

Abstract
The NADPH oxidases (NOXs) play a recognized role in the development and progression of inflammation-associated disorders, as well as cancer. To date, several NOX inhibitors have been developed, through either high throughput screening or targeted disruption of NOX interaction partners, although only a few have reached clinical trials. To improve the efficacy and bioavailability of the iodonium class NOX inhibitor diphenylene iodonium (DPI), we synthesized 36 analogs of DPI, focusing on improved solubility and functionalization. The inhibitory activity of the analogs was interrogated through cell viability and clonogenic studies with a colon cancer cell line (HT-29) that depends on NOX for its proliferative potential. Lack of altered cellular respiration at relevant iodonium analog concentrations was also demonstrated. Additionally, inhibition of ROS generation was evaluated with a luminescence assay for superoxide, or by Amplex Red® assay for H2O2 production, in cell models expressing specific NOX isoforms. DPI and four analogs (NSCs 740104, 751140, 734428, 737392) strongly inhibited HT-29 cell growth and ROS production with nanomolar potency in a concentration-dependent manner. NSC 737392 and 734428, which both feature nitro functional groups at the meta position, had >10-fold higher activity against ROS production by cells that overexpress dual oxidase 2 (DUOX2) than the other compounds examined (IC50 ≈ 200–400 nM). Based on these results, we synthesized and tested NSC 780521 with optimized potency against DUOX2. Iodonium analogs with anticancer activity, including the first generation of targeted agents with improved specificity against DUOX2, may provide a novel therapeutic approach to NOX-driven tumors.
Keywords: dual oxidase, NADPH oxidase, colon cancer, diphenylene iodonium, reactive oxygen
Graphical abstract

1. Introduction
The members of the NADPH oxidase (NOX) family—NOX1–5 and dual oxidases (DUOX) 1 and 2—are conserved transmembrane enzymes expressed in a variety of human tissues. The NOX isoforms share core structural elements, including a cytosolic NADPH binding domain and FAD binding site, and a heme-containing 6-transmembrane domain. Additionally, NOX5 and DUOX1/2 have cytosolic calmodulin-like Ca2+-binding functionality at the N-terminal, and the DUOX enzymes contain an extracellular peroxidase homology domain. By mediating the transport of electrons across the plasma membrane, NOXs are important for redox homeostasis in various non-malignant tissues [1]. The first isoform to be described, NOX2, was discovered in phagocytes (i.e., neutrophils and macrophages) where it catalyzes a respiratory burst in response to pathogens, growth factors, or cytokine exposure [1]. However, the generation of reactive oxygen species (ROS) can stimulate cellular proliferation, contributing to the pathogenesis of pre-malignant, chronic inflammatory conditions such as pancreatitis and Crohn’s disease [2–4]. Increased production of ROS has also been linked to enhanced invasiveness and angiogenesis in a variety of malignant conditions [5–7]. Certain NOX isoforms, in particular NOX1, NOX4, NOX5, and DUOX2, are highly expressed in specific epithelial malignancies and malignant melanoma; furthermore, genetic manipulation of NOX1 and NOX4 expression significantly alters the growth of human tumors in vitro and in vivo [8–12]. Consequently, the NOX isoforms constitute attractive targets for the development of anticancer therapeutics.
Favored characteristics for new agents targeting NOX-related ROS formation would include high potency and specificity, increased solubility, and limited toxicity. The preferred mechanism of action of these new compounds would be direct inhibition of NOX enzymatic activity, as opposed to modulation of signaling pathways upstream of NOX, or ROS scavenging. Recent drug screening and rational design efforts aiming to identify isoform-specific NOX inhibitors have uncovered several compounds of interest, although specificity for a single isoform has not yet been convincingly demonstrated. GKT136901 and GKT137831 have specific activity against NOX1, NOX4, and NOX5 [13–15]; ML171 is specific for NOX1 but at low potency [16]; VAS2878 and close derivative VAS3947 have demonstrated activity against NOX1, NOX2, NOX4, and NOX5; recently, peptides with more specific activity against NOX1 have been described [17–20]. However, because of the potential for off-target effects, low potency, and poor solubility [21], most of these agents have, to date, not demonstrated clinical utility as NOX inhibitors.
Historically, NOX function and activity have been interrogated in vitro through modulation with small molecule flavoenzyme inhibitors, such as diphenylene iodonium (DPI), di-2-thienyliodonium (DTI), and apocynin [22]. DPI and DTI inhibit the growth of NOX1-expressing colon cancer cells without altering mitochondrial respiration, if studied at nanomolar concentrations, so that antiproliferative effects may be attributed, at least in part, to a cyclin D1-dependent G1 block that is secondary to inhibition of NOX function [23, 24]. However, these inhibitors lack specificity for NOX enzymes as they have been shown to disrupt the activity of various flavoproteins [21, 25, 26], which limits the precision with which they can be utilized in a therapeutic context. DTI is very sparingly soluble in water and all common organic solvents used in biological experiments. Furthermore, apocynin possesses free radical scavenging activity, in addition to NOX inhibiting potential, that limits the interpretation of studies in which it is utilized [27].
In the current study, our goals were to elucidate the mechanisms whereby iodonium-class inhibitors alter ROS production by NOXs, and to optimize the pharmacological properties and NOX isoform selectivity of new iodonium analogs compared to the parent molecule, DPI. Initially, thirty-five compounds derived from DPI were screened for inhibitory effects on cellular proliferation and ROS production in HT-29 colon cancer cells. These iodonium analogs were also evaluated for effects on mitochondrial function, and for NOX isoform selectivity in cell-based assays. We identified four compounds with increased potency compared to DPI in cellular models of NOX activity, and derived a thirty-sixth compound, 521, with increased selectivity for DUOX2.
2. Materials and methods
2.1 Materials and compounds
Diphenylene iodonium (DPI; NSC 735294, Catalog number: 43088) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The 36 DPI analogs (Fig. 1A and Fig. 6A) were synthesized by the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis of the National Cancer Institute (Bethesda, MD, USA) following the general procedure shown in Fig. 1B. DPI and its analogs were dissolved in dimethylsulfoxide (DMSO, Catalog number: D2650; Sigma-Aldrich, St. Louis, MO, USA) at their maximum soluble concentration [23]. All compounds were fully characterized by 1H NMR. Using the techniques described below, all 36 analogs were screened for their ability to inhibit O2•− production and proliferation in HT-29 cells. The four initial candidate molecules that performed optimally on the basis of their solubility and their ability to inhibit tumor cell growth and ROS production were subsequently evaluated for their effects on mitochondrial function and ROS formation (as shown in the testing funnel; Fig. 1D), and then for their NOX isoform selectivity. A fifth analog (NSC 780521; described below) was prepared after evaluation of the first four to enhance interaction with DUOX2. Compound characterization details are shown below for the 5 lead compounds; data is available upon request for the other analogs.
Fig. 1. Development of DPI analogs.
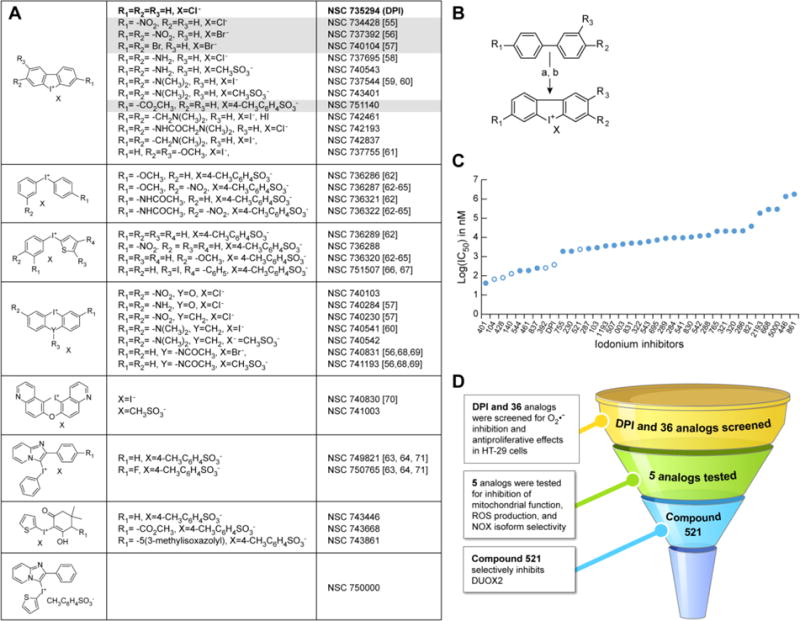
(A) Structures of DPI and 35 iodonium-class analogs. The structure for the thirty-sixth analog, compound NSC 780521 (521), is displayed in fig. 6A. DPI is shown in bold font, and the lead compounds described in the present study are highlighted in grey. (B) Synthetic pathway for the production of substituted DPI analogs. Reagents: a) I2, KIO3, H2SO4; b) KI. (C) IC50 values for iodonium compound inhibition of HT-29 cell proliferation assessed with the MTT assay at 48 h. Open circles indicate compounds described in the study. (D) Flowchart demonstrating the screening procedure for the identification of potent iodonium analogs.
Fig. 6. Compound 521.
