

Abstract
Bone-forming osteoblasts play critical roles in supporting bone marrow hematopoiesis. Pluripotent stem cells (PSCs), including embryonic stem (ES) cells and induced pluripotent stem (iPS) cells, are capable of differentiating into osteoblasts. To determine the capacity of stem cells needed to rescue aberrant skeletal development and bone marrow hematopoiesis in vivo, we employed a skeletal complementation model. Mice deficient in Runx2, a master transcription factor for osteoblastogenesis, fail to form a mineralized skeleton and bone marrow. Wild-type GFP+ ES and YFP+ iPS cells were introduced into Runx2-null blastocyst-stage embryos. We assessed GFP/YFP+ cell contribution by whole-mount fluorescence and histological analysis and found that the proportion of PSCs in the resulting chimeric embryos is directly correlated with the degree of mineralization in the skull. Moreover, PSC contribution to long bones successfully restored bone marrow hematopoiesis. We validated this finding in a separate model with diphtheria toxin A-mediated ablation of hypertrophic chondrocytes and osteoblasts. Remarkably, chimeric embryos harboring as little as 37.5% wild-type PSCs revealed grossly normal skeletal morphology, suggesting a near-complete rescue of skeletogenesis. In summary, we demonstrate that fractional contribution of PSCs in vivo is sufficient to complement and reconstitute an osteoblast-deficient skeleton and hematopoietic marrow. Further investigation using genetically modified PSCs with conditional loss of gene function in osteoblasts will enable us to address the specific roles of signaling mediators to regulate bone formation and hematopoietic niches in vivo.
Keywords: pluripotent stem cell, osteoblast, bone formation, hematopoietic niche
Graphical Abstract
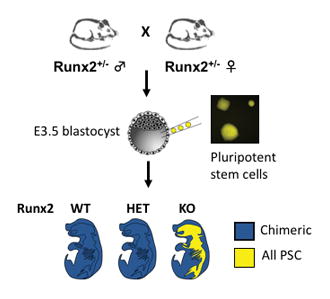
Schematic illustration of the skeletal complementation approach to demonstrate that pluripotent stem cells can contribute to formation of bone with hematopoietic bone marrow. Embryos lacking Runx2, an essential transcription factor for bone formation, cannot form bones. However, injection of Runx2−/− blastocysts with pluripotent stem cells results in chimeric embryos in which bone is derived from pluripotent stem cells, with rescue of the hematopoietic bone marrow.
Introduction
Within the bone marrow, hematopoiesis is critically dependent upon a supportive microenvironment, or niche, comprised of interactions between hematopoietic and non-hematopoietic cells. Among non-hematopoietic stromal cells, bone-forming osteoblasts of mesenchymal origin and their precursors are crucial components of the bone marrow hematopoietic niche. Growing evidence indicates that cells at each stage of differentiation from mesenchymal stem cells to fully mature osteoblasts serve distinct functions in supporting hematopoiesis [1–6].
The stage-specificity of hematopoietic support likely derives from the unique production of cytokines and growth factors by cellular populations at varying points of differentiation along the osteoblast lineage. However, clarification of the relative contributions of specific subsets of osteoblast lineage cells to hematopoietic niches is currently limited by two significant barriers: 1) the inability to prospectively distinguish osteoprogenitors, differentiating osteoblasts, and mature osteoblasts and to harvest them in large numbers, and 2) the lack of a rigorous in vivo model for assessment of osteogenic and hematopoietic-supporting potential of various cellular populations.
To circumvent these limitations, we have turned to pluripotent stem cells (PSCs) as a potential source of osteoblasts. PSCs are unique in their ability to both self-renew and give rise to differentiated tissues. They represent a potentially unlimited source of osteoblast lineage cells for localized bone repair and regeneration, as well as for disease modeling and drug screening for systemic skeletal diseases. One source of stem cell-derived osteoblasts is embryonic stem (ES) cells, which are derived from the inner cell mass of the blastocyst [7–9] and can contribute to any tissue. ES cells have been differentiated into several different tissue types including neurons [10–12], cardiomyocytes [13, 14], and pancreatic progenitors [15]. ES cells have also been differentiated into osteoblasts by several laboratories. A typical protocol to direct the differentiation of mouse [16, 17] or human [18] ES cells into osteoblasts involves formation of embryoid bodies (EB) that are subsequently disaggregated and plated in osteogenic medium containing ascorbic acid (AA) and β-glycerophosphate (βGP) (reviewed in [19]). The addition of factors such as dexamethasone, retinoic acid (RA), bone morphogenetic proteins (BMPs) and vitamin D3 (VD3) [20–24] as well as the use of 3-dimensional scaffolds [25–31] have been reported to enhance osteogenic differentiation. While the inability to derive patient-matched ES lines may hinder the use of ES cells in cellular transplantation, banks of ES lines to match various HLA-types could potentially be generated [32].
As an alternative approach, a combination of only 4 transcription factors – Oct3/4, Sox2, c-Myc, and Klf4 – can convert mouse fibroblasts into induced pluripotent stem (iPS) cells [33]. Human iPS cells can similarly be derived from human fibroblasts [34]. The ability to derive patient-matched iPS cells from accessible somatic cells offers great potential for understanding disease mechanisms, screening for novel therapeutics, and developing cell-based regenerative therapies. As with ES cells, both mouse [35, 36] and human [37] iPS cells have been differentiated into osteoblasts using similar protocols (reviewed in [19]). More recently, Kanke et al. have differentiated iPS cells into osteoblasts in a monolayer culture without EB formation via mesodermal intermediates using small molecule inhibitors [38]. ES cells and iPS cells therefore represent a renewable source of bone-forming osteoblasts, with significant clinical implications for understanding osteoblast support of hematopoiesis, as well as skeletal pathophysiology.
