

ABSTRACT
The development of effective inhibitors that block extended-spectrum β-lactamases (ESBLs) and restore the action of β-lactams represents an effective strategy against ESBL-producing Enterobacteriaceae. We evaluated the inhibitory effects of the diazabicyclooctanes avibactam and OP0595 against TLA-3, an ESBL that we identified previously. Avibactam and OP0595 inhibited TLA-3 with apparent inhibitor constants (Ki app) of 1.71 ± 0.10 and 1.49 ± 0.05 μM, respectively, and could restore susceptibility to cephalosporins in the TLA-3-producing Escherichia coli strain. The value of the second-order acylation rate constant (k2/K, where k2 is the acylation rate constant and K is the equilibrium constant) of avibactam [(3.25 ± 0.03) × 103 M−1 · s−1] was closer to that of class C and D β-lactamases (k2/K, <104 M−1 · s−1) than that of class A β-lactamases (k2/K, >104 M−1 · s−1). In addition, we determined the structure of TLA-3 and that of TLA-3 complexed with avibactam or OP0595 at resolutions of 1.6, 1.6, and 2.0 Å, respectively. TLA-3 contains an inverted Ω loop and an extended loop between the β5 and β6 strands (insertion after Ser237), which appear only in PER-type class A β-lactamases. These structures might favor the accommodation of cephalosporins harboring bulky R1 side chains. TLA-3 presented a high catalytic efficiency (kcat/Km) against cephalosporins, including cephalothin, cefuroxime, and cefotaxime. Avibactam and OP0595 bound covalently to TLA-3 via the Ser70 residue and made contacts with residues Ser130, Thr235, and Ser237, which are conserved in ESBLs. Additionally, the sulfate group of the inhibitors formed polar contacts with amino acid residues in a positively charged pocket of TLA-3. Our findings provide a structural template for designing improved diazabicyclooctane-based inhibitors that are effective against ESBL-producing Enterobacteriaceae.
KEYWORDS: diazabicyclooctane, avibactam, OP0595, TLA-3, extended-spectrum β-lactamase, crystal structure, ESBL
INTRODUCTION
The increasing prevalence of extended-spectrum β-lactamases (ESBLs) among pathogenic Enterobacteriaceae represents a major concern in clinical and veterinary settings, because these enzymes inactivate most β-lactams except cephamycins and carbapenems (1–3). Among the ESBLs, the CTX-M type and the TEM and SHV types are widely distributed worldwide and are regarded as pandemic ESBLs (4). Conversely, certain ESBLs, such as those of the TLA, BEL, PER, and GES types, have been reported only sporadically in Enterobacteriaceae (5). Specifically, the genes for TLA-1, TLA-2, and TLA-3 have been found mainly on plasmids in Enterobacteriaceae (6–8). We recently characterized TLA-3 produced by a Serratia marcescens clinical isolate (8).
New and effective inhibitors that block ESBLs and thereby restore the action of β-lactam antibiotics are powerful tools for overcoming the spread of ESBL-producing Enterobacteriaceae (9). Diazabicyclooctanes, such as avibactam (10), relebactam (MK-7655) (11), and OP0595 (12), which are effective non-β-lactam inhibitors of class A, C, and D β-lactamases, are currently under clinical development. Avibactam in combination with ceftazidime was recently approved by the U.S. Food and Drug Administration for treating infectious diseases for which no alternative therapeutic choices were available (13). The use of relebactam in combination with imipenem has now progressed to clinical trials. OP0595 differs from avibactam and relebactam because of three crucial properties: it is a β-lactamase inhibitor, an antibiotic agent that acts toward penicillin-binding proteins, and an enhancer of various β-lactam activities (12, 14). As the use of diazabicyclooctanes offers a promising strategy for the treatment of infectious diseases caused by ESBL producers, defining the mode of inhibition of β-lactamases by diazabicyclooctanes in detail is of considerable interest.
Three-dimensional structural studies have allowed extensive characterization of the inhibitory mechanism of these diazabicyclooctanes against major class A β-lactamases, such as SHV, CTX-M, and KPC (15, 16), but not against minor ESBLs, such as TLA, PER, and GES. In this study, we aimed to reveal the mode by which the aforementioned two new diazabicyclooctanes, avibactam and OP0595, inhibited a rare TLA-type ESBL, TLA-3, which is characterized by an inverted Ω loop and an extended loop between the β5 and β6 strands (insertion after Ser237). We also present the fine crystal structures of the avibactam–TLA-3 and OP0595–TLA-3 complexes.
RESULTS AND DISCUSSION
Purified TLA-3 has multiple N termini.
The TLA-3 β-lactamase contains a duplicate amino acid sequence (AKGTDSLKNS) in its N-terminal signal peptide (see Fig. S1 in the supplemental material). To determine the N terminus of mature TLA-3, we performed Edman N-terminal sequencing of purified TLA-3. This revealed multiple molecules with distinct N termini, including one containing the AKGTD sequence and another containing the TDSLK sequence (see Fig. S1 in the supplemental material).
TLA-3 hydrolyzes cephalosporins, whereas diazabicyclooctanes act as TLA-3 inhibitors.
