

ABSTRACT
Finafloxacin is a novel fluoroquinolone exhibiting enhanced activity under acidic conditions and a broad-spectrum antibacterial profile. The present study assessed the pharmacokinetic properties and the safety and tolerability of finafloxacin following intravenous infusions. In this mixed-parallel-group, crossover study, healthy male and female volunteers received single ascending doses (18 volunteers, 200 to 1,000 mg) or multiple ascending doses (40 volunteers, 600 to 1,000 mg) of finafloxacin or placebo. Plasma and urine samples were collected by a dense sampling scheme to determine the pharmacokinetics of finafloxacin using a noncompartmental approach. Standard safety and tolerability data were documented. Finafloxacin had a volume of distribution of 90 to 127 liters (range) at steady state and 446 to 550 liters at pseudoequilibrium, indicating the elimination of a large fraction before pseudoequilibrium was reached. Areas under the concentration-time curves and maximum plasma concentrations (geometric means) increased slightly more than proportionally (6.73 to 45.9 μg · h/ml and 2.56 to 20.2 μg/ml, respectively), the terminal elimination half-life increased (10.6 to 17.1 h), and the urinary recovery decreased (44.2% to 31.7%) with increasing finafloxacin doses (single doses of 200 to 1,000 mg). The pharmacokinetic profiles suggested multiphasic elimination by both glomerular filtration and saturable tubular secretion. The values of the parameters were similar for single and multiple administrations. The coefficient of variation for the between-subject variability of exposure ranged from 10% (≤600 mg) to 38% (>600 mg). Adverse events were mild and nonspecific, with no dependence of adverse events on dose or treatment (including placebo) being detected. Despite a relatively high interindividual variability at higher doses, the level of exposure following intravenous administration of finafloxacin appears to be predictable. Individual elimination processes should be evaluated in more detail. Finafloxacin exhibited a favorable safety and tolerability profile. (This study has been registered at ClinicalTrials.gov under registration no. NCT01910883.)
KEYWORDS: antimicrobial agents, clinical trials, finafloxacin, pharmacology
INTRODUCTION
Fluoroquinolones are antibacterial drugs which are widely used for the treatment of bacterial infections, such as complicated urinary tract infections (1–3); respiratory tract infections, including tuberculosis (4, 5); and numerous other infectious diseases. Albeit expanded-spectrum fluoroquinolones commonly exhibit high levels of efficacy and acceptable safety and tolerability profiles, the increasing rates of resistance development and the lack of activity under pathophysiologically relevant conditions, such as acidic pH, limit their use. Finafloxacin is a novel fluoroquinolone characterized by physiochemical properties which improve its efficacy over that of other fluoroquinolones at a slightly acidic pH, such as in urine and infected skin (6). This might be a key advantage over other fluoroquinolones (7). Its microbiological profile is as broad as that of other expanded-spectrum fluoroquinolones, such as moxifloxacin (8). The safety, tolerability, and pharmacokinetics of finafloxacin have so far been reported from a phase I clinical trial in which volunteers received oral doses (9). Finafloxacin was well tolerated as single or multiple oral doses of up to 800 mg. The pharmacokinetics of orally administered finafloxacin in humans showed rapid absorption, very minor accumulation with once-daily (q.d.) administration, and no distinct deviation from dose linearity under multiple-dose conditions for both cumulative and peak systemic exposure to the drug (9). An additional phase I clinical trial (ClinicalTrials.gov registration no. NCT01904162; data on file) was conducted to determine the effect of age and gender on the pharmacokinetics and tolerability of a single oral dose of 400 mg finafloxacin in healthy volunteers, indicating that finafloxacin is safe and well tolerated by individuals of different genders and ages. Proof-of-concept studies are ongoing. The objective of the present study was to evaluate the pharmacokinetics, safety, and tolerability of finafloxacin in healthy volunteers following intravenous administration.
(Part of the research reported in this paper was presented in a poster session at the 52nd Interscience Conference on Antimicrobial Agents and Chemotherapy, Washington, DC, September 2012 [10].)
RESULTS
Changes to trial design.
The individual predicted exposures and the variability between individual subjects were higher than expected in the first group receiving multiple ascending doses (group 3). Therefore, treatment with a dose level of 600 mg was repeated in group 4 and the dose in group 5 was reduced to 800 mg per day. An additional group receiving 1,000 mg per day (group 6) was subsequently evaluated to cover the entire dose range.
Baseline characteristics.
Fifty-eight subjects (18 subjects in part A, 40 subjects in part B) entered the study, and 56 subjects completed the study in accordance with the study protocol. The mean age and body mass index (BMI) were similar in parts A and B, with the mean age in the 1,000-mg group in part B being slightly higher (Table 1).
TABLE 1.
Demographic data for volunteers in all treatment groupsa
| Characteristic | Values |
||||
|---|---|---|---|---|---|
| Part A (single dose) |
Part B (multiple doses) |
||||
| Group 1 (n = 9) | Group 2 (n = 9) | Groups 3 and 4 (n = 16) | Group 5 (n = 8) | Group 6 (n = 8) | |
| Dose(s) (mg) | 200, 400, 600 | 800, 1,000 | 600 | 800 | 1,000 |
| Mean (SD) age (yr) | 38 (8.2) | 41 (7.5) | 37 (12.0) | 33 (12.8) | 53 (5.9) |
| Mean (SD) body wt (kg) | 79.7 (2.73) | 70.9 (14.5) | 79.4 (13.0) | 77.7 (9.11) | 72.4 (12.1) |
| Mean (SD) ht (cm) | 176 (2.8) | 167 (11.2) | 176 (9.5) | 179 (6.0) | 168 (13.8) |
| Mean (SD) BMI (kg/m2) | 25.7 (1.33) | 25.2 (2.50) | 25.7 (3.08) | 24.2 (2.15) | 25.7 (2.59) |
| Gender (no. of males/no. of females) | 9/0 | 5/4 | 15/1 | 8/0 | 5/3 |
Demographic data from n subjects are shown stratified by treatment group. Data for volunteers in groups 3 and 4 are summarized together since both groups received the same dose of finafloxacin.
