

Abstract
Recent advances in single-molecule fluorescence imaging have made it possible to perform measurements on microsecond time scales. Such experiments have the potential to reveal detailed information about conformational changes in biological macromolecules, including the reaction pathways and dynamics of the rearrangements involved in processes such as sequence-specific DNA ‘breathing’ and the assembly of protein-nucleic acid complexes. Because microsecond resolved single-molecule trajectories often involve ‘sparse’ data – i.e., they contain relatively few data points per unit time – they cannot be easily analyzed using the standard protocols that were developed for single-molecule experiments carried out with tens-of-millisecond time resolution and high ‘data density.’ We here describe a generalized approach, based on time correlation functions (TCFs), to obtain kinetic information from microsecond-resolved single-molecule fluorescence measurements. This approach can be used to identify short-lived intermediates that lie on reaction pathways connecting relatively long-lived reactant and product states. As a concrete illustration of the potential of this methodology for analyzing specific macromolecular systems, we accompany the theoretical presentation with a description of a specific biologically-relevant example drawn from studies of the reaction mechanisms of the assembly of the single-stranded DNA binding protein of the T4 bacteriophage replication complex onto a model DNA replication fork.
I. Introduction
During the past several years, significant advances have been made in the use of single-molecule fluorescence methods to monitor conformational changes in the structure and dynamics of fluorescently labeled macromolecular systems. Such studies can provide detailed information about the assembly and function of protein-DNA complexes.1–9 The recent development of sub-millisecond (tens-of-microseconds) single-molecule Förster resonance energy transfer (smFRET) experiments has opened the possibility to study relatively fast macromolecular processes, such as DNA ‘breathing’ and its role in the regulation of biochemical reactions,2, 10–11 which cannot be resolved on the time scales of most current single-molecule methods (~100 milliseconds). DNA breathing involves the thermal activation of segments of duplex DNA to form short-lived local ‘bubble-like’ states. Such locally disordered regions of DNA are thought to function as transient, secondary-structural motifs that can be bound by regulatory proteins as intermediate steps in the assembly and function of DNA-protein complexes. Microsecond-resolved smFRET experiments have the potential to reveal the mechanisms by which DNA-associated proteins can ‘harvest’ such specific thermally populated states in the course of carrying out reactions involved in the processes of genome expression.
Fast detection techniques, such as phase-synchronous single-photon-counting methods, can provide time-resolved data with tens-of-microsecond resolution.2 Such experiments rapidly detect individual fluorescence photons from a single molecule, and store information about the intervening time intervals and optical phase conditions associated with each detection event. Even under optimal conditions, microsecond-resolved single-molecule fluorescence experiments produce ‘sparse’ data sets, because the average interval between successively detected signal photons can greatly exceed the experimental time resolution. In order to extract sub-millisecond kinetic information from sparse data sets, certain experimental challenges must be overcome. For example, transient intermediates may be difficult to detect due to the limited signal integration period. Under such low-signal conditions, the signal-to-noise (S/N) ratio is often too small to construct single-molecule trajectories in which transitions between distinct ‘states’ can be unambiguously identified and state-to-state transition ‘pathways’ can be visualized. Thus, the analysis of sparse trajectories must be carried out in non-standard ways.
In this paper, we show how mechanistic information can be obtained from microsecond single-molecule fluorescence experiments by applying generalized concepts of time correlation functions (TCFs).12–21 TCFs provide a statistically meaningful way to characterize the time scales of stochastically fluctuating biochemical systems. Moreover, the time resolution of single molecule experiments can be maximized using TCFs, as demonstrated by Scherer and co-workers.22 By correlating the fluctuations of individual molecules as a function of time, one can learn about the pathways connecting the conformational states that are accessible to the system at equilibrium. A commonly used approach to analyze single-molecule trajectories is to directly visualize the transition steps within a finite data set by fitting to a so-called hidden Markov model (HMM).23 When utilized to their full advantage, TCFs constructed as a function of multiple time intervals can, in principle, provide more accurate and detailed information than HMM analyses.
In optimal situations, one can obtain several pieces of information from the analysis of single-molecule trajectories: (i) the number of conformational states reported by an experimental observation (such as a FRET measurement); (ii) the values of the observables associated with each state; and (iii) kinetic parameters associated with the inter-conversion between the states. When the experimental signal is especially noisy, as is the case for microsecond-resolved smFRET experiments, the application of HMM methods is inadequate to determine the above information. In contrast, TCFs provide an excellent approach to analyze the microsecond kinetics of macromolecular conformational transitions.
The situation can be described using the theory of Markov chains.24 We assume that the instantaneous state of the system is mapped onto an experimentally accessible stochastic variable A(t) that can be measured at discrete times. The distribution of A is characterized by its moments, and the time-dependent moments are the TCFs. In general, the nth-order TCF, C(n)(τ1, τ2, …, τn−1), can be written as the average product of n successive observations 〈A(t1)A(t2) … A(tn)〉, which depends on the n − 1 time intervals τ1 = t2 − t1, τ2 = t3 − t2, …, τn−1 = tn − tn−1. The complexity of information that is potentially available from a TCF depends on its order. For example, the 2nd-order (two-point) TCF, C(2)(τ) = 〈A(t1)A(t2)〉, is the average product of two successive observations written as a function of the time interval τ = t2 − t1. The 2nd-order TCF thus describes the average loss (or gain) in correlation of A over time, which can be used to obtain the average time scales of the fluctuations of the system. Nevertheless, 2nd-order TCFs do not provide information about ‘transition pathways’ – that is, whether a particular state-to-state transition must follow or precede another, or whether two such transitions occur independently. Such information is available through a higher-order TCF analysis. In the analysis that follows, we skip over 3rd-order TCFs and focus on the 4th-order (four-point) TCFs C(4)(τ1, τ2, τ3), because the latter contain more information and can handle reaction pathways that include a larger number of elementary steps. In principle, even higher-order TCFs (e.g., 5th-, 6th-order, etc.) could be employed, although this would require increasingly complex analyses that become more difficult due to the S/N limitations of finite data sets. We show, by performing a global analysis that includes 4th-order TCFs, that it is possible to characterize fundamental time scales of the system, including intervening (exchange) times that might be associated with short-lived chemical intermediates.
In addition, 4th-order TCFs are widely applied in molecular spectroscopy, such as two-dimensional (2D) NMR, 2D infra-red and 2D electronic spectroscopy.25–26 For example, the nonlinear optical response of a molecule can be formulated in terms of the 4th-order TCFs of the appropriately defined transition dipole moment operator.25 4th-order TCFs have also been applied to study the stochastic microscopic fluctuations of complex chemical systems,20–21 including protein reaction dynamics,14 protein diffusion in solution,15–16 liquid polymer diffusion,17, 27 and protein conformation fluctuations in Molecular Dynamics (MD) simulations.18
In spite of their advantages, higher-order TCFs have not been previously used to study conformational transition pathways of biological macromolecules. This may be because the underlying concepts of TCFs are relatively abstract, and there are few sources on this topic that are accessible to a general scientific audience. Here we seek to demonstrate the utility of TCFs to extract mechanistic information from single-molecule fluorescence experiments. We show that by using TCFs of sufficiently high order, it is possible to distinguish between macromolecular binding pathways of varying levels of complexity.
In a recent study from our laboratory,8 smFRET experiments were employed to analyze the cooperative binding of the single-stranded (ss) DNA binding protein of the T4 bacteriophage DNA replication complex (gp32) to single-stranded segments of primer-template (p/t) DNA constructs of varying lengths and polarities. These constructs can serve as models of DNA replication forks. Throughout this paper, we use a particular model experiment based on this study as an explicit molecular illustration of the principles and approaches developed in our analysis. As background, we note that gp32 protein molecules bind cooperatively and preferentially to ssDNA, with a binding site size of 7 nucleotide residues (nts, or DNA lattice positions) per gp32 molecule.28 We have shown8 that p/t DNA substrates with a ssDNA ‘tail’ region of 15 nts in length, which can cooperatively bind up to two gp32 proteins, can undergo stochastic fluctuations between 0-, 1- and 2-bound states (see Fig. 1A). In these experiments the ssDNA tail region was labeled on opposite ends with a FRET donor-acceptor chromophore pair that moves to longer inter-dye distances as gp32 molecules bind between them and thus increase the rigidity of the intervening ssDNA sequence. As a consequence, the sequential binding of gp32 molecules to the ssDNA tail can be monitored by tracking the changes in the FRET signal, as discussed further below.
Figure 1.

