

Abstract
Background
In Japan, selegiline has been approved for combination therapy with levodopa for Parkinson disease (PD). We conducted a trial of selegiline monotherapy for early PD.
Methods
In this 12-week controlled phase III trial, a total of 292 subjects were randomized to receive placebo (n = 146) (full analysis set 140) or selegiline (n = 146) (full analysis set 139). The primary outcome measure was the change in the Unified Parkinson Disease Rating Scale part I + II + III total score from baseline to the final visit. Other secondary measures and a safety profile were evaluated.
Results
Selegiline monotherapy reduced the primary outcome measure by −6.26 ± 7.86 compared with the placebo −3.14 ± 6.98 (mean ± SD, P = 0.0005 by analysis of covariance). There was no significant difference in the number of adverse events between the 2 groups (P > 0.05).
Conclusions
Selegiline monotherapy reduced the total Unified Parkinson Disease Rating Scale part I + II + III score and was well tolerated in Japanese patients with early PD.
Key Words: Parkinson disease, treatment, selegiline, monotherapy, UPDRS
Parkinson disease (PD) is one of the most common neurodegenerative disorders. In early de novo PD patients, the use of levodopa, dopamine agonists, and/or monoamine oxidase type B inhibitors (selegiline or rasagiline) is recommended.1–3 In our previous double-blind study in Japan regarding selegiline for PD, a significant improvement was obtained only for the combined use of selegiline and levodopa. Therefore, we conducted a phase III trial with selegiline monotherapy for de novo PD patients.
MATERIALS AND METHODS
This study was a 12-week, randomized, double-blind, parallel-group, placebo-controlled trial conducted between January 2012 and December 2013 at 50 Japanese sites. The study was approved by the institutional review boards in accordance with the principles described in the Declaration of Helsinki. After giving informed consent, patients from 20 to 75 years old and diagnosed with PD according to the UK Parkinson's Disease Society Brain Bank criteria were randomized (1:1) to receive selegiline (week 0–1, 2.5 mg once daily after breakfast; week 2–3, 2.5 mg twice daily; week 4–5, 5 mg and 2.5 mg; week 6–12, 5 mg all after breakfast and lunch) or placebo.
Enrolled patients had received no previous treatments and had exhibited motor symptoms for less than 5 years, a Hoehn and Yahr stage of 1 to 3, and the Unified Parkinson Disease Rating Scale (UPDRS) part III scores of 10 points or greater. Patients who had received anti-PD medication for less than 12 weeks and had not used anti-PD medications within 12 weeks before the first dose of the investigational drugs were also enrolled. Patients were excluded from the study if they received treatments, such as pethidine, tramadol, tricyclic antidepressants, selective serotonin reuptake inhibitors, serotonin-noradrenalin reuptake inhibitor, selective noradrenalin reuptake inhibitors, noradrenergic and specific serotonergic antidepressants, certain antihistamine drugs, or drugs possibly affecting PD symptoms, within 3 weeks before the first dose of the study drug. Patients who received anticancer agents and other investigational or unapproved drugs in Japan within 26 weeks before the first dose of the study drug were also excluded. Patients were also excluded if they had any of the following concomitant psychiatric symptoms: impaired consciousness, hallucinations, delusions, and abnormal disorders. Other patients were excluded if they received ongoing therapy for epilepsy, had serious subsequent complications (cardiovascular, renal, hepatic, or hematologic disorders), had past or current concomitant schizophrenia, or had past or current abuse of central nervous system stimulants, such as antihypnotic drugs or cocaine. Finally, patients were excluded if they were women who were pregnant or lactating, were willing to become pregnant during the trial, or who had participated in other past clinical trials of selegiline.
The primary outcome measure was the change in the total UPDRS part I + II + III score from the baseline to the final visit. Secondary outcome measures were the changes from baseline to the final visit: (1) total UPDRS part II + III score; (2) UPDRS part I, II, III, and IV scores; (3) proportions of responders who achieved more than a 20%, 25%, or 30% reduction in the total UPDRS part I + II + III scores; (4) modified Hoehn and Yahr scale; and (5) Clinical Global Impression of Improvement (CGI-I) at the final visit. Safety assessments included the number of adverse events, vital signs, electrocardiogram results, and laboratory tests.