We measured the kinetics parameters of TLA-3 against a variety of β-lactams (Table 1). TLA-3 showed a high catalytic efficiency (kcat/Km) against the cephalosporins cephalothin, cefuroxime, and cefotaxime but not against cefoxitin and meropenem. The enzymatic activity of TLA-3 was successfully inhibited by the diazabicyclooctanes avibactam and OP0595 (Fig. 1) and by the β-lactam-based inhibitors clavulanic acid, sulbactam, and tazobactam (Table 1). The apparent inhibitor constants (Ki app) of avibactam and OP0595 were 1.71 ± 0.10 and 1.49 ± 0.05 μM, respectively (Table 1). The Ki app of avibactam for TLA-3 was similar to that for KPC-2 (1.2 μM) (17) but ca. 10-fold lower than that for PER-2 (20 μM) (17) and ca. 100-fold higher than that for SHV-1 (0.022 μM) (18). Observed rate constant for inactivation (kobs) values were calculated on the basis of the fitting of progress curves for avibactam or OP0595 inhibition and were plotted against the inhibitor concentration to obtain the second-order acylation rate constant (k2/K, where k2 is the acylation rate constant and K is the equilibrium constant) value (Fig. 2A and B). Acylation of avibactam and OP0595 revealed k2/K values of (3.25 ± 0.03) × 103 M−1 · s−1 and (2.65 ± 0.26) × 103 M−1 · s−1, respectively. The acylation of avibactam by TLA-3 was similar to that by PER-2 (2.2 × 103 M−1 · s−1) (17) but was at least 1 magnitude lower than that by KPC-2 (19), SHV-1 (20), and CTX-M-15 (19) (k2/K range, 104 to 105 M−1 · s−1). The off-rate (koff) values of avibactam and OP0595 for TLA-3 were (9.57 ± 0.75) × 10−4 s−1 and (5.04 ± 1.03) × 10−4 s−1, respectively (Fig. 2C and D), resembling those of other class A β-lactamases (17, 19, 20). These low off rates indicate that diazabicyclooctanes can form stable complexes with TLA-3. In summary, the inhibitory action of TLA-3 against diazabicyclooctanes resembled that of PER-2 rather than that of other class A β-lactamases (KPC-2, TEM-1, and CTX-M-15).
TABLE 1.
Kinetic parameters of TLA-3
| Substrate | Km (μM)a | kcat (s−1)a | kcat/Km (μM −1 · s−1) | Ki app (μM)a |
|---|---|---|---|---|
| Nitrocefin | 37.6 ± 1.1 | 57.3 ± 0.7 | 1.5 | |
| Ampicillin | 61.1 ± 3.8 | 55.6 ± 2.9 | 0.9 | |
| Cephalothin | 29.9 ± 1.4 | 132.8 ± 2.3 | 4.4 | |
| Cefuroxime | 127.7 ± 16.2 | 246.0 ± 19.1 | 1.9 | |
| Ceftazidime | 177.9 ± 7.7 | 33.4 ± 1.0 | 0.2 | |
| Cefotaxime | 98.1 ± 5.3 | 156.3 ± 5.0 | 1.6 | |
| Cefepime | 197.8 ± 18.0 | 34.2 ± 1.9 | 0.2 | |
| Cefoxitin | NHb | NDc | ND | |
| Aztreonam | 105.6 ± 14.5 | 12.1 ± 0.7 | 0.1 | |
| Meropenem | NH | ND | ND | |
| Avibactam | 1.71 ± 0.10 | |||
| OP0595 | 1.49 ± 0.05 | |||
| Clavulanic acid | 0.41 ± 0.01 | |||
| Tazobactam | 0.14 ± 0.01 | |||
| Sulbactam | 0.57 ± 0.07 |
Determinations of kinetic parameters were performed in triplicate, and values are reported as means ± standard deviations.
NH, no measurable hydrolysis was detected at an enzyme concentration of 1 μM.
ND, not determined.
FIG 1.

Chemical structures of avibactam and OP0595.
FIG 2.

(A and B) Plots of kobs values against avibactam (A) and OP0595 (B) concentrations; these were used to obtain the corresponding k2/K values. (C and D) Recovery of nitrocefin hydrolysis rate by TLA-3 after inhibition by avibactam (C) and OP0595 (D). a.u., absorbance units.
Diazabicyclooctanes restore susceptibility in TLA-3-producing Escherichia coli.
The results of susceptibility testing are listed in Table 2. Production of TLA-3 could confer resistance to a penicillin (ampicillin), cephalosporins, and a monobactam (aztreonam) but not to a cephamycin (cefoxitin) and carbapenems. Diazabicyclooctanes (avibactam and OP0595) and β-lactam-based inhibitors (clavulanic acid, sulbactam, and tazobactam) could successfully restore the MICs of ceftazidime and cefotaxime for TLA-3-producing bacteria.
TABLE 2.
Result of susceptibility testing
| Agent | MIC (μg/ml) |
|
|---|---|---|
| E. coli DH5α(pBC-TLA-3b) | E. coli DH5α(pBC-SK+) | |
| Ampicillin | 128 | 2 |
| Cephalothin | 128 | 8 |
| Cefuroxime | 256 | 8 |
| Ceftazidime | 64 | 0.25 |
| Ceftazidime plus: | ||
| Avibactama | 0.25 | 0.25 |
| OP0595a | ≤0.13 | ≤0.13 |
| Clavulanic acida | 0.5 | 0.25 |
| Tazobactama | 1 | 0.25 |
| Sulbactama | 4 | 0.25 |
| Cefotaxime | 16 | ≤0.13 |
| Cefotaxime plus: | ||
| Avibactama | ≤0.13 | ≤0.13 |
| OP0595a | ≤0.13 | ≤0.13 |
| Clavulanic acida | ≤0.13 | ≤0.13 |
| Tazobactama | ≤0.13 | ≤0.13 |
| Sulbactama | 1 | ≤0.13 |
| Cefepime | 0.25 | ≤0.13 |
| Cefoxitin | 4 | 4 |
| Aztreonam | 8 | ≤0.13 |
| Imipenem | 0.25 | 0.25 |
| Meropenem | ≤0.13 | ≤0.13 |
| Avibactam | 32 | 32 |
| OP0595 | 4 | 4 |
The concentration was fixed at 4 μg/ml.
The pBC-TLA-3 vector was constructed in a previous study (8).
TLA-3 has two unique loops around the cephalosporin R1 side chain binding site.
