

Abstract
Ferroptosis is a form of regulated necrotic cell death controlled by glutathione peroxidase 4 (GPX4). At present, mechanisms that could predict sensitivity and/or resistance and that may be exploited to modulate this form of cell death are needed. We applied two independent approaches, a genome-wide CRISPR-based genetic screen and microarray analysis of ferroptosis-resistant cell lines to uncover acyl-CoA synthetase long-chain family member 4 (Acsl4) as an essential component for ferroptosis execution. Specifically, Gpx4/Acsl4 double knockout cells presented an unprecedented resistance to ferroptosis. Mechanistically, Acsl4 enriches cellular membranes with long polyunsaturated ω6 fatty acids. Moreover, Acsl4 is preferentially expressed in a panel of basal-like breast cancer cell lines and predicts their sensitivity to ferroptosis. We further demonstrate that pharmacological targeting of Acsl4 with the antidiabetic compound class, thiazolidinediones, ameliorates tissue demise in a murine model of ferroptosis, suggesting that Acsl4 inhibition is a viable therapeutic approach to prevent ferroptosis-related diseases.
Graphical abstract
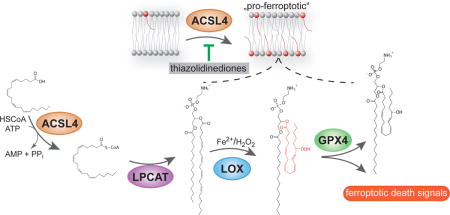
INTRODUCTION
Regulated necrotic cell death modalities are emerging as important players in (patho)physiological conditions1 Among these alternative forms of cell death particular attention has been paid to ferroptosis, a recently described form of regulated necrotic cell death implicated in tumor suppression, antiviral immunity, neurodegeneration and ischemia/reperfusion injury (IRI)2–6. Initially, ferroptosis was found to be induced by a set of small molecules identified in a screen for compounds able to selectively induce cell death in isogenic tumors carrying a mutated form of RAS7. Ferroptosis is distinct from other forms of cell death based on morphological, biochemical and genetic traits. An initial description of its molecular players has revealed that the small molecule erastin (ERA) induces ferroptosis via inhibition of system XC− 8. System XC− is the cystine/glutamate antiporter known to be essential for cells in culture by providing adequate levels of cystine (i.e. the oxidized form of cysteine) as an essential precursor for the synthesis of the tri-peptide glutathione (GSH)9. Recently, the requirement of GSH in inhibiting ferroptosis was demonstrated to be related to its specific requirement for proper functioning of glutathione peroxidase 4 (GPX4)4, a unique enzyme that prevents detrimental phospholipid oxidation10. In the study by Yang and colleagues, the first GPX4 inhibitor (1S, 3R)-RSL-3 (RSL3) was described4.
Studies on the induction of ferroptosis have provided evidence that some cell types are in fact refractory to this form of cell death11. Therefore, it is of utmost importance to identify and to understand the molecular mechanisms contributing to this resistance because this knowledge will prompt us to predict the therapeutic value of inducing ferroptosis as well as provide novel possibilities for prevention of this form of cell death in pathologically relevant scenarios such as I/R-induced tissue damage. Despite intense investigations on the molecular players involved in ferroptosis, only a few modulators have been implicated so far. Moreover, their relevance in these conditions appears to be drug- or cell type-specific, thus precluding their use as strong predictors of sensitivity in order to exploit them in the context of cytoprotective therapies8, 12–14.
Accounting for this still limited understanding of which factors determine ferroptosis resistance, we sought to identify factors that could be used first as markers of sensitivity, and second put forward as novel targets for cytoprotective strategies. Using two distinct independent approaches, we (i) generated cell lines resistant to ferroptosis and (ii) performed a genome-wide recessive genetic screen with a lentiviral CRISPR-guide RNA library. Both approaches independently identified acyl-CoA synthetase long-chain family member 4 (Acsl4) as a critical determinant of ferroptosis sensitivity. We now show that ferroptosis is specifically linked to Acsl4 and not to other Acsl family members. Lipidomic analysis demonstrates how Acsl4 shapes the lipid “make-up” required for ferroptosis execution. Intriguingly, Acsl4 also emerges to predict sensitivity in a subset of basal-like breast cancer cell lines as shown herein. Finally, we provide proof of concept that the pharmacological inhibition of Acsl4 holds great potential as a promising cytoprotective strategy aiming to preserve tissue homeostasis in an in vivo relevant ferroptotic setting.
RESULTS
Acsl4 is an essential pro-ferroptotic gene
In order to identify genes that are essential for ferroptosis induction, two approaches were employed. First, we generated cell lines intrinsically resistant to ferroptosis. Cell lines used for this study were previously described elsewhere15, where we demonstrated that these cells specifically undergo ferroptosis upon Gpx4 inactivation2. Importantly, no alternative cell death pathway is triggered when cells are treated with RSL32. The general approach to obtain the resistant cell lines is outlined in Supplementary Results, Supplementary Fig. 1a. Specifically, mouse embryonic fibroblasts carrying two floxed alleles for the ferroptosis regulator Gpx4 and stably expressing 4-hydroxytamoxifen (TAM)-inducible Cre recombinase (in the following referred to as Pfa1 cells15) were treated with a lethal concentration (100 nM) RSL3 (Supplementary Fig. 1b) for approximately 10 days. Single clones were then isolated and their specific resistance to ferroptosis was re-tested by genetic deletion of Gpx4 (Fig. 1a), thus excluding unspecific side effects due to potential drug resistance mechanisms. Subsequently, three resistant clones were selected and expression analysis was performed using Illumina chips. Fig. 1b depicts the most down-regulated genes. The full list of up- and down-regulated genes is provided in supplementary information (Supplementary Table 1).
Fig. 1. Identification of Acsl4 as an essential pre-requisite for ferroptosis execution.
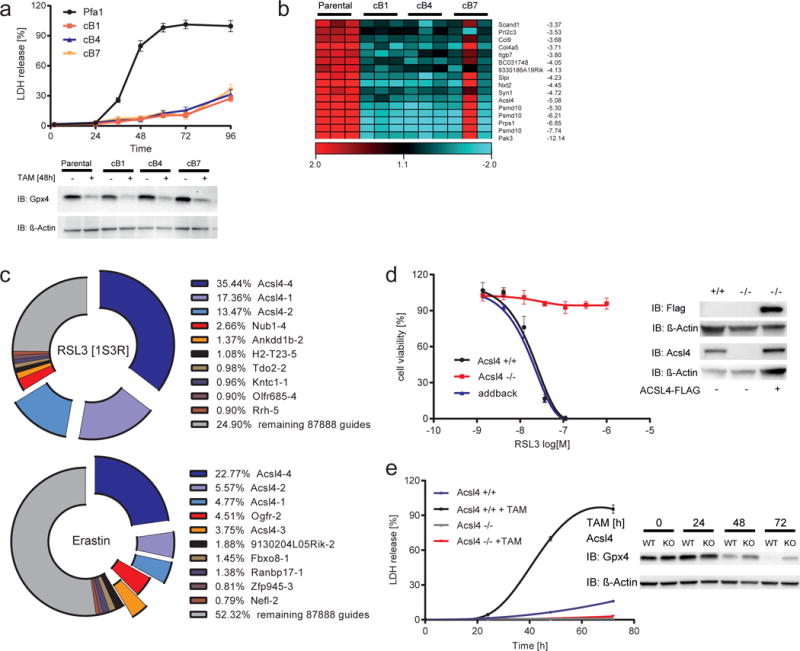
(a) Top, time-dependent increase of LDH release in Pfa1 cells and RSL3-resistant clones (cB1, cB3 and cB7) upon Gpx4 knockout induction. Below, Gpx4 immunoblot analysis 48 h after TAM treatment. (b) Heat maps of the 16 most down-regulated genes as detected by microarray analysis (Parental vs cB1/cB4/cB7 p<0.01). (c) Top 12 gRNAs identified in the population selected either with RSL3 (100 nM) or with ERA (1 μM). (d) Pfa1_Acsl4 KO (Acsl4−/−) cells are refractory to ferroptosis, which can be re-sensitized by Flag-tagged hACSL4 (ACSL4-FLAG) re-expression. Dose-dependent cytotoxicity of RSL3 was assessed 24 h after treatment using AquaBluer. Immunoblot depicts re-expression of ACSL4-FLAG in Pfa1_Acsl4 KO cells. (e) Time-dependent increase of LDH release was assayed in a 96 well plate using Pfa1_Acsl4 KO (Acsl4−/−) and Pfa1_Acsl4 WT (Acsl4 +/+) cells during KO induction. Immunoblot analysis of Gpx4 is shown at different time points after TAM treatment. Data shown represents the mean ± s.d. of n = 3 wells from a representative experiment performed independently two times (a), three times (d) or four times (e).