All efficacy analyses were performed on the full analysis set (FAS), which was defined as all randomized subjects who received at least 1 dose of the study drug and were assessed with any efficacy measurement after medication. If there were any missing values, then the last observation carried forward was applied. All the safety analyses were conducted on the safety population (SP), defined as subjects who received at least 1 dose of the study drug and were assessed with any safety measurements. The primary and secondary efficacy measurements (except for the modified Hoehn and Yahr scale, CGI-I, and proportions of responders) were compared between groups using an analysis of covariance. The modified Hoehn and Yahr scale or CGI-I was compared between groups using a Wilcoxon rank sum test and Fisher exact test, and the proportions of responders were compared using a Fisher exact test. The significance level was set at P < 0.05 (2-tailed test).
RESULTS
A total of 292 patients were randomized to either the selegiline or placebo group (both 146 patients). One patient from the selegiline and 2 patients from the placebo group were excluded before the initiation of treatment because of failure to meet the inclusion criteria or withdrawal of consent. Patients who violated the Good Clinical Practice guidelines after the initiation of treatment were excluded from the study (3 from each group). Another 3 patients from the selegiline group who met the exclusion criteria were excluded from the FAS. One patient in the placebo who violated Good Clinical Practice guidelines was excluded from the FAS but was included in the SP. Overall, 129 and 124 patients from the selegiline and placebo groups, respectively, completed the study. Patients who were discontinued after the initiation of therapy were included in the SP and FAS analyses (13 from the selegiline and 17 from the placebo groups). Therefore, 139 and 140 patients from the selegiline and placebo groups, respectively, were included in the FAS population. The SP population included 142 and 141 patients from the selegiline and placebo groups, respectively (Fig. 1). No significant differences between the 2 groups were noted in the baseline characteristics (Table 1).
FIGURE 1.
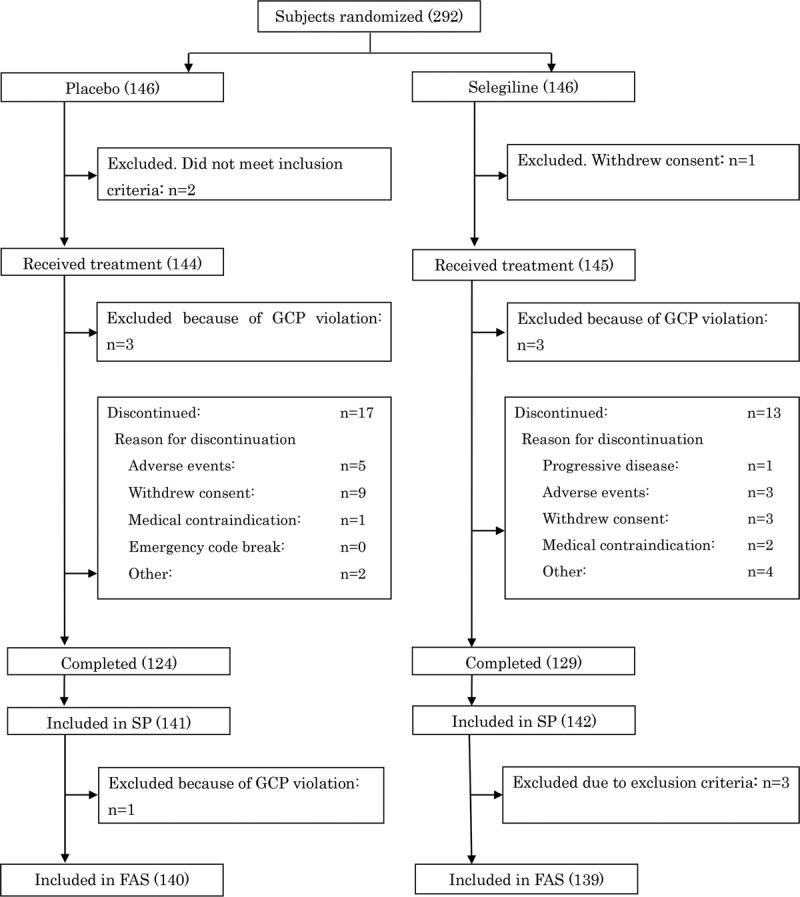
Study flow diagram (numbers in parentheses indicate the number of patients included in the respective categories).
