

Abstract
Alzheimer’s Disease (AD) features the accumulation of β-amyloid and Tau aggregates, which deposit as extracellular plaques and intracellular neurofibrillary tangles (NFTs), respectively. Neuronal Tau aggregates may appear early in life, in the absence of clinical symptoms. This occurs in the brainstem reticular formation and mostly within Locus Coeruleus (LC), which is consistently affected during AD. LC is the main source of forebrain norepinephrine (NE) and it modulates a variety of functions including sleep-waking cycle, alertness, synaptic plasticity, and memory. The iso-dendritic nature of LC neurons allows their axons to spread NE throughout the whole forebrain. Likewise, a prion-like hypothesis suggests that Tau aggregates may travel along LC axons to reach out cortical neurons. Despite this timing is compatible with cross-sectional studies, there is no actual evidence for a causal relationship between these events. In the present mini-review, we dedicate special emphasis to those various mechanisms that may link degeneration of LC neurons to the onset of AD pathology. This includes the hypothesis that a damage to LC neurons contributes to the onset of dementia due to a loss of neuroprotective effects or, even the chance that, LC degenerates independently from cortical pathology. At the same time, since LC neurons are lost in a variety of neuropsychiatric disorders we considered which molecular mechanism may render these brainstem neurons so vulnerable.
Keywords: neurofibrillary tangles, basal forebrain nuclei, phospho-Tau, amyloid, mild cognitive impairment, pre-clinical AD
Introduction
Alzheimer’s Disease (AD) affects more than 45 million of people worldwide (Alzheimer’s Disease International, 2015). Despite research efforts, key molecular mechanisms of disease remain uncertain. Milestones in AD pathology consist in alterations of the cytoskeleton-associated Tau protein along with abnormal β-amyloid (Aβ) depositions. A long-lasting time interval exists since the onset of early pathological alterations until the appearance of cognitive deterioration. In fact, a sub-clinical phase, in which cortical AD pathology occurs in the absence of a frank cognitive impairment is documented. Such a stage, in which a cognitive intact subject already bears AD markers is defined “pre-clinical asymptomatic at-risk for AD” (Dubois et al., 2014). We wish to emphasize that, despite being a fascinating hypothesis, the causal relationship between Tau pre-clinical pathology and later cognitive deterioration remains to be established. Cortical pathology consists of: (i) argyrophilic structures formed by abnormal intracellular aggregates of phospho-Tau (P-Tau) fibrils, known as neurofibrillary tangles (NFT); and (ii) extracellular aggregates of Aβ known as amyloid plaques (Hyman et al., 2012; Montine et al., 2012).
An updated hypothesis considers six stages of NFT pathology. At stage I, a low amount of Gallyas-positive NFT is present, mainly within the trans-entorhinal region, which corresponds to the peripheral entorhinal cortex (Braak and Braak, 1991 or the parahippocampal gyrus facing the fusiform gyrus with the collateral sulcus interposed (Taylor and Probst, 2008). At stage I scattered NFT can be detected also within entorhinal mesocortex, CA1, dorsomedial thalamus and basal forebrain nuclei (Braak and Braak, 1991). At stage II the trans-entorhinal cortex is more affected, while the entorhinal mesocortex is consistently affected along with CA1 and prosubiculum; dorsomedial thalamus and basal forebrain nuclei appear as at stage I. At stage III, NFT densely cluster in the superficial layer of both trans-entorhinal and entorhinal cortex, while hippocampal involvement extends towards subiculum. Isocortex (neocortex) is spared apart from a few NFT within associative ventral areas. At stage IV all layers of trans-entorhinal and entorhinal cortex are filled with NFT, while the CA1 region features ghost tangles, and the basal frontal and insular isocortex are involved. At this stage also the amygdala and endopiriform nucleus feature NFT. At stage V, the parasubiculum and the whole hippocampal formation are involved massively in a continuum with trans-entorhinal and entorhinal cortex. Remarkably, CA2 is mostly spared along with dentate gyrus (Dudek et al., 2016). This is reminiscent of hippocampal damage following seizures and ischemia (Giorgi et al., 2008). At this stage the associative isocortex is widely affected. Finally, at stage VI, primary sensory and motor areas are affected. In most patients stage I and II are rather asymptomatic, while during stage III memory impairment may appear.
Aβ aggregates follow an opposite spreading direction (Thal et al., 2002) since they first appear in the isocortex (phase 1) and later on within allocortex (phase 2), then downstream to diencephalon (phase 3) and furtherly towards brainstem nuclei including substantia nigra (phase 4). In the last step Aβ extends to caudal reticular nuclei including locus coeruleus (LC; phase 5).
The spreading of NFT through interconnected brain regions may occur trans-synaptically via a prion-like transmission of Tau (Mohamed et al., 2013; Goedert, 2015). These synaptic mechanisms of degeneration are supposed to start caudally within the iso-dendritic core of the reticular formation. In fact, LC and other reticular nuclei feature an impressive collateralization allowing a single axon to innervate multiple brain regions making these cells ideal spreading vectors. A variety of brainstem reticular nuclei features NFT. Similarly, when considering cortical neurons long-projecting pyramidal cells of layer V or hippocampal pyramidal cells appears more vulnerable to degeneration (Ovsepian et al., 2016; Schaeffer et al., 2017). The present review emphasizes the role of LC as a powerful brainstem nucleus, which projects mono-synaptically to all cortical regions (Nagai et al., 1981). Nonetheless, while writing this article we already feel the limits of such a LC-centered hypothesis, since other nuclei such as the rostral dorsal raphe, the parabrachial nuclei and the pedunculopontine nucleus are important as well. Thus, the LC should be regarded more as a paradigm, rather than the unique anatomical entity, which connects NFT pathology from the brainstem to the cortex.
The trans-synaptic spreading of Tau from LC to other brain regions may occur “a rebour” (i.e., in opposite directions) by hippocampal injections of Tau fibrils, which produce a frank pathology down to the LC (Iba et al., 2015).