Plasma pharmacokinetics.
Following single administrations of finafloxacin, the maximum concentration (Cmax) in plasma was attained at the end of the infusion for all subjects and all doses. Plasma concentrations appeared to decline in a multiphasic manner, with the apparent terminal elimination phase starting at 12 to 16 h after the start of the infusion (Fig. 1). For individual subjects, the terminal elimination half-life (t1/2) ranged from 5.55 to 23.9 h and tended to increase with an increase in the dose (Fig. 2). An exception was the long t1/2 of 45.9 h observed with a dose of 1,000 mg in one subject receiving multiple doses. The area under the concentration-time curve (AUC) from the start of infusion extrapolated to infinity (AUCinf), the area under the concentration-time curve from the start of infusion to the last quantifiable sampling point (AUC0–tlast), and Cmax appeared to increase slightly supraproportionally with an increase in the dose, with point estimates of the slopes being 1.16 (95% confidence interval [CI], 1.03, 1.29), 1.15 (95% CI, 1.03, 1.28), and 1.23 (95% CI, 1.11, 1.36), respectively. Clearances (CL) tended to decrease with an increase in the dose (mean CL, 488 to 375 ml/min), while no consistent relationship between the volume of distribution at steady state (Vss) and dose could be identified. The between-subject variability for AUCinf, AUC0–tlast, and Cmax was low at doses of 200 to 600 mg (geometric mean CV, 10% to 18%) and moderate to high at doses of 800 and 1,000 mg (geometric mean CV, 28% to 38%). Variability was the highest for t1/2 and the volumes of distribution at higher doses (Tables 2 and 3).
FIG 1.

Geometric mean plasma concentrations of finafloxacin following single and multiple intravenous doses. Geometric mean and standard deviation finafloxacin plasma concentrations on the last day of treatment following single (part A, left) or multiple (part B, right) 1-h intravenous infusions are shown on a semilogarithmic scale for all dose levels. Time is relative to the start of infusion. The dashed lines indicate the limit of quantification.
FIG 2.
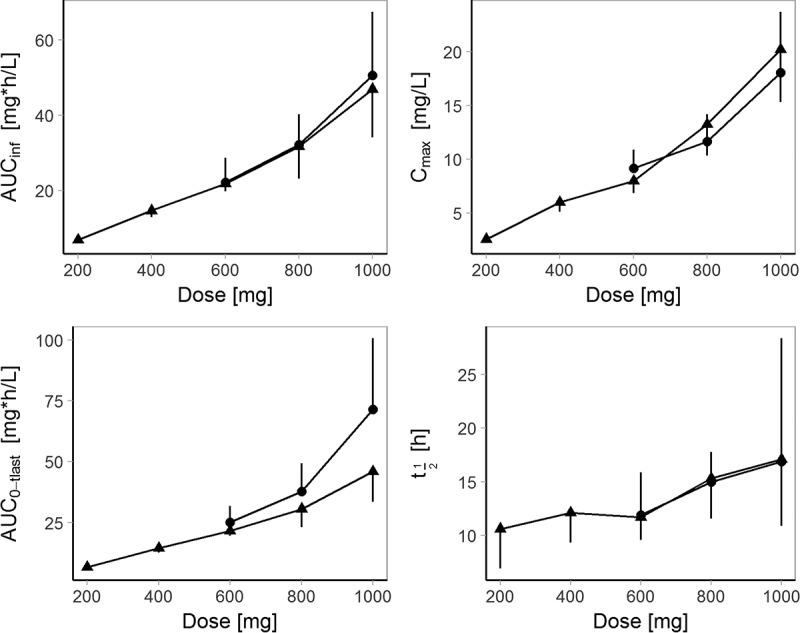
Relationship between dose and pharmacokinetic parameters of finafloxacin following single and multiple intravenous doses. Geometric means and standard deviations of AUCinf (top left; day 1 for single and multiple doses), AUC0–tlast (bottom left; 0 to 24 h postdosing on day 1 for single doses and 0 to 72 h postdosing on day 7 for multiple doses), Cmax (top right; day 1 for single doses and day 7 for multiple doses), and t1/2 (bottom right; day 1 for single doses and day 7 for multiple doses) following single (▲) and multiple (•) 1 h intravenous infusions of 200 to 1,000 mg finafloxacin. A deviation from dose linearity is apparent at higher doses. Bars indicating standard deviations point upwards for multiple administrations and downwards for single administrations.
TABLE 2.