(A) A hypothetical 3-state reaction scheme for the ssDNA binding protein gp32, which can bind up to two proteins to the p(dT)15 ‘tail’ region of a p/t DNA construct. FRET donor and acceptor chromophores (depicted as green and red circles) label the 3′ end of the ssDNA region and the p/t junction, respectively. The gp32 protein is shown in yellow. (B) The 0-, 1- and 2-bound states of the N = 3 system shown in Panel (A) are depicted as a linear reaction scheme, in which the reactant (state-0) and product (state-2) are coupled by a single intermediate (state-1). (C) The reaction is depicted as an N = 4 system, in which the conformational end-states are inter-connected by a ‘non-productive’ intermediate (state-1) and a ‘productive’ intermediate (state-1′). Stochastic transitions from state-i to state-j occur with probabilities determined by the rate constants kij, where i, j ∈ {0,1, …, N − 1}. (D) A simulated trajectory of the stochastic variable A(t) is shown for the N = 3 system. Here we have assigned the three states to the resolvable values A0 = 0.8, A1 = 0.5, and A2 = 0.2, and we have used the transition rates k01 = k21 = 5 s−1, and k01 = k12 = 10 s−1. An example of a four-point sequence of data points are shown corresponding to the time intervals τ1, τ2, and τ3. Figure partially adapted from reference 28.
While such experiments could detect the presence of distinct conformational sub-states of the ssDNA involved in association / dissociation events, the time-resolution of the experiments described in Lee et al.8 (~100 ms) was not sufficient to determine either the lifetimes of the short-lived singly-bound intermediates, or to directly observe their conversions to longer-lived end-states. Nevertheless, this model system can serve as a concrete illustration of the potential uses of the theoretical approaches developed here. We are currently applying these TCF methods to analyze new microsecond-resolved single-molecule experiments on this gp32 binding system.
II. Conformational Transition Pathways and the Role of Intermediates
We consider an equilibrium system composed of N discrete microscopic states. At any instant, the system can undergo a transition from state-i to state-j where i,j ∈ {0,1,…, N −1}. We assume that there exists an experimentally accessible stochastic variable A(t) that is coupled to the conformation of the system. For example, A might be a fluorescence signal from a single fluorophore or a collective signal from a FRET donor-acceptor pair that site-specifically labels a biological macromolecular complex and is sensitive to its local conformation or to a similar reaction coordinate. When the system occupies state-i, the variable A assumes a corresponding value Ai.
As indicated above, we illustrate our approach using the macromolecular system studied by Lee et al.,8 in which a ssDNA template interacts with the T4 bacteriophage gp32 binding protein (see Fig. 1A). The N = 3 reaction scheme (shown in Fig. 1B) is the simplest possible to describe the p(dT)15-(gp32)n system (with n = 0, 1, or 2), which involves 0-, 1- and 2-bound gp32 molecule states. Since the gp32 protein occludes 7 nts on the ssDNA template, there are nine possible binding conformations available to the 1-bound state (e.g., at positions 1 – 7, 2 – 8, …, and 9 – 15). This simplest model treats all 1-bound states as experimentally indistinguishable species that may lie on the accessible pathway connecting the reactant 0-bound state to the product 2-bound state. In this reaction scheme, we do not indicate direct transitions between 0- and 2-bound states, since it is known that gp32 does not directly bind to ssDNA as a dimer.29
Despite the appealing simplicity of the N = 3 scheme (Fig. 1B) for the p(dT)15-(gp32)n system, further consideration suggests that this mechanism cannot provide an adequate description of this gp32-binding model system because all of the 1-bound states on the 15 nts ssDNA ‘tail’ lattice cannot be treated as identical. Rather there are a number of ways in which a gp32 monomer might initially bind to the ssDNA template that would partially occlude the second binding site of 7 contiguous unoccupied nts, which is required to allow a second gp32 monomer to bind to the ssDNA tail of the p/t construct.30 Such 1-bound states that ‘overlap’ the potential second binding site represent ‘unproductive’ intermediates, and thus inhibit transitions between the 0-bound and 2-bound states. Clearly, the first gp32 protein can bind productively only at the four possible positions (1 – 7, 2 – 8, 8 – 14 or 9 – 15) to allow the ssDNA ‘tail’ sequence to retain a contiguous (7 nts) binding site that can accommodate a second gp32 monomer.31 These latter 1-bound states would function as ‘productive’ intermediates through which the 0-bound state can undergo transitions to the 2-bound states. The kinetics of a model of this type can be diagramed using the N = 4 scheme shown in Fig. 1C, in which we have labeled the ‘unproductive’ and ‘productive’ intermediates as state-1 and state-1′, respectively.
As pointed out above, the binding states of the ssDNA-(gp32)n system and their inter-conversion pathways can be studied using smFRET techniques.8 In the experiments by Lee et al.,8 which were performed using 100-ms time resolution, only two states – a 0-bound state and a 2-bound state – could be unambiguously observed, although indirect evidence for the existence of short-lived 1-bound states was also obtained. These results suggested that 1-bound states are present, but are too short-lived to be resolved in experiments conducted at 100-ms resolution. Because gp32 binding to ssDNA is known to be highly cooperative, 1-bound states are expected to be unstable in comparison to 2-bound states. A reasonable model for the assembly mechanism of the system might involve an initial singly bound gp32 molecule that either rapidly recruits a second gp32 protein to the ssDNA lattice to form a high affinity (cooperatively bound) dimer of gp32 molecules, or that rapidly dissociates from the ssDNA lattice. The relative probabilities of these competing scenarios should depend in part on the location of the initially bound gp32 protein, as described by the four state scheme of Fig. 1C. Indeed, a common situation for many single-molecule experiments is that intermediates can be very short-lived, and their observed signals might be degenerate. An idealized stochastic smFRET trajectory for the N = 3 scheme is shown in Fig. 1D, in which case A0, A1 and A2 are the values of the observable A(t) when the system is in states 0, 1 or 2, respectively.
To fully appreciate the kinetics of the ssDNA-(gp32)n system, one must properly account for the short-lived 1-bound intermediates, which may well give rise to indistinguishable signals. Experimentally, this requires making measurements at a higher time resolution than that used in the Lee et al. study.8 As the time resolution of a single-molecule fluorescence measurement approaches a few milliseconds, the signal will necessarily become too noisy to extract the state of the system through direct visualization of single-molecule trajectory data (e.g., by HMM analysis). Rather, we show below how equivalent information may be obtained through the application of the generalized concepts of TCFs.
III. Definitions of 2nd- and 4th-Order Time Correlation Functions
The 2nd-order TCF of A is the average product of two successive measurements, made at times t1 and t2, which are separated by the interval τ = t2 − t1
| (1) |
In Eq. (1), the angle brackets denote that the average has been performed over all possible starting times, according to . If the longest relaxation time of the system exceeds the duration of an individual data set, then the average two-point product is additionally integrated over a large number of single-molecule data sets. For a stochastic chemical system, C(2)(τ) decays from its maximum value 〈A2〉 at τ = 0 to its asymptotic minimum 〈A〉2 in the limit τ → ∞. For this reason, we define the fluctuation δA(t) = A(t) − 〈A〉, and its TCF:
| (2) |
The TCF C̄(2)(τ) defined by Eq. (2) decays from its maximum 〈δA2〉 to zero over the characteristic time scales of the system.
One can predict the form of C̄(2)(τ) for a given model using the theory of Markov chains, which assumes that the time interval between successive observations is long in comparison to ‘internal relaxation times,’ and that the probability that the system undergoes a transition from state-i to state-j depends only on its occupancy of state-i.24 This assumption ignores the possibility of memory effects, which become important if internal barriers associated with state-i influence the transition probability. The Markov chain expression for the 2nd-order TCF is:
| (3) |
In Eq. (3), is the equilibrium (time-independent) probability to observe the system in state-i, δAi is the value of the fluctuation observable associated with that state, and pji(τ) is the conditional probability that the system will be in state-j at a time τ after it was initially observed to be in state-i. Equation (3) shows that the 2nd-order TCF is the second moment of the time-dependent stochastic variable δA(t), which is the weighted average of all possible two-point products δAjδAi occurring within the time interval τ. It is instructive to note that when τ is short in comparison to the shortest transition time of the system, the two-point product is dominated by terms δAiδAi, such that . In contrast, for τ longer than the longest transition time, the two-point product is dominated by uncorrelated successive observations, such that . When the time interval τ is comparable to the time scale of a particular transition from state-i to state-j, the two-point product is dominated by terms δAjδAi, which reflect the weighted contributions of these particular transitions.
The information provided by the 2nd-order TCF alone cannot be used to determine whether the states visited during a single-molecule trajectory occur independently, or are connected through a ‘pathway’ of correlated sequential events. One can imagine that a particular fluctuation must occur first in order for a subsequent fluctuation to follow. For example, the N = 3 and N = 4 schemes depicted in Fig. 1B and Fig. 1C, respectively, illustrates the ssDNA-(gp32)n assembly pathways as a system of coupled elementary chemical steps in which the 0-bound and 2-bound states are inter-connected through ‘productive’ (and sometimes ‘unproductive’) intermediates. The 2nd-order TCF does not contain information, for example, about how a transition between any particular 1-bound and 2-bound state might be correlated to a preceding transition between the 0-bound and a 1-bound state. As we shall see, information about the preferred sequences of transitions that occur at equilibrium is contained in ‘higher-order’ TCFs.