Tau Pathology Occurs Early within Brainstem, Cortical-Projecting Nuclei
Recent studies indicate early impairment of LC with potential outcomes on preclinical staging and neurobiology of disease (Braak et al., 2011). In detail, stereology consistently evidenced that the number of LC neurons negatively correlates with AD pathology. Pioneer studies by Tomlinson et al. (1981), Bondareff et al. (1982) and Mann et al. (1982, 1984) described a reduction of LC neurons in AD patients. A few years later Chan-Palay and Asan (1989) proposed a rostro-caudal gradient of neuronal loss within LC of AD patients, differently from PD patients where LC is uniformly affected. In a large series of non-selected brains (N > 2300), Braak et al. (2011) by using P-Tau antibodies detected “pre-tangle material” (negative for Gallyas reaction) within LC of almost all the brains in the absence of Tau-related pathology in the trans-entorhinal region. This emphasizes the occurrence of subcortical deposition of Tau in the absence of NFT in any cortical area (less than stage I). Depending on the severity of brainstem involvement, a progressive staging (“a”, “b”, “c”) up to early cortical recruitment (stages “1a” and “1b”) is described (Table 1). These stages are age-related, being stages “a-c” present solely at age 20–30, while stages > I–II being detected only after the age of 40. These findings lead to the following statements: (i) LC features abnormal Tau deposits before a frank neuronal loss; (ii) Tau deposits in LC occur decades before the average onset of cortical AD pathology; (iii) P-Tau accumulation is likely to be key in the process of NFT formation; and (iv) Tau alterations anticipate dementia. The latter observation challenges quite directly the classic amyloid cascade hypothesis, according to which, an impairment in Aβ-pathway may trigger Tau pathology (Hardy and Selkoe, 2002; Jack et al., 2010). In contrast, one might hypothesize that early Tau pathology predisposes to Aβ accumulation, which in turn exacerbates Tau pathology. Very recently, Theofilas et al. (2017) applied up-to-date stereology to brains at various disease stages and showed a two-fold increase in P-Tau positive LC neurons from pre-stage 1 to stage I, with a positive correlation between the number of P-Tau positive cells in the LC and disease stage. They also found that volume in the rostral part of LC decreases already from pre-stage 1 to stage I. The number of LC neurons decreases form stage II onward. In another study in asymptomatic patients, Andrés-Benito et al. (2017) confirmed these data. On the other hand, these studies are grounded on static descriptions, where a dynamic mechanism remains at hypothetical level. The cross-sectional evidence reported in these studies does not represent a proof of principle for an actual spreading of pathology. Thus, casualty cannot be ruled out and a supreme vulnerability of LC neurons represents a sort of background noise occurring in a number of neurological disorders encompassing AD, PD, seizures, multiple system atrophy, Down syndrome and many others (Fornai, 2007; Phillips et al., 2016). In fact, the loss of brain norepinephrine (NE) levels characterizes a number of neurological patients and involves multiple brain areas (Gesi et al., 2000; Giorgi et al., 2003, 2004; Marien et al., 2004; Fornai et al., 2011; Ruffoli et al., 2011; Pifl et al., 2012, 2013) and various experimental neurological disorders (Fornai et al., 1995a,b, 1997a,b, 1998, 1999, 2007; Siciliano et al., 1999; Soldani and Fornai, 1999; Ferrucci et al., 2002, 2013; Fulceri et al., 2004; Giorgi et al., 2006; Weinshenker et al., 2008).
Table 1.
Staging of neurofibrillary tangles (NFT-related) pathology in Locus coeruleus (LC).
| Subcortical pretangles stages | a | P-Tau accumulation within the axon hillock of brainstem reticular neurons, mostly LC neurons. | |
| b | P-Tau accumulation extended further into LC cell bodies. | ||
| Pretangle Stages | c | Involvement of other reticular (or reticular-related) ascending nuclei (e.g., dorsal raphe nucleus, nuclei of the basal forebrain). | |
| Cortical pretangles stages | 1a | Pre-tangle material in LC axons within the trans-entorhinal and entorhinal regions. | |
| 1b | Pre-tangle inclusions within pyramidal cells of the trans-entorhinal region connected with NFT positive axons. |
The Neuroanatomy of LC in AD
LC is a tube-like shaped group of NE neurons placed in the upper part of the pons; it is composed of medium-sized neurons (Brodal, 1981). LC extends rostrocaudally for approximately 16 mm (German et al., 1988; Baker et al., 1989; Halliday, 2004). These neurons send their branched axons to innervate the entire cerebral cortex (Nagai et al., 1981). Thus, by releasing NE through “bouton en passage” (or synaptic varicosities), LC modulates the activity of several cortical areas. In fact, LC modulates sleep/wake cycle, learning and memory, early gene expression and neuroprotection (Fornai et al., 1990; Foote et al., 1991; Cirelli et al., 1996; Cirelli and Tononi, 2000; Giorgi et al., 2003, 2004; Aston-Jones, 2005; Aston-Jones et al., 2007; Fornai, 2007; Giorgi et al., 2008; Weinshenker et al., 2008; Sara, 2009; Fornai et al., 2011; Counts and Mufson, 2012; Ferrucci et al., 2013). LC mostly innervates limbic cortex compared with neocortex (here defined as isocortex, Jones and Yang, 1985; Loughlin et al., 1986; Giorgi et al., 2003). The ventral part of caudal LC sends projections even to lower medulla and spinal cord, and regulates autonomic functions (Ward and Gunn, 1976), while neurons which innervate the allocortex are placed in the rostral part of the nucleus (Ward and Gunn, 1976; Loughlin et al., 1986).
The projection of LC neurons to the cortex may occur monosynaptically (Fallon et al., 1978; Nagai et al., 1981; Harley, 1987), via the allothalamus (Krout et al., 2002; Garcia-Rill et al., 2013), or through basal cholinergic nuclei. These are severely involved in AD (Davies and Maloney, 1976; Coyle et al., 1983; Sassin et al., 2000). In particular, degeneration of Ch1 and Ch2 neurons is responsible for the loss of septo-hippocampal connections leading to memory impairment (Gertz et al., 1987; Brayda-Bruno et al., 2013). The Ch4 (nucleus basalis of Meynert) nuclear complex, which is divided into various subfields (Mesulam, 2013) and receives dense NE projections from LC degenerates as well (Mesulam et al., 1983a,b, 1984; Mesulam and Geula, 1988; Smiley and Mesulam, 1999; Haghdoost-Yazdi et al., 2009). The Ch4 sends a strong cholinergic input to allocortical and mesocortical areas (Mesulam et al., 1986). In line with this, the joint involvement of cholinergic and NE nuclei encoding motivational salience in spatial attention explains the loss of motivational information and spatial attention that is critically lost in dementia (Mohanty et al., 2008; Sara, 2015; Chandler, 2016). Another limbic region innervated by LC is the amygdala (Asan, 1998) which is involved in AD (Knafo, 2012). The entorhinal and trans-entorhinal cortex as well as the hippocampal formation are monosynaptically innervated by LC (Fallon et al., 1978; Room and Groenewegen, 1986; Harley, 1987). Entorhinal cortex is involved in memory consolidation and retrieval (Dolcos et al., 2005; Bott et al., 2016; Cho et al., 2016; Fayed et al., 2017). Within hippocampus, LC axons innervate the stratum radiatum of CA1 and stratum lucidum of CA3 (Melander et al., 1986; Moudy et al., 1993). Despite a preferential limbic innervation which is in line with AD pathology, a fine anatomical analysis of the cytotectonic pattern of LC innervation within allocortex does not provide any site-specificity which may justify a preferential damage to layer V long-projecting neurons which occurs in AD. In fact, the pattern of catecholamine innervation is rather uniform in the various layers of the allocortex (apart from the scarce density in layer I; Gaspar et al., 1989). This rules out a site-specific, disease-related synaptic contact with those cortical cells (pyramidal) which are mostly affected in AD. Thus, there is no anatomical overlapping between the fine pattern of NE innervation and the fine pattern of neuropathology within various cortical layers. Such a discrepancy also applies to NE innervation of isocortical (neocortical regions; Gaspar et al., 1989). Remarkably, albeit primary sensorimotor regions are affected only at latest AD stages, they possess the highest NE innervation among isocortical regions (Gaspar et al., 1989). Thus, we have to rule out a point-by-point matching between NE axons and degenerating cortical neurons, which in turn rules out a trans-synaptic disease spreading between NE fibers and cortical neurons. This calls for considering alternative mechanisms, which may link a damage to LC axons to cortical degeneration. In fact, when analyzing the pattern of NE release rather than a point-by-point connection between pre- and post-synaptic sites a massive extra-synaptic neurotransmitter diffusion takes place. In fact, NE release allows the amine to affect close glial cells and brain vessels (Stone and Ariano, 1989; Toussay et al., 2013).
