

Abstract
Post-translational modifications of histones by protein methyltransferases (PMTs) and histone demethylases (KDMs) play an important role in the regulation of gene expression and transcription and are implicated in cancer and many other diseases. Many of these enzymes also target various nonhistone proteins impacting numerous crucial biological pathways. Given their key biological functions and implications in human diseases, there has been a growing interest in assessing these enzymes as potential therapeutic targets. Consequently, discovering and developing inhibitors of these enzymes has become a very active and fast-growing research area over the past decade. In this review, we cover the discovery, characterization, and biological application of inhibitors of PMTs and KDMs with emphasis on key advancements in the field. We also discuss challenges, opportunities, and future directions in this emerging, exciting research field.
1. Introduction
Gene expression and transcription are critical for a variety of cellular processes and are controlled not only by DNA sequence and transcription factors but also by epigenetic regulation.1 This epigenetic regulation depends on the state of chromatin, which can be modified in a variety of ways, including DNA methylation, nucleosome remodeling histone variants, and post-translational modifications (PTMs) of histones.2 The proteins that are directly involved in PTMs of histones are divided into three categories: the enzymes that create these modifications (the “writers”), the proteins that recognize the modifications (the “readers”), and the enzymes that remove the modifications (the “erasers”). PTMs of histones include, but are not limited to methylation, acetylation, phosphorylation, sumoylation, ubiquitination, and glycosylation.3 Due to the crucial role of epigenetic regulation in important cellular processes, such as cell differentiation, proliferation, development, and maintaining the cell identity, epigenetic modifying enzymes have been increasingly recognized as potential therapeutic targets. Thus, there have been growing interests in the biomedical community to discover and develop selective small-molecule inhibitors of these enzymes. Many studies have already shown that these inhibitors are valuable chemical tools for investigating biological functions and disease association of the target enzymes and for assessing the potential of these enzymes as therapeutic targets.
Histone methylation is one of the most heavily investigated histone PTMs. It was first recognized in 20004 and was largely considered to be a permanent modification until the first histone demethylase was discovered in 2004.5 It is now appreciated that histone and nonhistone protein methylation and demethylation is a dynamic process that plays a key role in the regulation of gene expression and transcription and, in turn, is implicated in various cancers and numerous other diseases. Therefore, the discovery of selective small-molecule inhibitors of the enzymes that are responsible for the methylation and demethylation has become a very active and fast growing research area.6−21 The known methylation and demethylation sites for histone H3 and H4 tails and related enzymes are summarized in Figure 1. In this review, we focus on the enzymes that are responsible for the methylation and demethylation of histone and nonhistone proteins, namely, (1) protein methyltransferases (PMTs, also known as methyl writers) and (2) histone demethylases (KDMs, also known as methyl erasers). We comprehensively describe important past discoveries as well as current progress toward the discovery of small-molecule and peptide-based inhibitors of these methyl writers and erasers with the emphasis on small-molecule inhibitors. We also discuss future directions for developing inhibitors of these enzymes. It is our intention to thoroughly cover the inhibitors reported in the primary literature. However, it is beyond the scope of this review to include the inhibitors reported in the patent literature.
Figure 1.

Known methylation and demethylation sites for histone H3 and H4 tails and corresponding protein methyltransferases and histone demethylases.
2. Protein Methyltransferases
Histone methylation catalyzed by PMTs is one of the most important and highly studied PTMs due to its involvement in diverse biological processes, including heterochromatin formation and maintenance, transcriptional regulation, DNA repair, X-chromosome inactivation, and RNA maturation.22 PMTs have also been shown to target many nonhistone proteins.23,24 PMTs catalyze the transfer of the methyl group from the cofactor S-5′-adenosyl-l-methionine (SAM) to either lysine or arginine residues of histone and nonhistone substrates (Figure 2A). They are divided into two categories based on the type of residues they modify: protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs). While lysine residues can be mono-, di-, and/or trimethylated by PKMTs, the arginine guanidinium group can only be mono and/or dimethylated by PRMTs (Figure 2B).22 Dimethylation of terminal guanidino nitrogens following monomethylation of arginine (MMA) can occur on the same nitrogen resulting in asymmetrically dimethylated arginine (aDMA), or it can occur on two different guanidino nitrogens to give symmetrically dimethylated arginine (sDMA) (Figure 2B). Methylation of lysine or arginine residues does not alter the charge of these residues but modifies the bulkiness and hydrophobicity of the protein, consequently affecting the recognition of the methylated protein by methyl-lysine/arginine readers via protein–protein interactions. Each lysine or arginine methylation mark establishes a specific signal that is recognized by reader proteins.6
Figure 2.

(A) Methylation of lysine (K) and arginine (R) residues of histone tails and nonhistone proteins by protein methyltransferases. (B) Methylation states of K and R residues.
PKMTs and PRMTs bind lysine or arginine residues of substrate proteins via the substrate binding groove and SAM via the cofactor binding site.25 These two binding sites are linked by a narrow hydrophobic channel that brings the substrate and cofactor in close proximity to allow the transfer of the methyl group from the cofactor SAM to a lysine or arginine residue via an SN2 transition state. This process results in the methylation of the target residue and the release of the resulting cofactor product, S-5′-adenosyl-l-homocysteine (SAH). The process can occur successively to achieve higher states of lysine or arginine methylation (Figure 2B).
2.1. Protein Lysine Methyltransferases
Apart from DOT1L, all of the known PKMTs are composed of a conserved SET domain, approximately 130 amino acids in length.26−28 This domain was originally identified in three Drosophila genes. These genes include Su(var.)3–9 (the suppressor of position-effect variegation 3–9), En(zeste) (an enhancer of the eye color mutant zeste), and Trithorax (the homeotic gene regulator).26 PKMTs are divided into two classes: SET domain-containing PKMTs and non-SET domain-containing PKMTs, the latter of which DOT1L is the sole member. The SET domain folds into several small β-sheets that surround a knotlike structure, bringing together the two highly conserved motifs of the SET domain and forming an active site next to the SAM binding pocket.29 In addition, functional SET domain folds are usually flanked by pre-SET and post-SET domains that are crucial for enzymatic activity. SET domain-containing PKMTs are categorized according to their sequence similarities around the SET domain and divided into five major families: SUV, SET1, SET2, EZ, and RIZ.27,30 More recently, however, an alternative categorization and nomenclature has been suggested.31 This new classification aims to assign more generic names to histone-modifying enzymes according to the type of their enzymatic activity and the type of their target residue(s), since these enzymes have also been shown to target nonhistone proteins. As such, they were divided into eight major groups: KMT1 (lysine methyltransferases 1) to KMT8. It is worth noting that the SET domain is found in a large number of eukaryotic proteins and in several bacterial proteins. Thus, is not limited to PKMTs.32
Lysine methylation catalyzed by PKMTs has been recognized as a major mechanism in regulating gene expression and transcription.25,33 Histone lysine methylation can lead to either transcription activation or repression, depending on the site at which methylation occurred as well as the state of the methylation (e.g., mono-, di-, or trimethylation). While H3K4 (histone H3 lysine 4), H3K36, and H3K79 methylation are generally associated with transcription activation, H3K9 di- and trimethylation (H3K9me2 and H3K9me3) and H3K27 trimethylation (H3K27me3) are typically associated with repression.25,34−36
2.1.1. General Description
In the following sections, we organize inhibitors of PKMTs according to their histone methylation site(s) (Figure 2). In the case of PRMTs, we discuss each enzyme with its known inhibitors separately. We describe the structure, function, and disease relevance of these enzymes and then focus on the selective, small-molecule inhibitors published in the primary literature. We provide an account of in vitro characterization, selectivity, mechanism of action (MOA), and in vivo studies of reported inhibitors. A phylogenetic tree of protein methyltransferases is shown in Figure 3. It should be noted that the potencies of the most reversible protein methyltransferase inhibitors were reported as IC50 values, which are largely dependent on the assay conditions used. Consequently, this makes it difficult to compare and rank the inhibitors based on their potencies.
Figure 3.

Phylogenetic tree of PMTs. The PMTs with known inhibitors are indicated in boxes.
2.1.2. Inhibitors of H3K9 Methyltransferases
SUV39H1 (suppressor of variegation 3–9 homologue 1), SUV39H2, G9a (also known as euchromatic histone-lysine N-methyltransferase 2 (EHMT2)), GLP (G9a-like protein, or EHMT1), SETDB1 (SET domain, bifurcated 1), SETDB2, PRDM2 (PR domain containing 2, with ZNF domain), PRDM3, and PRDM16 are PKMT proteins that are known to govern H3K9 methylation.37 Dimethylation of H3K9 (H3K9me2) and trimethylation of H3K9 (H3K9me3) are repressive marks that result in localized transcriptional silencing, mediated by heterochromatin protein 1 (HP1).38 While inhibitors of H3K9 methyltransferases are highly desired due to the fact that these proteins are increasingly being implicated in a broad spectrum of human diseases, including various cancers, cocaine addiction, and HIV-1 latency, selective inhibitors have only been reported for SUV39H1, G9a, and GLP.39−43 These inhibitors are reviewed extensively in the following section.
SUV39H1 was the first histone lysine methyltransferase to be identified. The SET domain of SUV39H1 contains β-sheets that are packed alongside the protein’s pre-SET and post-SET domains.44 The post-SET domain is known to contain three conserved cysteine residues that play a crucial role in the domain’s enzymatic activity. SUV39H1 and SUV39H2 have been shown to play several biologically significant roles.45 In addition, it is believed that SUV39H1/2 may serve a tumor suppressor function by way of maintaining the H3K9 trimethylation mark at pericentric heterochromatin.46,47
It was the discovery of chaetocin, a fungal mycotoxin, as the first SUV39 methyltransferase inhibitor in 2005 that launched the pursuit of selective PKMT inhibitors.48 Screening a library of approximately 3000 compounds using a biochemical assay resulted in the identification of chaetocin, an epidithiodiketopiperazine (ETP) alkaloid, as a potent SUV39 inhibitor with an IC50 of 0.6 μM.48 Follow-up studies over the next several years have elucidated, however, that chaetocin is not a selective inhibitor.49−51 As such, it will not be covered further in this review.
The mono- and dimethylation of H3K9 is primarily catalyzed by the PKMTs G9a and GLP.52,53 These proteins both possess SET domains that share an approximately 80% sequence identity and have been demonstrated to form a heterodimer.53 It has also been shown that both G9a and GLP can catalyze the dimethylation of several nonhistone proteins. Most notably, these nonhistone substrates include lysine 373 on the tumor suppressor p53.54 Both G9a and GLP are of considerable biological importance and have been implicated in several human pathologies. For example, G9a has been shown to be overexpressed in a broad spectrum of cancers.41,54,55 As expected, knocking down G9a diminished cell growth and proliferation in cell-based models of prostate cancer,41 lung cancer,55 and leukemia,56 in addition to diminishing leukemia stem cell frequency and delaying acute myeloid leukemia (AML) progression in mouse models.57 G9a has also been implicated in a number of nononcological diseases, including HIV-1 latency,42 mental retardation,58 cocaine addiction,43,59 and inflammatory colitis.60 Additionally, G9a has been found to play an important role in diverse cellular processes, including coactivation leading to p21-mediated apoptosis.61 G9a has also been shown to help guide stem cell function, maintenance, differentiation, and reprogramming in various circumstances.62−65 For example, G9a plays a critical role in early embryonic development by way of guiding stem cell differentiation and H3K9me2 patterning that helps determine lineage specification in hematopoietic stem and progenitor cells (HSPCs).65 On the other hand, loss of function mutations in GLP has recently been shown to cause the 9q34 subtelomeric deletion, resulting in Kleefstra syndrome, which is characterized by intellectual disability.66,67 GLP was also recently shown to regulate brown adipose cell fate and thermogenesis by way of formation of the PRDM16 (PR domain containing protein 16) complex.68
Selective inhibitors of G9a and GLP were first reported by Kubicek and co-workers in 2007.69 High-throughput screening (HTS) of a 125000-compound library led to the discovery of BIX-01294 (Figure 4), a 2,4-diamino-6,7-dimethoxyquinazoline, as the first selective G9a and GLP inhibitor. It potently inhibited G9a (IC50 = 1.7 μM) and, to a lesser extent, GLP (IC50 = 38 μM) but did not inhibit other methyltransferases (PRMT1, SETD7, SETDB1, SUV39H1, and an H320R hyperactive SUV39H1 mutant) at up to 45 uM. A later study, however, found that BIX-01294 was actually more potent for GLP (IC50 = 0.7 μM) than G9a (IC50= 1.9 μM).70 In mechanism of action (MOA) studies, BIX-01294 was competitive with the peptide substrate and noncompetitive with the cofactor SAM.69 This mechanism of inhibition was confirmed by the crystal structure of the GLP SET domain in complex with BIX-01294 and the cofactor product SAH, which clearly shows that the inhibitor binds to the substrate-binding groove of GLP (PDB ID: 3FPD).70
Figure 4.