To distinguish between different mechanisms of coupled chemical transformations, we consider the information contained within 4th-order TCFs. The 4th-order TCF of δA is the average product of four sequential observations, separated by the three time intervals τ1 = t2 − t1, τ2 = t3 − t1, and τ3 = t4 − t3 (see Fig. 1D)
| (4) |
In Eq. (4), the angle brackets have the same meaning as those in Eqs. (1) and (2). The 4th-order TCF C(4)(τ1, τ2, τ3) depends on the probability of sampling each possible time-ordered sequence of δA. For the N = 4 scheme of Fig. 1C, for example, we might observe the sequence δA0δA0δA1, δA2 at the four times sampled. If, for a particular set of time intervals, we were to observe this sequence with greater frequency than sequences that contain sequential occurrences of δA0 followed by δA2, then we might conclude that direct transitions between state-0 and state-2 are unlikely, and must proceed through an intermediate state-1′. Because the timescales of transitions between the various states have definite values, certain sequences will be more prevalent for short time intervals, while others will occur with greater frequency for long time intervals. Thus, the information encoded in C(4)(τ1, τ2, τ3) provides direct insight into the kinetic scheme that defines the time-ordered fluctuations of a single-molecule trajectory.
It is helpful to visualize C(4)(τ1, τ2, τ3) as a series of two-dimensional (2D) contour plots, with horizontal and vertical axes given by the intervals τ1 and τ3. We present model calculations of C(4)(τ1, τ2, τ3) in the next section. Such plots are presented as a parametric function of the interval τ2, which is referred to as the waiting time. As mentioned above, the 4th-order TCF contains information about the presence of ‘higher-order temporal correlations’ between successive transitions, with the first transition occurring during τ1 and the second during τ3. By examining a series of 4th-order TCFs as a function of τ2, we can determine the average timescales over which successive transitions are correlated. In the absence of higher-order correlations, upstream and downstream transitions occur independently. In the limit that the waiting time τ2 becomes very long, or that higher-order correlations are short-lived, we see from Eq. (4) that limτ2→∞ C(4)(τ1, τ2, τ3) = 〈δA(0)δA(τ1)〉〈δA(0)δA(τ3)〉 = C̄(2)(τ1)C̄(2)(τ3). In this limit, the 4th-order TCF is equal to the square product of the 2nd-order TCF defined in Eq. (2). To isolate the effects of higher order correlations from those due to 2nd-order ‘background’ correlations, it is useful to define the 4th-order difference TCF
| (5) |
The 4th-order difference TCF C̄(4)(τ1, τ2, τ3) defined by Eq. (5) decays as a function of τ2 from its maximum value 〈δA(0)[δA(τ1)]2δA(τ3)〉 to zero over the characteristic time scales for which higher-order correlations exist.
The Markov chain expression for the 4th-order TCF can be written
| (6) |
where the conditional probability pji(τ) is defined similarly as in Eq. (3). Since the system may only occupy discrete states, the 4th-order TCF is the weighted sum of a finite number of four-point products δAlδAkδAjδAi. For the N = 3 example of Fig. 1B, each observation can take only one of three possible values: δA0, δA1, or δA2,. Thus for N = 3, the four-point product can acquire (3)(3)(3)(3) = 81 possible outcomes (or pathways). In general, the number of possible outcomes for an N-state system is N4, and the 4th-order TCF is composed of the weighted average of these outcomes as described by Eq. (6). In order to apply Eqs. (3) and (6) to a specific N-state system, one must solve for the conditional probabilities pji(τ). In the following sections, we show how the conditional probabilities may be obtained as the formal solution to a master equation for a system of N coupled differential equations that characterize the reaction pathway.
IV. Calculation of TCFs using Markov Chains
We apply the theory of Markov chains to relate the 2nd- and 4th-order TCFs defined in the previous sections to specific N-state models.24,32 Such analyses are generally useful for the interpretation of single-molecule trajectories in which stochastic transitions occur between a few discrete states. We write the memory-less master equation for an N-state system
| (7) |
In Eq. (7), p(t) is an N-dimensional vector containing the probabilities to find the system in each of its N states at time t, and K is the N×N rate matrix, with elements kij associated with the transitions from state-i to state-j. We constrain the diagonal elements of the rate matrix to enforce the mass action law, and we set the sum of the instantaneous state probabilities .
When constructing the rate matrix K, the elements kij must be chosen to satisfy the detailed balance condition, where is the stationary (equilibrium) occupancy of state-i. The detailed balance condition requires that in the long-time limit, the flow of probability from state-i to state-j is equal to the flow of probability from state-j to state-i. For coupled reactions that involve cyclical pathways, the requirements of the detailed balance condition lead to additional inter-dependencies of the rate constant matrix elements. In Fig. 2, we depict three reaction schemes as examples to illustrate this point. For a system that contains a single cyclical pathway (Fig. 2A), the product of rate constants moving along the clockwise path must equal the product of rate constants moving along the clockwise path must equal the product of rate constants moving along the counter-clockwise path; i.e. k30k32k21k10 = k30k01k12k23. Thus, a system that contains a single cyclical pathway leads to the constraint that one rate constant must depend on all others. This relationship ensures that the flow of probability in the clockwise direction is precisely balanced by the flow of occupancies in the counter-clockwise direction, as must be the case for an equilibrium system. In the absence of a cyclical pathway, the detailed balance condition can be satisfied locally for each successive step of the coupled chemical reaction (see Fig. 2B), so that the rate constants may be chosen independently of each other. When the system contains multiple cyclical pathways, such as the situation depicted in Fig. 2C, more complicated interrelationships between rate constants exist. The relationship between cyclical pathways in this instance leads to the requirement that two rate constants must be dependent on all others. An excellent description of enforcing detailed balance can be found in reference 33.
Figure 2.
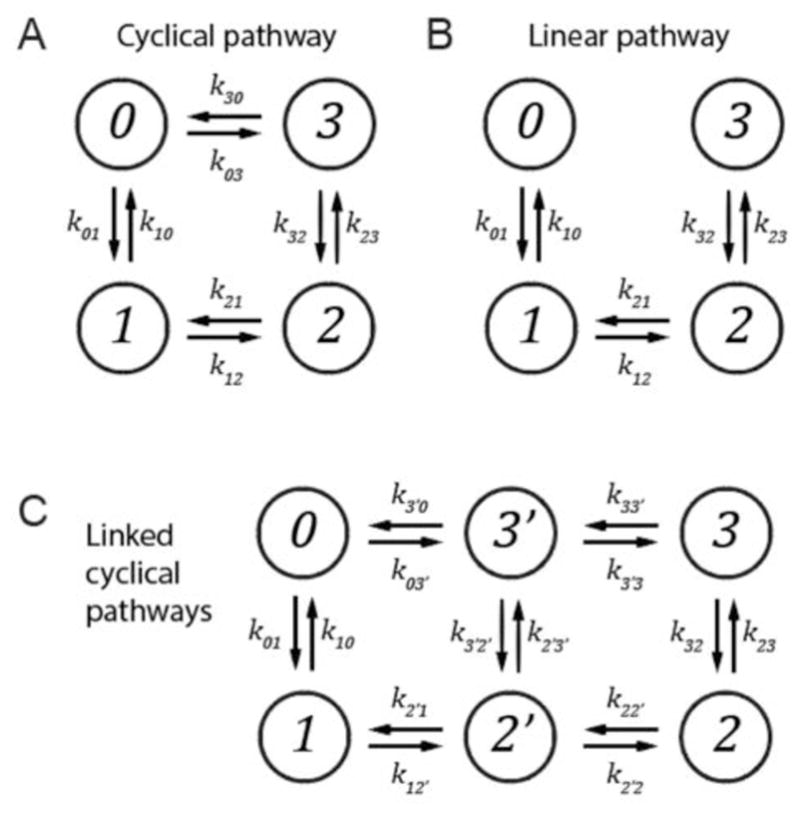
Example kinetic schemes for which the detailed balance condition requires different constraints to be applied to the rate constant relationships due to the presence or absence of cyclical pathways. (A) Single cyclical pathway. (B) Linear pathway. (C) Linked cyclical pathways.
Provided that a rate matrix K can be found to satisfy the detailed balance condition, a general solution of Eq. (7) can be obtained using the spectral decomposition method24
| (8) |
In Eq. (8), λi and vi are, respectively, the eigenvalues and the corresponding eigenvectors of the rate matrix of Eq. (7). We set the first eigenvalue λ0 = 0 to allow the time-dependent populations to decay to the constant equilibrium distribution c0v0 = peq. We may thus rewrite Eq. (8) explicitly in terms of the equilibrium distribution
| (9) |
The conditional probabilities pji(τ) needed for the evaluation of 2nd-order and 4th-order TCFs described by Eqs. (3) and (6) respectively, can be obtained using Eq. (9), with proper enforcement of the boundary conditions. For example, p21(τ) is the conditional probability that the system resides in state-2 at time τ, given that it was in state-1 at time zero. In this case, the initial condition is p1(0) = 1 and pi≠1(0) = 0. We may thus solve Eq. (9) for the set of expansion coefficients {c1, c2, …, cN−1}, and for the conditional probability p21(τ). We carry out a similar procedure for each conditional probability pji(τ) with i, j ∈ {0,1, …, N − 1}.
