

SUMMARY
Standard CRISPR-mediated gene disruption strategies rely on Cas9-induced DNA double-strand breaks (DSBs). Here, we show that CRISPR-dependent base editing efficiently inactivates genes by precisely converting four codons (CAA, CAG, CGA, and TGG) into STOP codons without DSB formation. To facilitate gene inactivation by induction of STOP codons (iSTOP), we provide access to a database of over 3.4 million single guide RNAs (sgRNAs) for iSTOP (sgSTOPs) targeting 97%–99% of genes in eight eukaryotic species, and we describe a restriction fragment length polymorphism (RFLP) assay that allows the rapid detection of iSTOP-mediated editing in cell populations and clones. To simplify the selection of sgSTOPs, our resource includes annotations for off-target propensity, percentage of isoforms targeted, prediction of nonsense-mediated decay, and restriction enzymes for RFLP analysis. Additionally, our database includes sgSTOPs that could be employed to precisely model over 32,000 cancer-associated nonsense mutations. Altogether, this work provides a comprehensive resource for DSB-free gene disruption by iSTOP.
In Brief
Billon et al. describe a CRISPR-based approach to inactivate genes by directly converting four codons into STOP codons. The induction of STOP codons (iSTOP) can be rapidly monitored using restriction enzymes. iSTOP can be employed to inactivate eukaryotic genes on a genome-wide scale and model cancer-associated nonsense mutations.
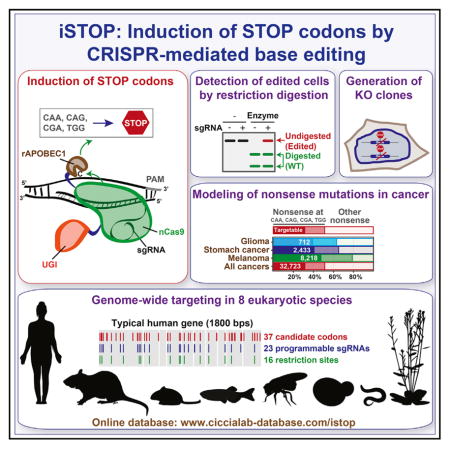
INTRODUCTION
CRISPR-Cas9 technology allows the precise modification of genomic sequences and interrogation of gene function at unprecedented speed (Barrangou and Doudna, 2016; Komor et al., 2017). This technology relies on the ability of CRISPR single guide RNAs (sgRNAs) to target the Cas9 endonuclease to precise genomic locations, where Cas9 introduces DNA double-strand breaks (DSBs) (Hsu et al., 2014). Current gene disruption strategies depend on the repair of Cas9-induced DSBs by non-homologous end joining (NHEJ) or homology-directed repair (HDR) (Jasin and Haber, 2016). During DSB repair, NHEJ occasionally introduces nucleotide insertions and deletions (indels) that result in frameshift mutations, thus disrupting gene open reading frames (ORFs), while HDR can lead to the integration of gene disruption cassettes at targeted loci (Ran et al., 2013). Gene disruption by NHEJ is efficient, although it does result in the generation of mosaic knockout (KO) alleles due to the variable number of nucleotides inserted or deleted prior to DSB end joining (van Overbeek et al., 2016). On the other hand, gene disruption by HDR is an accurate but inefficient process that requires exogenous DNA donor sequences as a template for DSB repair. One common limitation of both NHEJ- and HDR-dependent gene editing approaches is their reliance on the formation of DSBs, which are toxic DNA lesions that can cause genomic rearrangements and translocations, activate DNA damage checkpoints, and induce cell death (Aguirre et al., 2016; Choi and Meyerson, 2014; Frock et al., 2015; Ghezraoui et al., 2014; Roukos and Misteli, 2014; Torres et al., 2014). Furthermore, while NHEJ and HDR have both been exploited to modify precise genomic sequences with CRISPR-Cas9 technology (Cong et al., 2013; Mali et al., 2013), these gene editing approaches can occasionally alter non-targeted genomic loci due to Cas9-induced DSB formation at off-target sites (Hsu et al., 2013).
As an alternative to NHEJ- and HDR-dependent genome editing, CRISPR-dependent editing strategies that entail direct modification of DNA bases have recently been developed (Hess et al., 2016; Komor et al., 2016; Ma et al., 2016; Nishida et al., 2016; Plosky, 2016; Yang et al., 2016). Distinct from standard CRISPR-Cas9-dependent genome editing, CRISPR-mediated base editing avoids DSB formation and displays reduced genome-wide off-targeting (Kim et al., 2017a). CRISPR-dependent base editors consist of a catalytically inactive form of Cas9 or a Cas9 nickase mutant fused to cytidine deaminases, such as APOBEC1 or AID. For example, the CRISPR-dependent base editor BE3 is a fusion of rat APOBEC1 (rAPOBEC1), a uracil glycosylase inhibitor (UGI) and the Cas9-D10A nickase mutant (Komor et al., 2016). Following BE3 binding to target sites mediated by sgRNAs, rAPOBEC1 converts a targeted cytosine (C) into uracil (U) and UGI inhibits U removal by DNA glycosylases (Komor et al., 2016). The resulting G:U mismatch is then converted into an A:T base pair upon Cas9-mediated nicking of the G-containing DNA strand followed by DNA synthesis. This process generates permanent modifications of DNA bases within a window of high BE3 activity (13–17 nucleotides from the Cas9 protospacer adjacent motif or PAM) (Komor et al., 2016). Although recent studies have narrowed the activity window of BE3 variants from 5 to 1–2 nucleotides (Kim et al., 2017c), the presence of more than one cytosine within the BE3 activity window can result in the modification of multiple bases, causing potentially undesirable base substitutions (Kim et al., 2017c; Komor et al., 2016).
Base substitutions in the codons of ORFs can potentially cause amino acid substitutions (missense mutations) or generate premature STOP codons (nonsense mutations). Nonsense mutations can lead to the synthesis of truncated proteins or the degradation of mRNA transcripts by nonsense-mediated decay (NMD) and are often associated with human disease (Lykke-Andersen and Jensen, 2015). In particular, inherited nonsense mutations in tumor suppressor genes are observed in 10%–30% of patients that suffer from hereditary cancer syndromes (e.g., familial breast and ovarian cancer, hereditary non-polyposis colorectal cancer, familial adenomatous polyposis) (Bordeira-Carriço et al., 2012). In addition, nonsense mutations account for 12% of sporadic non-silent mutations in tumor suppressors, as reported by the catalog of somatic mutations in cancer (COSMIC) (Forbes et al., 2017). Despite their importance in the pathogenesis of human cancer, strategies to accurately and efficiently model tumor-associated nonsense mutations remain to be developed.
In this manuscript, we develop an application of CRISPR-mediated base editing for gene disruption studies. We show that CRISPR base editors efficiently inactivate human genes through the induction of STOP codons (iSTOP) in gene ORFs. This system relies on the ability of CRISPR-Cas9-dependent base editors to precisely convert four codons (CAA, CAG, CGA, and TGG) into STOP codons (TAG, TAA, or TGA) (Figures 1B, 1C, S1A, and S1B). The occurrence of iSTOP-mediated editing in sgRNA-transfected cellular populations or single clones can be rapidly monitored by restriction fragment length polymorphism (RFLP) assay using restriction enzymes that recognize bases targeted by iSTOP (Figures 2B and 3D). To facilitate the use of iSTOP, we have generated an online database (http://www.ciccialab-database.com/istop) of all sgRNAs for iSTOP (sgSTOPs) in eight eukaryotic species. Notably, 94%–99% of genes in the analyzed eukaryotic genomes can be targeted with multiple sgSTOPs, highlighting the genome-wide potential of iSTOP. Furthermore, iSTOP can be employed to model over 32,000 nonsense mutations observed in human cancer. Altogether, our studies establish iSTOP as a gene disruption technology to study gene function and model human disease.
Figure 1. Generation of STOP Codons Using CRISPR-Mediated Base Editing.
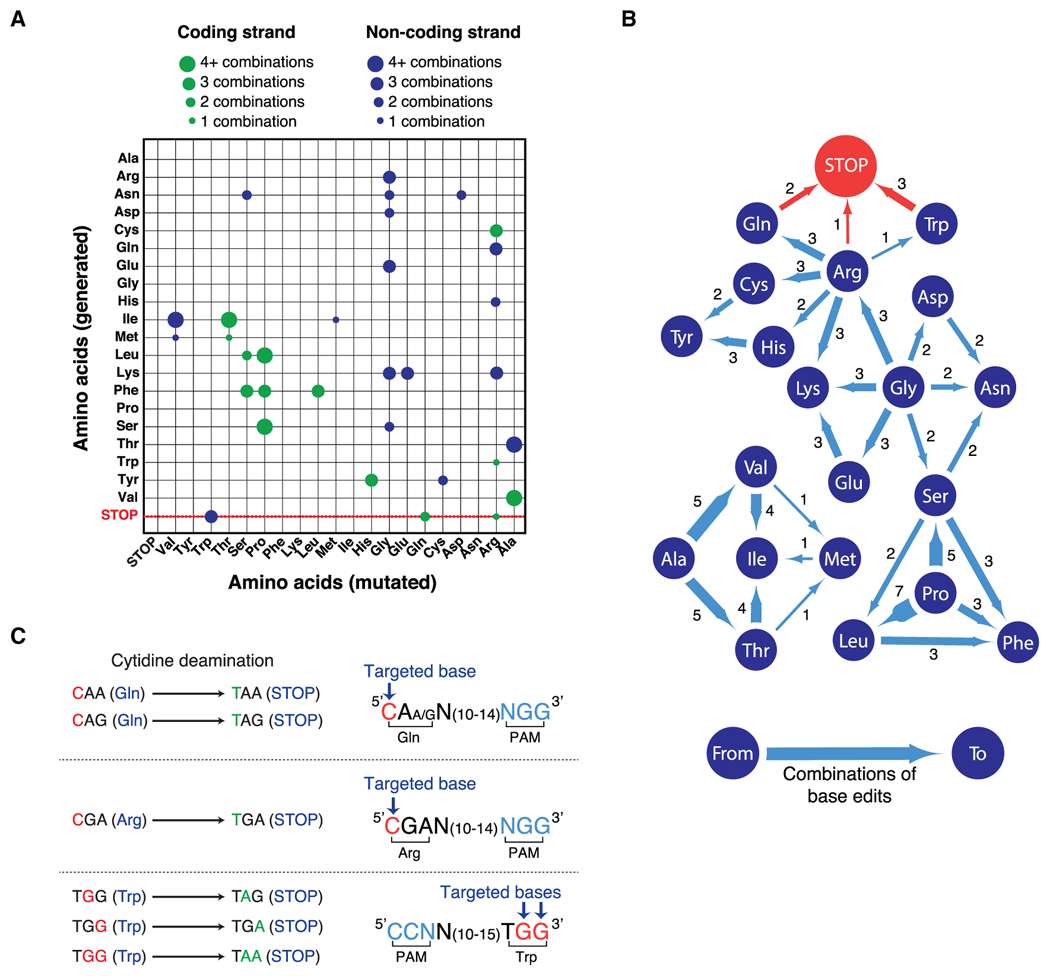
(A) Representation of the repertoire of mutations generated by cytidine deaminase-dependent CRISPR base editors. Mutated amino acids (x axis) and generated amino acids (y axis) either from coding (green) or non-coding (blue) strands are represented. The size of the circle indicates the number of combinations that generate each modification. Ala, alanine; Arg, arginine; Asn, asparagine; Asp, aspartic acid; Cys, cysteine; Gln, glutamine; Glu, glutamic acid; Gly, glycine; His, histidine; Ile, isoleucine; Met, methionine; Leu, leucine; Lys, lysine; Phe, phenylalanine; Pro, proline; Ser, serine; Thr, threonine; Trp, tryptophan; Tyr, tyrosine; Val, valine; STOP, STOP codon. See also Figures S1C and S1D and Table S1.