In parallel, we utilized a CRISPR/Cas9 screen in stably Cas9-expressing Pfa1 cells (Pfa1-CAS9) (Supplementary Fig. 1c). These cells were then transfected with a gRNA library covering the entire mouse genome (Supplementary Fig. 1d)16. We showed that Cas9 expression does not impact on ferroptosis sensitivity as this cell line efficiently underwent ferroptotic cell death induced by ERA, the Gpx4 inhibitor RSL3 and upon genetic Gpx4 deletion (Supplementary Fig. 1e,f). Prior to lentivirus-mediated transduction into the Pfa1-CAS9 cell line, the quality of the library per se was assessed by deep sequencing on an Ion torrent P1 chip, showing that 99.71 % gRNAs were found to be present in the lentiviral preparation (Supplementary Fig. 1g). Using this library, we conducted two recessive screens to identify genes that modulate susceptibility to ferroptosis induced by RSL3 and ERA. It was surprising that in both screens gRNAs targeting Acsl4 were represented by 66.38% and 36.95 % of the total sequenced guides in the RSL3- and ERA-selected cells, respectively (Fig. 1c). The list of other gRNAs identified in the screen is deposited in Supplementary Table 2. Therefore, both approaches remarkably pointed to Acsl4, thus supporting an essential role in ferroptosis execution.
Next, we validated these findings by generating an Acsl4 knockout (KO) cell line using an independent gRNA. To this end, Pfa1 cells were co-transfected with a plasmid expressing a gRNA targeting exon 1 of the Acsl4 gene and a plasmid encoding Cas9 with a nuclear localization signal. After transfection, cells were seeded at low cell densities and individual clones were allowed to grow. Individual clones were isolated, expanded and then retested for their sensitivity to RSL3. All clones resistant to RSL3 showed mutations in exon 1 of Acsl4, which corresponded to truncated versions of the protein as determined by immunoblotting against Acsl4 (Supplementary Fig. 1h,i). Tracking of insertions and deletions in the targeted Acsl4 locus of single cell clones using the tool TIDE: Tracking of Indels by DEcomposition (tide.nki.nl) confirmed out of frame mutations in all RSL3-resistant clones (Supplementary Fig. 1i). In accordance with this, Acsl4 null cells did not show any differences in GSH and Gpx4 levels and Gpx4-specific activity that could provide an indirect mechanism of resistance towards RSL3 (Supplementary Fig. 1j,k). Corroborating with the essential role of Acsl4 and unequivocally implicating the importance of Acsl4 in ferroptosis, the re-expression of human wildtype ACSL4_short (in the following referred to as ACSL4-FLAG) into the Acsl4 KO background regained full sensitivity to ferroptosis induction (Fig. 1d). Additionally, when Gpx4 was deleted in a TAM-inducible manner in the Acsl4 KO background, the double KO cells were fully viable after 72h upon TAM treatment (Fig. 1e) – this effect was not related to the kinetics of Gpx4 deletion. Moreover, the Acsl4/Gpx4 double KO cells could be passaged for a period of more than 10 days, a remarkable finding as to the best of our knowledge no single cell line, ex vivo cultured cells or explants have survived in the absence of Gpx4 so far3, 15, 17, 18
Lipid oxidation upon Gpx4 inhibition requires Acsl4
In order to understand how loss of Acsl4 provides its protective effects, we analyzed known critical events in ferroptosis execution, such as mitochondrial morphology and lipid oxidation2, 8. Electron microscopy studies of Acsl4 WT and KO cells revealed a marked increased resistance of mitochondria to RSL3-induced outer membrane rupture (Fig. 2a). Acsl4 is a member of larger family of enzymes consisting of five distinct and related isoforms, which can be further subdivided into two groups, Acsl1/Acsl5/Acsl6 and Acsl3/Acsl4, based on sequence homology19. We thus challenged the specificity of Acsl4 in ferroptosis by generating Pfa1 KO cell lines for all Acsl isoforms except Acsl6, which is not expressed in fibroblasts (Supplementary Table 3). Supplementary Fig. 2a illustrates that loss-of expression of other Acsl family members did not have any impact on ferroptosis sensitivity compared to Acsl4. A list of all single cell clones carrying well-characterized out-of-frame mutations for the different Acsl genes can be found in Supplementary Fig. 2b. Overexpression of rat Acsl1 or human ACSL3 (both tagged with GFP) failed to sensitize Pfal-CAS9_Acsl4−/− cells to undergo ferroptosis (Supplementary Fig. 2a). Corroborating the specific link to ferroptosis induction, we could further show that Acsl4 KO cells are also highly resistant to ferroptosis inducing agents other than RSL3 but not to alternative cell death inducing agents (Supplementary Fig. 2c).
Fig. 2. Acsl4 specifically contributes to ferroptotic cell death.
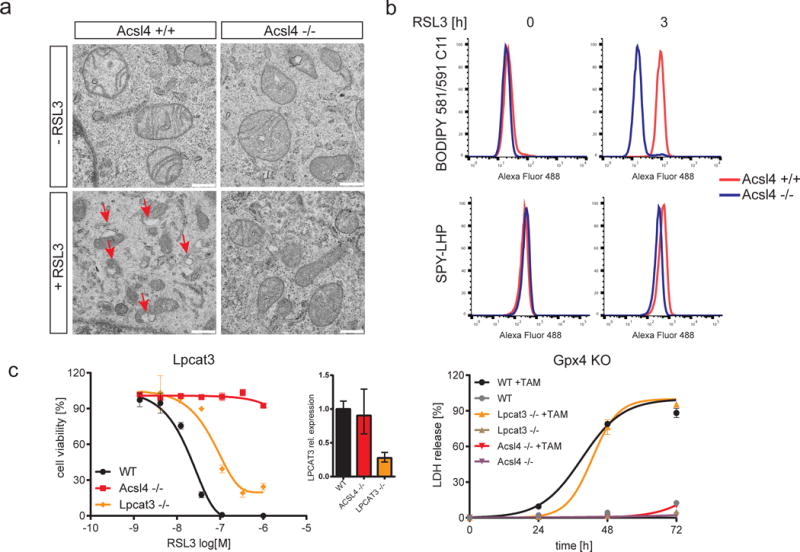
(a) Ultrastructural analysis revealed that upon RSL3 treatment Acsl4 WT cells unlike KO cells show outer membrane rupture (red arrows) as reported2 (Scale bars: 500 nm). (b) RSL3 induced lipid oxidation in WT Pfa1 cells (Acsl4 +/+) unlike in Pfa1_Acsl4 KO cells (Acsl4−/−) as determined by BODIPY 581/591 C11 and SPY-LHP oxidation at the indicated time points. (c) Knockout of Lpcat3 weakly protected against ferroptosis. Cell viability was determined 24 h after RSL3 treatment using AquaBluer. qPCR analysis indicates the relative expression of Lpcat3 in Pfa1 WT, Acsl4 −/− and Lpcat3 −/− cells. Time-dependent increase of LDH release in Pfa1_Acsl4 KO (Acsl4 −/−) and Pfa1_Acsl4 WT (Acls4 +/+) and Pfa1_Lpcat3 KO (Lpcat3 −/−) cells with or without TAM treatment. Data shown in and (c) represents the mean ± s.d. of n = 3 wells from a representative experiment performed independently three times.
Mechanistically, since lipid peroxidation is one hallmark of ferroptosis, we then estimated the levels of lipid peroxidation using first the lipid peroxidation-sensitive dye BODIPY 581/591 C11 (BODIPY)2. Although BODIPY is usually readily oxidized when ferroptosis is elicited by RSL3, in the Acsl4 KO cells no sign of probe oxidation was detectable upon RSL3 treatment (Fig. 2b). Additionally, we further validated this finding using another independent probe (Spy-LHP) that detects lipid hydroperoxides confirming the data obtained by BODIPY 581/591 C11 staining20 (Fig. 2b). As a most recent genetic screen in haploid cell pointed to lysophosphatidylcholine acyltransferase 3 (Lpcat3) as an important player for ferroptosis induction21, presumably by modulating arachidonic acid metabolism, we challenged the effect of Lpcat3 in ferroptosis induction in response to RSL3 treatment and in our genetic model of ferroptosis induction. As shown in Fig. 2c, only a mild protective effect could be observed in Lpcat3−/− cells in stark contrast to the protection offered by Acsl4. This suggests that Acsl4 has a more widespread role in ferroptosis and that the function of Lpcat3 is perhaps restricted to certain cellular subtypes.
