

Abstract
Schizophrenia is associated with alterations in sensory, motor, and cognitive functions that emerge prior to psychosis-onset; identifying pathogenic processes that can account for this multi-faceted phenotype remains a challenge. Accumulating evidence suggests that synaptic plasticity is impaired in schizophrenia. Given the role of synaptic plasticity in learning, memory, and neural circuit maturation, impaired plasticity may underlie many features of the schizophrenia syndrome. Here, we summarize the neurobiology of synaptic plasticity, review evidence that plasticity is impaired in schizophrenia, and explore a framework in which impaired synaptic plasticity interacts with brain maturation to yield the emergence of sensory, motor, cognitive, and psychotic features at different times during development in schizophrenia. Key gaps in the literature and future directions for testing this framework are discussed.
NEURODEVELOPMENT, SYNAPTIC PLASTICITY, AND THE ANTECEDENTS OF SCHIZOPHRENIA
Our understanding of the role of development in schizophrenia has undergone significant revision in recent decades. Early neurodevelopmental conceptualizations suggested that schizophrenia arose from pre- or peri-natal “lesions” that remained clinically silent until late adolescence or early adulthood [1,2]. However, many individuals who later develop schizophrenia show subclinical sensory, motor, and cognitive disturbances prior to psychosis-onset [e.g. 3,4]. Indeed, findings of altered trajectories of normal brain development in pre-schizophrenia individuals highlighted the potential role of progressive maturational disturbances [5-12]. Consequently, the field has moved towards a clinical staging model, in which the onset of psychotic symptoms in late adolescence or early adulthood is considered a “late-stage” of the disorder [13]. Identifying cognitive, behavioral, or neural markers that predict the later development of psychosis has thus become a prominent research goal. However, human development is a dynamic process, with individual sensory, motor, and cognitive functions following distinct trajectories of maturation. Markers that predict the later development of schizophrenia are therefore unlikely to be static throughout development. Clarifying how antecedents of schizophrenia relate to specific pathogenic processes and emerge over time is a key step that may enable us to identify those at risk and predict the onset of psychosis with greater precision.
Critically, the last decade has witnessed remarkable gains in our knowledge of the genetic architecture of schizophrenia. Schizophrenia appears to be a highly polygenic disorder involving hundreds, if not thousands of genes, with at least some common and rare genetic variants converging on a subset of biological pathways. In particular, genes involved in glutamatergic function and synaptic plasticity figure prominently among those associated with schizophrenia, suggesting that impaired synaptic plasticity may be a core pathogenic factor in the disorder. Importantly, while synaptic plasticity is critical for learning and memory across the lifespan, during development, synaptic plasticity is also critically involved in organizing neurons into the finely-tuned circuits that characterize a mature brain. The potential consequences of impaired plasticity across development may therefore provide a useful lens for understanding how and when the diverse signs and symptoms associated with schizophrenia emerge.
Here, we first briefly summarize the biological processes underlying synaptic plasticity. We then review the role of synaptic plasticity in learning and memory, and in normal brain maturation. Next, we review evidence implicating synaptic plasticity pathways in the pathogenesis of schizophrenia. We then propose a model in which genetically-determined impairments in synaptic plasticity interact with brain maturation to yield the progressive emergence of various signs and symptoms at different times across development. Finally, we suggest possible mechanisms through which impaired synaptic plasticity might lead to psychosis.
THE NEUROBIOLOGY OF SYNAPTIC PLASTICITY
The 100 billion neurons of the human brain communicate via synaptic connections (Box 1). These connections encode our experiences through patterns of activity, across populations of neurons [14]. Activity generated by salient inputs triggers changes that alter the synaptic weights between neurons. These changes are known as synaptic plasticity and reflect the fundamental capacity of the brain to change with environmental input and use.
TEXT BOX 1. Structural Components of the Synapse.
Neurons communicate via neurotransmitters that are released from a presynaptic neuron, diffuse across the synaptic cleft, and bind to receptors on a postsynaptic neuron. The "active zone" of presynaptic neurons is localized to axon terminals that contain actin cytoskeleton, scaffold proteins, and vesicles filled with neurotransmitter [16]. Scaffold proteins tether vesicles to the cytoskeleton near the presynaptic membrane, such that when an action potential arrives, vesicles can be docked by Soluble NSF-Attachment Protein Receptor (SNARE) proteins, fuse with the membrane, and release neurotransmitter into the synaptic cleft. The "active zone" of the majority of excitatory postsynaptic neurons is localized to dendritic spines, which are small membrane protuberances on the shaft of neuronal dendrites. Dendritic spines are found on most principal neurons in the brain, including pyramidal neurons of the cortex and hippocampus, medium spiny neurons of the striatum, and Purkinje cells of the cerebellum [17]. Dendritic spines contain an electron-rich postsynaptic density (PSD) comprised of neurotransmitter receptors, signaling molecules, cytoskeleton, and an abundant network of scaffold proteins. Mature dendritic spines are typically characterized by a spherical head and a thinner neck, which limits diffusion of molecules into and out of the head [18]. Actin filaments provide the structure for spine shape and scaffold proteins stabilize the PSD within the spine head by binding together the receptors, cell adhesion molecules, signaling enzymes, and cytoskeleton. Together, this compartmentalized structure facilitates the rapid translation of postsynaptic receptor activation into the intracellular signaling events, and ultimately local structural changes, that underlie the expression of synaptic plasticity.
Structurally, synaptic plasticity is characterized by the insertion or removal of amino-3-hydroxy-5-methyl-4-isoxazolepropionic receptors (AMPARs) into the postsynaptic membrane, and the enlargement or shrinkage of dendritic spines where the majority of excitatory synapses (~90%) are located (Figure 1; 15,16). Functionally, synaptic plasticity is expressed as long-term potentiation (LTP) or depression (LTD) of synaptic strength, reflecting changes in conductance through AMPARs in the postsynaptic membrane. During the induction of plasticity, N-methyl-D-aspartate receptor (NMDAR) activation allows Ca2+ to cross the postsynaptic membrane and initiate intracellular signaling cascades. These cascades trigger gene transcription, AMPAR trafficking via actin dynamics, reorganization of the cytoskeleton, and the enlargement and stabilization or elimination of dendritic spines. The integrity of the synaptic structure, AMPAR trafficking, and dendritic spine dynamics are all critical for generating lasting synaptic plasticity changes (Box 2; 16).
Figure 1. The induction and expression of synaptic plasticity.
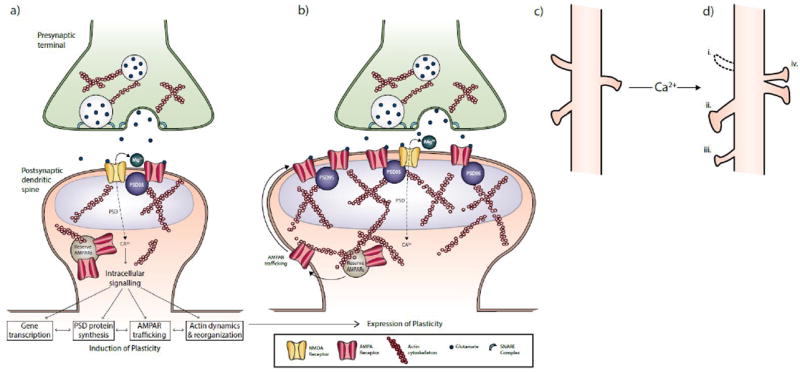
During the induction of plasticity at excitatory synapses (A), glutamate activates postsynaptic AMPARs and NMDARs located in the postsynaptic density (PSD) of dendritic spines. AMPARs generate a fast excitatory postsynaptic response via Na+, while NMDARs initiate intracellular signaling cascades via Ca2+ that trigger local gene transcription; phosphorylation and synthesis of new PSD proteins; AMPAR trafficking to and from the membrane via actin dynamics; and reorganization of the actin cytoskeleton. When synaptic activity is sufficient to induce long-term potentiation (LTP), these processes lead to the insertion of reserve AMPARs into the postsynaptic membrane and the enlargement of the PSD and dendritic spine (B). Scaffold proteins such as PSD-95 stabilize the new AMPARs by trapping them in the membrane and reducing their lateral mobility. Changes in the size and shape of dendritic spines from baseline (C) scale with changes in synaptic efficacy and include (D): the elimination of spines (i); enlargement of spine heads and necks (ii); growth of new spines (iii); and the splitting of single PSDs and spines into two synapses (iv).
TEXT BOX 2. Mechanics of Synaptic Plasticity Changes.
Excitatory neurotransmission is primarily mediated by glutamate at AMPARs and N-methyl-D-aspartate glutamate receptors (NMDARs). AMPARs and NMDARs are co-localized on dendritic spines, but have distinct properties that confer specialized roles for different phases of synaptic plasticity [16,19]. Thus, when glutamate binds to AMPARs, the AMPAR channel opens, allowing sodium (Na+) to flow across the postsynaptic membrane and generate a rapid excitatory current. Indeed, AMPARs are responsible for the majority of the excitatory postsynaptic response when a cell is close to its resting membrane potential. In contrast, NMDARs have a channel pore that is blocked by extracellular magnesium (Mg+) at resting potential. Due to this Mg+ block, the NMDAR channel only opens when concurrent binding of glutamate and glycine or D-serine is accompanied by sufficient postsynaptic depolarization from activation of non-NMDARs (i.e. largely AMPARs) to cause the Mg+ ion to disengage from the channel. This allows NMDARs to play the critical role of detecting coincident pre- and post-synaptic activity. NMDARs are additionally unique in that they allow not only Na+ to enter the cell, but also calcium (Ca2+), which activates signaling enzymes and subsequent intracellular signaling cascades. Thus, whereas AMPARs generate the rapid excitatory responses that permit NMDAR activation, NMDARs detect coincident pre- and post-synaptic activity and trigger the intracellular signaling cascades that ultimately generate synaptic plasticity changes [19].
Intracellular signaling initiated by NMDAR-mediated rises in Ca2+ ultimately leads to changes in the number of AMPARs incorporated into the PSD, which is the primary mechanism underlying the initial expression of LTP or LTD. Thus, during the induction of LTP or LTD, reserve AMPARs are trafficked to or from the postsynaptic membrane, respectively, via changes in actin dynamics [16]. Scaffold proteins facilitate the incorporation of new AMPARs into the membrane by trapping AMPARs in the membrane and reducing their lateral mobility. PSD-95 may be a particularly important scaffold protein for synaptic plasticity, as it is dynamically redistributed across synapses and appears to preferentially support the stability of AMPARs at potentiated synapses [20].
Experience-dependent changes in the number of AMPARs incorporated into the membrane are accompanied by structural changes in the size and shape of dendritic spines. Dendritic spine changes depend on actin reorganization, scale with synaptic efficacy, and include enlargement of spine heads and necks, growth of new spines, and the splitting of single PSDs and spines into two synapses [14,18]. Thus, as quickly as ~20 seconds after LTP induction, actin accumulates in the spine and begins polymerizing into filaments [21]. This polymerization is thought to generate a mechanical force against the membrane that induces a rapid spine volume increase [22], and together with AMPAR upregulation, characterizes the early phase of LTP [23]. Subsequent stabilization of spine enlargement requires the synthesis and transport of new proteins to the synapse. Thus, actin-related proteins supporting the assembly and stabilization of actin into branched filaments increase in the minutes following LTP induction, while concomitant increases in PSD proteins, including PSD-95 and NMDARs, increase over the following hours (i.e. corresponding to late phase LTP; [24-26]). Conversely, during LTD, actin de-polymerization leads to actin filament disassembly, destabilization of the PSD, and spine shrinkage [21]. As interfering with spine dynamics impairs LTP [16,27], both AMPAR trafficking and dendritic spine dynamics are critical for generating lasting plasticity changes.