Analytical expressions for the 2nd- and 4th-order TCFs for N = 2 and N = 3
We next consider analytical expressions for the 2nd and 4th-order TCFs that follow from Eq. (9) for common situations with N = 2 and N = 3. Although the expressions for N = 2 systems are trivial, we include them for completeness before examining the more complex situations with N = 3.
Two-state system
For an N = 2 scheme, the 2nd-order TCF described by Eq. (2) is a weighted average of 4 possible two-point product pathways, as shown schematically in Fig. 3A.
Figure 3. Transition pathway contributions to 2nd- and 4th-order TCFs for two-state (N = 2) system.
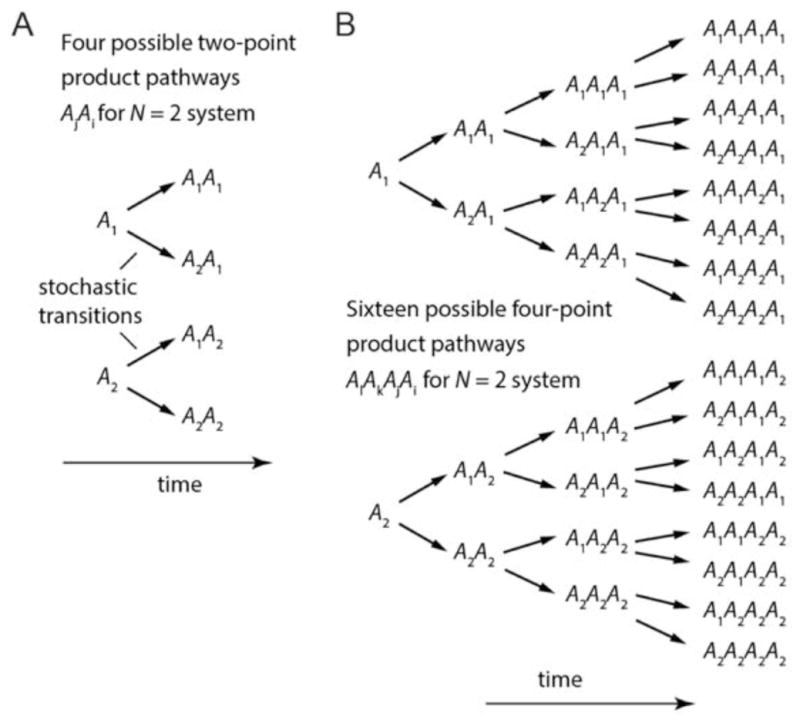
(A) There are N2 = 22 = 4 possible outcomes of a time-ordered two-point product of the observable A(t), which are used to construct the 2nd-order TCF C̄(2)(τ). (B) There are N4 = 24 = 16 such sequences for the four-point product that is used to construct the 4th-order TCF C̄(4)(τ1, τ2, τ3). The conditional probability pji(τ) that a stochastic transition will occur from state-i to state-j within the time interval τ is given by Eq. (10).
The master equation solution [Eq. (9)] specified for N = 2 yields the time-dependent conditional probabilities
| (10) |
where λ1 = k12 + k21 is the only non-zero eigenvalue. An analytical expression for the 2nd-order TCF follows from substitution of Eq. (10) into Eq. (3).
| (11) |
Equation (11) shows that the 2nd-order TCF for a two-state system decays exponentially with rate constant λ1 = k12 + k21.
For the N = 2 scheme, the 4th-order TCF described by Eq. (4) is a weighted average of 16 possible four-point product pathways, as shown schematically in Fig. 3B. Upon substitution of Eq. (10) into Eq. (6), it is straightforward to show that the 4th-order TCF for a two-state system has the form
| (12) |
where the constant 𝒜11 = 〈A2〉2. Equation (12) shows that the 4th-order TCF for an N = 2 system is simply the product of the 2nd-order TCFs C̄(2)(τ1) C̄(2)(τ3) for all values of τ2. This follows since there are no intermediates in an N = 2 scheme, and therefore no ‘higher-order’ transition pathways can exist. In this case, the 4th-order difference TCF C̄(4)(τ1, τ2, τ3), defined by Eq. (5), is equal to zero for all values of τ2.
Three-state system
We next consider the three-state scheme (N = 3) introduced in Fig. 1B, and redrawn for the following discussion in Fig. 4. In the redrawn scheme, we have allowed for the hypothetical transition between the 0-bound (reactant) state and the 2-bound (product) state, so that these might (or might not) be bridged by a 1-bound (intermediate) state. The 0 ⇄ 2 reaction pathway would require the binding of an appropriately pre-formed gp32 dimer directly from solution. This does not happen in the real system, but we include the possibility here to provide generality. Such schemes are the simplest that may exhibit higher-order temporal correlations, as reflected by the behavior of the 4th-order TCF. The derivations of the corresponding analytical expressions are straightforward, yet somewhat involved. We present the derivation here to illustrate how higher-order correlations emerge.
Figure 4.
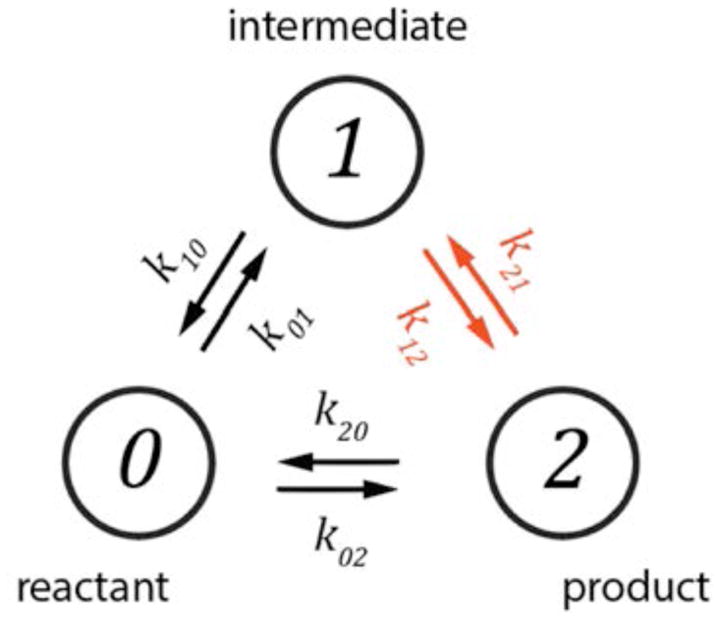
The N = 3 reaction redrawn from Fig. 1 as a cyclical scheme. This allows for the product state-2 to form either directly from the reactant state-0, or through the intermediate state-1. The ‘coupling step’ is indicated in red.
The master equation for an N = 3 system is specified, using Eq. (7), according to:
| (13) |
The general solution to Eq. (13) is
| (14) |
where the eigenvalues λ1 and λ2 and the eigenvectors and are functions of the rate constants (derivation given in Appendix 1). To satisfy detailed balance, one rate constant must depend on the others, such that k20 = k02k21k10/k12k01. The equilibrium populations are found by solving Eq. (13) with the boundary condition ṗ(t) = 0. These solutions must also satisfy completeness: . The above conditions lead to explicit forms for the component equilibrium populations and , which are explicit functions of the rate constants (see Appendix 1).
To determine the nine conditional probabilities pji(τ) with i, j ∈ {0,1,2}, we solve Eq. (14) for the expansion coefficients c1 and c2, while assuming the appropriate boundary conditions. We label each expansion coefficient with a superscript to indicate the boundary condition. For example, the expansion coefficient corresponds to the case when all population resides in state-0 at time zero, i.e. p0(0) = 1 and p1(0) = p2(0) = 0. This leads to closed form expressions for the six expansion coefficients: and (see Appendix 1). Upon substitution of these into Eq. (14), obtain the conditional probabilities
| (15) |
Substitution of Eq. (15) into Eqs. (3) and (6) provides analytical expressions for the 2nd- and 4th-order TCFs, respectively. Although these expressions are unwieldy to write in extended form, their solutions are readily obtained using a desktop computer. The 2nd-order TCF can be written succinctly
| (16) |
Equation (16) is composed of two exponentially decaying terms, with decay rates λ1 and λ2 and amplitudes 𝒜1 and 𝒜2, respectively. The constants λ1, λ2, 𝒜1 and 𝒜2 are polynomial functions of the six rate constants kij, with i,j ∈ {0,1,2} and i≠j.