The success of stem cell-derived osteoblasts in regenerative applications will depend largely on their ability to recapitulate osteoblast function. A major barrier to the development of a source of osteoblasts for regenerative purposes is the lack of a rigorous method for evaluating the osteogenic potential of mesenchymal progenitors or osteoblast precursors. The differentiation of ES and iPS cells into osteoblasts in culture is typically assayed by mineral deposition, alkaline phosphatase (ALP) activity, and expression of osteoblast genes [16–18, 21, 23, 24]. However, even under osteogenic conditions the differentiation of stem cells can result in a heterogeneous mix of multiple cellular lineages, and the above assays can yield positive results even when only a small percentage of osteoblasts are present. Furthermore, these assays do not accurately predict the ability of stem cell-derived osteoblasts to form bone in vivo [27, 39]. Therefore bone formation should be directly evaluated in vivo. Assays of osteogenic capacity typically rely on ectopic bone formation, for instance by implantation under the skin or kidney capsule of syngeneic or immunodeficient mice, often combined with a carrier material [20, 25, 27, 28, 39–43], or by healing of a critical-size calvarial defect [31, 44] or burr-hole fracture [25, 30]. However, these assays do not determine the ability of stem cell-derived osteoblasts to contribute to normal endogenous bone formation.
In mice, hematopoietic stem cells can reconstitute the entire hematopoietic system following lethal irradiation [45]. Analogous experiments to assess the regenerative capacity of stem cells that give rise to the skeleton, however, cannot been performed for several reasons – the skeleton is composed of greater than 200 distinct bones, it is not possible to ablate the postnatal skeleton in a manner compatible with survival, and mesenchymal stem cells introduced into the circulation have not been shown definitively to engraft in the skeleton [46]. Since a mineralized skeleton is dispensable for survival during embryogenesis, this developmental period affords an opportunity to investigate the skeletal contributions of PSCs via a blastocyst chimera assay.
We therefore sought to develop a more physiologic assessment of PSC contribution to skeletal function in vivo by organ complementation. Organ complementation has been used to demonstrate that β cell function in mutant mice lacking a pancreas can be restored by contribution of wild-type stem cells [47], and we have recently used a similar approach to examine the contribution of PSCs to cardiac development [48]. Here we report that PSCs can reconstitute skeletal elements and rescue bone marrow hematopoiesis in vivo. Based on cell ablation studies, we find that as little as 37.5% chimerism is sufficient to restore grossly normal skeletal development.
Materials and Methods
Experimental animals
Runx2−/− mice [49, 50] and Osx1-Cre mice [51] have been described. ROSA26eGFP-DTA mice were purchased from the Jackson Laboratory. All mice were examined at embryonic day 18.5 (E18.5) unless otherwise noted. Experimental animal protocols were approved by the Institutional Animal Care and Use Committee of Massachusetts General Hospital.
Skeletal complementation
The derivation of the ES and iPS lines has previously been reported [48]. Runx2+/− mice were mated to CD1 wild-type mice to generate F1 Runx2+CD1/− mice. F1 Runx2+CD1/− female mice were superovulated for timed matings to F1 Runx2+CD1/− male mice. Embryos were staged by vaginal plugging of the female, with noon on the day of appearance of the plug designated as embryonic day E0.5. E3.5 blastocysts were harvested for injection with ESCs or iPSCs at passage less than 25.
For the Osx:DTA study, Osx1-Cre/+ mice were mated to ROSA26eGFP-DTA (DTA) mice to generate eGFP+ embryos carrying either DTA/+ only (control) or Osx1-Cre/+;DTA/+ (ablated) alleles. Unlabeled wild-type ESCs were injected into eGFP+ blastocyst-stage embryos from each group to generate chimeras. P5 through P20 unlabeled wild-type ESCs were microinjected into E3.5 blastocysts from superovulated DTA/+ females that had been mated to Osx1-Cre/+ males. For both approaches, the injected blastocysts were subsequently transferred into the uterus of 2.5 days postcoitum pseudopregnant 6- to 8-week-old CD-1 foster mothers previously mated with vasectomized [52]. Chimeric embryos were recovered at E18.5.
Genotyping
Microsatellite markers are DNA loci with repeated short nucleotide sequences that can vary in length. We identified a microsatellite marker in the Runx2 locus that differed in length based on genetic background (CD1 vs C57BL/6) (Supplemental Figure 1A). We crossed Runx2+/− males to CD1 females, and intercrossed the resulting F1 heterozygous mice, in which the wild-type Runx2 allele is of CD1 background (Runx2+CD1/−). In contrast, the injected ES and iPS cells are of C57BL/6 origin. By PCR genotyping for the presence of Runx2 wild-type (CD1 or CD57BL/6 background), Runx2 null, and GFP/YFP alleles (Supplemental Figure 1B), we could distinguish the genotype of the original host blastocyst in resulting chimeric embryos with PSC contribution (Supplemental Figures 1C–D).
Genotyping was performed by polymerase chain reaction on tail genomic DNA. The following primers were used for genotyping: Runx2 wild-type allele forward 5’-AGCGACGTGAGCCCGGTGGT-3’, reverse 5’-CTCAATCGGGGCACTGCGGC-3’; Runx2 null allele forward 5’-TACGGTATCGCCGCTCCCGATTCG-3’, reverse 5’-ATGATCTCCACCATGGTGCGGTTG-3’; GFP forward 5’-TCATCTGCACCACCGGCAAGC-3’, reverse 5’-AGCAGGACCATGTGATCGCGC-3’; Cre forward 5’-CGCGGTCTGGCAGTAAAAACTATC-3’, reverse 5’-CCCACCGTCAGTACGTGAGATATC-3’. Microsatellite analysis was performed with forward 5’-TAGGTATTTTGCACGCGCGC-3’ and reverse 5’-GCGTAACCTCTGGTCCTCGA-3’ primers. PCR amplification was performed at 94°C for 3 min, 30 cycles of 94°C for 45 sec, 61°C for 30 sec, 72°C for 30 sec followed by 72°C for 6 min then hold at 4°C.