TABLE 1.
Patient Demographics at Baseline
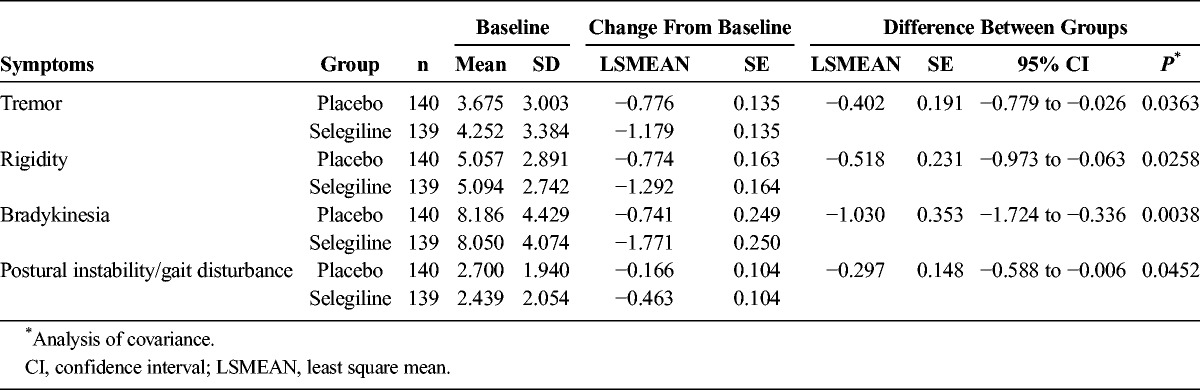
We observed a significant difference in the primary outcome, a change in total UPDRS part I + II + III, from baseline (mean ± SD; selegiline, 26.45 ± 11.16; placebo, 26.58 ± 11.53) to the final visit (selegiline, 20.19 ± 12.95; placebo, 23.44 ± 13.58) (difference, −3.12 ± 7.43; P = 0.0005; Fig. 2). The change in the total UPDRS part II + III score from baseline (selegiline, 25.69 ± 10.83; placebo, 25.95 ± 11.31) to the final visit (selegiline, 19.70 ± 12.60; placebo, 22.96 ± 13.41) was also significant (difference, −3.01 ± 7.25; P = 0.0006; Fig. 3A). No significant difference was noted between the groups regarding the change in the UPDRS part I score from baseline (Fig. 3B). The difference in the UPDRS part II score between groups from baseline (selegiline, 6.00 ± 3.37; placebo, 6.20 ± 3.95) to the final visit (selegiline, 4.87 ± 3.73; placebo, 5.91 ± 4.50) and the difference in the UPDRS part III score between the groups from baseline (selegiline, 19.69 ± 8.24; placebo, 19.75 ± 8.50) to the final visit (selegiline, 14.83 ± 9.47; placebo, 17.06 ± 10.24) were both significant (Figs. 3C, D). The difference between groups on the CGI-I scale (P < 0.0001, Fig. 4) and proportions of responders (P < 0.001, Table 2) were significant. The modified Hoehn and Yahr scale was not different between groups (Table 3). In the post hoc analyses, the efficacy regarding the individual PD symptoms is listed in Table 4.
FIGURE 2.