Pharmacokinetic parameters of finafloxacin following a single intravenous dosea
| Treatment group | Dose (mg) | AUC0–tlast (mg · h/liter) | AUCinf (mg · h/liter) | Cmax (mg/liter) | t1/2 (h) | CL (ml/min) | Vz (liters) | Vss (liter) |
|---|---|---|---|---|---|---|---|---|
| Group 1 | 200 (n = 6) | 6.73 (10.2) | 6.85 (10.2) | 2.56 (12.8) | 10.6 (54.0) | 486 (10.2) | 446 (53.7) | 127 (24.2) |
| 400 (n = 6) | 14.5 (14.4) | 14.7 (14.0) | 6.0 (17.6) | 12.1 (30.0) | 453 (13.2) | 473 (38.3) | 102 (24.4) | |
| 600 (n = 5) | 21.6 (10.4) | 21.8 (14.0) | 8.0 (16.7) | 11.7 (22.2) | 459 (10.1) | 465 (27.9) | 108 (25.2) | |
| Group 2 | 800 (n = 6) | 30.7 (32.7) | 31.7 (36.7) | 13.3 (27.9) | 15.3 (32.4) | 415 (36.2) | 550 (30.2) | 106 (45.0) |
| 1,000 (n = 6) | 45.9 (36.9) | 46.9 (37.7) | 20.2 (32.0) | 17.1 (57.4) | 355 (37.7) | 525 (60.9) | 90.2 (56.0) |
Data are presented as geometric means (coefficients of variation [in percent]) for the area under the concentration-time curve from the start of infusion to the last quantifiable sampling point (AUC0–tlast), the area under the concentration-time curve from the start of infusion extrapolated to infinity (AUCinf), the maximum concentration (Cmax) attained, the terminal elimination half-life (t1/2), elimination clearance (CL), the volume of distribution at pseudoequilibrium (Vz), and the volume of distribution at steady state (Vss) for n subjects receiving 1-h single intravenous infusions of finafloxacin (part A).
TABLE 3.
Pharmacokinetic parameters of finafloxacin following multiple intravenous dosesa
| Treatment group(s) | Day | Dose (mg) | AUC0–tau (mg · h/liter) | Cmax (mg/liter) | t1/2 (h) | CL (ml/min) | Vz (liter) | RAobs |
|---|---|---|---|---|---|---|---|---|
| Groups 3 and 4 | 1 | 600 (n = 16) | 21.80 (28.0) | 8.84 (20.9) | ||||
| 7 | 24.3 (26.1) | 9.15 (18.9) | 11.9 (33.3) | 410 (26.9) | 423 (54.3) | 1.11 (6.59) | ||
| Group 5 | 1 | 800 (n = 8) | 32.0 (22.9) | 11.5 (15.5) | ||||
| 7 | 35.4 (29.7) | 37.8 (30.6) | 15.0 (18.4) | 376 (29.7) | 490 (35.6) | 1.11 (11.4) | ||
| Group 6 | 1 | 1,000 (n = 8) | 49.8 (33.4) | 17.1 (23.6) | ||||
| 7 | 64.1 (38.5) | 18.1 (31.2) | 16.9 (68.0) | 260 (38.5) | 381 (74.8) | 1.29 (16.1) |
Data are presented as geometric means (coefficients of variation [in percent]) for the area under the concentration-time curve during one dosing interval (AUC0–tau), the maximum concentration (Cmax) attained, the terminal elimination half-life (t1/2), elimination clearance (CL), volume of distribution at pseudoequilibrium (Vz), and observed accumulation ratios (RAobs) for n subjects receiving multiple 1 h intravenous infusions of finafloxacin (part B).
Urine pharmacokinetics.
Following single administrations of finafloxacin, the mean fraction renally excreted unchanged ranged from 30% to 44% (lowest and highest mean fractions across dose levels) and tended to decrease with an increase in the dose (slope, 0.77; 95% CI, 0.67, 0.86). During the first 12 h after the start of the infusion, a major fraction (76 to 94% for single infusions, 77% to 88% for multiple infusions) of the renally eliminated finafloxacin was excreted. Individual renal clearances (CLR) ranged from 170 to 256 ml/min at a dose of 200 mg to 67.1 to 181 ml/min at a dose of 1,000 mg (Fig. 3). Further information is presented in Tables 4 and 5.
FIG 3.

Cumulative urinary excretion of finafloxacin following a single intravenous dose. Geometric means and standard deviations of the cumulative fractions of finafloxacin doses excreted in urine (fe) on the last day of treatment following single (part A; left) and multiple (part B; right) 1-h intravenous administrations are shown. Time is relative to the start of the infusion.
TABLE 4.
Urinary excretion of finafloxacin following a single intravenous dosea
| Treatment group | Dose (mg) | Ae0–48 (mg) | fe0–48 (%) | CLR 0–48 (ml/min) |
|---|---|---|---|---|
| Group 1 | 200 (n = 6) | 88.3 (15.5) | 44.2 (15.5) | 217 (19.0) |
| 400 (n = 6) | 150 (18.6) | 37.6 (18.6) | 173 (24.2) | |
| 600 (n = 5) | 210 (12.3) | 34.9 (12.3) | 162 (16.4) | |
| Group 2 | 800 (n = 6) | 239 (24.1) | 29.9 (24.1) | 130 (38.1) |
| 1,000 (n = 6) | 317 (17.5) | 31.7 (17.5) | 115 (39.9) |
Data are presented as geometric means (coefficients of variation [in percent]) of the amount of drug excreted in urine from 0 to 48 h after the start of the infusion (Ae0–48), the fraction of the dose excreted in urine from 0 to 48 h after the start of the infusion (fe0–48), and renal clearance from 0 to 48 h after the start of the infusion (CLR 0–48) in n subjects receiving single 1-h intravenous doses of finafloxacin (part A).
TABLE 5.