We determined the structure of native TLA-3 at a 1.6-Å resolution. The collected data and the refinement statistics are listed in Table S1. The TLA-3 crystal is in space group C2, with one molecule per asymmetric unit. The model includes 275 amino acids, from Gly32 to Lys306 (Fig. S2). TLA-3 forms one α/β domain and one α domain, with the active site being located at the interface between these domains (Fig. 3A), as in other class A β-lactamases (15, 16, 21). The active site is defined by motif I (Ser70-Thr71-Tyr72-Lys73), motif II (Ser130-Asp131-Asn132), motif III (Lys234-Thr235-Gly236), and the Ω loop (16 amino acids, Ala164 to Asn179) (Fig. S2). Compared with other class A β-lactamases, the most noticeable structural features of TLA-3 are two loops, the Ω loop (loop 1) and loop 2, which are predicted to be positioned around the R1 side chains of bound β-lactam antibiotics (22) (Fig. 3B and C). The overall configuration of the Ω loop (loop 1) in TLA-3 is most closely related to that of the Ω loop in PER-2 (21) but is inverted relative to the Ω loops in CTX-M-15 (16) and KPC-2 (15) (Fig. 3B). Loop 2 of TLA-3 (and PER-2) is extended by the insertion of additional amino acids between the β5 and β6 strands (insertion after Ser237) compared to the number of amino acids in CTX-M-15 and TEM-1 (Fig. S2). Consequently, the overall TLA-3 structure is considerably more similar to that of the PER-2 β-lactamase (C-α root mean square deviation [RMSD] value, 1.2 Å) than to the structures of other class A β-lactamases. Indeed, TLA-3 shares 42% amino acid sequence identity with PER-2. The opening between the Ω loop (loop 1) and loop 2 of TLA-3 (and PER-2) is slightly wider than the openings of CTX-M-15 and KPC-2 (Fig. 3B). Ruggiero et al. suggested that these configurations of the double loop structure in PER-2 (and TLA-3) yielded an expanded active site that could accommodate oxyiminocephalosporins harboring bulky R1 side chains (21). We predicted the binding mode between ceftazidime and TLA-3 through in silico analysis and revealed that TLA-3 had sufficient space to accommodate the bulky R1 side chains (Fig. 3C). This explains the high catalytic efficiencies (kcat/Km) of TLA-3 against oxyiminocephalosporins containing bulky R1 side chains, such as ceftazidime (0.2 μM−1 · s−1), cefotaxime (1.6 μM−1 · s−1), and cefepime (0.2 μM−1 · s−1) (Table 1).
FIG 3.

(A) Schematic representation of the overall structure of TLA-3. Cyan, α helices and loops; magenta, β strands; black circles, Ω loop (loop 1) and loop 2. (B) Superposition of the Ω loop (loop 1) and loop 2 from TLA-3 (cyan), PER-2 (PDB accession no. 4D2O; orange), CTX-M-15 (PDB accession no. 4HBU; gray), and KPC-2 (PDB accession no. 4ZBE; yellow). (C) Simulated interaction between ceftazidime and TLA-3. Cyan, two loops around the R1 side chain of ceftazidime; pink sticks, ceftazidime. Images were rendered using the PyMOL program.
Active site of TLA-3.
In the TLA-3 active site, the spatial positioning of Ser70, Lys73, Ser130, Asn132, Glu166, and Lys234, which are involved in β-lactam acylation/deacylation, is comparable to that in class A β-lactamases, including PER-2 (Fig. 4A and B). The TLA-3 active site is stabilized by a network of several hydrogen bonds: Ser70-Ser130 (3.0 Å), Ser70-Lys73 (2.7 Å), Lys73-Ser130 (2.8 Å), Lys73-Asn132 (2.8 Å), and Ser130-Lys234 (2.9 Å). Arg220, a unique amino acid present only in certain class A β-lactamases, such as TLA-, PER-, and KPC-type enzymes (Fig. S2), rigidly stabilizes the side chain of Ser237 (Fig. 4A). Ser237 in TLA-3 corresponds to Thr237 in PER-2, and the hydroxyl group of both residues is predicted to coordinate with the carboxylate group of substrate β-lactams (21). This stabilization of the Ser237 side chain by Arg220 potentially contributes to firm recognition of substrate β-lactams, which results in the increased β-lactam catalytic activity of TLA-3 (21). In fact, the substitutions at Arg220 and Thr237 deteriorate the enzymatic behavior of PER-1 (23). One water molecule (W1) is bound to Glu166, and this molecule is predicted to be involved in β-lactam deacylation.
FIG 4.

Schematic representation of the active sites of TLA-3 (A) and PER-2 (PDB accession no. 4D2O) (B). Cyan and orange sticks, carbon atoms of TLA-3 and PER-2, respectively; red and blue sticks, oxygen and nitrogen atoms, respectively; black dashed lines, hydrogen bonds (cutoff distance, 3.2 Å); red sphere, the water molecule (W1). Images were rendered using the PyMOL program.
Structural insights into the binding of avibactam to TLA-3.
Figures 5A to C show the avibactam–TLA-3 complex structure. The overall structure of the complex is highly similar to that of native TLA-3, with an RMSD of 0.2 Å and few conformational changes in the key residues around the active site. The avibactam molecule was bound to the active site and was clearly observed in an |Fobs|−|Fcalc| omit map contoured at 3σ (Fig. 5A). Bound avibactam assumes a form lacking the bond between the N-6 and C-7 atoms, similar to the form of typical hydrolyzed β-lactams, and adopts the chair conformation of a six-membered ring. Ser70 is covalently bound to avibactam. The oxyanion hole is formed by the main chain nitrogen atoms of Ser70 and Ser237 (Fig. 5B and C). The sulfate group binds to TLA-3 by forming polar contacts with the side chains of Ser130 (2.8 Å), Thr235 (3.0 Å), and Ser237 (2.7 Å). The N-6 atom of avibactam binds to Ser130 (3.2 Å), and the carbonyl oxygen of the carbamoyl group is hydrogen bonded to the side chain of Asn132 (2.9 Å) (Fig. 5B). The C-4 atom of the avibactam six-membered ring makes hydrophobic contact with the side chain of Trp105 (3.4 Å). These residues are conserved in class A β-lactamases, and thus, their contribution to the recognition of avibactam is also observed in other class A β-lactamase–avibactam complexes (Fig. 5D).