The Mechanisms Which May Link LC Degeneration to AD
In early studies LC neurons were quantified without stereology. This explains a great variability in LC cell counts even within control subjects.
Modern stereology confirms LC neuronal loss (Grudzien et al., 2007; Counts and Mufson, 2010; Kelly et al., 2017). P-Tau positive LC neurons are associated with a dramatic reduction of synaptophysin-positive perineural dots, which leads to a loss of synaptic connectivity, a sort of anatomical de-afferentiation of these neurons from activating inputs (Andrés-Benito et al., 2017). These authors found over-expression of α2-adrenergic receptors at stage I, while these receptors were reduced at stage IV in the hippocampus (Andrés-Benito et al., 2017).
Altogether these pieces of information suggest that P-Tau accumulates within LC very early, producing subtle, yet potentially relevant, functional alterations affecting NE release in target areas.
P-Tau pre-tangles accumulate within pyramidal cortical limbic cells after NE axon terminals are filled with pre-tangles themselves (pre-tangle stage 1b). Later on, the occurrence of NFT inclusions in trans-enthorinal cortical neurons parallels the increase of P-Tau inclusions within LC axons, and spreads to other cortical structures (Braak et al., 2011). At stage III, when prodromal form of AD may appear, P-Tau burdens in LC neurons are severe enough to cause neuronal death. Early pre-tangle impairment of LC leads to an altered pattern of NE release within target regions. In baseline conditions NE release is finely tuned, and it is regulated by strong afferents from different sensory inputs (Samuels and Szabadi, 2008) which are altered early in the course of disease (Andrés-Benito et al., 2017), making surviving LC cells less effective. The specific anatomy of LC neurons makes them ideal vectors throughout the CNS. In fact, Tau aggregates may pass trans-synaptically from LC axons to cortical neurons and from one cortical region to another and back again (Mohamed et al., 2013; Goedert, 2015).
At advanced stages, the loss of NE neurons enhances the accumulation of Aβ in cortical regions: this in line with experimental evidence. In fact, the neurotoxin DSP-4 (which causes selective degeneration of LC axons, Fornai et al., 1996) enhances Aβ plaque accumulation in the brain (Heneka et al., 2006). Thus, the loss of NE appears to directly foster Aβ plaque accumulation (Kalinin et al., 2007) and LC degeneration alters microglial activity, which may indirectly induce amyloid mismetabolism (Heneka et al., 2010). These changes correlate with cognitive impairment (Jardanhazi-Kurutz et al., 2011).
Again, a damage to LC produces a lack of neuroprotection. In fact β2-receptor stimulation is key in activating autophagy (Aránguiz-Urroz et al., 2011; Farah et al., 2014; Wauson et al., 2014) which removes P-Tau from the trans-Golgi network.
A recent study demonstrates an accumulation of P-Tau pyramidal cell bodies from AD patients correlates with alterations of the Golgi apparatus (Antón-Fernández et al., 2017).
A lack of β2-mediated autophagy stimulation both on pre-synaptic LC axons and post-synaptic cortical neurons might be key in triggering NFT (Figure 1). Similarly, the loss of β-receptors stimulation leads to a decrease in growth factors expression (Follesa and Mocchetti, 1993) and permanently alters immediate-early-genes expression (Cirelli and Tononi, 2000). Most directly, NE activation of neurotrophic pathways was shown to protect against neuronal amyloid toxicity (Counts and Mufson, 2010). Again, NE innervations, which occur following a volume transmission, may alter a variety of anatomical structures including astrocytes, glial cells and blood vessels which can be reached through a paracrine diffusion of NE (Stone and Ariano, 1989; Stone and John, 1991; Gesi et al., 2000; Marien et al., 2004). In this way, the neurovascular unit can be affected at multiple levels by a lack of NE innervation (Lecrux and Hamel, 2016). For instance, LC activation increases brain perfusion which triggers over-activity of multiple cortical cells (Toussay et al., 2013). This may contribute to explain why in vascular dementia Tau and amyloid accumulation occur as well (Mendel et al., 2010; Day et al., 2015; Saito et al., 2015). On the other hand, one should consider the chance that co-transmitters released in concomitancy with NE may participate to neuroprotective effects. This is the case of adenosine, which is known to be neuroprotective, as well as galanine, and others (Crawley, 1993, 1996; Stone et al., 2009; Huang et al., 2011). Just like NE these co-transmitters act by increasing autophagy, which removes both Tau and Aβ, while inducing a higher perfusion rate in the neurovascular unit and by modulating Ach release.
Figure 1.
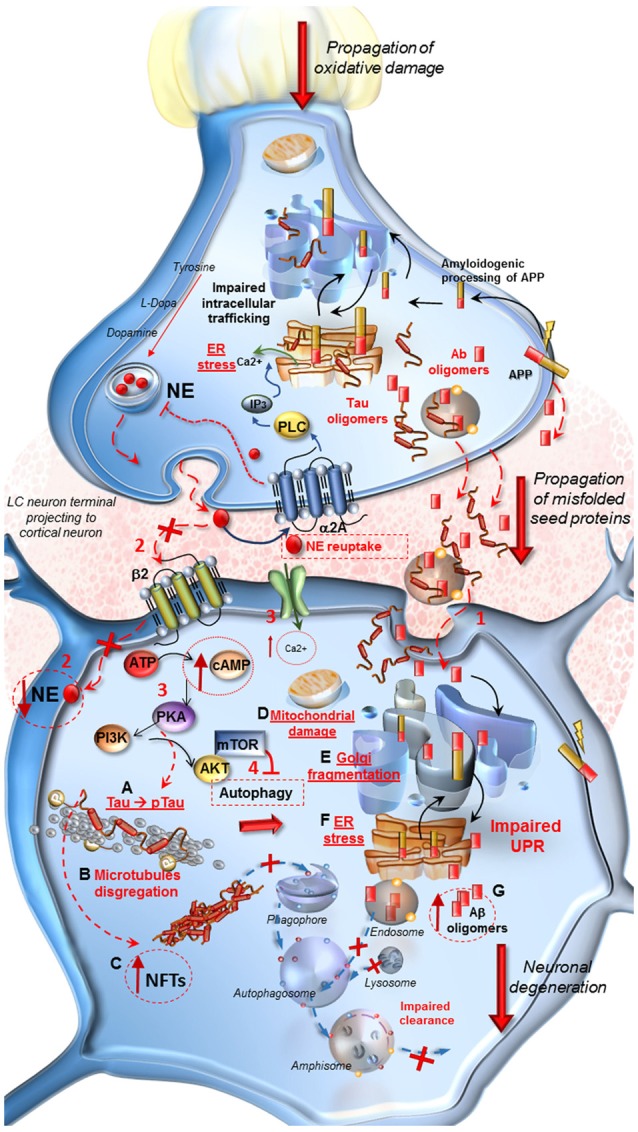
Molecular events occurring in Alzheimer’s Disease (AD) following locus coeruleus (LC) norepinephrine (NE) loss and autophagy impairment. At LC pre-synaptic terminal. Decreased expression of β2-adrenergic receptors in the pre-synaptic terminal leads to autophagy suppression. Oxidative stress, NE decrease and IP3 all contribute to endoplasmic reticulum stress and impairment of proteins processing, folding and trafficking in the trans-Golgi network. Aβ oligomers seeds, deriving from an amyloidogenic cleavage of APP, as well as Tau protein, are engulfed into vesicles and released into the axo-somatic synaptic cleft. At post-synaptic cortical neuron. (1) Aβ and Tau seeds are spread into the synaptic cleft and internalized into the cell body of the cortical neuron. (2) The loss of β2-adrenergic receptors (β2R) stimulation decreases autophagy. Thus the lack of NE, cannot exert its protective effect in the cell, it drives a cascade of detrimental intracellular effects instead. (3,4) Ca2+ entry into the cell, increase of cAMP levels, activation of PKA and PI3K/AKT/mTOR pathway lead to a further inhibition of autophagy. (A–G) PKA hyper-phosphorylates Tau protein (A) which leads to microtubules disgregation (B) and to the formation of neurofibrillary tangles (NFTs). (C) These effects translate into mitochondrial damage (D), Golgi fragmentation due to the inactivation of associated binding proteins (E) and ER stress (F). These events disrupt the Unfolded Protein Response (UPR) which does not provide for a proper intracellular trafficking, processing and sorting of misfolded Aβ, leading to further increase of harmful oligomers into the cell (G). Misfolded proteins are internalized into the endosomal compartment but autophagy impairment does not allow their removal and fosters trans-synaptic propagations.