ROLES OF SYNAPTIC PLASTICITY
Synaptic Plasticity in Learning and Memory
Numerous brain regions exhibit synaptic changes in response to environmental input and learning across the lifespan [14,27-33], with plasticity changes in distinct regions thought to underlie specific forms of learning. For example, synaptic plasticity appears to encode declarative and spatial learning at hippocampal and medial temporal lobe synapses [14,20], habit learning at striatal synapses [29,30], motor sequence learning at motor cortex synapses [31,32], and perceptual learning at sensory cortex synapses [33]. Thus, synaptic plasticity is a key mechanism for encoding distinct types of learning across many regions in the brain.
Synaptic Plasticity in Brain Maturation
On a wider scale, synaptic plasticity is also critically involved in the maturation of neural circuits. During early development, billions of cells proliferate, differentiate into neurons, and form connections. These events are programmed by intrinsic molecular cues and yield an over-proliferation of neurons with immature and redundant synapses [34]. As environmental input increases in postnatal life, competition for inputs yields the mass elimination of weak synapses (i.e. synaptic pruning), and the concurrent strengthening of remaining synapses [35]. This synaptic refinement helps establish the finely-tuned circuitry that characterizes a mature brain and depends on numerous mechanisms that parallel plasticity in the adult brain, including NMDAR-dependent AMPAR delivery to potentiated synapses, LTP, LTD, and the intact functioning of scaffold proteins [14,35-37]. Developmental synaptic refinement occurs in a series of regionally hierarchical, overlapping waves, and is accompanied by the increasing expression of cellular and molecular “brakes” that stabilize synapses into mature circuits (see Box 3).
TEXT BOX 3. Dampening the Brain’s Capacity for Plasticity During Development.
Neural circuits and synapses retain the capacity to be shaped by experience through adulthood, but relative to earlier periods in development, this capacity is greatly reduced. This shift is characterized by cellular and molecular changes that interact with synaptic pruning to gradually dampen the brain’s capacity for plasticity and stabilize experience-refined neural circuits [38]. These changes include maturation of GABAergic inhibitory circuits, particularly fast-spiking parvalbumin (PV)-containing interneurons which provide strong inhibition onto pyramidal neurons, and structural changes that inhibit neurite pruning and outgrowth, such as increased myelination and expression of perineuronal nets (PNNs; [38]). Indeed, maturation of GABAergic interneurons is more protracted than that of pyramidal neurons, and GABA maturation both shapes, and is shaped by, synaptic wiring and plasticity [39]. During development, GABA transmission is required for the onset of critical period plasticity [39] and GABA release influences the selective elimination of dendritic spines [40,41]. At the same time, PV cells have been found to adjust their synapses onto pyramidal neurons and reduce their production of PV and GAD67 in response to changes in pyramidal cell activity [39,42]. Thus, PV neurons appear to fine-tune their intrinsic excitability during maturation to produce a homeostatic balance of excitation/inhibition within neural circuits [42]. In addition, as PV interneurons mature, their cell bodies and neurites become ensheathed by PNNs, which are lattice-like extracellular matrix structures that appear to form a barrier between cells and molecular factors that regulate plasticity. PNN expression increases progressively until late adolescence or early adulthood, and is thought to contribute to the closure of critical developmental periods [38]. In the healthy brain, this dampening of plasticity “locks-in” neural circuitry that is optimally tuned to the environment, and occurs in regionally hierarchical, overlapping waves, consistent with the overall progression of brain maturation.
In schizophrenia, patients show reduced markers of GABA neuron function, particularly at PV interneurons in prefrontal cortex [43] and reduced density of PNNs [44-46], in addition to alterations in dendritic spines and markers of glutamatergic synapse function. Notably, PV interneurons in schizophrenia patients were recently found to receive fewer synaptic inputs from excitatory neurons than in control subjects, with excitatory input reductions predicting the downregulation of activity-dependent gene products in PV interneurons [47]. Thus, although reduced PV interneuron function and PNN expression could directly contribute to cortical disinhibition and circuit level dysfunction in schizophrenia [46,48,49], changes in PV interneurons and PNNs might arise during development as compensatory responses to reduced pyramidal cell activity.
Importantly, synapse and circuit maturation is thought to underlie the expression of emergent functions of the cortex. Over the first two decades of life, humans develop and refine many abilities. These range from basic sensory, motor, and associative learning functions that emerge within the first year of life, to higher-order cognitive functions such as those requiring planning, self-regulation, and/or the mental manipulation of complex information, which emerge and mature later [50,51]. Regional changes in gray matter density, which indexes neuronal cell bodies, dendrites, axons, and synaptic processes, parallel these milestones, and are accompanied by the integration of brain regions into increasingly specialized and functionally-connected neural circuits [52]. Structural magnetic resonance imaging (MRI) studies showed that gray matter thinning progresses from regions supporting primary sensory and motor functions in childhood (e.g. primary sensorimotor/dorsal parietal cortices, occipital pole, and frontal pole), to regions involved in spatial orientation and multisensory integration (e.g. parietal and temporal association cortices), and finally, to higher-order association regions involved in executive functions in late adolescence (e.g. prefrontal and superior temporal cortices; [53-55]). Indeed, developmental changes in prefrontal and parietal gray matter volume were found to predict improvements in information processing, working memory, and executive function from childhood to adulthood [56,57]. Similarly, functional MRI studies found that increased focal activation and functional integration within late-maturing regions predicted developmental improvements in higher-cognitive functions [52,58-61].
Human imaging studies cannot isolate changes at the synaptic level. Indeed, the extent to which developmental gray matter reductions reflect changes in neuronal and synaptic processes versus increased myelination, which likely reduces the ratio of gray matter detected in MRI relative to white matter, continues to be debated [62,63]. Nevertheless, gray matter thinning is generally considered a marker of regional maturation, and the regional heterochronicity of gray matter changes parallels that of spine density changes. For example, human post-mortem brain studies showed that primary visual and auditory cortices peak early in synaptic density around 3-4 months of age, and subsequently undergo a period of net synapse elimination that lasts until late childhood (i.e. 11-12 years). Conversely, prefrontal cortex does not peak in synaptic density until 15 month of age and shows a protracted period of synapse elimination that extends into late adolescence or adulthood [64-66; Figure 2A].
Figure 2. Synaptic Plasticity and Brain Maturation in the Healthy Brain and in Schizophrenia.
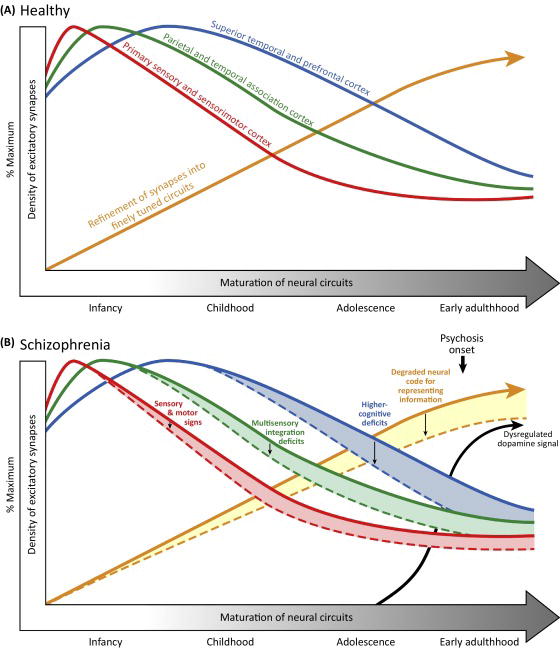
(A) Developmental trajectories of excitatory synapses in representative cortical regions in the healthy brain. Within each region, synapses are initially overproduced and undergo a period of net synaptic pruning in which weak synapses are eliminated and adaptive synapses are strengthened. These synaptic refinements are thought to contribute to the maturation of finely tuned and functionally organized neural circuits mediating sensory, motor, and cognitive functions. (B) Hypothesized consequences of impaired synaptic plasticity across development in schizophrenia. Impaired synaptic plasticity in schizophrenia is predicted to lead to increased synaptic pruning and suboptimal fine-tuning of neural circuits. Regional differences in the trajectories of synaptic refinement and circuit maturation predict the timing of the emergence of sensory, motor, and higher cognitive signs in schizophrenia, with the magnitude of the impairments being positively associated with the age of their appearance. Dysregulated dopamine signaling emerges in adolescence as a downstream consequence of impaired cortical synaptic plasticity, and ultimately interacts with suboptimal neural tuning to yield psychotic symptoms in late adolescence or early adulthood.
Together, the temporal convergence of regional synaptic pruning with the emergence and maturation of various sensory, motor, and cognitive functions has led to the hypothesis that plasticity-dependent synaptic refinement plays a key role in the maturation of local and distributed circuits, and the concurrent refinement of sensory, motor, and cognitive functions across development [50,67,68].
IMPAIRED SYNAPTIC PLASTICITY AS A PATHOGENIC FACTOR IN SCHIZOPHRENIA
Post-Mortem Evidence Implicating Synaptic Plasticity in Schizophrenia
Converging lines of evidence suggest that the cellular machinery underlying synaptic plasticity is impaired in schizophrenia. Early attention focused on the NMDAR, following findings that NMDAR antagonists, such as phencyclidine and ketamine, induced positive, negative, and cognitive symptoms in healthy individuals similar to those seen in schizophrenia [69,70]. However, post-mortem studies have since highlighted that schizophrenia is associated with aberrations that broadly affect dendritic spines and diverse aspects of the cellular machinery involved in synaptic plasticity. For example, dendritic spine density is reduced in multiple brain regions in schizophrenia, including prefrontal and temporal cortices [17,71-74]. Altered expression of numerous gene products involved in actin dynamics and spine stability has also been found in schizophrenia [75-79]. Schizophrenia subjects also showed altered expression of NMDAR and AMPAR subunits [74,81-85], and altered expression of proteins involved in NMDAR and AMPAR clustering and downstream signaling, including PSD-95 [86-89]. Further, schizophrenia subjects showed altered expression of presynaptic proteins, including components of the SNARE complex that facilitates synaptic vesicle fusion with the membrane [90,91]. Consistent with these findings, a recent proteomic study found that 143 out of ~700 PSD proteins showed differential expression in schizophrenia, with NMDAR-interacting proteins showing the most notable alterations [92].
Genetic Evidence Implicating Synaptic Plasticity in Schizophrenia
Similar to post-mortem findings, large-scale genetic studies have highlighted that schizophrenia is associated with common and rare genetic variants that appear to regulate components of the postsynaptic signaling complex involved in synaptic plasticity and dendritic spine stability. For example, copy number variant mutations associated with schizophrenia occur disproportionately at loci containing genes involved in synaptic function and the PSD, including genes encoding the MAGUK family of scaffold proteins, and the activity-regulated cytoskeleton protein (Arc) complex, which localizes to NMDAR-activated synapses and is involved in the remodeling and maintenance of plasticity-induced cytoskeleton changes [93-95]. Rare and de novo deleterious mutations in schizophrenia were similarly found to disproportionately affect genes involved in actin filament bundle assembly, the Arc complex, the NMDAR signaling complex, and MAGUK scaffold proteins [96,97]. Further, the largest genome-wide association study (GWAS) of schizophrenia, including over 36,000 patients, found that schizophrenia was associated with common variants near genes involved in glutamatergic neurotransmission and synaptic plasticity. Genes within risk loci included GRIN2A which encodes the NR2A NMDAR subunit; SRR, which encodes the enzyme that generates the NMDAR co-agonist D-serine from L-serine; and CNKSR2 which encodes the CNK2 scaffold/adaptor protein involved in coupling signal transduction to cytoskeletal remodeling in the PSD [98].