(B) Representation of amino acid substitutions generated by cytidine deaminase-dependent CRISPR base editors. Dark blue circles indicate amino acids, blue lines show the direction of amino acid substitutions induced by CRISPR-dependent base editing and the number of possible combinations to obtain the indicated substitutions. See also Figures S1C and S1D and Table S1.
(C) Representation of the cytidine deamination reactions induced by CRISPR-dependent base editors to generate STOP codons. The CRISPR base editor BE3 converts CAA, CAG, CGA, and TGG codons into STOP codons when the targeted base(s) (red) is at the correct distance (13–17 bps) from a protospacer adjacent motif (PAM, blue). See also Figures S1A and S1B and Table S1.
Figure 2. Restriction Fragment Length Polymorphism Assay to Detect iSTOP-Edited Cells.

(A) Schematic representation of the protocol utilized to disrupt genes by iSTOP. HEK293T cells are transfected with BE3 with or without sgSTOP, thus resulting in the generation of cells edited by iSTOP (green). The targeted locus is then amplified by PCR and digested with a restriction enzyme that recognizes a restriction site containing the base targeted by iSTOP or a restriction site generated by iSTOP-mediated base editing. Base editing by iSTOP results in PCR products refractory to restriction digestion (Site loss) or induces the formation of new restriction sites (Site gain).
(B) Conversion of CAG, CAA, TGG, and CGA codons into STOP codons by iSTOP in human cells. PCR products amplified from four different genomic loci (SPRTN, FANCM, CHEK2, and TIMELESS) edited by iSTOP were subjected to restriction digest with enzymes (PvuII, BsrGI, ApaI, and XhoI) that recognize sites containing the targeted bases. Products of the restriction digest reactions were run on polyacrylamide gels (left) and editing efficiency was determined by the percentage of undigested PCR amplicons (purple). Sequencing profiles of undigested PCR products (right) are compared to the wild-type genomic sequence, including the targeted base (blue arrow) and PAM (green). Editing of a cytosine that generates a missense mutation in the TIMELESS locus is indicated by red arrows. The sequencing profiles are representative of four to eight sequences for each targeted locus. Each base is colored according to the sequencing peaks (A in green, G in black, T in red, and C in blue). PAM, protospacer adjacent motif. See also Figures S2A and S2B.
(C) Detection of iSTOP-induced events by both loss and gain of restriction sites. The SPRTN locus was PCR-amplified and left undigested or digested with either PvuII or NheI (left). Sequencing profiles of PCR amplicons from sgSTOP-treated cells, including the targeted base (blue arrow) and PAM (green), are shown on the right. Editing efficiency was determined by the percentage of PCR amplicons refractory to PvuII digestion, as in (B), or by the percentage of NheI-digested PCR amplicons.
Figure 3. Generation of Knockout Human Cell Lines Using iSTOP.

(A) Editing of the SMARCAL1 locus by iSTOP. The SMARCAL1 locus was PCR-amplified after transfection of BE3 with or without SMARCAL1 sgSTOP. The SMARCAL1 amplicon was then digested with SfaNI, which recognizes a restriction site containing the base targeted by iSTOP and the products of the restriction digest reaction were run on a polyacrylamide gel (left). Editing efficiency at the SMARCAL1 locus was estimated by the percentage of SMARCAL1 PCR amplicons refractory to SfaNI digestion (purple), as indicated in Figure 2B. One sequencing profile representative of four sequences of undigested SMARCAL1 PCR products, including the targeted base, is indicated on the right inside.
(B) Schematic representation of the ATP1A1 co-selection strategy. HEK293T cells were transfected with BE3 with or without sgSTOPs targeting SMARCAL1 and/or ATP1A1. Cell populations were subsequently left untreated or treated with 1 μM ouabain, resulting in the enrichment of cells that had undergone genome editing at the ATP1A1 and SMARCAL1 loci.
(C) Polyacrylamide gel showing the SfaNI digestion products of SMARCAL1 PCR amplicons from HEK293T transfected with BE3 with or without sgSTOPs targeting SMARCAL1 and/or ATP1A1 and subjected to ouabain treatment, as represented in (B). Percentage of editing at the SMARCAL1 locus with or without ouabain treatment was assessed as indicated in (A).
(D) RFLP analysis of HEK293T clones edited by iSTOP in the SMARCAL1 locus. Clones (#16 and #17) were isolated from cells transfected with BE3 and sgSTOPs targeting SMARCAL1 and ATP1A1 and subjected to ouabain selection, as described in (B). Amplicons of the SMARCAL1 locus were digested by SfaNI and analyzed on polyacrylamide gel, as in (A). Restriction digest products of SMARCAL1 amplicons from wild-type (WT) cells, iSTOP-edited cellular pool and clones #16 and #17 are indicated. See also Figures S3E–S3G.
(E) Western blot analysis of SMARCAL1 protein levels in whole cellular extracts obtained from WT cells and clones #16 and #17, as shown in (D). GAPDH levels are used as loading controls.
RESULTS
CRISPR-Mediated Base Editing Precisely Converts Four Codons into STOP Codons
To study the mutagenic properties of cytidine deaminase-dependent CRISPR base editors, we investigated the repertoire of mutations that can be generated by these enzymes from the conversion of cytosine to thymine (C > T) on the coding or non-coding strand (Figures S1A and S1B; Table S1). This analysis revealed that 87.5% (56/64) of the codons can be mutated by CRISPR-dependent base editors in either the coding or non-coding strand and 27% (20/74) of the changes introduced by these gene editing enzymes generate silent mutations (Figure S1C; Table S1). Despite the fact that CRISPR-dependent base editors can only generate 7.6% (32/420) of all the possible 420 non-silent substitutions (Figures 1A, 1B, and S1D), these enzymes can convert four different codons (CAA, CAG, CGA, and TGG) encoding for 3 amino acids (glutamine [Gln], arginine [Arg], tryptophan [Trp]) exclusively into the TAA, TAG, and TGA STOP codons (Figures 1B, 1C, and S1D). CAA, CAG, and CGA triplets can be converted into STOP codons when modified by CRISPR-dependent base editors on the coding strand, while TGG can be converted into STOP codons if targeted on the non-coding strand (Figures 1C, S1A, and S1B).
CRISPR-Mediated Base Editing Efficiently Disrupts Human Genes through iSTOP
To determine whether CRISPR-mediated base editing could be effectively used in human cells to convert CAA, CAG, CGA, and TGG into STOP codons, we designed sgSTOPs to target these four codons in the SPRTN, FANCM, TIMELESS, and CHEK2 genes within the window of high activity (13–17 nucleotides from the PAM) of the CRISPR-dependent base editor BE3 (Komor et al., 2016) (Figure 1C). To simplify the detection of base editing, we targeted codons containing cytosines within the recognition sequences of restriction enzymes, thus allowing us to monitor base mutations that rendered restriction sites refractory to restriction digestion (Figure 2A). In these experiments, the sgSTOPs were transfected into HEK293T cells together with a DNA plasmid encoding the BE3 enzyme (Figure 2A). Three days after transfection, the genomic DNA of the transfected cell populations was extracted and the targeted genomic loci of SPRTN, FANCM, TIMELESS, and CHEK2 were PCR-amplified. The amplicons were subsequently digested with restriction enzymes recognizing the targeted sites (Figures 2A, 2B, S2A, and S2B). Remarkably, restriction fragment length polymorphism (RFLP) analysis revealed that 21%–35% of the PCR products specifically amplified from sgSTOP-transfected cell populations were insensitive to restriction enzyme digestion (Figure 2B). Complete digestion of PCR amplicons was observed upon incubation with restriction enzymes that did not recognize the targeted sites, confirming the specificity of this RFLP assay for the detection of sgSTOP-mediated editing (Figures S2A–S2C). Furthermore, a time course experiment showed that the incomplete digestion of PCR amplicons was not due to a limited incubation time of the reaction (Figure S3A). Cloning and sequencing of individual SPRTN, FANCM, TIMELESS, and CHEK2 PCR products refractory to restriction enzyme digestion confirmed the presence of STOP-inducing C > T transitions in each of the four targeted codons (Figure 2B). Sequencing of a TIMELESS amplicon resistant to restriction enzyme digestion also revealed editing of another cytosine located within the window of BE3 activity, as previously observed in other BE3-edited loci (Kim et al., 2017c; Komor et al., 2016) (Figure 2B).