FIG 5.

(A) |Fobs|−|Fcalc| omit map of avibactam, contoured at 3.0σ (gray mesh). Avibactam is illustrated using green (carbon), ocher (sulfur), red (oxygen), and blue (nitrogen) sticks. (B) Interactions between TLA-3 and avibactam. Amino acids are drawn as cyan (carbon), red (oxygen), and blue (nitrogen) sticks, and avibactam is shown as described in the legend to panel A. Red sphere, the water molecule (W1); black dashed lines, hydrogen bonds (cutoff distance, 3.2 Å). (C) Two-dimensional view of the binding between TLA-3 and avibactam. Green, avibactam; black, amino acids; dashed lines, hydrogen bonds (cutoff distance, 3.2 Å). The figure was rendered using ChemBioDraw (version 13.0) software. (D) Interactions between CTX-M-15 and avibactam (PDB accession no. 4HBU). Amino acids are drawn as gray (carbon), red (oxygen), and blue (nitrogen) sticks, and avibactam is shown as described in the legend to panel A. Red sphere, the water molecule (W1); black dashed lines, hydrogen bonds (cutoff distance, 3.2 Å). (E) Molecular surface representation of TLA-3, colored according to electrostatic charge (red, negative; blue, positive). Avibactam is shown in stick form as described in the legend to panel A. (F) Interactions between KPC-2 and avibactam (PDB accession no. 4ZBE). Amino acids are drawn as yellow (carbon), red (oxygen), and blue (nitrogen) sticks, and avibactam is shown as described in the legend to panel A. Red sphere, the water molecule (W1); black dashed lines, hydrogen bonds (cutoff distance, 3.2 Å).
The sulfate group of avibactam is located in a positively charged pocket (Fig. 5E), where the carboxylate group of β-lactams is normally positioned. Multiple contacts of the sulfate moiety with polar residues, such as Ser130 and Ser237, in the active-site pocket might partially explain why avibactam acts as an effective inhibitor across class A β-lactamases (Fig. 5B, D, and F), whereas the carboxylate groups of β-lactam-based inhibitors make comparatively fewer polar contacts with the active site. In the native TLA-3 structure, the sulfate ion provided by the reservoir solution used for crystallization is detected precisely where the sulfate group of avibactam is positioned (data not shown). Thus, the sulfate group is a key factor that endows avibactam with the ability to strongly inhibit TLA-3.
The catalytic water molecule (W1), which is probably involved in the deacylation of the carbamyl-enzyme complex, is bound to Glu166 (2.5 Å) (Fig. 5B), as in the native structure (Fig. 4A). In avibactam–CTX-M-15 complexes, the same catalytic water is also bound to Glu166 (Fig. 5D). Lahiri et al. determined the avibactam–CTX-M-15 complex structure at an ultrahigh resolution and concluded that Glu166 was protonated and neutral; consequently, the water molecule's orientation is disfavored and it becomes a weak nucleophile (16). Avibactam's decarbamylation might be disfavored partially due to the presence of protonated Glu166, and a similar mechanism might also apply to the avibactam–TLA-3 complex. Furthermore, prevention of decarbamylation results from the carbamyl carbon being originally less susceptible to nucleophilic attack than the ester of the acyl-enzyme complex (24). Therefore, carbamyl bond formation and the establishment of a considerably higher number of polar contacts through the sulfate group are common structural features that could explain avibactam's ability to effectively inhibit most class A β-lactamases.
The Ω-loop configuration of TLA-3 results in the creation of a relatively large space that accommodates cephalosporins possessing bulky R1 side chains. This is in spite of the larger spaces created around the carbamoyl group of avibactam's binding pocket being expected to diminish avibactam's inhibitory effect following the loss of certain interactions, such as the interaction of the Asn170-carbamoyl moiety in avibactam–CTX-M-15 complexes (Fig. 4D). As mentioned above, kinetic inhibition constants indicated that TLA-3 was more resistant to avibactam inhibition than representative class A β-lactamases. The Ω-loop configuration observed in TLA-3 (and also present in PER-2) may confer increased resistance to avibactam inhibition. However, avibactam binding largely depends on residues Ser70, Ser130, Thr235, and Ser(Thr)237, which are conserved in class A β-lactamases. As a result, avibactam could, with a greater or lesser degree, act as a universal and effective inhibitor across class A β-lactamases.
Structural insights into OP0595 binding and proposed role of the terminal chain in diazabicyclooctanes.
The crystal structure of the OP0595–TLA-3 complex was determined at a 2.0-Å resolution. OP0595 was bound to the active site and was clearly observed in an |Fobs|−|Fcalc| omit map contoured at 2.5σ (Fig. 6A). OP0595 bound to the active-site pocket through Ser70, Ser130, Asn132, Thr235, and Ser237 in a manner highly similar to that of avibactam (Fig. 5B) and that of Toho-1 (CTX-M-44) and OP0595 (Fig. 6B). The diazabicyclooctane core was located at almost the same position in OP0595- and avibactam-complexed structures (Fig. 6C) (12).
FIG 6.