The time window, lasting years, from the onset of P-Tau accumulation in the LC to the onset of LC neuronal loss, might be the best timing for trying to halt AD onset and progression (Mather and Harley, 2016; Ehrenberg et al., 2017). Thus, molecular mechanisms and sub-cellular sites, which first accumulate P-Tau under the effects of NE loss, need to be investigated. The high vulnerability of the isodendritic cells of the LC reticular neurons relies on the archaic nature of these cells which are preserved to transmit multimodal activities and do not develop effective protective mechanisms (Theofilas et al., 2015; Gambardella et al., 2017). In fact, these neurons are sensitive to a wide spectrum of stressful stimuli. This implies the convergency of a number of excitatory pathways making these cells a paradigm for critical overstimulation within the brain. A balanced stimulation of the autophagy machinery, which removes both Tau and Aβ could be an important step in relenting the neurobiology of disease.
Author Contributions
FSG wrote the article. LR contributed to the writing of the article and made artwork. RR provided a critical review of the literature. FB contributed to the artwork. FL contributed to the writing of the article and conceptualization. MF contributed the writing of the article. CLB contributed to the review of the literature. UB provided a review of the literature. FF wrote the article and provided a critical review of the whole manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Alzheimer’s Disease International (2015). The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Costs and Trends. London: World Alzheimer Report; Available online at: https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf [Google Scholar]
- Andrés-Benito P., Fernández-Dueñas V., Carmona M., Escobar L. A., Torrejón-Escribano B., Aso E., et al. (2017). Locus coeruleus at asymptomatic early and middle Braak stages of neurofibrillary tangle pathology. Neuropathol. Appl. Neurobiol. 43, 373–392. 10.1111/nan.12386 [DOI] [PubMed] [Google Scholar]
- Antón-Fernández A., Aparicio-Torres G., Tapia S., DeFelipe J., Muñoz A. (2017). Morphometric alterations of Golgi apparatus in Alzheimer’s disease are related to tau hyperphosphorylation. Neurobiol. Dis. 97, 11–23. 10.1016/j.nbd.2016.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aránguiz-Urroz P., Canales J., Copaja M., Troncoso R., Vicencio J. M., Carillo C., et al. (2011). Beta2-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim. Biophys. Acta 1812, 23–31. 10.1016/j.bbadis.2010.07.003 [DOI] [PubMed] [Google Scholar]
- Asan E. (1998). The catecholaminergic innervation of the rat amygdala. Adv. Anat. Embryol. Cell Biol. 142, 1–118. 10.1007/978-3-642-72085-7 [DOI] [PubMed] [Google Scholar]
- Aston-Jones G. (2005). Brain structures and receptors involved in alertness. Sleep Med. 6, S3–S7. 10.1016/s1389-9457(05)80002-4 [DOI] [PubMed] [Google Scholar]
- Aston-Jones G. S., Iba M., Clayton E., Rajkowski J., Cohen J. (2007). “Locus coeruleus and regulation of behavioral flexibility and attention: clinical implications,” in Brain Norepinephrine—Neurobiology and Therapeutics, eds Ordway G. A., Schwartz M. A., Frazer A. (Cambridge, MA: Cambridge University Press; ), 196–235. [Google Scholar]
- Baker K. G., Töerk I., Hornung J. P., Halasz P. (1989). The human locus coeruleus complex: an immunohistochemical and three-dimensional reconstruction study. Exp. Brain Res. 77, 257–270. 10.1007/bf00274983 [DOI] [PubMed] [Google Scholar]
- Bondareff W., Mountjoy C. Q., Roth M. (1982). Loss of neurons of origin of the adrenergic projection to cerebral cortex (nucleus locus ceruleus) in senile dementia. Neurology 32, 164–168. 10.1212/WNL.32.2.164 [DOI] [PubMed] [Google Scholar]
- Bott J. B., Héraud C., Cosquer B., Herbeaux K., Aubert J., Sartori M., et al. (2016). APOE-sensitive cholinergic sprouting compensates for hippocampal dysfunctions due to reduced entorhinal input. J. Neurosci. 36, 10472–10486. 10.1523/JNEUROSCI.1174-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H., Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. 10.1007/bf00308809 [DOI] [PubMed] [Google Scholar]
- Braak H., Thal D. R., Ghebremedhin E., Del Tredici K. (2011). Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J. Neuropathol. Exp. Neurol. 70, 960–969. 10.1097/NEN.0b013e318232a379 [DOI] [PubMed] [Google Scholar]
- Brayda-Bruno L., Mons N., Yee B. K., Micheau J., Abrous D. N., Nogues X., et al. (2013). Partial loss in septo-hippocampal cholinergic neurons alters memory-dependent measures of brain connectivity without overt memory deficits. Neurobiol. Dis. 54, 372–381. 10.1016/j.nbd.2013.01.010 [DOI] [PubMed] [Google Scholar]
- Brodal A. (1981). “The reticular formation and some related nuclei. The nucleus locus coeruleus,” in Neurological Anatomy in Relation to Clinical Medicine, ed. Brodal A. (New York, NY: Oxford University Press; ), 416–419. [Google Scholar]
- Chandler D. J. (2016). Evidence for a specialized role of the locus coeruleus noradrenergic system in cortical circuitries and behavioral operations. Brain Res. 1641, 197–206. 10.1016/j.brainres.2015.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Palay V., Asan E. (1989). Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson’s disease with and without dementia and depression. J. Comp. Neurol. 287, 373–392. 10.1002/cne.902870308 [DOI] [PubMed] [Google Scholar]
- Cho H., Choi J. Y., Hwang M. S., Lee J. H., Kim Y. J., Lee H. M., et al. (2016). Tau PET in Alzheimer’s disease and mild cognitive impairment. Neurology 87, 375–383. 10.1212/WNL.0000000000002892 [DOI] [PubMed] [Google Scholar]
- Cirelli C., Pompeiano M., Tononi G. (1996). Neuronal gene expression in the waking state: a role for the locus coeruleus. Science 274, 1211–1215. 10.1126/science.274.5290.1211 [DOI] [PubMed] [Google Scholar]
- Cirelli C., Tononi G. (2000). Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J. Neurosci. 20, 9187–9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts S. E., Mufson E. J. (2010). Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. J. Neurochem. 113, 649–660. 10.1111/j.1471-4159.2010.06622.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts S. E., Mufson E. J. (2012). “Locus coeruleus,” in The Human Nervous System, 3rd Edn. eds Mai J. K., Paxinos G. (San Diego, CA: Elsevier Academic Press; ), 425–438. [Google Scholar]