It is straightforward to show that the difference 4th-order TCF, which is given by Eq. (5), has the succinct form
| (17) |
Equation (17) is composed of four terms, each with an amplitude 𝒜mn [n,m ∈ {1,2}] that depends on the waiting time τ2. Similar to the 2nd-order TCF, the 4th-order TCF decays exponentially. For a fixed waiting time τ2, the decay of the 4th-order TCF occurs in two dimensions, corresponding to the time intervals τ1 and τ3. The characteristic decay rates of the 4th-order TCF are the same as those of the 2nd-order TCF. In Eq. (17), the two terms with amplitudes 𝒜11 and 𝒜22 designate global relaxation self-terms (i.e. terms that each depend on a single eigenvalue, λ1 or λ2, respectively), while the terms with amplitudes 𝒜12 and 𝒜21 designate inter-dependent cross-terms, which each depend on both decay constants, λ1 and λ2. For an equilibrium system, the detailed balance condition requires that 𝒜12 = 𝒜21.19 As we discuss further below, the self-term amplitudes, 𝒜11 and 𝒜22, indicate the relative weights of the global relaxation processes, while the sign and magnitude of the cross-term amplitudes, 𝒜12 and 𝒜21, indicate positive or negative 4th-order correlations that effectively couple these processes.
We now return to the example of the ssDNA-(gp32)2 assembly reaction, as depicted in Fig. 4. To illustrate how the local connectivity between states can affect the collective dynamics characterized by the 4th-order TCF, we present in Fig. 5A – 5D calculations for a specific case in which the rate constants k12 and k21 are varied while the remaining parameters are held fixed. For the purpose of this discussion, we have set the waiting time interval τ2 = 1 ms, and we have chosen plausible values for the rate constants k01 = 10 s−1, k10 = 20 s−1, k02 = 2 s−1, and k20 = 4 s−1 with signal observables A0 = 0.9, A1 = 0.3, and A2 = 0.1. This particular choice of parameters assumes that the time scales of exchange between reactant state-0 and intermediate state-1 are much faster than those between reactant and product state-2. It is worth noting that for time intervals in which four-point pathways are dominated by recurring observations of the end state-0 or state-2 (e.g., δA0δA0δA0δA0), the 4th-order TCF will tend to be high-valued. Alternatively, for intervals in which the majority of four-point pathways include observations of the intermediate state-1 (e.g., δA0δA0δA0δA0), the 4th-order TCF will tend to be low-valued. For this particular example with the given rates under the detailed balance condition, the symmetry of the system dictates that for all values of the rate constants k12 = k21, the equilibrium distribution of populations are given by , and .
Figure 5.

Calculated 4th-order TCFs (panels A – D) and associated two-dimensional (2D) rate spectra (panels E – H) for the cyclical N = 3 system shown in Fig. 4. Here we have taken the waiting time interval τ2 = 1 ms, and the rate constants k01 = 10 s−1, k10 = 20 s−1, k02 = 2 s−1, and k20 = 4 s−1. The TCFs are described by Eq. (17) and the 2D rate spectra by Eq. (18). The rate constants of the ‘coupling step,’ k12 = k21 are adjusted over the range (A and E) 0, (B and F) 16.7 s−1, (C and G) 33.3 s−1, and (D and H) 66.7 s−1. For each of these conditions, values of the self- and cross-term amplitudes 𝒜11, 𝒜22, 𝒜12 = 𝒜21, respectively, are given in the text.
We initially consider the case in which transitions between state-1 and state-2 are prohibitively slow (i.e., k12 = 0). The time scales of the local elementary chemical reaction steps 0 ⇄ 1 and 0 ⇄ 2 can be estimated by assuming that these transitions occur independently of one another. We thus estimate the time scale of ‘fast’ transitions between state-0 and state-1 as (k01 + k10)−1 = 33 ms, and that of ‘slow’ transitions between state-0 and state-2 as (k02 + k20)−1 = 167 ms. By solving the master equation for the coupled system [Eq. (13)], we determine the time scales of the global relaxations (eigenvalues) λ1 = 31 s−1 and λ2 = 5.2 s−1, which correspond to the times and , respectively. Because in this example there is a clear separation between fast and slow elementary chemical steps (i.e. 0 ⇄ 1 and 0 ⇄ 2), these time scales closely approximate those of the eigenvalues of the coupled system (1 ⇄ 0 ⇄ 2). In Fig. 5A, we plot the 4th-order TCF corresponding to these conditions. We note that this function slowly rises to a peak value close to the point τ1 = τ3 ~ 33 ms and then gradually decays to zero with increasing values of τ1 and τ3. This behavior reflects the fact that multi-step transitions occur only rarely on time scales shorter than the fastest exchange process of the system. For values of τ1 and τ3 that match the time scale of the fast 0 ⇄ 1 exchange process, the 4th-order TCF is heavily weighted by terms that involve successive observations of the reactant and intermediate states (e.g., δA0δA1δA1δA0). For values of τ1 and τ3 in which one or the other of these intervals approaches time scales comparable to the slow 0 ⇄ 2 exchange process, the 4th-order TCF is composed mostly of terms that include successive observations of all three states involved in both fast and slow local reactions (e.g., δA1δA0δA0δA2), which in turn cause the function to decay. The self- and cross-term amplitudes corresponding to these conditions are 𝒜11 = 1.08, 𝒜22 = 6.73, and 𝒜12 = 𝒜21 = −2.68, which indicates that the slow eigen-mode is dominant. We note that the negative sign of the cross-term amplitudes are responsible for the concave downward shape of the three-dimensional surface, and for its convex contours for values of τ1, τ3 > 32 ms. From the above analysis, we conclude that for this model, the 32 ms time scale serves as an experimental demarcation point. For short time intervals (τ1, τ3 ≈ 32 ms), the system primarily undergoes ‘fast’ exchange of population between state-0 and state-1, and for longer time intervals (τ1, τ3 > 32 ms), the system undergoes a combination of ‘fast’ and ‘slow’ processes that exchanges population between all three states.
We next examine the possibility that state-1 shown in Fig. 4 can function as an intermediate, so that the exchange reactions 1 ⇄ 2 (shown in red) can bridge the 0 ⇄ 1 and the 0 ⇄ 2 reactions (shown in black). We first outline our expectations based on qualitative arguments before examining the theoretical results of the model. Suppose, for example, that when a gp32 monomer binds to the ssDNA template to form state-1, that it might rapidly slide to a ‘productive’ site allowing for a second gp32 monomer to bind cooperatively, and thus to form a stable dimer. Were this the prevalent mechanism, it would be reflected by the occurrence of four-point pathways at short time intervals that lead to the assembly of the ssDNA-(gp32)2 product (e.g., δA0δA1δA1δA2). The resulting 4th-order TCF would then decay rapidly with increasing values of τ1, τ3, and exhibit a pattern of positive correlation between successive elementary steps 0 ⇄ 1 and 1 ⇄ 2, which collectively lead to the formation of product. In contrast, if the gp32 monomer state-1 were unstable (due to its presumably slow exchange with state-2), its rapid dissociation would block its ability to act as a ‘gateway’ intermediate along the assembly pathway. In this latter situation, the intermediate state-1 behaves as a competitive inhibitor to the direct formation of state-2, so that the 4th-order TCF would decay slowly and exhibit a pattern of negative correlation between the successive elementary steps 0 ⇄ 1 and 1 ⇄ 2 (or 0 ⇄ 2). Therefore, depending on whether the 1 ⇄ 2 exchange time scale is fast, slow or intermediate in comparison to the fastest local relaxation time of the system (in the current example, ~ 32 ms), the global rate of population exchange can either be sped up, slowed down, or left unaffected by the presence of the intermediate state-1. These three scenarios correspond to positive, negative, and zero 4th-order correlation, respectively, between successive elementary chemical steps. The signs and magnitudes of the cross-term amplitudes, 𝒜12 and 𝒜21 serve to characterize whether 4th-order correlation is positive, negative or zero.
We now consider the case in which the exchange rate constants between state-1 and state-2 are assigned to an intermediate value in comparison to the ‘fast’ and ‘slow’ local exchange processes (30 s−1 and 6 s−1, respectively) described for the case of k12 = 0. These conditions are expected to mimic the scenario of competitive inhibition described above. In Fig. 5B, we plot the 4th-order TCF using these parameters, which decays for all non-zero values of τ1 and τ3 with collective relaxation rates λ1 = 50 s−1 and . The introduction of the 1 ⇄ 2 step permits a new pathway for population exchange to occur between all three states, which leads to a dramatic speedup of the slow collective relaxation (i.e. the second eigenvalue λ2: 5.2 → 19 s−1). Under these conditions, the self-term amplitudes are determined to be 𝒜11 = 0.472, 𝒜22 = 4.94, and the cross-term amplitudes 𝒜12 = 𝒜21 = −1.52. As in the previous case, the convex contour lines exhibited by the 4th-order TCF are due to the negative cross-term amplitudes, which indicate the presence of kinetic ‘bottleneck’ states within the four-point pathways that lead to the exchange of population between all three states. Under these conditions, the reactant state-0 is much more likely to form the intermediate state-1 than to directly form the product state-2. However, once formed, the intermediate is much more likely to undergo the reverse dissociation reaction than to proceed to form product. Thus, for short intervals τ1 and τ3 (< 32 ms), the 4th-order TCF is most heavily weighted by the ‘fast’ exchange between state-0 and state-1. Only at longer time intervals does the 4th-order TCF decay due to the contributions of slower processes such as the coupling step from state-1 to state-2.