Urinary excretion of finafloxacin following multiple intravenous dosesa
| Treatment group(s) | Day | Dose (mg) | Ae0–48 (mg) | fe0–48 (%) | CLR 0–48 (ml/min) |
|---|---|---|---|---|---|
| Groups 3 and 4 | 1 | 600 (n = 16) | 209 (21.2) | 34.8 (21.2) | 160 (30.0) |
| 7 | 210 (22.7) | 35.0 (22.7) | 147 (30.0) | ||
| Group 5 | 1 | 800 (n = 8) | 249 (23.1) | 31.1 (23.1) | 130 (42.0) |
| 7 | 254 (28.5) | 31.8 (28.5) | 124 (38.7) | ||
| Group 6 | 1 | 1,000 (n = 8) | 270 (25.1) | 27.0 (25.1) | 90.5 (28.8) |
| 7 | 334 (33.5) | 33.4 (33.5) | 92.2 (21.4) |
Data are presented as geometric means (coefficients of variation [in percent]) of the amount of drug excreted in urine from 0 to 48 h after the start of the infusion (Ae0–48), the fraction of the dose excreted in urine from 0 to 48 h after the start of the infusion (fe0–48), and renal clearance from 0 to 48 h after the start of the infusion (CLR 0–48) on days 1 and 7 in n subjects receiving multiple 1-h intravenous doses of finafloxacin (part B).
Safety and tolerability.
In the single- and multiple-dose parts of the study, 11 and 74 treatment-emergent adverse events, respectively, were reported by 10 and 26 subjects, respectively; with 6 and 45 of these adverse events having suspected relationships to the investigated drug in the single- and multiple-dose parts of the study, respectively (Table 6). No serious adverse events were reported. The majority of the adverse events reported were mild in severity and resolved without treatment. The most frequently observed adverse events which were suspected to be drug related were disorders of the gastrointestinal system (abdominal discomfort, abdominal pain, and diarrhea) or the nervous system (headache) (Table 7). Pain in an extremity or chest discomfort was the only other drug-related adverse event occurring in more than one volunteer. In the clinical laboratory evaluations, no findings were of clinical significance at any dose level and no treatment- or dose-related effects could be observed. There was especially no evidence for any disturbance in liver function parameters. Electrocardiograms did not indicate prolongations of QTc intervals in part B, and no significant changes in the findings of physical examinations were observed over the study course.
TABLE 6.
Numbers and types of adverse events in subjects receiving single or multiple doses of finafloxacin or placeboa
| Study part and dose (mg) | NS | NS with AE | No. of AE | No. of AE related to study drug | Type of AE |
|||
|---|---|---|---|---|---|---|---|---|
| GI | RTI | NS | Other | |||||
| Part A | ||||||||
| 200 | 6 | 1 | 1 | 1 | 1 | 0 | 0 | 0 |
| 400 | 6 | 2 | 2 | 2 | 2 | 0 | 0 | 0 |
| 600 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 800 | 6 | 2 | 2 | 0 | 0 | 0 | 0 | 2 |
| 1,000 | 6 | 2 | 2 | 1 | 0 | 1 | 0 | 1 |
| Placebo | 15 | 3 | 4 | 2 | 1 | 1 | 0 | 2 |
| Part B | ||||||||
| 600 | 16 | 9 | 17 | 9 | 2 | 0 | 6 | 9 |
| 800 | 8 | 6 | 20 | 13 | 4 | 1 | 5 | 10 |
| 1,000 | 8 | 6 | 19 | 10 | 5 | 2 | 2 | 10 |
| Placebo | 8 | 5 | 18 | 13 | 5 | 0 | 3 | 10 |
The numbers and types of adverse events (AE) in subjects receiving single (part A) or multiple (part B) 1-h infusions of 200 to 1,000 mg finafloxacin are indicated. NS, number of subjects; GI, gastrointestinal; RTI, respiratory tract infections; NS, nervous system; Other, other types of AEs, including local reactions to intravenous catheters and skin, musculoskeletal, vascular, metabolic, psychiatric, and respiratory disorders.
TABLE 7.
Study drug-related adverse events reported for subjects receiving single and multiple doses of finafloxacin and placeboa
| Body system or disorder | Groups 1 and 2 (single dose) |
Groups 3 to 6 (multiple dose) |
||||||
|---|---|---|---|---|---|---|---|---|
| Finafloxacin (n = 18) |
Placebo (n = 15) |
Finafloxacin (n = 32) |
Placebo (n = 8) |
|||||
| No. (%) of subjects | No. of adverse events | No. (%) of subjects | No. of adverse events | No. (%) of subjects | No. of adverse events | No. (%) of subjects | No. of adverse events | |
| CNS | 0 (0) | 0 | 0 (0) | 0 | 9 (28) | 12 | 3 (38) | 3 |
| Gastrointestinal | 3 (17) | 3 | 1 (7) | 1 | 5 (16) | 8 | 4 (50) | 5 |
| Musculoskeletal | 0 (0) | 0 | 0 (0) | 0 | 2 (6) | 4 | 2 (25) | 3 |
| General disorders | 0 (0) | 0 | 0 (0) | 0 | 4 (13) | 4 | 0 (0) | 0 |
| Infections and infestations | 0 (0) | 0 | 0 (0) | 0 | 1 (3) | 1 | 0 (0) | 0 |
| Metabolism | 0 (0) | 0 | 0 (0) | 0 | 1 (3) | 1 | 0 (0) | 0 |
| Psychiatric | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 0 | 1 (13) | 1 |
| Respiratory | 0 (0) | 0 | 0 (0) | 0 | 1 (3) | 1 | 0 (0) | 0 |
| Skin | 1 (6) | 1 | 0 (0) | 0 | 0 (0) | 0 | 1 (13) | 1 |
| Vascular | 0 (0) | 0 | 0 (0) | 0 | 1 (3) | 1 | 0 (0) | 0 |
| Renal | 0 (0) | 0 | 1 (7) | 1 | 0 (0) | 0 | 0 (0) | 0 |
Study drug-related adverse events reported for subjects receiving single and multiple doses of finafloxacin and placebo are indicated. CNS, central nervous system.