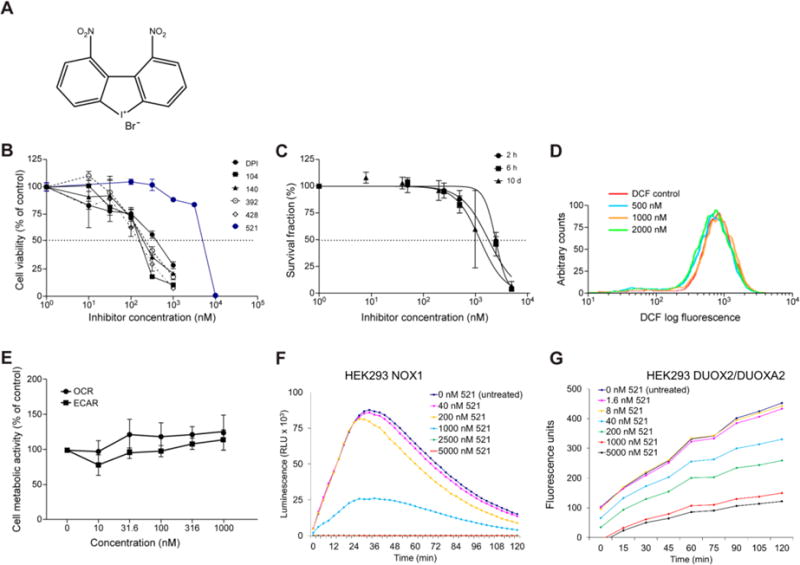
The inhibitory effects of 521 on HT-29 cell growth, whole-cell ROS production, cellular respiration, and extracellular ROS production were assessed using the same methods described above for the other DPI analogs. (A) Chemical structure of NSC 780521. (B, C) Concentration-dependent inhibition of HT-29 cell proliferation after 72-h exposure (B), measured by MTT assay; and of colony formation after 2 h, 6 h, or 10 days of HT-29 cell exposure to compound 521 (C). (D) Effect of 24-h treatment with 521 on intracellular ROS production in HT-29 cells, measured by analytical cytometry using the redox-sensitive dye CM-H2-DCF-DA. (E) Effect of compound 521 on cellular metabolism following 24-h exposure evaluated by measuring oxygen consumption rates (OCR) and extracellular acidification rate (ECAR), respectively, with the Seahorse Extracellular Flux Analyzer; (F, G) PMA-induced extracellular ROS production measured by luminescence assay and Amplex Red assay in NOX1 (baseline O2•− production rate = 1.37 × 10−2 RLU/min/cell) and DUOX2/DUOXA2 overexpression stable HEK293 cells (baseline H2O2 production rate = 1.0 × 10−4 RFU/min/cell), respectively, treated with 521 for 30 min. Data in panels B, C, and E represent the mean ± SD (error bars) of at least three experiments. RLU, relative light units; RFU, relative fluorescence units.
Dibenziodolium, 3,7-dibromo-, bromide (NSC 740104-T, 104)
Mp 202-205 °C (decomposes). 1H NMR, DMSO-d6: δ 8.69 (d, 2H); 8.37-8.36 (d, 2H); 8.03-8.01 (dd, 2H). Anal. Calc’d (C12H6Br2I∙Br) C,H,Br,I. Yield: 79 %.
Dibenz[b,d]iodolium, 3-(methoxycarbonyl)-, salt with 4-methyl-benzenesulfonic acid (1:1) (NSC 751140-P, 140)
Mp >250 °C (decomposes). 1H NMR, DMSO-d6: δ 8.84 (d, 1H); 8.64-8.62 (d, 1H); 8.61-8.59 (dd, 1H); 8.37-8.35 (dd, 1H); 8.30-8.28 (d, 1H); 7.94-7.90 (m, 1H); 7.82-7.78 (m, 1H); 7.50-7.49 (d, 2H); 7.13-7.11 (d, 2H); 3.96 (s, 3H); 2.30 (s, 3H). Anal. Calc’d (C14H10IO2∙C7H7O3S) C,H,S,I. Yield: 37 %.
Dibenziodolium, 3,7-dinitro-, bromide (NSC 737392-V, 392)
Mp >270 °C (decomposes). 1H NMR, DMSO-d 6: δ 9.41-9.40 (d, 2H); 8.78-8.76 (d, 2H); 8.65-8.62 (dd, 2H). Anal. Calc’d (C12H6IN2O4 ∙ Br) C,H,N,Br,I. Yield: 96 %.
Dibenz[b,d]iodolium, 3-nitro-, chloride (NSC 734428-Y, 428)
Mp 282–285 °C (decomposes). 1H NMR, DMSO-d6: δ 9.40-9.39 (d, 1H); 8.68-8.64 (dd, 2H); 8.59-8.57 (dd, 2H); 7.90-7.87 (t, 1H); 7.81-7.79 (t, 1H). Anal. Calc’d (C12H7INO2∙Cl) C,H,N,Cl,I. Yield: 94 %.
Dibenziodolium, 1,9-dinitro-, salt with bromide (1:1) (NSC 780521, 521)
MP 207–209 °C (decomposes). 1H NMR, DMSO-d6: δ 9.02-9.01 (d, 2H); 8.50-8.49 (d, 2H); 8.01-7.98 (t, 2H). Anal. Calc’d (C12H6IN2O4 Br) C,H,N,Br,I. Yield: 93%.
2.2 Cell culture
HT-29, HL-60, UACC-257, and HEK293 cell lines were obtained from ATCC (Manassas, VA, USA). Human HT-29 colon cancer cells were propagated in McCoy’s 5A medium supplemented with 10% FBS (Lonza, Walkersville, MD, USA). HL-60 and UACC-257 cells were grown in RPMI-1640 medium containing 10% FBS. The stable HEK293 cell line expressing both the human DUOX2 and DUOXA2 enzymes was kindly provided by Dr. William M. Nauseef (University of Iowa, Iowa City, IA, USA) and maintained in DMEM:F12 medium supplemented with 10% FBS, 800 μg/ml G418 (Catalog number: 5005; Teknova, Hollister, CA, USA) and 250 μg/ml Zeocin (Catalog number: 46-0509; Invitrogen, Carlsbad, CA, USA) [28]. HEK293 cell lines that stably express the human NOX1 (HEK293 NOX1) and NOX4 (HEK293 NOX4) enzymes were engineered in-house. Briefly, stable NOX1 NOXA1/NOXO1 cells were initiated by transfection of HEK293 cells with a pCMV-NOX1 (3 μg) plasmid using the Lonza system (Kit V, Program Q-001; Walkersville, MD, USA), followed by selection with 800 μg/ml G418 (Sigma, St. Louis, MO, USA). After stable clones were developed and evaluated, a single clone was selected for transfection with pCMV-NOXA1/NOXO1 (3 μg), and single clones were selected with 800 ng/ml puromycin. The final, active NOX1-overexpressing clonal cell line was maintained with 500 μg/ml G418 and 500 ng/ml puromycin. To produce a stable, clonal cell line overexpressing NOX4, 4 μg pCMV-MycDDK-NOX4 plasmid cDNA was transfected into HEK293 cells, again using the Lonza system (Kit V, Program Q-001; Walkersville, MD, USA). Resistant clones were selected with 750 μg/ml G418, and single clones were then maintained under G418 selection. To obtain polymorphonuclear (PMN) leukocytes, whole blood (200 ml) was drawn into BD Glass K3 EDTA tubes (Catalog number: 02-685-2B; Fisher Scientific, Hampton, NH, USA) from a healthy volunteer after written informed consent, in accordance with ethical guidelines at the Frederick National Laboratory for Cancer Research (Research Donor Protocol [RDP] 10-001). Within 2 h of drawing, PMNs were isolated using the PolymorphPrep™ system (Catalog number: AXS-1114683; Cosmo Bio USA, Carlsbad, CA, USA) and following manufacturer recommended protocol. All cell lines were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
2.3 MTT cell growth assay
HT-29 tumor cells were seeded into 96-well plates (Catalog number: 3596; Corning Inc., Vienna, VA, USA) at a density of 1 ×104 cells/well in McCoy’s 5A medium supplemented with 10% FBS for 24 h. Serial dilutions of DPI analogs in culture medium (0 to 100 μM) were added to the cells in 150 μl total volume, and incubated for various time intervals. Cell viability was then assayed by adding 50 μl of 2 mg/ml 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT, Life Technology, Eugene, OR, USA) in McCoy’s 5A medium. The cells were incubated with MTT for 1 h at 37°C; the medium was removed, and the blue pigment produced by viable cells was solubilized in 100 μl/well of 0.5% (w/v) SDS and 25 mM HCl in 90% (v/v) isopropyl alcohol [29]. The plates were shaken for 10 min, and the oxidized MTT was measured at A570nm with background measurement at A650nm on a SpectraMax M5 reader (Molecular Devices Corp., Sunnyvale, CA, USA) [30].
2.4 Cell proliferation assays
HT-29 cells (5 × 102) were seeded in human fibronectin-coated 6-well plates (Catalog number: 354402, Corning, Corning, NY, USA), and grown in McCoy’s 5A medium with 10% FBS for 24 h. The cells were treated with DPI or analogs over a range of concentrations (DPI, NSCs 740104, 751140, 737392, and 734428: 0–1000 nM; 780521: 0–5000 nM) for 2 h, 6 h, or 10 days. After short-term exposure (2 h or 6 h), the inhibitor solutions were removed and the cells were washed; fresh complete medium was added and colony formation was evaluated after 10 days of growth. Long-term exposure proceeded with cells cultured in complete medium containing iodonium-class inhibitors for 10 days. For evaluation, all colonies were washed and fixed with 4% paraformaldehyde (Catalog number: 15714-S; Electron Microscopy Sciences, Hatfield, PA, USA) for 1 h and stained with 0.02% crystal violet (Catalog number: V5265; Sigma-Aldrich, St. Louis, MO, USA) for 45 min. Subsequently, the staining solution was removed, and the cells were washed 4 times with PBS. Stained colonies were counted with an AccuCount 1000 Automated Colony Counter (BioLogics, Inc., Manassas, VA, USA).
To determine the effects of SOD and catalase on cell growth, trypsinized HT-29 cells were counted with a T4 cellometer (NexCelom Bioscience, Lawrence, MA, USA). Fifty thousand cells were plated in full media in 6-well tissue culture plates (Corning, Corning, NY, USA) in triplicate for each time point and condition. SOD (cat# S9549; Sigma Aldrich, St. Louis, MO, USA) and/or human catalase (cat# C1345-10 g; Sigma Aldrich, St. Louis, MO, USA) were added at concentrations ranging from 0 to 2000 U/ml. For the combination treatment, SOD and catalase were added simultaneously. After 96 h, the cells were trypsinized again and counted with the T4 Cellometer (NexCelom Bioscience, Lawrence, MA, USA). The growth of control-treated HT-29 cells was compared to the growth of SOD- and/or catalase-treated cells with the two-tailed Student’s t-test; values of p<0.05 were considered significant.