Structures of G9a/GLP inhibitors.
BIX-01294 was active in multiple cell-based assays. For example, it reduced global levels of H3K9me2 and concurrently increased unmodified H3K9, but did not alter H3K9me1, H3K9me3, H3K27, H3K36, or H4K20 methylation marks in mouse embryonic stem cells (ESCs).69 A global reduction of H3K9me2 was also observed when mouse embryonic fibroblasts (MEFs) and HeLa cells were treated with BIX-01294. Furthermore, BIX-01294 reduced the H3K9me2 mark at promoters of G9a target genes such as MAGEA2, BMIL, and SERAC1 but had no effect on the H3K9me2 mark at promoters of G9a nonresponsive genes such as MAGEB4 and TUBULIN.69
The second G9a/GLP selective inhibitor was discovered in 2009 by Liu and co-workers, who conducted structure–activity relationship (SAR) studies on the 2,4-diaminoquinazoline core of BIX-01294.71 Specifically, UNC0224 (Figure 4) was designed based on the cocrystal structure of the GLP-BIX-01294 complex70 to have a 7-dimethylaminopropoxy group to occupy the lysine-binding channel of G9a/GLP. Numerous biochemical assays revealed that UNC0224 indeed possessed significantly increased potency and selectivity, including a Kd of 23 ± 8 nM and 1000-fold selectivity for G9a and GLP over SETD7 and SETD8. The occupation of the G9a lysine-binding channel by UNC0224’s 7-dimethylaminopropoxy group was confirmed by a crystal structure of the G9a-UNC0224 complex (PDB ID: 3K5K) (Figure 5). The cocrystal structure also revealed additional key inhibitor/protein interactions that guided future inhibitor development, leading to the discovery of UNC0321, which is the most potent G9a inhibitor (Morrison Ki= 63 pM) to date and retains a similar selectivity profile as UNC0224.71
Figure 5.
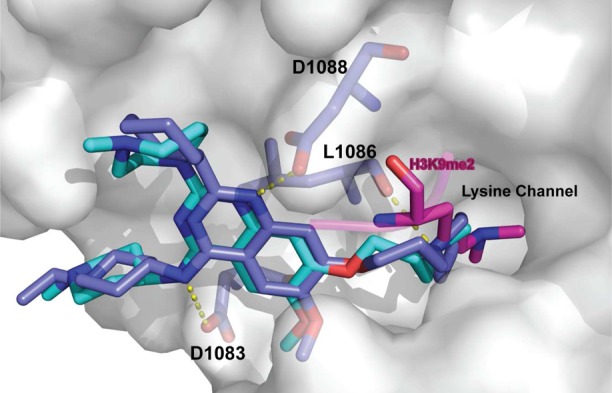
Co-crystal structures of UNC0224 (cyan) and UNC0638 (blue) in complex with G9a are superimposed (PDB ID: 3K5K and 3RJW, respectively). H3K9me2 peptide (magenta) overlaid for reference. Hydrogen bonds with G9a residues are represented as yellow dashed lines.
In 2010, based on the same crystal structure of the GLP-BIX-01294 complex, Chang and co-workers reported the discovery of E72 (Figure 4), a selective G9a/GLP inhibitor that also features a lysine mimic added onto the quinazoline scaffold.72 E72 was found to have a Kd of ca. 136 nM and an IC50 of 100 nM against GLP. In studies using NIH 3T3 cells, E72 reactivated the proapoptotic Fas gene after it was epigenetically silenced by K-Ras. Further, E72 showed less cytotoxicity than BIX-01294, likely due to its increased polarity and decreased cell membrane permeability.
The low cell membrane permeability of UNC0321 is also the likely reason that it displayed poor cellular activity despite its high potency in biochemical assays.73 This led Liu and co-workers to optimize physicochemical properties of this chemical series by increasing hydrophobicity while maintaining high potency for G9a/GLP. These efforts resulted in the discovery of UNC0638, a potent, selective, and cell-active inhibitor of G9a and GLP,74 with an IC50 of 19 nM for GLP and an IC50 less than 15 nM for G9a in biochemical assays.74 In MOA studies, UNC0638 was competitive with the peptide substrate (Ki= 3.0 ± 0.05 nM) but noncompetitive with the cofactor SAM. The MOA was confirmed with a crystal structure of G9a in complex with UNC0638 and SAH, which unambiguously indicates that UNC0638 binds the substrate binding groove of G9a (PDB ID: 3RJW) (Figure 5).
UNC0638 was thoroughly characterized in additional biochemical, biophysical, and cellular assays. It was more than 200-fold selective for G9a and GLP over 16 other methyltransferases and epigenetic targets.74 It was also at least 100-fold selective over more than 80 G protein-coupled receptors (GPCRs), kinases, transporters, and ion channels. UNC0638 reduced global H3K9me2 levels in MDA-MB-231 cells, a human breast carcinoma cell line, with significantly greater potency (IC50 = 81 ± 9 nM) than BIX-01294 (IC50 = 500 ± 43 nM).74 Interestingly, the abundances of G9a protein and mRNA transcripts in cells were unchanged following UNC0638 treatment, suggesting that reduction of H3K9me2 occurs purely from enzymatic inhibition. In addition, UNC0638 displayed considerably reduced cytotoxicity (EC50 = 11000 ± 710 nM) compared to BIX-01294 (EC50= 2700 ± 76 nM) in MDA-MB-231 cells. Thus, UNC0638 has a large separation of functional potency and cell toxicity (function/toxicity ratio = 138), whereas BIX-01294 has a relatively poor separation (function/toxicity = 5.6). UNC0638 also exhibited high cellular potency and low cell toxicity in six other cancer and normal cell lines. Furthermore, mass spectrometry (MS)-based proteomics was implemented to examine effects of UNC0638 treatment on H3K9me2 as well as 20 other common histone modifications. This study found that only H3K9me2 and H3K14ac levels were changed by UNC0638, suggesting (1) that UNC0638’s cellular actions are specific, and (2) that there may be potential crosstalk between H3K9me2 and H3K14ac marks. In addition, UNC0638 selectively reduced the H3K9me2 mark at promoters of G9a target genes, such as MAGEA1, TCB1D5, and MAGEA2 but did not change the H3K9me2 mark at promoters of G9a nonresponsive genes, such as MAGEB4.
UNC0638 also displayed significant phenotypic effects in cellular assays. For example, UNC0638 was shown to reduce MCF7 cell clonogenicity, but not that of MDA-MB-231 cells, suggesting that the phenotypic effect depends upon cell types and epigenetic states.74 UNC0638 was also found to induce differentiation of naive T cells into regulatory T cells and Th17 cells.60 In addition, UNC0638 induced leukemia stem cell differentiation thereby suppressing the proliferation of primary human AML cells,57 an effect that was similarly observed with G9a-conditional knockout AML cells. A key interaction between G9a and the leukemogenic transcription factor HoxA9, which regulates myeloid progenitor proliferation, was found to underlie the observed suppression of AML cell proliferation following UNC0638 treatment. This in turn, suggests potential clinical utility of pharmacological inhibition of G9a in the treatment of AML.
Despite its utility as a cell-based chemical probe, UNC0638 possessed poor pharmacokinetic (PK) properties in animals.74 To improve PK properties of UNC0638 and make it suitable for in vivo studies, Liu and co-workers conducted further optimization and discovered UNC0642 (Figure 4),75 which retained high in vitro potency for G9a and GLP (IC50 < 2.5 nM) and was >20000-fold selective for G9a/GLP over other methyltransferases (e.g., MLL1, SETD7, SETD8, SETDB1, PRMT3, PRMT5, SMYD2, SMYD3, SUV39H2, SUV420H1, SUV420H2, DOT1L, and DNMT1) and >300-fold selective over approximately 100 kinases, GPCRs, transporters, and ion channels. It also potently and selectively reduced the H3K9me2 mark, while maintaining low cell toxicity, in normal and tumor cell lines. Importantly, UNC0642 exhibited >3-fold higher exposure in plasma compared to UNC0638 in mouse PK studies, making it suitable for in vivo studies. Very recently, Kim and co-workers have shown that UNC0642 reactivated silent Prader-Willi Syndrome (PWS) candidate genes from the maternal allele in PWS cellular and mouse models, and the inhibitor significantly increased the growth and prolonged the survival of PWS mouse pups.76 In addition, a biotinylated chemical tool has been derived from UNC0638. This derivative had high in vitro potency for G9a and can precipitate G9a from whole-cell lysates and be used for exploring the localization of G9a on chromatin in chem-ChIP studies.77
In 2012, Yuan and co-workers synthesized and screened a focused library of 2-substituted benzimidazoles that structurally mimick SAM and discovered BRD9539 (Figure 4), a SAM-competitive inhibitor of G9a, and BRD4770 (Figure 4), the methyl ester of BRD9539, as a prodrug.78 While BRD9539 inhibited G9a with an IC50 of 6.3 μM, it also inhibited PRC2-EZH2 with a similar potency and NSD1 with a lower potency (IC50 = 40 μM). It was, however, selective over SUV39H1, SUV39H2, MLL1, SETD7, SETD8, PRMT1, PRMT3, PRMT5, DNMT1, and HDAC1–9. It is unclear as to whether or not BRD9539 also inhibits GLP, as this information was not reported. Regardless, BRD9539 appeared to be competitive with SAM, as increasing SAM concentrations result in a decline in G9a inhibition. BRD4770 significantly reduced H3K9me2 and H3K9me3 levels and concurrently increased H3K9me1 levels in cells. The cytotoxicity of BRD4770 was also tested by assessing its ability to induce cellular apoptosis, as monitored by caspase 3/7 activity. It had low cytotoxicity as it failed to induce caspase activity in PANC-1 cells. Of note, BRD4770-treated PANC-1 cells did appear to take on a senescence associated phenotype, namely flattened morphology and more intense β-galactosidase staining. Additionally, BRD4770 reduced the clonogenicity of PANC-1 cells.
Another G9a and GLP selective inhibitor was reported in 2014 by Sweis and co-workers.79 Their compound, A-366 (Figure 4), features a novel spiro(cyclobutane-1,3′-indol)-2′-amine core and potently inhibited G9a (IC50 = 3.3 nM) and GLP (IC50 = 38 nM). A-366 was competitive with the peptide substrate and noncompetitive with SAM. It was selective for G9a/GLP over 17 other methyltransferases. The crystal structure of A-366 in complex with G9a shows that A-366 binds G9a similarly as UNC0638 binds G9a (PDB ID: 4NVQ). Finally, A-366 reduced global levels of H3K9me2 but did not change H3K27me3 and H3K36me2 levels in PC3 cells, a prostate cancer cell line.
Several additional G9a/GLP inhibitors have been reported recently.80,81 These compounds have been shown to inhibit G9a/GLP in biochemical assays. However, extensive characterization of these inhibitors in additional assays was not reported.
While very closely related, GLP and G9a possess distinct physiological and pathophysiological functions. Therefore, GLP or G9a selective small-molecule inhibitors would be important tools to examine their individual biological functions. Among all the inhibitors discussed above, only BIX-01294 displayed limited selectivity for GLP (3–5 fold) over G9a. Very recently, Xiong and co-workers reported the discovery of potent and selective GLP inhibitor, MS012 (IC50 = 7 ± 2 nM), which is 140-fold selective for GLP over G9a (Figure 4).82 MS012 was also selective for GLP over a broad range of other PKMTs, PRMTs, DNMTs, and RNMTs. The direct binding of MS012 to GLP and G9a was shown by ITC. In addition, cocrystal structures of GLP and G9a in complex with the inhibitor were obtained (PDB ID: 5TTG and 5TTF) and showed that the inhibitor occupies the peptide-binding site, confirming the finding from MOA experiments that the inhibitor is competitive with the peptide substrate. Interestingly, X-ray structures revealed that MS012 binds to GLP and G9a in virtually identical binding modes, highlighting the challenges in structure-based design of selective inhibitors for these highly identical enzymes.
2.1.3. Inhibitors of H3K27 Methyltransferases
Methylation of H3K27 is catalyzed by a highly conserved, multisubunit protein complex, known as polycomb repressive complex 2 (PRC2).83−88 As its name suggests, PRC2 primarily functions to silence its target genes by trimethylating H3K27, resulting in their transcriptional repression. This process is important in a variety of physiological processes, including those involving differentiation and development.86 The core PRC2 complex consists of four subunits: (1) EZH1 (enhancer of zeste homologue 1, also known as KMT6B) or EZH2 (enhancer of zeste homologue 2, also known as KMT6A), (2) SUZ12 (suppressor of zeste 12), (3) EED (embryonic ectoderm development), and (4) RbAp46/48. The EZH1 or EZH2 subunit is the catalytic subunit of PRC2 and contains a SET domain at the C-terminal, which is necessary to exert the methyltransferase activity.84,88,89 While EZH1 and EZH2 share 96% sequence identity in their SET domains, they possess considerably different tissue distributions.90 For example, EZH1 exists in both differentiated and dividing cells, while EZH2 is found only in dividing cell populations.86,91 Yet another reported distinction between EZH1 and EZH2 is that the PRC2 complex containing EZH2 (PRC2-EZH2) possesses greater methyltransferase activity compared to the PRC2 complex with EZH1 (PRC2-EZH1).91 Nevertheless, both of these complexes have been demonstrated to carry out H3K27 methylation successively to produce the H3K27me3 repressive mark.86,91−93
Despite its status as the catalytic subunit of PRC2, EZH2, or EZH1 alone is not catalytically active. Instead, EZH2 or EZH1 requires at least two other subunits, EED and SUZ12, to have the methyltransferase activity.94−96 It has also been demonstrated that PRC2 can contain other protein subunits as well, including AEBP2, PCLs, and JARID2.97−100 Defects in EZH2 have been shown to result in a number of pathological outcomes. For example, point mutations at Y641 in the EZH2 C-terminal SET domain have been observed in 7% of follicular lymphomas and 22% of germinal center B-cell (GCB) and diffuse large B-cell lymphomas (DLBCLs).101 These Y641 point mutations are known to have a gain-of-function. They have a preference for H3K27me2 as the substrate, resulting in increased trimethylation of H3K27.102,103 Another EZH2 point mutation, A677G, has also recently been identified in lymphoma cell lines and primary tumor samples.104 Importantly, overexpression of EZH2 and hypertrimethylation of H3K27 have been associated with multiple human cancers,105,106 including breast,107,108 prostate,109 lymphoma,101,110 myeloma,111 and leukemia.112 While it has not yet been determined whether EZH1 overexpression is involved in B-cell malignancies, it has been shown that EZH1 and EZH2 can compensate for one another and are both required for maintaining cell proliferation and suppressing cell differentiation to sustain aggressive MLL-rearranged leukemias.113−115
A number of highly potent and selective inhibitors of PRC2 have been reported. The first selective PRC2 inhibitor targeting the EZH2 subunit was reported by Knutson and co-workers in 2012.116 A hit with an IC50 of 620 nM against PRC2 was identified through HTS of a 175000-compound library. Optimization of this hit resulted in EPZ005687 (Figure 6, IC50 = 54 ± 5 nM), which was competitive with the cofactor SAM (Ki = 24 ± 7 nM) and noncompetitive with the peptide substrate.116 It was postulated that EPZ005687 might bind the SAM binding pocket of EZH2 due to the above MOA finding and that EPZ005687 and SAH are mutually exclusive PRC2 inhibitors. Importantly, EPZ005687 was over 500-fold selective for the PRC2-EZH2 complex over 14 other methyltransferases. It was also approximately 50-fold selective for PRC2-EZH2 over PRC2-EZH1. While EPZ005687 displayed similar potencies for Y641 mutations compared to the wild-type EZH2, it was 5-fold more potent for the A677G mutation compared to the wild-type EZH2. EPZ005687 also had no appreciable affinity for a broad range of other protein targets, including 77 GPCRs and ion channels.
Figure 6.