Due to the function of Acsl4 in preferentially activating long PUFAs for phospholipid biosynthesis, we envisioned that they would be the most likely candidates to fuel the ferroptotic process. Therefore, we challenged this assumption by treating Acsl4 KO cells with a panel of fatty acids. We decided to use Acsl4 KO cells due to their inherent resistance to ferroptosis, which can be regained and modulated by supplementing with increasing concentrations of different FA. As such, this would help us to pinpoint critical fatty acids contributing to the ferroptotic process. To this end, Acsl4 KO cells were enriched with a mixture of BSA/fatty acids and, subsequently, ferroptosis was triggered using RSL3 to allow a well-defined kinetic of cell death induction. Surprisingly, ω6 fatty acids were able to further sensitize Acsl4 KO cells to undergo ferroptosis, whereas ω3 fatty acids only had a marginal or no effect (Supplementary Fig. 3a). Compared to ω3 fatty acids, the ω6 fatty acids were in general 5–10-fold more efficient in cell death induction, the only exception being docosahexaenoic acid. Moreover, this effect could be phenocopied by fatty acids containing 18 and 22 carbon long acyl chains. This is very noteworthy as the degree of unsaturation, which is directly related to the susceptibility to oxidative damage22, 23, appears to be less critical for ferroptosis induction compared to the position of the last double bond.
Insights into the underlying mechanisms for the protective effects conferred by Acsl4 came from an (oxi-)lipidomics approach using cells undergoing ferroptosis. A detailed analysis of potentially oxidized phospholipids generated during ferroptosis was performed in the accompanying manuscript by Kagan et al. This study pinpointed that arachidonic acid (AA)- and adrenic acid (AdA)-containing species in phosphatidylethanolamine (PE) represented preferred substrates for oxidation. The respective oxidized species were elevated in WT MEFs treated with RSL3 as well as in Gpx4 KO cells, in agreement with our results from fatty acid-induced sensitization of Acsl4 KO cells (Supplementary Fig. 3a).
Consistent with the key function of Acsl4 in the synthesis of long chain PUFA-CoA, particularly arachidonic acid-CoA (AA-CoA) and adrenic acid-CoA (AdA-CoA), the contents of AA-CoA and AdA-CoA were decreased approximately 4-fold (from 9.3 +/−1.2 to 2.4 +/− 0.8 pmol/μmol of phospholipids) and 16-fold (from 3.2+/−1.1 to 0.2 pmol/μmol of phospholipids) in Acsl4−/− versus control cells, respectively (accompanying manuscript Kagan et al.). The levels of free AA and AdA did not follow this pattern remaining essentially unchanged (Supplementary Fig. 3b). Supporting a function of Acsl4 to channel AA and AdA for lipid biosynthesis, we found that AA- and AdA-containing phosphatidylethanolamine (PE) species were markedly reduced in Acsl4 KO cells compared to WT cells (Fig. 3a,b). Significant reduction of the levels of AA-containing species along with a trend to a decreased level of AdA-containing species was also observed in phosphatidylinositol (PI) (Supplementary Fig. 3c). The effects were specific with regards to these anionic phospholipids as no significant decreases were revealed in phosphatidylcholine (PC) and in phosphatidylserine (PS) (Supplementary Fig. 3d,e). Finally, we compared the level of the recently described oxidized lipids formed in Acsl4 WT and KO cells treated with RSL3 (for details please see accompanying manuscript by Kagan et al.). Accordingly, genetic loss of Acsl4 suppressed the formation of doubly- and triply-oxidized AA- and AdA-containing PE species (Fig. 3c) in RSL3-treated samples. Overall, our data indicate that Acsl4 deficiency is accompanied by a significant and preferential decrease of AA- and AdA-containing PE species that are utilized as substrates to steer the ferroptotic cascade of events in the absence or inactivation of Gpx4.
Fig. 3. Acsl4 sensitizes to ferroptosis by specifically esterifying AA and AdA into PE.
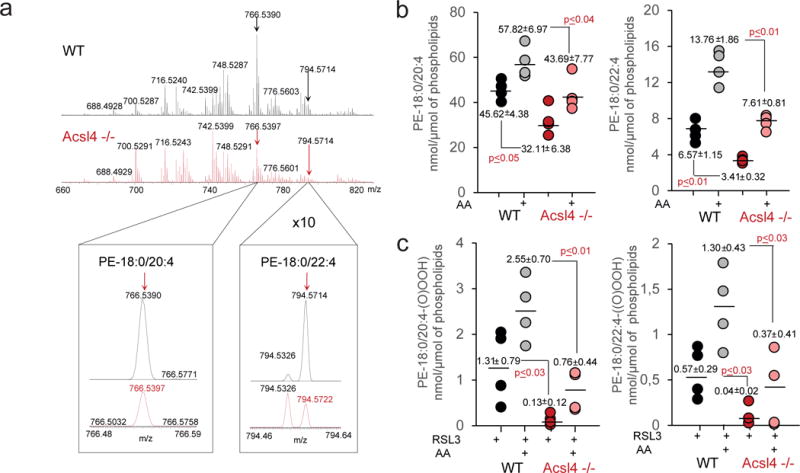
(a) Typical MS spectra of phosphatidylethanolamine (PE) from control (black) and Acsl4 −/− (red) Pfa1 cells. Inserts: MS spectra of PE molecular species (18:0/20:4) (left) and (18:0/22:4) (right) in the range of m/z 766.48–766.59 and 794.46–794.64, respectively. (b) Quantitative assessment of PE molecular species (18:0/20:4) (left panel) and (18:0/22:4) (right panel) in control WT and Acsl4 −/− cells in the absence and in the presence of AA. Cells were supplemented with arachidonic acid (AA) (2.5 μM, 16 hrs at 37°C). Data are mean ± s.d., n = 4. (c) Quantitative assessment of hydroperoxy-PE molecular species (18:0/20:4) (left panel) and (18:0/22:4) (right panel) in RSL3-exposed WT and Acsl4 −/− cells in presence and in the absence of AA. Cells were treated with AA for 16 hrs and exposed to RSL3 (100 nM) for 6 hrs at 37°C. Data represent the mean ± s.d., n = 4.
Acsl4 sensitizes breast cancer cells to ferroptosis
Recently, it was reported that a subset of breast cancer cell lines shows a marked dependence on glutamine metabolism necessary to fuel System XC− 24, thus hinting at a potential link to ferroptosis. We sought to challenge this by treating a panel of breast cancer cells with the Gpx4 inhibitor RSL3. Interestingly, the cell lines that were reported to be auxotrophic for glutamine presented a marked sensitivity to RSL3 (Fig. 4a and Supplementary Fig. 4a). Therefore, we next analyzed the expression of Acsl4 in these breast cancer cells. Intriguingly, Acsl4 was found to be preferentially expressed in a panel of basallike breast cancer cell lines (Fig. 4b), and its expression appeared to strongly correlate with the sensitivity to ferroptosis induction by RSL3. Most strikingly, only Acsl4 expression correlated to ferroptosis sensitivity as neither of the so far identified regulators of ferroptosis, namely intracellular glutathione (GSH) levels, System XC− and Gpx4 activity (Supplementary Fig. 4b) were shown to be down-regulated in the sensitive cell lines. Intriguingly, sensitive cells showed an even increased activity of System XC−, indicating a potential compensatory mechanism in susceptible cell lines. This was corroborated by the finding that TAM-inducible Gpx4−/− cells overexpressing the substrate specific subunit of System XC−, xCT (Slc7A11), are indeed protected from cell death (Supplementary Fig. 4c). Accordingly, this protection could be abolished by pharmacological inhibition of System XC− with ERA, further supporting the findings that double targeting of ferroptosis via System XC− and Gpx4 inhibition by RSL3 produced a dramatic additive effect in cells expressing ACSL4 in contrast to cells devoid of ACSL4 expression (Supplementary Fig. 4a). Interestingly, we could not detect a clear correlation between the levels of ACSL4 expression and ferroptosis sensitivity (Fig. 4a,b). This was further evidenced when we used Acsl4 KO MEFs carrying a Doxycycline (Dox)-inducible ACSL4 expression system. Fig. 4c illustrates that just minute amounts of the protein are required to re-sensitize Acsl4 KO MEFs to RSL3 induced ferroptosis and that after a certain threshold increased levels of Acsl4 do not markedly increase sensitivity, thus supporting an all or nothing kind of response of Acsl4 in ferroptosis.
Fig. 4. Acsl4 predicts sensitivity in a panel of basal-like breast cancer cell lines.