- Coyle J. T., Price D. L., DeLong M. R. (1983). Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science 219, 1184–1190. 10.1126/science.6338589 [DOI] [PubMed] [Google Scholar]
- Crawley J. N. (1993). Functional interactions of galanin and acetylcholine: relevance to memory and Alzheimer’s disease. Behav. Brain Res. 57, 133–141. 10.1016/0166-4328(93)90129-e [DOI] [PubMed] [Google Scholar]
- Crawley J. N. (1996). Minireview. Galanin-acetylcholine interactions: relevance to memory and Alzheimer’s disease. Life Sci. 58, 2185–2199. 10.1016/0024-3205(96)00093-8 [DOI] [PubMed] [Google Scholar]
- Davies P., Maloney A. J. (1976). Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 2:1403. 10.1016/s0140-6736(76)91936-x [DOI] [PubMed] [Google Scholar]
- Day R. J., Mason M. J., Thomas C., Poon W. W., Rohn T. T. (2015). Caspase-cleaved tau co-localizes with early tangle markers in the human vascular dementia brain. PLoS One 10:e0132637. 10.1371/journal.pone.0132637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B., Feldman H. H., Jacova C., Hampel H., Molinuevo J. L., Blennow K., et al. (2014). Advancing research diagnostic criteria for Alzheimer’s disease: the IWG-2 criteria. Lancet Neurol. 13, 614–629. 10.1016/S1474-4422(14)70090-0 [DOI] [PubMed] [Google Scholar]
- Dolcos F., LaBar K. S., Cabeza R. (2005). Remembering one year later: role of the amygdala and the medial temporal lobe memory system in retrieving emotional memories. Proc. Natl. Acad. Sci. U S A 102, 2626–2631. 10.1073/pnas.0409848102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek S. M., Alexander G. M., Farris S. (2016). Rediscovering area CA2: unique properties and functions. Nat. Rev. Neurosci. 17, 89–102. 10.1038/nrn.2015.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenberg A. J., Nguy A. K., Theofilas P., Dunlop S., Suemoto C. K., Di Lorenzo Alho A. T., et al. (2017). Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 43, 393–408. 10.1111/nan.12387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon J. H., Koziell D. A., Moore R. Y. (1978). Catecholamine innervation of the basal forebrain. II. Amygdala, suprarhinal cortex and entorhinal cortex. J. Comp. Neurol. 180, 509–532. 10.1002/cne.901800308 [DOI] [PubMed] [Google Scholar]
- Farah B. L., Sinha R. A., Wu Y., Singh B. K., Zhou J., Bay B. H., et al. (2014). β-Adrenergic agonist and antagonist regulation of autophagy in HepG2 cells, primary mouse hepatocytes and mouse liver. PLoS One 9:e98155. 10.1371/journal.pone.0098155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayed N., Modrego P. J., García-Martí G., Sanz-Requena R., Marti-Bonmatí L. (2017). Magnetic resonance spectroscopy and brain volumetry in mild cognitive impairment. A prospective study. Magn. Reson. Imaging 38, 27–32. 10.1016/j.mri.2016.12.010 [DOI] [PubMed] [Google Scholar]
- Ferrucci M., Gesi M., Lenzi P., Soldani P., Ruffoli R., Pellegrini A., et al. (2002). Noradrenergic loss enhances MDMA toxicity and induces ubiquitin-positive striatal whorls. Neurol. Sci. 23, S75–S76. 10.1007/s100720200077 [DOI] [PubMed] [Google Scholar]
- Ferrucci M., Giorgi F. S., Bartalucci A., Busceti C. L., Fornai F. (2013). The effects of locus coeruleus and norepinephrine in methamphetamine toxicity. Curr. Neuropharmacol. 11, 80–94. 10.2174/157015913804999522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follesa P., Mocchetti I. (1993). Regulation of basic fibroblast growth factor and nerve growth factor mRNA by beta-adrenergic receptor activation and adrenal steroids in rat central nervous system. Mol. Pharmacol. 43, 132–138. [PubMed] [Google Scholar]
- Foote S. L., Berridge C. W., Adams L. M., Pineda J. A. (1991). “Electrophysiological evidence for the involvement of the locus coeruleus in alerting, orienting and attending,” in Progress in Brain Research, (Vol. 88) eds Barnes C. D., Pompeiano O., (Amsterdam, NL: Elsevier; ), 521–532. [DOI] [PubMed] [Google Scholar]
- Fornai F. (2007). “Norepinephrine in neurological disorders,” in Brain Norepinephrine—Neurobiology and Therapeutics, eds Ordway G. A., Schwartz M. A., Frazer A. (Cambridge, MA: Cambridge University Press; ), 436–471. [Google Scholar]
- Fornai F., Alessandrì M. G., Fascetti F., Vaglini F., Corsini G. U. (1995a). Clonidine suppresses 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced reductions of striatal dopamine and tyrosine hydroxylase activity in mice. J. Neurochem. 65, 704–709. 10.1046/j.1471-4159.1995.65020704.x [DOI] [PubMed] [Google Scholar]
- Fornai F., Bassi L., Torracca M. T., Scalori V., Corsini G. U. (1995b). Norepinephrine loss exacerbates methamphetamine-induced striatal dopamine depletion in mice. Eur. J. Pharmacol. 283, 99–102. 10.1016/0014-2999(95)00313-a [DOI] [PubMed] [Google Scholar]
- Fornai F., Alessandrì M. G., Torracca M. T., Bassi L., Corsini G. U. (1997a). Effects of noradrenergic lesions on MPTP/MPP+ kinetics and MPTP-induced nigrostriatal dopamine depletions. J. Pharmacol. Exp. Ther. 283, 100–107. [PubMed] [Google Scholar]
- Fornai F., Bassi L., Bonaccorsi I., Giorgi F., Corsini G. U. (1997b). Noradrenaline loss selectivity exacerbates nigrostriatal toxicity in different species of rodents. Funct. Neurol. 12, 193–198. [PubMed] [Google Scholar]
- Fornai F., Alessandrì M. G., Torracca M. T., Bassi L., Scalori V., Corsini G. U. (1998). Noradrenergic modulation of methamphetamine-induced striatal dopamine depletion. Ann. N Y Acad. Sci. 844, 166–177. 10.1111/j.1749-6632.1998.tb08231.x [DOI] [PubMed] [Google Scholar]
- Fornai F., Bassi L., Torracca M. T., Alessandrì M. G., Scalori V., Corsini G. U. (1996). Region- and neurotransmitter-dependent species and strain differences in DSP-4-induced monoamine depletion in rodents. Neurodegeneration 5, 241–249. 10.1006/neur.1996.0032 [DOI] [PubMed] [Google Scholar]
- Fornai F., Blandizzi C., del Tacca M. (1990). Central alpha-2 adrenoceptors regulate central and peripheral functions. Pharmacol. Res. 22, 541–554. 10.1016/s1043-6618(05)80046-5 [DOI] [PubMed] [Google Scholar]
- Fornai F., di Poggio A. B., Pellegrini A., Ruggieri S., Paparelli A. (2007). Noradrenaline in Parkinson’s disease: from disease progression to current therapeutics. Curr. Med. Chem. 14, 2330–2334. 10.2174/092986707781745550 [DOI] [PubMed] [Google Scholar]