2.5 Measurement of intracellular reactive oxygen production by flow cytometry
HT-29 cells were trypsinized, seeded in 6-well plates, and treated with DPI or DPI analogs for 24 h at 37°C. To remove large multi-cellular groups, cells (1 × 106) were passed into a tube through a cell-strainer cap (Catalog number: 352235, BD Biosciences, San Jose, CA, USA) and collected by centrifugation. Pelleted cells were resuspended in 0.5 ml PBS containing 0.1% BSA, and 5 μM redox-sensitive dye CM-H2-DCF-DA (Catalog number: C6827, Invitrogen, Carlsbad, CA, USA) and incubated in the dark for 30 min at 37°C with shaking. Then the dye solution was removed, and 0.5 ml PBS containing 0.1% BSA was added to each pellet. Fluorescence intensity of the cells was immediately measured on a FACScalibur (BD Bioscience, San Jose, CA, USA) cytometer, acquired using the data file acquisition program CellQuest (BD Bioscience, San Jose, CA, USA), and analyzed using the FlowJo® Software (FlowJo LLC, Ashland, OR, USA). For measurements of mitochondrial superoxide production, HT-29 cells were pre-treated with lower concentrations of DPI (100 or 200 nM) for 24 h or with a high concentration of DPI (1 μM) for 2 h, and prepared as described above; pelleted cells were resuspended in 1 ml of PBS containing 3 μM MitoSox™ red mitochondrial superoxide indicator (Catalog number: M36008, Molecular Probe), and incubated in the dark for 30 min at 37°C with shaking. After removing the solution of MitoSox™ red mitochondrial superoxide indicator, the cells were resuspended in 0.5 mL PBS. Red fluorescence intensity of the cells was measured as described above.
2.6 Measurement of oxygen consumption rate and extracellular acidification rate
Oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) of HT-29 colon cancer cells were measured in real time using a Seahorse Bioscience XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA) as described [31–35]. To achieve ~85% confluence, the optimum number of cells/well was determined in preliminary studies. Seahorse assay media (Catalog number 102365-100, Seahorse Bioscience, North Billerica, MA, USA) containing 5 mM glucose, 2 mM L-glutamine, and 1 mM sodium pyruvate (pH 7.4) was freshly prepared for all experiments. For short exposure times (30 min and 60 min), HT-29 cells (7 × 104 cells/well) were plated in XF24 V7 24-well cell culture plates (Catalog number: 100850-001, Seahorse Bioscience, North Billerica, MA, USA) and incubated overnight at 37°C with 5% CO2; culture media was removed the following day, and fresh Seahorse assay media was added to the cells under study at 37°C in a CO2-absent environment for 30 min. DPI, KCN (2 mM), NSCs 740104, 751140, 737392, 734428, or 780521 were added to each well to make final concentrations of 0 nM to 1000 nM just prior to bioenergetic measurements. For the 24-h treatment, HT-29 cells (4 × 104/well) were plated in 24-well plates and incubated overnight at 37°C. The cells were then exposed to DPI or analog for 24 h. Subsequently, the growth media was removed from each well, cells were rinsed with Seahorse assay media, and fresh assay media was added to each well at 37°C in the absence of CO2 for 30 min prior to assay measurements. The results represent the mean ± S.D. of triplicate or quadruplicate wells tested at each concentration. To compare the impact of KCN and a high concentration of DPI on HT-29 cellular metabolism, 2 mM KCN or 2 μM DPI were added to each well prior to measurements, or 2 μM DPI was added to KCN-treated cells 40 minutes after initiation of the experiment. The OCR value represents the maximal mitochondrial respiratory capacity of the cells as a fraction of its control (pre-treatment) value after DPI or analog exposure. Similarly, the ECAR value corresponds to the maximal glycolytic activity as a fraction of its control value following exposure to an iodonium inhibitor.
2.7 Superoxide anion assay
Superoxide anion (O2•−), the major ROS product of HEK293 NOX1 cells, differentiated HL-60 cells (expressing NOX2), polymorphonuclear leukocytes (PMNs; expressing NOX2) and UACC-257 cells (expressing NOX5), was detected using a luminol-based superoxide anion assay kit (Catalog number: CS-1000; Sigma, St. Louis, MO, USA). HL-60 cells were fully differentiated in complete medium containing 2% DMSO for 3 days before the start of each experiment. Briefly, cells in log-phase were harvested and washed with PBS; HEK293 NOX1 (2 × 105cells/well), HL-60 (2 × 105 cells/well), PMNs (2 × 105 cells/well), and UACC-257 (2 × 105 cells/well) cells were resuspended in 96-well plates (Catalog number: 136101; Thermo Scientific, Rockford, IL, USA) with 100 μl assay medium containing a serial dilution of DPI or analogs (0 μM to 100 μM), or superoxide dismutase (4 U). The cells were incubated at 37°C for 30 min. One hundred microliters of assay buffer mix containing 3 μl of luminol solution, 3 μl of enhancer solution, with or without 200 nM of phorbol 12-myristate 13-acetate (PMA; Catalog number: P8139, Sigma-Aldrich, St. Louis, MO, USA) was added to each well. Luminescence was immediately detected at 37°C using a GloMax® Microplate Luminometer (Promega BioSciences, San Luis Obispo, CA, USA), with measurements taken every 3 min for HEK293 NOX1 and HL-60 cells, and every 30 seconds for UACC-257 cells, up to a 2 h period of observation. The data for each condition represents a minimum of three independent experiments. The rate of change of luminescence (in relative luminescence units, RLU) produced by untreated cells from time 0 min to time 30 min (~ peak luminescence) was determined empirically as a proxy for the baseline rate of O2•− production (unit: RLU/min/number of cells). For PMNs, the rate of change of luminescence from 0 to 10 min (~ peak luminescence) was used as a proxy for the baseline rate of O2•− production in these cells.
2.8 Extracellular H2O2 measurement using Amplex Red®
The Amplex Red® Hydrogen Peroxide/Peroxidase Assay Kit (Catalog number: A22188; Invitrogen, Carlsbad, CA, USA) was used to detect extracellular H2O2 release by HEK293 cells stably overexpressing NOX4 or DUOX2/DUOXA2. NOX4 and DUOX2/DUOXA2 clonal cells in log-phase growth were trypsinized, washed, and dispersed thoroughly. Following trypsinization, the cells (3 × 104) were resuspended in 100 μl of 1X Krebs-Ringer phosphate glucose (KRPG) buffer containing various concentrations of DPI or DPI analog and incubated at 37°C for 30 min. Cells were collected by centrifugation, and the inhibitor solution was discarded. The cells were washed once, and then resuspended in 100 μl of 1X KRPG buffer. Twenty microliters of the suspension containing 3 × 104 cells were added per well of a 96-well plate (Catalog number: 3596; Corning Inc., Corning, NY, USA) and mixed with 100 μl of Amplex Red® reagent solution containing 50 μM Amplex Red and 0.1 units/ml of HRP in KRPG buffer; each assay condition was evaluated in triplicate. The fluorescence of the oxidized 10-acetyl-3,7-dihydroxyphenoxazine was measured in real-time at 37°C every 15 min at an excitation wavelength of 530 nm and an emission wavelength of 590 nm using a SpectraMax M5 Multiplate reader (Molecular Devices, Sunnyvale, CA, USA). The rate of change of fluorescence (in relative fluorescence units, RFU) produced by untreated cells from time 0 min to time 60 min (~ peak fluorescence) was determined empirically as a proxy for the baseline rate of H2O2 production (unit: RFU/min/number of cells).
2.9 Formation and characterization of FAD-iodonium analog adducts
For adduct identification, a mixture of 5 nmol FAD and 10 nmol DPI or DPI analog was exposed to 25 nmol sodium dithionate in 200 μl PBS at room temperature for at least 5 min prior to analysis. Product compounds from a 2 μl aliquot of the reaction mixture were separated by HPLC using an ACE Excel5 C18 column, (150 × 2.1 mm; MAC-MOD Analytical Inc., Chadds Ford, PA, USA) at a flow rate of 250 μl/min. The mobile phase consisted of 26 mM ammonium acetate (pH 6) and acetonitrile. Compounds were eluted using an acetonitrile gradient as follows: 0–2 min, 5% acetonitrile; 2–13 min, linear ramp to 90% acetonitrile; 13–18 min, 90% acetonitrile. Product compounds were analyzed by mass spectrometry on a Q-Exactive MS system (Thermo Scientific, Rockford, IL, USA) using a heated electrospray source operated in positive ion mode. Mass spectra were collected over a range of 100 to 1500 m/z at a nominal resolution of 70,000.
3. Results
3.1 Synthesis of DPI analogs and functional selection
To improve the solubility, potency, and specificity of the iodonium-class flavoprotein inhibitor DPI, a library of thirty-six analogous compounds was developed by varying the identity of the substituent groups on the phenylene moieties of DPI, as well as that of the associated counter ion. Figs. 1A, B illustrate the range of molecules produced, and the general reaction sequences for the synthesis of these analogs. Following a preliminary functional screen (Fig. 1C), four analogs were retained for further biochemical characterization and evaluation against the parent compound, DPI. These included: dibenziodolium-3,7-dibromo bromide (NSC 740104-T, 104), dibenz[b,d]iodolium-3-(methoxycarbonyl)-, salt with 4-methyl-benzenesulfonic acid (1:1) (NSC 751140-P, 140), dibenziodolium-3,7-dinitro bromide (NSC 737392-V, 392), and dibenz[b,d]iodolium-3-nitro chloride (NSC 734428-Y, 428). Overall yields ranged from 37% (compound 140) to 96% (compound 392). A few analogs demonstrated better potency than DPI against HT-29 cell proliferation at 48 h, but these compounds either were poorly soluble (NSC 737544, 544), or did not outperform the parent molecule in inhibiting whole-cell ROS production (NSC 743401, 401; NSC 742461, 461) or HT-29 colony formation (NSC 742837, 837), and were therefore not retained for further analysis (data not shown).
