Recognit. 2003;16:121–124. doi: 10.1002/jmr.615. [DOI] [PubMed] [Google Scholar]
- 230.Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, Liddington RC, Salvesen GS. Cell. 2001;104:791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- 231.Schroeder T, Barandun J, Flutsch A, Briand C, Mittl PR, Grutter MG. Structure. 2013;21:277–289. doi: 10.1016/j.str.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 232.Fang B, Boross PI, Tozser J, Weber IT. J Mol Biol. 2006;360:654–666. doi: 10.1016/j.jmb.2006.05.041. [DOI] [PubMed] [Google Scholar]
- 233.Fang B, Fu G, Agniswamy J, Harrison RW, Weber IT. Apoptosis. 2009;14:741–752. doi: 10.1007/s10495-009-0333-y. [DOI] [PubMed] [Google Scholar]
- 234.Kang HJ, Lee YM, Jeong MS, Kim M, Bae KH, Kim SJ, Chung SJ. Biosci Rep. 2012;32:305–313. doi: 10.1042/BSR20120009. [DOI] [PubMed] [Google Scholar]
- 235.Becker JW, Rotonda J, Soisson SM, Aspiotis R, Bayly C, Francoeur S, Gallant M, Garcia-Calvo M, Giroux A, Grimm E, et al. J Med Chem. 2004;47:2466–2474. doi: 10.1021/jm0305523. [DOI] [PubMed] [Google Scholar]
- 236.Feeney B, Pop C, Swartz P, Mattos C, Clark AC. Biochemistry. 2006;45:13249–13263. doi: 10.1021/bi0611964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Walters J, Swartz P, Mattos C, Clark AC. Arch Biochem Biophys. 2011;508:31–38. doi: 10.1016/j.abb.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Walters J, Schipper JL, Swartz P, Mattos C, Clark AC. Biosci Rep. 2012;32:401–411. doi: 10.1042/BSR20120037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ganesan R, Jelakovic S, Mittl PR, Caflisch A, Grutter MG. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67:842–850. doi: 10.1107/S1744309111018604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Erlanson DA, Lam JW, Wiesmann C, Luong TN, Simmons RL, DeLano WL, Choong IC, Burdett MT, Flanagan WM, Lee D, et al. Nat Biotechnol. 2003;21:308–314. doi: 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- 241.Du JQ, Wu J, Zhang HJ, Zhang YH, Qiu BY, Wu F, Chen YH, Li JY, Nan FJ, Ding JP, et al. J Biol Chem. 2008;283:30205–30215. doi: 10.1074/jbc.M803347200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Havran LM, Chong DC, Childers WE, Dollings PJ, Dietrich A, Harrison BL, Marathias V, Tawa G, Aulabaugh A, Cowling R, et al. Bioorg Med Chem. 2009;17:7755–7768. doi: 10.1016/j.bmc.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 243.Lee D, Long SA, Adams JL, Chan G, Vaidya KS, Francis TA, Kikly K, Winkler JD, Sung CM, Debouck C, et al. J Biol Chem. 2000;275:16007–16014. doi: 10.1074/jbc.275.21.16007. [DOI] [PubMed] [Google Scholar]
- 244.Yamamoto M, Miyai M, Matsumoto Y, Tsuboi R, Hibino T. J Biol Chem. 2012;287:32825–32834. doi: 10.1074/jbc.M112.357467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Krumschnabel G, Sohm B, Bock F, Manzl C, Villunger A. Cell Death Differ. 2009;16:195–207. doi: 10.1038/cdd.2008.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Bouchier-Hayes L, Green DR. Cell Death Differ. 2012;19:51–57. doi: 10.1038/cdd.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.James KE, Asgian JL, Li ZZ, Ekici OD, Rubin JR, Mikolajczyk J, Salvesen GS, Powers JC. J Med Chem. 2004;47:1553–1574. doi: 10.1021/jm0305016. [DOI] [PubMed] [Google Scholar]
- 248.Graybill TL, Prouty CP, Speier G, Hoyer D, Dolle RE, Helaszek CT, Ator MA, Uhl J, Strasters J. Bioorg Med Chem Lett. 1997;7:41–46. [Google Scholar]
- 249.Van Noorden CJ. Acta Histochem. 2001;103:241–251. doi: 10.1078/0065-1281-00601. [DOI] [PubMed] [Google Scholar]
- 250.Gregoli PA, Bondurant MC. J Cell Physiol. 1999;178:133. doi: 10.1002/(SICI)1097-4652(199902)178:2<133::AID-JCP2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 251.Melnikov VY, Faubel S, Siegmund B, Lucia MS, Ljubanovic D, Edelstein CL. J Clin Invest. 2002;110:1083. doi: 10.1172/JCI15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Apoptosis. 2003;8:345. doi: 10.1023/a:1024116916932. [DOI] [PubMed] [Google Scholar]
- 253.Chauvier D, Ankri S, Charriaut-Marlangue C, Casimir R, Jacotot E. Cell Death Differ. 2007;14:387. doi: 10.1038/sj.cdd.4402044. [DOI] [PubMed] [Google Scholar]
- 254.Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- 255.Thornberry NA, Molineaux SM. Protein Sci. 1995;4:3–12. doi: 10.1002/pro.5560040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, Chartrain NA, Schmidt JA. Proc Natl Acad Sci USA. 1989;86:5227–5231. doi: 10.1073/pnas.86.14.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Dolle RE, Prasad CV, Prouty CP, Salvino JM, Awad MM, Schmidt SJ, Hoyer D, Ross TM, Graybill TL, Speier GJ, et al. J Med Chem. 1997;40:1941–1946. doi: 10.1021/jm9701637. [DOI] [PubMed] [Google Scholar]