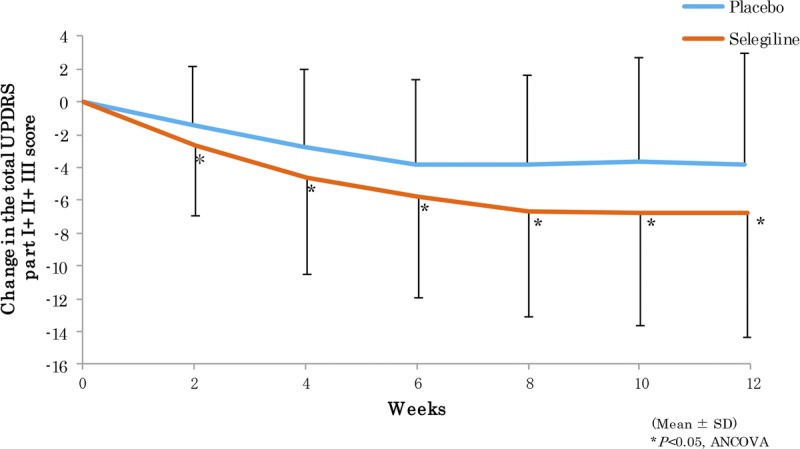
Mean changes from baseline in the total UPDRS part I + II + III scores.
FIGURE 3.
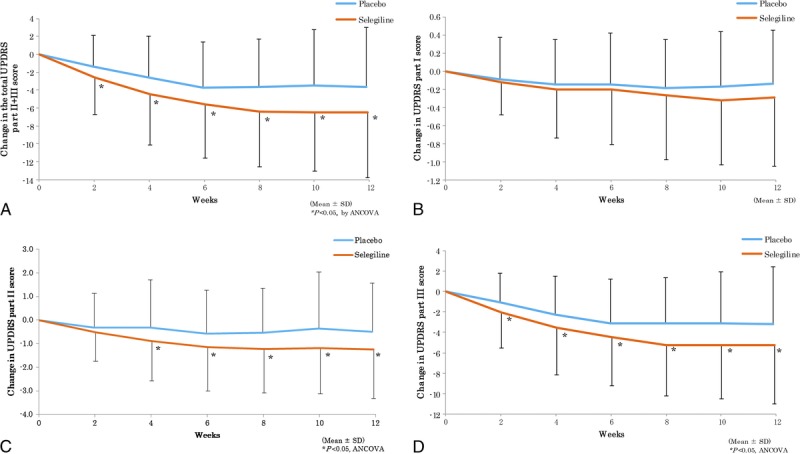
Mean changes from baseline in the UPDRS part II + III total scores and part I, II, and III scores.
FIGURE 4.
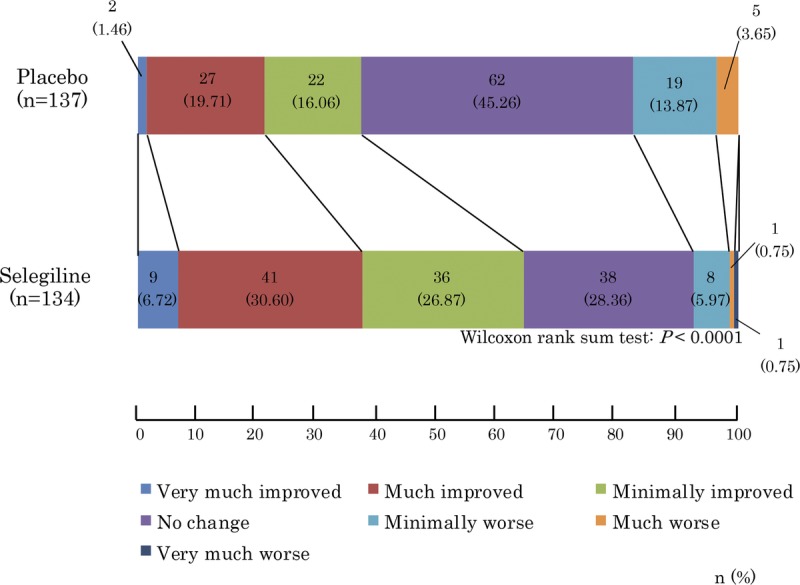
Comparison of CGI-I scores between placebo and selegiline.
TABLE 2.