Structures of EZH2/EZH1 inhibitors.
EPZ005687 has displayed promising activities in cellular assays. For example, it potently reduced H3K27me3 levels in OCI-LY19 cells, a wild-type EZH2 lymphoma cell line, and WSU-DLCL2 cells, a Y641F mutant lymphoma cell line.116 EPZ005687 also significantly blocked the growth of WSU-DLCL2 and Pfeiffer cells (an A677G mutant cell line) but not OCI-LY19 cells, suggesting that hypertrimethylation of H3K27 is important for the proliferation of EZH2-mutated lymphomas. Subsequent gene set enrichment analysis (GSEA) studies confirmed an expected derepression of known EZH2 target genes in WSU-DLCL2 cells following EZH2 inhibition.
In 2012, McCabe and co-workers reported GSK126, a SAM-competitive EZH2 selective inhibitor.117 Similar to the discovery of EPZ005687, a hit was identified by HTS and subsequent optimization resulted in the discovery of GSK126 (Figure 6),117 as well as other potent and selective EZH2 inhibitors, such as GSK34390 and GSK503.118 These EZH2 inhibitors share a similar core structure with EPZ005687; however, GSK126 bears an indole group as opposed to the indazole group of EPZ005687. GSK126 potently inhibited both wild-type and mutant forms of EZH2 (Ki = 0.5–3 nM) and was over 1000-fold selective for EZH2 over 20 other methyltransferases.117 Remarkably, it was also over 150-fold selective for EZH2 over EZH1, despite EZH2 and EZH1 sharing 96% sequence identity in their SET domains. It also showed almost no affinity for a large variety of kinases, GPCRs, ion channels, transporters, and other chromatin modifiers.
GSK126 was assessed for its effect on cell proliferation in a panel of B-cell lymphoma cell lines. Six of the seven DLBCL cell lines that were the most sensitive to GSK126 did in fact contain either Y641N, Y641F, or A677G mutations, while the least sensitive DLBCL cell lines had no mutations.117 In the most sensitive DLBCL lines, GSK126 treatment resulted in transcriptional activation and gene expression changes that closely matched those of EZH2 knockdown. Subsequent ChIP-Seq analysis of these sensitive DLBCL lines demonstrated that H3K27me3 levels were enriched prior to GSK126 treatment and diminished post-treatment, suggesting that EZH2 target genes are repressed by the H3K27me3 mark and that this repressive effect is relieved by GSK126 treatment.
GSK126 was also studied in Pfeiffer and KARPAS-422 tumor xenograft mouse models.117 A significant reduction in tumor volume and improvement in survival against the more aggressive KARPAS-422 xenograft was observed when mice received intraperitoneal (IP) administration of GSK126 at 50 mg/kg once daily, 150 mg/kg once daily, or 300 mg/kg twice per week over the course of 35 days. Importantly, a dose-dependent decrease in H3K27me3 and a corresponding increase in EZH2 target gene expression were observed in both xenograft models. Of note, GSK126 was also well-tolerated by the treated mice. This inhibitor entered a Phase I clinical trial in 2014 as GSK2816126 in patients with relapsed DLBCL, transformed follicular lymphoma, other Non-Hodgkin’s Lymphomas, solid tumors, and multiple myeloma.119 However, this inhibitor was not orally bioavailable and was given to patients as an intravenous infusion twice weekly. As of the time of this writing, there have been no study results reported.
Shortly after the publication of EPZ005687 and GSK126, Qi and co-workers reported the discovery of EI1 (Figure 6), another EZH2 selective inhibitor.120 EI1, which bears the pyridone and indole/indazole core common to EPZ005687 and GSK126, was SAM-competitive (Ki = 13 ± 3 nM) and potently inhibited both wild-type EZH2 (IC50 = 15 ± 2 nM) and the Y641F mutant (13 ± 3 nM). Furthermore, it was approximately 90-fold selective for EZH2 over EZH1 and 10000-fold selective for EZH2 over other 10 methyltransferases. EI1 in a concentration-dependent manner reduced the H3K27me3 and H3K27me2 marks but did not change other histone marks such as H3K27me1 and di- and trimethylation marks on H3K4, H3K9, H3K36, and H3K79 in DLBCL cells and a rhabdoid tumor cell line, G4001.120 In addition, EI1 activated EZH2 target genes including p16, whose expression increased 20-fold after 5 days of treatment. Phenotypically, EI1 inhibited the proliferation of EZH2 gain-of-function mutated DLBCL lines, including WSU-DLCL2, SU-DHL6, KARPAS-422, DB, and SU-DHL4. It was also demonstrated that EI1 blocked cell cycle progression and induced apoptosis in these EZH2 mutant DLBCL cells. On the other hand, EI1 only weakly inhibited, if at all, the growth of wild-type EZH2 cell lines, including OCI-LY19, GA10, and Toledo. Results of EI1 in tumor xenograft mouse models were not reported.
In 2013, Konze and co-workers reported UNC1999 (Figure 6), an orally bioavailable, potent, selective, and cell-active inhibitor of EZH2 and EZH1.121 This inhibitor was developed based on EPZ005687 and GSK126 but has more desirable physicochemical properties. UNC1999 was competitive with SAM (Ki = 4.6 ± 0.8 nM) and noncompetitive with the peptide substrate. Interestingly, it was only about 10-fold selective for EZH2 over EZH1. On the other hand, UNC1999 was >1000-fold selective for EZH2 and EZH1 over 15 other methyltransferases. With the exception of sigma2, it was also selective over a broad panel of 90 kinases, GPCRs, transporters, and ion channels. UNC1999 was characterized in numerous cell-based studies. In MCF10A cells, UNC1999 potently reduced H3K27me3 levels (IC50 = 124 ± 11 nM), while displaying low cell toxicity (EC50 = 19200 ± 1200 nM).121 In MCF7 cells, UNC1999 effectively removed the H3K27me3 mark and did not affect EZH2 protein levels. In DB cells, a DLBCL cell line harboring the EZH2 gain-of-function mutant Y641N, UNC1999 also effectively reduced the H3K27me3 mark, did not change EZH2 protein levels, and potently and concentration-dependently inhibited cell proliferation. In MLL-AF9-transformed murine leukemia progenitor cells, UNC1999, which is a dual EZH2 and EZH1 inhibitor, effectively blocked cell proliferation.115 On the other hand, GSK126, which is a potent EZH2 but weak EZH1 inhibitor, had little antiproliferative activity. These results support the earlier findings uncovered via genetic approaches that in addition to EZH2, EZH1 plays an important role in MLL-AF9 AML progression113 and suggest that the methyltransferase activity of PRC2 is critical for maintaining proliferation of these tumor cells. Furthermore, UNC1999 was orally bioavailable in mouse PK studies.121 It was the first reported orally bioavailable EZH2 inhibitor. Importantly, oral administration of UNC1999 at 50 mg/kg twice daily prolonged the survival of mice with MLL-AF9-induced leukemia,115 suggesting that pharmacological inhibition of both EZH2 and EZH1 may provide an emerging therapeutic approach for treating MLL-rearranged leukemia. The same research group also recently identified several moieties of UNC1999 that play an important role in selectivity between EZH2 and EZH1 via a comprehensive SAR study.122 They also developed a biotinylated derivative of UNC1999 for chemiprecipitation of EZH2 and EZH1 and a cell-penetrant dye conjugate based on UNC1999 for live cell imaging studies.121
In 2013, shortly after UNC1999 was published, another orally active EZH2 inhibitor, named EPZ-6438 (Figure 6), was reported by Knutson and co-workers.123 Notably, EPZ-6438 displayed an improved potency and pharmacokinetic profile compared to their earlier compound, EPZ005687. While this newer compound contains the same pyridone core as previous EZH2 inhibitors, it lacks the indole/indazole moiety. EPZ-6438 was competitive with SAM and noncompetitive with the peptide substrate. It potently inhibited wild-type EZH2 (Ki = 2.5 ± 0.5 nM) as well as EZH2 mutants, including Y641F, Y641C, Y641H, Y641N, Y641S, and A677G. In addition, EPZ-6438 was about 35-fold selective for EZH2 over EZH1 and over 4500-fold selective for EZH2 over 14 other methyltransferases.
Several human cancers have been found to contain specific inactivating mutations in subunits of the chromatin remodeling complex SWI/SNF (switch/sucrose nonfermentable).124 For example, nearly all malignant rhabdoid tumors (MRTs) contain an inactivated SMARCB1 subunit. Coincidentally, EZH2 expression has been noted to be elevated in these SMARCB1-deficient tumors.123,125 EPZ-6438 has shown remarkable promise in these tumor types, displaying antiproliferative effects at nanomolar potencies while not affecting cells with wild-type SMARCB1 (RD cells). In cell-based studies with a SMARCB1-deficient MRT line (G401), EPZ-6438 selectively reduced H3K27me3, H3K27me2, and H3K27me1 marks in a concentration-dependent manner.123 Treatment with EPZ-6438 in G401 cells resulted in G1 arrest and apoptosis. On the other hand, the same treatment in RD cells did not affect the cell cycle and did not induce apoptosis, suggesting that the proliferation of SMARCB1-deficient MRT cells depends on EZH2 overexpression and H3K27 hypertrimethylation. Twice daily oral dosing of EPZ-6438 at 250 or 500 mg/kg over the course of 28 days was well-tolerated and completely eliminated G401 xenografts in mice with severe combined immunodeficiency (SCID).123 In addition, the tumor regression was maintained for 32 days after the cessation of treatment. Biopsy and analysis of certain tumors in this study revealed a strong correlation between H3K27me3 inhibition and antitumor activity, as expected.
Currently, EPZ-6438 is in a two-part Phase 1/2 study, under the name E7438.126 The compound is in Phase 1 for B-cell lymphomas and advanced solid tumors and is in Phase 2 for DLBCL and follicular lymphoma. The Phase 1 portion of the study seeks to establish dosing, the effects of food intake on bioavailability, and possible drug–drug interactions, with E7438 being orally administered twice daily. The phase 2 portion is examining the safety and efficacy of E7438 in histologically confirmed DLBCL and follicular lymphoma patients with wild-type or mutated EZH2. The same compound is also in additional clinical trials.127−130
In 2013, Garapaty-Rao and co-workers reported compound 1 (Figure 6), an EZH2-selective inhibitor with a new tetramethylpiperidinyl benzamide scaffold, differing from the pyridone indole/indazole scaffold of previously reported EZH2 inhibitors.131 Compound 1 was discovered via HTS of a 150000-compound library and subsequent optimization of the hit identified. Compound 1 was competitive with the cofactor SAM, potent for wild-type EZH2 (IC50 = 21 ± 4 nM), and slightly less potent for the Y641N mutant EZH2 (IC50 = 197 ± 14 nM).132 This inhibitor was about 10-fold selective for EZH2 over EZH1 and selective for EZH2 over 5 other methyltransferases. In HeLa cells, compound 1 reduced H3K27me3 and H3K27me2 levels with modest potency (EC50 = 7 μM), while not affecting the levels of H3K27me1, H3K4me3, H3K9me3, H3K36me3, EZH2, EZH1, SUZ12, or EED.131 Global reduction of H3K27me3 and H3K27me2 marks was confirmed using mass spectrometry-based proteomics studies in two germinal center B cell-like (GCB) DLBCL lines, HT (wild-type EZH2) and SUDHL6 (mutant EZH2), treated with compound 1. This inhibitor blocked the proliferation of Pfeiffer cells but not OCI-LY19 cells with wild-type EZH2, despite the fact that H3K27me3 and H3K27me2 levels were reduced in both cell lines. Further analysis of the Pfeiffer cells revealed that compound 1 concentration dependently increased transcription levels of genes known to be regulated by EZH2 but either did not affect or downregulated genes that play a role in cell cycle progression. Interestingly, it was also observed that PC3 and DU145 prostate cancer cells treated with compound 1 displayed lowered H3K27me3 levels but did not display altered cell proliferation or transcript levels of EZH2 target genes, suggesting that their proliferation occurs independent of EZH2 methyltransferase activity and H3K27 hypertrimethylation.
In 2014, Bradley and co-workers reported a series of SAM-competitive inhibitors of EZH2, which also contain the pyridone headgroup and displayed high potency, selectivity, and cellular activity.133 More recently, in late 2016, the discovery of CPI-1205 (Figure 6), an optimal compound in this series, was reported.134 CPI-1205 is a highly potent EZH2 inhibitor (biochemical IC50 = 2.0 nM, cellular EC50 = 32 nM). It showed high selectivity against a panel of 30 other histone or DNA methyltransferases, while it inhibited EZH1 with an IC50 of 52 ± 11 nM. Furthermore, it did not inhibit a panel of 54 physiologically relevant receptors, transporters, and ion channels at 10 μM by more than 50%. CPI-1205 displayed good oral bioavailability in both rats and dogs (45% F in rats and 46% F in dogs) but exhibited relatively high clearance in both species. Importantly, it exhibited robust antitumor effects in a KARPAS-422 xenograft model when dosed at 160 mg/kg twice daily. In toxicology studies, CPI-1205 was well-tolerated for 28 days and any findings were reversible over the recovery period. CPI-1205 is the third EZH2 inhibitor advanced into human clinical trials. This compound is currently in a Phase 1 clinical trial for B-cell lymphomas and is being tested for pharmacodynamic and pharmacokinetic properties in lymphoma tissue, bone marrow, and skin. Additionally, safety and dosing are being evaluated in patients with B-cell lymphomas (NCT02395601).
In 2015, Campbell and co-workers reported EPZ011989 (Figure 6), a potent, selective, and orally bioavailable EZH2 inhibitor.135 This compound was discovered by modifications of the pyran moiety in EPZ-6438. It potently inhibited both wild-type (Ki < 3 nM) and Y641-mutated (Ki < 3 nM) EZH2 in biochemical assays and reduced the H3K27me3 mark with an IC50 = 94 ± 48 nM in a cellular ELISA assay. EPZ011989 was >15-fold selective for EZH2 over EZH1 and >3000-fold selective for EZH2 over 20 other methyltransferases tested. It was metabolically stable in human and rat liver microsomal stability studies. In addition, cell-based studies in the Y641F-mutated human lymphoma line, WSU-DLCL2, demonstrated that EPZ011989 reduced H3K27 methylation with a sub-100 nM potency, while maintaining a lowest cytotoxic concentration (LCC) of 208 nM.135 One of the most significant improvements seen in EPZ011989 compared to its predecessors was its pharmacokinetic and pharmacodynamic properties, as assessed in a 7-day PK study with PD measurement of the H3K27me3 mark in bone marrow following various doses of the compound twice daily (BID).135 The compound was well-tolerated, and at a dose of 500 mg/kg, it appeared to achieve complete coverage over the predicted efficacious plasma level. Further, at this dose, the H3K27me3 mark in bone marrow was completely eliminated by the end of the 7-day treatment. The antitumor activity of EPZ011989 was also assessed in vivo using KARPAS-422 human DLBCL xenografts in mice. Treatment with 250 and 500 mg/kg suspensions of EPZ011989 administered BID for 21 days resulted in significant tumor regression at both doses while not affecting body weight. Further, a robust reduction of the H3K27me3 mark was observed in these tumors after treatment.
Early in 2016, Song and co-workers reported ZLD1039 (Figure 6), a potent, selective, and orally bioavailable EZH2 inhibitor.136 This inhibitor was developed based on the core scaffold of EPZ-6438. ZLD1039 potently inhibited wild-type (IC50 = 5.6 ± 0.4 nM), Y641F mutant (IC50 = 15 ± 0.5 nM), and A677G mutant (IC50 = 4.0 ± 0.3 nM) EZH2. It was 14-fold selective for EZH2 over EZH1 and >10000-fold selective for EZH2 over 10 other methyltransferases tested. In MOA studies, ZLD1039 was competitive with SAM and noncompetitive with the peptide substrate. ZLD1039 reduced the H3K27me3 mark in MCF7 and MDA-MB-231 breast cancer cell lines, while other histone methyl marks and EZH2 levels were unaffected.136 In antiproliferative growth assays with several breast cancer cell lines, MCF7 and ZR-75-1 cells were most sensitive to ZLD1039, displaying IC50 values of 0.99 ± 0.23 μM and 0.089 ± 0.019 μM, respectively. Additionally, ZLD1039 was found to induce apoptosis, evidenced by decreased levels of Bcl-2 and elevated levels of Bcl-2-associated X protein (BAX), cleaved-caspase 3, and cleaved-caspase 9. Finally, ZLD1039 was assessed in vivo in three different breast tumor xenograft models (MCF7, MDA-MB-231, and 4T1). Oral dosing of ZLD1039 was well-tolerated, inhibited cell cycle-associated proteins and cell proliferation, and induced apoptosis. Shortly after the publication of ZLD1039, the same group reported ZLD1122, another potent and selective inhibitor of EZH2 and EZH1, which is structurally similar to ZLD1039.137 ZLD1122 significantly reduced the H3K27me3 mark without affecting H3K9me3 and H3K4me3 marks in cell-based assays. It induced G0/G1 phase arrest in DLBCL cells and inhibited DLBCL cell growth.
In late 2015, Jiao and Liu reported the first crystal structures of an active PRC2 complex from the yeast Chaetomium thermophilum, which contain EZH2, EED, and SUX12-VEFS in complex with inhibiting H3K27 M peptide and SAH (PDB ID: 5CH1 and 5CH2) (Figure 7A).138 In this work, an EZH2 loop region that migrates away from the EED surface and extends to the back of the SET domain of the catalytic moiety is referred to as an SET activation loop (SAL) (Figure 7A). It has been shown that SAL and SET regions together compose a split catalytic domain of EZH2. Shortly after, in 2016, Justin and co-workers published the structure of the human PRC2 complex (PDB ID: 5HYN).900 Concurrently, Brooun and co-workers reported the crystal structure of a small-molecule inhibitor in complex with the wild-type and Y641N-mutated PRC2 complex, consisting of human EED, human SUZ12-VEFS, and engineered American chameleon (Anolis carolinensis) EZH2 (AcEZH2) subunits (PDB ID: 5IJ7 and 5IJ8).139 Most recently, a cocrystal structure of an analog of CPI-1205 bound to human PRC2 was also obtained (PDB ID: 5LS6).134 The crystal structure revealed that the pyridone motif forms two hydrogen bonds with the protein backbone of W624 and is constrained in an aromatic cage generated by F665, F686, and W624 (Figure 7B). These crucial interactions with the pyridone motif explain the importance of this functional group for its high affinity binding, its dominance in almost all reported EZH2 inhibitors, and the difficulty in finding suitable pyridone replacements. These structures represent a landmark in this field. The PRC2 complex used by Brooun and co-workers was a functional complex, with robust methyltransferase activity on the H3 peptide. This same study also examined the subunit architecture of AcEZH2. It was discovered that the architecture of the N-terminal region of AcEZH2 depended on its interactions with the EED subunit. Further, residues 108–124 in the N-terminal appeared to be important for the activation of the SET domain. Hence, it was termed the “activation loop”, which is the equivalent stretch of residues in CtEZH2 referred to as SAL (Figure 7A).139 Given that previous studies of the EZH2 SET domain have shown it to be inactive when isolated, as well as the fact that EZH2 is known to require EED and SUZ12-VEFS to form a catalytically active complex, it was suggested that perhaps EED and SUZ12-VEFS allosterically activated the SET domain. Structural analysis of other SET domain containing methyltransferases suggests that the activation loop is a conserved structural feature, although it differs in their primary sequence. In PRC2, EED is reported to play a clear role in shaping the activation loop through its extensive interactions with the EZH2 N-terminal segment.139 Closer examination of the activation loop revealed that it occupies the interface of the SET domain, EED, and VEFS. Further modeling suggested that the EED-mediated structuring of the EZH2 activation loop induces a conformational change in the I-SET region, consisting of residues 643–681. It is believed then that the I-SET conformation change, in turn, allows for the recognition of cofactor and inhibitor binding to occur.
Figure 7.
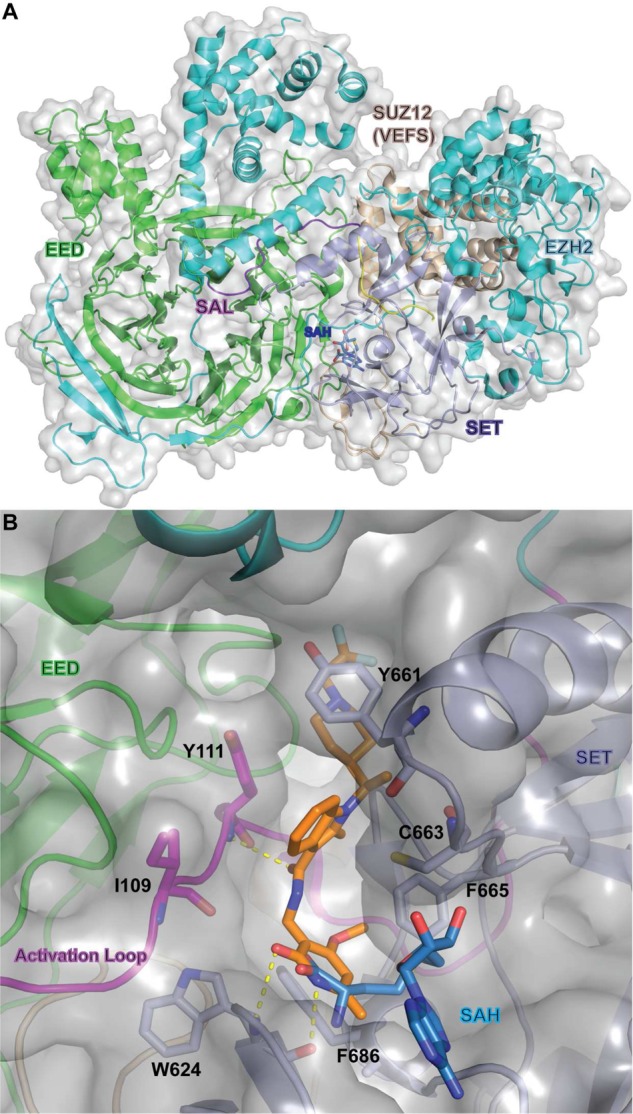
(A) X-ray structure of Ct-PRC2 complex (PDB ID: 5CH1). (B) Co-crystal structure of PRC2 with a derivative of CPI-1205 (PDB ID: 5LS6). The key residues and interactions are indicated. SAH (blue) is overlaid for reference (PDB ID: 5HYN). Hydrogen bonds are represented as yellow dashed lines.
Studies to understand inhibitor recognition were conducted using the engineered Ac/human PRC2 complex.139 On the basis of previous modeling, it was expected that the pyridone-containing PRC2 inhibitors would target the SAM-binding pocket. In these studies, however, it was determined using Hydrogen–Deuterium Exchange-Mass Spectrometry (HDX-MS) that compound recognition lowered deuterium exchange around both the SET domain and for the protein backbone near Y111 residue of the activation loop.139 A crystal structure of an inhibitor in complex with the engineered PRC2 elucidated that its binding was distinct from that of SAM (Figure 7B). More specifically, it was observed to be virtually orthogonal to the SAM-binding mode, with the pyridone moiety anchoring the rest of the compound to the backbone of the W624 contained in the GXG motif of the SET domain via hydrogen bonding. Indeed, this mode of binding, with the partial overlap of cofactor and inhibitor binding sites, is still consistent with the previously observed fact that these pyridone-containing inhibitors are SAM-competitive. Given the clearly important anchoring role played by the pyridone group, it is also believed that the other pyridone-containing EZH2 inhibitors bind in a very similar manner.
One final aspect of PRC2 inhibition addressed by this study was that of mechanisms of acquired resistance that have appeared in various lymphoma cancer cell lines following treatment with various EZH2-selective pyridone-containing inhibitors.139 Most notably, it has previously been demonstrated that growing KARPAS-422 and Pfeiffer cells with EI1 and EPZ-6438 results in acquired resistance through mutation.140,141 One such documented mutation was Y661, located in the I-SET region. Other documented mutations include I109 K, Y111D, and Y111L, which are all found in the activation loop and may influence the inhibitor-binding potency of the pyridone-containing inhibitors through both changes in electrostatic interactions and, in certain instances, steric conflict. For example, one model predicts that the Y111L mutation may induce between a 100–1000-fold shift in the IC50 values. Thus, this provides a compelling argument that mutations in Y661 and Y111, which lead to decreased inhibitor potencies, are closely linked to the PRC2 activation mechanism.
The inhibitor that was cocrystallized with the PRC2 complex by Brooun and co-workers differed from most of the other pyridone-containing inhibitors. It features a substituted phenyl ring joined to a dimethylpyridone moiety via an amide linkage.139 Very recently, Kung and co-workers reported the design and synthesis of this new series of pyridone inhibitors, which led to the discovery of compound 2 (Figure 6).142 Compound 2 is a potent EZH2 inhibitor with an IC50 of <5 nM and a Ki of 0.7 nM in biochemical assays. It exhibited cellular potencies in KARPAS-422 cells that are comparable to previously reported EZH2 inhibitors. MOA studies revealed that 2 was competitive with SAM. It was selective for EZH2 over other methyltransferases and protein kinases tested. Target engagement of this inhibitor was demonstrated by its ability to modulate the expression of TNFRSF21 and PRDM1, two PRC2 target genes that have previously been shown to be repressed by H3K27me3. Both of these genes were upregulated in KARPAS-422 cells containing the EZH2 Y641N mutant. On the other hand, minimal changes of target gene expression were observed in the OCI-LY19 cell line, which contains wild-type EZH2. In mouse PK studies, relatively high doses of 2 were required to maintain reasonable mouse plasma exposures. The tumor growth inhibition was performed to demonstrate in vivo efficacy. Treatment with compound 2 at 200 and 300 mg/kg twice daily for 20 days was well-tolerated with less than 10% body weight loss observed. Compound 2 demonstrated tumor stasis and regression at the 200 and 300 mg/kg dose levels, and tumor growth inhibition was sustained for at least another 3 weeks after the last dose. The H3K27me3 mark was reduced by more than 50% and PRC2 target genes TNFRSF21 and PRDM1 were both upregulated in the collected tumor samples.
In 2016, Souroullas and co-workers reported that the EZH2 Y641F mutation induced lymphoma and melanoma through a reorganization of chromatin structure, altering both repression and activation of polycomb-regulated loci.143 In this study a previously unpublished pyridone inhibitor JQEZ5, which has a very similar structure to the aforementioned pyridone inhibitors, was introduced and used. This inhibitor was about 10-fold selective for EZH2 over EZH1.
In 2014, astemizole, an FDA-approved drug, was identified as a small-molecule inhibitor of the EZH2-EED interaction.144 This inhibitor destabilized the PRC2 complex and inhibited its methyltransferase activity in cancer cells. This report demonstrated that the EZH2-EED interaction could be perturbed by a small molecule. Recently published PRC2 crystal structures revealed an important role for EED in the enzymatic activity of the PRC2 complex, and very recently, several EED inhibitors that target the EED component of the PRC2 complex have been published.145−148
Lastly, it is worth noting that Kim and co-workers in 2013 developed a stapled EZH2 peptide (SAH-EZH2), which disrupted the EZH2-EED interaction and reduced EZH2 protein levels and the H3K27me3 mark.149 Extensive cell-based studies were conducted using SAH-EZH2. In MLL-AF9 leukemia cells, SAH-EZH2 inhibited cell proliferation and induced monocyte-macrophage differentiation. The induction of differentiation was observed by monitoring certain PRC2-regulated biomarker genes. For example, following SAH-EZH2 treatment, the expressions of monocyte- and macrophage lineage-specific markers, such as ADAM8, FCER1A, and ACE, were observed to be upregulated, while markers associated with hematopoietic stem cells, such as CD133, were suppressed.
2.1.4. Inhibitors of H3K4 and H3K36 Methyltransferases
SETD7, SETD1A, SETD1B, the MLL family proteins (MLL1–5), SETMAR, SMYD1, SMYD2, SMYD3, ASH1L, as well as PRDM7, and PRDM9, all catalyze the methylation of H3K4 in humans.150−155 H3K4 trimethylation is associated with transcriptional activation.6
SETD7 [SET domain containing (lysine methyltransferase) 7, also known as KMT7, SET7, SET9, and SET7/9] monomethylates H3K4 N-terminal peptides in vitro, but displays limited activity on nucleosomal substrates, thereby rendering it an unlikely enzyme for in vivo methylation of H3K4.156,157 On the other hand, studies have shown that SETD7 targets many nonhistone proteins and transcriptional regulators, such as p53, ERα, pRb, STAT3, HIF-α, FoxO3, and DNMT1 and, in turn, plays a role in transcriptional regulation and differentiation.24,158−164 SETD7 has been associated with the regulation of NF-κB, which occurs by monomethylation of p65.165,166 It has also been proposed as a potential target for the treatment of diabetes.167 Given its suggested roles in important biological functions, there is a need for the discovery of SETD7 selective inhibitors as chemical tools for investigating its physiological and pathophysiological functions.
In 2014, Barsyte-Lovejoy and co-workers reported (R)-PFI-2 (Figure 8) as the first potent, selective, and cell-active inhibitor of SETD7.168 This compound was discovered by HTS of a 150000-compound collection and subsequent optimization of the hit identified. (R)-PFI-2 was highly potent for SETD7 with an IC50 of 2.0 ± 0.2 nM and a Morrison Ki of 0.33 ± 0.04 nM, while its enantiomer, (S)-PFI-2, was about 500-fold less potent and was therefore used as a negative control for cell-based studies. (R)-PFI-2 exhibited >1000-fold selectivity for SETD7 over 18 other methyltransferases and was also selective for SETD7 over 134 GPCRs, ion channels, and other enzyme targets. The X-ray crystal structure of SETD7 in complex with (R)-PFI-2 (PDB ID: 4JLG) showed this compound occupying the substrate-binding groove and revealed the key ligand/protein interactions, which was verified by site-directed mutagenesis experiments (Figure 9). In addition, the cocrystal structure revealed that the inhibitor induced conformational modifications in the post-SET loop and made hydrophobic interactions with the methyl group of SAM (Figure 9). The SPR experiments confirmed compound binding to SETD7 but only in the presence of SAM. Kinetic experiments measuring IC50 values with varying SAM and peptide concentrations suggested that (R)-PFI-2 exhibited a cofactor-dependent and substrate-competitive MOA. In other words, inhibition of SETD7 by (R)-PFI-2 is not purely substrate-competitive and SAM plays a significant role in the binding of the inhibitor to SETD7. This result is consistent with earlier reports that suggested that SAM binding to SET domain-containing methyltransferases has an important role in the folding and stabilization of the post-SET loop169 and also in an ordered binding mechanism for SETD7, whereby peptide binding follows SAM binding.170 The direct interaction of (R)-PFI-2 with SETD7 in cells was demonstrated by pull-down studies using a biotinylated derivative of (R)-PFI-2, and by that, (R)-PFI-2 increased the stability of SETD7 in a cellular thermal shift assay (CETSA). The inhibitor exhibited good physicochemical properties and showed no observable cell toxicity at up to 50 μM in various cell lines tested. Furthermore, (R)-PFI-2 increased nuclear localization of the transcriptional coactivator Yes-Associated Protein (YAP) in a concentration-dependent manner and induced expression of YAP-silenced genes in cells. The effect of (R)-PFI-2 was consistent with the genetic deletion of SETD7. Overall, (R)-PFI-2 is the most potent, selective, and cell-active small-molecule inhibitor of SETD7 to date.
Figure 8.