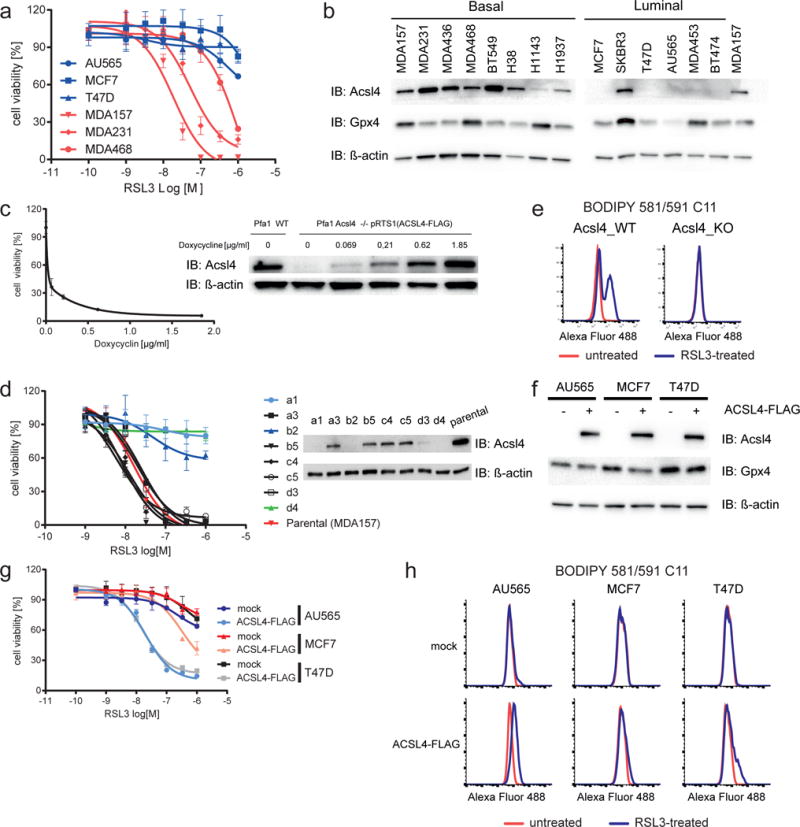
(a) Basallike breast cancer cells are susceptible to ferroptosis induction by RSL3 as determined by AquaBluer 48 h after treatment. For an extended panel of breast cancer cell lines please see Supplementary Fig. 4a. (b) Immunoblot analysis of ACSL4 and GPX4 in a panel of breast cancer cell lines. (c) Pfa1_Acsl4−/− cells stably transfected with a doxycycline-inducible ACSL4-FLAG expression vector (pRTS1(ACSL4-FLAG)) (right) demonstrate that minute amounts of ACSL4 are required for RSL3-induced ferroptosis as assessed using the AquaBluer assay (left). (d) ACSL4 KO in MDA-MB-157 confers resistance to RSL3-triggered ferroptosis. Single cell clones selected after transient transfection with Cas9 and a gRNA targeting ACSL4 were tested for their sensitivity to RSL3 and KO was confirmed by immunoblotting. (e) MDA157_Acsl4 KO cells are refractory to RSL3-triggered lipid peroxidation (100 nM) using BODIPY 581/591 C11 staining after 3h.(f) Expression of FLAG-tagged human ACSL4 (ACSL4-FLAG) in breast cancer cells devoid of endogenous ACSL4 sensitizes to RSL3-induced ferroptosis (g). (h) Cells expressing Flag_hACSL4 show increased levels of BODIPY 581/591 C11 oxidation upon treatment with RSL3 (100 nM – 4h). Data shown represents the mean ± s.d. of n ≥ 3 wells from representative experiments performed independently two times (d), three times (c, g) or four times (a).
Additionally, to further corroborate the critical role of ACSL4, we abrogated ACSL4 expression in MDA-MB-157 cells using CRISPR/Cas9-mediated KO (Fig. 4d). Analysis of single cell clones transfected with Cas9 and a gRNA targeting ACSL4 showed that in all clones resistant to RSL3 ACSL4 expression was successfully abrogated (Fig. 4d). This resistance was not related to any general resistance mechanism as the ACSL4 KO tumor cells did not show any increased resistance in response to a broad panel of cell death inducers besides ferroptosis inducing agents (Supplementary Fig. 4d). Moreover, lipid peroxidation analysis using BODIPY 581/591 C11 again showed that these cells became refractory to lipid peroxidation (Fig. 4e). Finally, overexpression of ACSL4-FLAG in a subset of breast cancer cell lines that endogenously lack ACSL4 expression (Fig. 4f) was sufficient to sensitize them to undergo cell death and lipid peroxidation induced by RSL3 (Fig. 4g,h). Importantly, the reexpression of ACSL4-FLAG specifically sensitized to RSL3-induced ferroptosis as this form of cell death was shown to be prevented by canonical ferroptosis inhibitors, such as iron chelators (Ciclopirox olamine) and Liproxstatin-1 (Supplementary Fig. 4e). Additionally, the pan-caspase apoptosis inhibitor ZVAD.fmk and the necroptosis inhibitor Nec1-s did not show any protective effect (Supplementary Fig. 4e), thus providing the rationale for eliciting ferroptosis in tumor cells expressing ACSL4.
Pharmacological Acsl4 inhibition prevents ferroptosis
We next sought to interrogate the possibility of pharmacologically blocking Acsl4 as a viable therapeutical approach in diseases with a ferroptotic signature. Early reports have shown that the thiazolidinedione (TZD) class of peroxisome proliferator-activated receptor gamma (PPARγ) agonist also selectively inhibits Acsl4 over other Acsl isoforms25. Therefore, by using distinct TZDs, namely rosiglitazone (ROSI), pioglitazone (PIO) and troglitazone (TRO), we found that all compounds prevented ferroptosis (Fig. 5a), as well as lipid peroxidation (Fig. 5b) induced by RSL3 treatment of cells. Interestingly, TRO was shown to be the most protective one despite being a less potent Acsl4 inhibitor25. This is most likely due to the chromanol ring (which provides tocopherols their antioxidant activity) and its inherent antioxidant activity (Supplementary Fig. 5a). As such it is envisioned that a combination of Acsl4 inhibitors with anti-ferroptotic compounds, such as Liproxstatin-1, would synergize. In fact, this was demonstrated in Supplementary Fig. 5b, suggesting that dual function inhibitors could be effective anti-ferroptotic agents. Moreover, since ROSI, PIO and TRO have been marketed as PPARγ agonists26, we asked whether the protective effects are at least in part related to PPARγ-mediated gene transcription. In fact, treating cells with non-thiazolinediones PPARγ agonists, such as GW1929 and Azelaoyl PAF, failed to rescue cells from RSL3-induced ferroptosis (Supplementary Fig. 5c), whereas ROSI protected against RSL3-induced ferroptosis in a manner like already observed in Acsl4 KO cells (Fig. 5a). Importantly, ROSI maintained its anti-ferroptotic effects even in Pparγ KO cells (Supplementary Fig. 5d,e).
Fig. 5. Thiazolidinediones protect from ferroptosis through inhibition of Acsl4.