- 258.Dolle RE, Prouty CP, Prasad CV, Cook E, Saha A, Ross TM, Salvino JM, Helaszek CT, Ator MA. J Med Chem. 1996;39:2438–2440. doi: 10.1021/jm9601516. [DOI] [PubMed] [Google Scholar]
- 259.Prasad CV, Prouty CP, Hoyer D, Ross TM, Salvino JM, Awad MM, Graybill TL, Schmidt SJ, Osifo IK, Dolle RE, et al. Bioorg Med Chem Lett. 1995;5:315–318. [Google Scholar]
- 260.Margolin N, Raybuck SA, Wilson KP, Chen W, Fox T, Gu Y, Livingston DJ. The J Biol Chem. 1997;272:7223–7228. doi: 10.1074/jbc.272.11.7223. [DOI] [PubMed] [Google Scholar]
- 261.Mjalli AMM, Chapman KT, Maccoss M, Thornberry NA. Bioorg Med Chem Lett. 1993;3:2689–2692. [Google Scholar]
- 262.Harter WG, Albrect H, Brady K, Caprathe B, Dunbar J, Gilmore J, Hays S, Kostlan CR, Lunney B, Walker N. Bioorg Med Chem Lett. 2004;14:809–812. doi: 10.1016/j.bmcl.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 263.Dolle RE, Hoyer D, Prasad CV, Schmidt SJ, Helaszek CT, Miller RE, Ator MA. J Med Chem. 1994;37:563–564. doi: 10.1021/jm00031a003. [DOI] [PubMed] [Google Scholar]
- 264.Dolle RE, Singh J, Rinker J, Hoyer D, Prasad CV, Graybill TL, Salvino JM, Helaszek CT, Miller RE, Ator MA. J Med Chem. 1994;37:3863–3866. doi: 10.1021/jm00049a001. [DOI] [PubMed] [Google Scholar]
- 265.Dolle RE, Singh J, Whipple D, Osifo IK, Speier G, Graybill TL, Gregory JS, Harris AL, Helaszek CT, Miller RE, et al. J Med Chem. 1995;38:220–222. doi: 10.1021/jm00002a002. [DOI] [PubMed] [Google Scholar]
- 266.Thornberry NA, Peterson EP, Zhao JJ, Howard AD, Griffin PR, Chapman KT. Biochemistry. 1994;33:3934–3940. doi: 10.1021/bi00179a020. [DOI] [PubMed] [Google Scholar]
- 267.Boxer MB, Quinn AM, Shen M, Jadhav A, Leister W, Simeonov A, Auld DS, Thomas CJ. ChemMedChem. 2010;5:730–738. doi: 10.1002/cmdc.200900531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Graybill TL, Dolle RE, Helaszek CT, Miller RE, Ator MA. Int J Pept Protein Res. 1994;44:173–182. doi: 10.1111/j.1399-3011.1994.tb00573.x. [DOI] [PubMed] [Google Scholar]
- 269.Galatsis P, Caprathe B, Downing D, Gilmore J, Harter W, Hays S, Kostlan C, Linn K, Lunney E, Para K, et al. Bioorg Med Chem Lett. 2010;20:5089–5094. doi: 10.1016/j.bmcl.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 270.Loser R, Abbenante G, Madala PK, Halili M, Le GT, Fairlie DP. J Med Chem. 2010;53:2651–2655. doi: 10.1021/jm901790w. [DOI] [PubMed] [Google Scholar]
- 271.Mjalli AMM, Chapman KT, Zhao JJ, Thornberry NA, Peterson EP, MacCoss M. Bioorg Med Chem Lett. 1995;5:1405–1408. [Google Scholar]
- 272.Mjalli AMM, Zhao JJ, Chapman KT, Thornberry NA, Peterson EP, MacCoss M, Hagmann WK. Bioorg Med Chem Lett. 1995;5:1409–1414. [Google Scholar]
- 273.Shahripour AB, Plummer MS, Lunney EA, Albrecht HP, Hays SJ, Kostlan CR, Sawyer TK, Walker NP, Brady KD, Allen HJ, et al. Bioorg Med Chem. 2002;10:31–40. doi: 10.1016/s0968-0896(01)00250-4. [DOI] [PubMed] [Google Scholar]
- 274.Ullman BR, Aja T, Deckwerth TL, Diaz JL, Herrmann J, Kalish VJ, Karanewsky DS, Meduna SP, Nalley K, Robinson ED, et al. Bioorg Med Chem Lett. 2003;13:3623–3626. doi: 10.1016/s0960-894x(03)00755-8. [DOI] [PubMed] [Google Scholar]
- 275.Linton SD, Karanewsky DS, Ternansky RJ, Wu JC, Pham B, Kodandapani L, Smidt R, Diaz JL, Fritz LC, Tomaselli KJ. Bioorg Med Chem Lett. 2002;12:2969–2971. doi: 10.1016/s0960-894x(02)00629-7. [DOI] [PubMed] [Google Scholar]
- 276.Linton SD, Karanewsky DS, Ternansky RJ, Chen N, Guo X, Jahangiri KG, Kalish VJ, Meduna SP, Robinson ED, Ullman BR, et al. Bioorg Med Chem Lett. 2002;12:2973–2975. doi: 10.1016/s0960-894x(02)00630-3. [DOI] [PubMed] [Google Scholar]
- 277.Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, Decker C, Charifson P, Weber P, Germann UA, et al. J Pharmacol Exp Ther. 2007;321:509–516. doi: 10.1124/jpet.106.111344. [DOI] [PubMed] [Google Scholar]
- 278.Golec JM, Mullican MD, Murcko MA, Wilson KP, Kay DP, Jones SD, Murdoch R, Bemis GW, Raybuck SA, Luong YP, et al. Bioorg Med Chem Lett. 1997;7:2181–2186. [Google Scholar]
- 279.Semple G, Ashworth DM, Baker GR, Batt AR, Baxter AJ, Benzies DW, Elliot LH, Evans DM, Franklin RJ, Hudson P, et al. Bioorg Med Chem Lett. 1997;7:1337–1342. doi: 10.1016/s0960-894x(98)00136-x. [DOI] [PubMed] [Google Scholar]
- 280.Semple G, Ashworth DM, Batt AR, Baxter AJ, Benzies DW, Elliot LH, Evans DM, Franklin RJ, Hudson P, Jenkins PD, et al. Bioorg Med Chem Lett. 1998;8:959–964. doi: 10.1016/s0960-894x(98)00136-x. [DOI] [PubMed] [Google Scholar]
- 281.Siegmund B, Zeitz M. IDrugs. 2003;6:154–158. [PubMed] [Google Scholar]