Structures of SETD7 inhibitors.
Figure 9.
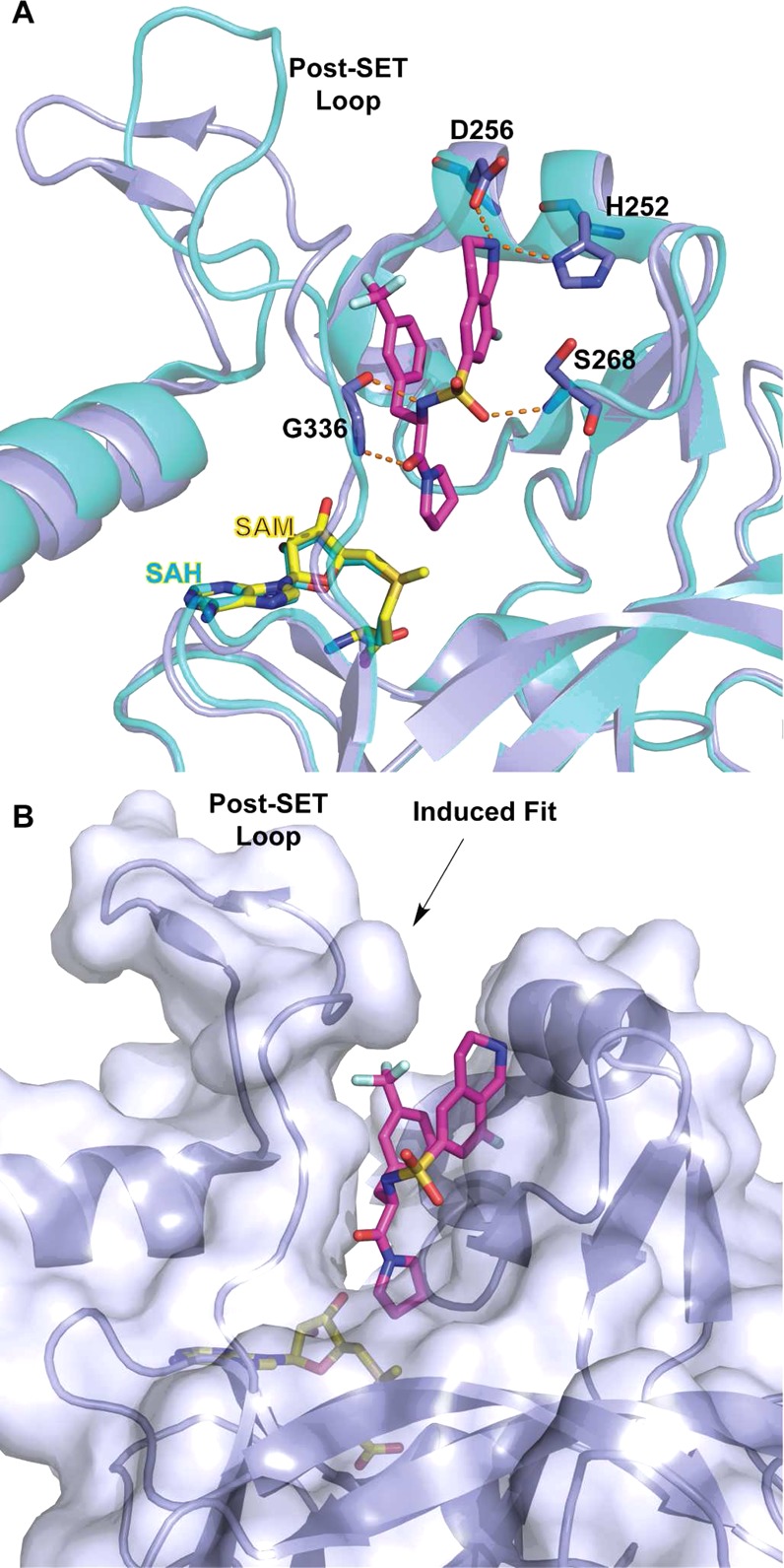
(A) SETD7 (light blue) in complex with (R)-PFI-2 (magenta) and SAM (yellow) (PDB ID: 4JLG) is superimposed with SETD7 structure in complex with SAH (1O9S) depicting the conformational variability of the post-SET loop. Hydrogen bonds are represented as orange dashed lines with key residues. (B) Surface representation of (R)-PFI-2-bound SETD7 highlighting an induced conformational modification of the post-SET loop.
There have been several additional studies in recent years toward the discovery of SETD7 inhibitors.171−174 In 2015, Meng and co-workers utilized a pharmacophore- and docking-based virtual screening approach and performed SAR studies.173 DC-S239 (Figure 8), a SETD7 inhibitor with an IC50 of 4.6 μM in biochemical assays, was identified. This inhibitor showed selectivity for SETD7 over 7 other methyltransferases. It inhibited the proliferation of MCF7, HL60, and MV4–11 cells in a concentration-dependent manner with micromolar potencies, while displaying no cellular cytotoxicity against HCT116 and DHL4 cells. Altogether, this inhibitor may be used as a starting point for further optimization and development of more potent SETD7 inhibitors in the future. More recently, Takemoto and co-workers identified cyproheptadine (Figure 8), a known antagonist of histamine H1 and serotonin 5-HT2A receptors and a clinically approved antiallergy drug, as an inhibitor of SETD7 (IC50 = 1.0 μM).174 It was selective for SETD7 over G9a, SUV39H1, SETD8, and DOT1L. Kinetic experiments and a cocrystal structure showed that it was noncompetitive with SAM but competitive with the peptide substrate. Cyproheptadine reduced the expression of ERα in a concentration- and time-dependent manner in MCF7 cells, to a similar extent as seen in SETD7 knockdown. While the effect of cyproheptadine on ERα activity was shown to be independent of its known antagonistic effect mediated by H1 and 5-HT2A receptors, its value as a chemical tool for exploring SETD7 biological functions is limited to non-neuronal cells that do not express H1 and 5-HT2A receptors.
The SMYD (SET and MYND domain-containing) family of proteins, SYMD1–5, possess a unique SET domain that is divided into two fragments by a zinc ion-binding domain MYND (myeloid translocation protein-8, Nervy, and DEAF-1) and is followed by a cysteine-rich post-SET domain.4,151 This family of proteins is considered to be critical regulators of development, as disruption of the SMYD1 gene results in impaired cardiomyocyte maturation, flawed cardiac morphogenesis, and embryonic lethality in mice.175 While SMYD3 (also known as KMT3E) was initially described as an H3K4 methyltransferase,176 H3K4 methylation by SMYD3 has not been clearly verified by subsequent studies.177,178 On the other hand, SMYD3 has been shown to directly methylate both histone H4K5 and MAP3K2 (also known as MEKK2) at K260, although the latter has exhibited a 100-fold increase in catalytic efficiency as a substrate and seems to be the major target of SMYD3.177,178 A recent report showed that the methylation of MAP3K2 by SMYD3 increases MAP kinase signaling and promotes the formation of Ras-driven carcinomas.177 SMYD3 has also been implicated in the regulation of gene transcription and signal transduction pathways critical for cell survival in multiple cancer models.176,178−180 In addition, SMYD3 has been shown to be involved in cancer cell proliferation and overexpressed in most hepatocellular and colorectal carcinomas, as well as most breast cancers.176,180 Recently, Sarris and co-workers reported their insights into the mechanism of nuclear SMYD3 function in vivo. They studied this mechanism by generating and experimenting with SYMD3-KO mice that were subjected to chemically induced liver or colon carcinogenesis.181 As studies suggested, SMYD3 might promote cancer through multiple mechanisms, dependent or independent of its enzymatic activity. Nevertheless, SMYD3 is a promising target for cancer, and thus, the development of selective and cell-active SMYD3 inhibitors will help elucidate its mechanism and roles in cancer and test therapeutic hypotheses.
Mitchell and co-workers reported the first selective small-molecule inhibitors of SYMD3, EPZ0330456, and EPZ031686 (Figure 10).182 Screening of a proprietary compound library resulted in the identification of an initial hit. A cocrystal structure of this hit in a ternary complex with SMYD3 and SAM (PDB ID: 5CCL) provided structural insights and guidance for hit optimization, which led to the discovery of EPZ0330456 and EPZ031686. Both compounds potently inhibited SMYD3 (IC50 < 5 nM) in biochemical assays. They were highly selective for SYMD3 over 16 other methyltransferases, including the highly homologous SMYD2. In a MAP3K2 In-Cell-Western assay, EPZ0330456 and EPZ031686 showed IC50s of 48 and 36 nM, respectively. A cocrystal structure of SMYD3 in complex with EPZ030456 (PDB ID: 5CCM) revealed that the inhibitor makes only a few specific interactions with the protein beyond the oxindole headgroup and amide linker, even though it binds with low nanomolar affinity (Figure 11A). EPZ030456 displayed mixed-type inhibition with respect to SAM with a Ki = 4.7 ± 1.8 nM and was noncompetitive with MAP3K2 with a Ki = 1.3 ± 0.1 nM. EPZ031686, however, displayed noncompetitive inhibition with respect to both SAM and MAP3K2 with Ki = 1.2 ± 0.1 and 1.1 ± 0.1 nM, respectively. Although EPZ030456 occupied the lysine-binding pocket in the cocrystal structure, suggesting that these compounds would be competitive with MAP3K2, the observed mechanism was noncompetitive. It was postulated that the significant binding affinity to MAP3K2 stems from interactions with SMYD3 outside of the lysine-binding pocket (i.e., exosite binding) in such a way that these inhibitors cannot displace the protein substrate in competition assays. Noncompetitive inhibition by active site-directed small-molecule inhibitors is a well-established phenomenon for enzymes acting on macromolecular substrates and is also observed with PRMT inhibitors.183 This will be described in later sections pertaining to PRMTs. In addition, EPZ031686 was orally bioavailable in mouse PK studies, making it suitable for in vivo efficacy studies. Overall, the oxindole sulfonamide EPZ031686 and the oxindole sulfamide EPZ030456 are the first potent, selective, and cell-active SMYD3 inhibitors and are valuable chemical tools to investigate biological functions of SMYD3 and validate SMYD3 as a potential therapeutic target.
Figure 10.