Flow cytometry
Liver and spleen were homogenized to obtain single cells. Following addition of propidium iodide to gate out dead cells, flow cytometric cell counting was performed on a FACSCalibur or FACSAria II flow cytometer (BD Biosciences) using CellQuest v3.3 software (BD Biosciences, San Jose, CA). Doublet discrimination and exclusion was performed by gating cells according to their FSC-H versus FSC-W and SSC-H versus SSC-W distributions. To determine the proportion of eGFP+/eGFP− cells in each tissue sample, the data was analyzed with FlowJo v7.6 software (Tree Star, Ashland, OR).
Skeletal preparations
Skeletons were fixed in 95% ethanol, then stained overnight in 0.015% alcian blue in acetic acid/ethanol. Soft tissues were cleared in 1% KOH, then stained overnight in 0.01% alizarin red. % mineralization was calculated by measuring the area of alizarin red staining using Fiji image analysis software [53].
Histology and immunohistochemistry
Mouse limbs were snap frozen in liquid nitrogen and stored at −80°C until used. Undecalcified frozen bones were mounted on OCT medium and 10μm slices were cut on a Leica CM1850 cryostat. Frozen sections were fixed in 4% paraformaldehyde for 10 minutes at room temperature (RT). After washing with PBS, endogenous biotin was blocked using the Avidin/Biotin Blocking System (BioLegend). Sections were blocked in PBS with 10% normal donkey serum for 1 hour at RT, and then stained overnight at 4°C with chicken anti-GFP (Aves) and rat anti-CD45 biotin (BioLegend) antibodies. After washing with PBS, sections were stained for 1 hour at RT with donkey anti-chicken CF488A (Sigma-Aldrich) and Alexa Fluor 647 Streptavidin (BioLegend). Nuclei were counterstained with DAPI (Sigma-Aldrich). Images were taken with Zeiss LSM780 confocal microscope.
Results
To evaluate the ability of PSCs to contribute to skeletal development during embryogenesis, we used an organ complementation approach. Runx2/Cbfa1 is a transcription factor that is absolutely required for osteoblast maturation [54]; Runx2−/− mice fail to form a mineralized skeleton [50, 55]. In chimeric embryos resulting from the introduction of GFP+ or YFP+ wild-type PSCs into Runx2−/− blastocysts, we reasoned that only PSCs expressing wild-type Runx2 could contribute to the formation of a mineralized skeleton (Figure 1A). To determine the genotype of the host blastocyst, we developed a microsatellite assay to distinguish the origin of wild-type alleles (host versus injected PSCs) (Supplemental Figure 1). While Runx2 is not required for survival during early embryogenesis, Runx2−/− mice die of perinatal respiratory failure [50, 55]. Because we anticipated a small number of Runx2-deficient embryos with substantial PSC contribution and did not want to lose these mice to perinatal lethality, we therefore analyzed all embryos prior to delivery at E18.5. We tested several lines of GFP/YFP-labeled ES and iPS cell lines (Table 1), and found the highest contributions with V6.5 SA-eGFP+ ES cells (67% embryos with contribution) and TTF-R21-6 YFP+ iPS cells (34.9% embryos with contribution). The overall frequency of Runx2−/− embryos was 22.9–23.8% (Table 1), as expected based on Mendelian inheritance.
Figure 1. Skeletal complementation with pluripotent stem cells.

(A) In chimeric embryos resulting from Runx2−/− blastocysts injected with wild-type stem cells, the mineralized skeleton will be entirely derived from stem cells. (B) Skeletal preparation of WT (left), Runx2−/− (KO, right) and ESC-injected KO blastocyst (KO + ES) E18.5 embryos (i). WT (left panels) embryo reveals normal mineralization of the head (ii), ribs and vertebrae (iii), forelimb (iv) and hindlimb (v) as highlighted by alizarin red staining. KO embryos (right panels) exhibit minimal mineralization of the ulna, radius, and tibia (arrowheads in i). In chimeric embryos derived from KO blastocysts injected with WT ES cells, patchy mineralization is observed in the skull, maxilla, mandible (arrows, panel ii), vertebrae (arrows, panels ii and iii), humerus (arrow, panel iv) and femur (arrow, panel v).
Table 1.