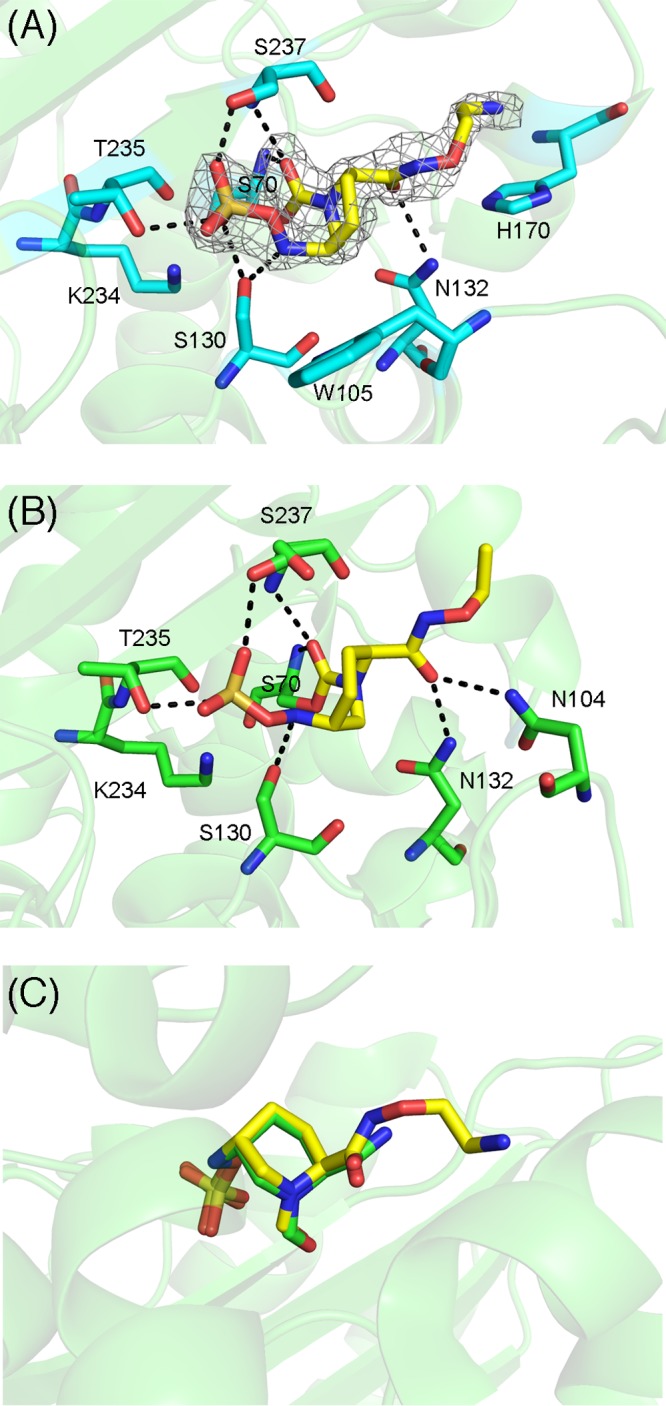
(A) Interactions between TLA-3 and OP0595. Amino acids are drawn as cyan (carbon), red (oxygen), and blue (nitrogen) sticks, whereas OP0595 is depicted by yellow sticks. Black dashed lines, hydrogen bonds (cutoff distance, 3.2 Å). The figure shows the |Fobs|−|Fcalc| omit map of OP0595, contoured at 2.5σ (gray mesh). (B) Interactions between Toho-1 (CTX-M-44) and OP0595 (PDB accession no. 4X69). Amino acids are drawn as green (carbon), red (oxygen), and blue (nitrogen) sticks, whereas OP0595 is depicted by yellow sticks. Black dashed lines, hydrogen bonds (cutoff distance, 3.2 Å). (C) Superposition of avibactam and OP0595 in TLA-3. Avibactam and OP0595 are represented by green and yellow sticks, respectively. Images were rendered using the PyMOL program.
In OP0595, the terminal alkyl amine moiety is linked to the carbamoyl group side chain, and this moiety may mediate affinity to the penicillin-binding proteins (12, 25). This terminal moiety on the OP0595 side chain does not contact the amino acid residues in TLA-3; thus, extension of the terminal side chain does not enhance the inhibitory effect of OP0595 relative to that of avibactam (Table 1). Because the His170 contained in the unique Ω loop (loop 1) is positioned near the tip of the terminal alkyl amine moiety, this residue may represent a favorable target for enhancing the inhibitory effect of modifications to the side chain structure of diazabicyclooctane-based inhibitors. Indeed, relebactam (MK-7655) contains a terminal piperidine ring moiety and might bind to His170, making it an inhibitor superior to avibactam. The side chain structure of diazabicyclooctanes is thus a key target for enhancing inhibition against ESBLs, such as TLA-3.
Conclusions.
In this paper, we report the crystal structure of the ESBL TLA-3. The active site of TLA-3 contains residues comparable to those of other class A β-lactamases, such as Ser70, Lys73, Ser130, Glu166, and Lys234, which are involved in the acylation and deacylation of β-lactams. Conversely, TLA-3 presents two structural features, an inverted Ω loop and an extended loop between the β5 and β6 strands (insertion after Ser237), that have been observed only in PER-type β-lactamases. These two loops might enable TLA-3 to readily accommodate cephalosporins harboring a bulky R1 side chain.
In addition, we determined the kinetics of TLA-3 inactivation by the diazabicyclooctanes avibactam and OP0595. TLA-3 displayed a lower affinity for diazabicyclooctanes than other class A β-lactamases evaluated so far. This characteristic may be partially due to the two unique loop structures in TLA-3; however, we found that avibactam and OP0595 could successfully restore cephalosporin susceptibility in TLA-3-producing cells, as well as other class A β-lactamase producers (25, 26). Thus, diazabicyclooctanes might be the compounds of choice for targeting ESBL producers, regardless of the exact ESBL type. Our findings provide a structural basis for designing improved diazabicyclooctane-based inhibitors with increased effectiveness against ESBL-producing Enterobacteriaceae.
MATERIALS AND METHODS
Protein expression and purification.