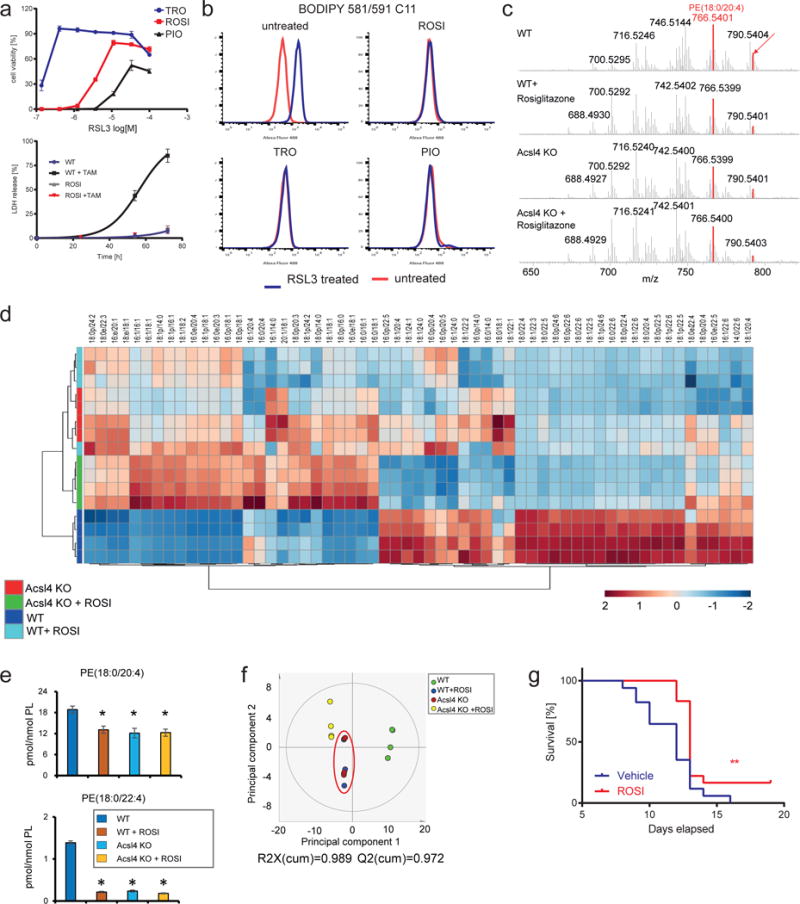
(a) Top: the thiazolidinedione, troglitazones (TRO), rosiglitazone (ROSI) and pioglitazone (PIO), protect from RSL3-induced cell death. Bottom: ROSI treatment phenocopies Acsl4 knockout measured by LDH release upon Gpx4 deletion. Data shown represents the mean ± s.d. of n = 3 wells of a 96-well plate from a representative experiment performed independently 2 times. (b) Thiazolidinediones prevent BODIPY 581/591 C11 oxidation induced by 125 nM RSL3 for 3h. (c) MS spectra of PE species at the indicated conditions. Highlighted in red are the molecular species of PE decreased by the treatment with ROSI and Acsl4 KO. (d) Heatmap of all major PE species with hierarchical clustering of the groups WT, WT+ROSI, Acsl4 KO, Acsl4 KO+ROSI. Each PE species was normalized to the corresponding mean value. (e) Effects of ROSI and Acsl4 KO on two major molecular species of PE(18:0/20:4) and PE(18:0/22:4) representing substrates for oxygenation during ferroptosis. Data represents the mean ± s.d. of n = 4 samples, *p<0.05 vs WT. (f) Principal component analysis (PCA) score plot of the first two principal components of PE between four groups. PE data were mean-centered and UV-scaled. The red circle highlights the close similarity of the effects of ROSI and Acsl4 KO on the resulting PE profiles. (g) ROSI treatment delays mortality due to acute renal failure in TAM-inducible Gpx4 knockout mice. Drinking water treatment started 1 week prior to TAM injection.
Subsequently, we expanded these findings and interrogated whether the protective effects conferred by ROSI is due to changes in the lipidome by mimicking Acsl4 deficiency. We thus analyzed all major PE species in WT and Acsl4 KO MEFs treated with ROSI (Fig. 5c,d). ROSI treatment of WT MEFs phenocopied the lipidomic changes of PE triggered by Acsl4 depletion as shown by a decrease of AA and AdA containing PE species to the levels of Acsl4 KO cells (Fig. 5e). Moreover, treatment of Acsl4 KO cell with ROSI did not lead to any further decrease in AA and AdA containing PE, thus supporting the specific activity of ROSI on Acsl4 inhibition (Fig. 5d,e). Supporting the inhibitory effect of ROSI on Acsl4, principal component analysis clustered together Acsl4 KO and ROSI treated WT cells (Fig. 5f)25.
To further evaluate if pharmacological inhibition of Acsl4 is a potential therapeutic opportunity to halt ferroptosis in vivo, we used a conditional KO model of Gpx4-deficiency, leading to acute renal failure and early death of mice approx. 10–12 days after knockout induction by TAM injection2. This mouse model represents one of few known in vivo models of ferroptosis, thus making it a suitable and reliable model to assess the in vivo efficacy of small molecules with anti-ferroptotic properties. Supporting the use of Acsl4 inhibitors to halt ferroptosis in vivo, animals treated with ROSI had a significantly prolonged overall survival compared to vehicle treated animals (Fig. 5g).
DISCUSSION
Results provided here identify and establish an essential regulatory role for Acsl4 in the ferroptotic cell death process. Ferroptosis is a form of regulated necrotic cell death that is tightly controlled by Gpx4 as demonstrated by us and others2, 4. The inactivation of Gpx4 is believed to lead to increased levels of uncontrolled lipid peroxidation culminating in cell death, in vitro and in vivo, although critical players and metabolic constraints regulating this event have remained unknown.
In two independent functional screens we now demonstrate that Acsl4 is responsible for shaping the cellular lipidome by acting as an important node that determines sensitivity versus resistance to this form of cell death. Interestingly, during finalization of this manuscript, a study using haploid screens has also pinpointed to ACSL4 as an important player for RSL3-induced cell death, although the cellular mechanism and its functional implication in ferroptosis has not been addressed21. Enzymatically, Acsl4 is responsible for the esterification of CoA to free fatty acids in an ATP dependent manner. Formation of Acyl-CoA activates the corresponding fatty acids for fatty acid oxidation or lipid biosynthesis. Specifically, Acsl4 has a marked preference for long polyunsaturated fatty acids, such as AA and AdA. Additionally, two isoforms are found in man and mouse. A short form of Acsl4 is localized to the inner side of the plasma membrane including microvilli and was also present in the cytosol. The long form has an additional N-terminal hydrophobic region that is exclusively expressed in neurons being located at the endoplasmic reticulum and on lipid droplets27.
Accordingly, experiments using exogenous supplementation of long chain fatty acids with different degrees of saturation and positions of the last double bond (i.e. ω3, ω6 ω9) have supported these findings showing that ferroptosis induction is in fact exacerbated in the presence of longer and more unsaturated fatty acids (Supplementary Fig. 3a). Interestingly, exogenous supplementation of AA/AdA (and other long PUFAs) is able to sensitize Acsl4 KO cells to ferroptosis. This is rationalized based on the kinetical characteristic of different Acsl enzymes. For instance, the only two enzymes which efficiently use AA are Acsl3 (Vmax/Kd=452*) and Acsl4 (Vmax/Kd=420*). Yet, overexpression of Acsl3 failed to sensitize cells to ferroptosis. This is most likely explained by the promiscuity of Acsl3 to other fatty acids, for instance Acsl3 also efficiently uses oleic acid, whereas Acsl4 does not (Vmax/Kd=470* and 35* respectively)28. As such, it is envisioned that at low AA/AdA and in the presence of other fatty acids Acsl4 will preferentially utilize AA/AdA for phospholipid synthesis and other fatty acids would outcompete AA/AdA usage by other Acsl. This notion is physiologically relevant as AA levels in plasma (mol% of total fatty acids) are at least one to two orders of magnitude lower than that of other fatty acids, such as oleic acid. For instance, in plasma free AA is believed to be in the low nanomolar range29. Therefore, Acsl4 appears to be a privileged enzyme utilizing arachidonic acid that can have its function being compensated in KO cells by poor AA utilizing Acsl enzymes when flooded with AA.
Particularly intriguing is the finding that up until now no other cell type such as cortical neurons15, fibroblasts15, vascular cells17, T cells and erythroid cells18, have been able to be cultured in the absence of Gpx4 under normal cell culture conditions. By stark contrast, Acsl4 and Gpx4 double KO cells were viable and proliferated normally in cell culture for an extended period of time, underscoring an important functional interplay between Gpx4 and Acsl4.
In addition to the unprecedented significance of Acsl4 in ferroptosis induction, we expanded this finding showing that Acsl4 is preferentially expressed in a subset of triple-negative breast cancer cell lines. Remarkably, Acsl4 expression clearly correlates with sensitivity to ferroptosis induction. This discovery provides intriguing consequences as some studies have already correlated Acsl4 expression levels with a more aggressive phenotype of cancer cells including increased invasiveness and proliferation30–33. The recognition that this subset of breast cancer is among the most difficult to treat34 highlights our findings that this group is in fact sensitive to ferroptosis induction, which stresses the urgency to develop in vivo efficacious ferroptosis inducing agents to specifically treat these otherwise poorly treatable tumor entities.
Moreover, data presented here show that ferroptosis can be efficiently inhibited by thiazolidinediones, a class of hypoglycemic drugs, and that this effect is mediated through inhibition of Acsl4 and not by their effect on Pparγ. Interestingly, these compounds have been already reported to ameliorate pathological conditions, where ferroptosis has been proposed to play a significant role including neurodegeneration, such as Huntington’s disease and dementia35, 36, liver necrosis37, 38 and in ischemia/reperfusion damage in tissues including brain, kidney, liver and heart39–43. This is in fact an intriguing finding as it may shed light on previous studies, where the beneficial activity of TZD could not be explained solely on their direct effects on PPARγ function. Interestingly, in all of these conditions TZD appeared to work preventively rather than therapeutically, meaning that they were efficacious when the treatment was initiated prior to the development of pathology. This is further supported by our in vivo and in vitro models experiments using ROSI showing that protection is achieved only when the treatment is applied before ferroptosis is elicited, thus allowing sufficient time for proper phospholipid remodeling.