3.2 Iodonium analogs inhibit the proliferation of HT-29 colon cancer cells
In a previous study, we demonstrated that DPI inhibits NOX1-dependent ROS production in HT-29 human colon cancer cells, resulting in growth inhibition [23]. To evaluate the potency of the new iodonium analogs compared to the parent compound, we measured their effects on the growth of NOX1-expressing HT-29 human colon cancer cells using the MTT assay. This colorimetric assay allowed for a quantitative assessment of the number of viable, metabolically active cells remaining following 72-h incubation with iodonium analogs at concentrations ranging from 0 to 1000 nM. As shown in Fig. 2A, compounds 104, 140, 392, and 428 inhibited the proliferation of HT-29 cells with IC50 values ranging from 150–250 nM, in contrast to the DPI IC50 of ~ 450 nM. Subsequently, we investigated the time-dependent antiproliferative activity of the DPI analogs in HT-29 cells at a low concentration (100 nM) and a high concentration (500 nM). At the lower concentration, only compounds 104 and 428 achieved 50% growth inhibition of HT-29 cells within the 72-h duration of the experiment (Fig. 2B). At the high concentration, the novel iodonium inhibitors all reached their IC50 by 16 h, whereas DPI reached its IC50 after 24 h of exposure (Fig. 2C). Hence, the addition of bromine (compound 104), methoxycarbonyl- (140), or nitro- (392 and 428) functional groups on DPI enhanced antiproliferative activity in HT-29 cells.
Fig. 2. DPI and iodonium analogs inhibit HT-29 cell proliferation and colony formation in a concentration- and time-dependent manner.

(A) HT-29 cells were treated for 72 h with concentrations of the iodonium analogs ranging from 0 to 1000 nM, or (B) with 100 nM iodonium analogs for the indicated times, or (C) with 500 nM iodonium analogs for different time intervals ranging from 8 to 72 h. Cell viability was assessed with the MTT assay. The inhibition of the colony-forming abilities of HT-29 cells was also assessed after iodonium analog treatment. (D, E) HT-29 cells were treated with DPI (D), or 392 (E) at 0, 8, 40, 200, 500, and 1000 nM for 6 h. (F, G) HT-29 cells treated with compound 104 (F), or compound 428 (G) for 2 h, 6 h, or 10 days. All data represent the mean ± SD of at least three experiments. (H) HT-29 cells were treated for 96 h with 0–2000 U/ml SOD (black bars), 0–2000 U/ml catalase (dark grey bars), or the combination of SOD and catalase at 0–2000 U/ml (light grey bars). SOD, catalase, or the combination had significantly decreased proliferation compared to control-treated cells (for catalase alone or in combination with SOD at concentrations ≥ 500 U/ml: ***P< 0.001, df=4; for SOD alone: **P< 0.01, df=4 at 500 U/ml SOD; **P< 0.01, df=3 at 1000 U/ml SOD; ***P< 0.001, df=4 at SOD concentrations ≥1500 U/ml SOD).
Furthermore, an evaluation of the colony-forming ability of HT-29 cells following treatment with iodonium analogs revealed that, in contrast to DPI and compound 104, which required high nanomolar concentrations for 6 h (Figs. 2D, F and Table 1) or longer exposure to appreciably reduce clonal proliferation, the nitro-substituted compounds 392 and 428 noticeably inhibit the clonal formation of HT-29 cells at low nanomolar concentrations over short exposure times (Figs. 2E, G and Table 1). Additionally, 96 h treatment with ROS detoxifying enzymes superoxide dismutase (SOD) and/or catalase similarly reduced the proliferation of HT-29 cells compared to control-treated cells (Fig. 2H; for catalase alone or in combination with SOD at concentrations ≥ 500 U/ml: ***P< 0.001, df=4; for SOD alone: **P< 0.01, df=4 at 500 U/ml SOD; **P< 0.01, df=3 at 1000 U/ml SOD; ***P< 0.001, df=4 at SOD concentrations ≥1500 U/ml SOD), supporting the hypothesis that ROS production may contribute to the growth of HT-29 cells.
Table 1.
Effect of DPI analogs on colony formation in HT-29 cells. Compound concentration for 2 h, 6 h, or 10-day treatment that reduced the number of HT-29 colonies formed by 50% compared to pretreated values. The data represent the mean ± standard deviation (SD) of 3 independent experiments.
| Iodonium analog | 2-h treatment IC50 (nM) | 6-h treatment IC50 (nM) | 10-day treatment IC50 (nM) |
|---|---|---|---|
| DPI | 9,257 ± 3,920 | 799 ± 180 | 39 ± 23 |
| 104 | 1,218 ± 1,124 | 536 ± 635 | 9 ± 2 |
| 140 | 5,716 ± 1,249 | 1,268 ± 290 | 40 ± 32 |
| 392 | 180 ± 157 | 116 ± 108 | 27 ± 28 |
| 428 | 77 ± 12 | 29 ± 23 | 11 ± 6 |
| 521 | 5,716 ± 1,249 | 2,046 ± 1,125 | 488 ± 101 |
3.3 Iodonium-class analogs modulate the production of intracellular ROS in HT-29 cells
To probe whether the iodonium analogs might affect the proliferation of HT-29 cells by inhibiting ROS production, HT-29 cells were treated with analog concentrations ranging from 10 to 1000 nM for 24 h. Whole cell intracellular ROS production was measured with the cell-permeant indicator 5- and 6-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2-DCF-DA) and quantified by analytical cytometry, as described previously [36]. Pre-treatment of HT-29 cells with 50 nM DPI, 104, 392, or 428 produced a left-shift (decrease) in DCF fluorescence compared to vehicle-treated control cells (Fig. 3A). However, in contrast to DPI, and to a lesser extent compound 428, which both produced increases in intracellular ROS following 500 nM drug exposure for 24 h, compounds 104, 140, and 392 did not demonstrate this effect. Using the MitoSox reagent which is specific for mitochondrial ROS, we found that at concentrations ≤ 200 nM DPI had no effect on mitochondrial ROS production (Fig. 3B; brown and blue lines). A concentration of 1 μM, DPI did produce a right shift in MitoSox fluorescence (green line). Enhanced intracellular ROS levels (as measured by DCF fluorescence) were abolished by the addition of mitochondrial electron transport chain inhibitors KCN and rotenone (Fig. 3C; upper panel). Similar results were found for compound 428 (Fig. 3C; lower panel). These results suggest that at nanomolar concentrations the iodonium analogs inhibit nonmitochondrial, intracellular ROS.
Fig. 3. Inhibition of whole-cell ROS production by iodonium analogs in HT-29 cells.
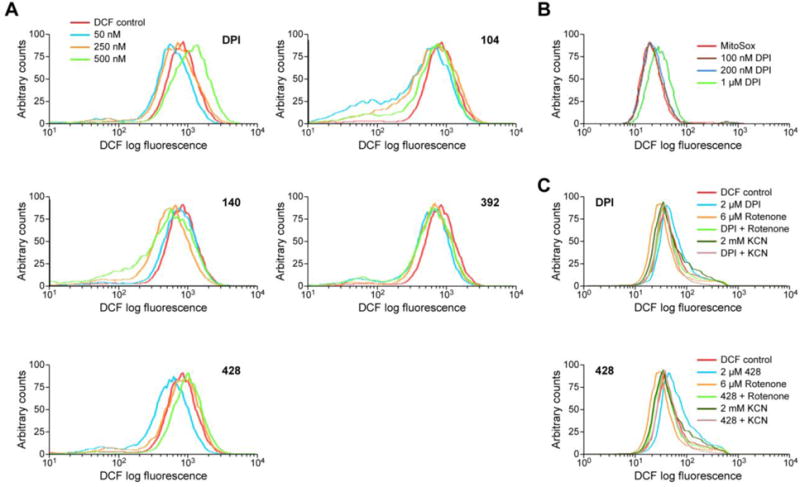
(A) HT-29 cells were treated with DPI, 104, 140, 392, or 428 for 24 h, and intracellular ROS production was detected by analytical cytometry using the redox-sensitive dye CM-H2-DCF-DA. (B) The effects of 24 h treatment with different concentrations of DPI on mitochondrial ROS levels in HT-29 cells were evaluated with the MitoSox™ reagent. (C) Upper panel: HT-29 cells received 2 h treatment with 2 μM DPI (blue line), alone or with 6 μM rotenone for 2 h (light green line), or with 2 mM KCN for 40 min (dark green line). The same experiments with 428 are displayed in the lower panel. Representative graphs over multiple experiments are shown.
To assess the direct effect of the iodonium analogs on cellular respiration and cell metabolism, we used the Seahorse metabolic analyzer to determine IC50 values for inhibition of oxygen consumption rates (OCR) in HT-29 cells. We also measured the corresponding concentration-dependent extracellular acidification rates (ECAR) following 24-h treatment with the inhibitors; representative results are displayed in Fig. 4A, B. For the 24-h treatment, the OCR of HT-29 cells began to decrease following exposure to 31.6 nM DPI, and the ECAR increased at 316 nM DPI, compared to control metabolic activity (Fig. 4A). In contrast, compound 392 produced no adverse effects on cellular metabolic activity when applied for 24 h at concentrations ≤ 316 nM (Fig. 4B). The analog concentrations required to inhibit cellular respiration by 50 % in HT-29 cells following 30-min, 60-min, or 24-h treatment are shown in Table 2. For a 30-min exposure, the analogs demonstrated IC50 values > 1 μM, except for compound 140. Following a 60-min treatment time, the IC50 for cellular respiration in HT-29 cells remained in the micromolar range, except for compounds 140 and 428. The 24 h IC50’s were considerably lower, except for compound 392, which had an IC50 of ~ 800 nM for inhibition of cellular respiration after 24-h exposure (Fig. 4B and Table 2). To directly compare the immediate effects of DPI on cellular respiration to that of a well-known inhibitor of mitochondrial electron transport, KCN, we measured OCR following the addition of 2 μM DPI to HT-29 cells. At this concentration, DPI rapidly decreased oxygen consumption by 80%, similar to KCN (Fig. 4C). Furthermore, addition of DPI to KCN-treated HT-29 cells after 40 min of KCN exposure (black arrow), yielded an additional decrease in OCR (as well as an increase in extracellular acidification; Fig. 4D). These results support our hypothesis that nanomolar concentrations of DPI produce limited adverse effects on mitochondrial metabolism.