- 282.Lauffer DJ, Mullican MD. Bioorg Med Chem Lett. 2002;12:1225–1227. doi: 10.1016/s0960-894x(02)00107-5. [DOI] [PubMed] [Google Scholar]
- 283.Oppong KA, Ellis CD, Laufersweiler MC, O’Neil SV, Wang Y, Soper DL, Baize MW, Wos JA, De B, Bosch GK, et al. Bioorg Med Chem Lett. 2005;15:4291–4294. doi: 10.1016/j.bmcl.2005.06.050. [DOI] [PubMed] [Google Scholar]
- 284.Wang Y, O’Neil SV, Wos JA, Oppong KA, Laufersweiler MC, Soper DL, Ellis CD, Baize MW, Fancher AN, Lu W, et al. Bioorg Med Chem. 2007;15:1311–1322. doi: 10.1016/j.bmc.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 285.Ellis CD, Oppong KA, Laufersweiler MC, O’Neil SV, Soper DL, Wang Y, Wos JA, Fancher AN, Lu W, Suchanek MK, et al. Bioorg Med Chem Lett. 2006;16:4728–4732. doi: 10.1016/j.bmcl.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 286.Soper DL, Sheville JX, O’Neil SV, Wang Y, Laufersweiler MC, Oppong KA, Wos JA, Ellis CD, Baize MW, Chen JJ, et al. Bioorg Med Chem. 2006;14:7880–7892. doi: 10.1016/j.bmc.2006.07.056. [DOI] [PubMed] [Google Scholar]
- 287.O’Neil SV, Wang Y, Laufersweiler MC, Oppong KA, Soper DL, Wos JA, Ellis CD, Baize MW, Bosch GK, Fancher AN, et al. Bioorg Med Chem Lett. 2005;15:5434–5438. doi: 10.1016/j.bmcl.2005.08.109. [DOI] [PubMed] [Google Scholar]
- 288.Ng SL, Yang PY, Chen KY, Srinivasan R, Yao SQ. Org Biomol Chem. 2008;6:844–847. doi: 10.1039/b718304f. [DOI] [PubMed] [Google Scholar]
- 289.Howley B, Fearnhead HO. J Cell Mol Med. 2008;12:1502–1516. doi: 10.1111/j.1582-4934.2008.00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Snipas SJ, Drag M, Stennicke HR, Salvesen GS. Cell Death Differ. 2008;15:938–945. doi: 10.1038/cdd.2008.23. [DOI] [PubMed] [Google Scholar]
- 291.Berger AB, Sexton KB, Bogyo M. Cell Res. 2006;16:961–963. doi: 10.1038/sj.cr.7310112. [DOI] [PubMed] [Google Scholar]
- 292.Murphy DJ, Walker B, Ryan CA, Martin SL. Biochem Biophys Res Commun. 2010;402:483–488. doi: 10.1016/j.bbrc.2010.10.054. [DOI] [PubMed] [Google Scholar]
- 293.Goode DR, Sharma AK, Hergenrother PJ. Org Lett. 2005;7:3529–3532. doi: 10.1021/ol051287d. [DOI] [PubMed] [Google Scholar]
- 294.Asgian JL, James KE, Li ZZ, Carter W, Barrett AJ, Mikolajczyk J, Salvesen GS, Powers JC. J Med Chem. 2002;45:4958–4960. doi: 10.1021/jm025581c. [DOI] [PubMed] [Google Scholar]
- 295.Ekici OD, Gotz MG, James KE, Li ZZ, Rukamp BJ, Asgian JL, Caffrey CR, Hansell E, Dvorak J, McKerrow JH, et al. J Med Chem. 2004;47:1889–1892. doi: 10.1021/jm049938j. [DOI] [PubMed] [Google Scholar]
- 296.Han Y, Giroux A, Colucci J, Bayly CI, McKay DJ, Roy S, Xanthoudakis S, Vaillancourt J, Rasper DM, Tam J, et al. Bioorg Med Chem Lett. 2005;15:1173–1180. doi: 10.1016/j.bmcl.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 297.Ullman BR, Aja T, Chen N, Diaz JL, Gu X, Herrmann J, Kalish VJ, Karanewsky DS, Kodandapani L, Krebs JJ, et al. Bioorg Med Chem Lett. 2005;15:3632–3636. doi: 10.1016/j.bmcl.2005.05.036. [DOI] [PubMed] [Google Scholar]
- 298.Linton SD, Aja T, Allegrini PR, Deckwerth TL, Diaz JL, Hengerer B, Herrmann J, Jahangiri KG, Kallen J, Karanewsky DS, et al. Bioorg Med Chem Lett. 2004;14:2685–2691. doi: 10.1016/j.bmcl.2003.12.106. [DOI] [PubMed] [Google Scholar]
- 299.Linton SD, Aja T, Armstrong RA, Bai X, Chen LS, Chen N, Ching B, Contreras P, Diaz JL, Fisher CD, et al. J Med Chem. 2005;48:6779–6782. doi: 10.1021/jm050307e. [DOI] [PubMed] [Google Scholar]
- 300.Guo Z, Xian M, Zhang W, McGill A, Wang PG. Bioorg Med Chem. 2001;9:99–106. doi: 10.1016/s0968-0896(00)00222-4. [DOI] [PubMed] [Google Scholar]
- 301.Newton AS, Gloria PM, Goncalves LM, dos Santos DJ, Moreira R, Guedes RC, Santos MM. Eur J Med Chem. 2010;45:3858–3863. doi: 10.1016/j.ejmech.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 302.Yang W, Guastella J, Huang JC, Wang Y, Zhang L, Xue D, Tran M, Woodward R, Kasibhatla S, Tseng B, et al. J Pharmacol. 2003;140:402–412. doi: 10.1038/sj.bjp.0705450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Wang Y, Huang JC, Zhou ZL, Yang W, Guastella J, Drewe J, Cai SX. Bioorg Med Chem Lett. 2004;14:1269–1272. doi: 10.1016/j.bmcl.2003.12.065. [DOI] [PubMed] [Google Scholar]
- 304.Cai SX, Guan L, Jia S, Wang Y, Yang W, Tseng B, Drewe J. Bioorg Med Chem Lett. 2004;14:5295–5300. doi: 10.1016/j.bmcl.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 305.Wang Y, Guan L, Jia S, Tseng B, Drewe J, Cai SX. Bioorg Med Chem Lett. 2005;15:1379–1383. doi: 10.1016/j.bmcl.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 306.Han Y, Giroux A, Grimm EL, Aspiotis R, Francoeur S, Bayly CI, McKay DJ, Roy S, Xanthoudakis S, Vaillancourt JP, et al. Bioorg Med Chem Lett. 2004;14:805–808. doi: 10.1016/j.bmcl.2003.10.064. [DOI] [PubMed] [Google Scholar]