Pluripotent stem cell lines
| Embryonic stem cell lines | ||||
|---|---|---|---|---|
| Injected line | No. injected | No. with stem cell contribution (%) | No. Runx2−/− (%) | No. Runx2−/− with contribution (%) |
| RN23 | 22 | 0 | 4 (18.2) | 0 |
| RN21G3 | 5 | 0 | 2 (40) | 0 |
| YFP ES | 10 | 1 (10) | 3 (30) | 0 |
| eYFP4 | 10 | 0 | 3 (30) | 0 |
| eYFP6 | 17 | 1 (5.9) | 2 (11.8) | 0 |
| eYFP8 | 1 | 0 | 1 (100) | 0 |
| eYFP11 | 18 | 0 | 3 (16.7) | 0 |
| eYFP12 | 101 | 32 (31.7) | 26 (25.7) | 8 (7.9) |
| SA-GFP | 91 | 61 (67) | 19 (20.9) | 15 (16.5) |
| Total | 275 | 95 (34.5) | 63 (22.9) | 23 (8.4) |
| Induced pluripotent stem cell lines | ||||
| Injected line | No. injected | No. with stem cell contribution (%) | No. Runx2−/− (%) | No. Runx2−/− with contribution (%) |
| Nkx-z35 | 37 | 4 (10.8) | 2 (5.4) | 0 |
| Nk5-2 | 28 | 0 | 6 (21.4) | 0 |
| YFP iPS | 17 | 2 (11.8) | 2 (11.8) | 0 |
| iYFPLz2 | 85 | 29 (34.1) | 11 (12.9) | 3 (3.5) |
| TTF-R21-6 | 341 | 119 (34.9) | 100 (29.3) | 38 (11.1) |
| Total | 508 | 154 (30.3) | 121 (23.8) | 41 (8.1) |
We first examined Runx2−/− blastocysts injected with eGFP+ ES cells (Figure 1B). Alizarin staining of skeletal preparations highlights areas of mineralization. In the control embryo by E18.5 there was abundant mineralization of the skull, ribs, vertebral column and long bones. In contrast, the Runx2−/− embryo exhibited minimal mineralization only in the mid-shaft of distal long bones (radius/ulna, tibia/fibula) due to mineralization of chondrocytes (Figures 1Bi, iv, v); as previously reported, no mineralization was seen in the skull, mandible, humerus or femur [55]. In a partially rescued chimeric embryo derived from a Runx2−/− host, patchy mineralization was observed in the skull, mandible, maxilla, vertebrae and proximal long bones (humerus and femur) (Figure 1B). We observed similar patchy contribution of YFP+ iPS cells in chimeric embryos (data not shown).
To quantitate the contribution of GFP+ ES cells to chimeric embryos, we performed flow cytometry analysis for GFP expression in the liver and the spleen. There was good correlation between the frequency of GFP+ ES-derived cells in liver and spleen (R2 = 0.955, Figure 2A), and increasing GFP frequency correlated with increasing GFP visualized in the skin by whole-mount fluorescence (Figure 2B). Moreover, as stem cell contribution increased, skeletal morphology and mineralization improved (R2 = 0.801, Figure 2C–D). Similar findings were observed with chimeras derived from iPS injections (R2 = 0.747, Figure 3).
Figure 2. ES cell contribution in chimeric embryos.

(A) The frequency of GFP+ cells in the liver by flow cytometry correlates with the frequency of GFP+ in the spleen in chimeric embryos. (B) Representative flow cytometry profiles for % GFP expression in the liver, with corresponding whole mount images of GFP fluorescence shown below each panel. (C) Skeletal preparations of the skull reveal increasing rescue of skull mineralization with increasing ES cell contribution. A control skull from a Runx2 wild-type embryo is shown for reference. (D) Quantitation of the % mineralized area of each skull as a function of % GFP contribution in the liver.
Figure 3. iPS cell contribution in chimeric embryos.

(A) Representative flow cytometry profiles for % YFP expression in the liver, with corresponding whole mount images of YFP fluorescence shown below each panel. (B) Skeletal preparations of the skull reveal increasing rescue of skull mineralization with increasing iPS cell contribution. A control skull from a Runx2 wild-type embryo is shown for reference. (C) Quantitation of the % mineralized area of each skull as a function of % GFP contribution in the liver.
During embryonic development hematopoiesis initiates in the yolk sac, then migrates to the aorta-gonad-mesonephros region, fetal liver and spleen before ultimately taking up residence in the bone marrow perinatally [56]. Runx2−/− mice lack a bone marrow cavity, and therefore hematopoiesis is sustained in extramedullary sites such as the spleen [57]. In the humerus of Runx2−/−, the entire skeletal element consists only of chondrocytes with no bone marrow cavity (Figure 4A). In contrast, in a chimeric embryo with approximately 15% contribution (estimated proportion of GFP+ cells) from stem cells, hematopoietic cells were identifiable within the bone marrow cavity (Figure 4B). Stem cell contribution exceeding 50% appears to restore a near-normal bone marrow cavity in Runx2−/− blastocyst-derived chimeric embryos (Figures 4C–D). To confirm that hematopoietic cells are present with the bone marrow cavities of chimeric embryos, we performed immunohistochemical staining for the hematopoietic cell marker CD45 (Figure 4E). We found that the majority of CD45+ hematopoietic cells are derived from the host blastocyst, although stem cell-derived (GFP+) hematopoietic cells ranged in frequency from 5–40%. The frequency of GFP+ hematopoietic cells did not appear to correlate with the overall degree of stem cell contribution as assessed by GFP frequency in the liver (Figure 4F). Thus the presence of a fraction of PSC-derived progeny can rescue the bone marrow hematopoietic microenvironment in vivo.
Figure 4. Rescue of bone marrow hematopoiesis by stem cell-derived osteoblasts.

(A) Skeletal preparations and hematoxylin- and eosin-stained sections of the proximal humerus of E18.5 control (upper) and Runx2−/− (lower) embryos. Note the absence of hematopoietic bone marrow in the Runx2−/− humerus. Scale bar = 1 mm (middle panels) or 200 μm (right panels). (B) Skull skeletal preparation demonstrating partial rescue in a Runx2−/− host blastocyst injected with stem cells. Histological analysis reveals the formation of a bone marrow cavity. GFP expression confirms chimeric contribution of stem cells. Scale bar = 200 μm. (C) Histological analyses of humeri of Runx2−/− blastocysts injected with ES cells. ES contribution (%) as determined by flow cytometry is provided under each panel. Scale bar = 200 μm. (D) Histological analyses of humeri of Runx2−/− blastocysts injected with iPS cells. iPS contribution (%) as determined by flow cytometry is provided under each panel. Scale bar = 200 μm. (E) Immunohistochemical staining for CD45 (white), GFP (green) and nuclei (DAPI, blue) reveals CD45+ hematopoietic cells in the bone marrow. (F) The frequency of CD45+ hematopoietic cells that are stem cell-derived (GFP+) does not correlate with the % GFP contribution in the liver.