Although many individual post-mortem findings await replication and the specific genes and corresponding mechanisms that map onto many genetic loci associated with schizophrenia require further clarification (e.g. see [99] for discussion), these findings suggest a convergence of aberrations that broadly affect the postsynaptic signaling complex and dendritic spines in schizophrenia. In concert, these data support the idea that impaired synaptic plasticity is a critical pathogenic process in schizophrenia.
CONSEQUENCES OF IMPAIRED SYNAPTIC PLASTICITY IN SCHIZOPHRENIA DURING DEVELOPMENT
A Heuristic Model for the Signs and Symptoms of Schizophrenia
In adulthood, schizophrenia is characterized by diverse abnormalities in cognitive, motor, and sensory domains (Box 4). However, many of these features appear prior to psychosis-onset. Given the role of synaptic plasticity in developmental synaptic refinement, if synaptic plasticity is disrupted in schizophrenia, the age at which many sensory, motor, and cognitive signs, and their underlying neural correlates emerge may parallel the timing of regional synaptic and circuit maturation (Figure 2B). In addition, because neural circuit refinement progresses hierarchically, with later-maturing circuits building upon inputs from lower-level circuits, the consequences of impaired plasticity may be amplified through development. Subtle deficits in sensory and motor functions would therefore be expected to appear early in development, while more pronounced deficits in higher-cognitive functions (e.g. working memory, selective attention, cognitive control) would not reliably appear until later in development. It is worth noting here that deficits in higher-cognitive functions would not be expected to be absent altogether in early development, but rather, that deficits in these functions and their corresponding neural correlates should be detected less reliably at younger ages given their protracted maturation. Conversely, given the fundamental role of synaptic plasticity for learning across the lifespan, robust deficits reflecting learning and memory functions should be evident early in development.
TEXT BOX 4. The Diverse Signs and Symptoms of Adult Schizophrenia.
In its chronic form, schizophrenia is characterized by diverse clinical features affecting cognitive, motor, and sensory domains. For example, schizophrenia patients show impaired general intellectual functioning, learning and memory, working memory, attention, and language ability [100,101]. Patients also show deficits in motor coordination, motor skill learning, and psychomotor speed [102-104]. Additionally, schizophrenia patients show deficits in higher-level sensory processing, including impaired tone and phoneme discrimination, sound localization, visual form organization and discrimination, perception of visual motion, and olfactory identification [105-115]. In chronic patients, deficits appear largest in declarative memory and attention, with relatively smaller deficits in perception and motor skills [116,117]. In patients with recent onset of psychosis, deficits appear largest in immediate verbal memory, with smaller but still large deficits in nonverbal memory, working memory, attention, and processing speed, and medium deficits in motor skills [100,118].
Consistent with this phenotype, schizophrenia patients show abnormalities on functional imaging indices implicating disturbances in both lower-level sensory cortices and higher-order association cortices. For example, schizophrenia patients show altered electroencephalograph (EEG) event related potentials (ERPs) in response to auditory and visual stimuli, as well as altered neural activation in sensory cortices during fMRI studies of sensory processing [105,119,120]. Schizophrenia patients also show altered activation across many individual brain regions and altered connectivity within large-scale brain networks during a variety of sensory and cognitive tasks, although, relative to sensory regions, activation and connectivity involving association cortices may be more disrupted [121-123]. Similarly, schizophrenia patients show altered neural oscillations, as measured by EEG and magnetoencephalography (MEG), during sensory and cognitive tasks, as well as at rest [123,124], with gamma-band oscillations thought to show particular disturbance. While these imaging measures are generally agnostic to potential mechanisms contributing to differences in signal amplitude or connectivity, it is notable that schizophrenia patients do show deficits on functional indices of synaptic plasticity. For example, schizophrenia patients show reduced LTP-like effects following high-frequency sensory [125,126] or transcranial magnetic stimulation [127], as measured by EEG. Similarly, schizophrenia patients show reduced brain responses during the mismatch negative task (MMN; [128,129]), in which a stream of “standard” stimuli are interrupted by periodic “deviant” stimuli. Brain responses to deviant stimuli are thought to represent an error prediction signal during perceptual learning that depends on synaptic plasticity [130] and human and animal studies showed that the LTP and MMN tasks are NMDAR-dependent [131-133].
We thus suggest the following 3 specific hypotheses regarding the timing of appearance of different features of schizophrenia during the premorbid and prodromal phases:
Subtle deficits in sensory and motor function emerge early in development, as impaired synaptic plasticity disrupts the optimal refinement of local sensory and motor circuits.
Given the fundamental role of synaptic plasticity for diverse forms of learning, deficits reflecting learning and memory are also detected early in development.
Large deficits in higher-cognitive functions emerge later in development, as impaired synaptic refinement progresses to distributed circuits that integrate input from local circuits and involve prefrontal and/or temporal association cortices.
In this next section, we consider what is known about the timing of emergence of these broader antecedents of schizophrenia. We focus on behavioral and cognitive signs and structural MRI findings given that the majority of research in pre-schizophrenia individuals has been limited to these measures; however, we briefly consider the limited literature on functional imaging measures as well. This section is not intended as a comprehensive review of all studies examining potential antecedents of schizophrenia, but to highlight relevant studies and current gaps in the literature.
Motor and Sensory Signs
Consistent with early refinements in sensorimotor cortices, the earliest impairments documented in individuals who later develop schizophrenia are motor and sensorimotor deficits. In prospective studies of infants at genetic risk for schizophrenia and population-based studies, infants who later develop schizophrenia show reliable delays attaining motor milestones, such as delayed head lifting, crawling, and walking [4,134-137]. Similarly, children who later develop schizophrenia show impaired motor skills at 3, 5, and 9 years of age [138], motor coordination at 4 and 7 years [139], and perceptual-motor performance at 10-13 years relative to healthy controls [140,141].
Compared to sensorimotor function, fewer studies examined high-level visual, auditory, or olfactory function in schizophrenia, and the extant literature largely involves adult patients. This difference is likely due to practical limitations such as a lack of widely available standardized tests of higher-level perceptual functions, or similarly, a lack of consensus on best-practice paradigms [112]. Nevertheless, the extant literature suggests that sensory processing impairments exist early-on in individuals who later develop schizophrenia. For example, children at genetic risk for schizophrenia showed poorer sound discrimination compared to healthy controls [142] and children who later developed schizophrenia showed a higher frequency and/or severity of broadly defined visual dysfunction at 4, 7, and 11-13 years of age [143,144]. Impaired visual processing was also found in adolescents and young adults at clinical or genetic high risk for schizophrenia, including altered illusion susceptibility [145,146] and impaired visual target detection [147]. Furthermore, deficits in olfactory identification were found in adolescents and young adults who later developed schizophrenia [148]. Interestingly, pre-schizophrenia individuals also show deficits in reading that may appear during childhood and increase across adolescence [4,149,150]. As normal reading ability depends on the complex integration of visual and auditory processing, the initial reading impairment in schizophrenia may partially reflect primary deficits in unisensory processing, with the later decline over adolescence reflecting deficits in multisensory integration as heteromodal association cortices mature [151]. Additional studies in infants and children who later develop schizophrenia are needed to clarify the precise ages at which unisensory and multisensory deficits emerge. However, these findings suggest that subtle deficits in high-level sensory functions are present prior to psychosis-onset and are broadly consistent with early disruptions in early maturing motor and sensory cortices in individuals who develop schizophrenia.
Cognitive Signs
Children and adolescents who later develop schizophrenia have been consistently found to have impaired general intellectual functioning relative to their healthy peers [4,138,152-155]. The size of impairment in childhood (i.e. ~0.5 SD) is smaller than that in adulthood (i.e.~1.0 SD) and appears to increase over development [4,156-159].
Regarding specific cognitive functions, few birth cohort or genetic risk studies directly assessed learning and memory in children who later developed schizophrenia. This limitation may reflect the absence of subtests for learning and memory in common tests of general intellectual functioning for children (e.g. the Stanford-Binet and Wechsler Intelligence Scale for Children). Nevertheless, young children who later develop schizophrenia show deficits in language and knowledge acquisition, which could conceivably reflect learning and memory deficits. For example, pre-schizophrenia individuals show reliable delays in speech acquisition in infancy [4,160], receptive language deficits at ages 3, 5, 7, and 9 [3,138,161], and expressive language, verbal and visual knowledge, and academic achievement impairments at age 7 [3,4,156,160-166, but see 167]. Additionally, child and adolescent relatives of patients with schizophrenia show deficits on various learning and memory tests [167-170] and nearly all studies that directly assessed learning and memory in adolescents and young adults who later developed schizophrenia found that learning/memory was indeed impaired [159,171-178].
Conversely, studies assessing higher-cognitive functions in individuals who later develop schizophrenia suggest that working memory and attention deficits may be less robust during childhood and adolescence. For example, although some birth cohort and genetic high risk studies detected attention or working memory deficits among children or young adolescents who later developed schizophrenia [141,155] others studies did not detect deficits [163,166]. Similarly, some studies found deficits in working memory and selective attention in adolescents and young adults at clinical high risk for schizophrenia who later developed psychosis [173,174,177], but several others did not [158,171,176,178]. Interestingly, multiple studies found that executive function deficits were less predictive of later psychosis than deficits in learning and memory [171-176,179].
As large deficits in both learning/memory and higher cognitive functions are present by first psychotic episode [100], the variable detection of higher cognitive deficits during childhood and adolescence may indicate that these deficits do not reliably emerge until later in development. Indeed, a longitudinal study of over 1,000 individuals found that while children who later developed schizophrenia showed deficits in verbal and visual knowledge acquisition at the first assessment time-point (i.e. age 7) that remained stable thereafter, deficits in processing speed, attention, and working memory were not evident until early adolescence, with pre-schizophrenia individuals showing a lag in performance gains compared to healthy peers [159,180].
Given that sensory, motor, and cognitive functions emerge at different ages and mature at different rates, assessing these functions with uniform sensitivity across development remains an unresolved challenge. Discrepancies in the detection of individual deficits could therefore reflect differences in the psychometric properties and/or discriminative power between tasks across development. Additionally, many of the above-cited studies were cross-sectional, contained relatively small numbers of individuals who developed schizophrenia, and/or utilized neuropsychological tests that depend on several cognitive functions, limiting their ability to isolate specific cognitive functions. Nevertheless, this pattern is intriguingly consistent with early deficits in learning and memory in individuals who ultimately develop schizophrenia, and a progressive emergence of broader deficits in motor, sensory, and higher-cognitive functions. Interestingly, continuity has been found between early motor and language delays and later cognitive deficits in individuals who develop schizophrenia, providing support for a common etiological mechanism [136,180,181].
Neural Signs
Consistent with the broad phenotype of schizophrenia, subtle deficits in gray matter and spine density have been found across the brain in adult schizophrenia. Meta-analyses indicate that gray matter deficits are largest in frontal (-4.8%) and temporal (-4.1%) cortices, with pronounced decreases in the late maturing prefrontal cortex (-6.1%) and superior temporal gyrus (-8.2%; 182). However, subtler decreases also exist in the earlier maturing orbitofrontal cortex (-3.0%) which contains secondary and tertiary olfactory cortex, occipital cortex (-2.7%), and parietal cortex (-2.5%). Interestingly, large decreases are seen in primary auditory cortex (-7.7%). Postmortem studies similarly indicate that in addition to spine deficits in frontal and temporal association regions, spine density may be reduced in primary auditory, visual, somatosensory, and motor cortices in schizophrenia [17,71-74]. Studies quantifying spine density in multiple cortical regions suggest that deficits are subtler in some sensory and motor cortices relative to temporal and frontal association cortices [71,72]; however, few studies examined spine density across multiple regions simultaneously. The presence of pervasive gray matter and spine density deficits that are generally most pronounced in late-maturing regions could reflect the cumulative consequences of impaired synaptic plasticity over development, as synaptic refinement in late-maturing cortices build upon inputs from earlier-maturing sensory, motor, and association cortices.