- 307.Mellon C, Aspiotis R, Black CW, Bayly CI, Grimm EL, Giroux A, Han Y, Isabel E, McKay DJ, Nicholson DW, et al. Bioorg Med Chem Lett. 2005;15:3886–3890. doi: 10.1016/j.bmcl.2005.05.116. [DOI] [PubMed] [Google Scholar]
- 308.Colantonio P, Leboffe L, Bolli A, Marino M, Ascenzi P, Luisi G. Biochem Biophys Res Commun. 2008;377:757–762. doi: 10.1016/j.bbrc.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 309.Ferrucci A, Leboffe L, Agamennone M, Di Pizio A, Fiocchetti M, Marino M, Ascenzi P, Luisi G. Amino Acids. 2015;47:153–162. doi: 10.1007/s00726-014-1855-3. [DOI] [PubMed] [Google Scholar]
- 310.Leyva MJ, Degiacomo F, Kaltenbach LS, Holcomb J, Zhang N, Gafni J, Park H, Lo DC, Salvesen GS, Ellerby LM, et al. Chem Biol. 2010;17:1189–1200. doi: 10.1016/j.chembiol.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Leyva MJ, DeGiacomo F, Kaltenbach LS, Holcomb J, Zhang NZ, Gafni J, Park H, Lo DC, Salvesen GS, Ellerby LM, et al. Chemistry & Biology. 2010;17:1189–1200. doi: 10.1016/j.chembiol.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Allen DA, Pham P, Choong IC, Fahr B, Burdett MT, Lew W, DeLano WL, Gordon EM, Lam JW, O’Brien T, et al. Bioorg Med Chem Lett. 2003;13:3651–3655. doi: 10.1016/j.bmcl.2003.08.024. [DOI] [PubMed] [Google Scholar]
- 313.Isabel E, Black WC, Bayly CI, Grimm EL, Janes MK, McKay DJ, Nicholson DW, Rasper DM, Renaud J, Roy S, et al. Bioorg Med Chem Lett. 2003;13:2137–2140. doi: 10.1016/s0960-894x(03)00390-1. [DOI] [PubMed] [Google Scholar]
- 314.Karanewsky DS, Bai X, Linton SD, Krebs JF, Wu J, Pham B, Tomaselli KJ. Bioorg Med Chem Lett. 1998;8:2757–2762. doi: 10.1016/s0960-894x(98)00498-3. [DOI] [PubMed] [Google Scholar]
- 315.Linton S, Karanewsky DS. 5,968,927. United State Patent. 1999
- 316.Deckwerth TL, Adams LM, Wiessner C, Allegrini PR, Rudin M, Sauter A, Hengerer B, Sayers RO, Rovelli G, Aja T, et al. Drug Develop Res. 2001;52:579–586. [Google Scholar]
- 317.Micale N, Vairagoundar R, Yakovlev AG, Kozikowski AP. J Med Chem. 2004;47:6455–6458. doi: 10.1021/jm049248f. [DOI] [PubMed] [Google Scholar]
- 318.Vazquez J, Garcia-Jareno A, Mondragon L, Rubio-Martinez J, Perez-Paya E, Albericio F. ChemMedChem. 2008;3:979–985. doi: 10.1002/cmdc.200800020. [DOI] [PubMed] [Google Scholar]
- 319.Choong IC, Lew W, Lee D, Pham P, Burdett MT, Lam JW, Wiesmann C, Luong TN, Fahr B, DeLano WL, et al. J Med Chem. 2002;45:5005–5022. doi: 10.1021/jm020230j. [DOI] [PubMed] [Google Scholar]
- 320.Ueno H, Kawai M, Shimokawa H, Hirota M, Ohmi M, Sudo R, Ohta A, Arano Y, Hattori K, Ohmi T, et al. Bioorg Med Chem Lett. 2009;19:199–202. doi: 10.1016/j.bmcl.2008.10.117. [DOI] [PubMed] [Google Scholar]
- 321.Kasperkiewicz P, Gajda AD, Drag M. Biol Chem. 2012;393:843–851. doi: 10.1515/hsz-2012-0167. [DOI] [PubMed] [Google Scholar]
- 322.Vickers CJ, Gonzalez-Paez GE, Litwin KM, Umotoy JC, Coutsias EA, Wolan DW. ACS Chem Biol. 2014;9:2194–2198. doi: 10.1021/cb5004256. [DOI] [PubMed] [Google Scholar]
- 323.Lee D, Long SA, Adams JL, Chan G, Vaidya KS, Francis TA, Kikly K, Winkler JD, Sung CM, Debouck C, et al. J Biol Chem. 2000;275:16007–16014. doi: 10.1074/jbc.275.21.16007. [DOI] [PubMed] [Google Scholar]
- 324.Chen YH, Zhang YH, Zhang HJ, Liu DZ, Gu M, Li JY, Wu F, Zhu XZ, Li J, Nan FJ. J Med Chem. 2006;49:1613–1623. doi: 10.1021/jm050896o. [DOI] [PubMed] [Google Scholar]
- 325.Kim S-G, Jung Y-S, Kong J-Y, Park W-K. 7,009,053 B2. United State Patent. 2006
- 326.Scott CW, Sobotka-Briner C, Wilkins DE, Jacobs RT, Folmer JJ, Frazee WJ, Bhat RV, Ghanekar SV, Aharony D. J Pharmacol Exp Ther. 2003;304:433–440. doi: 10.1124/jpet.102.039651. [DOI] [PubMed] [Google Scholar]
- 327.Hazel BA, Baum C, Kalf GF. Stem Cells. 1996;14:730–742. doi: 10.1002/stem.140730. [DOI] [PubMed] [Google Scholar]
- 328.Scott CW, Sobotka-Briner C, Wilkins DE, Jacobs RT, Folmer JJ, Frazee WJ, Bhat RV, Ghanekar SV, Aharony D. J Pharmacol Exp Ther. 2003;304:433–440. doi: 10.1124/jpet.102.039651. [DOI] [PubMed] [Google Scholar]
- 329.Lee D, Long SA, Murray JH, Adams JL, Nuttall ME, Nadeau DP, Kikly K, Winkler JD, Sung CM, Ryan MD, et al. J Med Chem. 2001;44:2015–2026. doi: 10.1021/jm0100537. [DOI] [PubMed] [Google Scholar]
- 330.Chu W, Zhang J, Zeng C, Rothfuss J, Tu Z, Chu Y, Reichert DE, Welch MJ, Mach RH. J Med Chem. 2005;48:7637–7647. doi: 10.1021/jm0506625. [DOI] [PubMed] [Google Scholar]
- 331.Kopka K, Faust A, Keul P, Wagner S, Breyholz HJ, Holtke C, Schober O, Schafers M, Levkau B. J Med Chem. 2006;49:6704–6715. doi: 10.1021/jm051217c. [DOI] [PubMed] [Google Scholar]