The gene blaTLA-3 (GenBank accession no. AP014611) was amplified with primers NdeI-TLA3-F (5′-GGG AAT TCC ATA TGA AAA AAC ATC TTA TTG-3′) and BamHI-TLA3-R (5′-CGG GAT CCT TAC TAT TTC CCA TCC TTA ACT AGA T-3′) (where the underlined nucleotides represent restriction endonuclease sequences) using PrimeSTAR HS DNA polymerase (TaKaRa), digested with NdeI and BamHI, and ligated into the vector pET-29a. The recombinant plasmid (pET-TLA3) was introduced into Escherichia coli BL21(DE3)pLysS. Transformants were grown at 37°C in Luria-Bertani broth until the optical density at 610 nm reached 0.6, at which point isopropyl-β-d-1-thiogalactopyranoside was added to a final concentration of 0.5 mM. After incubation at 37°C for another 3 h, bacterial cells were harvested, suspended in 50 mM HEPES buffer (pH 7.5) containing 0.1 M NaCl, and disrupted by sonication. The supernatant obtained after ultracentrifugation (100,000 × g, 1 h) was loaded onto a HiTrap SP HP column (GE Healthcare) and eluted using a linear gradient of 0.1 to 0.5 M NaCl in HEPES buffer. The eluted protein was dialyzed against 50 mM Tris-HCl (pH 7.5) containing 0.1 M NaCl and 2.0 M ammonium sulfate, loaded onto a HiTrap phenyl HP column (GE Healthcare), and eluted with 50 mM Tris-HCl (pH 7.5) containing 0.1 M NaCl. Finally, the protein was loaded onto a HiLoad 16/60 Superdex 200 pg column (GE Healthcare) and then eluted with 20 mM Tris-HCl (pH 7.4) containing 0.1 M NaCl. The concentration of the eluted protein was determined using a Pierce bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Protein purity was estimated by SDS-PAGE and Coomassie brilliant blue staining. The N-terminal sequence of purified TLA-3 was obtained by Edman degradation.
Kinetic parameters.
Steady-state kinetic parameters were determined using a V-730Bio spectrophotometer (Jasco Corporation). The assay was performed with 100 mM phosphate buffer (pH 7.0) at 30°C. The Km and kcat values were obtained by fitting the data to a Michaelis-Menten equation (equation 1) using KaleidaGraph (version 4.5) software.
| (1) |
where v is the velocity of the reaction and [S] is concentration of the substrate. The interaction between TLA-3 and the inhibitors was evaluated as described previously (17, 27).
where E represents TLA-3, I represents the inhibitors, k1 represents the association rate constant, k−1 represents the disassociation rate constant, k2 represents the acylation (carbamylation) rate constant, and k−2 represents the decarbamylation (recyclization) rate constant for diazabicyclooctanes. Apparent inhibitor constant (Ki app) values were determined using a direct competition assay under steady-state conditions: nitrocefin (120 μM) was used as a reporter substrate, TLA-3 was kept at 1 nM, and the concentration of each inhibitor was varied. The velocity (v0) was obtained by fitting the data to equation 2:
| (2) |
The inverse initial steady-state velocity (1/v0) was plotted against the inhibitor concentration ([I]), and Ki app was obtained by dividing the y-intercept value by the slope of the line. Finally, the Ki app (corrected) value was determined by considering the concentration and affinity for nitrocefin of TLA-3 according to equation 3:
| (3) |
To determine the observed rate constant for inactivation (kobs), progress curves obtained by mixing 1 nM TLA-3 with the inhibitor at various concentrations and 120 μM nitrocefin as a substrate were fitted to equation 4, and k2/K was determined from equation 5:
| (4) |
| (5) |
where A is the absorbance, vf is the final velocity, t is time, v0 is the initial velocity, and A0 is the initial absorbance at 482 nm. kobs was plotted against the inhibitor concentration ([I]). k2/K (corrected) was determined by considering the concentration and affinity for nitrocefin of TLA-3 according to equation 6:
| (6) |
The off-rate (koff) value was obtained as follows: 1 μM TLA-3 and 15 μM avibactam or OP0595 were mixed and incubated for 5 min. The mixture was diluted (1:2,000), and the hydrolysis of 120 μM nitrocefin was monitored. Progress curves were fitted to equation 4.
Susceptibility testing.
MICs were determined using the microdilution method following Clinical and Laboratory Standards Institute guidelines (28). The following antimicrobial agents and β-lactamase inhibitors tested in the present study were obtained from the indicated sources: cephalothin, cefuroxime, and cefoxitin were from Sigma-Aldrich; ampicillin, clavulanic acid, cefotaxime, and meropenem were from Wako Pure Chemical Industries; ceftazidime and aztreonam were from Tokyo Chemical Industry; tazobactam and sulbactam were from LKT Laboratories; imipenem was from Apollo Scientific; cefepime was from the United States Pharmacopeial Convention; avibactam was from MedKoo Biosciences; and OP0595 was synthesized by Meiji Seika Pharma.
Crystallization.
To obtain native crystals, 1 μl of 15 mg/ml TLA-3 was mixed with 1 μl of reservoir solution (0.1 M sodium citrate, 2.4 to 3.0 M ammonium sulfate) and incubated at 20°C using the sitting vapor diffusion method. Crystals suitable for the collection of diffraction data were obtained within 1 week.
TLA-3 crystals complexed with avibactam and OP0595 were prepared as follows: sodium avibactam and OP0595 were dissolved in the reservoir solution to a final concentration of 10 mg/ml; next, 1 μl of each inhibitor was added to drops of solution containing the native crystals, and the mixture was incubated for another 24 h before collection of the diffraction data.
Data collection and refinement.
Crystals were frozen in liquid nitrogen with 20% glycerol as a cryoprotectant. X-ray diffraction data were collected at the BL2S1 beamline of the Aichi Synchrotron Radiation Center (Aichi, Japan) (29), processed, and scaled using iMosfilm/SCALA software (30, 31). Structures were solved by performing molecular replacement in the MOLREP program (32), using a modified PER-1 structure solved at a 1.9-Å resolution (Protein Data Bank [PDB] accession no. 1E25) (33). The PER-1 structure was modified by removing water molecules and replacing amino acids that did not match those of TLA-3 with alanine. Models were built and refined using the Coot (34) and REFMAC5 (35) programs, respectively, and the quality of the final models was assessed using the RAMPAGE program (36).