Conclusively, we identify and characterize Acsl4-dependent modulation of phospholipids, specifically that of PE, as critical determinants of sensitivity to ferroptosis. Moreover, the expression of ACSL4 appears to be a predictive marker for ferroptosis sensitivity in different cellular contexts. Finally, the pharmacological inhibition of Acsl4 (for instance by TZD) might represent an attractive pharmacological intervention that could also explain some of the cytoprotective effects of TZD in different tissue pathologies, which certainly deserves further exploration and encourages revisiting some of the studies where TZD was demonstrated to confer beneficial effects.
ONLINE METHODS
Chemicals
All chemicals were purchased from Sigma-Aldrich unless stated otherwise. Fatty acids were purchased from Cayman Chemicals.
Cell lines
The TAM-inducible Gpx4−/− cells (Pfa1) were described previously15. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and cultured in a 37°C incubator with humidified atmosphere of 5% CO2 (Binder). Human breast cancer cell lines (MDA-MB-157, MDA-MB-231, MDA-MB-436, MDA-MB-453, MDA-MB-468, BT-549, HCC38, HCC1143, HCC1937, MCF7, T-47D, AU565, BT-474) were purchased from ATCC and cultured according to ATCC guidelines. SK-BR-3 cells were a kind gift from Dr. Gabriele Multhoff (Technical University Munich).
Assessment of lipid peroxidation using C11-BODIPY(581/591) and SPY-LHP staining and flow cytometry
150,000 cells/well were seeded in 6-well dishes (Nunc) one day prior to the experiment. On the next day, cells were treated with indicated concentrations of (1S, 3R)-RSL3 to induce ferroptosis. Cells were incubated with C11-BODIPY(581/591) (1 μM) or SPY-LHP (10 μM; Dojindo EU GmbH, Munich, Germany) for 30 min at 37°C in a tissue culture incubator before harvest by trypsinisation. Subsequently, cells were resuspended in 500 μL of fresh PBS (DPBS, Gibco) strained through a 35 μM cell strainer (Falcon tube with cell strainer CAP) and analyzed using the 488-nm laser of flow cytometer (FACS Canto II, BD Biosciences) for excitation. Data were collected from the FL1 detector (C11-BODIPY/Spy-LHP) with a 502LP and 530/30 BP filter. At least 10,000 cells were analyzed per sample. Data analysis was conducted using the FlowJo Software.
Cell viability assays
Aquabluer method
Cells were seeded onto 96-well plates (2000 cells per well) and treated with the compounds ((1S,3R)-RSL3, erastin, L-buthionine sulfoximine [BSO], dimethyl sulfoxide [DMSO], menadione, tert-butylhydroperoxide [tBOOH], carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone [FCCP], rotenone, nocodazole, sulphoraphane, etoposide, phenylarsine oxide [PAO], tunicamycin, staurosporine, irinotecan, mitoxanrone, paclitaxel, rapamycin, auranofin, vinoblastine, cytochalasin-D, rosiglitazone, pioglitazone, troglitazone, triacsin C GW1929, azelaoy PAF) after plating. Cell viability was assessed at different time points after treatment (usually 24 h, unless stated otherwise) using AquaBluer as an indicator of viable cells according to the manufacturer’s recommendations (MultiTarget Pharmaceuticals, LLC).
LDH release method
Pfa1 cells and derived cell clones were seeded onto 6-well plates (30,000 cells per well) and treated with 1 μM 4-OH-tamoxifen (TAM) after plating to induce the knockout of Gpx4. Cell death was quantified by measuring released lactate dehydrogenase (LDH) activity after different time points using the Cytotoxicity Detection Kit (LDH) according to the manufacturer’s instructions (Roche Diagnostics Deutschland).
Determination of intracellular GSH levels
Cells were seeded on a 6-well plate (1×105 cells per well) and cultured for 24 h as outlined above. Cells were washed three times with ice-cold PBS, extracted with 5% trichloroacetic acid, and then treated with ether to remove the trichloroacetic acid. Total glutathione content in the aqueous layer was measured using an enzymatic method, based on the catalytic action of glutathione in the reduction of 5,50-dithiobis (2-nitrobenzoic acid) by the glutathione reductase system44.
Thiol measurement
To assay total mercaptans released into the medium, 200,000 cells were plated onto a 6-well plate and cultured overnight. Subsequently, the medium were removed and cells washed two times with PBS and covered in serum-free and phenol redfree media. Aliquots were subsequently collected at desired time points. Total mercaptans secreted into the cell culture medium were determined as described previously45 using GSH as a standard.
Gpx4-specific activity assay
Gpx4-specific activity using phosphatidylcholine hydroperoxide as substrate was measured following the protocol previously described in46.
CRISPR/Cas9 genome-wide recessive screen
In a similar approach as used in Koike-Yusa et al., Pfa1 cells stably expressing Cas9 were transduced by a lentiviral CRISPR-guide RNA library16 pseudotyped with the ecotropic envelope. This library contained 87,897 mouse gRNAs targeting 19,150 mouse protein coding genes. Transduction efficiency was adjusted to an MOI of 0.3 to avoid multiple infections with different gRNAs. Two days after infection, cells were selected with increasing concentrations of (1S, 3R)-RSL3 (25–50 nM) or ERA (3 μM) for 11 days. Genomic DNA was extracted from selected and unselected cells pools. Sample preparation was performed with primers designed to bind to the pKLV-U6gRNA (Bbsl)-PGKpuro2ABFP library amplifying a product of 194 Bp encompassing the variable region (coding for the gRNA). By using different barcode sequences in the forward primer it was possible to combine all PCR products in a unimolecular ratio to form a next generation sequencing (NGS) library suitable for sequencing on an Ion Torrent P1 chip (PrimBio Research Institute, LLC, Exton, PA). Raw sequence results were provided as separate FASTQ files for each barcode. The necessary sgRNA guide information was extracted from single reads of the FASTQ file by counting the number of each sgRNA sequenced per sample.
Primer sequences for sample preparation:
-
-Erastin selection:
- Forward: CCATCTCATCCCTGCGTGTCTCCGACTCAGTAAGGAGAACGGCTTTATATATCTTGTGGAAAGGACG
- Reverse: CCTCTCTATGGGCAGTCGGTGATAGCACCGACTCGGTGCCACTTTTTCAA
-
-RSL3 selection:
- Forward: CCATCTCATCCCTGCGTGTCTCCGACTCAGTACCAAGATCGGCTTTATATATCTTGTGGAAAGGACG
- Reverse: CCTCTCTATGGGCAGTCGGTGATAGCACCGACTCGGTGCCACTTTTTCAA
-
-Unselected control:
- Forward: CCATCTCATCCCTGCGTGTCTCCGACTCAGCAGAAGGAACGGCTTTATATATCTTGTGGAAAGGACG
- Reverse: CCTCTCTATGGGCAGTCGGTGATAGCACCGACTCGGTGCCACTTTTTCAA
CRISPR/Cas9-mediated knockout of individual genes
Single sgRNA guides were designed targeting critical exons using the online tool http://crispr.mit.edu. Guides were cloned using annealed oligonucleotides (Invitrogen) with specific overhangs complementary to the BbsI-digested pKLV-U6gRNA(BbsI)-PGKpuro2aBFP vector.
Transient expression of the CRISPR/Cas9 system
Cells were seeded on a 6-well plate (200,000) and cultured for 24 h. On the next day, cells were co-transfected by lipofection at a 1:1 ratio mix (usually 1 μg each) of a Cas9 expressing plasmid (pCAG-Flag-Nls-Cas9-Nls47) and the desired sgRNA expressing plasmid (pKLV-U6gRNA-PGKpuro2aBFP) using the X-tremeGENE HP agent according to the manufacturer’s recommendations (Roche Diagnostics, Mannheim, Germany). After 2 days cells were plated onto a 15 cm plate at very low density (100–500 cells per 15 cm plate) and colonies were allowed to form during the course of 10 days. Single cell clones were picked and genotypes analyzed using sequence validation of the corresponding genomic locus. CRISPR/Cas9 induced deletion/insertions were detected using the TIDE: Tracking of Indels by DEcomposition (https://tide.nki.nl/) online tool48. Single clones with out-of-frame mutations were used for further analysis (Western blot/qPCR).