Proportions of Responders Achieving a 20%, 25%, 30%, or More Reduction in the Total UPDRS Part I + II + III Scores
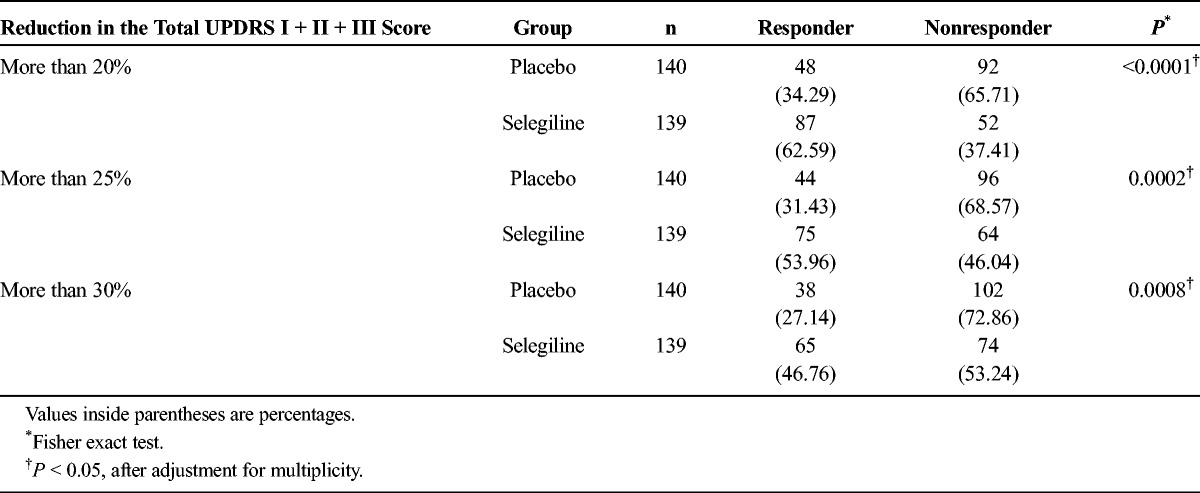
TABLE 3.
Comparison of the Modified Hoehn and Yahr Staging Scale in the Selegiline and Placebo Groups at the Final Assessment
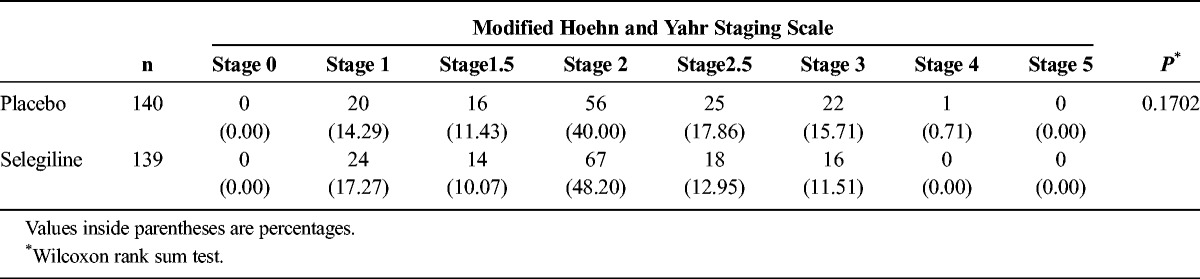
TABLE 4.