Molecular modeling of TLA-3 with ceftazidime.
The crystal structure of avibactam bound to TLA-3 was analyzed and prepared by the protein preparation wizard module of Schrodinger (37). After ionizing acylated ceftazidime, tautomers and stereoisomers were generated by using the OPLS2003 force field in the LigPrep (37). Low-energy conformations were used for the docking. Using the crystal structure of avibactam bound to TLA-3, ceftazidime was covalently docked in the grid space created around the active site of avibactam using the CovDoc (37). A thorough pose prediction was performed for covalent docking with default parameters and the reaction type set to nucleophilic addition to a double bond.
Accession number(s).
The atomic coordinates and structure factors of native TLA-3, avibactam–TLA-3, and OP0595–TLA-3 have been deposited in the PDB database under accession no. 5GS8, 5GWA, and 5X5G, respectively.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the staff of the Division for Medical Research Engineering, Nagoya University Graduate School of Medicine, for their technical support. We also thank the staff at the Aichi Synchrotron Radiation Center (Aichi, Japan).
This study was supported by grants from the Japan Society for the Promotion of Science (JSPS KAKENHI grant number JP15H05672, Wakate A), the Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) of the Japan Agency for Medical Research and Development (AMED) (grant number fm0108014h), and the Association for Research on Lactic Acid Bacteria.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00501-17.
REFERENCES
- 1.Lukac PJ, Bonomo RA, Logan LK. 2015. Extended-spectrum β-lactamase-producing Enterobacteriaceae in children: old foe, emerging threat. Clin Infect Dis 60:1389–1397. doi: 10.1093/cid/civ020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oteo J, Perez-Vazquez M, Campos J. 2010. Extended-spectrum β-lactamase producing Escherichia coli: changing epidemiology and clinical impact. Curr Opin Infect Dis 23:320–326. doi: 10.1097/QCO.0b013e3283398dc1. [DOI] [PubMed] [Google Scholar]
- 3.Michael GB, Kaspar H, Siqueira AK, de Freitas Costa E, Corbellini LG, Kadlec K, Schwarz S. 2017. Extended-spectrum β-lactamase (ESBL)-producing Escherichia coli isolates collected from diseased food-producing animals in the GERM-Vet monitoring program 2008-2014. Vet Microbiol 200:142–150. doi: 10.1016/j.vetmic.2016.08.023. [DOI] [PubMed] [Google Scholar]
- 4.Canton R, Gonzalez-Alba JM, Galan JC. 2012. CTX-M enzymes: origin and diffusion. Front Microbiol 3:110. doi: 10.3389/fmicb.2012.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naas T, Poirel L, Nordmann P. 2008. Minor extended-spectrum β-lactamases. Clin Microbiol Infect 14(Suppl 1):42–52. doi: 10.1111/j.1469-0691.2007.01861.x. [DOI] [PubMed] [Google Scholar]
- 6.Silva J, Aguilar C, Ayala G, Estrada MA, Garza-Ramos U, Lara-Lemus R, Ledezma L. 2000. TLA-1: a new plasmid-mediated extended-spectrum β-lactamase from Escherichia coli. Antimicrob Agents Chemother 44:997–1003. doi: 10.1128/AAC.44.4.997-1003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Girlich D, Poirel L, Schluter A, Nordmann P. 2005. TLA-2, a novel Ambler class A expanded-spectrum β-lactamase. Antimicrob Agents Chemother 49:4767–4770. doi: 10.1128/AAC.49.11.4767-4770.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jin W, Wachino J, Kimura K, Yamada K, Arakawa Y. 2015. New plasmid-mediated aminoglycoside 6′-N-acetyltransferase, AAC(6′)-Ian, and ESBL, TLA-3, from a Serratia marcescens clinical isolate. J Antimicrob Chemother 70:1331–1337. doi: 10.1093/jac/dku537. [DOI] [PubMed] [Google Scholar]
- 9.Bush K. 2015. A resurgence of β-lactamase inhibitor combinations effective against multidrug-resistant Gram-negative pathogens. Int J Antimicrob Agents 46:483–493. doi: 10.1016/j.ijantimicag.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Bonnefoy A, Dupuis-Hamelin C, Steier V, Delachaume C, Seys C, Stachyra T, Fairley M, Guitton M, Lampilas M. 2004. In vitro activity of AVE1330A, an innovative broad-spectrum non-β-lactam β-lactamase inhibitor. J Antimicrob Chemother 54:410–417. doi: 10.1093/jac/dkh358. [DOI] [PubMed] [Google Scholar]
- 11.Blizzard TA, Chen H, Kim S, Wu J, Bodner R, Gude C, Imbriglio J, Young K, Park YW, Ogawa A, Raghoobar S, Hairston N, Painter RE, Wisniewski D, Scapin G, Fitzgerald P, Sharma N, Lu J, Ha S, Hermes J, Hammond ML. 2014. Discovery of MK-7655, a β-lactamase inhibitor for combination with Primaxin(R). Bioorg Med Chem Lett 24:780–785. doi: 10.1016/j.bmcl.2013.12.101. [DOI] [PubMed] [Google Scholar]
- 12.Morinaka A, Tsutsumi Y, Yamada M, Suzuki K, Watanabe T, Abe T, Furuuchi T, Inamura S, Sakamaki Y, Mitsuhashi N, Ida T, Livermore DM. 2015. OP0595, a new diazabicyclooctane: mode of action as a serine β-lactamase inhibitor, antibiotic and β-lactam ‘enhancer.’ J Antimicrob Chemother 70:2779–2786. doi: 10.1093/jac/dkv166. [DOI] [PubMed] [Google Scholar]
- 13.Falcone M, Paterson D. 2016. Spotlight on ceftazidime/avibactam: a new option for MDR Gram-negative infections. J Antimicrob Chemother 71:2713–2722. doi: 10.1093/jac/dkw239. [DOI] [PubMed] [Google Scholar]