Stable expression of the CRISPR/Cas9 system
Pfa1 cells were infected with ecotropic lentivirus particles containing the LentiCas9-Blast construct in principle as described in49. After 48 h cells were selected with 10 μg/ml blasticidin for 5 days. Blasticidin-resistant cells were used for a second infection with ecotropic lentivirus particles containing the desired sgRNA expressing plasmid (pKLV-U6gRNA-PGKpuro2ABFP). Two days after infection, cells were sorted on a BD Bioscience FACSARIA II using Blue Fluorescent Protein (BFP) as a marker. The BFP-positive cell population was seeded onto a 15 cm dish at very low density (100–500 cells per 15 cm plate) and colonies were allowed to form in the course of 10 days. For single clone analysis see protocol above.
qPCR analysis
RNA was extracted from exponentially growing cells using the RNeasy kit Mini Kit from Qiagen according to manufacturer’s instructions. 2 μg of total RNA was put into reverse transcription reactions using random primers for amplification (iScript™ cDNA Synthesis Kit-Bio-Rad). qPCR reactions were performed on the 7900HT Fast Real-Time PCR System using gene specific TaqMan probes or specific primers in a SYBR GREEN reaction to detect expression levels. Hypoxanthine-guanine phosphoribosyltransferase (HPRT) expression was used for normalization. The following TaqMan probes were used: Acsl3: Mm01255804_m1; Lpcat3: Mm00520147_m1; Pparγ: Mm00440940_m1; Hprt: Mm00446968_m1. The following primers were used for Acsl5 (Forward: 5′-CTGATCTGCCTCCTGACGTTTGGAA-3′; Reverse: 5′-ACAACGTCTTGGCGTCTGAGAAGT-3′) and Hprt (Forward: 5′-AGCTACTGTAATGATCAGTCAACG-3′; Reverse: 5′-AGAGGTCCTTTTCACCAGCA-3′).
Immunoblotting
Western blot analysis of cell lysates was performed essentially as described previously15, by using antibodies against Gpx4 (1:1000; no. ab125066, Abcam), Acsl1 (1:1000; no. 9189, Cell Signaling Technologies), Acsl4 (1:200; no. sc-271800, Santa Cruz), GFP (1:1000; no. gfp-1020, Aves) and β-actin (1:10000; no. A5441, Sigma).
Microarray analysis
300 ng of high quality total RNA were amplified using the Illumina TotalPrep RNA Amplification kit (Ambion). Amplified cRNA was hybridised to Mouse Ref-8 v2.0 Expression BeadChips (Illumina, San Diego, CA, USA). Staining and scanning were done according to the Illumina expression protocol. Data was processed using the GenomeStudioV2010.1software (gene expression module version 1.6.0) in combination with the MouseRef-8_V2_0_R3_11278551_A.bgx annotation file. The background subtraction option was used and an offset to remove remaining negative expression values was introduced. In the heat map, each column represents a value of a single experiments performed three times independently.
RNAseq analysis
Transcriptome analysis, quantitative library preparation and enrichment were performed as described in50. RNA libraries were assessed for quality and quantity with the Agilent 2100 BioAnalyzer and the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies) and sequenced as 100 bp paired-end runs on an Illumina HiSeq2500 platform.
Transmission electron microscopy of Acsl4 WT and KO cells
Cell pellets were fixed in 2.5% electron microscopy grade glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4 (Science Services, Munich, Germany), postfixed in 2% aqueous osmium tetraoxide, dehydrated in gradual ethanol (30–100%) and propylene oxide, embedded in Epon (Merck, Darmstadt, Germany) and cured for 24 h at 60°C. Semithin sections were prepared and stained with toluidine blue. Ultrathin sections (50 nm) were placed onto 200 mesh copper grids, stained with uranyl acetate and lead citrate before transmission electron microscopy analysis (Zeiss Libra 120 Plus, Carl Zeiss NTS GmbH, Oberkochen, Germany). Pictures were taken using Slow Scan CCD-camera and iTEM software (Olympus Soft Imaging Solutions, Münster, Germany).
Fatty acid-dependent sensitization of ferroptosis
Pfa1_Acsl4−/− cells were seeded onto 96-well plates (2000 cells per well). After cells adhered to the cell culture dish (approximately 6h after plating), they were treated for 16h with different concentrations of fatty acids (archidic acid, oleic acid, eicosaeonic acid, (11, 14) eicosadienoic acid, (5, 8, 11) eicosatrienoic acid, (11, 14, 17) eicosatrienoic acid, (8, 11, 14) eicosatrienoic acid, γ-linoleic acid, α-linolenic acid, linoleic acid, AA, AdA, docosahexaenoic acid, eicosapentaenoic acid) solved in 10% fatty acid-free BSA. Subsequently, cells where treated with/without 100 nM (1S, 3R)-RSL3 and viability was assessed 4h thereafter using AquaBluer as described above.
Doxycycline-inducible expression of ACSL4-FLAG
Human ACSL4-FLAG was cloned in the doxycyclin inducible expression vector pRTS151, kindly provided by Dr. G. W. Bornkamm. Acsl4 KO PFa1 cells were electroporated with the cloned construct and selected with hygromycin (600 μg/ml) for at least 3 weeks. Single cell clones were selected and the doxycycline-inducible ACSL4-FLAG expression was verified by western blot after cells were treated for 24 h with 1 μM doxycycline. In order to determine the amount of ACSL4-FLAG necessary for ferroptosis execution, 96-well plates were seeded with 2000 cells per well, treated overnight with increasing concentrations of doxycycline, and subsequently ferroptosis was induced by addition of 120 nM RSL3. On the next day, the cell viability was assessed using the Aquabluer assay. In parallel 6-well plates containing 100,000 cells were treated overnight with the same concentrations of doxycycline to determine the ACSL4 expression levels via western blot.
Measurement of antioxidant/radical trapping activity
Substances (rosiglitazone, pioglitazone, troglitazone, triacsin C, α-tocopherol, DMSO) were diluted in methanol and pipetted in a 96-well plate in triplicates. Di(phenyl)-(2,4,6-trinitrophenyl)iminoazanium (DPPH) solution (final concentration 200 μM) was added to start the reaction. After the equilibrium settled (in generally after 1h) the absorbance was measured at 520 nm using a SpectraMax M5 instrument.
Mass spectrometry analysis
Redox-lipidomics analysis was performed as previously described2 and as further detailed in the accompanying manuscript by Kagan et al.
Inducible disruption of Gpx4 in mice
The generation of mice with a TAM-inducible full body deletion of Gpx4 except brain (referred to as CreERT2;Gpx4fl/fl; fl, floxed) was detailed previously2. In brief, TAM treatment of these mice causes loss of Gpx4 expression in kidney (and other tissues), acute renal failure and death at approx. 12 days post induction. To achieve inducible disruption of the floxed Gpx4 allele(s), mice (>8 weeks of age; male and female) were injected twice with 0.5 mg 4-OH-Tamoxifen (TAM) dissolved in Miglyol.
In vivo treatment of TAM-inducible Gpx4−/− mice with rosiglitazone
Animals included in the treatment study of TAM-inducible Gpx4−/− mice were equally distributed between sex and weight, with typically 8–10 weeks of age. The average weight within the groups was between 22 and 24 g. Groups were formed to have comparable numbers of females/males of the same age. Animal weight was arranged to have a similar distribution between females and males. For the pharmacological inhibitor experiments, CreERT2;Gpx4fl/fl mice were injected on day 0 and 2 with 0.5 mg TAM dissolved in Miglyol. ROSI was administrated in the drinking water (0.0125 mg/ml) along with vehicle control (1% dimethylsulfoxide (DMSO) in water) 1 week prior to the TAM injections; drinking water was replaced every second day continually during the whole experiment. Survival analysis was performed using the GraphPad Prism software and statistical analysis was done according to the log-rank (Mantel_Cox) test. The compounds, vehicle and ROSI, were both odorless and colorless ensuring no detectable bias. Daily animal assessment was performed in a blinded fashion. When animals showed terminal signs, they were euthanized. No statistical method was used to predetermine sample size for the treatment of the Gpx4−/− mice. Mice were kept under standard conditions with food and water ad libitum (ssniff). All experiments were performed in compliance with the German Animal Welfare Law and have been approved by the institutional committee on animal experimentation and the government of Upper Bavaria.
Data presentation and statistical analyses
Data are presented as mean ± s.d. unless stated otherwise. As a general rule for the cell-based experiments, the graphs show the mean ± s.d. of n = x wells (x values are given in the figure legends) representative of a single experiment performed independently y times (y value is given in figure legends) for reproducibility. Statistical analysis was performed using GraphPad Prism 5.0 software.