Fig. 4. Effect of DPI analogs on cellular respiration and cell metabolism in HT-29 cells over 24 h.
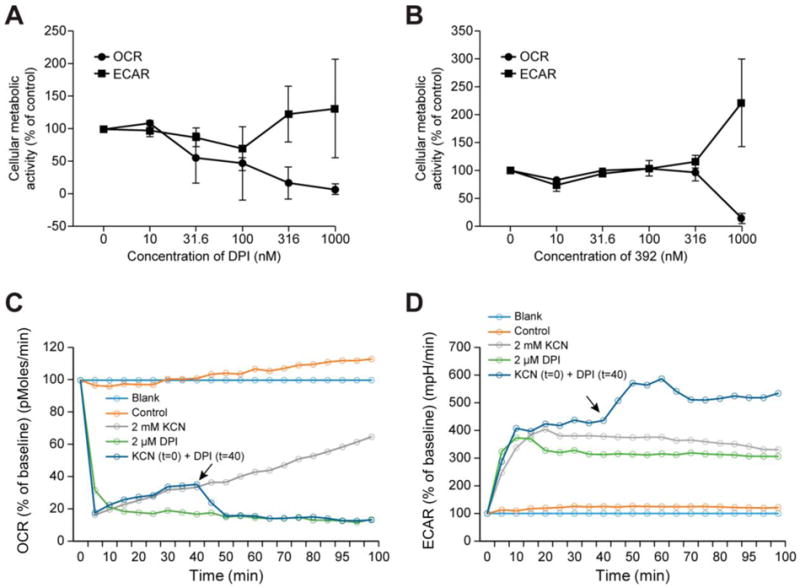
(A, B) Concentration-dependent changes in cellular respiration and glycolytic activity following 24-h exposure to DPI (A) or compound 392 (B) were evaluated by measuring oxygen consumption rates (OCR) and extracellular acidification rate (ECAR), respectively, with the Seahorse Extracellular Flux Analyzer. Baseline OCR and ECAR were 383.00 ± 73.65 pMoles/min and 1.19 ± 0.31 mpH/min (A); and 485.00 ± 120.81 pMoles/min and 0.53 ± 0.09 mpH/min (B), respectively. All data represent the mean ± SD of at least three experiments. (C, D) The short-term effects of KCN (2 mM) and a high concentration DPI (2 μM) on OCR (C) and ECAR (D) was also measured in HT-29 cells over 100 min with the Seahorse analyzer. When added to KCN-treated HT-29 cells after 40 min of KCN exposure, DPI (black arrow) recapitulated the effect of KCN.
Table 2.
Effect of iodonium analogs on cellular respiration in HT-29 cells. The data represent the mean ± standard deviation (SD) of 3 independent experiments.
| Iodonium analog | 30-min treatment IC50 (nM) | 60-min treatment IC50 (nM) | 24-h treatment IC50 (nM) |
|---|---|---|---|
| DPI | 8,283 ±1,627 | 4,571 ± 745 | 200 ± 245 |
| 104 | 5,964 ±2,816 | 1,472 ± 660 | 74 ± 54 |
| 140 | 678 ±201 | 475 ± 116 | 76 ± 21 |
| 392 | 5,090 ±1,439 | 2,182 ± 410 | 801 ± 194 |
| 428 | 1,538 ±1,450 | 211 ± 33 | 45 ± 23 |
| 521 | 1,667 ±81 | 1,000 ± 14 | No effect |
3.4 Screening for selectivity of iodonium analogs against NADPH oxidase isoforms
The specificity of the DPI analogs to inhibit different NOX isoforms was characterized by evaluating the respective concentrations required to decrease ROS production (O2•− or H2O2) by 50 % in cellular models that specifically express a single NOX family gene product. We measured PMA-induced O2•− production by luminescence assay in HEK293 cells expressing all necessary components to generate a functional NOX1 complex, in fully differentiated HL-60 cells and polymorphonuclear leukocytes (PMNs) expressing active NOX2 [37], and in UACC-257 melanoma cells that express active NOX5 [12]. Because the luminescence assay detects any ROS including, but not limited to superoxide, we confirmed the identity of the detected ROS as superoxide by treating a subset of cells with superoxide dismutase (SOD). Similarly, we measured H2O2 formation by Amplex Red assay from NOX4 or DUOX2/DUOXA2 proteins stably overexpressed in HEK293 cells. Representative plots are shown in Figs. 5A–D. The cells were pre-treated with iodonium analogs for 30 min before exposure to PMA. In HEK293 cells that exclusively expressed NOX1, compound 104 was the most potent inhibitor of superoxide anion production (IC50 [mean ± SD] 70 ± 57 nM), outperforming the parent molecule, DPI (IC50 = 233 ± 122 nM) (Table 3). Interestingly, 104 also displayed high potency against NOX5 (IC50 = 44 ± 21 nM). Compound 428, in which a single nitro group was added to DPI in the meta position, had equivalent or improved potency in all five cellular models compared to DPI. This compound achieved the lowest IC50 values in UACC-257 cells (NOX5) and in HL-60 cells (NOX2). DPI demonstrated low inhibitory activity against the DUOX2 isoform (IC50 = 6488 ± 1626 nM). The meta-substitution of two nitro groups on both benzene rings of DPI to yield compound 392 greatly improved potency in HEK293 DUOX2/DUOXA2 cells (IC50 = 225 ± 24 nM) compared to the parent compound.
Fig. 5. Concentration-dependent, selective inhibition of NOX1- and NOX2-related superoxide formation, and DUOX2- and NOX4-related H2O2 production by compound 392 in human cancer cell lines.
(A) PMA-induced superoxide anion production was measured by superoxide anion assay in HEK293 NOX1 cells treated with different concentrations of compound 392 or SOD (4 U) for 30 min (baseline O2•− production rate = 9.88 × 10−2 RLU/min/cell). (B) Hydrogen peroxide production in relative fluorescence units was measured by Amplex Red assay in HEK293 DUOX2/DUOXA2 cells (baseline H2O2 production rate = 1.0 × 10−4 RFU/min/cell). (C) PMA-induced superoxide anion production was measured by superoxide anion assay in NOX2-expressing HL-60 human promyelocytic leukemia cells (baseline O2•− production rate = 1.01 RLU/min/cell). (D) Hydrogen peroxide production in relative fluorescence units was measured by Amplex Red assay in HEK293 NOX4 cells (baseline H2O2 production rate = 2.0 × 10−4 RFU/min/cell). Cells were exposed to PMA immediately prior to assay measurements. RLU, relative light units; RFU, relative fluorescence units.
Table 3.
Effects of DPI analogs on superoxide and H2O2 production in human cells. PMA-induced superoxide production was measured by luminescence assay in HEK293 NOX1, PMNs (NOX2), HL-60 (NOX2), and UACC-257 (NOX5) cells. Hydrogen peroxide production was measured by Amplex Red assay of DPI or analog-treated NOX4 and DUOX2/DUOXA2 overexpression cell lines. The cells were incubated with DPI analogs for 30 min prior to assay measurement. The data represent the mean ± standard deviation (SD) of at least 3 independent experiments.
| Superoxide (O2•−) | Hydrogen peroxide (H2O2) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| HEK293 NOX1 | PMN (NOX2) | HL-60 (NOX2) | UACC-257 (NOX5) | HEK293 NOX4 | HEK293 DUOX2/DUOX A2 | |
| Baseline ROS producti on rate | 9.88 × 10−2 RLU/min/c ell | 0.55 RLU/min/c ell | 1.01 RLU/min/c ell | 1.34 × 10−2 RLU/min/c ell | 2.0 × 10−4 RFU/min/c ell | 1.0 × 10−4 RFU/min/cell |
|
| ||||||
| DPI IC50 (nM) | 233 ± 122 | 180 ± 36 | 105 ± 55 | 42 ± 27 | 880 ± 214 | 6,488 ± 1,626 |
| 104 IC50 (nM) | 70 ± 57 | 481 ± 44 | 127 ± 20 | 44 ± 21 | 161 ± 153 | 4,136 ± 1,724 |
| 140 IC50 (nM) | 249 ± 116 | 177 ± 60 | 71 ± 15 | 55 ± 20 | 127 ± 110 | 1,990 ± 1,690 |
| 392 IC50 (nM) | 357 ± 268 | 850 ± 197 | 310 ± 53 | 208 ± 129 | 139 ± 143 | 225 ± 24 |
| 428 IC50 (nM) | 136 ± 81 | 324 ± 144 | 57 ± 35 | 35 ± 21 | 260 ± 254 | 412 ± 236 |
| 521 IC50 (nM) | 1,019 ± 361 | 1049 ± 126 | 1,470 ± 308 | 229 ± 123 | 5,533 ± 2,681 | 773 ± 362 |
PMN, polymorphonuclear leukocytes.