If deficits in gray matter and spine density reflect disruptions in plasticity-dependent synaptic refinement, deficits in primary sensory and motor cortices should emerge earlier than deficits in temporal and frontal association cortices. To our knowledge, no published studies investigated gray matter volumes in children who later developed schizophrenia. However, it is interesting to note that in adolescence, offspring of parents with schizophrenia showed reduced volumes in the earlier maturing amygdala and hippocampus, but not in the prefrontal cortex [183-185]. Similarly, significantly reduced temporal volume was detected in an older adolescent sample at genetic risk for schizophrenia [186] but not in a younger adolescent sample [185]. Interestingly, multiple studies of adolescents and young adults at clinical high risk for schizophrenia found that the proximal development of a psychotic disorder was associated with exaggerated cortical thinning in superior temporal and frontal regions relative to healthy controls [5-12]. One study in adolescents and young adults also found that baseline reductions in insular cortex volume and the rate of progressive insular reductions predicted later psychosis [187]. The emergence of full-blown psychosis in adolescents and young adults therefore appears to frequently coincide with exaggerated pruning in late-maturing prefrontal and temporal cortical regions. This has been interpreted by some to suggest that temporal and frontal cortex deficits may be required for psychosis; however, it is interesting to note that in longitudinal studies of patients with childhood-onset schizophrenia (i.e. psychosis-onset prior to age 13), deficits in temporal and frontal association gray matter were not detectable until late adolescence [188,189]. Indeed, among these childhood-onset patients, deficits in prefrontal and temporal regions were only observed after psychotic symptom onset. Conversely, gray matter deficits that were evident in early adolescence included parietal, motor, and frontal pole deficits that progressed over time [188,189]. These findings suggest that the robust prefrontal and temporal gray matter deficits seen in adult schizophrenia may not be required for psychosis per se, and instead, may be late-manifesting markers of a broader pathological process of disrupted synaptic plasticity. Regardless, developmental stage may be an important mediator for the detectability of regional volume and gray matter deficits in schizophrenia, parallel to behavioral signs and symptoms.
Additional studies examining the temporal emergence of regional gray matter deficits in individuals who later develop schizophrenia are greatly needed, as are functional MRI and electrophysiological studies examining the age at which neurobiological correlates of impaired plasticity and broader sensory, motor, and higher-cognitive deficits emerge. For example, given that electrophysiological signatures during the MMN and LTP tasks are thought to index synaptic plasticity processes, deficits on these tasks should be detectable early in development. Conversely, abnormalities in gamma oscillations or brain activation and connectivity involving late-maturing regions (e.g. prefrontal cortex) would not be expected to be reliably observed until later in adolescence or early adulthood in pre-schizophrenia individuals, given their protracted development [54,190]. An initial study identified deficits in the MMN response among adolescents and young adults who subsequently developed schizophrenia [191]. However, to our knowledge, no published reports shed light on the age at which indices of abnormal brain activation and connectivity or neural oscillations emerge in pre-schizophrenia individuals.
PATHWAYS FROM IMPAIRED SYNAPTIC PLASTICITY TO PSYCHOSIS
Given evidence suggesting that impaired synaptic plasticity could give rise to the sequential appearance of sensory, motor and cognitive impairments over childhood and early adolescence in pre-schizophrenia individuals, could a similar mechanism account for the onset of psychotic symptoms in late adolescence or early adulthood? The following ideas regarding the neural substrate for psychosis are considered from the perspective of impaired synaptic plasticity.
Impaired Neural Tuning
If impaired synaptic plasticity disrupts the stabilization of adaptive synapses, this is likely to lead to increased synaptic pruning during development, and possibly impaired tuning of neural networks overall. Feinberg [192] was the first to suggest that aberrant synaptic pruning in adolescence may lead to faulty “neuronal integration” in schizophrenia, and ultimately, psychotic symptoms, possibly through disordered neural feedback regarding self-generated thoughts and movements (i.e. impaired “corollary discharge”; 193). Others have expanded on this hypothesis (e.g. [194]) and provided interpretations that incorporate advances in our understanding of how the brain codes information. For example, Krystal et al., [49] described the critical role of balance between glutamatergic excitation and GABAergic inhibition in generating finely-tuned neural circuits that efficiently represent information. They postulated that genetically-driven deficits in glutamatergic synaptic development may ultimately lead to impaired tuning of neural circuits; imprecise representations of information; and cognitive symptoms, delusions, and hallucinations. The notion that psychotic symptoms are an outcome of degraded code for representing and distinguishing between stimuli is an attractive conceptualization, and may be consistent with evidence that aberrant gamma oscillations (which reflect interactions between cortical pyramidal neurons and PV interneurons) predict both psychotic symptoms [195-197], and deficits in processing and discriminating between stimuli in schizophrenia [197-198].
Circuit Dysfunction and Dopamine
Separate lines of evidence have highlighted a role for dopamine in psychotic symptoms, particularly excessive dopamine function in the associative striatum. For example, schizophrenia patients show higher dopamine synthesis in the striatum that correlates with positive symptoms [199] and increases during the transition to psychosis among prodromal patients [200,201]; antipsychotic medications exert therapeutic effects via the dopamine system [202], with a threshold of striatal D2 receptor blockade required for antipsychotic effect [203]; and reduced cortico-striatal connectivity is associated with poor response to antipsychotics and longer duration of untreated psychosis [204], whereas positive response to antipsychotics is associated with increases in functional connectivity between the striatum and prefrontal and limbic cortices [205]. As dopamine is critically involved in assigning salience to internal and external stimuli, the role of dopamine in psychosis has been most commonly explained within a framework of aberrant salience [206]. Specifically, the aberrant salience model postulates that dysregulated dopamine release leads to the misattribution of salience to innocuous stimuli, and ultimately delusions, as individuals form narratives over time to explain their experiences [199,206]. Aberrant salience is consistent with the common experience of emotional and sensory overload during the prodromal period of schizophrenia, and/or the increasing belief that innocuous stimuli or events have important meaning [207].
The appearance of excessive striatal dopamine could be a downstream consequence of impaired synaptic plasticity in schizophrenia. As described by others, dopamine neurons are regulated by prefrontal pyramidal neuron projections to midbrain dopamine nuclei (i.e. the ventral tegmental area and substantia nigra; VTA/SN), which then project to the striatum. This suggests that dopamine transmission is vulnerable to alterations in prefrontal cortical drive that may emerge as prefrontal circuits mature in adolescence [48]. Critically, animal studies recently demonstrated that genetically-driven deficits in plasticity-induced spine remodeling can produce the age-dependent emergence of various cognitive, behavioral, and neural signs, including elevated striatal dopamine and psychotic-like symptoms in adulthood that are ameliorated by antipsychotic administration [208,209]. Indeed, in these animals, impaired plasticity-induced spine remodeling was found to lead to a loss of cortical dendritic spines in adulthood, and ultimately, elevated striatal dopamine via aberrant frontal cortex projections to the dopamine-producing neurons of the VTA/SN. This provides compelling evidence that genetic alterations that impair synaptic plasticity can produce the age-dependent emergence of not only dendritic spine deficits, but also excessive striatal dopamine via downstream circuit-level dysfunction [210].
Impaired Synaptic Plasticity and the Pathogenesis of Psychosis
In concert, the currently available data provide support for the concept that impaired synaptic plasticity may manifest as impaired stability of spines/synapses in schizophrenia, the consequences of which are propagated and amplified through development as neural circuit refinement progresses hierarchically across different neural circuits. In the face of these impairments, compensatory responses (such as homeostatic scaling of inhibitory activity; [211]) may mitigate the effects of increasingly altered glutamatergic synaptic neurotransmission. However, the net effect might be sub-optimally tuned cortical circuits that generate faulty representations of stimuli and experiences, and disrupted prefrontal inputs that alter subcortical dopamine neurotransmission in late adolescence, ultimately leading to psychosis.
CONCLUDING REMARKS
Disentangling the pathogenesis of schizophrenia to account for its multi-faceted phenotype remains a challenge. However, accumulating evidence suggests that impaired synaptic plasticity may be a key pathogenic factor in the illness. Here, we reviewed the biological processes involved in synaptic plasticity; considered post-mortem and genetic evidence that plasticity is impaired in schizophrenia; and presented a framework for considering how impaired synaptic plasticity could interact with brain maturation to yield the progressive emergence of diverse signs and symptoms associated with schizophrenia across development. Although future studies are needed to test this model directly (see Outstanding Questions Box), available neurocognitive and structural imaging data suggest that impaired synaptic plasticity that may be detectable as early and fundamental deficits in learning and memory, and the sequential emergence of broader deficits in sensory, motor, and higher-cognitive functions that parallel normal brain maturation.
Importantly, this model suggests that synaptic plasticity pathways may be a promising treatment target in schizophrenia. Some studies have attempted to target synaptic plasticity in adult schizophrenia patients, largely by targeting NMDAR function, with some positive effects on specific plasticity measures (e.g., [212,213] but see [214]). However, benefits have generally not extended to broader aspects of cognition [212,215]. If impaired synaptic plasticity disrupts the optimal tuning of local and long-range circuits, targeting plasticity deficits with pharmacological interventions and/or behavioral interventions intended to strengthen adaptive synapses earlier in development (e.g., support for school/extracurricular involvement, cognitive enhancement training, cognitive behavioral therapy) may provide more effective options for ameliorating the emergence of subsequent cognitive and/or psychotic symptoms [216–219]. Indeed, meta-analyses suggest that behavioral interventions can delay or prevent conversion to psychosis among some clinical high risk individuals [220,221], although sample sizes have been modest, effects on cognitive and neural functioning have been rarely investigated, and the neural mechanisms of treatment efficacy are unclear.
As the field increasingly moves towards a clinical staging model of schizophrenia, hope for our ability to identify individuals early in the course of illness, and ultimately, to alter disease trajectory, is an increasingly salient and conceivable goal. Incorporating a developmental perspective and testing specific hypotheses regarding how and when specific pathogenic factors, such as impaired synaptic plasticity, interact with neurodevelopment to yield the broad signs and symptoms associated with schizophrenia is a crucial next step.
Trends Box.
The broad phenotype of schizophrenia includes sensory, motor, and cognitive signs and symptoms that emerge prior to psychosis.
Evidence from post-mortem and large-scale genetic studies suggests that synaptic plasticity is disrupted in schizophrenia and may be a key pathogenic factor in the disease.
Given the role of synaptic plasticity in learning, memory, and the maturation of neural circuits, the consequences of impaired plasticity through development may explain the emergence of many signs and symptoms of the broader schizophrenia phenotype.
Extant literature is consistent with manifestations of impaired synaptic plasticity that may be detectable as early and fundamental deficits in learning and memory, and the progressive emergence of broader sensory, motor, cognitive, and psychotic features.
OUTSTANDING QUESTIONS BOX.
1. At what age do deficits in higher-level auditory, visual, and olfactory sensory processing emerge in children and adolescents who later develop schizophrenia?
2. At what age do regionally-specific gray matter deficits emerge in children and adolescents who later develop schizophrenia?
3. At what age do abnormalities in neural indices of synaptic plasticity such as altered MMN or LTP deficits, or neurobiological correlates of neurocognitive deficits, such as altered dorsolateral prefrontal cortex activation and connectivity during higher-order cognitive tasks, emerge in individuals who later develop schizophrenia?