- Fornai F., Giorgi F. S., Alessandrì M. G., Giusiani M., Corsini G. U. (1999). Effects of pretreatment with N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) on methamphetamine pharmacokinetics and striatal dopamine losses. J. Neurochem. 72, 777–7784. 10.1046/j.1471-4159.1999.0720777.x [DOI] [PubMed] [Google Scholar]
- Fornai F., Ruffoli R., Giorgi F. S., Paparelli A. (2011). The role of locus coeruleus in the antiepileptic activity induced by vagus nerve stimulation. Eur. J. Neurosci. 33, 2169–2178. 10.1111/j.1460-9568.2011.07707.x [DOI] [PubMed] [Google Scholar]
- Fulceri F., Biagioni F., Ferrucci M., Lazzeri G., Bartalucci A., Galli V., et al. (2004). Abnormal involuntary movements (AIMs) following pulsatile dopaminergic stimulation: severe deterioration and morphological correlates following the loss of locus coeruleus neurons. Brain Res. 1135, 219–229. 10.1016/j.brainres.2006.12.030 [DOI] [PubMed] [Google Scholar]
- Gambardella S., Ferese R., Biagioni F., Busceti C. L., Campopiano R., Griguoli A. M. P., et al. (2017). The monoamine brainstem reticular formation as a paradigm for re-defining various phenotypes of Parkinson’s disease owing genetic and anatomical specificity. Front. Cell. Neurosci. 11:102. 10.3389/fncel.2017.00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rill E., Kezunovic N., Hyde J., Simon C., Beck P., Urbano F. J. (2013). Coherence and frequency in the reticular activating system (RAS). Sleep Med. Rev. 17, 227–238. 10.1016/j.smrv.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar P., Berger B., Febvret A., Vigny A., Henry J. P. (1989). Catecholamine innervation of the human cerebral cortex as revealed by comparative immunohistochemistry of tyrosine hydroxylase and dopamine-beta-hydroxylase. J. Comp. Neurol. 279, 249–271. 10.1002/cne.902790208 [DOI] [PubMed] [Google Scholar]
- German D. C., Walker B. S., Manaye K., Smith W. K., Woodward D. J., North A. J. (1988). The human locus coeruleus: computer reconstruction of cellular distribution. J. Neurosci. 8, 1776–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertz H. J., Cervos-Navarro J., Ewald V. (1987). The septo-hippocampal pathway in patients suffering from senile dementia of Alzheimer’s type. Evidence for neuronal plasticity? Neurosci. Lett. 76, 228–232. 10.1016/0304-3940(87)90720-8 [DOI] [PubMed] [Google Scholar]
- Gesi M., Soldani P., Giorgi F. S., Santinami A., Bonaccorsi I., Fornai F. (2000). The role of the locus coeruleus in the development of Parkinson’s disease. Neurosci. Biobehav. Rev. 24, 655–668. 10.1016/s0149-7634(00)00028-2 [DOI] [PubMed] [Google Scholar]
- Giorgi F. S., Blandini F., Cantafora E., Biagioni F., Armentero M. T., Pasquali L., et al. (2008). Activation of brain metabolism and fos during limbic seizures: the role of locus coeruleus. Neurobiol. Dis. 30, 388–399. 10.1016/j.nbd.2008.02.008 [DOI] [PubMed] [Google Scholar]
- Giorgi F. S., Ferrucci M., Lazzeri G., Pizzanelli C., Lenzi P., Alessandrì M. G., et al. (2003). A damage to locus coeruleus neurons converts sporadic seizures into self-sustaining limbic status epilepticus. Eur. J. Neurosci. 17, 2593–2601. 10.1046/j.1460-9568.2003.02692.x [DOI] [PubMed] [Google Scholar]
- Giorgi F. S., Mauceli G., Blandini F., Ruggieri S., Paparelli A., Murri L., et al. (2006). Locus coeruleus and neuronal plasticity in a model of focal limbic epilepsy. Epilepsia 47, 21–25. 10.1111/j.1528-1167.2006.00872.x [DOI] [PubMed] [Google Scholar]
- Giorgi F. S., Pizzanelli C., Biagioni F., Murri L., Fornai F. (2004). The role of norepinephrine in epilepsy: from the bench to the bedside. Neurosci. Biobehav. Rev. 28, 507–524. 10.1016/j.neubiorev.2004.06.008 [DOI] [PubMed] [Google Scholar]
- Goedert M. (2015). Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349:1255555. 10.1126/science.1255555 [DOI] [PubMed] [Google Scholar]
- Grudzien A., Shaw P., Weintraub S., Bigio E., Mash D. C., Mesulam M. M. (2007). Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging 28, 327–335. 10.1016/j.neurobiolaging.2006.02.007 [DOI] [PubMed] [Google Scholar]
- Haghdoost-Yazdi H., Pasbakhsh P., Vatanparast J., Rajaei F., Behzadi G. (2009). Topographical and quantitative distribution of the projecting neurons to main divisions of the septal area. Neurol. Res. 31, 503–513. 10.1179/174313208X353712 [DOI] [PubMed] [Google Scholar]
- Halliday G. (2004). “Substantia nigra and locus Coeruleus,” in The Human Nervous System—II Edition, eds Paxinos G., Mai J. K. (San Diego, CA: Elsevier Academic Press; ), 449–463. [Google Scholar]
- Harley C. W. (1987). A role for norepinephrine in arousal, emotion and learning?: limbic modulation by norepinephrine and the Kety hypothesis. Prog. Neuropsychopharmacol. Biol. Psychiatry 11, 419–458. 10.1016/0278-5846(87)90015-7 [DOI] [PubMed] [Google Scholar]
- Hardy J. A., Selkoe D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. 10.1126/science.1072994 [DOI] [PubMed] [Google Scholar]
- Heneka M. T., Nadrigny F., Regen T., Martinez-Hernandez A., Dumitrescu-Ozimek L., Terwel D., et al. (2010). Locus coeruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc. Natl. Acad. Sci. U S A 107, 6058–6063. 10.1073/pnas.0909586107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T., Ramanathan M., Jacobs A. H., Dumitrescu-Ozimek L., Bilkei-Gorzo A., Debeir T., et al. (2006). Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 26, 1343–1354. 10.1523/JNEUROSCI.4236-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. L., Yang J. M., Wang K. C., Lee Y. C., Lin Y. L., Yang Y. C., et al. (2011). Gastrodia elata prevents huntingtin aggregations through activation of the adenosine A2A receptor and ubiquitin proteasome system. J. Ethnopharmacol. 138, 162–168. 10.1016/j.jep.2011.08.075 [DOI] [PubMed] [Google Scholar]