In the skull the degree of mineralization seemed to correlate with the proportion of PSCs up until approximately 50% contribution, above which skull mineralization appeared grossly normal (Figures 2C and 3B). Ossification of calvarial bones occurs by intramembranous bone formation, in which mesenchymal progenitors give rise directly to osteoblasts. In contrast, bones of the axial and appendicular skeleton are formed by endochondral ossification, in which skeletal elements form via a cartilage template intermediate [58]. In the ribs of Runx2−/− embryos injected with either ES or iPS cells, we noticed that rescue of rib mineralization always appeared to initiate in the same proximal location, and extended only for limited distances (Figure 5). During endochondral bone formation, chondrocyte differentiation and hypertrophy are followed by apoptosis of hypertrophic chondrocytes, which in turn triggers vascular invasion bringing osteoprogenitors that will form trabecular and cortical bone [58, 59]. Runx2 is expressed in prehypertrophic chondrocytes and required for chondrocyte hypertrophy and vascular invasion [60, 61]. In the ribs, vascular invasion initiates at the proximal end between E15.5 and E16.5, at the same location where we found partial mineralization in PSC-injected Runx2−/− chimeric embryos (Supplemental Figure 2). Our observations in the ribs (Figure 5) raised the possibility that persistence of Runx2-deficient chondrocytes that are unable to hypertrophy and undergo apoptosis might hinder the expansion of PSC-derived osteoblasts in endochondral bones. Since osteoblast progenitors have migratory potential only during early differentiation [62], any obstacle to migration might limit the domain of mature osteoblasts and ossification within a skeletal element.
Figure 5. Stereotypical rescue of rib mineralization by skeletal complementation.
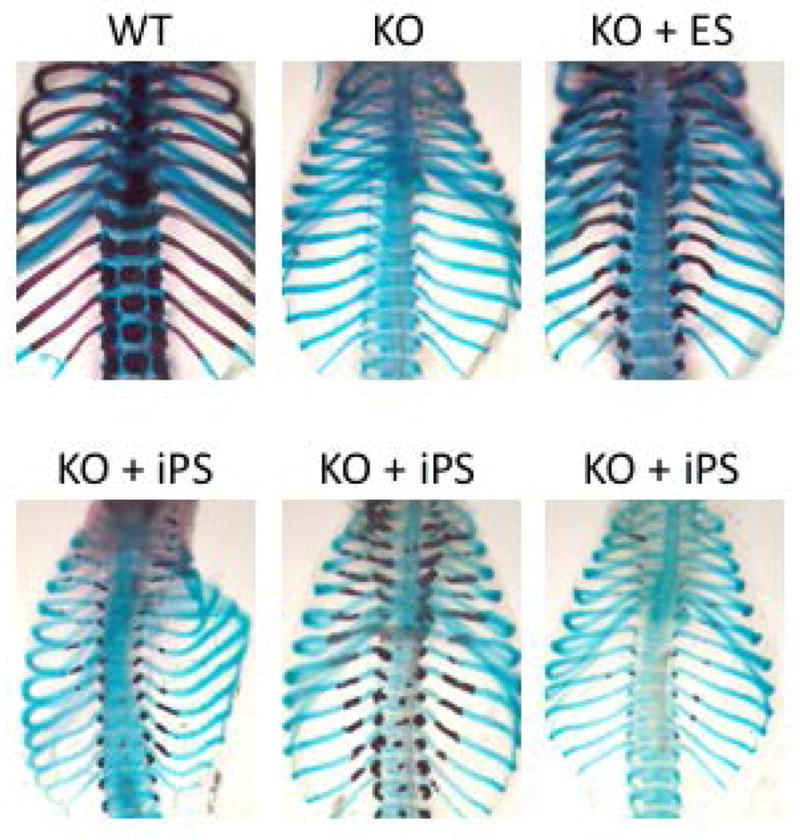
Skeletal preparations of WT and KO ribs. In ribs of chimeric embryos rescued with ES or iPS cell injection, mineralization is detected in proximal ends of thoracic ribs.
To determine whether clearing skeletal elements of Runx2-deficient late hypertrophic chondrocytes and osteoprogenitors might enable wild-type PSCs to contribute to a greater proportion of the skeleton, we developed a cellular ablation model. Osx1-Cre mice expressing Cre recombinase in late hypertrophic chondrocytes and osteoprogenitors under control of the osterix (Osx) promoter [51] were crossed to ROSA26-flox-eGFP-flox-diptheria toxin A (DTA) mice (Figure 6A). In response to Osx-driven Cre expression during skeletal development, expression of DTA will ablate Osx-expressing cells. In mice carrying both Osx1-Cre and DTA transgenes, skeletal elements were absent in the hindlimb (Figure 6B). We screened E3.5 blastocysts for the presence of the DTA allele by GFP expression, of which 50% are expected to also carry the Osx1-Cre transgene, and injected these with wild-type (GFP-negative) ES cells. In the resulting chimeric mice the contribution of stem cells can be estimated by the percent of GFP-negative cells in the liver. In Osx1-Cre/+; DTA/+ double transgenic mice, increasing rescue of skeletal formation was noted with increased stem cell contribution. In the skeleton, ES contributions of 20% or less resulted in minimal skeletogenesis, while contributions above 37.5% resulted in significant restoration of the skeleton as assessed by skeletal preparations (Figure 6C). Therefore ES- and iPS-derived osteoblasts can reconstitute the hematopoietic niche in vivo, and above a contribution threshold of ~40%, can restore near normal gross skeletal morphology.