Supplementary Material
Acknowledgments
We would like to thank Dr. Brent R. Stockwell for providing (1S, 3R)-RSL-3 and Dr. Fulvio Ursini and Dr. Matilde Maiorino for phosphatidylcholine hydroperoxide. We would also like to thank Dr. Anton Berns, The Netherlands Cancer Institute, Amsterdam, for providing the ROSA26-CreERT2 mouse line. This work was in part supported by grants from the Deutsche Forschungsgemeinschaft (DFG) CO 291/2-3 and CO 291/5-1 to M.C., a fellowship from the Japan Society for the Promotion of Science (JSPS) to S.K., the Human Frontier Science Program (HFSP) RGP0013 to M.C. and V.E.K., and by the m4 Award (Bavarian Ministry of Economic Affairs) to J.A.S. and M.C.
Footnotes
URLs
Competing financial interests
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
J.P.F.A., S.D. and M.C. conceived the study. M.A. and A.W carried out and analysed electron microscopy studies. Y.Y.T., G.M., F.Q., H.B. and V.E.K. performed oxi-lipidomics analysis and data interpretation. M.I., J.B., H.P. and D.T conducted microarray analysis, deep-sequencing and analysis. S.D., B.P., E.P., S.K., I.I. and J.P.F.A. performed in vitro and in vivo experiments. J.F., C.S., and W.W. provided reagents and participated in the discussion. S.D., J.A.S., J.P.F.A., and M.C. performed evaluation and interpretation of the in vitro data. Paper was written by J.P.F.A. and M.C. All authors read and agreed on the content of the paper.
References
- 1.Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedmann Angeli JP, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsushita M, et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212:555–568. doi: 10.1084/jem.20140857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang WS, et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell. 2014;156:317331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linkermann A, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111:16836–16841. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang L, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3:285–296. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 8.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishii T, Sugita Y, Bannai S. Regulation of glutathione levels in mouse spleen lymphocytes by transport of cysteine. J Cell Physiol. 1987;133:330–336. doi: 10.1002/jcp.1041330217. [DOI] [PubMed] [Google Scholar]
- 10.Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211. doi: 10.1016/0005-2760(82)90150-3. [DOI] [PubMed] [Google Scholar]
- 11.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixon SJ, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Louandre C, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732–1742. doi: 10.1002/ijc.28159. [DOI] [PubMed] [Google Scholar]
- 14.Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23:270–278. doi: 10.1038/cdd.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seiler A, et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008;8:237–248. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 16.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32:267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 17.Wortmann M, et al. Combined deficiency in glutathione peroxidase 4 and vitamin e causes multiorgan thrombus formation and early death in mice. Circ Res. 2013;113:408417. doi: 10.1161/CIRCRESAHA.113.279984. [DOI] [PubMed] [Google Scholar]
- 18.Canli O, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–148. doi: 10.1182/blood-2015-06-654194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soupene E, Fyrst H, Kuypers FA. Mammalian acyl-CoA:lysophosphatidylcholine acyltransferase enzymes. Proc Natl Acad Sci U S A. 2008;105:88–93. doi: 10.1073/pnas.0709737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamanaka K, et al. A novel fluorescent probe with high sensitivity and selective detection of lipid hydroperoxides in cells. RSC Advances. 2012;2:7894–7900. [Google Scholar]
- 21.Dixon SJ, et al. Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol. 2015;10:1604–1609. doi: 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chemical reviews. 2011;111:5944–5972. doi: 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- 23.Pratt DA, Mills JH, Porter NA. Theoretical calculations of carbon-oxygen bond dissociation enthalpies of peroxyl radicals formed in the autoxidation of lipids. J Am Chem Soc. 2003;125:5801–5810. doi: 10.1021/ja034182j. [DOI] [PubMed] [Google Scholar]
- 24.Timmerman LA, et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013;24:450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JH, Lewin TM, Coleman RA. Expression and characterization of recombinant rat Acyl-CoA synthetases 1, 4, and 5. Selective inhibition by triacsin C and thiazolidinediones. J Biol Chem. 2001;276:24667–24673. doi: 10.1074/jbc.M010793200. [DOI] [PubMed] [Google Scholar]
- 26.Gale EA. Lessons from the glitazones: a story of drug development. Lancet. 2001;357:1870–1875. doi: 10.1016/S0140-6736(00)04960-6. [DOI] [PubMed] [Google Scholar]
- 27.Kuch EM, et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta. 2014;1841:227–239. doi: 10.1016/j.bbalip.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 28.Van Horn CG, et al. Characterization of recombinant long-chain rat acyl-CoA synthetase isoforms 3 and 6: identification of a novel variant of isoform 6. Biochemistry. 2005;44:1635–1642. doi: 10.1021/bi047721l. [DOI] [PubMed] [Google Scholar]
- 29.Brash AR. Arachidonic acid as a bioactive molecule. J Clin Invest. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orlando UD, et al. Acyl-CoA synthetase-4, a new regulator of mTOR and a potential therapeutic target for enhanced estrogen receptor function in receptor-positive and -negative breast cancer. Oncotarget. 2015;6:42632–42650. doi: 10.18632/oncotarget.5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu X, et al. ACSL4 promotes prostate cancer growth, invasion and hormonal resistance. Oncotarget. 2015;6:44849–44863. doi: 10.18632/oncotarget.6438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X, et al. Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS One. 2013;8:e77060. doi: 10.1371/journal.pone.0077060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monaco ME, et al. Expression of Long-chain Fatty Acyl-CoA Synthetase 4 in Breast and Prostate Cancers Is Associated with Sex Steroid Hormone Receptor Negativity. Translational oncology. 2010;3:91–98. doi: 10.1593/tlo.09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist. 2011;16(Suppl 1):1–11. doi: 10.1634/theoncologist.2011-S1-01. [DOI] [PubMed] [Google Scholar]
- 35.Jin J, et al. Neuroprotective effects of PPAR-gamma agonist rosiglitazone in N171-82Q mouse model of Huntington’s disease. J Neurochem. 2013;125:410–419. doi: 10.1111/jnc.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heneka MT, Fink A, Doblhammer G. Effect of pioglitazone medication on the incidence of dementia. Ann Neurol. 2015;78:284–294. doi: 10.1002/ana.24439. [DOI] [PubMed] [Google Scholar]
- 37.Belfort R, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 38.Aithal GP, et al. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008;135:1176–1184. doi: 10.1053/j.gastro.2008.06.047. [DOI] [PubMed] [Google Scholar]
- 39.Han L, et al. Rosiglitazone Promotes White Matter Integrity and Long-Term Functional Recovery After Focal Cerebral Ischemia. Stroke. 2015;46:2628–2636. doi: 10.1161/STROKEAHA.115.010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Culman J, et al. Treatment of rats with pioglitazone in the reperfusion phase of focal cerebral ischemia: a preclinical stroke trial. Exp Neurol. 2012;238:243–253. doi: 10.1016/j.expneurol.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Rennings AJ, et al. Rosiglitazone reduces ischaemia-reperfusion injury in patients with the metabolic syndrome. Eur Heart J. 2010;31:983. doi: 10.1093/eurheartj/ehp562. [DOI] [PubMed] [Google Scholar]
- 42.Wu JS, et al. Ligand-activated peroxisome proliferator-activated receptor-gamma protects against ischemic cerebral infarction and neuronal apoptosis by 14-3-3 epsilon upregulation. Circulation. 2009;119:1124–1134. doi: 10.1161/CIRCULATIONAHA.108.812537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuboki S, et al. Peroxisome proliferator-activated receptor-gamma protects against hepatic ischemia/reperfusion injury in mice. Hepatology. 2008;47:215–224. doi: 10.1002/hep.21963. [DOI] [PubMed] [Google Scholar]
- 44.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 45.Bannai S, Ishii T. Transport of cystine and cysteine and cell growth in cultured human diploid fibroblasts: effect of glutamate and homocysteate. J Cell Physiol. 1982;112:265–272. doi: 10.1002/jcp.1041120216. [DOI] [PubMed] [Google Scholar]
- 46.Roveri A, Maiorino M, Ursini F. Enzymatic and immunological measurements of soluble and membrane-bound phospholipid-hydroperoxide glutathione peroxidase. Methods Enzymol. 1994;233:202–212. doi: 10.1016/s0076-6879(94)33023-9. [DOI] [PubMed] [Google Scholar]
- 47.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mannes AM, Seiler A, Bosello V, Maiorino M, Conrad M. Cysteine mutant of mammalian GPx4 rescues cell death induced by disruption of the wild-type selenoenzyme. FASEB J. 2011;25:2135–2144. doi: 10.1096/fj.10-177147. [DOI] [PubMed] [Google Scholar]
- 50.Haack TB, et al. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2013;93:211–223. doi: 10.1016/j.ajhg.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bornkamm GW, et al. Stringent doxycycline-dependent control of gene activities using an episomal one-vector system. Nucleic Acids Res. 2005;33:e137. doi: 10.1093/nar/gni137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.