In Fig. 5C, we plot the 4th-order TCF for the case . Under these conditions the rate constants for the 1 ⇄ 2 exchange reactions closely match those of the 0 ⇄ 1 process discussed above for the k12 = 0 ms−1 case. The 4th-order TCF decays for all values of τ1 and τ3 with collective relaxation rates λ1 = 81 s−1 and , and with self- and cross-term amplitudes 𝒜11 = 0.0001, 𝒜22 = 2.26, and 𝒜12 = 𝒜21 = 0.017, respectively. Under these conditions, only the slower of the two collective relaxation processes carries significant amplitude, and the curvature of the 4th-order TCF is neither convex nor concave. From Eq. (17), we see that in the absence of cross-term amplitude (i.e., for 𝒜12 = 𝒜21 ≈0), a cross-section of the 4th-order TCF along a vertical slice (with respect to τ3 and, for example, setting τ1 = 0) decays at precisely half the rate as does the decay along the diagonal line (with respect to τ1+τ3, and setting τ1 = τ3), so that the contours of the 2D surfaces are straight anti-diagonal lines. The absence of 4th-order correlation can be understood as a consequence of the close matching of time scales between the 1 ⇄ 2 and 0 ⇄ 1 exchange processes. Because population can readily exchange between all three states via the intermediate state-1, successive elementary reaction steps may occur in an uncorrelated manner.
By further increasing the 1 ⇄ 2 exchange rate constants to the value , we model the situation of enhanced kinetic exchange between the intermediate and product states, as described above. In Fig. 5D, we plot the 4th-order TCF for these conditions, which decays for all non-zero values of τ1 and τ3 with characteristic relaxation rates λ1 = 146 s−1 and , and with self- and cross-term amplitudes 𝒜11 = 0.155, 𝒜22 = 1.16, and 𝒜12 = 𝒜21 = 0.419, respectively. In this case, the 1 ⇄ 2 exchange rate constants are much faster than those of the 32 ms 0 ⇄ 1 process. This leads the 4th-order TCF to decay much more rapidly than in any of the previous situations, and to exhibit concave surface contours as a consequence of the positive-valued cross-term amplitudes. The concave surface curvature indicates that under these conditions, the intermediate state-1 functions as a ‘gateway’ species whose presence enhances the formation of the product state.
It is often useful to represent Eq. (17) as a two-dimensional (2D) rate domain spectrum through the inverse Laplace transform (ILT) – i.e.,
| (18) |
The 2D rate spectrum is a sum of four delta functions, which are defined in the k1,k3-plane. Comparison between Eq. (17) and Eq. (18) shows that exponentially decaying terms in the 4th-order TCF are represented as delta functions centered at values corresponding to the collective relaxation rates, λ1 and λ2 (see Figs. 5E – 5H). The two terms positioned along the ‘diagonal’ line (k1 = k3), which occur at the positions (k1,k3) = (λ1,λ1) and (λ2,λ2), respectively, correspond to the self-terms with amplitudes 𝒜11 and 𝒜22. The cross-terms with amplitudes 𝒜12 and 𝒜21 occur above and below the diagonal, at the positions (k1,k3) = (λ1,λ2) and (λ2,λ1), respectively. These self- and cross-term features of the 2D rate spectrum represent the same amplitudes discussed above for the 4th-order TCF, and thus serve as an equivalent representation of the collective dynamics of the coupled cyclical N = 3 system.
Such 2D rate spectra are made in analogy to the often-used frequency domain spectra of 2D Fourier transform spectroscopy.17, 25–27 The diagonal and off-diagonal terms generally decay as a function of the waiting time τ2. Cross-term amplitudes indicate the ‘exchange’ of populations between states involved in collective relaxation processes, and these terms decay on time scales that match the exchange dynamics. For situations in which the cross-term amplitudes are zero, the collective relaxation processes (defined by the eigenvectors v1and v2) are independent as depicted in Fig. 5G. Negative or positive cross-term amplitudes (as depicted in Figs. 5F and 5H, respectively) indicate that such processes are negatively or positively correlated, which is possible for pathways with N ≥ 3. As discussed in the context of our model calculations, the N = 3 scheme shown in Fig. 4, in which the intermediate state-1 functions as a rate-limiting ‘bottleneck’ (i.e., with k01,k10 ≫ k12 ≈ k21 ≫ k02,k20), exhibits negative 4th-order correlation. In contrast, the same scheme in which the intermediate functions as a ‘gateway’ species (i.e., with k01,k10 ≪ k12 ≈ k21 ≫ k02,k20) exhibits positive 4th-order correlation. For display purposes, we have artificially broadened the diagonal and off-diagonal features in our 2D rate spectra in Figs. 5E – 5H using a Gaussian convolution.
As previously mentioned, both TCF and HMM analyses can, in principle, provide similar information about the states and kinetics of a stochastically fluctuating chemical system. To illustrate this point, we plot in Fig. 6 the so-called ‘transition density plot’ (TDP) alongside the corresponding 4th-order TCFs and 2D rate spectra. A TDP is a useful way to present the information about transition pathways that is potentially available from an HMM analysis.23 The TDP describes the time-integrated joint distribution pji(Aj,τ;Ai,0) of molecules that are initially in state-i with observable value Ai, and which at a later time τ undergo a transition directly to state-j with observable value Aj. The weights of the TDP are given by the expression
Figure 6.

Model calculations for (A) the 4th-order TCFs, (B) the 2D rate spectra, and (C) the transition density plots (TDPs) as a function of time. For these calculations, we have used the linear N = 3 kinetic scheme diagrammed in Fig. 1B, with values A0 = 0.9, A1 = 0.3, and A2 = 0.1, and the rate constants k01 = k21 = 10 s−1, and k10 = k12 = 20 s−1.
| (19) |
(see Appendix 2 for derivation). Thus, a time-dependent TDP contains information about the direct state-to-state transitions that occur within the time interval τ, and such information could be useful, in principle, to infer assignments to the various states involved within a transition pathway. We note that in the long-time limit, the joint distribution must be a symmetric function, i.e., with , which is necessary to satisfy detailed balance. Nevertheless, this symmetry need not be valid at short or intermediate times, since the various state-to-state transitions may occur on entirely different time scales. Only in the limit of very long time intervals (i.e., longer than the slowest relaxation of the system) are the forward and backward flow of state occupancies along all inter-connected transition paths expected to be equal.
In Fig. 6, we present model calculations for the linear N = 3 scheme (shown in Fig. 1B) of the 4th-order TCFs, the 2D rate spectra, and the TDPs as a function of the waiting period τ2. For these calculations, we have chosen the rate constants k01 = k21 = 10 s−1 and k10 = k12 = 20 s−1, with signal observables A0 = 0.9, A1 = 0.3, and A2 = 0.1 (see Fig. 1B). The collective relaxation rates of the system are λ1 = 50 s−1 and , and the equilibrium distribution of populations is given by , and . This system has the interesting property that it crosses over from a regime of negative 4th-order correlation at short waiting intervals (τ2 < 27 ms) to one of positive 4th-order correlation at long waiting intervals (τ2 > 27 ms). The time-dependent crossover is evident from the shapes of the contour lines of the 4th-order TCFs (Fig. 6A) and the signs of the cross-term amplitudes of the 2D rate spectra (Fig. 6B). This is due to the fact that for waiting periods less than 27 ms, the four-point pathways are heavily weighted by transitions leading away from the intermediate state-1, either in the backward direction toward the reactant state-0, or in the forward direction toward the product state-2. An initial step in either direction will tend to inhibit the successive step in the opposite direction, thereby inhibiting the global exchange of population between all three states. An example four-point pathway for a short waiting time is δA1δA0· τ2,short · δA0δA1. In contrast, for waiting time intervals greater than 27 ms, the four-point pathways will tend to be dominated by sequences in which an initial fast step in the direction away from the intermediate state-1 (towards state-0 or state-2) will, after an intervening waiting time that exceeds the fast process, be positively correlated to a subsequent fast step in the opposite direction. An example four-point pathway such a waiting time is δA1δA0···τ 2,long ···δA1δA2. This example illustrates that the time-dependences of the 4th-order TCFs, 2D rate spectra, and the TDPs can provide information about the connectivity of a chemical network, its rate constants, and the observable values A0, A1and A2.