Structures of SMYD3 inhibitors.
Figure 11.
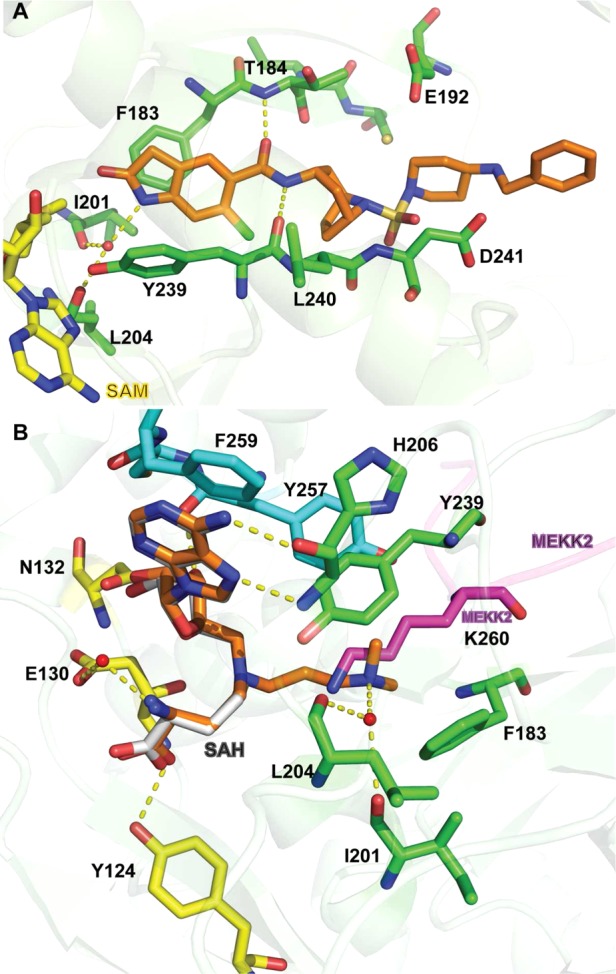
(A) Co-crystal structure of EPZ030456 (orange) in complex with SMYD3 (green) and SAM (yellow) (PDB ID: 5CCM). (B) Co-crystal structure of GSK2807 (orange) (PDB ID: 5HI7) in complex with SMYD3. MEKK2 (MAP3K2) peptide (magenta) and SAH (gray) is overlaid for reference (PDB ID: 5HQ8). Selected SET (green), SET-I (yellow), and post-SET (aqua) residues are indicated. Hydrogen bonds are represented as yellow dashed lines and water molecules as red spheres.
In 2016, Van Aller and co-workers reported the structure-based design of a novel SMYD3 inhibitor.184 A cocrystal structure of SMYD3 and a MAP3K2 peptide (residues YDNPIFEKFGKGGTY) was solved (PDB ID: 5HQ8), and a previously unidentified ternary complex composed of SMYD3, substrate, and SAH was observed. It was postulated that the methyl transfer reaction catalyzed by SMYD3 proceeds through a ternary complex mechanism and a compound that contains portions of both SAH and MAP3K2 K260 could provide a useful bisubstrate inhibitor. Therefore, SAH analogs with basic amino side chains extending into the lysine-binding channel were designed and synthesized, resulting in the discovery of GSK2807 (Figure 10), which possesses a propyl dimethylamino side chain. GSK2807 showed good potency for SMYD3, with an IC50 of 130 nM in biochemical assays. A cocrystal structure of SMYD3 in complex with GSK2807 confirmed that it occupied the cofactor-binding site (PDB ID: 5HI7). GSK2807 and SAH shared key interactions in the cofactor-binding site formed by the SET-I, SET, and post-SET domains (Figure 11B). As predicted, the propyl dimethylamino side chain of GSK2807 extended into the lysine-binding channel of SMYD3 and formed a hydrogen bond with a highly conserved water molecule that interacts with the backbone carbonyls of I201 and L204 (Figure 11B). GSK2807 was 24-fold selective for SMYD3 over SMYD2 [Ki = 14 ± 6 nM (SMYD3) and 345 ± 36 nM (SMYD2)] and highly selective for SMYD3 over 8 other methyltransferases. In MOA studies, GSK2807 was competitive with SAM, consistent with the cocrystal structure; however, it was noncompetitive with both full-length MAP3K2 protein and peptide substrate. This result was interesting since the propyl dimethylamino side chain of GSK2807 occupies a portion of the lysine-binding channel where the substrate lysine of MAP3K2 binds. However, GSK2807 has poor cell membrane permeability, making it unsuitable for cellular studies.
Another SMYD3 inhibitor, BCI-121, was discovered via virtual screening. It was reported that this compound reduced global H3K4me3/me2 and H4K5me levels in colorectal cancer cells.185 It should be noted that the observation of reduction in H3K4me3/me2 levels is inconsistent with other reports, indicating no methylation activity of SYMD3 on H3K4.177,178 Thorough characterization of this compound in biochemical and biophysical assays have not been reported. Therefore, caution should be taken while attributing the results in this study to pharmacological inhibition of SYMD3 by this compound.
MLL [also known as lysine (K)-specific methyltransferase 2A (KMT2A), TRX1, and MLL1] is a large multidomain (several N-terminal DNA domains and a C-terminal SET domain with an essential post-SET region) protein186 that is specific for H3K4 mono-, di-, and trimethylation.187−189 Chromosomal rearrangements associated with MLL have been shown to cause acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), or mixed lineage leukemia (MLL).190 More than 50 functionally diverse MLL-fusion proteins have been identified in human leukemias, with AF4, AF9, AF10, AF6, and ENL being the most common MLL fusion partners in MLL-rearranged leukemias.188,191 MLL was reported to be crucial for homeotic gene regulation and embryonic development via regulation of Hox gene expression in mice.192 Interestingly, differing from most SET domain-containing methyltransferases, MLL1 itself reveals poor methyltransferase activity.193 The crystal structures of the MLL1 SET domain (PDB ID: 2W5Y and 2W5Z) display an open conformation, which is ineffective in catalyzing the transfer of the methyl from SAM to the target lysine residue.193,194 The optimal methyltransferase activity of MLL1 requires additional components, WDR5 (WD repeat-containing protein 5), RBBP5 (retinoblastoma-binding protein 5), and ASH2L (ASH2 like histone lysine methyltransferase complex subunit), which are evolutionarily conserved from yeast to humans and core components of all MLL complexes.193,194 While selective small-molecule direct inhibitors of MLLs have not yet been reported, small molecules that perturb protein–protein interactions of MLL with its partners have been discovered. For example, small-molecule inhibitors of WDR5, which disrupt MLL activity in an indirect manner, have recently been reported.195−198 In addition, inhibitors selectively targeting the menin-MLL protein–protein interaction have been discovered.199−202
Like H3K4 methylation, H3K36 methylation is a hallmark that is associated with transcriptional activation. The human genome encodes at least eight methyltransferases containing a SET domain, that are responsible for H3K36 methylation: NSD1, MMSET (NSD2), WHSC1L1 (NSD3), SETD2, SETD3, ASH1L, SETMAR, and SMYD2. While these proteins have all been reported to methylate H3K36, they differ based on the state of methylation and whether or not they methylate additional substrates. NSD1, MMSET, WHSC1L1, ASH1L, and SETD2 have closely related catalytic SET domains and show H3K36 methylation specificity in vitro and in vivo, while SMYD2, SETMAR, and SETD3 have less similarity in their SET domains with less well-characterized activities toward H3K36.203 For example, SMYD2 (also known as KMT3C) was reported to methylate H3K4 as well as H3K36 in vitro,152 and SMYD2-mediated H3K36me2 was reported to repress the transcription of pro-inflammatory cytokines IL-6 and TNF-α in macrophages.204 However, inhibition or knockdown of SMYD2 does not affect global levels of mono-, di-, or trimethylation of H3K4 or H3K36, and most of SMYD2 is found in the cytoplasm, suggesting minimal SMYD2 activity on chromatin.151,152,205 A variety of nonhistone substrates of SMYD2 including p53,206 Rb,207 heat shock protein 90AB1 (HSP90),208 poly(ADP-ribose) polymerase 1 (PARP1),209 and ERα210 have been reported, thus implicating effects on transcriptional regulation, protein homeostasis, apoptosis, and the DNA damage response.203 Very recently, SMYD2 was demonstrated to be highly expressed in pediatric acute lymphoblastic leukemia and constitutes a poor prognostic factor.211 Overexpression of SMYD2 was also connected to tumor cell proliferation and resulted in malignant esophageal squamous cell carcinoma.212 As a result, there is a growing interest in developing SMYD2 inhibitors.
In 2011, Ferguson and co-workers reported the discovery of AZ-505 (Figure 12), and four cocrystal structures of SMYD2 in complex with a p53 substrate, product peptides, or AZ-505 (PDB IDs: 3S7J, 3S7B, 3S7F, 3S7D).213 The structural information obtained in this study was very valuable and provided insight and guidance for the development of ensuing SMYD2 inhibitors. AZ-505 was discovered by a HTS campaign. It displayed an IC50 of 0.12 μM in a biochemical assay. The direct binding of the inhibitor to the protein was shown by ITC, and a Kd of 0.5 μM was observed. AZ-505 was around 700-fold selective for SMYD2 over 6 other PKMTs, including the closely related SMYD3. Kinetic experiments revealed that AZ-505 was competitive with a peptide substrate (361–380 of the C-terminal regulatory domain of p53) and uncompetitive with SAM. No significant conformational changes were observed in the protein upon binding with the p53 peptide substrate or with the inhibitor.213 Cocrystal structures in complex with p53 peptides revealed that K370 occupies a hydrophobic, mostly aromatic pocket, and that the amino group of lysine is oriented by the hydroxyl groups of the highly conserved Y240 and Y258 residues and numerous main chain carbonyls. The mutation of Y240 has been shown to eradicate the catalytic activity of SMYD2.151 The cocrystal structure of SMYD2 with AZ-505 is consistent with it being a substrate-competitive inhibitor (Figure 13). AZ-505 features three distinctive functional groups: benzooxazinone, cyclohexyl, and dichlorophenethyl moieties (Figure 12). The benzooxazinone moiety lies deep in the lysine-binding channel, interacting with both Y258 as well as SAM (Figure 13). The cyclohexyl group is placed in the interface of the core SET and I-SET domains. The dichlorophenethyl moiety extends across the peptide-binding groove and interacts with a secondary hydrophobic pocket. The inhibitor binding mode, together with the partially overlapping p53 peptide and AZ505 binding site, creates opportunities for designing the next generation of SMYD2 inhibitors. In fact, Ferguson and co-workers suggested possible adjustments for improving potency and selectivity of AZ-505. However, cellular activities of this inhibitor were not reported. Very recently, the same research group disclosed full account of their studies for the development of SMDY2 inhibitors, which facilitated additional studies resulting in the discovery of SMDY2 inhibitors discussed below.214
Figure 12.