DISCUSSION
The pharmacokinetics of finafloxacin are subject to high interindividual variability, and renal excretion seems to be saturable, thereby introducing some limited nonlinearity to systemic pharmacokinetics. Finafloxacin was well tolerated by the volunteers.
The elimination of finafloxacin was found to be multiphasic, which might indicate a complex distribution pattern, and apparent terminal elimination half-lives were slightly dose dependent. Measures of exposure, such as AUC0–inf, AUC0–tlast, and Cmax, were found to increase slightly more than proportionally with an increase in the dose, which was accompanied by a dose-dependent decrease in the fraction excreted renally. Accordingly, the renal clearance decreased with an increase in the dose. When the fraction of unbound drug in plasma (approximately 75%), which is the fraction subject to glomerular filtration, is taken into account, the renal clearance of finafloxacin was generally greater than the typical glomerular filtration rate in humans (approximately 125 ml/min for a standard body surface area of 1.73 m2) (11). This suggests the active secretory transport of finafloxacin in the kidneys. While glomerular filtration is a passive process, tubular secretion involves drug transporters, which have a limited capacity and are susceptible to competition and/or inhibition by other substances (12). The observed nonlinearity suggests that transporter saturation may be approached in the presence of the finafloxacin concentrations that occur after the administration of higher doses. The remaining extrarenal clearance appears to be relatively constant across doses. The extensive distribution and binding of finafloxacin to tissues is proposed by the high steady-state volume of distribution, which was greater than the typical volume of total body water in humans (11). The estimated volume of distribution at pseudoequilibrium (Vz) was found to be approximately 5-fold higher than the volume of distribution at steady state. This reflects the fact that a large fraction of the drug is already eliminated before pseudoequilibrium is reached (13). Trough samples indicated that steady state was generally reached by day 4, with a low level of accumulation, which was only 1.1- to 1.3-fold, being observed until day 7. This is less than the theoretically expected accumulation ratios (accumulation at steady state compared to accumulation on the first day of treatment) of 1.32 to 1.80 (14), given a terminal elimination half-life of 11.8 to 20.5 h.
The pharmacokinetics following intravenous administrations are largely comparable to those after oral administrations. Reported mean AUCinf values from single oral administrations ranged from 4.08 to 29.2 mg · h/liter (for doses of 200 to 800 mg) (9), which indicates that bioavailability ranges from approximately 60% to essentially complete bioavailability. Apparent elimination half-lives ranged from about 4.6 to 10.5 h (for doses of 200 to 800 mg) for oral administration (9) and therefore tended to be shorter than those for intravenous administration. However, neither systematic relationships to dose levels nor significant differences were evident when the reported variability was considered. Slight deviations from dose proportionality have been observed and confirmed statistically for AUCinf in subjects receiving single oral doses (9). This was confirmed by our analysis, which revealed slightly more than proportional increases in AUCinf, AUC0–tlast, and Cmax for single intravenous administrations and AUC during one dosing interval (AUC0–tau) and Cmax for multiple intravenous administrations. The fraction of unchanged finafloxacin recovered in urine following administration of intravenous doses (approximately 30 to 45%) is also similar to that after oral administration (approximately 30%) (9).
In most investigated aspects, the pharmacokinetics of finafloxacin were similar to those of other fluoroquinolones. The bioavailability of finafloxacin is in the upper range of the bioavailabilities of other fluoroquinolones, which typically have bioavailabilities of about 30% (15) to nearly 100% (16). High volumes of distribution, indicating distribution to and accumulation in some tissues, have been reported for common fluoroquinolones (17, 18) and are largely similar to those of finafloxacin. Both a high volume of distribution and a high bioavailability have previously been attributed to the lipophilic cyclopropyl group at position N-1 (7), which finafloxacin and some other fluoroquinolones (ciprofloxacin, moxifloxacin) have in common. Typical terminal elimination half-lives range from about 3 h (norfloxacin, ciprofloxacin) to 8 h (levofloxacin) and 15 h (moxifloxacin) (17–19). An increased bulkiness of the substituent at C-7 (7) has especially been identified to increase elimination half-lives, which might partly explain the slow terminal elimination half-life of finafloxacin. Moxifloxacin, which has a similarly bulky substituent, has a half-life which is comparable to that of finafloxacin, while norfloxacin, which has a distinctly smaller substituent, has the shortest half-life among common fluoroquinolones. The fraction of finafloxacin excreted in urine (about 30% to 45%) is comparable to that of norfloxacin (20) and ciprofloxacin (21), slightly higher than that of moxifloxacin (19), and distinctly lower than that of ofloxacin and levofloxacin (22, 23). This indicates that the elimination of finafloxacin might be similar to that of the previously mentioned fluoroquinolones, which are eliminated by glomerular filtration (15), tubular secretion (24, 25), and metabolism. For many of these fluoroquinolones, renal clearance has been shown to exceed the glomerular filtration rate, which represents tubular secretion. The involvement of organic anion transporters (OAT) and the saturability of tubular secretion have been shown for several fluoroquinolones, including ciprofloxacin (25), norfloxacin (24), gatifloxacin (26), fleroxacin (27), and enoxacin (28). This is additionally supported by the results of in vitro and in vivo experiments, which showed that ciprofloxacin and ofloxacin are substrates for OATs and other fluoroquinolones are inhibitors of OATs (29, 30). Thus, it is likely that the renal elimination of finafloxacin at least partly depends on related transporters, which is in accordance with its pharmacokinetic characteristics described above. Both renal impairment and transporter-based drug-drug interactions may affect finafloxacin pharmacokinetics. The current data are not suitable to fully assess the role of a saturable elimination component and the role of covariates on finafloxacin pharmacokinetics. Therefore, more extensive evaluations in patients and of transporter activities will be useful.