V. Optimization of N-State Kinetic Models to Sub-Millisecond Single-Molecule Fluorescence Data
In the preceding discussion, we have shown that analytical expressions for the TCFs of discrete stochastic systems with N = 2 or 3 can be readily obtained. For systems of higher complexity (N ≥ 4), it is often practical to solve Eq. (8) numerically. These solutions can be used to rapidly generate: (i) the 2nd-order TCF C̄(2) (τ); (ii) the 4th-order TCF C̄(4) (τ1,τ2,τ3) and its corresponding 2D rate spectrum; (iii) the equilibrium distribution of states ; and (iv) the time-dependent joint distribution of states (i.e. the time-dependent transition density plot, TDP) pji(Aj,τ;Ai,0). By applying the algorithms discussed in Section IV to calculate quantities (i) – (iv), we may implement a multi-parameter optimization strategy to obtain the simplest kinetic scheme that can accurately represent the experimental behavior of single-molecule fluorescence data.
As indicated above, conventional single-molecule fluorescence experiments performed on discrete-state systems often employ 100-ms time resolution. Such measurements can provide useful kinetic information on this time scale through direct visual inspection, or by using hidden Markov model (HMM) analyses to obtain idealized single-molecule trajectories.23 Single-molecule experiments with sub-millisecond time resolution provide only sparse trajectory data2 that are not strictly amenable to direct visual inspection or HMM analyses. This is mostly due to the influence of stochastic noise – i.e., when a fixed number (n) of data points is measured over a short period of time, the signal-to-noise ratio (S/N) during this interval has a lower bound of . We therefore turn to the analysis described in this work, which is based on the use of TCFs and state distribution functions to extract detailed and useful kinetic information about multi-step transition pathways.
Here we prescribe a step-by-step protocol to analyze sparse single-molecule trajectory data. This approach is based on multi-parameter optimization algorithms that have been widely applied in numerous experimental contexts.34–36 We must first consider the 2nd-order TCF, which is constructed from individual single-molecule trajectories as described by Eq. (2). Each TCF may vary from trajectory-to-trajectory, depending on system heterogeneity, the experimental S/N, and on the number of data-points included in the calculation. The characteristic relaxation times are reflected by the decay of the 2nd-order TCF. These are limited in range by the time-resolution of the measurement and by the maximum duration of a single-molecule trajectory. To reduce the effects of stochastic noise, the TCFs constructed from many individual trajectories should be averaged together.17 By fitting this decay to a model multi-exponential function, one determines the minimum number (N − 1) of relaxation components necessary to represent the system. The value of N so determined represents the minimum number of states, since the presence of additional relaxation components might be difficult to detect due to relatively small contributing amplitudes, or to the presence of eigen-mode degeneracy – i.e. the possibility that multiple relaxation components share the same (or nearly the same) relaxation time (eigenvalue).
Information about forward and backward rate constants associated with individual steps along the reaction pathway is contained within the 4th-order TCF. The 4th-order TCF is constructed from experimental trajectories using Eq. (5). The time intervals τ1 and τ3 must be varied over a range that spans the individual decay components present in the 2nd-order TCF, while the waiting time interval τ2 must be varied over a range that spans slow exchange time dynamics of the system.
Simulated expressions for the 2nd- and 4th-order TCFs, and the equilibrium distribution of states, are calculated using the N-state master equation that is described by Eq. (7). An optimized solution can be determined by minimizing the difference between the experimentally derived functions, and the simulated functions while varying the input parameters specified by the rate constants kij and the observable values Ai. We thus achieve a globally optimized solution to the kinetic problem of the N-state system.
VI. Conclusions
We have shown how the analysis of 2nd- and 4th-order TCFs of single-molecule trajectories can be used to learn about the roles of short-lived intermediates in biochemical reactions. In principle, 6th-order and higher TCFs could be used to study the details of even more complex biochemical reactions than the relatively simple N = 3 and N = 4 schemes examined here. The implementation of higher dimensionality TCFs is, of course, limited by S/N and data availability. Nevertheless, with the steady improvements that are currently underway to single-molecule methodology and detector technologies, such applications of generalized TCFs to elucidate complex biochemical pathways are now feasible.
The implementation of a generalized TCF analysis to microsecond-resolved single-molecule fluorescence measurements can be a powerful way to extract detailed information when the signal is too noisy to warrant analysis by direct visualization methods (e.g., HMM). However, unlike HMM, generalized TCFs are rarely utilized for such experiments. This is likely because the theory surrounding this analysis is relatively abstract and not easily approached by a general biophysical audience. In this manuscript, we have outlined the theoretical foundations to apply a generalized TCF approach to analyze single-molecule data, and we illustrated these ideas in the context of the ssDNA- (gp32)n binding system shown in Fig. 1A.
While the generalized concepts of TCF have not yet been widely applied to the analysis of single-molecule fluorescence measurements, they hold great promise for future microsecond kinetic studies, and for experiments carried out under low signal conditions. Since many important bio-molecular interactions occur on sub-millisecond timescales, we anticipate that the application of TCF methodology can help to provide new insights to understand these dynamics, which have thus far proven difficult to access experimentally.
Acknowledgments
A.H.M. acknowledges useful discussions with Prof. Mark Berg and Prof. Marina Guenza. The authors are grateful to our laboratory colleagues, in particular Dr. Wonbae Lee and John Gillies, for many useful discussions. This work was supported by grants from the National Institutes of Health/National Institute of General Medical Sciences Grant (GM-15792 - to P.H.v.H. and A.H.M.) and from the National Science Foundation, Chemistry of Life Processes Program (CHE-1608915 - to A.H.M.). C.P. was supported on NIH-NIGMS Ruth L. Kirschstein Postdoctoral Individual Research Service Award (1F32GM-109614). B.I. was supported as a predoctoral trainee on NIH-NIGMS Institutional Research Service Award in Molecular Biology and Biophysics (GM-07759). P.H.v.H. is an American Cancer Society Research Professor of Chemistry.
Appendix 1 Analytical Expression for N = 3 System
The general solution to Eq. (13) is
| (A1) |
where the eigenvalues are given by λ1 = a + b and λ2 = a − b with and , and the eigenvectors are given by and with , and .
To satisfy detailed balance, one rate constant must depend on the others, such that k20 = k02k21k10/k12k01. The equilibrium populations are found by solving Eq. (13) with the boundary condition ṗ(t) = 0. These solutions must also satisfy completeness: . This gives , and .
To determine the nine conditional probabilities pji(τ) with i,j ∈ {0,1,2}, we solve Eq. (A1) for the expansion coefficients c1 and c2, while assuming the appropriate boundary conditions. We label each expansion coefficient with a superscript to indicate the boundary condition. For example, the expansion coefficient corresponds to the case when all population resides in state-0 at time zero, i.e. p0(0) = 1 and p1(0) = p2(0) = 0. This leads to the following expressions for the expansion coefficients: , and . Upon substitution of these into Eq (A1), we obtain the conditional probabilities described by Eq. (15) of the text.