Structures of SMYD2 inhibitors.
Figure 13.
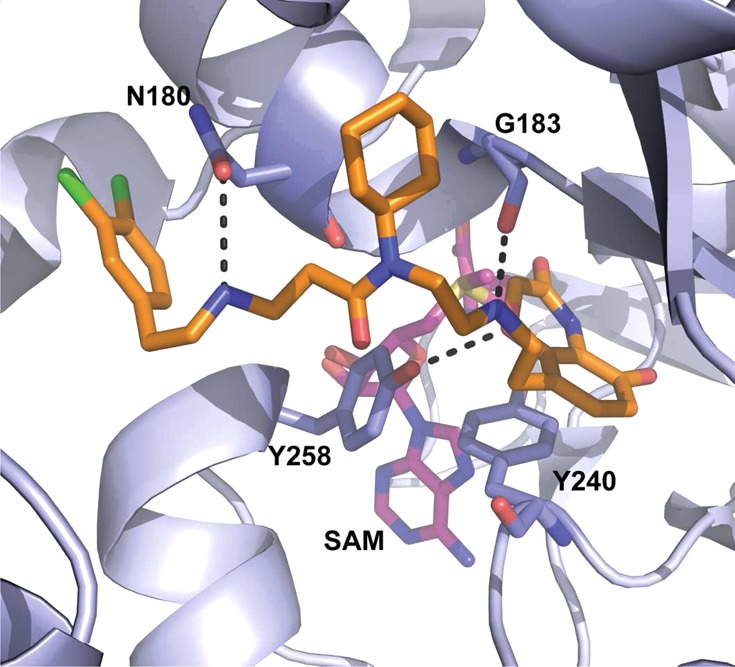
X-ray cocrystal structure of the SMYD2-AZ-505 complex (PDB ID: 3S7B).
In 2015, Sweis and co-workers reported a detailed SAR study of AZ-505, particularly on the three aforementioned main regions of the molecule.215 The analysis of the benzoxazinone region occupying the lysine-binding channel revealed the importance of several key hydrogen bond interactions with the protein. The SAR studies resulted in the installation of a secondary alcohol in the linker region of the molecule and led to the discovery of A-893 with >80-fold improvement in potency (IC50 = 2.8 nM) over the parent compound AZ-505 (IC50 = 120 nM) (Figure 12). A-893 was also a substrate-competitive inhibitor and exhibited a high selectivity over a panel of 30 additional methyltransferases, including PKMTs, PRMTs, and DNMTs. The interaction of the newly installed hydroxyl group with a complex network of hydrogen bonds around the lysine-binding pocket was observed upon analysis of the cocrystal structure of the SMYD2-A-893 complex (PDB ID: 4YND). Importantly, A-893 was cell-active and reduced p53 methylation levels by 42% in A549 lung cancer cells, which are known to express high levels of SMYD2, while overall p53 levels were unaltered. In summary, potency of AZ-505 was improved by this SAR study, resulting in the discovery of A-893, which is a valuable chemical probe for studying SMYD2 biology.
LLY-507 (Figure 12), another potent, selective, and cell-active inhibitor of SMYD2, was discovered by Nguyen and co-workers just a few months before the discovery of A-893.205,216 LLY-507 does not share a common scaffold with AZ-505 or A-893; however, it was designed by applying the structural insights reported by Ferguson and co-workers in the AZ-505 cocrystal structure, whereby a polar group was extended into the substrate lysine-binding channel and two lipophilic pockets were occupied, resulting in increased affinity. LLY-507 inhibited SMYD2 with an IC50 of <15 nM and was >100-fold selective over 21 other methyltransferases including SMYD2. It was also inactive against nonepigenetic targets including 454 kinases, 35 GPCRs, and 14 nuclear hormone receptors. A cocrystal structure of SMYD2 in complex with LLY-507 (PDB ID: 4WUY) showed that the inhibitor occupies the substrate binding site of SMYD2, with the pyrrolidine group occupying the lysine-binding pocket. It reduced monomethylation of p53 K370 in several cell systems (IC50 = 0.6 μM in U2OS cells). In addition, the antiproliferative activity of LLY-507 was shown in tumor types in which SMYD2 is amplified and/or overexpressed [in various breast cancer, hepatocellular carcinoma (HCC), and esophageal squamous cell carcinoma (ESCC) cell lines]. However, analysis of the relationship between SMYD2 expression and LLY-507 IC50 values revealed that sensitivity to LLY-507 was not dependent on the SMYD2 protein levels. Additionally, the relationship between p53 or Rb mutation status and the antiproliferative effect of LLY-507 indicated that there was also no correlation between the presence of wild-type p53 and/or Rb and sensitivity to LLY-507. These results suggested that the methylation of p53 or Rb by SMYD2 is not the principal driver of SMYD2-mediated cancer cell growth and that other SMYD2 substrates or a second genetic or epigenetic driver may be involved in the process. In summary, LLY-507 is the first potent, selective, and cell-active inhibitor of SMYD2 and is another valuable chemical probe to elucidate the role of SMYD2 in cancer and other diseases.
An HTS campaign resulted in the discovery of pyrazoline-containing tractable hits, which were further optimized through SAR and structural studies to give enantiomerically pure (S)- BAY-598 (Figure 12) as an inhibitor of SMYD2 with an IC50 of 27 ± 7 nM in a biochemical assay.217 It was >10-fold selective for SMYD2 over SMYD3 (IC50 = ∼ 3 μM), the most closely related methyltransferase and >100-fold selective for SMYD2 over 31 other methyltransferases. It was also highly selective against kinases and other primary molecular targets, including several CNS targets. The kinetic studies demonstrated that the inhibitor was competitive with the peptide substrate (Ki = 8 ± 1 nM) but was uncompetitive with SAM. BAY-598 preferentially binds to the SMYD2–SAM complex, suggesting an ordered sequential Bi Bi mode of substrate binding, where SAM is required to bind before the peptide substrate. Treating HEK293T cells with BAY-598 decreased p53 methylation levels in a concentration-dependent manner (IC50 = 58 nM) but did not alter the total protein levels of p53. This well-established assay has been previously used to characterize LLY-507.205 The neuroblast differentiation-associated protein AHNAK was identified as a novel substrate of SMYD2.218,219 The strong AHNAK methylation signal was used to set up an In-Cell-Western assay for cellular optimization. IC50 values determined through this cellular assay for aminopyrazoline-based inhibitors correlated well with potency in the biochemical assay and the p53 methylation assay.
To explore potential effects of BAY-598 on cell proliferation, a panel of 240 different cancer cell lines was tested. SMYD2 inhibition by BAY-598 displayed partial antiproliferative effects only in a small subset of cancer cell lines tested.217 It was previously hypothesized that the methylation of p53 by SMYD2 would cause the suppression of apoptosis. To test this hypothesis, the effect of BAY-598 in combination with an apoptotic stimulus was investigated. KYSE-150, U2OS, and A2780 cell lines were pretreated with BAY-598 or its inactive derivative as a negative control (demethylation phase), followed by treatment with doxorubicin (apoptotic stimulus). BAY-598, but not the negative control, significantly improved caspase 3/7 activation in all tested cell lines. Therefore, it was concluded that SMYD2 inhibition could enhance apoptotic responses.
In vivo inhibition of SMYD2 by BAY-598 was also examined using mice-bearing subcutaneous tumor xenografts (tumor tissues derived from the SMYD2-overexpressing KYSE-150 cell line). Mice were treated orally with different doses once daily for 3 days. After the treatment period, tumors were harvested and analyzed ex vivo for methylation of AHNAK by blotting. BAY-598 drastically reduced the methylation at doses starting from 30 mg/kg, with the most significant effects seen in the 100 mg/kg treated group. Furthermore, it was shown that SMYD2 inhibition could enhance the efficacy of doxorubicin in vivo, which confirmed the observation of higher caspase 3/7 activation in cellular assays. However, only moderate effects were observed in initial in vivo studies with BAY-598 on xenografted tumors. In addition, high doses of BAY-598 were needed to achieve effects on methylation in vivo. Therefore, the possibility that additional, unknown activities of SMYD2 might be responsible for the observed effects cannot be excluded. Nevertheless, BAY-598 is a valuable chemical tool for further exploration of complex SMYD2 biology in both cellular and in vivo studies.
SETD2 (SET domain containing 2, also known as KMT3A and SET2) is a SET domain-containing methyltransferase that is responsible for H3K36 methylation. It has been shown to be a tumor suppressor associated with p53-dependent gene regulation, as well as transcription elongation and intron-exon splicing.220−222 Evidence for the tumor suppressor role of SETD2 in human breast cancer was also provided.221 SETD2 mutations have frequently been identified in renal cell carcinoma223,224 and nonsmall cell lung cancer.225 For example, loss-of-function SETD2 mutations disrupting H3K36 trimethylation has been proposed to cause hemispheric high-grade gliomas (HGGs) in older children and young adults.226 It has also been suggested that the disruption of SETD2-H3K36 trimethylation pathway is a distinct mechanism for leukemia development.227
N-propyl sinefungin (Pr-SNF), a N-alkyl derivative of sinefugin (Figure 14), was discovered as a selective inhibitor of SETD2 (IC50 = 0.8 ± 0.02 μM) by Zheng and co-workers.228 Sinefugin is a close analog of the cofactor SAM and a nonselective inhibitor of methyltransferases.7 On the other hand, Pr-SNF is highly selective for SETD2 over 14 other methyltransferases. However, it displayed only modest selectivity over SETD7 (IC50 = 2.2 ± 0.4 μM, 2.8-fold), CARM1 (IC50 = 3.0 ± 0.3 μM, 3.8-fold), and PRMT1 (IC50 = 9.5 ± 0.4 μM, 12-fold). The selectivity of Pr-SNF for SETD2 over 14 other methyltransferases is remarkable, considering the overall structural similarity between SET-domain-containing PKMTs. Therefore, cocrystal structures of SETD2’s catalytic domain [pre-SET (also known as AWS), SET, and post-SET motifs] with SAH (Figure 15, panelss A and B) and Pr-SNF (Figure 15, panels C and D) (PDB IDs: 4H12 and 4FMU) were solved to elucidate the structural basis of the selectivity of the Pr-SNF for SETD2. In these structures, SAH occupied a deep pocket formed between the SET and post-SET domains of SETD2, as in all PKMTs. In the SETD2–SAH binary complex that differs from other PKMTs, an autoinhibitory post-SET loop, which is positioned to prevent substrate binding, was identified. A characteristic R1670 residue of this loop was located in the pocket that is otherwise occupied by the substrate lysine. A similar autoinhibitory topology has also been reported for NSD1 and ASH1L, two closely related homologues of SETD2, and was proposed to regulate the access of substrates to the enzymes. Although the overall structure of SETD2 with Pr-SNF was similar to that with SAH, the Pr-SNF’s propyl group partially extended into the lysine-binding pocket, causing SETD2 to reorient this R1670 residue away from the lysine-binding pocket and, in turn, flip the otherwise autoinhibitory post-SET loop (Figure 15). Overall, the structural analysis revealed that the catalytic domain of SETD2 could adopt at least two alternative conformations by flipping its post-SET loop: an autoinhibitory closed conformation and a substrate-accessible open conformation. In this study Pr-SNF was used as a structure probe through its preferential interaction with the latter. Kinetic studies together with structural analysis suggested that the SETD2-catalyzed methylation goes through a random sequential mechanism and inhibition occurs via either a Pr-SNF-SETD2 binary complex or a Pr-SNF-SETD2-substrate ternary complex. To date, Pr-SNF is the only SETD2 inhibitor; however, no cellular studies were reported for this inhibitor.
Figure 14.