Single and multiple intravenous doses of finafloxacin were considered to be safe and well tolerated by male and female subjects when it was administered at dose levels of 200 to 1,000 mg q.d. and 600 to 1,000 mg q.d. for 7 days, respectively. In comparison to oral administration (9), single intravenous doses of finafloxacin appeared to be related to a lower rate of adverse events, while there was no relevant difference for multiple doses. The previously reported increase in alanine aminotransferase (ALT) levels in subjects receiving multiple oral doses (9) was not confirmed for intravenous administration. Since the increases in ALT levels were observed only in healthy carriers of Helicobacter pylori, the possibility that the effect on the ALT levels was a result of the H. pylori infection cannot be excluded.
Conclusion.
Despite a moderate to high interindividual variability at higher doses, exposure following intravenous administration of finafloxacin appears to be predictable. Individual elimination processes should be evaluated in more detail. Finafloxacin exhibited favorable safety and tolerability.
MATERIALS AND METHODS
Trial design.
This was a randomized, double-blind, placebo-controlled, dose-escalating trial in healthy adult male and female volunteers receiving single or multiple intravenous administrations of finafloxacin or placebo. Single doses of 200 to 1,000 mg or multiple doses of 600 to 1,000 mg finafloxacin were administered by intravenous short-term infusions (infusion duration, 1 h). In part A of the study, subjects were assigned to one of two treatment groups (groups 1 and 2). Each subject was randomized to receive two out of three single doses of finafloxacin (group 1, 200, 400, and 600 mg; group 2, 800, 1,000, and 1,200 mg) and one dose of placebo (crossover design). The initially planned dose of 1,200 mg in group 2 was not studied on the basis of intermediate pharmacokinetic results. In part B of the study, each subject participated in a single treatment period in which a daily dose was administered for 7 days (groups 3 and 4, 600 mg; group 5, 800 mg; group 6, 1,000 mg). Each subject in part B was randomized to receive finafloxacin or placebo, with eight subjects receiving finafloxacin and two receiving placebo in each treatment period. Doses were escalated on the basis of safety, tolerability, and intermediate pharmacokinetic results, with a minimum washout period of 12 days taking place between dose escalations. The trial design was adjusted if needed. All volunteers received information on the investigated drug, the conduct of the study, and potential risks and gave written informed consent. The study was conducted in accordance with guidelines on good clinical practice, the current revision of the Declaration of Helsinki, and International Conference on Harmonization (ICH) guidelines and was carried out at the Covance Clinical Research Unit in Leeds, United Kingdom. The investigation (ClinicalTrial.gov registration no. NCT01910883) was approved by the Yorkshire, United Kingdom, Independent Research Ethics Committee.
Evaluation of volunteers.
Volunteers of both genders were screened not earlier than 28 days before the first administration of finafloxacin. The screening included evaluation of inclusion/exclusion criteria, physical and laboratory examinations, 12-channel electrocardiography (ECG), and inquiries about demographic and biometric data. From 24 h before the first administration to 48 h after the last administration, volunteers resided at the clinical research unit site. Poststudy examinations of the volunteers, including physical and laboratory examinations and a 12-channel ECG, were conducted 5 to 7 days after the administration of the last dose of finafloxacin. Female volunteers 50 years of age or younger were included only if they were surgically sterile or postmenopausal (defined as at least 2 years after the cessation of menses and/or a follicular stimulating hormone level of >40 mU/ml and a serum estradiol level of <110 pmol/liter).
Sampling scheme.
In part A, venous blood samples for the determination of finafloxacin plasma concentrations, blood chemistry, and hematology safety parameters were drawn before dosing and at 0.33, 0.67, 1, 1.17, 1.33, 1.67, 2, 3, 4, 6, 8, 12, 16, 24, 36, and 48 h after the start of the infusion. Urine was collected before dosing and in fractions of 0 to 4, 4 to 8, 8 to 12, 12 to 24, and 24 to 48 h after the start of the infusion. In part B, on day 1 blood samples were taken before dosing and at 0.33, 0.67, 1, 1.17, 1.33, 1.67, 2, 3, 4, 6, 8, 12, and 16 h after the start of the infusion. On days 2, 4, and 6, only predosing samples were taken, and on day 7, samples were taken before dosing and at 0.33, 0.67, 1.0, 1.17, 1.33, 1.67, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, and 72 h after the start of the infusion. For part B, urine was collected on day 1 before dosing (spot sample) and in fractions of 0 to 12 and 12 to 24 h after the start of the infusion, on days 2 to 6 in fractions of 0 to 24 h after the start of the infusion, and on day 7 in fractions of 0 to 12, 12 to 24, 24 to 48, and 48 to 72 h after the start of the infusion.
Bioanalytical quantification.
A liquid chromatography/tandem mass spectrometry (LC-MS/MS) method was validated to quantify finafloxacin concentrations in plasma and urine samples. The lower limit of quantification was 5 and 100 ng/ml for plasma and urine concentration measurements, respectively (ClinicalTrials.gov registration no. NCT01910883). Inaccuracy and precision were better than 2.7% to 11.1%, respectively, for plasma samples (concentration range, 15 to 20,000 ng/ml) and 3.9% to 7.2%, respectively, for urine samples (concentration range, 300 to 90,000 ng/ml) (ClinicalTrials.gov registration no. NCT01910883).