- 332.Krause-Heuer AM, Lidia Matesic NRH, Dhand G, Young EL, Burgess L, Jiang CD, Lengkeek NA, Fookes ChJR, Pham TQ, Sobrio F, et al. MedChemComm. 2013:347–352. [Google Scholar]
- 333.Chu W, Rothfuss J, d’Avignon A, Zeng C, Zhou D, Hotchkiss RS, Mach RH. J Med Chem. 2007;50:3751–3755. doi: 10.1021/jm070506t. [DOI] [PubMed] [Google Scholar]
- 334.Chen DL, Zhou D, Chu W, Herrbrich PE, Jones LA, Rothfuss JM, Engle JT, Geraci M, Welch MJ, Mach RH. Nucl Med Biol. 2009;36:651–658. doi: 10.1016/j.nucmedbio.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Smith G, Glaser M, Perumal M, Nguyen QD, Shan B, Arstad E, Aboagye EO. J Med Chem. 2008;51:8057–8067. doi: 10.1021/jm801107u. [DOI] [PubMed] [Google Scholar]
- 336.Jiang Y, Hansen TV. Bioorg Med Chem Lett. 2011;21:1626–1629. doi: 10.1016/j.bmcl.2011.01.110. [DOI] [PubMed] [Google Scholar]
- 337.Podichetty AK, Faust A, Kopka K, Wagner S, Schober O, Schafers M, Haufe G. Bioorg Med Chem. 2009;17:2680–2688. doi: 10.1016/j.bmc.2009.02.048. [DOI] [PubMed] [Google Scholar]
- 338.Podichetty AK, Wagner S, Schroer S, Faust A, Schafers M, Schober O, Kopka K, Haufe G. J Med Chem. 2009;52:3484–3495. doi: 10.1021/jm8015014. [DOI] [PubMed] [Google Scholar]
- 339.Limpachayaporn P, Schafers M, Schober O, Kopka K, Haufe G. Bioorg Med Chem. 2013;21:2025–2036. doi: 10.1016/j.bmc.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 340.Limpachayaporn P, Riemann B, Kopka K, Schober O, Schafers M, Haufe G. Eur J Med Chem. 2013;64:562–578. doi: 10.1016/j.ejmech.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 341.Wang Q, Mach RH, Reichert DE. J Chem Inf Model. 2009;49:1963–1973. doi: 10.1021/ci900144x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Kopka K, Faust A, Keul P, Wagner S, Breyholz HJ, Holtke C, Schober O, Schafers M, Levkau B. J Med Chem. 2006;49:6704–6715. doi: 10.1021/jm051217c. [DOI] [PubMed] [Google Scholar]
- 343.Chu WH, Rothfuss J, d’Avignon A, Zeng CB, Zhou D, Hotchkiss RS, Mach RH. J Med Chem. 2007;50:3751–3755. doi: 10.1021/jm070506t. [DOI] [PubMed] [Google Scholar]
- 344.Smith G, Glaser M, Perumal M, Nguyen QD, Shan B, Arstad E, Aboagye EO. J Med Chem. 2008;51:8057–8067. doi: 10.1021/jm801107u. [DOI] [PubMed] [Google Scholar]
- 345.Havran LM, Chong DC, Childers WE, Dollings PJ, Dietrich A, Harrison BL, Marathias V, Tawa G, Aulabaugh A, Cowling R, et al. Bioorg Med Chem. 2009;17:7755–7768. doi: 10.1016/j.bmc.2009.09.036. [DOI] [PubMed] [Google Scholar]
- 346.Kamal A, Ramakrishna G, Nayak VL, Raju P, Rao AVS, Viswanath A, Vishnuvardhan MVPS, Ramakrishna S, Srinivas G. Bioorg Med Chem. 2012;20:789–800. doi: 10.1016/j.bmc.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 347.Liu D, Tian Z, Yan Z, Wu L, Ma Y, Wang Q, Liu W, Zhou H, Yang C. Bioorg Med Chem. 2013;21:2960–2967. doi: 10.1016/j.bmc.2013.03.075. [DOI] [PubMed] [Google Scholar]
- 348.Wu L, Lu M, Yan Z, Tang X, Sun B, Liu W, Zhou H, Yang C. Bioorg Med Chem. 2014;22:2416–2426. doi: 10.1016/j.bmc.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 349.Li ZH, Pan Y, Zhong WL, Zhu YP, Zhao YL, Li LX, Liu W, Zhou HG, Yang C. Bioorg Med Chem. 2014;22:6735–6745. doi: 10.1016/j.bmc.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 350.Guo Z, Yan Z, Zhou X, Wang Q, Lu M, Liu W, Zhou H, McClain EJ. Med Chem Res. 2014;24:1814–1829. [Google Scholar]
- 351.Kravchenko DV, Kuzovkova YA, Kysil VM, Tkachenko SE, Maliarchouk S, Okun IM, Balakin KV, Ivachtchenko AV. J Med Chem. 2005;48:3680–3683. doi: 10.1021/jm048987t. [DOI] [PubMed] [Google Scholar]
- 352.Kravchenko DV, Kysil VV, Ilyn AP, Tkachenko SE, Maliarchouk S, Okun IM, Ivachtchenko AV. Bioorg Med Chem Lett. 2005;15:1841–1845. doi: 10.1016/j.bmcl.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 353.Kravchenko DV, Kysil VM, Tkachenko SE, Maliarchouk S, Okun IM, Ivachtchenko AV. Farmaco. 2005;60:804–809. doi: 10.1016/j.farmac.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 354.Kravchenko DV, Kysil VM, Tkachenko SE, Maliarchouk S, Okun IM, Ivachtchenko AV. Eur J Med Chem. 2005;40:1377–1383. doi: 10.1016/j.ejmech.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 355.Okun I, Malarchuk S, Dubrovskaya E, Khvat A, Tkachenko S, Kysil V, Ilyin A, Kravchenko D, Prossnitz ER, Sklar L, et al. J Biomol Screen. 2006;11:277–285. doi: 10.1177/1087057105285048. [DOI] [PubMed] [Google Scholar]
- 356.Zhu Q, Gao L, Chen Z, Zheng S, Shu H, Li J, Jiang H, Liu S. Eur J Med Chem. 2012;54:232–238. doi: 10.1016/j.ejmech.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 357.Tsukuda E, Tanaka T, Ochiai K, Kondo H, Yoshida M, Agatsuma T, Saitoh Y, Teshiba S, Matsuda Y. J Antibiot (Tokyo) 1995;49:333–339. doi: 10.7164/antibiotics.49.333. [DOI] [PubMed] [Google Scholar]