- Hyman B. T., Phelps C. H., Beach T. G., Bigio E. H., Cairns N. J., Carrillo M. C., et al. (2012). National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 8, 1–13. 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iba M., McBride J. D., Guo J. L., Zhang B., Trojanowski J. Q., Lee V. M. (2015). Tau pathology spread in PS19 tau transgenic mice following locus Coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol. 130, 349–362. 10.1007/s00401-015-1458-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C. R., Jr., Knopman D. S., Jagust W. J., Shaw L. M., Aisen P. S., Weiner M. W., et al. (2010). Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. 10.1016/S1474-4422(09)70299-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardanhazi-Kurutz D., Kummer M. P., Terwel D., Vogel K., Thiele A., Heneka M. T. (2011). Distinct adrenergic system changes and neuroinflammation in response to induced locus ceruleus degeneration in APP/PS1 transgenic mice. Neuroscience 176, 396–407. 10.1016/j.neuroscience.2010.11.052 [DOI] [PubMed] [Google Scholar]
- Jones B. E., Yang T. Z. (1985). The efferent projections from the reticular formation and the locus coeruleus studies by anterograde and retrograde axonal transport in the rat. J. Comp. Neurol. 242, 56–92. 10.1002/cne.902420105 [DOI] [PubMed] [Google Scholar]
- Kalinin S., Gavrilyuk V., Polak P. E., Vasser R., Zhao J., Heneka M. T., et al. (2007). Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiol. Aging 28, 1206–1214. 10.1016/j.neurobiolaging.2006.06.003 [DOI] [PubMed] [Google Scholar]
- Kelly S. C., He B., Perez S. E., Ginsberg S. D., Mufson E. J., Counts S. E. (2017). Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 5:8. 10.1186/s40478-017-0411-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knafo S. (2012). “Amygdala in Alzheimer’s disease,” in The Amygdala—A Discrete Multitasking Manager, ed. Ferry B. (Rijeka: InTech; ), 375–384. [Google Scholar]
- Krout K. E., Belzer R. E., Loewy A. D. (2002). Brainstem projections to midline and intralaminar thalamic nuclei of the rat. J. Comp. Neurol. 448, 53–101. 10.1002/cne.10236 [DOI] [PubMed] [Google Scholar]
- Lecrux C., Hamel E. (2016). Neuronal networks and mediators of cortical neurovascular coupling responses in normal and altered brain states. Philos. Trans. R. Soc. Lond. B Biol. Sci. 371:20150350. 10.1098/rstb.2015.0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin S. E., Foote S. L., Bloom F. E. (1986). Efferent projections of nucleus locus coeruleus: topographic organization of cells of origin demonstrated by three-dimensional reconstruction. Neuroscience 18, 291–306. 10.1016/0306-4522(86)90155-7 [DOI] [PubMed] [Google Scholar]
- Mann D. M., Yates P. O., Hawkes J. (1982). The noradrenergic system in Alzheimer and multi-infarct dementias. J. Neurol. Neurosurg. Psychiatry 45, 113–119. 10.1136/jnnp.45.2.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann D. M., Yates P. O., Marcyniuk B. (1984). A comparison of changes in the nucleus basalis and locus caeruleus in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 47, 201–203. 10.1136/jnnp.47.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marien M. R., Colpaert F. C., Rosenquist A. C. (2004). Noradrenergic mechanisms in neurodegenerative diseases: a theory. Brain Res. Rev. 45, 38–78. 10.1016/j.brainresrev.2004.02.002 [DOI] [PubMed] [Google Scholar]
- Mather M., Harley C. W. (2016). The locus coeruleus: essential for maintaining cognitive function and the aging brain. Trends Cogn. Sci. 20, 214–226. 10.1016/j.tics.2016.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melander T., Staines W. A., Rökaeus A. (1986). Galanin-like immunoreactivity in hippocampal afferents in the rat, with special reference to cholinergic and noradrenergic inputs. Neuroscience 19, 223–240. 10.1016/0306-4522(86)90017-5 [DOI] [PubMed] [Google Scholar]
- Mendel T., Bertrand E., Szpak G. M., Stepień T., Wierzba-Bobrowicz T. (2010). Complications of severe cerebral amyloid angiopathy in the course of dementia with Lewy bodies. A case report. Folia Neuropathol. 48, 293–299. [PubMed] [Google Scholar]
- Mesulam M. M. (2013). Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J. Comp. Neurol. 521, 4124–4144. 10.1002/cne.23415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam M. M., Geula C. (1988). Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J. Comp. Neurol. 275, 216–240. 10.1002/cne.902750205 [DOI] [PubMed] [Google Scholar]
- Mesulam M. M., Mufson E. J., Levey A. I., Wainer B. H. (1983a). Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol. 214, 170–197. 10.1002/cne.902140206 [DOI] [PubMed] [Google Scholar]
- Mesulam M. M., Mufson E. J., Wainer B. H., Levey A. I. (1983b). Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1–Ch6). Neuroscience 10, 1185–1201. 10.1016/0306-4522(83)90108-2 [DOI] [PubMed] [Google Scholar]
- Mesulam M. M., Mufson E. J., Levey A. I., Wainer B. H. (1984). Atlas of cholinergic neurons in the forebrain and upper brainstem of the macaque based on monoclonal choline acetyltransferase immunohistochemistry and acetylcholinesterase histochemistry. Neuroscience 12, 669–686. 10.1016/0306-4522(84)90163-5 [DOI] [PubMed] [Google Scholar]
- Mesulam M. M., Mufson E. J., Wainer B. H. (1986). Three-dimensional representation and cortical projection topography of the nucleus basalis (Ch4) in the macaque: concurrent demonstration of choline acetyltransferase and retrograde transport with a stabilized tetramethylbenzidine method for horseradish peroxidase. Brain Res. 367, 301–308. 10.1016/0006-8993(86)91607-0 [DOI] [PubMed] [Google Scholar]
- Mohamed N. V., Herrou T., Plouffe V., Piperno N., Leclerc N. (2013). Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur. J. Neurosci. 37, 1939–1948. 10.1111/ejn.12229 [DOI] [PubMed] [Google Scholar]
- Mohanty A., Gitelman D. R., Small D. M., Mesulam M. M. (2008). The spatial attention network interacts with limbic and monoaminergic systems to modulate motivation-induced attention shifts. Cereb. Cortex 18, 2604–2613. 10.1093/cercor/bhn021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine T. J., Phelps C. H., Beach T. G., Bigio E. H., Cairns N. J., Dickson D. W., et al. (2012). National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moudy A. M., Kunkel D. D., Schwartzkroin P. A. (1993). Development of dopamine-beta-hydroxylase-positive fiber innervation of the rat hippocampus. Synapse 15, 307–318. 10.1002/syn.890150407 [DOI] [PubMed] [Google Scholar]
- Nagai T., Satoh K., Immamoto K., Maeda T. (1981). Divergent projections of catecholamine neurons of the locus coeruleus as revealed by fluorescent retrograde double labeling technique. Neurosci. Lett. 23, 117–123. 10.1016/0304-3940(81)90027-6 [DOI] [PubMed] [Google Scholar]