Figure 6. Skeletal complementation in an osteoprogenitor ablation model.

(A) In chimeric embryos resulting from blastocysts expressing both Osx1-Cre and DTA alleles and injected with wild-type PSCs, the mineralized skeleton will be entirely derived from PSC progenies. (B) Skeletal preparation of E18.5 hindlimb from DTA/+ (left) and OsxCre/+;DTA/+ embryos demonstrates near ablation of skeletal elements in the latter. (C) Increased stem cell contribution is associated with increasing normalization of the mineralized skeleton. Two control skeletons are shown for reference.
Discussion
Osteoblasts are essential component of skeletal tissues, vital for bone formation and repair. Osteoblast lineage cells are also essential to the support of hematopoiesis in the bone marrow microenvironment. Because osteoblasts are situated in mineralized tissue, the ability to harvest and investigate large populations of osteoblast lineage cells at defined stages of differentiation has so far been limited. Here we demonstrate with skeletal complementation models that PSCs can contribute robustly to skeletal development during embryogenesis, and can compensate for the absence of osteoblasts to restore the hematopoietic niche.
We demonstrate that in Runx2−/− mice, which fail to form a mineralized skeleton or hematopoietic bone marrow, introduction of wild-type PSCs at the blastocyst stage partially rescues development of skeletal elements, and can support the formation of hematopoietic bone marrow as demonstrated by the expression of CD45. As expected, the resulting hematopoietic cells are chimeric, and can be of either host blastocyst or injected stem cell origin. Due to the limited numbers of chimeric embryos with >40% PSC contribution to Runx-deficient embryos, the functional nature of the hematopoietic cells was not evaluated. Additional studies are needed to more carefully examine the phenotype and function of rescued hematopoietic cells by flow cytometry, colony forming assays, and/or transplantation. Also of interest is whether substantial PSC contribution results in survival of Runx2-deficient mice past the perinatal period.
PSCs can contribute to both endochondral and intramembranous bone formation by skeletal complementation of Runx2-deficient embryos. Intramembranous ossification occurs without a cartilage intermediate, and the mineralization of the skull clearly correlates with the degree of PSC contribution. In contrast, in the ribs, which develop by endochondral ossification, our findings of limited rescue of mineralization in the ribs might reflect persistent Runx2-deficient cartilage template. Future studies will examine in detail whether the efficiency of skeletal rescue differs in endochondral vs intramembranous bones.
To examine the contribution of PSCs in the absence of a persistent cartilage template, we developed a second model to ablate both hypertrophic chondrocytes and osteoblasts. In this DTA ablation model, we find that, as in the calvariae, contribution of >50% stem cells is sufficient to restore a grossly normal pattern of skeletal mineralization, while chimerism in the range of 20–50% is associated with partial rescue. These are in line with our findings with organ complementation of cardiac development, where we have found in the heart that 40–50% contribution was sufficient to restore normal cardiac development [48]. Therefore both the heart and the skeleton can tolerate loss of up to 50% of contributing cells without gross defects in organogenesis.
There are several potential uses for these models. They can be used to examine the abilities of osteoblast lineage cells at defined stages of differentiation to contribute to the formation of a mineralized skeletal. We predict that while osteoprogenitors may contribute broadly to skeletal development, mature osteoblasts may have more restricted contribution due to their limited proliferative potential. Furthermore, since osteoblast lineage cells have unique roles in supporting hematopoiesis at specific stages of differentiation, we can now examine in vivo whether reconstitution of the hematopoietic marrow favors certain hematopoietic lineages depending on the stage of osteoblasts introduced. Future studies will explore these possibilities in greater detail and determine the relative efficiency of stem cell-derived osteoblast lineage cells to contribute to skeletal development.
Supplementary Material
Acknowledgments
This worked was funded by the Harvard Stem Cell Institute (to S.M.W. and J.Y.W.) and by National Institute of Health grants DP2OD004411 to S.M.W. and DP2OD008466 to J.Y.W.
We thank the Harvard Stem Cell Institute flow cytometry core and the MGH Center for Comparative Medicine staff for care of the mice. This worked was funded by the Harvard Stem Cell Institute (to S.M.W. and J.Y.W.) as well as by National Institute of Health grants DP2OD004411 to S.M.W. and DP2OD008466 to J.Y.W.
Footnotes
The authors declare no conflicts of interest.