- 358.Tanaka T, Tsukuda E, Uosaki Y, Matsuda Y. J Antibiot (Tokyo) 1996;49:1085–1090. doi: 10.7164/antibiotics.49.1085. [DOI] [PubMed] [Google Scholar]
- 359.Koizumi F, Agatsuma T, Ando K, Kondo H, Saitoh Y, Matsuda Y, Nakanishi S. J Antibiot (Tokyo) 2003;56:891–898. doi: 10.7164/antibiotics.56.891. [DOI] [PubMed] [Google Scholar]
- 360.Tanaka T, Tsukuda E, Ochiai K, Kondo H, Teshiba S, Matsuda Y. J Antibiot (Tokyo) 1996;49:1073–1078. doi: 10.7164/antibiotics.49.1073. [DOI] [PubMed] [Google Scholar]
- 361.Uosaki Y, Agatsuma T, Tanaka T, Saitoh Y. J Antibiot (Tokyo) 1996;49:1079–1084. doi: 10.7164/antibiotics.49.1079. [DOI] [PubMed] [Google Scholar]
- 362.Koizumi F, Matsuda Y, Nakanishi S. J Antibiot (Tokyo) 2003;56:464–469. doi: 10.7164/antibiotics.56.464. [DOI] [PubMed] [Google Scholar]
- 363.Koizumi F, Ishiguro H, Ando K, Kondo H, Yoshida M, Matsuda Y, Nakanishi S. J Antibiot (Tokyo) 2003;56:603–609. doi: 10.7164/antibiotics.56.603. [DOI] [PubMed] [Google Scholar]
- 364.Tsukuda E, Tanaka H, Ochiai K, Kondo H, Yoshida M, Agatsuma T, Saitoh Y, Teshiba S, Matsuda Y. J Antibiot. 1995;49:333–339. doi: 10.7164/antibiotics.49.333. [DOI] [PubMed] [Google Scholar]
- 365.Koizumi F, Hasegawa A, Ochiai K, Ando K, Kondo H, Yoshida M, Matsuda Y, Nakanishi S. J Antibiot (Tokyo) 2003;56:985–992. doi: 10.7164/antibiotics.56.985. [DOI] [PubMed] [Google Scholar]
- 366.Renz JF, Kalf GF. Blood. 1991;78:938–944. [PubMed] [Google Scholar]
- 367.Niculescu R, Bradford HN, Colman RW, Kalf GF. Chem Biol Interact. 1995;98:211–222. doi: 10.1016/0009-2797(95)03647-4. [DOI] [PubMed] [Google Scholar]
- 368.Salvatore MJ, Hensens OD, Zink DL, Liesch J, Dufresne C, Ondeyka JG, Jurgens TM, Borris RP, Raghoobar S, McCauley E, et al. J Nat Prod. 1994;57:755–760. doi: 10.1021/np50108a011. [DOI] [PubMed] [Google Scholar]
- 369.Matsumoto T, Ishiyama A, Yamaguchi Y, Masuma R, Ui H, Shiomi K, Yamada H, Omura S. J Antibiot (Tokyo) 1999;52:754–757. doi: 10.7164/antibiotics.52.754. [DOI] [PubMed] [Google Scholar]
- 370.Bonavida B, Khineche S, Huerta-Yepez S, Garban H. Drug Resist Updat. 2006;9:157–173. doi: 10.1016/j.drup.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 371.Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Biochem Biophys Res Commun. 2001;282:1075–1079. doi: 10.1006/bbrc.2001.4670. [DOI] [PubMed] [Google Scholar]
- 372.Rossig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mulsch A, Dimmeler S. J Biol Chem. 1999;274:6823–6826. doi: 10.1074/jbc.274.11.6823. [DOI] [PubMed] [Google Scholar]
- 373.Fiorucci S, Santucci L, Cirino G, Mencarelli A, Familiari L, Soldato PD, Morelli A. J Immunol. 2000;165:5245–5254. doi: 10.4049/jimmunol.165.9.5245. [DOI] [PubMed] [Google Scholar]
- 374.Fiorucci S, Antonelli E, Tocchetti P, Morelli A. Cardiovasc Drug Rev. 2004;22:135–146. doi: 10.1111/j.1527-3466.2004.tb00136.x. [DOI] [PubMed] [Google Scholar]
- 375.Sengupta S, Rao GV, Dubey PK. Indian J Chem B. 2011;50:901–905. [Google Scholar]
- 376.Perry DK, Smyth MJ, Stennicke HR, Salvesen GS, Duriez P, Poirier GG, Hannun YA. J Biol Chem. 1997;272:18530–18533. doi: 10.1074/jbc.272.30.18530. [DOI] [PubMed] [Google Scholar]
- 377.Takahashi A, Goldschmidt-Clermont PJ, Alnemri ES, Fernandes-Alnemri T, Yoshizawa-Kumagaya K, Nakajima K, Sasada M, Poirier GG, Earnshaw WC. Exp Cell Res. 1997;231:123–131. doi: 10.1006/excr.1996.3459. [DOI] [PubMed] [Google Scholar]
- 378.Nobel CS, Burgess DH, Zhivotovsky B, Burkitt MJ, Orrenius S, Slater AF. Chem Res Toxicol. 1997;10:636–643. doi: 10.1021/tx970006a. [DOI] [PubMed] [Google Scholar]
- 379.Nobel CS, Kimland M, Nicholson DW, Orrenius S, Slater AF. Chem Res Toxicol. 1997;10:1319–1324. doi: 10.1021/tx970131m. [DOI] [PubMed] [Google Scholar]
- 380.Edgington LE, Verdoes M, Bogyo M. Curr Opin Chem Biol. 2011;15:798–805. doi: 10.1016/j.cbpa.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Kidd D, Liu Y, Cravatt BF. Biochemistry. 2001;40:4005–4015. doi: 10.1021/bi002579j. [DOI] [PubMed] [Google Scholar]
- 382.Liu Y, Patricelli MP, Cravatt BF. Proc Natl Acad Sci USA. 1999;96:14694–14699. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Kam CM, Abuelyaman AS, Li Z, Hudig D, Powers JC. Bioconjug Chem. 1993;4:560–567. doi: 10.1021/bc00024a021. [DOI] [PubMed] [Google Scholar]
- 384.Serim S, Haedke U, Verhelst SH. ChemMedChem. 2012;7:1146–1159. doi: 10.1002/cmdc.201200057. [DOI] [PubMed] [Google Scholar]