4. Do environmental processes, such as chronic stress associated with migration/ethnic minority status and/or withdrawal from structured activities during the prodromal phase further compound gray matter loss or exacerbate reductions in synapses encoding adaptive pathways?
4. Do genetic animal models targeting synaptic plasticity pathways recapitulate this pattern of magnitude and developmental age of emergence of schizophrenia-like signs and symptoms?
5. Do all or only a subset of individuals with schizophrenia have a genetic risk profile that disproportionately affects synaptic plasticity pathways? Similarly, does this pattern of the magnitude and age of emergence of signs and symptoms exist only for a subset of patients with a genetic risk profile affecting synaptic plasticity?
6. Can we distinguish the role of impaired synaptic plasticity in generating diverse signs and symptoms associated with schizophrenia from other hypothesized pathogenic processes?
7. Is impaired synaptic plasticity a primary pathogenic process that contributes to and/or can partially explain other neurobiological markers seen in schizophrenia (e.g. alterations in GABAergic interneurons, PNNs, excitatory/inhibitory balance, and dopamine transmission)?
Glossary
- Actin
An abundant protein that dynamically assembles and disassembles in long filaments to form part of the cytoskeleton. During the induction of plasticity, dynamic changes in actin polymerization underlie morphological changes in the size and shape of dendritic spines and actin filaments traffic AMPA receptors to and from the postsynaptic membrane.
- Arc protein
Activity-regulated cytoskeleton-associated protein an early gene product that localizes to NMDAR-activated synapses following the induction of synaptic plasticity and is involved in the remodeling and maintenance of synaptic changes.
- Common genetic variant
Genetic variants present in more than 1 percent of the population.
- Copy number variant mutation
Deletions or duplications of stretches of DNA in a chromosomal region.
- De novo genetic mutation
A genetic variant that is present for the first time in a family member as a result of mutation (i.e. not found in either parent).
- Cytoskeleton
A cellular meshwork comprised of microtubules, actin filaments, and intermediate filaments that provides structure and shape to cells through linkages to itself, the membrane, and internal organelles.
- MAGUK proteins
Membrane associated guanylate kinase proteins a large family of scaffold proteins that are highly enriched at synapses and form protein-protein interactions with cytoskeleton proteins and molecules involved in signal transduction.
- PSD
Postsynaptic density; an electron-dense structure that arises from a network of hundreds of proteins lying immediately below the membrane of glutamatergic synapses and includes neurotransmitter receptors, signaling molecules, cytoskeleton, and scaffold proteins.
- PSD-95
Postsynaptic density protein 95; a MAGUK protein that associates with glutamate receptors is the major scaffolding protein of the postsynaptic density, and critically regulates synaptic plasticity.
- Rare genetic variant
Genetic variants with a minor allele that occurs in less than 1 percent of the population.
- Scaffold proteins
Proteins that interact with multiple members within a signaling pathway, tethering them into complexes.
- SNARE proteins
Soluble NSF-Attachment Protein Receptor proteins a group of proteins that form a complex and mediate the fusion of vesicles and the membrane.
- Synapse
The junction between two neurons through which signals are passed from one neuron to the next, most commonly via neurotransmitters.
- Synaptic plasticity
The capacity of synapses to undergo structural and functional changes following environmental input or use that strengthen or weaken their strength.
- Synaptic weight
The strength of connection between two neurons, and consequently, the extent to which the activity of one neuron influences the activity of the next.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatr. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 2.Murray RM, Lewis SW. Is schizophrenia a neurodevelopmental disorder? BMJ. 1987;295:681–682. doi: 10.1136/bmj.295.6600.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welham J, et al. The antecedents of schizophrenia: a review of birth cohort studies. Schizophr Bull. 2009;35:603–623. doi: 10.1093/schbul/sbn084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones P, et al. Child developmental risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet. 1994;344:1398–1402. doi: 10.1016/s0140-6736(94)90569-x. [DOI] [PubMed] [Google Scholar]
- 5.Pantelis C, et al. Structural brain imaging evidence for multiple pathological processes at different stages of brain development in schizophrenia. Schizophr Bull. 2005;31:672–696. doi: 10.1093/schbul/sbi034. [DOI] [PubMed] [Google Scholar]
- 6.Gogtay N, et al. Age of onset of schizophrenia: perspectives from structural neuroimaging studies. Schizophr Bull. 2011;37:504–513. doi: 10.1093/schbul/sbr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pantelis C, et al. Neuroanatomical abnormalities before and after onset of psychosis: a cross-sectional and longitudinal MRI comparison. Lancet. 2003;361:281–288. doi: 10.1016/S0140-6736(03)12323-9. [DOI] [PubMed] [Google Scholar]
- 8.Sun D, et al. Progressive brain structural changes mapped as psychosis develops in “at risk” individuals. Schizophr Res. 2009;108:85–92. doi: 10.1016/j.schres.2008.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Job DE, et al. Grey matter changes over time in high risk subjects developing schizophrenia. Neuroimage. 2005;25:1023–1030. doi: 10.1016/j.neuroimage.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi T, et al. Progressive gray matter reduction of the superior temporal gyrus during transition to psychosis. Arch Gen Psychiatry. 2009;66:366–376. doi: 10.1001/archgenpsychiatry.2009.12. [DOI] [PubMed] [Google Scholar]
- 11.Cannon TD, et al. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry. 2015;77:147–157. doi: 10.1016/j.biopsych.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun D, et al. Brain surface contraction mapped in first-episode schizophrenia: a longitudinal magnetic resonance imaging study. Mol Psychiatry. 2009;14:976–986. doi: 10.1038/mp.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McGorry PD, et al. Clinical staging of psychiatric disorders: a heuristic framework for choosing earlier, safer and more effective interventions. Aust N Z J Psychiatry. 2006;40:616–622. doi: 10.1080/j.1440-1614.2006.01860.x. [DOI] [PubMed] [Google Scholar]
- 14.Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- 15.Derkach VA, et al. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- 16.Ho VM, et al. The cell biology of synaptic plasticity. Science. 2011;334:623–628. doi: 10.1126/science.1209236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Glausier JR, Lewis DA. Dendritic spine pathology in schizophrenia. Neuroscience. 2013;251:90–107. doi: 10.1016/j.neuroscience.2012.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tønnesen J, Nägerl UV. Dendritic spines as tunable regulators of synaptic signals. Front Psychiatry. 2016;7:101. doi: 10.3389/fpsyt.2016.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lüscher C, Malenka RC. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD) Cold Spring Harb Perspect Biol. 2012;4:a005710. doi: 10.1101/cshperspect.a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kessels HW, Malinow R. Synaptic AMPA receptor plasticity and behavior. Neuron. 2009;61:340–350. doi: 10.1016/j.neuron.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okamoto KI, et al. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci. 2004;7:1104–1112. doi: 10.1038/nn1311. [DOI] [PubMed] [Google Scholar]
- 22.Honkura N, et al. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron. 2008;57:719–729. doi: 10.1016/j.neuron.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 23.Kasai H, et al. Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 2010;33:121–129. doi: 10.1016/j.tins.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 24.Bosch M, et al. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron. 2014;82:444–459. doi: 10.1016/j.neuron.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer D, et al. Balance and stability of synaptic structures during synaptic plasticity. Neuron. 2014;82:430–443. doi: 10.1016/j.neuron.2014.02.031. [DOI] [PubMed] [Google Scholar]
- 26.Watt AJ, et al. A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat Neurosci. 2004;7:518–524. doi: 10.1038/nn1220. [DOI] [PubMed] [Google Scholar]
- 27.Caroni P, et al. Structural plasticity upon learning: regulation and functions. Nat Rev Neurosci. 2012;13:478–490. doi: 10.1038/nrn3258. [DOI] [PubMed] [Google Scholar]
- 28.Johansen JP, et al. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grueter BA, et al. Integrating synaptic plasticity and striatal circuit function in addiction. Curr Opin Neurobiol. 2012;22:545–551. doi: 10.1016/j.conb.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin HH, et al. Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nature Neurosci. 2009;12:333–341. doi: 10.1038/nn.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu M, et al. Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature. 2012;483:92–95. doi: 10.1038/nature10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu X, Zuo Y. Spine plasticity in the motor cortex. Curr Opin Neurobiol. 2011;21:169–174. doi: 10.1016/j.conb.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci. 2009;32:33–55. doi: 10.1146/annurev.neuro.051508.135516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tau GZ, Peterson BS. Normal development of brain circuits. Neuropsychopharmacology. 2010;35:147–168. doi: 10.1038/npp.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang ZW, et al. Essential role of postsynaptic NMDA receptors in developmental refinement of excitatory synapses. Proc Natl Acad Sci USA. 2013;110:1095–1100. doi: 10.1073/pnas.1212971110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi T, et al. Experience strengthening transmission by driving AMPA receptors into synapses. Science. 2003;299:1585–1588. doi: 10.1126/science.1079886. [DOI] [PubMed] [Google Scholar]
- 37.Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takesian AE, Hensch TK. Balancing plasticity/stability across brain development. Prog Brain Res. 2013;207:3–34. doi: 10.1016/B978-0-444-63327-9.00001-1. [DOI] [PubMed] [Google Scholar]
- 39.Le Magueresse C, Monyer H. GABAergic interneurons shape the functional maturation of the cortex. Neuron. 2013;77:388–405. doi: 10.1016/j.neuron.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 40.Wu X, et al. GABA signaling promotes synapse elimination and axon pruning in developing cortical inhibitory interneurons. J Neurosci. 2012;32:331–343. doi: 10.1523/JNEUROSCI.3189-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hayama T, et al. GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat Neurosci. 2013;16:1409–1416. doi: 10.1038/nn.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dehorter N, et al. Tuning neural circuits by turning the interneuron knob. Curr Opin Neurobiol. 2017;42:144–151. doi: 10.1016/j.conb.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Lewis DA. Inhibitory neurons in human cortical circuits: substrate for cognitive dysfunction in schizophrenia. Curr Opin Neurobiol. 2014;26:22–26. doi: 10.1016/j.conb.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mauney SA, et al. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry. 2013;74:427–435. doi: 10.1016/j.biopsych.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Enwright JF, et al. Reduced Labeling of Parvalbumin Neurons and Perineuronal Nets in the Dorsolateral Prefrontal Cortex of Subjects with Schizophrenia. Neuropsychopharmacology. 2016;41:2206–2214. doi: 10.1038/npp.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bitanihirwe BKY, et al. Weaving a Net of Neurobiological Mechanisms in Schizophrenia and Unraveling the Underlying Pathophysiology. Biol Psychiatry. 2016;80:589–598. doi: 10.1016/j.biopsych.2016.03.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung DW, et al. Pathological Basis for Deficient Excitatory Drive to Cortical Parvalbumin Interneurons in Schizophrenia. Am J Psychiatry. 2016;173:1131–1139. doi: 10.1176/appi.ajp.2016.16010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis DA, Gonzalez-Burgos G. Pathophysiologically based treatment interventions in schizophrenia. Nat Med. 2006;12:1016–1022. doi: 10.1038/nm1478. [DOI] [PubMed] [Google Scholar]