Pharmacokinetics.
The pharmacokinetic analysis was conducted using standard noncompartmental procedures in WinNonlin (version 5.2) software (Pharsight Corporation, Mountain View, CA, USA) and SAS (version 8.2) software (SAS Institute Inc., Cary, NC, USA). Maximum concentrations (Cmax) in plasma and the times at which Cmax was reached (Tmax) were obtained directly from the concentration-time profiles. Areas under the concentration-time curves (AUC) were calculated from 0 h to 24 h after dosing (AUC0–24) and from 0 h to the last quantifiable point (AUC0–tlast) using the linear and logarithmic trapezoidal method for increasing and decreasing concentrations, respectively. Elimination rate constants were estimated by least-squares regression on the terminal portion of log-transformed concentration-time curves (λz), and the respective AUCs were extrapolated to infinity (AUCinf). Elimination clearance (CL), the volume of distribution at pseudoequilibrium during the terminal elimination phase (Vz; calculated as CL/λz), the volume of distribution at steady state (Vss), and accumulation ratios were calculated by standard approaches. The cumulative amounts of finafloxacin excreted in urine (Ae) from 0 to 48 h after dosing were calculated in part A, and Ae from 0 h to the end of the dosing interval on day 1 and from 0 to 72 h on day 7 were calculated in part B. Average renal clearances were calculated from Ae and the AUC of the respective time interval. To assess the dose proportionality of pharmacokinetic parameters, a statistical analysis using a power model and, if the assumption of dose linearity was discarded, an analysis of variance (ANOVA) of log-transformed dose-normalized parameters was conducted. Pharmacokinetic stationarity, i.e., the time dependence of the pharmacokinetic parameters, was evaluated using a mixed linear model approach in part B. Treatment, day, and their interaction were treated as fixed effects, and subjects were treated as random effects with respect to log-transformed AUCs, Cmax, and Ae. Subsequently, differences between days 1 and 7 were evaluated by calculating the respective geometric least-squares means and 90% confidence intervals.
ACKNOWLEDGMENTS
This work was supported by a grant from MerLion Pharmaceuticals GmbH, Berlin, Germany, to the Covance Clinical Research Unit, Leeds, United Kingdom.
M. Lückermann and C. Fischer were employed by or currently work for MerLion Pharmaceuticals GmbH.
REFERENCES
- 1.Kobayashi M, Shapiro DJ, Hersh AL, Sanchez GV, Hicks LA. 2016. Outpatient antibiotic prescribing practices for uncomplicated urinary tract infection in women in the United States, 2002–2011. Open Forum Infect Dis 3:ofw159. doi: 10.1093/ofid/ofw159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pärn T, Mäkelä M, Lyytikäinen O. 2016. Urinary tract infections and antimicrobial use among Finnish home care clients, April-September 2014. Am J Infect Control 44:1390–1392. doi: 10.1016/j.ajic.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Koningstein M, van der Bij AK, de Kraker MEA, Monen JC, Muilwijk J, de Greeff SC, Geerlings SE, van Hall MAL, on behalf of ISIS-AS Study Group. 2014. Recommendations for the empirical treatment of complicated urinary tract infections using surveillance data on antimicrobial resistance in the Netherlands. PLoS One 9:e86634. doi: 10.1371/journal.pone.0086634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blasi F, Tarsia P, Cosentini R, Cazzola M, Allegra L. 2003. Therapeutic potential of the new quinolones in the treatment of lower respiratory tract infections. Expert Opin Investig Drugs 12:1165–1177. doi: 10.1517/13543784.12.7.1165. [DOI] [PubMed] [Google Scholar]
- 5.Thee S, Garcia-Prats AJ, Donald PR, Hesseling AC, Schaaf HS. 2015. Fluoroquinolones for the treatment of tuberculosis in children. Tuberculosis 95:229–245. doi: 10.1016/j.tube.2015.02.037. [DOI] [PubMed] [Google Scholar]
- 6.Schneider LA, Korber A, Grabbe S, Dissemond J. 2007. Influence of pH on wound-healing: a new perspective for wound-therapy? Arch Dermatol Res 298:413–420. doi: 10.1007/s00403-006-0713-x. [DOI] [PubMed] [Google Scholar]
- 7.Bartoletti R, Cai T, Perletti G, Wagenlehner FME, Bjerklund Johansen TE. 2015. Finafloxacin for the treatment of urinary tract infections. Expert Opin Investig Drugs 24:957–963. doi: 10.1517/13543784.2015.1052401. [DOI] [PubMed] [Google Scholar]
- 8.Stubbings W, Leow P, Yong GC, Goh F, Körber-Irrgang B, Kresken M, Endermann R, Labischinski H. 2011. In vitro spectrum of activity of finafloxacin, a novel, pH-activated fluoroquinolone, under standard and acidic conditions. Antimicrob Agents Chemother 55:4394–4397. doi: 10.1128/AAC.00833-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel H, Andresen A, Vente A, Heilmann H-D, Stubbings W, Seiberling M, Lopez-Lazaro L, Pokorny R, Labischinski H. 2011. Human pharmacokinetics and safety profile of finafloxacin, a new fluoroquinolone antibiotic, in healthy volunteers. Antimicrob Agents Chemother 55:4386–4393. doi: 10.1128/AAC.00832-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lückermann M, Patel H, Mooney L, Wohlert S-E, Fischer C, Bentley C, Vente A. 2012. A phase I study to determine safety, tolerability and pharmacokinetics (PK) of intravenous doses of finafloxacin HCl in healthy subjects, poster A-1960. Abstr 52nd Intersci Conf Antimicrob Agents Chemother, American Society for Microbiology, Washington, DC. [Google Scholar]