Besides causing restriction site loss, iSTOP-mediated editing can create new restriction sites, resulting in the generation of new DNA fragments upon digestion (Figure 2A). To test whether restriction site gain could be observed in iSTOP-edited cells, HEK293T were transfected with the SPRTN sgSTOP indicated above, which causes the simultaneous loss of a PvuII site and gain of an NheI site. Notably, a fraction of SPRTN PCR amplicons from SPRTN sgSTOP-transfected cells was digested by NheI, while being refractory to PvuII digestion, revealing that restriction site gain could be employed to monitor iSTOP-mediated editing (Figure 2C). As shown in Figure 2C, a lower percentage of editing was observed by NheI digestion compared to PvuII digestion (30.7% versus 36.2%). This observation could depend on the fact that BE3-induced deamination of a second cytosine within the NheI restriction site (Figure 2C) could result in the disruption of NheI-mediated digestion. Collectively, these experiments demonstrate the feasibility of introducing nonsense mutations into the human genome by CRISPR-mediated base editing of CAA, CAG, CGA, and TGG codons and define a rapid RFLP assay to monitor the occurrence of base editing within populations of modified cells.
iSTOP Allows the Efficient Generation of Knockout Human Cell Lines
Next, we determined whether our iSTOP approach is an efficient strategy to create human knockout cell lines. To this end, we targeted SMARCAL1, a gene mutated in Schimke immunoosseous dysplasia that has been implicated in the DNA damage response (Bansbach et al., 2009; Ciccia et al., 2009; Postow et al., 2009; Yuan et al., 2009; Yusufzai et al., 2009). To disrupt the SMARCAL1 gene, we utilized an sgSTOP that efficiently converts a CAG triplet located within the first exon of the SMARCAL1 locus into a STOP codon (Figure 3A). To further increase iSTOP efficiency, we determined whether cells edited by iSTOP in the SMARCAL1 locus could be enriched by using a marker-free co-selection strategy previously employed to select cells that have undergone genome editing at Cas9- or Cpf1-induced DSBs (Agudelo et al., 2017). This strategy relies on the simultaneous targeting of a gene of interest and the sodium-potassium channel ATP1A1, which when mutated in its first extracellular loop renders cells resistant to ouabain treatment (Agudelo et al., 2017). Using this approach, HEK293T cells were transfected with BE3 in combination with sgSTOPs targeting the first exon of SMARCAL1 and the extracellular loop sequence of ATP1A1 and then cultured in the presence or absence of ouabain (Figure 3B). Interestingly, the number of cells edited by iSTOP in the SMARCAL1 locus increased from 24% to 38.8% upon ouabain selection, as determined by RFLP analysis (Figure 3C). To isolate SMARCAL1 KO clones, ouabain-resistant cell populations transfected with SMARCAL1 and ATP1A1 sgSTOPs were seeded at single-cell density and isolated cell clones were screened by RFLP analysis (Figures 3D and S3E). From this analysis, we retrieved 10/19 (52.6%) clones with at least one edited SMARCAL1 allele, with 2/19 (10.5%) clones (#16 and #17) being homozygously edited (Figure S3E). DNA sequencing and protein blot analysis of the two SMARCAL1 homozygously edited clones confirmed the presence of the desired STOP codon within the SMARCAL1 gene and the absence of SMARCAL1 protein (Figures 3E and S3F). Sanger sequencing also confirmed the genotype of the heterozygous clones that we observed using the RFLP assay (Figure S3G). Although ouabain treatment causes the accumulation of indels in the ATP1A1 locus (Figure S3D) that can result in the generation of gain-of-function ATP1A1 alleles (Agudelo et al., 2017), we only observed 1/11 modified clones with indels in the SMARCAL1 locus (Figure S3G, see clone #18). This observation could be explained by the fact that indel formation depends on the DNA nickase activity of BE3, which is significantly less efficient than the cytidine deaminase activity of BE3 (Komor et al., 2016), thus rendering the probability of ouabain to select for an infrequent event at two independent sites remarkably rare. As further validation of the above co-selection strategy, we also observed enrichment in iSTOP-edited cells upon co-targeting of the SPRTN and ATP1A1 genes, as determined by both restriction site loss and gain analysis and DNA sequencing of SPRTN amplicons from ouabain-resistant cell populations (Figures S3B and S3C). Taken together, these findings establish iSTOP as an efficient CRISPR-mediated base editing approach to disrupt human genes and demonstrate that iSTOP is compatible with co-selection enrichment methods.
iSTOP Enables the Disruption of Human Genes on a Genome Scale
As mentioned above, the use of iSTOP is restricted to four codons located at a specific distance from PAM sequences (Figure 1C). To determine whether, despite this limitation, iSTOP could be utilized to disrupt genes on a genome-wide scale, we identified sgSTOPs for all 69,180 ORFs reported in the human reference genome GRCh38 (STAR Methods, Data and Software Availability). Using CDS coordinates from the UCSC genome browser, we first identified all CAA, CAG, CGA, and TGG codons and PAMs for all validated BE3 variants located at the correct distance from the targeted base (Kim et al., 2017c; Komor et al., 2016) (Figure 4A). This genome-wide analysis revealed that 62.5% of the CAA, CAG, CGA, and TGG codons in the human reference genome can be targeted by iSTOP, thus allowing the possibility to precisely convert 523,340 codons into STOP codons (Figures 4C and S4C). Remarkably, 98.6% of human ORFs, corresponding to 99.7% of human genes, can be targeted by one or more sgSTOPs, with over 80% of ORFs targetable within the first 20% of their sequence, 99.2% of ORFs targetable within the first 100 codons, and 94.7% of ORFs targetable at a position predicted to cause nonsense-mediated decay (Popp and Maquat, 2016) (Figures 4C, 4D, and S4E). An sgSTOP can be designed for approximately one out of every 26 codons such that the typical gene of 600 amino-acid residues can potentially be targeted at 23 different codons (Figures 4B and S4D). From this analysis, we observed that only 68 (0.35%) human genes cannot be targeted by iSTOP due to unavailable CAA, CAG, CGA, or TGG codons or PAM sequences (Figures S4A and S4B). However, 24/68 of the non-targetable human genes have eukaryotic orthologs that can be targeted by iSTOP, thus allowing the study of these genes in model organisms (Figure S4A; Table S2). To determine the off-target propensity of iSTOP guides, we performed a genome-wide search for sequences similar to each sgSTOP. When allowing up to 2 mismatches outside of the guide’s seed sequence, we observe that 74% of NGG PAM-based sgSTOPs map uniquely to the genome (Figure S4F). To aid in selecting sgSTOPs with low off-target propensity, we identified putative off-target sites in the human genome for each sgSTOP. This analysis demonstrates that almost all human ORFs can be targeted by iSTOP, thus highlighting iSTOP as a promising application of CRISPR-mediated base editing for genome-wide gene disruption studies in human cells.
Figure 4. Comprehensive Detection of iSTOP Targets in Eukaryotic Genomes.
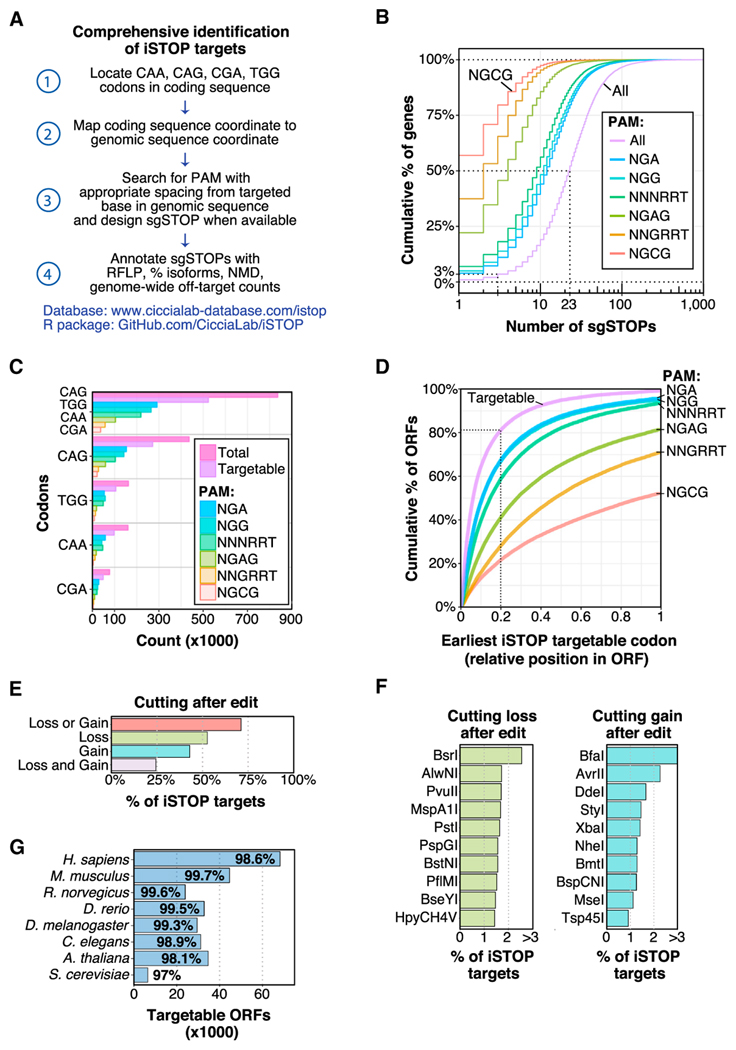
(A) Workflow utilized to identify iSTOP targetable sites in all ORFs with CDS coordinates available from the UCSC genome browser (https://genome.ucsc.edu/). Targetable sites were identified by first locating all CAA, CAG, CGA, and TGG codons in each coding sequences, then mapping the coordinates of the targeted base(s) in each codon to a genomic coordinate (steps 1–2). In total, 150 bases of genomic sequence flanking the targeted site were used to search for an appropriately spaced PAM (13–17 nucleotides from a targeted base) for all validated BE3 variants and for unique cutting of restriction enzymes (steps 3–4). Targeted isoforms and NMD predictions were determined as described in the STAR Methods.
(B) Cumulative distribution of the number of sgSTOPs designed per gene. The number of sgSTOPs for an average ORF (50%) in the human genome is indicated by a dotted line. Distributions for distinct PAM specificities (NGA, NGG, NNNRRT, NGAG, NNGRRT, and NGCG) are also shown.
(C) Number of CAG, TGG, CAA, and CGA codons in the GRCh38 human reference genome targetable by iSTOP using BE3 variants with distinct PAM specificities (NGA, NGG, NNNRRT, NGAG, NNGRRT, and NGCG).
(D) Relative position of the earliest iSTOP codon targetable in human ORFs (cumulative percentage) by BE3 variants with distinct PAM specificities (NGA, NGG, NNNRRT, NGAG, NNGRRT, and NGCG). The purple line takes into account all iSTOP targetable codons.