Appendix 2. Analytical Description of Time-Dependent Transition Density Plots (TDPs)
Consider an N-state Markov system at equilibrium for which stochastic transitions may occur from state-i to state-j. At any instant in time, the probability to observe the system in state-i is given by the rate expression
| (A2) |
which decays according to the general solution
| (A3) |
where A is an integration constant. The elements of the time-dependent TDP are described by the probabilities that a transition occurs from state-i to state-j within a time interval τ. By integrating Eq. (A3) over this time interval, we obtain:
| (A4) |
In the limit of very long times (τ → ∞), we expect the transition probability pij(τ → ∞) to depend on the equilibrium probability that the system resides in state-i, according to . Taking the long-time limit of Eq. (A4), we obtain pij (τ → ∞) = A/kij. Solving for A, and substitution into Eq. (A4) gives the expression for the elements of the time-dependent TDP:
| (A5) |
References
- 1.Lee W, Jose D, Phelps C, Marcus AH, von Hippel PH. A Single-Molecule View of the Assembly Pathway, Subunit Stoichiometry and Unwinding Activity of the Bacteriophage T4 Primosome (Helicase-Primase) Complex. Biochemistry. 2013;52:3157–3170. doi: 10.1021/bi400231s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phelps C, Lee W, Jose D, von Hippel PH, Marcus AH. Single-Molecule Fret and Linear Dichroism Studies of DNA ‘Breathing’ and Helicase Binding at Replication Fork Junctions. Proc Natl Acad Sci U S A. 2013;110:17320–17325. doi: 10.1073/pnas.1314862110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myong SM, Bruno M, Pyle AM, Ha T. Spring-Loaded Mechanism of DNA Unwinding by Hepatitis C Virus Ns3 Helicase. Science. 2007;317:513–16. doi: 10.1126/science.1144130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy MC, Rasnik I, Cheng W, Lohman TM, Ha T. Probing Single-Stranded DNA Conformational Flexibility Using Fluorescence Spectroscopy. Biophys J. 2004;86:2530–2537. doi: 10.1016/S0006-3495(04)74308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasnik I, Myong S, Cheng W, Lohman TM, Ha T. DNA-Binding Orientation and Domain Conformation of the E. Coli Rep Helicase Monomer Bound to a Partial Duplex Junction: Single-Molecule Studies of Fluorescently Labeled Enzymes. J Mol Biol. 2004;336:395–408. doi: 10.1016/j.jmb.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 6.Rasnik I, Myong S, Ha T. Unraveling Helicase Mechanisms One Molecule at a Time. Nucleic Acids Res. 2006;34:4225–4231. doi: 10.1093/nar/gkl452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morten MJ, Peregrina JR, Figueira-Gonzalez M, Ackermann K, Bode BE, White MF, Penedo JC. Binding Dynamics of a Monomeric Ssb Protein to DNA: A Single-Molecule Multi-Process Approach. Nucleic Acids Res. 2015:1–18. doi: 10.1093/nar/gkv1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee W, Gillies JP, Jose D, Israels BA, von Hippel PH, Marcus AH. Single-Molecule Fret Studies of the Cooperative and Non-Cooperative Binding Kinetics of the Bacteriophage T4 Single-Stranded DNA Binding Protein (Gp32) to Ssdna Lattices at Replication Fork Junctions. Nucleic Acids Res. 2016:1–20. doi: 10.1093/nar/gkw863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santoso Y, Joyce CM, Potapova O, Le Reste L, Hohlbein J, Torella JP, Grindley NDF, Kapanidis AN. Conformational Transitions in DNA Polymerase I Revealed by Single-Molecule Fret. Proc Natl Acad Sci U S A. 2010;107:715–720. doi: 10.1073/pnas.0910909107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Hippel PH, Johnson NP, Marcus AH. 50 Years of DNA ‘Breathing’: Reflections on Old and New Approaches. Biopolymers. 2013;99:923–954. doi: 10.1002/bip.22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung HS, McHale K, Louis JM, Eaton WA. Single-Molecule Fluorescence Experiments Determine Protein Folding Transition Path Times. Science. 2012;335:981–984. doi: 10.1126/science.1215768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heuer A. Information Content of Multitime Correlation Functions for the Interpretation of Structural Relaxation in Glass-Forming Systems. Phys Rev E. 1997;56:730–740. [Google Scholar]
- 13.Yang S, Cao J. Two-Event Echos in Single-Molecule Kinetics: A Signature of Conformational Fluctuations. J Phys Chem B. 2001;105:6536–6549. [Google Scholar]
- 14.Barsegov V, Chernyak V, Mukamel S. Multitime Correlation Functions for Single Molecule Kinetics with Fluctuating Bottlenecks. J Chem Phys. 2002;116:4240–4251. [Google Scholar]
- 15.Senning EN, Lott GA, Fink MC, Marcus AH., II Kinetic Pathways of Switching Optical Conformations in Dsred by 2d Fourier Imaging Correlation Spectroscopy. J Phys Chem B. 2009;113:6854–6860. doi: 10.1021/jp901542b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Senning EN, Marcus AH. Subcellular Dynamics and Protein Conformation Fluctuations Measured by Fourier Imaging Correlation Spectroscopy. Annu Rev Phys Chem. 2010;61:111–128. doi: 10.1146/annurev.physchem.012809.103500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verma SD, Vanden Bout DA, Berg MA. When Is a Single Molecule Heterogeneous? A Multidimensional Answer and Its Application to Dynamics near the Glass Transition. J Chem Phys. 2015;143:024110. doi: 10.1063/1.4926463. [DOI] [PubMed] [Google Scholar]
- 18.Ono J, Takada S, Saito S. Couplings between Hierarchical Conformational Dynamics from Multi-Time Correlation Functions and Two-Dimensional Lifetime Spectra: Application to Adenylate Kinase. J Chem Phys. 2015;142:212404. doi: 10.1063/1.4914328. [DOI] [PubMed] [Google Scholar]
- 19.Qian H, Elson EL. Fluorescence Correlation Spectroscopy with High-Order and Dual-Color Correlation to Probe Nonequilibrium Steady States. Proc Natl Acad Sci U S A. 2004;101:2828–2833. doi: 10.1073/pnas.0305962101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kryvohuz M, Mukamel S. Nonlinear Response Theory in Chemical Kinetics. J Chem Phys. 2014;140:034111. doi: 10.1063/1.4861588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kryvohuz M, Mukamel S. Multidimensional Measures of Response and Fluctuations in Stochastic Dynamical Systems. Phys Rev A. 2012;86:043818. doi: 10.1103/PhysRevA.86.043818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pelton M, Smith G, Scherer NF, Marcus RA. Evidence for a Diffusion-Controlled Mechanism for Fluorescence Blinking of Colloidal Quantum Dots. Proc Natl Acad Sci U S A. 2007;104:14249–14254. doi: 10.1073/pnas.0706164104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKinney SA, Joo C, Ha T. Analysis of Single-Molecule Fret Trajectories Using Hidden Markov Modeling. Biophys J. 2006;91:1941–1951. doi: 10.1529/biophysj.106.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reichl LE. A Modern Course in Statistical Physics. 2. John Wiley & Sons, Inc; New York: 1998. [Google Scholar]
- 25.Mukamel S. Principles of Nonlinear Optical Spectroscopy. Oxford University Press; Oxford: 1995. [Google Scholar]
- 26.Mukamel, Abramavicius SD, Yang L, Zhuang W, Schweigert IV, Voronine DV. Coherent Multidimensional Optical Probes for Electron Correlations and Exciton Dynamics: From Nmr to X-Rays. Acc Chem Res. 2009;42:553–562. doi: 10.1021/ar800258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khurmi C, Berg MA. Parallels between Multiple Population-Period Trasient Spectroscopy and Multidimensional Coherence Spectroscopies. J Chem Phys. 2008;129:064504-1-17. doi: 10.1063/1.2960589. [DOI] [PubMed] [Google Scholar]
- 28.Kowalczykowski SC, Lonberg N, Newport JW, von Hippel PH. Interactions of Bacteriophage T4-Coded Gene 32 Protein with Nucleic Acids. J Mol Biol. 1981;145:75–104. doi: 10.1016/0022-2836(81)90335-1. [DOI] [PubMed] [Google Scholar]
- 29.Jose D, Weitzel SE, Baase WA, von Hippel PH. Mapping the Interactions of the Single-Stranded DNA Binding Protein of Bacteriophage T4 (Gp32) with DNA Lattices at Single Nucleotide Resolution: Gp32 Monomer Binding. Nucleic Acids Res. 2015;43:9276–9290. doi: 10.1093/nar/gkv817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGhee JD, von Hippel PH. Theoretical Aspects of DNA-Protein Interactions: Cooperative and Non-Cooperative Binding of Large Ligands to a One-Dimensional Homogeneous Lattice. J Mol Biol. 1974;86:469–489. doi: 10.1016/0022-2836(74)90031-x. [DOI] [PubMed] [Google Scholar]
- 31.Epstein IR. Cooperative and Non-Cooperative Binding of Large Ligands to a Finite One-Dimensional Lattice. A Model for Ligand-Oligonucleotide Interactions. Biophys Chem. 1978;8:327–339. doi: 10.1016/0301-4622(78)80015-5. [DOI] [PubMed] [Google Scholar]
- 32.Noé F, Fischer S. Transition Networks for Modeling the Kinetics of Conformational Change in Macromolecules. Curr Opin Struct Biol. 2008;18:154–162. doi: 10.1016/j.sbi.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 33.Colquhoun D, Dowsland KA, Beato M, Plested AJR. How to Impose Microscopic Reversibility in Complex Reaction Mechanisms. Biophys J. 2004;86:3510–3518. doi: 10.1529/biophysj.103.038679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perdomo-Ortiz A, Widom JR, Lott GA, Aspuru-Guzik A, Marcus AH. Conformation and Electronic Population Transfer in Membrane Supported Self-Assembled Porphyrin Dimers by Two-Dimensional Fluorescence Spectroscopy. J Phys Chem B. 2012;116:10757–10770. doi: 10.1021/jp305916x. [DOI] [PubMed] [Google Scholar]
- 35.Steinbach PJ, Ionescu R, Matthews CR. Anaysis of Kinetics Using a Hybrid Maximum-Entropy/Nonlinear-Least-Squares Method: Application to Protein Folding. Biophys J. 2002;82:2244–2255. doi: 10.1016/S0006-3495(02)75570-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byrd RH, Nocedal J, Waltz RA. Knitro: An Integrated Package for Nonlinear Optimization. In: Pillo G, Roma M, editors. Large-Scale Nonlinear Optimization. Springer-Verlag; Berlin, Germany: 2006. pp. 35–59. [Google Scholar]