Efficacy Regarding Individual PD Symptoms
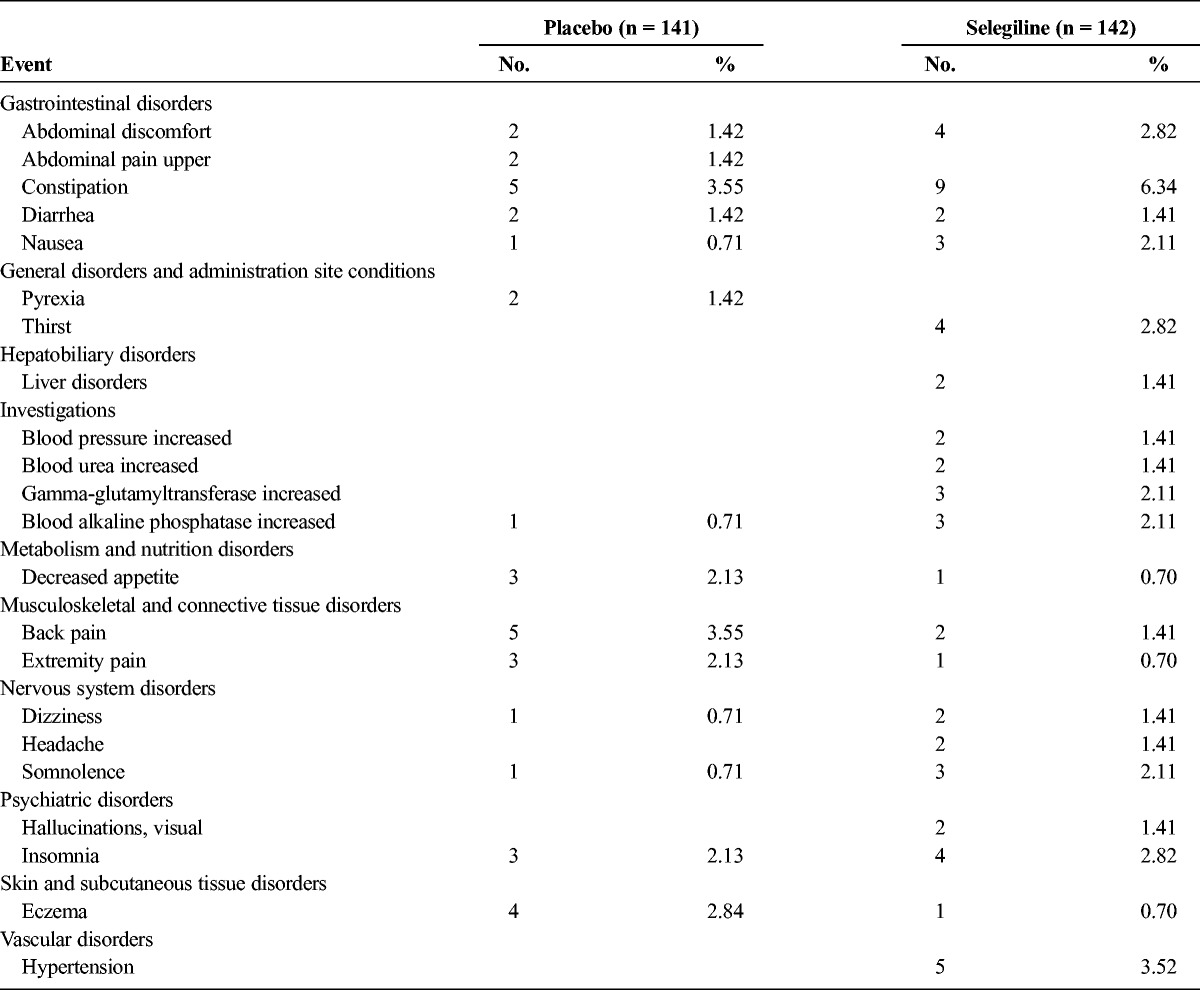
The total incidence of adverse events in the placebo and selegiline groups was 90 and 100 cases, respectively. The total number of adverse drug reactions was 41 and 53 cases, respectively. These differences were not significant (P > 0.05). The only adverse drug reaction that occurred in more than 5% of patients was constipation in the selegiline group, but all of these incidences were mild or moderate in intensity. The adverse drug reactions that were reported to occur in more than 1% of patients are shown in Table 5. Serious adverse events were observed in 4 patients treated with selegiline (1 had pneumonia, 1 had gallstones and acute cholecystitis, 1 had akinesia, and 1 had obstructive jaundice, pancreatic duct obstruction, and pancreatic cancer). However, these adverse events were not judged to be related to the study drug. No clinically relevant changes from the baseline were observed in the laboratory results, vital signs, or electrocardiogram results.