(E) Percentage of human iSTOP sites verifiable by RFLP analysis using restriction enzymes that cut only once within a genomic region of ±50 bps flanking the targeted site. Bars indicate percentage of sites that can be verified by restriction enzyme cutting loss and/or gain.
(F) Top ten restriction enzymes that can be utilized to validate iSTOP targets in the human genome by loss or gain of cutting within a genomic region of ±150 bps flanking the targeted site. The complete enzyme list is available in Table S3.
(G) Number and percentage of iSTOP targetable ORFs in eight different eukaryotic species. See also Figures S4C–S4E.
To determine whether the RFLP assay described above could be generalized to monitor the efficacy of multiple sgSTOPs, we identified restriction sites of enzymes that recognize DNA bases targetable by iSTOP in the human genome (Table S3). This analysis revealed that over 70% of targetable sites, corresponding to 98% of human genes, can be monitored by either restriction site loss or gain using enzymes that do not cut within ±50 nucleotides from the targeted base (Figure 4E). Furthermore, this analysis predicted BsrI and BfaI as two of the most common restriction enzymes for detecting iSTOP-edited targets (>15,000 sgSTOPs) in the human genome by restriction site loss or gain, respectively (Figure 4F). We validated the use of BsrI in the RFLP assay by targeting the PARP4 gene with an sgSTOP that edits a BsrI restriction site (Figure S2C). These studies indicate that the RFLP assay described here allows rapid assessment of base editing activity for the majority of sgSTOPs, thus greatly simplifying the use of iSTOP to interrogate gene functions in human cells.
iSTOP Is Compatible with Genome-wide Gene Disruption Studies in Eukaryotic Model Organisms
To determine whether iSTOP could be utilized for genome-wide studies in other eukaryotic species, we identified sgSTOPs for all ORFs of 7 other eukaryotic model organisms, ranging from S. cerevisiae to M. musculus. An additional 2,490,479 codons can be targeted in these species, with the possibility to design sgSTOPs approximately every 26 codons in each organism, with the exception of A. thaliana and S. cerevisiae, which can be targeted approximately once every 32 codons (Figure S4C). Moreover, we found that the percentage of ORFs that can be targeted by iSTOP is between 97% (S. cerevisiae) and 99.7% (M. musculus), demonstrating the robustness of this method to target almost any ORF of various organismal models (Figure 4G). To facilitate the use of iSTOP, we provide a full list of sgSTOP sequences, genomic coordinates of each targeted base and targeted codons for any gene in the examined eukaryotic species (STAR Methods, Data and Software Availability). In addition, this database contains information on the number and percentage of alternatively spliced isoforms targeted by each sgSTOP, a prediction of nonsense-mediated decay, counts of putative off-target sites for each sgSTOP and a list of restriction enzymes that can be used to monitor iSTOP-mediated editing. This database contains a total of 3,483,549 targetable gene coordinates and is available online at http://www.ciccialab-database.com/istop.
iSTOP Enables the Modeling of Cancer-Associated Nonsense Mutations
Nonsense mutations are frequently associated with human diseases. In cancer, nonsense mutations account for 4%–5% of the total number of observed mutations in coding sequences. Cytidine deaminases of the APOBEC protein family are frequently overexpressed in cancer and are responsible for up to 68% of all nonsense and missense mutations observed in certain cancer types (Roberts and Gordenin, 2014). To determine whether iSTOP could be utilized to model cancer-associated nonsense mutations due to cytidine deamination, we determined the prevalence of C > T and G > A base transitions occurring at CAA, CAG, CGA, and TGG codons observed in COSMIC (Figures 5A–5C). When considering each mutation site once, this analysis revealed that over 50% of nonsense mutation sites across all cancers occur at CAA, CAG, CGA, and TGG codons, with 32,723 (61%) of these nonsense mutation sites being directly reproducible using iSTOP (Figure 5A). We then determined which genes within each cancer type incurred nonsense mutations in CAA, CAG, CGA, and TGG codons more often than expected—we refer to these genes as frequent iSTOPers (Figures 5B, 5C, and S5A). The top 100 iSTOPers include 37 tumor suppressor genes (TSGs) listed in the Cancer Gene Census (e.g., TP53, PTEN, VHL, APC, ATM, and SETD2) and an additional 13 putative TSGs identified by previously published methods, such as TUSON, MutSigCV, and 20/20+ (Davoli et al., 2013; Lawrence et al., 2013; Tokheim et al., 2016) (Figure S5B). Of the 120 frequent iSTOPers (q < 0.001), 118 have at least one cancer-associated nonsense mutation that can be directly modeled with iSTOP (Table S4). Using iSTOP, we successfully modeled the most recurrent nonsense mutation (R348*; 26 cases in COSMIC) in the iSTOPer PIK3R1, which is known to promote tumorigenesis (Jaiswal et al., 2009) (Figure S5C). This work establishes iSTOP as a promising approach to model cancer-associated nonsense mutations for functional studies.
Figure 5. Modeling of Cancer-Associated Nonsense Mutations by iSTOP.

(A) Percentage of unique nonsense coordinates in cancer types, as observed in COSMIC. The percentage of base substitutions in CAA, CAG, CGA, or TGG codons that result in nonsense mutations in each cancer type is indicated. The total number of iSTOP targetable sites in each cancer type is annotated in white text.
(B) Genes with frequently observed nonsense mutations at CAA, CAG, CGA, and TGG codons (iSTOPers) and their prevalence in different cancer types. The size and opacity of each circle represents the percentage of possible CAA, CAG, CGA, and TGG codons in the gene that were observed mutated to nonsense in each cancer type. See also Figures S5A and S5B.
(C) Percentage of CAA, CAG, CGA, and TGG codons observed mutated to nonsense in the genes shown in (B) across all cancers. The total number of iSTOP targetable sites in each gene is annotated in white text. See also Figures S5A and S5B.
(D) Maps of three iSTOPers (ATM, SETD2, and EZH2) indicating locations of (1) nonsense base substitutions in cancer (red tick marks), (2) CAA, CAG, CGA, and TGG codons (black tick marks), (3) iSTOP targetable codons (blue tick marks), and (4) iSTOP targetable codons that are verifiable via RFLP (green tick marks). The largest isoform for each gene is shown with exon numbers indicated below the gene. The size of the maps is not proportional to the length of the ORFs.
DISCUSSION
In this study, we describe iSTOP as a CRISPR-mediated base editing methodology that converts CAA, CAG, CGA and TGG codons into STOP codons, thus enabling gene disruption and facile generation of KO cells. Moreover, we describe an RFLP assay for easy monitoring of the efficiency of base editing in cell populations and clones. Additionally, we provide access to an online resource containing 3,483,549 targetable gene coordinates in eight different eukaryotic species. This resource includes useful annotations to aid in searching and selecting sgSTOPs. These annotations include off-target propensity, nonsense-mediated decay prediction, relative position in the gene, percentage of isoforms targeted and a list of restriction enzymes that can be used to monitor iSTOP-mediated editing. Finally, we provide software to design base-editing guides for any annotated genome and any PAM. Collectively, these studies greatly expand the targeting range recently described for iSTOP-based approaches (Kuscu et al., 2017).
Advantages of iSTOP over Standard CRISPR-Dependent Base Editing and Gene Disruption Strategies
iSTOP exhibits several properties that distinguish it from standard CRISPR-mediated base editing applications for correcting/inserting targeted missense mutations. Given that iSTOP causes the disruption of genes and not the creation/correction of precise missense gene variants, (1) iSTOP is not affected by potentially undesired cytosine deamination events catalyzed by BE3 within its activity window. Furthermore, whereas standard CRISPR-mediated approaches of genome base editing can generate distinct missense mutations from the same targeted codon (Table S1), (2) iSTOP induces the precise conversion of four codons exclusively into STOP codons. These properties, combined with the fact that over 3 million targetable codons in eight eukaryotic species can be precisely converted into STOP codons by iSTOP to allow targeting of 97%–99.7% of eukaryotic genes, render iSTOP the first CRISPR-mediated base editing application compatible with genome-scale analyses.
In addition, iSTOP presents several advantages compared to traditional CRISPR-Cas9-mediated gene disruption technology. Indeed, (1) iSTOP does not induce DSBs, whose formation is particularly deleterious in human cells (Kuscu et al., 2017) and during organismal development (Harrison et al., 2014). By avoiding DSB formation, iSTOP leads to reduced cell death compared to conventional CRISPR-Cas9-based methods (Kuscu et al., 2017), thus potentially facilitating the generation of KO animals, as recently shown for CRISPR-mediated strategies of genome base editing in mouse embryos (Kim et al., 2017b). Furthermore, (2) iSTOP disrupts genes by precisely editing DNA bases, without relying on NHEJ-induced frameshift mutations, which create a mosaic population of distinct KO alleles. Absence of mosaicism has in fact been observed in mice subjected to CRISPR-mediated base editing (Kim et al., 2017b). Moreover, (3) iSTOP does not require the use of synthetic DNA donor molecules employed for HDR-dependent genome editing, thus rendering iSTOP-gene disruption strategies simpler and more efficient than HDR-dependent approaches, particularly for the genetic engineering of plants, for which the use of DNA donor templates is limited by technical and legislative obstacles (Jones, 2015). Recent studies have indeed shown that CRISPR-dependent approaches of genome base editing are highly efficient in rice, wheat, tomatoes, and maize (Shimatani et al., 2017; Zong et al., 2017).
Limitations of iSTOP
At present, one of the main limitations of iSTOP arises from the restrictive rules for designing sgSTOPs. Unlike canonical Cas9/sgRNAs, which can be programmed to generate DSBs adjacent to any PAM sequence, BE3/sgSTOPs require the presence of CAA, CAG, CGA, and TGG codons and a PAM located 13–17 bps away from the targeted base(s). Although ~60% of all available CAA, CAG, CGA, and TGG codons are targetable by iSTOP, the remainder cannot currently be targeted due to the absence of a nearby PAM sequence (Figures S4B and S4C). We anticipate that the generation of additional BE3 variants and the conversion of other RNA-guided DNA nickases into base editors that recognize different PAM sequences will increase the number of codons targetable by iSTOP. An additional limitation of iSTOP is due to the inability of BE3 to efficiently edit cytosines with a G on the immediate 5′ side, as recently shown by Komor et al. (2016). As a result, iSTOP is predicted to have reduced editing efficiency at 24% of targetable sites, although future improvements in the catalytic activity of BE3 may mitigate this effect.