3.5 Compound 521
To investigate the selectivity for DUOX2 that the addition of nitro substituent groups appeared to confer to DPI, we synthesized dibenziodolium, 1,9-dinitro-, salt with bromide (1:1) (NSC 780521, 521), an isomer of compound 392 (Fig. 6A). Changing the configuration of 392 by moving the nitro groups around the DPI aromatic rings to yield compound 521 markedly decreased the antiproliferative effect of the compound on HT-29 cells (Figs. 6B–G, Tables 1–2). However, compound 521 exhibited enhanced selectivity for DUOX2-related ROS production in HEK293 DUOX2/DUOXA2 cells and decreased potency for NOX isoforms other than NOX5 (Table 3).
3.6 Proof of concept of the formation of FAD-iodonium analog adducts
To determine whether DPI analogs target the NOX enzymes by disrupting the flow of electrons from NADPH to O2 via FAD and heme redox centers, as was believed to be the case for DPI [38], we sought to observe the formation of an adduct between the FAD cofactor and iodonium-class inhibitors. In our experiment, we mimicked the activity of the NOX enzyme, which catalyzes the reduction of FAD by NADPH, by pre-incubating free FAD with the reductive agent sodium dithionate before exposure to iodonium inhibitors. We found that DPI and all the analogs formed adducts with reduced FAD (Fig. 7A), as demonstrated by HPLC and mass spectrometry analysis (Figs. 7B–C and Table 4). All the analogs gave multiple peaks, suggesting the formation of isomeric adducts resulting from the alkylation of different sites on FAD. These results suggest that the previously hypothesized mode of action for DPI applies to the iodonium-class flavoenzyme inhibitors we have developed. In the context of an enzyme-free environment, the FAD adducts achieved have masses corresponding to the calculated value for each unique product.
Fig. 7. Reduced FAD forms adducts with DPI and the iodonium analogs.
(A) Illustration highlighting possible sites where adduct formation may occur. (B) Ion Mass tracing of m/z 1066.09–1066.19 from an aliquot of the reaction with reduced FAD and DPI, inset; mass spectrum of the M+H region. (C) Ion Mass tracing of m/z 1156.09–1156.13 from an aliquot of the reaction with reduced FAD and 521, inset; mass spectrum of the M+H region.
Table 4.
Adduct formation of FAD with DPI and analogs compounds.
| Iodonium analog | Chemical Formula (Product) | Major Peaks Observed | Calculated Mass | Observed Mass |
|---|---|---|---|---|
| IDP | C33H40N9O15P2 | 2 | 864.21 | 864.21 |
| DPI | C39H43IN9O15P2 | 4 | 1066.14 | 1066.14 |
| 104 | C39H41Br2IN9O15P2 | 4 | 1221.96 | 1223.96* |
| 140 | C41H45IN9O17P2 | 6 | 1124.15 | 1124.15 |
| 392 | C39H41IN11O19P2 | 4 | 1156.11 | 1156.11 |
| 428 | C39H42IN10O17P2 | 4 | 1111.13 | 1111.13 |
| 521 | C39H41IN11O19P2 | 6 | 1156.11 | 1156.11 |
+2 m/z units due to a Br isotope effect
4. Discussion
Because of the role of NOX-induced ROS in the etiology of inflammation-related pathologies, the development of NOX inhibitors has been actively pursued; however, the elaboration of specific inhibitors with clinical utility has not yet been fully achieved. Although widely recognized and employed as an inhibitor of NOXs, DPI has broad specificity for flavoenzymes, including xanthine oxidase, cytochrome P450 reductase, NADH:ubiquinone oxidoreductase, and nitric oxide synthase, thus confounding our understanding of its mechanism of growth inhibition [25, 38–42]. In this study, our goals were to identify inhibitors of NOXs derived from DPI that possessed improved solubility, potency, and selectivity for specific NOX isoforms when compared to the parent molecule.
We have described here the development and validation of 5 analogs of DPI, 4 of which exhibited improved inhibitory profiles, including greater antiproliferative effects at nanomolar concentrations, against HT-29 human colon cancer cells (Figs. 2A–C), a tumor cell line that depends on NOX activity, and not reactive nitrogen species, for its proliferative potential [10, 24]. In this cell line, the iodonium analogs produced an inhibitory effect on cell proliferation, similar to that observed following genetic inhibition (knockdown) of NOX1 activity [43]. Additionally, compounds 104, 392, and 428 consistently outperformed DPI with respect to the inhibition of HT-29 colony formation at both short- and long-term exposure times (Figs. 2D–G and Table 1). The hypothesis that diminished HT-29 proliferation was achieved, at least in part, by inhibiting the NOX1-associated production of reactive oxygen was supported by the observed decrease in intracellular (Fig. 3A) and extracellular (Fig. 5 and Table 3) ROS caused by our DPI analogs. Although high concentrations of DPI and 428 produced a right-shift in DCF fluorescence—suggestive of increased electron leak from the mitochondria [41, 42, 44]—we demonstrated that the concentrations of DPI and analogs 104, 140, 392 and 428 required for inhibition of ROS production and cell proliferation in HT-29 cells are distinctly lower than the concentrations that induce mitochondrial toxicity (Fig. 3B, Fig. 4A–D, and Table 2). Moreover, 104, 140, 392, and 428 were 2- to 5-fold more potent in HL-60 leukemia line compared to PMNs, which also express the NOX2 isoform, thus displaying some selectivity for malignant cells (Table 3). At these nanomolar concentrations, we postulate that inhibition of cell growth by our DPI analogs may, in part, be due to a similar mechanism as described in the case of genetic inhibition of NOX1 activity; that is, a decrease in NOX1-associated ROS production coupled to an arrest in the G1 phase of the cell cycle due to greatly decreased MAPK signaling, leading to downregulation of cyclin D1 [43]. Consistently, treatment with the antioxidant enzymes SOD and catalase also appreciably decreased HT-29 cell proliferation after a 96-h treatment (Fig. 2H). The latter has been shown previously to alter proliferative signaling through the MAPK and AKT pathways in fibroblasts [45, 46] and human leukemia cell lines [47].
In addition to improved inhibitory potency, the development of NOX inhibitors with well-characterized NOX-isoform selectivity is a critical feature to be considered for clinical use. Indeed, the expression profiles of NOX isoforms and their impact on cellular outcome vary by cancer histology and pre-malignant condition [8]. For instance, NOX1 and DUOX2 are both known to be significantly overexpressed in colon cancer and inflammatory bowel disease compared to matched normal tissues at the transcriptional and translational levels [48]. NOX4 and NOX5 are both overexpressed in human malignant melanomas [11, 12]. NOX2 specifically promotes the survival of human leukemia cells [49], and DUOX2 is implicated in pancreatic and thyroid cancer progression and chronic pancreatitis [9]. Thus, the availability of NOX inhibitors targeted against both single and multiple NOX isoforms in vivo will be required for the most favorable clinical application of this class of compounds in oncology. Herein, we have examined, using genetically engineered cell lines, the differential effects of our inhibitors in cells that exclusively produce a specific ROS (H2O2 or O2•− which was measured) due to the presence of a single isoform (Table 3).
The optimization of targeted inhibitors is best achieved through structure-based, rational design approaches [50]; however, the lack of experimentally-determined three-dimensional structures for NOX protein domains makes targeted inhibitor development difficult. In this investigation, we used the structure of DPI as a scaffold for developing NOX enzyme inhibitors, and we assessed the changes in selectivity produced by the addition of substituent groups on the phenylene moieties. The identity and positioning of substituent groups had quantifiable effects on inhibitor potency; interestingly, the addition of bromine (compound 104) or nitro (compounds 392 and 428) groups manifestly decreased the IC50 for HT-29 proliferation and colony formation when these new compounds are compared with the parent molecule DPI. On the other hand, 521, an isomer of 392, lacked potency against NOX1 activity (Fig. 6 and Tables 1–3) and HT-29 proliferation. Moreover, although all three nitro-substituted compounds potently inhibited ROS production in DUOX2-expressing HEK293 DUOX2/DUOXA2 cells compared to the less potent DPI, 104, and 140, compound 521 exhibited selectivity for inhibiting NOX5 (IC50 = 229 ± 123 nM in UACC-257 cells) and DUOX2 (IC50 = 773 ± 362 nM in HEK293 DUOX2/DUOXA2 cells), compared to other NOX isoforms. These results suggest that compound 521 could be used to probe NOX5 or DUOX2 functions at concentrations that would not have an adverse impact on phagocyte NOX. The observed differences in selectivity outlined herein may be the result of the accessibility of and/or features within the FAD/NADPH binding pocket of the NOX/DUOX enzymes [51].
Consistent with the observations described by O’Donnell et al. and with the hypothesis that iodonium-class flavoprotein inhibitors decrease ROS production by preventing the transfer of electrons from FAD [38], we have shown that DPI and the 5 lead analogs react with reduced FAD to form stable adducts under cell-free conditions, as confirmed by mass spectrometry (Fig. 7). Additionally, the interaction of the inhibitors with the enzymatic binding pocket is probably favored by non-covalent π-stacking with aromatic side chains (Phe, Trp, Tyr), as has been described for other protein-aromatic ligand interactions [52, 53]. Substituent groups may act as modifiers of the strength and stability of this interaction [53, 54]. Structural characterization of the covalent adducts by NMR spectroscopy would further inform compound optimization for better reactivity against FAD, and consequently, more potent NOX inhibition. For instance, the selective potency of the nitro-substituted analogs for DUOX2 may be attributed to a favorable interaction between the nitro substituents and the binding site in the DUOX2 enzymes. Furthermore, the spatial arrangement of the nitro groups dictates the level of affinity of the small molecule for the NOX isoform, as demonstrated by the differences between the inhibitory activities of the isomers 392 and 521. We are developing a new series of compounds inspired by the nitro-substituted DPI analogs to better define the molecular characteristics that confer selective potency.