- 11.Davies B, Morris T. 1993. Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. doi: 10.1023/A:1018943613122. [DOI] [PubMed] [Google Scholar]
- 12.Masereeuw R, Russel FGM. 2001. Mechanisms and clinical implications of renal drug excretion. Drug Metab Rev 33:299–351. doi: 10.1081/DMR-120000654. [DOI] [PubMed] [Google Scholar]
- 13.Toutain PL, Bousquet-Melou A. 2004. Volumes of distribution. J Vet Pharmacol Ther 27:441–453. doi: 10.1111/j.1365-2885.2004.00602.x. [DOI] [PubMed] [Google Scholar]
- 14.Gabrielsson J, Weiner D. 2000. Multiple dosing, p 46–48. In Pharmacokinetic and pharmacodynamic data analysis: concepts and applications, 4th ed. Swedish Pharmaceutical Press, Stockholm, Sweden. [Google Scholar]
- 15.Stein GE. 1987. Review of the bioavailability and pharmacokinetics of oral norfloxacin. Am J Med 82:18–21. [DOI] [PubMed] [Google Scholar]
- 16.Fish DN, Chow AT. 1997. The clinical pharmacokinetics of levofloxacin. Clin Pharmacokinet 32:101–119. doi: 10.2165/00003088-199732020-00002. [DOI] [PubMed] [Google Scholar]
- 17.Rodvold KA, Neuhauser M. 2001. Pharmacokinetics and pharmacodynamics of fluoroquinolones. Pharmacotherapy 21:233S–252S. doi: 10.1592/phco.21.16.233S.33992. [DOI] [PubMed] [Google Scholar]
- 18.Pickerill KE, Paladino JA, Schentag JJ. 2000. Comparison of the fluoroquinolones based on pharmacokinetic and pharmacodynamic parameters. Pharmacotherapy 20:417–428. doi: 10.1592/phco.20.5.417.35062. [DOI] [PubMed] [Google Scholar]
- 19.Stass H, Kubitza D. 1999. Pharmacokinetics and elimination of moxifloxacin after oral and intravenous administration in man. J Antimicrob Chemother 43:83–90. [DOI] [PubMed] [Google Scholar]
- 20.Swanson BN, Boppana VK, Vlasses PH, Rotmensch HH, Ferguson RK. 1983. Norfloxacin disposition after sequentially increasing oral doses. Antimicrob Agents Chemother 23:284–288. doi: 10.1128/AAC.23.2.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Höffken G, Lode H, Prinzing C, Borner K, Koeppe P. 1985. Pharmacokinetics of ciprofloxacin after oral and parenteral administration. Antimicrob Agents Chemother 27:375–379. doi: 10.1128/AAC.27.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norrby SR. 1999. Levofloxacin. Expert Opin Pharmacother 1:109–119. doi: 10.1517/14656566.1.1.109. [DOI] [PubMed] [Google Scholar]
- 23.Farinotti R, Trouvin JH, Bocquet V, Vermerie N, Carbon C. 1988. Pharmacokinetics of ofloxacin after single and multiple intravenous infusions in healthy subjects. Antimicrob Agents Chemother 32:1590–1592. doi: 10.1128/AAC.32.10.1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shimada J, Yamaji T, Ueda Y, Uchida H, Kusajima H, Irikura T. 1983. Mechanism of renal excretion of AM-715, a new quinolonecarboxylic acid derivative, in rabbits, dogs, and humans. Antimicrob Agents Chemother 23:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landersdorfer CB, Kirkpatrick CMJ, Kinzig M, Bulitta JB, Holzgrabe U, Jaehde U, Reiter A, Naber KG, Rodamer M, Sörgel F. 2010. Competitive inhibition of renal tubular secretion of ciprofloxacin and metabolite by probenecid. Br J Clin Pharmacol 69:167–178. doi: 10.1111/j.1365-2125.2009.03564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakashima M, Uematsu T, Kosuge K, Kusajima H, Ooie T, Masuda Y, Ishida R, Uchida H. 1995. Single- and multiple-dose pharmacokinetics of AM-1155, a new 6-fluoro-8-methoxy quinolone, in humans. Antimicrob Agents Chemother 39:2635–2640. doi: 10.1128/AAC.39.12.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiba K, Saito A, Shimada J, Hori S, Kaji M, Miyahara T, Kusajima H, Kaneko S, Saito S, Ooie T. 1990. Renal handling of fleroxacin in rabbits, dogs, and humans. Antimicrob Agents Chemother 34:58–64. doi: 10.1128/AAC.34.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wijnands WJ, Vree TB, Baars AM, van Herwaarden CL. 1988. Pharmacokinetics of enoxacin and its penetration into bronchial secretions and lung tissue. J Antimicrob Chemother 21(Suppl B):67–77. [DOI] [PubMed] [Google Scholar]
- 29.Burckhardt G, Burckhardt BC. 2011. In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy, p 29–104. Springer, Berlin, Germany. [DOI] [PubMed] [Google Scholar]
- 30.Vanwert AL, Srimaroeng C, Sweet DH. 2008. Organic anion transporter 3 (Oat3/Slc22a8) interacts with carboxyfluoroquinolones, and deletion increases systemic exposure to ciprofloxacin. Mol Pharmacol 74:122–131. doi: 10.1124/mol.107.042853. [DOI] [PMC free article] [PubMed] [Google Scholar]