Similar to other CRISPR-related technologies, the efficacy of iSTOP could be affected by the occurrence of off-target mutations. Notably, initial work that examined the specificity of the cytidine deaminase activity of BE3 using a BE3 variant lacking the UGI domain (BE3ΔUGI) concluded that BE3 causes fewer off-targets than Cas9 (Kim et al., 2017a). Additional studies on BE3-dependent off-targets led to the development of a high-fidelity BE3 enzyme (HF-BE3) with reduced off-target base editing activity (Rees et al., 2017). Despite these important studies, it remains to be determined whether the nickase activity of BE3 might also contribute to the generation of off-targets. To limit the use of sgSTOPs with potentially higher off-target activity, we have estimated the off-target propensity of all our predicted sgSTOPs and have designed filters in our online database that allow users to exclude the most promiscuous sgSTOPs targeting their genes of interest.
Potential iSTOP Applications
iSTOP provides possible strategies to address currently challenging biological questions. Indeed, by disrupting gene ORFs without altering gene structures, (1) iSTOP could allow the separation of coding from non-coding functions of genes, such as ASCC3 (Williamson et al., 2017), that encode for both proteins and long non-coding RNAs (lncRNAs), and lncRNAs that contain putative alternative short ORFs (Andrews and Rothnagel, 2014). In addition, (2) iSTOP could be employed to incorporate modified or non-natural amino acids into proteins using tRNAs that suppress the newly introduced STOP codons (Elsässer et al., 2016). Moreover, by enabling the modeling of nonsense mutations at a genome-wide level, (3) iSTOP could allow genome-wide studies to investigate eukaryotic gene functions and (4) facilitate the identification of pathogenic variants in cancer through large-scale functional studies of cancer-associated mutations. Recent work has indeed confirmed the promising potential of iSTOP-related approaches for pooled screening applications (Kuscu et al., 2017). Altogether, our work establishes iSTOP as a robust and efficient gene disruption technology compatible with genome-wide studies to investigate eukaryotic gene functions and model human diseases.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SMARCAL1 (A-2) | Santa Cruz | Cat#sc-376377; RRID: AB_10987841 |
| Mouse monoclonal anti-GAPDH | Novus | Cat#NB300-221; RRID: AB_10077627 |
| Anti-Mouse IgG, HRP-linked | GE Healthcare | Cat#NA931-1ML; RRID: AB_772210 |
| Bacterial and Virus Strains | ||
| Subcloning Efficiency DH5α | Life Technologies | Cat#18265-017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Transfection reagent: TransIT-293 | Mirus | Cat#MIR 2700 |
| Carbenicillin | Goldbio | Cat#C-103-25 |
| Ouabain octahydrate | Sigma | Cat#03125-1G |
| Q5 High-Fidelity DNA polymerase | NEB | Cat#M0491L |
| Critical Commercial Assays | ||
| Quick Extract DNA Extraction solution | Epicenter | Cat#QE09050 |
| Zero BLUNT II TOPO PCR Cloning kit | Life Technologies | Cat#450245 |
| SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#34087 |
| Deposited Data | ||
| Unprocessed images of gels and blots | This paper; Mendeley Data | http://dx.doi.org/10.17632/jw5rjmypy2.1 |
| Annotated sgSTOPs for 8 organisms and 6 PAMs | This paper; Mendeley Data | http://www.ciccialab-database.com/istop; http://dx.doi.org/10.17632/xbdtvf6bvj.1 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | CRL-11268 |
| Oligonucleotides | ||
| Primers for PCR | This paper | Table S5 |
| Oligonucleotides for sgRNA cloning | This paper | Table S5 |
| Recombinant DNA | ||
| Plasmid: eSpCas9(1.1) | Addgene | Addgene Plasmid #71814 |
| Plasmid: MLM3636 | Addgene | Addgene Plasmid #43860 |
| Plasmid: B52 (containing 2 empty sgRNAs-expressing cassettes) | This paper | Available in Addgene |
| Plasmid: B270 (containing sgRNA targeting ATP1A1 + empty sgRNA-expressing cassette) | This paper | Available in Addgene |
| Plasmid: pCMV-BE3 | Addgene | Addgene Plasmid #73021 |
| Software and Algorithms | ||
| iSTOP R package 0.1.0 | This paper | https://github.com/CicciaLab/iSTOP/tree/0.1.0 |
| iSTOP reproducible analysis 0.1.1 | This paper | https://github.com/CicciaLab/iSTOP-paper/tree/0.1.1 |
| R 3.4.1 | The R project for statistical computing | https://www.r-project.org |
| RStudio Desktop IDE 1.0.143 | RStudio | https://www.rstudio.com |
| Bioconductor R packages 3.5 | Bioconductor | http://bioconductor.org |
| Tidyverse R packages 1.1.1 | CRAN | https://cran.r-project.org/web/packages/tidyverse/index.html |
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
| SnapGene Viewer | SnapGene | www.snapgene.com/products/snapgene_viewer/ |
| Li-COR Odyssey | N/A | https://www.licor.com/bio/products/imaging_systems/odyssey |
| Other | ||
| Catalogue of Somatic Mutations in Cancer v80; Cancer gene census tumor suppressor annotations April 2017 | Forbes et al., 2017 | http://cancer.sanger.ac.uk/cosmic |
| Tumor suppressor predictions | Tokheim et al., 2016 | http://www.pnas.org/content/suppl/2016/11/22/1616440113.DCSupplemental/pnas.1616440113.sd04.xlsx |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alberto Ciccia (ac3685@cumc.columbia.edu). Plasmids have been deposited in Addgene.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T cells (ATCC) were cultured in DMEM supplemented with 10% Fetalgro bovine growth serum (BGS, RMBIO) and 1X penicillin-streptomycin (Life Technologies). Cells were grown at 37°C with 5% CO2. HEK293T cells were tested for mycoplasma.
METHOD DETAILS
Transfection
HEK293T cells were seeded at 50%–70% confluency into 24-well plates and reverse transfected by mixing 0.5 μg of sgRNA and 1 μg of BE3 plasmid into 100 μL of DMEM without Fetalgro BGS and antibiotics. 4.5 μL of TransIT-293 (Mirus) was added to the DNA, mixed and incubated for 15 min at room temperature. The DNA-transfection reagent mix was added dropwise to the cells and incubated at 37°C with 5% CO2 for 3 days.
Vector construction
The B52 plasmid used to express sgRNAs in human cells is a derivative of the MLM3636 construct (Addgene #43860). B52 contains a second sgRNA-expressing cassette, which was PCR amplified from the plasmid eSpCas9 (1.1) (Addgene #71814) using the primers PB96 and PB97 (Table S5) and cloned into the MLM3636 plasmid using the restriction enzymes HindIII-HF (NEB #R3104S) and XhoI (NEB #R0146S). The final B52 plasmid contains a ColE1 origin, an ampicillin resistance gene and two sgRNA expression cassettes with either BsmBI or BbsI sites for sgRNA cloning (see below). The B52 plasmid has been further modified to contain EcoRI and XbaI sites for cloning additional sgRNA-expressing cassettes. This plasmid is available in Addgene.
sgSTOP design and cloning
sgSTOPs were synthesized as oligonucleotide pairs (IDT) compatible with BbsI and BsmBI restriction sites. The oligonucleotide sequences were designed as follows: 5′-ACACCG(N)20G-3′ and 5′-AAAAC(N)20CG-3′ for BsmBI restriction sites; 5′-CACCG(N)20-3′ and 5′AAAC(N)20C-3′ for BbsI restriction sites, where “(N)20” corresponds to each sgRNA sequence. The following oligonucleotides were used for sgSTOP cloning: SPRTN (PB551 and PB552), SMARCAL1 (PB580 and PB581), CHEK2 (PB732 and PB733), PARP4 (PB734 and PB735), FANCM (PB736 and PB737), TIMELESS (PB738 and PB739), and PIK3R1 (PB776 and PB777). Oligonucleotide sequences are available in Table S5. Oligonucleotide pairs were resuspended in TE (100 μM final concentration) and annealed in the following reaction buffer: 6.5 μL water, 2 μL 5X T4 ligase buffer (Life Technologies), 0.5 μL T4 PNK (NEB #M0201L) and 0.5 μL of each oligonucleotide (100 μM; IDT). The reaction was conducted for 1 hr at 37°C followed by incubation for 5 min at 95°C and gradual temperature decrease from 95°C to 15°C. sgSTOP oligonucleotides were cloned into the B52 plasmid, which was digested with either BbsI-HF (NEB #R3539L) or BsmBI (NEB #R0580L) in a 20 μL reaction according to the manufacturer’s recommendations. 0.5 μL of recombinant shrimp alkaline phosphatase (NEB #M0371L) were then added to the digestion reaction and incubated for 1hr at 37°C to dephosphorylate the plasmid. The linearized plasmid was gel purified and incubated with the sgSTOP oligonucleotides for 1 hr at room temperature in the following ligation reaction: 50 ng of digested plasmid, 0.5 μL phosphorylated and annealed sgRNAs (diluted 1/100 in water), 1 μL 5X ligase buffer (Life Technologies) and 0.25 μL of T4 ligase. Ligation products were sequenced to confirm the integration of the sgRNA using the oligonucleotides PB518 or PB573 (Table S5) for BsmBI or BbsI cloning, respectively.