In conclusion, this study uncovered structural characteristics of iodonium-class inhibitors that affect target selectivity, and these characteristics may serve as a starting point for future rational design of selective NOX inhibitors. The building of high-fidelity homology models for the NOX cytosolic domains—specifically the FAD/NADPH binding pocket of DUOX2—would allow for further exploration of the molecular interaction with the inhibitors. Previous studies have demonstrated a favorable therapeutic index for the parent molecule DPI against human colon cancer xenografts in vivo [24], suggesting that the DPI concentrations reached in vivo may be high enough to inhibit NOX1 but insufficient to cause mitochondrial toxicity. Further investigations of the pharmacodynamic effects and pharmacokinetics of these analogs in vivo aided by xenograft models, will allow for a better understanding and differentiation of these compounds, and will inform the potential applicability of this class of compounds as anticancer therapeutics in the clinic.
Acknowledgments
The authors would like to acknowledge Dr. William M. Nauseef at the University of Iowa for providing the HEK293 DUOX2/DUOXA2 cell line; and Mr. Francis Owusu at the Frederick National Laboratory for Cancer Research for providing the PMNs. This project has been funded in whole or in part with federal funds from the Center for Cancer Research, the Division of Cancer Treatment and Diagnosis, and NCI Contract No. HHSN261200800001E, National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- NOX
NADPH oxidase
- DUOX
dual oxidase
- ROS
reactive oxygen species
- DPI
diphenylene iodonium
- DTI
di-2-thienyliodonium chloride
- DMSO
dimethyl sulfoxide
- FMOC
fluorenylmethyloxycarbonyl
- IC50
inhibitory concentration 50%
- RLU
relative light units
Footnotes
Classification: Antibiotics and Chemotherapeutics
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflicts of interest to disclose.
Chemical compounds studied in this article: diphenylene iodonium, DPI (PubChem CID:3101); diphenyliodonium, IDP (PubChem CID: 12877); di-2-thienyliodonium chloride, DTI (PubChem CID: 162551); FAD (PubChem CID: 643975).
References
- 1.Lambeth JD. Nox enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 2.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 3.Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med. 2007;43:332–347. doi: 10.1016/j.freeradbiomed.2007.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamata T. Roles of Nox1 and other Nox isoforms in cancer development. Cancer Sci. 2009;100:1382–1388. doi: 10.1111/j.1349-7006.2009.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–85. doi: 10.1158/0008-5472.CAN-07-5259. [DOI] [PubMed] [Google Scholar]
- 7.Coso S, Harrison I, Harrison CB, Vinh A, Sobey CG, Drummond GR, Williams ED, Selemidis S. NADPH oxidases as regulators of tumor angiogenesis: current and emerging concepts. Antioxid Redox Signal. 2012;16:1229–1247. doi: 10.1089/ars.2011.4489. [DOI] [PubMed] [Google Scholar]
- 8.Juhasz A, Ge Y, Markel S, Chiu A, Matsumoto L, Van Balgooy J, Roy K, Doroshow JH. Expression of NADPH oxidase homologues and accessory genes in human cancer cell lines, tumours and adjacent normal tissues. Free Radic Res. 2009;43:523–532. doi: 10.1080/10715760902918683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu YZ, Antony S, Hewitt SM, Jiang GJ, Yang SX, Meitzler JL, Juhasz A, Lu JM, Liu H, Doroshow JH, Roy K. Functional activity and tumor-specific expression of dual oxidase 2 in pancreatic cancer cells and human malignancies characterized with a novel monoclonal antibody. Int J Oncol. 2013;42:1229–1238. doi: 10.3892/ijo.2013.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang R, Dashwood WM, Nian H, Lohr CV, Fischer KA, Tsuchiya N, Nakagama H, Ashktorab H, Dashwood RH. NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) Int J Cancer. 2011;128:2581–2590. doi: 10.1002/ijc.25610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaura M, Mitsushita J, Furuta S, Kiniwa Y, Ashida A, Goto Y, Shang WH, Kubodera M, Kato M, Takata M, Saida T, Kamata T. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G(2)-M cell cycle progression. Cancer Res. 2009;69:2647–2654. doi: 10.1158/0008-5472.CAN-08-3745. [DOI] [PubMed] [Google Scholar]
- 12.Antony S, Wu Y, Hewitt SM, Anver MR, Butcher D, Jiang G, Meitzler JL, Liu H, Juhasz A, Lu J, Roy KK, Doroshow JH. Characterization of NADPH oxidase 5 expression in human tumors and tumor cell lines with a novel mouse monoclonal antibody. Free Radic Biol Med. 2013;65:497–508. doi: 10.1016/j.freeradbiomed.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schildknecht S, Weber A, Gerding HR, Pape R, Robotta M, Drescher M, Marquardt A, Daiber A, Ferger B, Leist M. The NOX1/4 inhibitor GKT136901 as selective and direct scavenger of peroxynitrite. Curr Med Chem. 2014;21:365–376. doi: 10.2174/09298673113209990179. [DOI] [PubMed] [Google Scholar]
- 14.Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J Med Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- 15.Somanna NK, Valente AJ, Krenz M, Fay WP, Delafontaine P, Chandrasekar B. The Nox1/4 dual inhibitor GKT137831 or Nox4 knockdown inhibits angiotensin-II-induced adult mouse cardiac fibroblast proliferation and migration. AT1 physically associates with Nox4. J Cell Physiol. 2016;231:1130–1141. doi: 10.1002/jcp.25210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM, Rosen H. A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem Biol. 2010;5:981–993. doi: 10.1021/cb100219n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vander M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S. Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation. Cardiovasc Res. 2006;71:331–341. doi: 10.1016/j.cardiores.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 18.Altenhofer S, Kleikers PWM, Radermacher KA, Scheurer P, Hermans JJR, Schiffers P, Ho HD, Wingler K, Schmidt HHHW. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seredenina T, Chiriano G, Filippova A, Nayernia Z, Mahiout Z, Fioraso-Cartier L, Plastre O, Scapozza L, Krause KH, Jaquet V. A subset of N-substituted phenothiazines inhibits NADPH oxidases. Free Radic Biol Med. 2015;86:239–249. doi: 10.1016/j.freeradbiomed.2015.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Mousslim M, Pagano A, Andreotti N, Garrouste F, Thuault S, Peyrot V, Parat F, Luis J, Culcasi M, Thetiot-Laurent S, Pietri S, Sabatier JM, Kovacic H. Peptide screen identifies a new NADPH oxidase inhibitor: impact on cell migration and invasion. Eur J Pharmacol. 2017;794:162–172. doi: 10.1016/j.ejphar.2016.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Altenhofer S, Radermacher KA, Kleikers PWM, Wingler K, Schmidt HHHW. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Sign. 2015;23:406–427. doi: 10.1089/ars.2013.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stuehr DJ, Fasehun OA, Kwon NS, Gross SS, Gonzalez JA, Levi R, Nathan CF. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J. 1991;5:98–103. doi: 10.1096/fasebj.5.1.1703974. [DOI] [PubMed] [Google Scholar]
- 23.Doroshow JH, Juhasz A, Ge Y, Holbeck S, Lu J, Antony S, Wu Y, Jiang G, Roy K. Antiproliferative mechanisms of action of the flavin dehydrogenase inhibitors diphenylene iodonium and di-2-thienyliodonium based on molecular profiling of the NCI-60 human tumor cell panel. Biochem Pharmacol. 2012;83:1195–207. doi: 10.1016/j.bcp.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doroshow JH, Gaur S, Markel S, Lu J, van Balgooy J, Synold TW, Xi B, Wu X, Juhasz A. Effects of iodonium-class flavin dehydrogenase inhibitors on growth, reactive oxygen production, cell cycle progression, NADPH oxidase 1 levels, and gene expression in human colon cancer cells and xenografts. Free Radic Biol Med. 2013;57:162–75. doi: 10.1016/j.freeradbiomed.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab. 2008;9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- 26.Jaquet V, Scapozza L, Clark RA, Krause KH, Lambeth JD. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- 27.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–7. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 28.Meitzler JL, Hinde S, Banfi B, Nauseef WM, Ortiz de Montellano PR. Conserved cysteine residues provide a protein-protein interaction surface in dual oxidase (DUOX) proteins. J Biol Chem. 2013;288:7147–57. doi: 10.1074/jbc.M112.414797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S, Leppla SH. Cell surface tumor endothelium marker 8 cytoplasmic tail-independent anthrax toxin binding, proteolytic processing, oligomer formation, and internalization. J Biol Chem. 2003;278:5227–34. doi: 10.1074/jbc.M210321200. [DOI] [PubMed] [Google Scholar]