Structures of Sinefungin and Pr-Sinefungin (Pr-SNF).
Figure 15.

(A) Co-crystal structure of SETD2 in complex with SAH (PDB ID: 4H12). SET domain (light orange), N-SET domain (light blue), pre-SET domain (cyan), and post-SET motif (red) are highlighted. (B) The SAH binding pocket between SET and Post-SET domains of SETD2. (C) Co-crystal structure of SETD2 in complex with Pr-SNF (PDB ID: 4FMU). (D) The key residues stabilizing the alternative configuration of the post-SET loop and interacting with Pr-SNF’s N-propyl chain.
Very recently, Tisi and co-workers obtained a crystal structure of the MMSET SET domain.229 Analyses of crystal structures of the SET domains of NSD1 and the closely related protein SETD2 in complex with SAH and Pr-SNF have provided valuable insights into the architecture of the cofactor and substrate binding sites, as well as conformational changes required for substrate binding as was previously discussed.228,230 The NSD family primarily mono- and dimethylate H3K36 in vivo.231,232 MMSET has been associated with multiple myeloma (MM)233,234 and lymphoid malignancies.235 Overexpression of MMSET has been reported in solid tumors of the lung, prostate, and bladder, as well as neuroblastomas.236,237 On the basis of the close homology between SETD2 and MMSET, N-substituted sinefungin analogs were explored as potential inhibitors of MMSET and Pr-SNF was identified as an MMSET inhibitor with an IC50 of 3.3 ± 1.0 μM (IC50 against SETD2 in the same assay conditions was 0.49 μM) while N-sec-butyl sinefungin was slightly more potent with an IC50 of 1.8 ± 0.4 μM. It should be noted that the inhibitory activity of Pr-SNF was not determined against ASH1L, NSD1, or MMSET in previous studies.228 The direct binding of Pr-SNF to the MMSET SET domain was demonstrated by ITC. However, a cocrystal structure of the MMSET SET domain in complex with Pr-SNF could not be obtained. To date, no potent and selective MMSET inhibitors have been reported. We expect that the insights provided by the crystal structure of the MMSET SET domain, together with the existing structural knowledge gained by the NSD1 and SETD2 structures, would facilitate the development of selective inhibitors for these closely related SET domain-containing enzymes.
2.1.5. Inhibitors of H4K20 Methyltransferases
Methylation of H4K20 is catalyzed by the SUV420H1, SUV420H2, and SETD8 [SET domain containing (lysine methyltransferase) 8] protein methyltransferases in humans.238 The latter, also known as SET8, PR-SET7, and KMT5A, is the sole methyltransferase that catalyzes monomethylation of H4K20.238−240 Monomethylation of H4K20 has been associated with both activation and repression, and it has been implicated in regulating important biological processes, including the DNA damage response, DNA replication, and mitotic condensation.238,241,242 In addition to H4K20, SETD8 methylates many nonhistone substrates, including the tumor suppressor p5323 and proliferating cell nuclear antigen (PCNA).243 The monomethylation of p53 (K382) by SETD8 represses p53 target genes.23 Furthermore, the monomethylation of PCNA by SETD8 at K248 stabilizes the PCNA protein and increases the interaction between PCNA and the flap endonuclease FEN1, promoting the proliferation of cancer cells.243 SETD8 has been shown to be overexpressed in various types of cancer, including nonsmall cell and small cell carcinoma, hepatocellular carcinoma, pancreatic cancer bladder cancer, as well as chronic mylogenous leukemia. Therefore, as the sole enzyme capable of H4K20 monomethylation, there has been a growing interest in the development of selective, small-molecule inhibitors of SETD8 that can advance our understanding of its mechanism and disease associations.
The first reported inhibitor of SETD8 was a marine natural product, nahuoic acid A (Figure 16), which displayed an IC50 of 6.5 ± 0.5 μM in a biochemical assay.244 Nahuoic acid A was determined to be a competitive inhibitor with respect to the cofactor SAM (Ki = 2.0 ± 0.3 μM) and noncompetitive with respect to the peptide substrate. It was selective for SETD8 over 10 other methyltransferases. In a more recent report, nahuoic acid A and its derivatives were further characterized. Nahuoic acid A and its penta-acetate analogue inhibited the proliferation of several cancer cell lines with relatively poor potencies.245 For example, exposure of U2OS cells to nahuoic acid A or to its penta-acetate resulted in a decrease in cell proliferation with an IC50 of 65 ± 2 and 39 ± 4 μM, respectively. A similar effect was also observed in SUM159 (IC50 = 45 μM) and MDA-MB-436 (IC50 = 85 μM) breast cancer cell lines for the nahuoic acid A penta-acetate. Nahuoic acid A is the only SAM-competitive, selective SETD8 inhibitor known to date.
Figure 16.

Structures of SETD8 inhibitors.
In 2014, Ma and co-workers reported the first substrate-competitive, selective inhibitor of SETD8, UNC0379 (Figure 16).246,247 This small-molecule inhibitor was discovered by cross-screening approximately 150 quinazoline-based compounds against SETD8. UNC0379 displayed inhibitory activity against SETD8 with micromolar potency in multiple biochemical and biophysical assays. It was selective for SETD8 over 15 other methyltransferases, including G9a and GLP. Very recently, Veschi and co-workers have identified SETD8 as a crucial regulator of growth and differentiation in high-risk neuroblastoma (NB).248 Knockdown of SETD8 via siRNA rescued the proapoptotic and cell-cycle arrest functions of p53 by decreasing p53K382me1, leading to activation of the p53 canonical pathway in NB cells. Pharmacological inhibition of SETD8 by UNC0379 phenocopied SETD8 knockdown and effectively inhibited the proliferation of neuroblastoma cells in cellular and ex vivo models.
Butler and co-workers recently reported a more potent derivative of UNC0379.249 Drawing parallels from the discovery of G9a/GLP inhibitors, the installation of an aminoalkyl group to the 7-position of UNC0379 was proposed as a way to improve potency. Indeed, the installation of an aminoethyl group at this position resulted in a significant improvement in potency. The resulting inhibitor, MS2177 (Figure 16), displayed an IC50 of 1.9 μM in a biochemical assay. Direct binding of MS2177 to SETD8 was confirmed by ITC with a Kd of 1.3 μM, which was greater than that of UNC0379 (Kd = 18 μM). In MOA studies, MS2177 was competitive with the H4 peptide but noncompetitive with the cofactor SAM. A cocrystal structure of MS2177 in complex with SETD8 (PDB ID: 5T5G), which is the first crystal structure of SETD8 in complex with a small-molecule inhibitor, revealed important structural insights regarding the binding of the inhibitor to SETD8 (Figure 17A). The cocrystal structure also revealed that C311 is near the inhibitor binding site, presenting an opportunity to develop a covalent inhibitor of SETD8. Therefore, MS453 (Figure 16), an analog of MS2177 containing an electrophilic acrylamide group, was designed and synthesized to specifically react with the thiol group of C311. Indeed, it has been clearly demonstrated that MS453 covalently modified C311 but not other cysteine residues of SETD8 by MS-based analyses using wild-type, C311S, and other mutant SETD8. As expected for a covalent inhibitor, MS453 displayed time-dependent inhibition. It exhibited an IC50 of 804 nM following an extended preincubation period. The covalent modification of SETD8 by MS453 was not affected by the presence of 5 mM glutathione (GSH), which mimics cellular conditions. In addition, MS453 was incubated with other protein methyltransferases, such as PRC2, SMYD2, and SMYD3, and no covalent adduct was observed by MS analysis, suggesting that the covalent modification by MS453 is specific to SETD8. Furthermore, MS453 was selective for SETD8 over 29 other methyltransferases in biochemical assays. The crystal structure of MS453 in complex with SETD8 (PDB ID: 5TH7) confirmed that C311 was covalently modified by MS453 (Figure 17B). Interestingly, the cocrystal structure revealed that the inhibitor was flipped out from the active site and occupied the active site of the other subunit of a SETD8 homodimer. However, while cellular activities of MS2177 have not been reported, MS453 has poor cell membrane permeability and high efflux ratio, thus, is not suitable for cellular studies.
Figure 17.
(A) Co-crystal structure of MS2177 (orange) in complex with SETD8 (PDB ID: 5T5G). Hydrogen bonds are represented as magenta dashed lines and water molecule as red sphere. (B) Co-crystal structure of MS453 (green and cyan) in complex with SETD8 homodimer (subunits depicted in cyan and green, PDB ID: 5TH7).
Another report on irreversible, small-molecule inhibitors of SETD8 was published by Blum and co-workers.250 Screening of more than 5000 commercial compounds resulted in the discovery of three SETD8 inhibitors: SPS8I1 (also known as NSC663284, IC50 = 0.21 ± 0.03 μM), SPS8I2 (also as known as BVT948, IC50 = 0.50 ± 0.20 μM), and SPS8I3 (also known as ryuvidine, IC50 = 0.70 ± 0.20 μM) (Figure 18).250 The selectivity of these inhibitors were evaluated against SETD2, GLP, G9a, SMYD2, SETD7, PRMT1, PRMT3, and CARM1. SPS8I1 was only 2.5-fold selective for SETD8 over SMYD2 and >6-fold selective over other PMTs tested. Similarly, SPS8I2 showed modest selectivity over SETD2, G9a, SMYD2, CARM1, and PRMT3, while SPS8I3 was less selective. Given that all of these inhibitors shared a common quinone motif, which can react with cysteine residues, it was postulated that these compounds irreversibly inhibited SETD8. Indeed, SPS8I1 and SPS8I2 modified C311 of SETD8, while SPS8I3 targeted cysteine residues in a nonspecific manner. In HEK293T cells, treatment with the inhibitors resulted in reduction of the H4K20me1 mark, while other histone marks such as H4K20me2/3 and H3K9me were not altered. However, off-target effects on other PMTs (SPS8I1 for SMYD2 and SPS8I3 for PRMT3 and SETD2) and other cellular targets (SPS8I1: inhibition of Cdc25; SPS8I2: inhibition of cyclin-dependent kinase 4 and 2 (CDK4/2); and SPS8I3: inhibition of protein tyrosine phosphatase PTB1B) were reported. Overall, SPS8I1, SPS8I2, and SPS8I3 are irreversible, modestly selective inhibitors of SETD8 that showed some activity in cells.
Figure 18.