RFLP assay and DNA sequencing
The occurrence of iSTOP-mediated editing was monitored three days after transfection of HEK293T cells with BE3 and sgSTOPs, as indicated above. Cells were recovered by trypsin (Life Technologies) and washed with PBS. The cell pellet was resuspended in the Quick Extract DNA Extraction Solution (Epicenter) and heated sequentially at 65°C for 5 min and 95°C for 5 min to isolate genomic DNA (gDNA). The isolated gDNA was quantified using Nanodrop, diluted in water and stored at −20°C or directly used in PCR reactions. The following genomic loci were targeted by sgSTOPs for SPRTN (chr1+ 231338561), SMARCAL1 (chr2+ 216414804), CHEK2 (chr22- 28734431), TIMELESS (chr12- 56431493) and FANCM (chr14+ 45164491). PCR primers were designed by Primer 3 (http://bioinfo.ut.ee/primer3-0.4.0/) using the default parameters with the following changes: Mispriming library = “HUMAN,” Primer size “min = 25, Opt = 27, Max = 30,” Primer Tm “Min= 57.0, Opt = 60.0, Max = 63.0.” PCR reactions were prepared in a 25 μL reaction volume containing: 1 μM forward and reverse primers, 0.1 nM dNTPs (NEB #N0447L), 1X Q5 buffer (NEB), 200–300 ng gDNA, 1 unit Q5 polymerase (NEB) and water. PCR reactions were conducted as follows: 95°C for 1 min, 30 cycles of 95°C for 10 s, melting temperature (Tm) for 10 s and 72°C for 45 s and a final step at 72°C for 1 min. The following oligonucleotides were utilized for this reaction: SPRTN (PB571 and PB572, Tm = 64°C), SMARCAL1 (PB590 and PB591, Tm = 60°C), ATP1A1 (PB711 and PB712, Tm = 63°C), CHEK2 (PB740 and PB741, Tm = 62°C), PARP4 (PB742 and PB743, Tm = 64°C), FANCM (PB744 and PB745, Tm = 63°C), TIMELESS (PB746 and PB747, Tm = 61°C), and PIK3R1 (PB782 and PB783, Tm = 60°C). The sequence of each oligonucleotide is available in Table S5. 4 μL of PCR products were digested until completion with the indicated restriction enzymes (NEB) in a 20 μL reaction. For BsrI digestions, PCR products were purified on columns before digestion. In the case of SfaNI, reactions were heated at 65°C for 30 min to denaturate the enzyme after digestion. After digestion, 5 μL of 6X loading buffer (NEB) was added to each reaction and 2 μL of the mixture was loaded on a 6% TBE polyacrylamide gel. Gels were run at 160 V in 1X TBE buffer, then washed in 1X TBE and stained for 5 min using SybrGold diluted in 1X TBE buffer. Gels were developed using LI-COR Odyssey. To confirm base editing by Sanger sequencing, 6 to 8 PCR reactions were performed as indicated above, pooled together, purified on columns and digested in 20 μL reaction using the respective restriction enzymes. DNA fragments refractory to digestion were gel purified and cloned using the pCR-BLUNT II-TOPO kit (Life Technologies). The cloned products were sequenced by Eton Bioscience using the universal T7 promoter primer (5′-TAATACGACTCACTATAGGG-3′). Sanger sequencing data were analyzed using Serial Cloner and SnapGene Viewer. The sequencing profiles shown in this manuscript were generated by SnapGene Viewer.
ATP1A1 co-selection strategy
To conduct the marker-free co-selection strategy based on ATP1A1 targeting (Agudelo et al., 2017), we generated the plasmid B270 expressing an sgRNA against ATP1A1 and an empty sgRNA-expressing cassette. B270 was derived from B52 upon cloning of ATP1A1 sgRNA oligonucleotides (PB609 and PB610) into the BsmBI restriction site, as detailed above. sgRNA oligonucleotides for SMARCAL1 (PB580 and PB581) or SPRTN (PB551 and PB552) were introduced into the B270 plasmid using BbsI cloning sites. Transfection of HEK293T cells was conducted as follows: 6-well plates were seeded at approximately 70% cell confluency and HEK293T cells were reverse transfected by mixing 1 μg of sgRNA and 2 μg of BE3 plasmid into 600 μL of DMEM without Fetalgro BGS and antibiotics. Four days after transfection, the cells were split in 3 × 6-well plates and treated with 1 μM ouabain. Untreated cells were plated in 12-well plates and grown in parallel. 48 hr after treatment, ouabain was removed from the medium and the surviving cells were cultured in regular DMEM medium for 2 days, prior to a new 1 μM ouabain treatment for 2 additional days. The surviving cells were amplified over the course of 2 weeks and untreated cells were grown in parallel as controls.
Western blot
HEK293T cell pellets were resuspended in 30 μL of PBS and 30 μL of 2X lysis buffer (100mM Tris pH 6.8,4% SDS and 1.716M2-mercaptoethanol) and incubated for 10 min at 95°C. Cell extracts were then diluted in 1X loading buffer (NuPage, Life Technologies) and heated for 5 min at 95°C. Samples were run on an 8% polyacrylamide gel at 160 V in Tris-glycine buffer. Gels were subsequently transferred onto nitrocellulose membranes, which were stained with ponceau solution (Sigma) and then incubated for 30 min with 5% milk (Bio Basic) in TBS 0.1% Tween 20 (TBS-T). Membranes were then incubated with primary antibodies in TBS-T supplemented with 1% milk for 2 hr at room temperature. The following antibodies were used for western blotting: anti-SMARCAL1 (1/500) and anti-GAPDH (1/1000) diluted in TBS-T with 1% milk (Bio-Rad). Membranes were then washed three times in TBS-T and incubated for 1 hr with anti-mouse IgG secondary antibodies coupled with HRP at 1/5000 dilution in TBS-T/1% milk. Membranes were subsequently washed three more times in TBS-T and the HRP signal was detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and autoradiography films (Southern Labware).
Genome-wide iSTOP analysis
To identify iSTOP targetable sites, coding sequence was constructed from genomes using available CDS coordinates. Each coding sequence was verified to have a start and STOP codon, no internal STOP codons and a sequence length evenly divisible by three. Coding sequence coordinates for CAA, CAG, CGA and TGG codons were then mapped to genome sequence coordinates to then extract the genomic sequence context of each targeted base. sgSTOPs were designed by searching for each PAM 13 to 17 bps downstream of each targeted C. To count putative off-target sites, all sgSTOPs (including PAMs) were aligned to the genome, allowing up to two mismatches in the first eight bases of the guide sequence. Alignment criteria were chosen as a compromise between computational efficiency and the reported mismatch tolerance of Cas9 (Cho et al., 2014). Nonsense-mediated decay prediction was determined based on whether the targeted base was 55 nucleotides upstream of the final exon-exon junction (Popp and Maquat, 2016). All genomes and CDS coordinates are from the UCSC genome browser (https://genome.ucsc.edu), with the exception of the A. thaliana genome, which originates from TAIR (https://www.arabidopsis.org), and A. thaliana CDS coordinates, which originate from Ensembl BioMart (http://www.biomart.org). All computational analysis was performed in R (https://www.r-project.org) via RStudio (https://www.rstudio.com), making extensive use of Tidyverse (http://www.tidyverse.org/), and Bioconductor (https://bioconductor.org) R packages. A step-by-step guide to reproduce the figures and computational analyses found in this paper is available on GitHub (https://github.com/CicciaLab/iSTOP-paper).
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of iSTOP-mediated editing
The percentage of iSTOP-mediated editing (% editing) was estimated by quantifying digested and undigested bands in RFLP assays using ImageJ. iSTOP-mediated editing resulting in the loss of a restriction site was estimated by the following formula: intensity of the undigested band divided by the combined intensity of digested and undigested bands. iSTOP-mediated editing resulting in the gain of a restriction site was quantified as follows: intensity of the digested bands divided by the combined intensity of digested and undigested bands.
Analysis of frequent iSTOPers
Genes observed to incur frequent cancer-associated nonsense mutations at CAA, CAG, CGA and TGG codons (iSTOPers) were scored using a one-tailed binomial test. For this test, the probability of “success” for each gene is estimated based on the number of nonsense mutations observed at CAA, CAG, CGA and TGG codons in a given cancer type and the number of CAA, CAG, CGA and TGG codons present in the gene. The number of “successes” is the number of CAA, CAG, CGA and TGG codons in the gene observed to incur nonsense mutations in a given cancer type. Mutated coordinates in the COSMIC database were only counted once to avoid bias from duplicated entries. We report unadjusted p-values and FDR corrected q-values to account for multiple tests (Benjamini and Hochberg, 1995). An “All cancers” category is included as a summary of the lowest q-value observed for a gene across all cancer types. High scoring iSTOPers are shown in Figures 5B and 5C. Genes/cancer type combinations with a q-value less than 0.001 are included in Table S4.
DATA AND SOFTWARE AVAILABILITY
All source code necessary to reproduce figures and computational analyses is available on GitHub (https://github.com/CicciaLab/iSTOP-paper). An R package that enables iSTOP detection for any DNA sequence with annotated CDS coordinates and customizable PAM/codon/spacing parameters is available on GitHub (https://github.com/CicciaLab/iSTOP). Detailed installation and usage instructions for both of these projects are available on GitHub. An interface for convenient search of sgSTOPs is available online (http://www.ciccialab-database.com/istop). Comprehensive datasets of pre-designed and annotated sgSTOPs for 8 organisms and 6 PAMs are available on Mendeley Data (http://dx.doi.org/10.17632/xbdtvf6bvj.1). Plasmids created in this study will be made available from Addgene. Raw gel images are available on Mendeley Data (http://dx.doi.org/10.17632/jw5rjmypy2.1).
Supplementary Material
Figure S1 (Related to Figure 1). Repertoire of amino acid substitutions generated by CRISPR-mediated base editing on coding or non-coding strands
(A) Schematic representation of the BE3 CRISPR-dependent base editor, consisting of rAPOBEC1 (brown), nCas9 (green) and UGI (red), in the process of targeting a cytosine located on the coding strand. By converting C to T in CAA, CAG and CGA triplets on the coding strand, BE3 generates STOP codons.
(B) Schematic representation of BE3 targeting a cytosine located on the non-coding strand. This process results in G>A transitions on the coding strand. By converting G to A in TGG triplets on the coding strand, BE3 generates STOP codons.
(C) Percentage and number of targetable and untargetable codons and type of mutations generated on the 64 codons by targeting cytidine deaminase-dependent CRISPR base editors on either coding or non-coding strands. Codons untargetable by CRISPR base editors are indicated in white. Codons that when targeted cause silent or possible missense mutations are indicated in green and orange, respectively.
(D) Number of amino acids that can be converted into other amino acids/STOP codons (orange) or generated from different amino acids (green) by cytidine deaminase-dependent CRISPR base editors, as shown in Figure 1B. Ala = Alanine, Arg = Arginine, Asn = Asparagine, Asp = Aspartic acid, Cys = Cysteine, Gln = Glutamine, Glu = Glutamic acid, Gly = Glycine, His = Histidine, Ile = Isoleucine, Met = Methionine, Leu = Leucine, Lys = Lysine, Phe = Phenylalanine, Pro = Proline, Ser = Serine, Thr = Threonine, Trp = Tryptophan, Tyr = Tyrosine, Val = Valine and STOP = STOP codon. See also Table S1.