- 385.Haberkorn U, Kinscherf R, Krammer PH, Mier W, Eisenhut M. Nucl Med Biol. 2001;28:793–798. doi: 10.1016/s0969-8051(01)00247-5. [DOI] [PubMed] [Google Scholar]
- 386.Uttamchandani M, Li J, Sun H, Yao SQ. ChemBioChem. 2008;9:667–675. doi: 10.1002/cbic.200700755. [DOI] [PubMed] [Google Scholar]
- 387.Speers AE, Cravatt BF. ChemBioChem. 2004;5:41–47. doi: 10.1002/cbic.200300721. [DOI] [PubMed] [Google Scholar]
- 388.Evans MJ, Cravatt BF. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 389.Kuzelova K, Grebenova D, Hrkal Z. Cytometry A. 2007;71:605–611. doi: 10.1002/cyto.a.20415. [DOI] [PubMed] [Google Scholar]
- 390.Smolewski P, Bedner E, Du L, Hsieh TC, Wu JM, Phelps DJ, Darzynkiewicz Z. Cytometry. 2001;44:73–82. doi: 10.1002/1097-0320(20010501)44:1<73::aid-cyto1084>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 391.Rozman-Pungercar J, Kopitar-Jerala N, Bogyo M, Turk D, Vasiljeva O, Stefe I, Vandenabeele P, Bromme D, Puizdar V, Fonovic M, et al. Cell Death Differ. 2003;10:881–888. doi: 10.1038/sj.cdd.4401247. [DOI] [PubMed] [Google Scholar]
- 392.Schotte P, Declercq W, Van Huffel S, Vandenabeele P, Beyaert R. FEBS Lett. 1999;442:117–121. doi: 10.1016/s0014-5793(98)01640-8. [DOI] [PubMed] [Google Scholar]
- 393.Pozarowski P, Huang X, Halicka DH, Lee B, Johnson G, Darzynkiewicz Z. Cytometry A. 2003;55:50–60. doi: 10.1002/cyto.a.10074. [DOI] [PubMed] [Google Scholar]
- 394.Puri AW, Broz P, Shen A, Monack DM, Bogyo M. Nat Chem Biol. 2012;8:745–747. doi: 10.1038/nchembio.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Kato D, Verhelst SH, Sexton KB, Bogyo M. Org Lett. 2005;7:5649–5652. doi: 10.1021/ol052275v. [DOI] [PubMed] [Google Scholar]
- 396.Kato D, Boatright KM, Berger AB, Nazif T, Blum G, Ryan C, Chehade KA, Salvesen GS, Bogyo M. Nat Chem Biol. 2005;1:33–38. doi: 10.1038/nchembio707. [DOI] [PubMed] [Google Scholar]
- 397.Sexton KB, Kato D, Berger AB, Fonovic M, Verhelst SH, Bogyo M. Cell Death Differ. 2007;14:727–732. doi: 10.1038/sj.cdd.4402074. [DOI] [PubMed] [Google Scholar]
- 398.Faust A, Wagner S, Law MP, Hermann S, Schnockel U, Keul P, Schober O, Schafers M, Levkau B, Kopka K. Q J Nucl Med Mol Imaging. 2007;51:67–73. [PubMed] [Google Scholar]
- 399.Zhou D, Chu W, Chen DL, Wang Q, Reichert DE, Rothfuss J, D’Avignon A, Welch MJ, Mach RH. Org Biomol Chem. 2009;7:1337–1348. doi: 10.1039/b819024k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Nguyen QD, Smith G, Glaser M, Perumal M, Arstad E, Aboagye EO. Proc Natl Acad Sci USA. 2009;106:16375–16380. doi: 10.1073/pnas.0901310106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Zhou D, Chu W, Rothfuss J, Zeng C, Xu J, Jones L, Welch MJ, Mach RH. Bioorg Med Chem Lett. 2006;16:5041–5046. doi: 10.1016/j.bmcl.2006.07.045. [DOI] [PubMed] [Google Scholar]
- 402.Nguyen QD, Challapalli A, Smith G, Fortt R, Aboagye EO. Eur J Cancer. 2012;48:432–440. doi: 10.1016/j.ejca.2011.11.033. [DOI] [PubMed] [Google Scholar]
- 403.Methot N, Vaillancourt JP, Huang J, Colucci J, Han Y, Menard S, Zamboni R, Toulmond S, Nicholson DW, Roy S. J Biol Chem. 2004;279:27905–27914. doi: 10.1074/jbc.M400247200. [DOI] [PubMed] [Google Scholar]
- 404.Podichetty AK, Faust A, Kopka K, Wagner S, Schober O, Schafers M, Haufe G. Bioorg Med Chem. 2009;17:2680–2688. doi: 10.1016/j.bmc.2009.02.048. [DOI] [PubMed] [Google Scholar]
- 405.Podichetty AK, Wagner S, Schroer S, Faust A, Schafers M, Schober O, Kopka K, Haufe G. J Med Chem. 2009;52:3484–3495. doi: 10.1021/jm8015014. [DOI] [PubMed] [Google Scholar]
- 406.Hight MR, Cheung YY, Nickels ML, Dawson ES, Zhao P, Saleh S, Buck JR, Tang D, Washington MK, Coffey RJ, et al. Clin Cancer Res. 2014;20:2126–2135. doi: 10.1158/1078-0432.CCR-13-2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Lee H, Akers WJ, Cheney PP, Edwards WB, Liang K, Culver JP, Achilefu S. J Biomed Opt. 2009;14:040507. doi: 10.1117/1.3207156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Mizukami S, Takikawa R, Sugihara F, Hori Y, Tochio H, Walchli M, Shirakawa M, Kikuchi K. J Am Chem Soc. 2008;130:794–795. doi: 10.1021/ja077058z. [DOI] [PubMed] [Google Scholar]
- 409.Mizukami S. Chem Pharm Bull (Tokyo) 2011;59:1435–1446. doi: 10.1248/cpb.59.1435. [DOI] [PubMed] [Google Scholar]
- 410.Kikuchi K. Adv Biochem Eng Biotechnol. 2010;119:63–78. doi: 10.1007/10_2008_42. [DOI] [PubMed] [Google Scholar]
- 411.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 412.Thornberry NA. Methods Enzymol. 1994;244:615–631. doi: 10.1016/0076-6879(94)44045-x. [DOI] [PubMed] [Google Scholar]