Author contributions:
Rhiannon Chubb: collection and/or assembly of data, data analysis and interpretation
James Oh: collection and/or assembly of data, data analysis and interpretation
Alyssa K. Riley: collection and/or assembly of data, data analysis and interpretation
Takaharu Kimura: collection and/or assembly of data, data analysis and interpretation
Sean M. Wu: conception and design, financial support, provision of study material, data analysis and interpretation, manuscript writing
Joy Y. Wu: conception and design, financial support, provision of study material, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript
References
- 1.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495(7440):231–5. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ding L, Saunders TL, Enikolopov G, et al. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fulzele K, Krause DS, Panaroni C, et al. Myelopoiesis is regulated by osteocytes through Gsalpha-dependent signaling. Blood. 2013;121(6):930–9. doi: 10.1182/blood-2012-06-437160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenbaum A, Hsu YM, Day RB, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495(7440):227–30. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panaroni C, Fulzele K, Saini V, et al. PTH Signaling in Osteoprogenitors Is Essential for B-Lymphocyte Differentiation and Mobilization. J Bone Miner Res. 2015;30(12):2273–86. doi: 10.1002/jbmr.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rankin EB, Wu C, Khatri R, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell. 2012;149(1):63–74. doi: 10.1016/j.cell.2012.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292(5819):154–6. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 8.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78(12):7634–8. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–7. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 10.Carpenter MK, Inokuma MS, Denham J, et al. Enrichment of neurons and neural precursors from human embryonic stem cells. Exp Neurol. 2001;172(2):383–97. doi: 10.1006/exnr.2001.7832. [DOI] [PubMed] [Google Scholar]
- 11.Schuldiner M, Eiges R, Eden A, et al. Induced neuronal differentiation of human embryonic stem cells. Brain Res. 2001;913(2):201–5. doi: 10.1016/s0006-8993(01)02776-7. [DOI] [PubMed] [Google Scholar]
- 12.Zhang SC, Wernig M, Duncan ID, et al. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19(12):1129–33. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- 13.Kehat I, Gepstein A, Spira A, et al. High-resolution electrophysiological assessment of human embryonic stem cell-derived cardiomyocytes: a novel in vitro model for the study of conduction. Circ Res. 2002;91(8):659–61. doi: 10.1161/01.res.0000039084.30342.9b. [DOI] [PubMed] [Google Scholar]
- 14.Xu C, Police S, Rao N, et al. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91(6):501–8. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- 15.Assady S, Maor G, Amit M, et al. Insulin production by human embryonic stem cells. Diabetes. 2001;50(8):1691–7. doi: 10.2337/diabetes.50.8.1691. [DOI] [PubMed] [Google Scholar]
- 16.Buttery LD, Bourne S, Xynos JD, et al. Differentiation of osteoblasts and in vitro bone formation from murine embryonic stem cells. Tissue Eng. 2001;7(1):89–99. doi: 10.1089/107632700300003323. [DOI] [PubMed] [Google Scholar]
- 17.Phillips BW, Belmonte N, Vernochet C, et al. Compactin enhances osteogenesis in murine embryonic stem cells. Biochem Biophys Res Commun. 2001;284(2):478–84. doi: 10.1006/bbrc.2001.4987. [DOI] [PubMed] [Google Scholar]
- 18.Sottile V, Thomson A, McWhir J. In vitro osteogenic differentiation of human ES cells. Cloning Stem Cells. 2003;5(2):149–55. doi: 10.1089/153623003322234759. [DOI] [PubMed] [Google Scholar]
- 19.Wu JY. Pluripotent stem cells and skeletal regeneration - promise and potential. Curr Osteoporos Rep. 2015;13(5):342–50. doi: 10.1007/s11914-015-0285-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bielby RC, Boccaccini AR, Polak JM, et al. In vitro differentiation and in vivo mineralization of osteogenic cells derived from human embryonic stem cells. Tissue Eng. 2004;10(9–10):1518–25. doi: 10.1089/ten.2004.10.1518. [DOI] [PubMed] [Google Scholar]
- 21.Bourne S, Polak JM, Hughes SP, et al. Osteogenic differentiation of mouse embryonic stem cells: differential gene expression analysis by cDNA microarray and purification of osteoblasts by cadherin-11 magnetically activated cell sorting. Tissue Eng. 2004;10(5–6):796–806. doi: 10.1089/1076327041348293. [DOI] [PubMed] [Google Scholar]
- 22.Camargos BM, Tavares RL, Del Puerto HL, et al. BMP-4 increases activin A gene expression during osteogenic differentiation of mouse embryonic stem cells. Growth Factors. 2015;33(2):133–8. doi: 10.3109/08977194.2014.984805. [DOI] [PubMed] [Google Scholar]
- 23.zur Nieden NI, Kempka G, Ahr HJ. In vitro differentiation of embryonic stem cells into mineralized osteoblasts. Differentiation. 2003;71(1):18–27. doi: 10.1046/j.1432-0436.2003.700602.x. [DOI] [PubMed] [Google Scholar]
- 24.zur Nieden NI, Price FD, Davis LA, et al. Gene profiling on mixed embryonic stem cell populations reveals a biphasic role for beta-catenin in osteogenic differentiation. Mol Endocrinol. 2007;21(3):674–85. doi: 10.1210/me.2005-0438. [DOI] [PubMed] [Google Scholar]
- 25.Alfred R, Taiani JT, Krawetz RJ, et al. Large-scale production of murine embryonic stem cell-derived osteoblasts and chondrocytes on microcarriers in serum-free media. Biomaterials. 2011;32(26):6006–16. doi: 10.1016/j.biomaterials.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 26.Evans ND, Gentleman E, Chen X, et al. Extracellular matrix-mediated osteogenic differentiation of murine embryonic stem cells. Biomaterials. 2010;31(12):3244–52. doi: 10.1016/j.biomaterials.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 27.Kuznetsov SA, Cherman N, Robey PG. In vivo bone formation by progeny of human embryonic stem cells. Stem Cells Dev. 2011;20(2):269–87. doi: 10.1089/scd.2009.0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marolt D, Campos IM, Bhumiratana S, et al. Engineering bone tissue from human embryonic stem cells. Proc Natl Acad Sci U S A. 2012;109(22):8705–9. doi: 10.1073/pnas.1201830109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rutledge K, Cheng Q, Pryzhkova M, et al. Enhanced differentiation of human embryonic stem cells on extracellular matrix-containing osteomimetic scaffolds for bone tissue engineering. Tissue Eng Part C Methods. 2014;20(11):865–74. doi: 10.1089/ten.tec.2013.0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taiani JT, Krawetz RJ, Yamashita A, et al. Embryonic stem cells incorporate into newly formed bone and do not form tumors in an immunocompetent mouse fracture model. Cell Transplant. 2013;22(8):1453–62. doi: 10.3727/096368912X658755. [DOI] [PubMed] [Google Scholar]