- 49.Krystal JH, et al. Impaired Tuning of Neural Ensembles and the Pathophysiology of Schizophrenia: A Translational and Computational Neuroscience Perspective. Biol Psychiatry. doi: 10.1016/j.biopsych.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson MH. Functional brain development in humans. Nat Rev Neurosci. 2001;2:475–483. doi: 10.1038/35081509. [DOI] [PubMed] [Google Scholar]
- 51.Best JR, Miller PH. A developmental perspective on executive function. Child Dev. 2010;81:1641–1660. doi: 10.1111/j.1467-8624.2010.01499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menon V. Developmental pathways to functional brain networks: emerging principles. Trends Cogn Sci. 2013;17:627–640. doi: 10.1016/j.tics.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 53.Amso D, Casey BJ. Beyond what develops when neuroimaging may inform how cognition changes with development. Curr Dir Psychol Sci. 2006;15:24–29. [Google Scholar]
- 54.Gogtay N, et al. Dynamic mapping of human cortical development during childhood through early adulthood. Proc Natl Acad Sci USA. 2004;101:8174–8179. doi: 10.1073/pnas.0402680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaw P, et al. Neurodevelopmental trajectories of the human cerebral cortex. J Neurosci. 2008;28:3586–3594. doi: 10.1523/JNEUROSCI.5309-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breukelaar IA, et al. Cognitive control network anatomy correlates with neurocognitive behavior: A longitudinal study. Hum Brain Mapp. 2016;38:631–643. doi: 10.1002/hbm.23401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tamnes CK, et al. Longitudinal working memory development is related to structural maturation of frontal and parietal cortices. J Cogn Neurosci. 2013;25:1611–1623. doi: 10.1162/jocn_a_00434. [DOI] [PubMed] [Google Scholar]
- 58.Luna B, et al. What has fMRI told us about the Development of Cognitive Control through Adolescence? Brain Cogn. 2010/2;72:101–113. doi: 10.1016/j.bandc.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koolschijn PCMP, et al. A three-year longitudinal functional magnetic resonance imaging study of performance monitoring and test-retest reliability from childhood to early adulthood. J Neurosci. 2011;31:4204–4212. doi: 10.1523/JNEUROSCI.6415-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finn AS, et al. Longitudinal evidence for functional specialization of the neural circuit supporting working memory in the human brain. J Neurosci. 2010;30:11062–11067. doi: 10.1523/JNEUROSCI.6266-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bunge SA, Wright SB. Neurodevelopmental changes in working memory and cognitive control. Curr Opin Neurobiol. 2007;17:243–250. doi: 10.1016/j.conb.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 62.Paus T, et al. Why do many psychiatric disorders emerge during adolescence? Nat Rev Neurosci. 2008;9:947–957. doi: 10.1038/nrn2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tamnes CK, et al. Brain maturation in adolescence and young adulthood: regional age-related changes in cortical thickness and white matter volume and microstructure. Cereb Cortex. 2010;20:534–548. doi: 10.1093/cercor/bhp118. [DOI] [PubMed] [Google Scholar]
- 64.Petanjek Z, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci USA. 2011;108:13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 66.Huttenlocher PR. Morphometric study of human Cereb. Cortex development. Neuropsychologia. 1990;28:517–527. doi: 10.1016/0028-3932(90)90031-i. [DOI] [PubMed] [Google Scholar]
- 67.Gogtay N, Thompson PM. Mapping gray matter development: implications for typical development and vulnerability to psychopathology. Brain Cognition. 2010;72:6–15. doi: 10.1016/j.bandc.2009.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Selemon LD. A role for synaptic plasticity in the adolescent development of executive function. Transl Psychiatry. 2013;3:e238. doi: 10.1038/tp.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Luby ED, et al. Study of a new schizophrenomimetic drug—Sernyl. AMA Arch Neurol. 1959;81:363–369. doi: 10.1001/archneurpsyc.1959.02340150095011. [DOI] [PubMed] [Google Scholar]
- 70.Krystal JH, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 71.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 72.Garey L. When cortical development goes wrong: schizophrenia as a neurodevelopmental disease of microcircuits. J Anat. 2010;217:324–333. doi: 10.1111/j.1469-7580.2010.01231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sweet RA, et al. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.MacDonald ML, et al. Altered glutamate protein co-expression network topology linked to spine loss in the auditory cortex of schizophrenia. Biol Psychiatry. 2015;77:959–968. doi: 10.1016/j.biopsych.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hill JJ, et al. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- 76.Ide M, Lewis DA. Altered cortical CDC42 signaling pathways in schizophrenia: implications for dendritic spine deficits. Biol Psychiatry. 2010;68:25–32. doi: 10.1016/j.biopsych.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Datta D, et al. Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biol Psychiatry. 2015;78:775–785. doi: 10.1016/j.biopsych.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shelton MA, et al. Loss of microtubule-associated protein 2 immunoreactivity linked to dendritic spine loss in schizophrenia. Biol Psychiatry. 2015;78:374–385. doi: 10.1016/j.biopsych.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Datta D, et al. Altered Expression of ARP2/3 Complex Signaling Pathway Genes in Prefrontal Layer 3 Pyramidal Cells in Schizophrenia. Am J Psychiatry. 2017;174:163–171. doi: 10.1176/appi.ajp.2016.16020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao Z, et al. Transcriptome sequencing and genome-wide association analyses reveal lysosomal function and actin cytoskeleton remodeling in schizophrenia and bipolar disorder. Mol Psychiatry. 2015;20:563–572. doi: 10.1038/mp.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akbarian S, et al. Selective alterations in gene expression for NMDA receptor subunits in prefrontal cortex of schizophrenics. J Neurosci. 1996;16:19–30. doi: 10.1523/JNEUROSCI.16-01-00019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- 83.Kristiansen LV, et al. Changes in NMDA receptor subunits and interacting PSD proteins in dorsolateral prefrontal and anterior cingulate cortex indicate abnormal regional expression in schizophrenia. Mol Psychiatry. 2006;11:737–747. doi: 10.1038/sj.mp.4001844. [DOI] [PubMed] [Google Scholar]
- 84.Emamian ES, et al. Decreased phosphorylation of NMDA receptor type 1 at serine 897 in brains of patients with Schizophrenia. J Neurosci. 2004;24:1561–1564. doi: 10.1523/JNEUROSCI.4650-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of AMPA receptor trafficking and signaling molecule transcripts in the prefrontal cortex in schizophrenia. Synapse. 2006;60:585–598. doi: 10.1002/syn.20329. [DOI] [PubMed] [Google Scholar]
- 86.Pennington K, et al. Prominent synaptic and metabolic abnormalities revealed by proteomic analysis of the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder. Mol Psychiatry. 2008;13:1102–1117. doi: 10.1038/sj.mp.4002098. [DOI] [PubMed] [Google Scholar]
- 87.Funk AJ, et al. Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia. Neuroreport. 2009;20:1019. doi: 10.1097/WNR.0b013e32832d30d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Funk AJ. Abnormal activity of the MAPK-and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology. 2012;37:896–905. doi: 10.1038/npp.2011.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li W, et al. Synaptic Proteins in the Hippocampus Indicative of Increased Neuronal Activity in CA3 in Schizophrenia. Am J Psychiatry. 2015;4:373–382. doi: 10.1176/appi.ajp.2014.14010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sawada K, et al. Hippocampal complexin proteins and cognitive dysfunction in schizophrenia. Arch Gen Psychiatry. 2005;62:263–272. doi: 10.1001/archpsyc.62.3.263. [DOI] [PubMed] [Google Scholar]
- 91.Maycox PR, et al. Analysis of gene expression in two large schizophrenia cohorts identifies multiple changes associated with nerve terminal function. Mol Psychiatry. 2009;14:1083–1094. doi: 10.1038/mp.2009.18. [DOI] [PubMed] [Google Scholar]
- 92.Föcking M, et al. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol Psychiatry. 2015;20:424–432. doi: 10.1038/mp.2014.63. [DOI] [PubMed] [Google Scholar]
- 93.Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marshall C, et al. A contribution of novel CNVs to schizophrenia from a genome-wide study of 41, 321 subjects. bioRxiv. 2016:040493. [Google Scholar]
- 95.CNV and Schizophrenia Working Groups of the Psychiatric Genomics Consortium and Psychosis Endophenotypes International Consortium. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35. doi: 10.1038/ng.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Purcell SM, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506:185–190. doi: 10.1038/nature12975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McCarroll SA, et al. Genome-scale neurogenetics: methodology and meaning. Nat Neurosci. 2014;17:756–763. doi: 10.1038/nn.3716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mesholam-Gately RI, et al. Neurocognition in first-episode schizophrenia: a meta-analytic review. Neuropsychology. 2009;23:315. doi: 10.1037/a0014708. [DOI] [PubMed] [Google Scholar]
- 101.Schaefer J, et al. The global cognitive impairment in schizophrenia: consistent over decades and around the world. Schizophr Res. 2013;150:42–50. doi: 10.1016/j.schres.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Walther S, Strik W. Motor symptoms and schizophrenia. Neuropsychobiology. 2012;66:77–92. doi: 10.1159/000339456. [DOI] [PubMed] [Google Scholar]
- 103.Exner C, et al. Reduced size of the pre-supplementary motor cortex and impaired motor sequence learning in first-episode schizophrenia. Schizophr Res. 2006;84:386–396. doi: 10.1016/j.schres.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 104.Morrens M. Psychomotor slowing in schizophrenia. Schizophr Bull. 2007;33:1038–1053. doi: 10.1093/schbul/sbl051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Javitt DC, Freedman R. Sensory processing dysfunction in the personal experience and neuronal machinery of schizophrenia. Am J Psychiatry. 2014;172:17–31. doi: 10.1176/appi.ajp.2014.13121691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Javitt DC, Sweet RA. Auditory dysfunction in schizophrenia: integrating clinical and basic features. Nat Rev Neurosci. 2015;16:535–550. doi: 10.1038/nrn4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kugler BT, Caudrey DJ. Phoneme discrimination in schizophrenia. Br J Psychiatry. 1983;142:53–59. doi: 10.1192/bjp.142.1.53. [DOI] [PubMed] [Google Scholar]
- 108.Holcomb HH, et al. Tone discrimination performance in schizophrenic patients and normal volunteers: impact of stimulus presentation levels and frequency differences. Psychiatry Res. 1995;57:75–82. doi: 10.1016/0165-1781(95)02270-7. [DOI] [PubMed] [Google Scholar]
- 109.Perrin MA, et al. Spatial localization deficits and auditory cortical dysfunction in schizophrenia. Schizophr Res. 2010;124:161–168. doi: 10.1016/j.schres.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brenner CA, et al. Psychometrically matched visual-processing tasks in schizophrenia spectrum disorders. J Abnorm Psychol. 2003;112:28–37. [PubMed] [Google Scholar]
- 111.Butler PD, et al. Visual perception and its impairment in schizophrenia. Biol Psychiatry. 2008;64:40–47. doi: 10.1016/j.biopsych.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Silverstein SM, Keane BP. Perceptual organization impairment in schizophrenia and associated brain mechanisms: review of research from 2005 to 2010. Schizophr Bull. 2011;37:690–699. doi: 10.1093/schbul/sbr052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen Y. Abnormal visual motion processing in schizophrenia: a review of research progress. Schizophr Bull. 2011;37:709–715. doi: 10.1093/schbul/sbr020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Brewer WJ, et al. Stability of olfactory identification deficits in neuroleptic-naive patients with first-episode psychosis. Am J Psychiatry. 2001;158:107–115. doi: 10.1176/appi.ajp.158.1.107. [DOI] [PubMed] [Google Scholar]