- Ovsepian S. V., Blazquez-Llorca L., Freitag S. V., Rodrigues E. F., Herms J. (2016). Ambient glutamate promotes paroxysmal hyperactivity in cortical pyramidal neurons at amyloid plaques via presynaptic mGluR1 receptors. Cereb. Cortex [Epub ahead of print]. 10.1093/cercor/bhw267 [DOI] [PubMed] [Google Scholar]
- Phillips C., Fahimi A., Das D., Mojabi F. S., Ponnusamy R., Salehi A. (2016). Noradrenergic system in down syndrome and Alzheimer’s disease a target for therapy. Curr. Alzheimer Res. 13, 68–83. 10.2174/1567205012666150921095924 [DOI] [PubMed] [Google Scholar]
- Pifl C., Hornykiewicz O., Blesa J., Adánez R., Cavada C., Obeso J. A. (2013). Reduced noradrenaline, but not dopamine and serotonin in motor thalamus of the MPTP primate: relation to severity of parkinsonism. J. Neurochem. 125, 657–662. 10.1111/jnc.12162 [DOI] [PubMed] [Google Scholar]
- Pifl C., Kish S. J., Hornykiewicz O. (2012). Thalamic noradrenaline in Parkinson’s disease: deficits suggest role in motor and non-motor symptoms. Mov. Disord. 27, 1618–1624. 10.1002/mds.25109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Room P., Groenewegen H. J. (1986). Connections of the parahippocampal cortex in the cat. II. Subcortical afferents. J. Comp. Neurol. 251, 451–473. 10.1002/cne.902510403 [DOI] [PubMed] [Google Scholar]
- Ruffoli R., Giorgi F. S., Pizzanelli C., Murri L., Paparelli A., Fornai F. (2011). The chemical neuroanatomy of vagus nerve stimulation. J. Chem. Neuroanat. 42, 288–296. 10.1016/j.jchemneu.2010.12.002 [DOI] [PubMed] [Google Scholar]
- Saito S., Yamamoto Y., Ihara M. (2015). Mild cognitive impairment: at the crossroad of neurodegeneration and vascular dysfunction. Curr. Alzheimer Res. 12, 507–512. 10.2174/1567205012666150530202508 [DOI] [PubMed] [Google Scholar]
- Samuels E. R., Szabadi E. (2008). Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr. Neuropharmacol. 6, 235–253. 10.2174/157015908785777229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara S. J. (2009). The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 10, 211–223. 10.1038/nrn2573 [DOI] [PubMed] [Google Scholar]
- Sara S. J. (2015). Locus Coeruleus in time with the making of memories. Curr. Opin. Neurobiol. 35, 87–94. 10.1016/j.conb.2015.07.004 [DOI] [PubMed] [Google Scholar]
- Sassin I., Schultz C., Thal D. R., Rüb U., Arai K., Braak E., et al. (2000). Evolution of Alzheimer’s disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol. 100, 259–269. 10.1007/s004019900178 [DOI] [PubMed] [Google Scholar]
- Schaeffer E. L., Catanozi S., West M. J., Gattaz W. F. (2017). Stereological investigation of the CA1 pyramidal cell layer in untreated and lithium-treated 3xTg-AD and wild-type mice. Ann. Anat. 209, 51–60. 10.1016/j.aanat.2016.10.002 [DOI] [PubMed] [Google Scholar]
- Siciliano R., Fornai F., Bonaccorsi I., Domenici L., Bagnoli P. (1999). Cholinergic and noradrenergic afferents influence the functional properties of the postnatal visual cortex in rats. Vis. Neurosci. 16, 1015–1028. 10.1017/s0952523899166045 [DOI] [PubMed] [Google Scholar]
- Smiley J. F., Mesulam M.-M. (1999). Cholinergic neurons of the nucleus basalis of Meynert receive cholinergic, catecholaminergic and GABAergic synapses: an electron microscopic investigation in the monkey. Neuroscience 88, 241–255. 10.1016/s0306-4522(98)00202-4 [DOI] [PubMed] [Google Scholar]
- Soldani P., Fornai F. (1999). The functional anatomy of noradrenergic neurons in Parkinson’s disease. Funct. Neurol. 14, 97–109. [PubMed] [Google Scholar]
- Stone E. A., Ariano M. A. (1989). Are glial cells targets of the central noradrenergic system? A review of the evidence. Brain Res. Rev. 14, 297–309. 10.1016/0165-0173(89)90015-5 [DOI] [PubMed] [Google Scholar]
- Stone T. W., Ceruti S., Abbracchio M. P. (2009). Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb. Exp. Pharmacol 193, 535–587. 10.1007/978-3-540-89615-9_17 [DOI] [PubMed] [Google Scholar]
- Stone E. A., John S. M. (1991). Further evidence for a glial localization of rat cortical beta-adrenoceptors: studies of in vivo cyclic AMP responses to catecholamines. Brain Res. 549, 78–82. 10.1016/0006-8993(91)90601-q [DOI] [PubMed] [Google Scholar]
- Taylor K. I., Probst A. (2008). Anatomic localization of the transentorhinal region of the perirhinal cortex. Neurobiol. Aging 29, 1591–1596. 10.1016/j.neurobiolaging.2007.03.024 [DOI] [PubMed] [Google Scholar]
- Thal D. R., Rüb U., Orantes M., Braak H. (2002). Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. 10.1212/wnl.58.12.1791 [DOI] [PubMed] [Google Scholar]
- Theofilas P., Dunlop S., Heinsen H., Grinberg L. T. (2015). Turning on the light within: subcortical nuclei of the isodentritic core and their role in Alzheimer’s disease pathogenesis. J. Alzheimers Dis. 46, 17–34. 10.3233/jad-142682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theofilas P., Ehrenberg A. J., Dunlop S., Di Lorenzo Alho A. T., Nguy A., Leite R. E., et al. (2017). Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: a stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement. 13, 236–246. 10.1016/j.jalz.2016.06.2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson B. E., Irving D., Blessed G. (1981). Cell loss in the locus coeruleus in senile dementia of Alzheimer type. J. Neurol. Sci. 49, 419–428. 10.1016/0022-510x(81)90031-9 [DOI] [PubMed] [Google Scholar]
- Toussay X., Basu K., Lacoste B., Hamel E. (2013). Locus coeruleus stimulation recruits a broad cortical neuronal network and increases cortical perfusion. J. Neurosci. 33, 3390–3401. 10.1523/jneurosci.3346-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward D. G., Gunn C. G. (1976). Locus coeruleus complex: elicitation of a pressor response and a brain stem region necessary for its occurrence. Brain Res. 107, 401–406. 10.1016/0006-8993(76)90236-5 [DOI] [PubMed] [Google Scholar]
- Wauson E. M., Dbouk H. A., Ghosh A. B., Cobb M. H. (2014). G protein-coupled receptors and the regulation of autophagy. Trends Endocrinol. Metab. 25, 274–282. 10.1016/j.tem.2014.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D., Ferrucci M., Busceti C. L., Biagioni F., Lazzeri G., Liles L. C., et al. (2008). Genetic or pharmacological blockade of noradrenaline synthesis enhances the neurochemical, behavioral, and neurotoxic effects of methamphetamine. J. Neurochem. 105, 471–483. 10.1111/j.1471-4159.2007.05145.x [DOI] [PMC free article] [PubMed] [Google Scholar]