- 30.Szabo C, Zingarelli B, O’Connor M, Salzman AL. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci USA. 1996;93:1753–8. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Issaq SH, Teicher BA, Monks A. Bioenergetic properties of human sarcoma cells help define sensitivity to metabolic inhibitors. Cell Cycle. 2014;13:1152–61. doi: 10.4161/cc.28010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Modis K, Gero D, Erdelyi K, Szoleczky P, DeWitt D, Szabo C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem Pharmacol. 2012;83:633–43. doi: 10.1016/j.bcp.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–36. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 34.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, Darley-Usmar VM. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51:1621–35. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: the impact of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2010;48:905–14. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y, Antony S, Juhasz A, Lu J, Ge Y, Jiang G, Roy K, Doroshow JH. Up-regulation and sustained activation of Stat1 are essential for interferon-gamma (IFN-gamma)-induced dual oxidase 2 (Duox2) and dual oxidase A2 (DuoxA2) expression in human pancreatic cancer cell lines. J Biol Chem. 2011;286:12245–56. doi: 10.1074/jbc.M110.191031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Teufelhofer O, Weiss RM, Parzefall W, Schulte-Hermann R, Micksche M, Berger W, Elbling L. Promyelocytic HL60 cells express NADPH oxidase and are excellent targets in a rapid spectrophotometric microplate assay for extracellular superoxide. Toxicol Sci. 2003;76:376–83. doi: 10.1093/toxsci/kfg234. [DOI] [PubMed] [Google Scholar]
- 38.O’Donnell BV, Tew DG, Jones OT, England PJ. Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J. 1993;290(Pt 1):41–9. doi: 10.1042/bj2900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cross AR, Jones OT. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochem J. 1986;237:111–6. doi: 10.1042/bj2370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O’Donnell VB, Smith GC, Jones OT. Involvement of phenyl radicals in iodonium inhibition of flavoenzymes. Mol Pharmacol. 1994;46:778–85. [PubMed] [Google Scholar]
- 41.Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–9. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 42.Riganti C, Gazzano E, Polimeni M, Costamagna C, Bosia A, Ghigo D. Diphenyleneiodonium inhibits the cell redox metabolism and induces oxidative stress. J Biol Chem. 2004;279:47726–31. doi: 10.1074/jbc.M406314200. [DOI] [PubMed] [Google Scholar]
- 43.Juhasz A, Markel S, Gaur S, Liu H, Lu J, Jiang G, Wu X, Antony S, Wu Y, Melillo G, Meitzler JL, Haines DC, Butcher D, Roy K, Doroshow JH. NADPH oxidase 1 supports proliferation of colon cancer cells by modulating reactive oxygen species-dependent signal transduction. J Biol Chem. 2017;292:7866–7887. doi: 10.1074/jbc.M116.768283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hutchinson DS, Csikasz RI, Yamamoto DL, Shabalina IG, Wikstrom P, Wilcke M, Bengtsson T. Diphenylene iodonium stimulates glucose uptake in skeletal muscle cells through mitochondrial complex I inhibition and activation of AMP-activated protein kinase. Cell Signal. 2007;19:1610–1620. doi: 10.1016/j.cellsig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 45.Burdon RH, Alliangana D, Gill V. Hydrogen peroxide and the proliferation of BHK-21 cells. Free Radic Res. 1995;23:471–86. doi: 10.3109/10715769509065268. [DOI] [PubMed] [Google Scholar]
- 46.Preston TJ, Muller WJ, Singh G. Scavenging of extracellular H2O2 by catalase inhibits the proliferation of HER-2/Neu-transformed rat-1 fibroblasts through the induction of a stress response. J Biol Chem. 2001;276:9558–64. doi: 10.1074/jbc.M004617200. [DOI] [PubMed] [Google Scholar]
- 47.Gao N, Rahmani M, Dent P, Grant S. 2-Methoxyestradiol-induced apoptosis in human leukemia cells proceeds through a reactive oxygen species and Akt-dependent process. Oncogene. 2005;24:3797–809. doi: 10.1038/sj.onc.1208530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rokutan K, Kawahara T, Kuwano Y, Tominaga K, Sekiyama A, Teshima-Kondo S. NADPH oxidases in the gastrointestinal tract: a potential role of Nox1 in innate immune response and carcinogenesis. Antioxid Redox Signal. 2006;8:1573–82. doi: 10.1089/ars.2006.8.1573. [DOI] [PubMed] [Google Scholar]
- 49.Maraldi T, Prata C, Vieceli Dalla Sega F, Caliceti C, Zambonin L, Fiorentini D, Hakim G. NAD(P)H oxidase isoform Nox2 plays a prosurvival role in human leukaemia cells. Free Radic Res. 2009;43:1111–21. doi: 10.1080/10715760903186132. [DOI] [PubMed] [Google Scholar]
- 50.Huggins DJ, Sherman W, Tidor B. Rational approaches to improving selectivity in drug design. J Med Chem. 2012;55:1424–44. doi: 10.1021/jm2010332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meitzler JL, Antony S, Wu Y, Juhasz A, Liu H, Jiang G, Lu J, Roy K, Doroshow JH. NADPH oxidases: a perspective on reactive oxygen species production in tumor biology, Antioxid. Redox Signal. 2014;20:2873–89. doi: 10.1089/ars.2013.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stornaiuolo M, De Kloe GE, Rucktooa P, Fish A, van Elk R, Edink ES, Bertrand D, Smit AB, de Esch IJ, Sixma TK. Assembly of a pi-pi stack of ligands in the binding site of an acetylcholine-binding protein. Nat Commun. 2013;4:1875. doi: 10.1038/ncomms2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mukherjee P, Cinelli MA, Kang S, Silverman RB. Development of nitric oxide synthase inhibitors for neurodegeneration and neuropathic pain. Chem Soc Rev. 2014;43:6814–38. doi: 10.1039/c3cs60467e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hwang J, Li P, Carroll WR, Smith MD, Pellechia PJ, Shimizu KD. Additivity of substituent effects in aromatic stacking interactions. J Am Chem Soc. 2014;136:14060–7. doi: 10.1021/ja504378p. [DOI] [PubMed] [Google Scholar]
- 55.Wasylewsky A, Brown RK, Sandin RB. Preparation of 4-Nitrodiphenyleneiodonium Chloride. J Am Chem Soc. 1950;72:1038–1039. [Google Scholar]
- 56.Beringer FM, Kravetz L, Topliss GB. Iodonium Salts Containing Heterocyclic Iodine. J Org Chem. 1965;30:1141–1148. [Google Scholar]
- 57.Hou Z, Huang W. Synthesis of 3,6-Dihalodibenzocycloiodonium Salts, Lanzhou Daxue Xuebao. Ziran Kexueban. 1984;4:67–70. [Google Scholar]
- 58.Ding Z, Hou Z, Huang W. Improved Synthesis of 3,6-Diaminobiphenyl Fused Iodine-Containing Heterocycles, Lanzhou Daxue Xuebao. Ziran Kexueban. 1984;20:169. [Google Scholar]
- 59.Huang WK, Chang CC. Heterocyclic compounds containing iodine. IV. Synthesis of 3,6-dialkylaminodiphenyl heterocyclic compounds. K’O Hsueh T’ung Pao. 1963;11:47. [Google Scholar]
- 60.Hwang WK. Studies on Heterocyclic Compounds Containing Iodine. II. The Synthesis of 3,6-Di[Dimethylamino]dibenzopyriodonium Salts (III) and Their Properties. Huaxue Xuebao. 1957;23:438. [Google Scholar]
- 61.Blatchly JM, Mcomie JFW, Watts ML. Biphenylenes .7. Synthesis of 2,3-Dimethoxybiphenylene. J Chem Soc. 1962:5085–5090. [Google Scholar]
- 62.Moriarty RM, Penmasta R, Prakash I. Novel Pentafluorophenyl Hypervalent Iodine Reagents. Tetrahedron Lett. 1987;28:877–880. [Google Scholar]
- 63.Togo H, Nabana T, Yamaguchi K. Preparation and reactivities of novel (diacetoxyiodo)arenes bearing heteroaromatics. J Org Chem. 2000;65:8391–8394. doi: 10.1021/jo001186n. [DOI] [PubMed] [Google Scholar]
- 64.Nabana T, Togo H. Reactivities of novel [hydroxy(tosyloxy)iodo]arenes and [hydroxy(phosphoryloxy)iodo]arenes for alpha-tosyloxylation and alpha-phosphoryloxylation of ketones. J Org Chem. 2002;67:4362–4365. doi: 10.1021/jo0200670. [DOI] [PubMed] [Google Scholar]
- 65.Mckillop A, Kemp D. Further Functional-Group Oxidations Using Sodium Perborate. Tetrahedron. 1989;45:3299–3306. [Google Scholar]
- 66.Johnson AL. Synthesis of 3 Isomeric Ortho-Substituted Phenylthienyl Benzoic-Acids. J Org Chem. 1976;41:1320–1324. [Google Scholar]
- 67.Kellogg RM, Schaap AP, Harper ET, Wynberg H. Acid-Catalyzed Brominations Deuterations Rearrangements and Debrominations of Thiophenes under Mild Conditions. J Org Chem. 1968;33:2902–2909. [Google Scholar]
- 68.Kehrmann F, Baumgartner E. The acetylation of derivatives of diphenylamine with acetic acid anhydride and zinc chloride. Helv Chim Acta. 1926;9:673–675. [Google Scholar]
- 69.Smith PAS, Brown BB, Putney RK, Reinisch RF. The Synthesis of Heterocyclic Compounds from Aryl Azides .3. Some 6-Membered Rings and Some Azidobiaryls. J Am Chem Soc. 1953;75:6335–6337. [Google Scholar]
- 70.Zhang GT, Zhen, Li Yulin. Synthesis of the 8-oxadiquino[5,6-b;6′,5′-e]pyriodonium compound. Yingyong Huaxue. 1986;1:48. [Google Scholar]
- 71.Enguehard C, Renou JL, Collot V, Hervet M, Rault S, Gueiffier A. Reactivity of 3-iodoimidazo[1,2-a]pyridines using a Suzuki-type cross-coupling reaction. J Org Chem. 2000;65:6572–6575. doi: 10.1021/jo000698z. [DOI] [PubMed] [Google Scholar]
- 72.Cragoe EJJ, Woltersdorf Otto W., Jr (1-Oxo-2-aryl or thienyl-2-substituted-5-indanyloxy (or thio) alkanoic acids, and derivatives thereof. US Patent (Ed) USA. 1978 [Google Scholar]
- 73.Beringer FM, Bachofner HE, Falk RA, Leff M. Diaryliodonium Salts .7. 2,2′-Dithienyliodonium and Phenyl-2-Thienyliodonium Salts. J Am Chem Soc. 1958;80:4279–4281. [Google Scholar]
- 74.Stang PJ, Tykwinski R, Zhdankin VV. Preparation of Bis(Heteroaryl)Iodonium Salts Via an Iodonium Transfer-Reaction between Di(Cyano)Iodonium Triflate and Organostannanes. J Heterocycl Chem. 1992;29:815–818. [Google Scholar]
- 75.DAuria M, Esposito V, Mauriello G. Photochemical reactivity of aromatic and heteroaromatic nitroderivatives in the presence of arylalkenes. Tetrahedron. 1996;52:14253–14272. [Google Scholar]
- 76.Kazmierczak P, Skulski L. A Short-Cut Synthesis of Diaryliodonium Bromides Followed by Oxidative Anion Metatheses. Synthesis (Stuttg) 1995:1027–1032. [Google Scholar]