- 115.Turetsky BI, et al. Scents and non-sense: olfactory dysfunction in schizophrenia. Schizophr Bull. 2009;35:1117–1131. doi: 10.1093/schbul/sbp111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reichenberg A. The assessment of neuropsychological functioning in schizophrenia. Dialogues Clin Neurosci. 2010;12:383–392. doi: 10.31887/DCNS.2010.12.3/areichenberg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Heinrichs RW, Zakzanis KK. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology. 1998;12:426–445. doi: 10.1037//0894-4105.12.3.426. [DOI] [PubMed] [Google Scholar]
- 118.Reichenberg A, et al. Neuropsychological function and dysfunction in schizophrenia and psychotic affective disorders. Schizophr Bull. 2009;35:1022–1029. doi: 10.1093/schbul/sbn044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Javitt DC. When doors of perception close: bottom-up models of disrupted cognition in schizophrenia. Annu Rev Clin Psychol. 2009;5:249–275. doi: 10.1146/annurev.clinpsy.032408.153502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sehatpour P, et al. Impaired visual object processing across an occipital-frontal-hippocampal brain network in schizophrenia: an integrated neuroimaging study. Arch Gen Psychiatry. 2010;67:772–782. doi: 10.1001/archgenpsychiatry.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yang GJ, et al. Functional hierarchy underlies preferential connectivity disturbances in schizophrenia. Proc Natl Acad Sci U S A. 2016;113:E219–28. doi: 10.1073/pnas.1508436113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Barch DM, Ceaser A. Cognition in schizophrenia: core psychological and neural mechanisms. Trends Cogn Sci. 2012;16:27–34. doi: 10.1016/j.tics.2011.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Uhlhaas PJ. Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Curr Opin Neurobiol. 2013;23:283–290. doi: 10.1016/j.conb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 124.Sun Y, et al. Gamma oscillations in schizophrenia: Mechanisms and clinical significance. Brain Res. 2011;1413:98–114. doi: 10.1016/j.brainres.2011.06.065. [DOI] [PubMed] [Google Scholar]
- 125.Cavu I, et al. Impaired Visual Cortical Plasticity in Schizophrenia. Biol Psychiatry. 2012;71:512–520. doi: 10.1016/j.biopsych.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mears RP, Spencer KM. Electrophysiological assessment of auditory stimulus-specific plasticity in schizophrenia. Biol Psychiatry. 2012;71:503–511. doi: 10.1016/j.biopsych.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhou D, et al. Altered Motor-Striatal Plasticity and Cortical Functioning in Patients with Schizophrenia. Neurosci Bull. 2016 doi: 10.1007/s12264-016-0079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Erickson MA, et al. A Meta-Analysis of Mismatch Negativity in Schizophrenia: From Clinical Risk to Disease Specificity and Progression. Biol Psychiatry. 2016;79:980–987. doi: 10.1016/j.biopsych.2015.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005;76:1–23. doi: 10.1016/j.schres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 130.Stephan KE, et al. Dysconnection in Schizophrenia: From Abnormal Synaptic Plasticity to Failures of Self-monitoring. Schizophr Bull. 2009;35:509–527. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Javitt DC, et al. Role of cortical N-methyl-D-aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci U S A. 1996;93:11962–11967. doi: 10.1073/pnas.93.21.11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Forsyth JK, et al. Augmenting NMDA receptor signaling boosts experience-dependent neuroplasticity in the adult human brain. Proc Natl Acad Sci U S A. 2015;112:15331–15336. doi: 10.1073/pnas.1509262112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Clapp WC, et al. Translating long-term potentiation from animals to humans: a novel method for noninvasive assessment of cortical plasticity. Biol Psychiatry. 2012;71:496–502. doi: 10.1016/j.biopsych.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Walker EF, et al. Neuromotor precursors of schizophrenia. Schizophr Bull. 1994;20:441–451. doi: 10.1093/schbul/20.3.441. [DOI] [PubMed] [Google Scholar]
- 135.Sørensen HJ, et al. Early developmental milestones and risk of schizophrenia: a 45-year follow-up of the Copenhagen Perinatal Cohort. Schizophr Res. 2010;118:41–47. doi: 10.1016/j.schres.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Murray GK, et al. Infant motor development and adult cognitive functions in schizophrenia. Schizophr Res. 2006;81:65–74. doi: 10.1016/j.schres.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 137.Isohanni M, et al. Early developmental milestones in adult schizophrenia and other psychoses. A 31-year follow-up of the Northern Finland 1966 Birth Cohort. Schizophr Res. 2001;52:1–19. doi: 10.1016/s0920-9964(00)00179-1. [DOI] [PubMed] [Google Scholar]
- 138.Cannon M, et al. Evidence for early-childhood, pan-developmental impairment specific to schizophreniform disorder: results from a longitudinal birth cohort. Arch Gen Psychiatry. 2002;59:449–456. doi: 10.1001/archpsyc.59.5.449. [DOI] [PubMed] [Google Scholar]
- 139.Rosso IM, et al. Childhood neuromotor dysfunction in schizophrenia patients and their unaffected siblings: a prospective cohort study. Schizophr Bull. 2000;26:367. doi: 10.1093/oxfordjournals.schbul.a033459. [DOI] [PubMed] [Google Scholar]
- 140.Rakhshan P, et al. Childhood pegboard task predicts adult-onset psychosis-spectrum disorder among a genetic high-risk sample. Schizophr Res. 2016;178:68–73. doi: 10.1016/j.schres.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cannon M, et al. Neuropsychological performance at the age of 13 years and adult schizophreniform disorder. Br J Psychiatry. 2006;189:463–464. doi: 10.1192/bjp.bp.105.020552. [DOI] [PubMed] [Google Scholar]
- 142.Hallett S, Green P. Possible defects of interhemispheric integration in children of schizophrenics. J Nerv Ment Dis. 1983;171:421–425. doi: 10.1097/00005053-198307000-00005. [DOI] [PubMed] [Google Scholar]
- 143.Schubert EW, et al. A prospective study of offspring of women with psychosis: visual dysfunction in early childhood predicts schizophrenia-spectrum disorders in adulthood. Acta Psychiatr Scand. 2005;112:385–393. doi: 10.1111/j.1600-0447.2005.00584.x. [DOI] [PubMed] [Google Scholar]
- 144.Schiffman J, et al. Premorbid childhood ocular alignment abnormalities and adult schizophrenia-spectrum disorder. Schizophr Res. 2006;81:253–260. doi: 10.1016/j.schres.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 145.Mittal VA, et al. Visual context processing dysfunctions in youth at high risk for psychosis: Resistance to the Ebbinghaus illusion and its symptom and social and role functioning correlates. J Abnorm Psychol. 2015;124:953. doi: 10.1037/abn0000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Koethe D, et al. Binocular depth inversion as a paradigm of reduced visual information processing in prodromal state, antipsychotic-naive and treated schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2009;259:195–202. doi: 10.1007/s00406-008-0851-6. [DOI] [PubMed] [Google Scholar]
- 147.Lieb K, et al. Preattentive information processing as measured by backward masking and texton detection tasks in adolescents at high genetic risk for schizophrenia. Schizophr Res. 1996;21:171–182. doi: 10.1016/0920-9964(96)00025-4. [DOI] [PubMed] [Google Scholar]
- 148.Brewer WJ, et al. Impairment of olfactory identification ability in individuals at ultra-high risk for psychosis who later develop schizophrenia. Am J Psychiatry. 2003;160:1790–1794. doi: 10.1176/appi.ajp.160.10.1790. [DOI] [PubMed] [Google Scholar]
- 149.Fuller R, et al. Longitudinal assessment of premorbid cognitive functioning in patients with schizophrenia through examination of standardized scholastic test performance. Am J Psychiatry. 2002;159:1183–1189. doi: 10.1176/appi.ajp.159.7.1183. [DOI] [PubMed] [Google Scholar]
- 150.Crow TJ, et al. Chidhood precursors of psychiosis as clues to its evolutionary orgins. Eur Arch Psychiatry Clin Nuerosci. 1995;245:61–69. doi: 10.1007/BF02190732. [DOI] [PubMed] [Google Scholar]
- 151.Revheim N, et al. Reading deficits in schizophrenia and individuals at high clinical risk: relationship to sensory function, course of illness, and psychosocial outcome. Am J Psychiatry. 2014;171:949–959. doi: 10.1176/appi.ajp.2014.13091196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cannon TD. Childhood cognitive functioning in schizophrenia patients and their unaffected siblings: a prospective cohort study. Schizophr Bull. 2000;26:379. doi: 10.1093/oxfordjournals.schbul.a033460. [DOI] [PubMed] [Google Scholar]
- 153.Reichenberg A, et al. A population-based cohort study of premorbid intellectual, language, and behavioral functioning in patients with schizophrenia, schizoaffective disorder, and nonpsychotic bipolar disorder. Am J Psychiatry. 2002;159:2027–2035. doi: 10.1176/appi.ajp.159.12.2027. [DOI] [PubMed] [Google Scholar]
- 154.Khandaker GM, et al. A quantitative meta-analysis of population-based studies of premorbid intelligence and schizophrenia. Schizophr Res. 2011;132:220–227. doi: 10.1016/j.schres.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Seidman LJ, et al. Neuropsychological performance and family history in children at age 7 who develop adult schizophrenia or bipolar psychosis in the New England Family Studies. Psychol Med. 2013;43(01):119–131. doi: 10.1017/S0033291712000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Woodberry KA, et al. Premorbid IQ in schizophrenia: a meta-analytic review. Am J Psychiatry. 2008;165:579–587. doi: 10.1176/appi.ajp.2008.07081242. [DOI] [PubMed] [Google Scholar]
- 157.Bilder RM, et al. Neuropsychology of first-episode schizophrenia: initial characterization and clinical correlates. Am J Psychiatry. 2000;157:549–559. doi: 10.1176/appi.ajp.157.4.549. [DOI] [PubMed] [Google Scholar]
- 158.Cosway R, et al. Neuropsychological change in young people at high risk for schizophrenia: results from the first two neuropsychological assessments of the Edinburgh High Risk Study. Psychol Med. 2000;30:1111–1121. doi: 10.1017/s0033291799002585. [DOI] [PubMed] [Google Scholar]
- 159.Meier MH, et al. Neuropsychological decline in schizophrenia from the premorbid to the postonset period: evidence from a population-representative longitudinal study. Am J Psychiatry. 2014;171:91–101. doi: 10.1176/appi.ajp.2013.12111438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Nicolson R, et al. Premorbid speech and language impairments in childhood-onset schizophrenia: association with risk factors. Am J Psychiatry. 2000;157:794–800. doi: 10.1176/appi.ajp.157.5.794. [DOI] [PubMed] [Google Scholar]
- 161.Bearden CE, et al. A prospective cohort study of childhood behavioral deviance and language abnormalities as predictors of adult schizophrenia. Schizophr Bull. 2000;26:395–410. doi: 10.1093/oxfordjournals.schbul.a033461. [DOI] [PubMed] [Google Scholar]
- 162.Dickson H, et al. Meta-analyses of cognitive and motor function in youth aged 16 years and younger who subsequently develop schizophrenia. Psychol Med. 2012;42:743–755. doi: 10.1017/S0033291711001693. [DOI] [PubMed] [Google Scholar]
- 163.Niendam TA, et al. A prospective study of childhood neurocognitive functioning in schizophrenic patients and their siblings. Am J Psychiatry. 2003;160:2060–2062. doi: 10.1176/appi.ajp.160.11.2060. [DOI] [PubMed] [Google Scholar]
- 164.Seidman LJ, et al. Intellectual decline in schizophrenia: evidence from a prospective birth cohort 28 year follow-up study. J Clin Exp Neuropsychol. 2006;28:225–242. doi: 10.1080/13803390500360471. [DOI] [PubMed] [Google Scholar]
- 165.Davidson M, et al. Behavioral and intellectual markers for schizophrenia in apparently healthy male adolescents. Am J Psychiatry. 1999;156:1328–1335. doi: 10.1176/ajp.156.9.1328. [DOI] [PubMed] [Google Scholar]