- 31.Xin X, Jiang X, Wang L, et al. A Site-Specific Integrated Col2. 3GFP Reporter Identifies Osteoblasts Within Mineralized Tissue Formed In Vivo by Human Embryonic Stem Cells. Stem Cells Transl Med. 2014;3(10):1125–37. doi: 10.5966/sctm.2013-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamanaka S. A fresh look at iPS cells. Cell. 2009;137(1):13–7. doi: 10.1016/j.cell.2009.03.034. [DOI] [PubMed] [Google Scholar]
- 33.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 34.Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–20. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- 35.Li F, Bronson S, Niyibizi C. Derivation of murine induced pluripotent stem cells (iPS) and assessment of their differentiation toward osteogenic lineage. J Cell Biochem. 2010;109(4):643–52. doi: 10.1002/jcb.22440. [DOI] [PubMed] [Google Scholar]
- 36.Tashiro K, Inamura M, Kawabata K, et al. Efficient adipocyte and osteoblast differentiation from mouse induced pluripotent stem cells by adenoviral transduction. Stem Cells. 2009;27(8):1802–11. doi: 10.1002/stem.108. [DOI] [PubMed] [Google Scholar]
- 37.Lian Q, Zhang Y, Zhang J, et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation. 2010;121(9):1113–23. doi: 10.1161/CIRCULATIONAHA.109.898312. [DOI] [PubMed] [Google Scholar]
- 38.Kanke K, Masaki H, Saito T, et al. Stepwise differentiation of pluripotent stem cells into osteoblasts using four small molecules under serum-free and feeder-free conditions. Stem Cell Reports. 2014;2(6):751–60. doi: 10.1016/j.stemcr.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Phillips MD, Kuznetsov SA, Cherman N, et al. Directed differentiation of human induced pluripotent stem cells toward bone and cartilage: in vitro versus in vivo assays. Stem Cells Transl Med. 2014;3(7):867–78. doi: 10.5966/sctm.2013-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bilousova G, Jun du H, King KB, et al. Osteoblasts derived from induced pluripotent stem cells form calcified structures in scaffolds both in vitro and in vivo. Stem Cells. 2011;29(2):206–16. doi: 10.1002/stem.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Peppo GM, Marcos-Campos I, Kahler DJ, et al. Engineering bone tissue substitutes from human induced pluripotent stem cells. Proc Natl Acad Sci U S A. 2013;110(21):8680–5. doi: 10.1073/pnas.1301190110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin GZ, Kim TH, Kim JH, et al. Bone tissue engineering of induced pluripotent stem cells cultured with macrochanneled polymer scaffold. J Biomed Mater Res A. 2013;101(5):1283–91. doi: 10.1002/jbm.a.34425. [DOI] [PubMed] [Google Scholar]
- 43.Kim K, Zhao R, Doi A, et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011;29(12):1117–9. doi: 10.1038/nbt.2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jukes JM, Both SK, Leusink A, et al. Endochondral bone tissue engineering using embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105(19):6840–5. doi: 10.1073/pnas.0711662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spangrude GJ, Heimfeld S, Weissman IL. Purification and characterization of mouse hematopoietic stem cells. Science. 1988;241(4861):58–62. doi: 10.1126/science.2898810. [DOI] [PubMed] [Google Scholar]
- 46.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4(3):206–16. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Kobayashi T, Yamaguchi T, Hamanaka S, et al. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142(5):787–99. doi: 10.1016/j.cell.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 48.Sturzu AC, Rajarajan K, Passer D, et al. Fetal Mammalian Heart Generates a Robust Compensatory Response to Cell Loss. Circulation. 2015;132(2):109–21. doi: 10.1161/CIRCULATIONAHA.114.011490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirai T, Kobayashi T, Nishimori S, et al. Bone Is a Major Target of PTH/PTHrP Receptor Signaling in Regulation of Fetal Blood Calcium Homeostasis. Endocrinology. 2015;156(8):2774–80. doi: 10.1210/en.2014-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 51.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 52.Hogan BL, Beddington RS, Constantini F, et al. Manipulating the Mouse Embryo. A Laboratory Manual. 2. Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 53.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ducy P, Zhang R, Geoffroy V, et al. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 55.Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 56.Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132(4):631–44. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deguchi K, Yagi H, Inada M, et al. Excessive extramedullary hematopoiesis in Cbfa1-deficient mice with a congenital lack of bone marrow. Biochem Biophys Res Commun. 1999;255(2):352–9. doi: 10.1006/bbrc.1999.0163. [DOI] [PubMed] [Google Scholar]
- 58.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 59.Maes C, Kobayashi T, Selig MK, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19(2):329–44. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Takarada T, Hinoi E, Nakazato R, et al. An analysis of skeletal development in osteoblast-specific and chondrocyte-specific runt-related transcription factor-2 (Runx2) knockout mice. J Bone Miner Res. 2013;28(10):2064–9. doi: 10.1002/jbmr.1945. [DOI] [PubMed] [Google Scholar]
- 61.Takeda S, Bonnamy JP, Owen MJ, et al. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001;15(4):467–81. doi: 10.1101/gad.845101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maes C, Kobayashi T, Kronenberg HM. A novel transgenic mouse model to study the osteoblast lineage in vivo. Ann N Y Acad Sci. 2007;1116:149–64. doi: 10.1196/annals.1402.060. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.