TABLE 5.
Adverse Drug Reactions With an Incidence Rate of 1% or Higher
DISCUSSION
According to the Parkinson Study Group, selegiline (10 mg) for early de novo PD delayed the onset of disability associated with early PD.4,5 In the final report of the study, they found the observed benefit of selegiline in delaying disability to be partly related to a symptomatic amelioration, because of worsening of the UPDRS motor scores during the 2 months after withdrawal of test drugs. They also described adverse events during the test trial in detail. There was no serious adverse event in 4 arms of the treatments.5
However, this effect was mainly symptomatic. It is well established that initial treatment with selegiline delays the need for levodopa in PD.6–9 In addition, in patients treated with levodopa or bromocriptine, selegiline might be partially neuroprotective in addition to its symptomatic effect.10–15
In 1996, we published a double-blind study using selegiline for Japanese patients with PD. Concomitant use of selegiline with levodopa was significantly better than placebo with levodopa (30.2% vs 15.3%, P < 0.002, n = 159 vs 157). Use of selegiline without levodopa did not reach statistical significance (27.5% vs 18.8%, P = 0.231, n = 69 vs 64). In this study, we investigated the effects of selegiline in de novo PD patients who were not taking any anti-PD drugs.
In the present study, the primary outcome measure (UPDRS I + II + III) in patients treated with selegiline improved by −6.26 ± 7.86 points (placebo, −3.14 ± 6.98 points). The difference between the groups was significant and supports the clinical efficacy of selegiline monotherapy for untreated patients with early PD. The adverse event rate was not significantly different between the 2 groups. The −3.12 point difference between the selegiline and placebo groups in the UPDRS I + II + III score after 3 months of treatment was similar to previous studies (−2.6 to −5.8).4,5,7,9,11,13,15 In the secondary analyses, selegiline was superior regarding the UPDRS part II + III, UPDRS part II, and UPDRS part III scores. The efficacy of selegiline was considered to be clinically meaningful with the improvements on the UPDRS scores (UPDRS part I + II + III [−6.26 ± 7.86], part II [−1.13 ± 2.18], and part III [−4.86 ± 5.94]), because these scores were greater than the score defined as a minimal clinically important change as reported before (−3.5, −0.7, and −2.4 points, respectively).16 Post hoc analyses revealed the efficacy of selegiline monotherapy for all cardinal symptoms of PD.
The safety analysis revealed no significant differences in the rates of adverse events and adverse drug reactions between the 2 groups. Although serious adverse events were observed only in the selegiline group, none of these events were judged by the investigators to be related to selegiline.
One of the weaknesses of our study was the duration of the treatment. Twelve weeks of observation may be too short. Currently, an open-label long-term study using selegiline is being conducted to determine the long-term efficacy and adverse effects.
In conclusion, selegiline monotherapy is efficacious in Japanese patients with early PD and is associated with acceptable, tolerable adverse effects.
ACKNOWLEDGMENT
The authors thank the following additional members who participated in the study as investigators: Kazunori Itoh, Sanae Honma, Masahiko Tomiyama, Tetsuya Maeda, Takashi Abe, Takafumi Hasegawa, Hiroyuki Enomoto, Takao Hashimoto, Ken-ichi Fujimoto, Kyoichi Nomura, Hideki Shimura, Fusako Yokochi, Masahiko Suzuki, Shin-ichiro Kubo, Hidehiro Mizusawa, Hideki Takubo, Satoshi Orimo, Kazuko Hasegawa, Fumihito Yoshii, Ichiro Imafuku, Hitoshi Yamada, Noriko Kawashima, Tsuyoshi Uchiyama, Tetsushi Atsumi, Masahisa Katsuno, Kotaro Tanaka, Hideyuki Sawada, Takahiko Tokuda, Yoshihisa Tatsuoka, Sadayuki Matsumoto, Yoshiyuki Mitsui, Yasuhiro Hiwatani, Ken-ichi Kashihara, Yoshito Nagano, Junji Yoshinaga, Yoshihiko Nishida, Yasushi Osaki, Noriko Nishikawa, Yoshio Tsuboi, Akira Sato, Nobuo Araki, Hideo Mori, Hideto Miwa, Noriyuki Matsukawa, Makio Takahashi, Takanori Hazama, Toshihiko Suenaga.