- 166.Sørensen HJ, et al. Premorbid neurocognitive functioning in schizophrenia spectrum disorder. Schizophr Bull. 2006;32:578–583. doi: 10.1093/schbul/sbj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Myles-Worsley M, et al. The Palau Early Psychosis Study: neurocognitive functioning in high-risk adolescents. Schizophr Res. 2007;89:299–307. doi: 10.1016/j.schres.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 168.Wagshal D, et al. Deficits in probabilistic classification learning and liability for schizophrenia. Psychiatry Res. 2012;200:167–172. doi: 10.1016/j.psychres.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Maziade M, et al. Verbal and visual memory impairments among young offspring and healthy adult relatives of patients with schizophrenia and bipolar disorder: selective generational patterns indicate different developmental trajectories. Schizophr Bull. 2011;37:1218–1228. doi: 10.1093/schbul/sbq026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Seidman LJ, et al. Neuropsychological functioning in adolescents and young adults at genetic risk for schizophrenia and affective psychoses: results from the Harvard and Hillside Adolescent High Risk Studies. Schizophr Bull. 2006;32:507–524. doi: 10.1093/schbul/sbj078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Whyte MC, et al. Neuropsychological performance over time in people at high risk of developing schizophrenia and controls. Biol Psychiatry. 2006;59:730–739. doi: 10.1016/j.biopsych.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 172.Johnstone EC, et al. Predicting schizophrenia: findings from the Edinburgh high-risk study. Br J Psychiatry. 2005;186:18–25. doi: 10.1192/bjp.186.1.18. [DOI] [PubMed] [Google Scholar]
- 173.Seidman LJ, et al. Neuropsychology of the prodrome to psychosis in the NAPLS consortium: relationship to family history and conversion to psychosis. Arch Gen Psychiatry. 2010;67:578–588. doi: 10.1001/archgenpsychiatry.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Seidman LJ, et al. Association of neurocognition with transition to psychosis: baseline functioning in the second phase of the North American Prodrome Longitudinal Study. JAMA Psychiatry. 2016;73:1239–1248. doi: 10.1001/jamapsychiatry.2016.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Lencz T, et al. Generalized and specific neurocognitive deficits in prodromal schizophrenia. Biol Psychiatry. 2006;59:863–871. doi: 10.1016/j.biopsych.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 176.Brewer WJ, et al. Memory impairments identified in people at ultra-high risk for psychosis who later develop first-episode psychosis. Am J Psychiatry. 2005;162:71–78. doi: 10.1176/appi.ajp.162.1.71. [DOI] [PubMed] [Google Scholar]
- 177.Bartók E, et al. Cognitive functions in prepsychotic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:621–625. doi: 10.1016/j.pnpbp.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 178.Becker HE, et al. Neurocognitive functioning before and after the first psychotic episode: does psychosis result in cognitive deterioration? Psychol Med. 2010;40:1599–1606. doi: 10.1017/S0033291710000048. [DOI] [PubMed] [Google Scholar]
- 179.Francey SM, et al. Sustained attention in young people at high risk of psychosis does not predict transition to psychosis. Schizophr Res. 2005;79:127–136. doi: 10.1016/j.schres.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 180.Reichenberg A, et al. Static and Dynamic Cognitive Deficits in Childhood Precede Adult Schizophrenia: A 30-Year Study. Schizophr Res. 2010;117:175. doi: 10.1176/appi.ajp.2009.09040574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Isohanni M, et al. The persistence of developmental markers in childhood and adolescence and risk for schizophrenic psychoses in adult life. A 34-year follow-up of the Northern Finland 1966 birth cohort. Schizophr Res. 2004;71:213–225. doi: 10.1016/j.schres.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 182.Haijma SV, et al. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull. 2013;39:1129–1138. doi: 10.1093/schbul/sbs118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Keshavan MS, et al. Decreased left amygdala and hippocampal volumes in young offspring at risk for schizophrenia. Schizophr Res. 2002;58:173–183. doi: 10.1016/s0920-9964(01)00404-2. [DOI] [PubMed] [Google Scholar]
- 184.Schreiber H, et al. Brain morphology in adolescents at genetic risk for schizophrenia assessed by qualitative and quantitative magnetic resonance imaging. Schizophr Res. 1999;40:81–86. doi: 10.1016/s0920-9964(99)00026-2. [DOI] [PubMed] [Google Scholar]
- 185.Şişmanlar G, et al. The volumetric differences of the fronto-temporal region in young offspring of schizophrenic patients. Eur Child Adolesc Psychiatry. 2010;19:151–157. doi: 10.1007/s00787-009-0052-5. [DOI] [PubMed] [Google Scholar]
- 186.Rajarethinam R, et al. Reduced superior temporal gyrus volume in young offspring of patients with schizophrenia. Am J Psychiatry. 2004;161:1121–1124. doi: 10.1176/appi.ajp.161.6.1121. [DOI] [PubMed] [Google Scholar]
- 187.Takahashi T, et al. Insular cortex gray matter changes in individuals at ultra-high-risk of developing psychosis. Schizophr Res. 2009;111:94–102. doi: 10.1016/j.schres.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 188.Thompson PM, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Natl Acad Sci USA. 2001;98:11650–11655. doi: 10.1073/pnas.201243998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Greenstein D, et al. Childhood onset schizophrenia: cortical brain abnormalities as young adults. J Child Psychol Psychiatry. 2006;47:1003–1012. doi: 10.1111/j.1469-7610.2006.01658.x. [DOI] [PubMed] [Google Scholar]
- 190.Cho RY, et al. Development of sensory gamma oscillations and cross-frequency coupling from childhood to early adulthood. Cereb Cortex. 2015;25:1509–1518. doi: 10.1093/cercor/bht341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Perez VB, et al. Automatic auditory processing deficits in schizophrenia and clinical high-risk patients: forecasting psychosis risk with mismatch negativity. Biol Psychiatry. 2014;75:459–469. doi: 10.1016/j.biopsych.2013.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 1982;17:319–334. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- 193.Feinberg I. Efference copy and corollary discharge: implications for thinking and its disorders. Schizophr Bull. 1978;4:636–640. doi: 10.1093/schbul/4.4.636. [DOI] [PubMed] [Google Scholar]
- 194.Stephan KE, et al. Dysconnection in Schizophrenia: From Abnormal Synaptic Plasticity to Failures of Self-monitoring. Schizophr Bull. 2009;35:509–527. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mulert C, et al. Long-range synchrony of gamma oscillations and auditory hallucination symptoms in schizophrenia. Int J Psychophysiol. 2011/1;79:55–63. doi: 10.1016/j.ijpsycho.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Spencer KM, et al. Left auditory cortex gamma synchronization and auditory hallucination symptoms in schizophrenia. BMC Neurosci. 2009;10:85. doi: 10.1186/1471-2202-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sun L, et al. Evidence for dysregulated high-frequency oscillations during sensory processing in medication-naïve, first episode schizophrenia. Schizophr Res. 2013;150:519–525. doi: 10.1016/j.schres.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 198.Spencer KM, Ghorashi S. Oscillatory dynamics of Gestalt perception in schizophrenia revisited. Front Psychol. 2014;5:68. doi: 10.3389/fpsyg.2014.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Howes OD, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry. 2012;69:776–786. doi: 10.1001/archgenpsychiatry.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Howes O, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry. 2011;16:885–886. doi: 10.1038/mp.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Egerton A, et al. Presynaptic striatal dopamine dysfunction in people at ultra-high risk for psychosis: findings in a second cohort. Biol Psychiatry. 2013;74:106–112. doi: 10.1016/j.biopsych.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 202.Seeman P, Lee T. Antipsychotic drugs: direct correlation between clinical potency and presynaptic action on dopamine neurons. Science. 1975;188:1217–1219. doi: 10.1126/science.1145194. [DOI] [PubMed] [Google Scholar]
- 203.Kapur S, et al. Relationship Between Dopamine D2 Occupancy, Clinical Response, and Side Effects: A Double-Blind PET Study of First-Episode Schizophrenia. AJP. 2000;157:514–520. doi: 10.1176/appi.ajp.157.4.514. [DOI] [PubMed] [Google Scholar]
- 204.Sarpal DK, et al. Relationship between Duration of Untreated Psychosis and Intrinsic Corticostriatal Connectivity in Patients with Early Phase Schizophrenia. Neuropsychopharmacology. 2017:55. doi: 10.1038/npp.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sarpal DK, et al. Antipsychotic treatment and functional connectivity of the striatum in first-episode schizophrenia. JAMA Psychiatry. 2015;72:5–13. doi: 10.1001/jamapsychiatry.2014.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- 207.Winton-Brown TT, et al. Dopaminergic basis of salience dysregulation in psychosis. Trends Neurosci. 2014;37:85–94. doi: 10.1016/j.tins.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 208.Kim IH, et al. Disruption of Arp2/3 results in asymmetric structural plasticity of dendritic spines and progressive synaptic and behavioral abnormalities. J Neurosci. 2013;33:6081–6092. doi: 10.1523/JNEUROSCI.0035-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kim IH, et al. Spine pruning drives antipsychotic-sensitive locomotion via circuit control of striatal dopamine. Nat Neurosci. 2015;18:883–891. doi: 10.1038/nn.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Yan Z, et al. Synaptic Actin Dysregulation, a Convergent Mechanism of Mental Disorders? J Neurosci. 2016;36:11411–11417. doi: 10.1523/JNEUROSCI.2360-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lewis DA, et al. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Cain CK, et al. d-Cycloserine augmentation of cognitive remediation in schizophrenia. Schizophr Res. 2014;153:177–183. doi: 10.1016/j.schres.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kantrowitz JT, et al. Neurophysiological mechanisms of cortical plasticity impairments in schizophrenia and modulation by the NMDA receptor agonist D-serine. Brain. 2016;139:3281–3295. doi: 10.1093/brain/aww262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Forsyth JK, et al. Effects of Augmenting N-Methyl-D-Aspartate Receptor Signaling on Working Memory and Experience-Dependent Plasticity in Schizophrenia: An Exploratory Study Using Acute d-cycloserine. Schizophr Bull. 2017 doi: 10.1093/schbul/sbw193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Iwata Y, et al. Effects of glutamate positive modulators on cognitive deficits in schizophrenia: a systematic review and meta-analysis of double-blind randomized controlled trials. Mol Psychiatry. 2015;20:1151–1160. doi: 10.1038/mp.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.McGorry PD. Early intervention in psychosis: obvious, effective, overdue. J Nerv Ment Dis. 2015;203:310–318. doi: 10.1097/NMD.0000000000000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Pantelis C, et al. Cognitive intervention in early psychosis — preserving abilities versus remediating deficits. Current Opinion in Behavioral Sciences. 2015;4:63–72. [Google Scholar]
- 218.Seidman LJ, Nordentoft M. New Targets for Prevention of Schizophrenia: Is It Time for Interventions in the Premorbid Phase? Schizophr Bull. 2015;41:795–800. doi: 10.1093/schbul/sbv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Millan MJ, et al. Altering the course of schizophrenia: progress and perspectives. Nat Rev Drug Discov. 2016;15:485–515. doi: 10.1038/nrd.2016.28. [DOI] [PubMed] [Google Scholar]
- 220.van der Gaag M, et al. Preventing a first episode of psychosis: meta-analysis of randomized controlled prevention trials of 12 month and longer-term follow-ups. Schizophr Res. 2013;149:56–62. doi: 10.1016/j.schres.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 221.Stafford MR, et al. Early interventions to prevent psychosis: systematic review and meta-analysis. BMJ. 2013;346:f185. doi: 10.1136/bmj.f185. [DOI] [PMC free article] [PubMed] [Google Scholar]