- 14.Morinaka A, Tsutsumi Y, Yamada K, Takayama Y, Sakakibara S, Takata T, Abe T, Furuuchi T, Inamura S, Sakamaki Y, Tsujii N, Ida T. 2016. In vitro and in vivo activities of OP0595, a new diazabicyclooctane, against CTX-M-15-positive Escherichia coli and KPC-positive Klebsiella pneumoniae. Antimicrob Agents Chemother 60:3001–3006. doi: 10.1128/AAC.02704-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishnan NP, Nguyen NQ, Papp-Wallace KM, Bonomo RA, van den Akker F. 2015. Inhibition of Klebsiella β-lactamases (SHV-1 and KPC-2) by avibactam: a structural study. PLoS One 10:e0136813. doi: 10.1371/journal.pone.0136813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lahiri SD, Mangani S, Durand-Reville T, Benvenuti M, De Luca F, Sanyal G, Docquier JD. 2013. Structural insight into potent broad-spectrum inhibition with reversible recyclization mechanism: avibactam in complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-lactamases. Antimicrob Agents Chemother 57:2496–2505. doi: 10.1128/AAC.02247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruggiero M, Papp-Wallace KM, Taracila MA, Mojica MF, Bethel CR, Rudin SD, Zeiser ET, Gutkind G, Bonomo RA, Power P. 2017. Exploring the landscape of diazabicyclooctane inhibition: avibactam inactivation of PER-2 β-lactamase. Antimicrob Agents Chemother 61:e02476-16. doi: 10.1128/AAC.02476-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Winkler ML, Papp-Wallace KM, Taracila MA, Bonomo RA. 2015. Avibactam and inhibitor-resistant SHV β-lactamases. Antimicrob Agents Chemother 59:3700–3709. doi: 10.1128/AAC.04405-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Durand-Reville TF, Lahiri S, Thresher J, Livchak S, Gao N, Palmer T, Walkup GK, Fisher SL. 2013. Kinetics of avibactam inhibition against class A, C, and D β-lactamases. J Biol Chem 288:27960–27971. doi: 10.1074/jbc.M113.485979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci U S A 109:11663–11668. doi: 10.1073/pnas.1205073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruggiero M, Kerff F, Herman R, Sapunaric F, Galleni M, Gutkind G, Charlier P, Sauvage E, Power P. 2014. Crystal structure of the extended-spectrum β-lactamase PER-2 and insights into the role of specific residues in the interaction with β-lactams and β-lactamase inhibitors. Antimicrob Agents Chemother 58:5994–6002. doi: 10.1128/AAC.00089-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamski CJ, Cardenas AM, Brown NG, Horton LB, Sankaran B, Prasad BV, Gilbert HF, Palzkill T. 2015. Molecular basis for the catalytic specificity of the CTX-M extended-spectrum β-lactamases. Biochemistry 54:447–457. doi: 10.1021/bi501195g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouthors AT, Delettre J, Mugnier P, Jarlier V, Sougakoff W. 1999. Site-directed mutagenesis of residues 164, 170, 171, 179, 220, 237 and 242 in PER-1 β-lactamase hydrolysing expanded-spectrum cephalosporins. Protein Eng 12:313–318. doi: 10.1093/protein/12.4.313. [DOI] [PubMed] [Google Scholar]
- 24.King DT, King AM, Lal SM, Wright GD, Strynadka NC. 2015. Molecular mechanism of avibactam-mediated β-lactamase inhibition. ACS Infect Dis 1:175–184. doi: 10.1021/acsinfecdis.5b00007. [DOI] [PubMed] [Google Scholar]
- 25.Livermore DM, Mushtaq S, Warner M, Woodford N. 2015. Activity of OP0595/β-lactam combinations against Gram-negative bacteria with extended-spectrum, AmpC and carbapenem-hydrolysing β-lactamases. J Antimicrob Chemother 70:3032–3041. doi: 10.1093/jac/dkv239. [DOI] [PubMed] [Google Scholar]
- 26.Karlowsky JA, Biedenbach DJ, Kazmierczak KM, Stone GG, Sahm DF. 2016. Activity of ceftazidime-avibactam against extended-spectrum- and AmpC β-lactamase-producing Enterobacteriaceae collected in the INFORM Global Surveillance Study from 2012 to 2014. Antimicrob Agents Chemother 60:2849–2857. doi: 10.1128/AAC.02286-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papp-Wallace KM, Winkler ML, Gatta JA, Taracila MA, Chilakala S, Xu Y, Johnson JK, Bonomo RA. 2014. Reclaiming the efficacy of β-lactam–β-lactamase inhibitor combinations: avibactam restores the susceptibility of CMY-2-producing Escherichia coli to ceftazidime. Antimicrob Agents Chemother 58:4290–4297. doi: 10.1128/AAC.02625-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clinical and Laboratory Standards Institute. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard, 9th ed. Document M07-A9 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 29.Watanabe N, Nagae T, Yamada Y, Tomita A, Matsugaki N, Tabuchi M. 2017. Protein crystallography beamline BL2S1 at the Aichi Synchrotron. J Synchrotron Radiat 24:338–343. doi: 10.1107/S1600577516018579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. 2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 32.Vagin A, Teplyakov A. 2010. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66:22–25. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 33.Tranier S, Bouthors AT, Maveyraud L, Guillet V, Sougakoff W, Samama JP. 2000. The high resolution crystal structure for class A β-lactamase PER-1 reveals the bases for its increase in breadth of activity. J Biol Chem 275:28075–28082. [DOI] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Lovell SC, Davis IW, Arendall WB III, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. 2003. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins 50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 37.Schrödinger. 2016. Schrödinger release 2016-1. Schrödinger, LLC, New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.