Footnotes
Conflicts of Interest and Source of Funding: This study was funded by FP Pharmaceutical corporation.
REFERENCES
- 1.Schapira AH, Olanow CW. Drug selection and timing of initiation of treatment in early Parkinson's disease. Ann Neurol 2008;64(Suppl 2):S47–S55. [DOI] [PubMed] [Google Scholar]
- 2.NICE. Parkinson's Disease in Over 20s: Diagnosis and Management [NICE Web site]. June 2006. Available at: https://www.nice.org.uk/guidance/cg35. Accessed January 16, 2017.
- 3.Fox SH, Katzenschlager R, Lim SY, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: treatments for the motor symptoms of Parkinson's disease. Mov Disord 2011;26(Suppl 3):S2–S41. [DOI] [PubMed] [Google Scholar]
- 4.The Parkinson Study Group. Effect of deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 1989;321(20):1364–1371. [DOI] [PubMed] [Google Scholar]
- 5.Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disability in early Parkinson's disease. N Engl J Med 1993;328(3):176–183. [DOI] [PubMed] [Google Scholar]
- 6.Tetrud JW, Langston JW. The effect of deprenyl (selegiline) on the natural history of Parkinson's disease. Science 1989;245(4917):519–522. [DOI] [PubMed] [Google Scholar]
- 7.Pålhagen S, Heinonen EH, Hägglund J, et al. Selegiline delays the onset of disability in de novo parkinsonian patients. Swedish Parkinson Study Group. Neurology 1998;51(2):520–525. [DOI] [PubMed] [Google Scholar]
- 8.Myllylä VV, Sotaniemi KA, Vuorinen JA, et al. Selegiline as initial treatment in de novo parkinsonian patients. Neurology 1992;42(2):339–343. [DOI] [PubMed] [Google Scholar]
- 9.Allain H, Pollak P, Neukirch HC. Symptomatic effect of selegiline in de novo Parkinsonian patients. The French Selegiline Multicenter Trial. Mov Disord 1993;8(Suppl 1):S36–S40. [DOI] [PubMed] [Google Scholar]
- 10.Calzetti S, Negrotti A, Cassio A. L-deprenyl as an adjunct to low-dose bromocriptine in early Parkinson's disease: a short-term, double-blind, and prospective follow-up study. Clin Neuropharmacol 1995;18(3):250–257. [DOI] [PubMed] [Google Scholar]
- 11.Olanow CW, Hauser RA, Gauger L, et al. The effect of deprenyl and levodopa on the progression of Parkinson's disease. Ann Neurol 1995;38(5):771–777. [DOI] [PubMed] [Google Scholar]
- 12.Przuntek H, Conrad B, Dichgans J, et al. SELEDO: a 5-year long-term trial on the effect of selegiline in early Parkinsonian patients treated with levodopa. Eur J Neurol 1999;6(2):141–150. [DOI] [PubMed] [Google Scholar]
- 13.Larsen JP, Boas J, Erdal JE. Does selegiline modify the progression of early Parkinson's disease? Results from a five-year study. The Norwegian-Danish Study Group. Eur J Neurol 1999;6(5):539–547. [DOI] [PubMed] [Google Scholar]
- 14.Shoulson I, Oakes D, Fahn S, et al. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson's disease: a randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann Neurol 2002;51(5):604–612. [DOI] [PubMed] [Google Scholar]
- 15.Pålhagen S, Heinonen E, Hägglund J, et al. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 2006;66(8):1200–1206. [DOI] [PubMed] [Google Scholar]
- 16.Hauser RA, Auinger P. Determination of minimal clinically important change in early and advanced Parkinson's disease. Mov Disord 2011;26(5):813–818. [DOI] [PubMed] [Google Scholar]