- 413.Axelrod D. Proc Natl Acad Sci USA. 1980;77:4823–4827. doi: 10.1073/pnas.77.8.4823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Sadaghiani AM, Verhelst SH, Bogyo M. Curr Opin Chem Biol. 2007;11:20–28. doi: 10.1016/j.cbpa.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 415.Yamin TT, Ayala JM, Miller DK. J Biol Chem. 1996;271:13273–13282. doi: 10.1074/jbc.271.22.13273. [DOI] [PubMed] [Google Scholar]
- 416.Faleiro L, Kobayashi R, Fearnhead H, Lazebnik Y. EMBO J. 1997;16:2271–2281. doi: 10.1093/emboj/16.9.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Fearnhead HO, McCurrach ME, O’Neill J, Zhang K, Lowe SW, Lazebnik YA. Genes Dev. 1997;11:1266–1276. doi: 10.1101/gad.11.10.1266. [DOI] [PubMed] [Google Scholar]
- 418.Martins LM, Kottke T, Mesner PW, Basi GS, Sinha S, Frigon N, Jr, Tatar E, Tung JS, Bryant K, Takahashi A, et al. J Biol Chem. 1997;272:7421–7430. doi: 10.1074/jbc.272.11.7421. [DOI] [PubMed] [Google Scholar]
- 419.Saunders PA, Cooper JA, Roodell MM, Schroeder DA, Borchert CJ, Isaacson AL, Schendel MJ, Godfrey KG, Cahill DR, Walz AM, et al. Anal Biochem. 2000;284:114–124. doi: 10.1006/abio.2000.4690. [DOI] [PubMed] [Google Scholar]
- 420.Shelton SN, Dillard CD, Robertson JD. J Biol Chem. 2010;285:40525–40533. doi: 10.1074/jbc.M110.167635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421.Henzing AJ, Dodson H, Reid JM, Kaufmann SH, Baxter RL, Earnshaw WC. J Med Chem. 2006;49:7636–7645. doi: 10.1021/jm060385h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Lathia US, Ornatsky O, Baranov V, Nitz M. Anal Biochem. 2010;398:93–98. doi: 10.1016/j.ab.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Lathia US, Ornatsky O, Baranov V, Nitz M. Anal Biochem. 2011;408:157–159. doi: 10.1016/j.ab.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Greenbaum D, Baruch A, Hayrapetian L, Darula Z, Burlingame A, Medzihradszky KF, Bogyo M. Mol Cell Proteomics. 2002;1:60–68. doi: 10.1074/mcp.t100003-mcp200. [DOI] [PubMed] [Google Scholar]
- 425.Patricelli MP, Giang DK, Stamp LM, Burbaum JJ. Proteomics. 2001;1:1067–1071. doi: 10.1002/1615-9861(200109)1:9<1067::AID-PROT1067>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 426.Gabriel D, Zuluaga MF, van den Bergh H, Gurny R, Lange N. Curr Med Chem. 2011;18:1785–1805. doi: 10.2174/092986711795496782. [DOI] [PubMed] [Google Scholar]
- 427.Terai T, Nagano T. Curr Opin Chem Biol. 2008;12:515–521. doi: 10.1016/j.cbpa.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 428.Edgington LE, Bogyo M. Curr Protoc Chem Biol. 2013;5:25–44. doi: 10.1002/9780470559277.ch120235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Bedner E, Smolewski P, Amstad P, Darzynkiewicz Z. Exp Cell Res. 2000;259:308–313. doi: 10.1006/excr.2000.4955. [DOI] [PubMed] [Google Scholar]
- 430.Darzynkiewicz Z, Pozarowski P, Lee BW, Johnson GL. Methods Mol Biol. 2011;682:103–114. doi: 10.1007/978-1-60327-409-8_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Smolewski P, Grabarek J, Lee BW, Johnson GL, Darzynkiewicz Z. Cytometry. 2002;47:143–149. doi: 10.1002/cyto.10062. [DOI] [PubMed] [Google Scholar]
- 432.Teodori L, Grabarek J, Smolewski P, Ghibelli L, Bergamaschi A, De Nicola M, Darzynkiewicz Z. Cytometry. 2002;49:113–118. doi: 10.1002/cyto.10160. [DOI] [PubMed] [Google Scholar]
- 433.Valero A, Merino F, Wolbers F, Luttge R, Vermes I, Andersson H, van den Berg A. Lab Chip. 2005;5:49–55. doi: 10.1039/b415813j. [DOI] [PubMed] [Google Scholar]
- 434.Luthra S, Fardin B, Dong J, Hertzog D, Kamjoo S, Gebremariam S, Butani V, Narayanan R, Mungcal JK, Kuppermann BD, et al. Vis Sci. 2006;47:5569–5575. doi: 10.1167/iovs.06-0333. [DOI] [PubMed] [Google Scholar]
- 435.Jadhav U, Ezhilarasan R, Vaughn SF, Berhow MA, Mohanam S. Int J Mol Med. 2007;19:353–361. [PMC free article] [PubMed] [Google Scholar]
- 436.Morgan MJ, Thorburn A. Cell Death Differ. 2001;8:38–43. doi: 10.1038/sj.cdd.4400800. [DOI] [PubMed] [Google Scholar]
- 437.Kaiser CL, Chapman BJ, Guidi JL, Terry CE, Mangiardi DA, Cotanche DA. Hear Res. 2008;240:1–11. doi: 10.1016/j.heares.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Darzynkiewicz Z, Bedner E, Smolewski P, Lee BW, Johnson GL. Methods Mol Biol. 2002;203:289–299. doi: 10.1385/1-59259-179-5:289. [DOI] [PubMed] [Google Scholar]
- 439.Luan Y, Yang Q, Xie Y, Duan S, Cai S, Forrest ML. Drug Discov Ther. 2011;5:220–226. doi: 10.5582/ddt.2011.v5.5.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Vickers CJ, Gonzalez-Paez GE, Wolan DW. ACS Chem Biol. 2013;8:1558–1566. doi: 10.1021/cb400209w. [DOI] [PubMed] [Google Scholar]
- 441.Bauer C, Bauder-Wuest U, Mier W, Haberkorn U, Eisenhut M. J Nucl Med. 2005;46:1066–1074. [PubMed] [Google Scholar]