Figure S2 (Related to Figure 2). Specificity of the RFLP assay utilized to detect iSTOP-mediated editing
(A) BsrGI-, MfeI- and SacI-mediated digestion of PCR products of the FANCM locus targeted with an sgSTOP that edits a BsrGI restriction site. A schematic map of the FANCM locus is indicated above.
(B) ApaI-, NcoI- and SacI-mediated digestion of PCR products of the CHEK2 locus targeted with an sgSTOP that edits an ApaI restriction site. A schematic map of the CHEK2 locus is indicated above.
(C) BamHI-, BsrI-, PvuII- and NcoI-mediated digestion of PCR products of the PARP4 locus targeted with an sgSTOP that edits BamHI and BsrI restriction sites. A schematic map of the PARP4 locus is indicated above. One sequencing profile representative of 4 sequences of PARP4 amplicons refractory to BsrI digestion is shown on the right inside.
Figure S3 (Related to Figure 3). Co-selection strategy to enrich for iSTOP-mediated editing and detection by RFLP assay
(A) PvuII restriction digest of PCR products of the SPRTN locus targeted with an sgSTOP that edits a PvuII restriction site. The reaction was terminated after 1, 30 or 60 min and the products of the reaction were run on a polyacrylamide gel.
(B) Digestion of SPRTN amplicons from cells transfected with sgSTOPs targeting SPRTN and/or ATP1A1 with or without 1 μM ouabain selection using the restriction enzymes PvuII and NheI. Editing efficiency was monitored by loss of PvuII cutting and gain of NheI cutting, as indicated in Figure 2C.
(C) Sequencing profile of the targeted SPRTN locus amplified from cell populations transfected with sgSTOPs targeting SPRTN and/or ATP1A1. The targeted base that creates a STOP codon (blue arrow) and other targeted bases that generate missense mutations (red arrows) in the SPRTN locus are indicated by asterisks (*).
(D) Alignment of sequences of ATP1A1 alleles from cells targeted with an ATP1A1 sgSTOP and selected with ouabain. The ATP1A1 sgSTOP target sequence is represented in red. The symbol “#” indicates a mismatch between the WT genomic sequence (above) and the genomic sequence isolated from ouabain resistant cells (below).
(E) RFLP analysis of the SMARCAL1 locus from 19 single cell clones derived from HEK-293T transfected with sgSTOPs targeting SMARCAL1 and/or ATP1A1 and selected with ouabain. SMARCAL1 amplicons derived from the above clones were digested with SfaNI, as indicated in Figure 3D.
(F) Sequencing profiles of SMARCAL1 loci from the SMARCAL1 KO clones #16 and #17 shown in (E). The targeted base is indicated by a blue arrow.
(G) Sequencing profiles of the SMARCAL1 locus in SMARCAL1 WT and heterozygous mutant clones shown in (E). The targeted base is indicated by a blue arrow and indels are indicated by a red arrow.
Figure S4 (Related to Figure 4). Extended genomic analysis of iSTOP targetable sites in the genomes of 8 eukaryotic species
(A) Human genes untargetable by iSTOP that have targetable orthologs in other eukaryotic species. All homologs reported by Ensembl (www.ensemble.org) were considered. The complete list of untargetable human genes is available in Table S2.
(B) Number of untargetable ORFs in all species considered in this study. Percentage of all ORFs that are untargetable due to an unavailable PAM are indicated in black text.
(C) Number of all CAA, CAG, CGA and TGG codons in each species, and whether they are targetable with iSTOP. Percentage of each category is annotated on each bar.
(D) Number of iSTOP targetable codons per ORF length in the human genome.
(E) Number of genes that are targetable in all or at least one (1+) isoform. Percentage of total number of genes considered is annotated in text on each bar.
(F) Distribution of the number of mapped sites in the human genome for NGG sgSTOPs. Each NGG sgSTOP was mapped to all matching locations in the genome allowing up to two mismatches outside of the guide’s seed sequence (positions 1 through 8). Each guide is expected to map once in the genome. More than 1 mapped site indicates potential for off-target binding.
Figure S5 (Related to Figure 5). Analysis of genes with frequent nonsense mutations at CAA, CAG, CGA and TGG codons in cancer and modeling of a recurrent cancer-associated nonsense mutation by iSTOP
(A) Genes with frequent nonsense mutations in COSMIC observed at CAA, CAG, CGA and TGG codons (iSTOPers). The frequent iSTOPer −log10(q) score is derived from an FDR adjusted one-tailed binomial test (STAR Methods, Analysis of frequent iSTOPers). “All cancers” is the highest score observed across all cancer types.
(B) Comparison to tumor suppressor annotation and prediction methods. The top 100 iSTOPers from “All cancers” were compared to Tumor Suppressor Genes (TSGs) annotated by the Cancer Gene Census, the top 100 TSGs from the 20/20+ prediction method, the top 100 TSGs from the TUSON prediction method and the top 100 significantly mutated genes from the MutSigCV method. Genes included in this figure were either annotated as a TSG by the Cancer Gene Census, or were considered a TSG by at least 3 of the 4 TSG prediction methods. The complete list of iSTOPers (q < 0.001) is available in Table S4.
(C) TaqαI-mediated digestion of PCR products of the PIK3R1 locus targeted with an sgSTOP that edits a TaqαI restriction site to generate the cancer associated nonsense mutation R348*. One sequencing profile representative of 4 sequences of PIK3R1 amplicons refractory to TaqαI digestion is shown on the right inside.
Highlights.
CRISPR-mediated base editing allows the conversion of four codons into STOP codons
Induction of STOP codons (iSTOP) can be monitored using restriction enzymes
97%–99% of genes in eight eukaryotic species are targetable by iSTOP
iSTOP can be employed to model over 32,000 cancer-associated nonsense mutations
Acknowledgments
We thank Yannick Doyon for critical reading of the manuscript and for kindly communicating unpublished data. pCMV-BE3 was a gift from David Liu (Addgene plasmid #73021), MLM3636 was a gift from Keith Joung (Addgene plasmid #43860), and eSpCas9(1.1) was a gift from Feng Zhang (Addgene plasmid #71814). Plasmids generated in this study have been deposited in Addgene. This work was supported by NIH T32 training grants T32CA009503 and T32GM008798 (to E.E.B.), R01CA197774 and R01GM117064 (to A.C.), and R35GM118180 (to R.R.).
Footnotes
Supplemental Information includes five figures and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.08.008.
AUTHOR CONTRIBUTIONS
P.B. and A.C. conceived the project and designed the experiments. P.B. performed the experiments helped by T.S.N. and S.B.H. E.E.B. performed the genome-scale analysis and wrote the iSTOP R package. S.A.J. designed the iSTOP website. R.R. supervised E.E.B. P.B., E.E.B., and A.C. wrote the manuscript. P.B., E.E.B., T.S.N., S.A.J., S.B.H., R.R., and A.C. edited and approved the manuscript.
References
- Agudelo D, Duringer A, Bozoyan L, Huard CC, Carter S, Loehr J, Synodinou D, Drouin M, Salsman J, Dellaire G, et al. Markerfree coselection for CRISPR-driven genome editing in human cells. Nat Methods. 2017;14:615–620. doi: 10.1038/nmeth.4265. [DOI] [PubMed] [Google Scholar]
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016;6:914–929. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet. 2014;15:193–204. doi: 10.1038/nrg3520. [DOI] [PubMed] [Google Scholar]
- Bansbach CE, Bétous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Doudna JA. Applications of CRISPR technologies in research and beyond. Nat Biotechnol. 2016;34:933–941. doi: 10.1038/nbt.3659. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Bordeira-Carriço R, Pêgo AP, Santos M, Oliveira C. Cancer syndromes and therapy by stop-codon readthrough. Trends Mol Med. 2012;18:667–678. doi: 10.1016/j.molmed.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi PS, Meyerson M. Targeted genomic rearrangements using CRISPR/Cas technology. Nat Commun. 2014;5:3728. doi: 10.1038/ncomms4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsässer SJ, Ernst RJ, Walker OS, Chin JW. Genetic code expansion in stable cell lines enables encoded chromatin modification. Nat Methods. 2016;13:158–164. doi: 10.1038/nmeth.3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–D783. doi: 10.1093/nar/gkw1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol. 2015;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Piganeau M, Renouf B, Renaud JB, Sallmyr A, Ruis B, Oh S, Tomkinson AE, Hendrickson EA, Giovannangeli C, et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell. 2014;55:829–842. doi: 10.1016/j.molcel.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Jenkins BV, O’Connor-Giles KM, Wildonger J. A CRISPR view of development. Genes Dev. 2014;28:1859–1872. doi: 10.1101/gad.248252.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess GT, Frésard L, Han K, Lee CH, Li A, Cimprich KA, Montgomery SB, Bassik MC. Directed evolution using dCas9-targeted somatic hypermutation in mammalian cells. Nat Methods. 2016;13:1036–1042. doi: 10.1038/nmeth.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal BS, Janakiraman V, Kljavin NM, Chaudhuri S, Stern HM, Wang W, Kan Z, Dbouk HA, Peters BA, Waring P, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16:463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Haber JE. The democratization of gene editing: insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst) 2016;44:6–16. doi: 10.1016/j.dnarep.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones HD. Regulatory uncertainty over genome editing. Nat Plants. 2015;1:14011. doi: 10.1038/nplants.2014.11. [DOI] [PubMed] [Google Scholar]
- Kim D, Lim K, Kim ST, Yoon SH, Kim K, Ryu SM, Kim JS. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol. 2017a;35:475–480. doi: 10.1038/nbt.3852. [DOI] [PubMed] [Google Scholar]
- Kim K, Ryu SM, Kim ST, Baek G, Kim D, Lim K, Chung E, Kim S, Kim JS. Highly efficient RNA-guided base editing in mouse embryos. Nat Biotechnol. 2017b;35:435–437. doi: 10.1038/nbt.3816. [DOI] [PubMed] [Google Scholar]
- Kim YB, Komor AC, Levy JM, Packer MS, Zhao KT, Liu DR. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol. 2017c;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168:20–36. doi: 10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuscu C, Parlak M, Tufan T, Yang J, Szlachta K, Wei X, Mammadov R, Adli M. CRISPR-STOP: gene silencing through base-editing-induced nonsense mutations. Nat Methods. 2017;14:710–712. doi: 10.1038/nmeth.4327. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol. 2015;16:665–677. doi: 10.1038/nrm4063. [DOI] [PubMed] [Google Scholar]
- Ma Y, Zhang J, Yin W, Zhang Z, Song Y, Chang X. Targeted AID-mediated mutagenesis (TAM) enables efficient genomic diversification in mammalian cells. Nat Methods. 2016;13:1029–1035. doi: 10.1038/nmeth.4027. [DOI] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, Mochizuki M, Miyabe A, Araki M, Hara KY, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science. 2016;353:aaf8729. doi: 10.1126/science.aaf8729. [DOI] [PubMed] [Google Scholar]
- Plosky BS. CRISPR-mediated base editing without DNA double-strand breaks. Mol Cell. 2016;62:477–478. doi: 10.1016/j.molcel.2016.05.006. [DOI] [PubMed] [Google Scholar]
- Popp MW, Maquat LE. Leveraging rules of nonsense-mediated mRNA decay for genome engineering and personalized medicine. Cell. 2016;165:1319–1322. doi: 10.1016/j.cell.2016.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees HA, Komor AC, Yeh WH, Caetano-Lopes J, Warman M, Edge ASB, Liu DR. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat Commun. 2017;8:15790. doi: 10.1038/ncomms15790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014;14:786–800. doi: 10.1038/nrc3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukos V, Misteli T. The biogenesis of chromosome translocations. Nat Cell Biol. 2014;16:293–300. doi: 10.1038/ncb2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimatani Z, Kashojiya S, Takayama M, Terada R, Arazoe T, Ishii H, Teramura H, Yamamoto T, Komatsu H, Miura K, et al. Targeted base editing in rice and tomato using a CRISPR-Cas9 cytidine deaminase fusion. Nat Biotechnol. 2017;35:441–443. doi: 10.1038/nbt.3833. [DOI] [PubMed] [Google Scholar]
- Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, Karchin R. Evaluating the evaluation of cancer driver genes. Proc Natl Acad Sci USA. 2016;113:14330–14335. doi: 10.1073/pnas.1616440113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres R, Martin MC, Garcia A, Cigudosa JC, Ramirez JC, Rodriguez-Perales S. Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat Commun. 2014;5:3964. doi: 10.1038/ncomms4964. [DOI] [PubMed] [Google Scholar]
- van Overbeek M, Capurso D, Carter MM, Thompson MS, Frias E, Russ C, Reece-Hoyes JS, Nye C, Gradia S, Vidal B, et al. DNA repair profiling reveals nonrandom outcomes at Cas9-mediated breaks. Mol Cell. 2016;63:633–646. doi: 10.1016/j.molcel.2016.06.037. [DOI] [PubMed] [Google Scholar]
- Williamson L, Saponaro M, Boeing S, East P, Mitter R, Kantidakis T, Kelly GP, Lobley A, Walker J, Spencer-Dene B, et al. UV irradiation induces a non-coding RNA that functionally opposes the protein encoded by the same gene. Cell. 2017;168:843–855. doi: 10.1016/j.cell.2017.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Briggs AW, Chew WL, Mali P, Guell M, Aach J, Goodman DB, Cox D, Kan Y, Lesha E, et al. Engineering and optimising deaminase fusions for genome editing. Nat Commun. 2016;7:13330. doi: 10.1038/ncomms13330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong Y, Wang Y, Li C, Zhang R, Chen K, Ran Y, Qiu JL, Wang D, Gao C. Precise base editing in rice, wheat and maize with a Cas9-cytidine deaminase fusion. Nat Biotechnol. 2017;35:438–440. doi: 10.1038/nbt.3811. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 (Related to Figure 1). Repertoire of amino acid substitutions generated by CRISPR-mediated base editing on coding or non-coding strands
(A) Schematic representation of the BE3 CRISPR-dependent base editor, consisting of rAPOBEC1 (brown), nCas9 (green) and UGI (red), in the process of targeting a cytosine located on the coding strand. By converting C to T in CAA, CAG and CGA triplets on the coding strand, BE3 generates STOP codons.
(B) Schematic representation of BE3 targeting a cytosine located on the non-coding strand. This process results in G>A transitions on the coding strand. By converting G to A in TGG triplets on the coding strand, BE3 generates STOP codons.
(C) Percentage and number of targetable and untargetable codons and type of mutations generated on the 64 codons by targeting cytidine deaminase-dependent CRISPR base editors on either coding or non-coding strands. Codons untargetable by CRISPR base editors are indicated in white. Codons that when targeted cause silent or possible missense mutations are indicated in green and orange, respectively.
(D) Number of amino acids that can be converted into other amino acids/STOP codons (orange) or generated from different amino acids (green) by cytidine deaminase-dependent CRISPR base editors, as shown in Figure 1B. Ala = Alanine, Arg = Arginine, Asn = Asparagine, Asp = Aspartic acid, Cys = Cysteine, Gln = Glutamine, Glu = Glutamic acid, Gly = Glycine, His = Histidine, Ile = Isoleucine, Met = Methionine, Leu = Leucine, Lys = Lysine, Phe = Phenylalanine, Pro = Proline, Ser = Serine, Thr = Threonine, Trp = Tryptophan, Tyr = Tyrosine, Val = Valine and STOP = STOP codon. See also Table S1.
Figure S2 (Related to Figure 2). Specificity of the RFLP assay utilized to detect iSTOP-mediated editing
(A) BsrGI-, MfeI- and SacI-mediated digestion of PCR products of the FANCM locus targeted with an sgSTOP that edits a BsrGI restriction site. A schematic map of the FANCM locus is indicated above.
(B) ApaI-, NcoI- and SacI-mediated digestion of PCR products of the CHEK2 locus targeted with an sgSTOP that edits an ApaI restriction site. A schematic map of the CHEK2 locus is indicated above.
(C) BamHI-, BsrI-, PvuII- and NcoI-mediated digestion of PCR products of the PARP4 locus targeted with an sgSTOP that edits BamHI and BsrI restriction sites. A schematic map of the PARP4 locus is indicated above. One sequencing profile representative of 4 sequences of PARP4 amplicons refractory to BsrI digestion is shown on the right inside.
Figure S3 (Related to Figure 3). Co-selection strategy to enrich for iSTOP-mediated editing and detection by RFLP assay
(A) PvuII restriction digest of PCR products of the SPRTN locus targeted with an sgSTOP that edits a PvuII restriction site. The reaction was terminated after 1, 30 or 60 min and the products of the reaction were run on a polyacrylamide gel.
(B) Digestion of SPRTN amplicons from cells transfected with sgSTOPs targeting SPRTN and/or ATP1A1 with or without 1 μM ouabain selection using the restriction enzymes PvuII and NheI. Editing efficiency was monitored by loss of PvuII cutting and gain of NheI cutting, as indicated in Figure 2C.
(C) Sequencing profile of the targeted SPRTN locus amplified from cell populations transfected with sgSTOPs targeting SPRTN and/or ATP1A1. The targeted base that creates a STOP codon (blue arrow) and other targeted bases that generate missense mutations (red arrows) in the SPRTN locus are indicated by asterisks (*).
(D) Alignment of sequences of ATP1A1 alleles from cells targeted with an ATP1A1 sgSTOP and selected with ouabain. The ATP1A1 sgSTOP target sequence is represented in red. The symbol “#” indicates a mismatch between the WT genomic sequence (above) and the genomic sequence isolated from ouabain resistant cells (below).
(E) RFLP analysis of the SMARCAL1 locus from 19 single cell clones derived from HEK-293T transfected with sgSTOPs targeting SMARCAL1 and/or ATP1A1 and selected with ouabain. SMARCAL1 amplicons derived from the above clones were digested with SfaNI, as indicated in Figure 3D.
(F) Sequencing profiles of SMARCAL1 loci from the SMARCAL1 KO clones #16 and #17 shown in (E). The targeted base is indicated by a blue arrow.
(G) Sequencing profiles of the SMARCAL1 locus in SMARCAL1 WT and heterozygous mutant clones shown in (E). The targeted base is indicated by a blue arrow and indels are indicated by a red arrow.
Figure S4 (Related to Figure 4). Extended genomic analysis of iSTOP targetable sites in the genomes of 8 eukaryotic species
(A) Human genes untargetable by iSTOP that have targetable orthologs in other eukaryotic species. All homologs reported by Ensembl (www.ensemble.org) were considered. The complete list of untargetable human genes is available in Table S2.
(B) Number of untargetable ORFs in all species considered in this study. Percentage of all ORFs that are untargetable due to an unavailable PAM are indicated in black text.
(C) Number of all CAA, CAG, CGA and TGG codons in each species, and whether they are targetable with iSTOP. Percentage of each category is annotated on each bar.
(D) Number of iSTOP targetable codons per ORF length in the human genome.
(E) Number of genes that are targetable in all or at least one (1+) isoform. Percentage of total number of genes considered is annotated in text on each bar.
(F) Distribution of the number of mapped sites in the human genome for NGG sgSTOPs. Each NGG sgSTOP was mapped to all matching locations in the genome allowing up to two mismatches outside of the guide’s seed sequence (positions 1 through 8). Each guide is expected to map once in the genome. More than 1 mapped site indicates potential for off-target binding.
Figure S5 (Related to Figure 5). Analysis of genes with frequent nonsense mutations at CAA, CAG, CGA and TGG codons in cancer and modeling of a recurrent cancer-associated nonsense mutation by iSTOP
(A) Genes with frequent nonsense mutations in COSMIC observed at CAA, CAG, CGA and TGG codons (iSTOPers). The frequent iSTOPer −log10(q) score is derived from an FDR adjusted one-tailed binomial test (STAR Methods, Analysis of frequent iSTOPers). “All cancers” is the highest score observed across all cancer types.
(B) Comparison to tumor suppressor annotation and prediction methods. The top 100 iSTOPers from “All cancers” were compared to Tumor Suppressor Genes (TSGs) annotated by the Cancer Gene Census, the top 100 TSGs from the 20/20+ prediction method, the top 100 TSGs from the TUSON prediction method and the top 100 significantly mutated genes from the MutSigCV method. Genes included in this figure were either annotated as a TSG by the Cancer Gene Census, or were considered a TSG by at least 3 of the 4 TSG prediction methods. The complete list of iSTOPers (q < 0.001) is available in Table S4.
(C) TaqαI-mediated digestion of PCR products of the PIK3R1 locus targeted with an sgSTOP that edits a TaqαI restriction site to generate the cancer associated nonsense mutation R348*. One sequencing profile representative of 4 sequences of PIK3R1 amplicons refractory to TaqαI digestion is shown on the right inside.