Structures of additional SETD8 inhibitors.
Recently, Judge and co-workers proposed that the replacement of K20 of the H4 peptide (16–23 residues) with a more hydrophobic residue could deliver a peptide inhibitor of SETD8 through a more energetically favorable interaction with the lysine-binding channel of SETD8.251 A modeling study based on the crystal structure of SETD8 in complex with the H4 peptide and SAH (PDB ID: 1ZKK) was conducted by manually substituting the side chain of K20 in the H4 peptide with various natural and unnatural amino acids. The substitution of the unnatural amino acid norleucine for K20 of the H4 (16–23) peptide resulted in a potent peptide inhibitor 3 (Figure 18) with a Ki of ∼50 nM and an IC50 of 0.33 μM. Further modifications, including additional residue substitutions in the N- or C-terminal regions and truncation of the N- and C-terminals of the norleucine peptide, did not improve potency. As expected, the norleucine-containing peptide is a substrate-competitive inhibitor. It also showed good selectivity for SETD8 over a panel of 32 methyltransferases. However, this peptide inhibitor is not cell-permeable and not suitable for cell-based studies.
SUV420H1 and SUV420H2 are two highly homologous methyltransferases that di- and trimethylate H4K20. Loss of H4K20me3 has been reported as a common hallmark of human cancer.252 A-196 (Figure 19) was recently discovered as the first and only potent, selective, and cell-active inhibitor of SUV420H1 and SUV420H2.253 A-196 inhibited SUV420H1 and SUV420H2 with IC50 values of 25 ± 5 and 144 ± 21 nM, respectively. Direct binding of A-196 to SUV420H1 was established by ITC in the presence and absence of the cofactor SAM. In MOA studies, A-196 inhibited SUV420H1 in a peptide-competitive manner. In addition, the cocrystal structure of SUV420H1 in complex with A-196 and SAM was determined, confirming the results of the MOA studies (PDB ID: 5CPR). A-196 was selective for SUV420H1 and SUV420H2 over other methyltransferases, epigenetic readers, chromatin binders and a broad range of nonepigenetic targets. Reduction of H4K20me2 and H4K20me3 levels and increase of H4K20me1 levels were observed throughout the cell cycle in cells treated with this inhibitor and no significant cell toxicity was observed. Therefore, A-196 is a valuable chemical probe of SUV4–20H1/2 that can be used to study biological functions of these enzymes in a cellular context.
Figure 19.
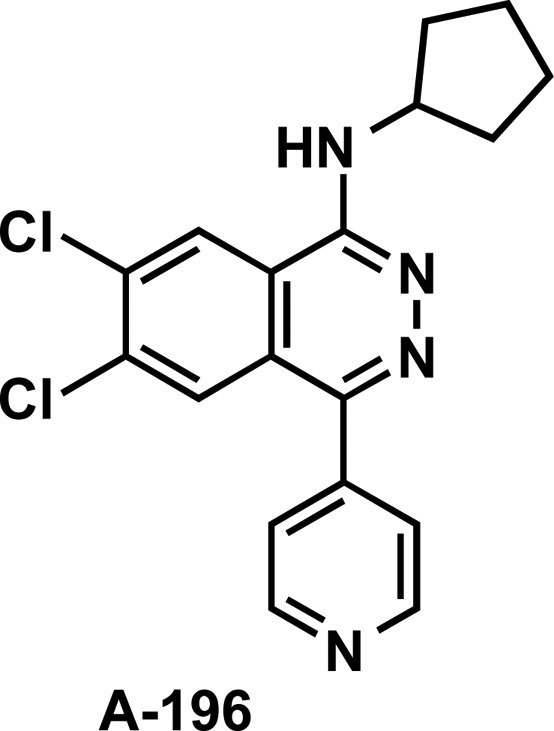
Structure of A-196, an inhibitor of SUV420H1 and SUV420H2.
2.1.6. Inhibitors of H3K79 Methyltransferases
DOT1L (disruptor of telomeric silencing 1-like, also known as KMT4) distinguishes itself from the other identified human PKMTs by the lack of the SET domain.254 DOT1L contains a non-SET catalytic domain, which adopts a folding topology that is also observed in PRMTs and DNMTs.255,256 It is therefore more closely related to these families of methyltransferases. It has been shown that DOT1L is responsible for mono-, di-, and trimethylation of H3K79.254,257 DOT1L has been thought to be the sole methyltransferase acting on H3K79; however, a recent report suggested that the MMSET isoform RE-IIBP (interleukin-5 response element II binding protein), which contains a SET domain, methylates this mark as well.258 In addition, no demethylase activity on H3K79 has been reported to date. Methylation of H3K79, which is generally correlated with transcriptional activation, has been associated with transcriptional regulation, DNA repair, embryonic development, cell cycle regulation, hematopoiesis, and cardiac function.259−261 It has also been reported that DOT1L interacts with AF4, AF9, AF10, AF6, and ENL, the most commonly seen MLL fusion proteins in MLL-rearranged leukemias.262−266 DOT1L interacts with these MLL fusion proteins and is recruited to their target genes, including HOXA9 and MEIS1 and are critical for leukemia.259 These interactions result in abnormal methylation that drives leukemogenesis. Therefore, DOT1L has been studied increasingly as a potential therapeutic target for the treatment of MLL-rearranged leukemia.7,267
In 2011, Daigle and co-workers reported the first selective DOT1L inhibitor, EPZ004777, with very high in vitro potency (IC50 = 400 ± 100 pM) (Figure 20).268 EPZ004777 was designed and synthesized based on the cofactor SAM and the crystal structure of the enzyme active site. It was more than 1000-fold selective for DOT1L over 9 other methyltransferases, despite its structural similarity to the cofactor SAM. As expected, in MOA studies, EPZ004777 was competitive with SAM and noncompetitive with the peptide substrate. It also exhibited very high binding affinity (Ki = 300 pM).269 In December 2012, Yu and co-workers reported the cocrystal structure of the DOT1L-EPZ004777 complex, revealing the remodeling of the catalytic site (Figure 21, panels A–C),270 consistent with the ligand-induced conformational adaptation reported by Basavapathruni and co-workers.269 A global reduction in H3K79me2 levels was observed in several leukemia cell lines treated with EPZ004777, while no significant reduction of other histone methylation marks was observed, suggesting cellular specificity for EPZ004777.268 Furthermore, EPZ004777 displayed a concentration-dependent inhibition of the expression of HOXA9 and MEIS1, overexpression of which is considered a hallmark of MLL-rearranged leukemia.268 In addition, it displayed an antiproliferative effect at low micromolar potencies in MLL-rearranged leukemia cell lines but was essentially ineffective in non-MLL-rearranged leukemia cell lines. Importantly, EPZ004777 was utilized to show for the first time that pharmacological inhibition of the methyltransferase activity of DOT1L had antitumor activity in animal models of MLL-rearranged leukemia.268,271
Figure 20.

Structures of DOT1L inhibitors.
Figure 21.

(A) Cofactor binding site of DOT1L (PDB ID: 1NW3). (B) Crystal structure of DOT1L in complex with EPZ004777 (PDB ID: 4ER3). Conformational rearrangements of DOT1L create a cavity to accommodate the t-butylphenyl group. (C) Overlay of DOT1L-SAM and DOT1L-EPZ004777 showing conformational rearrangement of substrate-binding and activation loop residues (magenta and yellow, respectively). (D) Overlay of DOT1L-EPZ004777 (PDB ID: 4ER3) and DOT1L-EPZ-5676 (PDB ID: 4HRA) complexes. Key hydrogen bonds are represented as green dashed lines between EPZ004777 (blue) and DOT1L and EPZ-5676 is depicted in orange.
On the basis of the cocrystal structure of the DOT1L-EPZ004777 complex, SGC0946 (Figure 20), a chemical probe of DOT1L with improved in vitro and cellular potencies, was developed by exploiting the hydrophobic cleft near the 7-position of the deazaadenosine of EPZ004777. SGC0946, which contains a bromo substitution at the 7-position of the deazaadenosine ring, was more potent than EPZ004777 in biochemical and biophysical assays (e.g., Kd = 0.06 nM versus 0.25 nM in SPR) with a similar selectivity profile.270 SGC0946 was also almost 10-fold more potent at reducing H3K79 methylation levels (IC50 = 8.8 ± 1.6 nM) than EPZ004777 (IC50 = 84 ± 20 nM) in MCF10A cells.
Another major advancement in the discovery of DOT1L inhibitors came in 2013, when EPZ-5676 was disclosed by Daigle and co-workers (Figure 20).272 EPZ-5676 (also known as pinometostat) was the first PMT inhibitor advanced to the clinic, the first major breakthrough in the PMT inhibitor field. Phase 1 clinical trials for this inhibitor were recently completed for the treatment of patients with MLL-r, a genetically defined acute leukemia.273,274
The cocrystal structure of EPZ-5676 in complex with DOT1L (PDB ID: 4HRA) confirmed the binding of the inhibitor to the cofactor-binding site as well as the ligand-induced conformational changes in DOT1L (Figure 21). EPZ-5676 inhibited DOT1L with a Morrison Ki of 0.08 ± 0.03 nM, a higher potency than EPZ004777 (Morrison Ki = 0.3 ± 0.02 nM). It exhibited remarkable selectivity for DOT1L (>37000-fold) over 16 other methyltransferases. EPZ-5676 reduced H3K79me2 levels in MV4–11 cells (an acute leukemia cell line expressing MLL-AF4) with an IC50 of 3 nM and in HL-60 cells (a non-MLL-rearranged cell line) with a similar potency.272 Treatment with EPZ-5676 resulted in greater than 90% reduction of H3K79me2 levels in 3–4 days as well as reduction of H3K79me1 levels. EPZ-5676 did not significantly reduce other common histone methylation marks, thus confirming the high selectivity of the compound in biochemical assays. Moreover, EPZ-5676 inhibited HOXA9 and MEIS1 mRNA levels in MV4–11 cells in a concentration-dependent manner. The antiproliferative effect of EPZ-5676 in MV4–11 cells (IC50 = 3.5 nM) was observed as early as 4 days but reached a maximum at day 7. This delayed onset of activity was attributed to a cascade of epigenetic events, including depletion of the H3K79me2 mark, inhibition of MLL-fusion target gene expression, and reversal of leukemogenic gene expression.268,275,276 In addition, EPZ-5676 exhibited nanomolar antiproliferative activity against most of the other MLL-rearranged cell lines that were tested but displayed weaker potencies against non-MLL-rearranged cell lines. Importantly, the complete elimination of established subcutaneous (SC) MV4–11 tumors in immunocompromised rats was achieved via continuous intravenous (IV) infusion of EPZ-5676 dosed at 70 mg/kg per day for 21 days. The tumor regression was sustained for more than 30 days after the termination of the compound treatment. All doses were well-tolerated by the test animals. Furthermore, H3K79me2 levels, as well as HOXA9 and MEIS1 mRNA levels, were significantly reduced in MV4–11 SC xenograft tissue harvested from rats dosed by continuous IV infusion for 14 days. Taken together, these results suggest that EPZ-5676 displayed on-target activity in vivo with minimal toxicity. Recently, PK properties of EPZ-5676 in rats, dogs, and humans were also reported.277
Several other inhibitors of DOT1L have also been reported in the literature. For example, shortly after the publication of EPZ004777, Yao and co-workers reported the discovery of a selective, covalent inhibitor of DOT1L.278 Compound 4 (Figure 20) had high in vitro potency for DOT1L (IC50 = 38 nM) and was >29-fold selective for DOT1L over CARM1, PRMT1, G9a, and SUV39H1. It was postulated that compound 4 would undergo an intramolecular cyclization to form a reactive aziridinium intermediate, which would in turn react with the ε-NH2 group of lysine 79 to covalently link to H3K79. In addition, the NH2 of the adenosine moiety (6-NH2) forms only one hydrogen bond with the enzyme, as opposed to SET domain-containing PKMTs, such as G9a, which form two hydrogen bonds with the 6-NH2 group. Analysis of the cocrystal structure of the DOT1L-SAM complex also revealed that a larger hydrophobic binding pocket exists for this amino group. Therefore, 6-N-methyl SAH was subsequently synthesized and was determined to be a highly selective inhibitor of DOT1L (Ki = 290 nM), achieving excellent selectivity over CARM1, PRMT1, G9a, and SUV39H1 (Ki > 20000 nM).278 The activities of these DOT1L inhibitors in cell-based assays were not reported. The same research group performed extensive SAR studies and reported the discovery of compound 5 (Figure 20) in 2012.279 Compound 5 had very high in vitro potency (Ki = 0.46 nM) and was >4500-fold selective for DOT1L over CARM1, PRMT1, and SUV39H1. Again, adding an alkyl group such as methyl, allyl, or benzyl group onto the 6-amino group (Ki = 0.76, 12, and 22 nM, respectively) was tolerated and led to high selectivity. Compound 5 inhibited the proliferation of MV4–11 cells with a slow onset of activity but did not affect the proliferation of wild-type MLL NB4 cells. Two other studies explored the 5-position of the adenosine moiety of SAH and led to the discovery of DOT1L selective inhibitors Br-SAH and CN-SAH.280,281
Yi and co-workers developed a set of assays, including a cell-based imaging assay to assess chemical tools for DOT1L in a miniaturized format.282 Affinity ligands were designed for the development of these assays. With the use of these assays and structural information, several inhibitors with increased cellular potency (IC50 values ∼10 nM) and excellent selectivity for DOT1L were identified. With the improved potency in the cellular assay, the relative potencies of these compounds were evaluated by immunoblotting for the H3K79me2 mark in MLL cells (MOLM-13). The only mark affected by these compounds was H3K79me2. Decreased expressions of MLL target genes, HOXA9 and MEIS1, were observed after 7 days of incubation with these inhibitors. These measurements also correlated with an antiproliferative effect in treated MV4–11 cells. Overall, this study resulted in the discovery of compounds more potent than EPZ004777. Recently, another study described additional affinity probes of DOT1L and used them to identify potential off-targets of SAM-based inhibitors.283
In June 2016, Scheufler and co-workers reported a new series of DOT1L inhibitors that differ structurally from all previously published SAM-based inhibitors.284 These new inhibitors interact with an induced pocket adjacent to the SAM binding site but do not bind the SAM binding site. This new class of DOT1L inhibitors was obtained by optimization of a weak fragment-based screening hit (IC50 = 320 μM) that displayed suboptimal interactions in the induced binding pocket. After careful analysis of the cocrystal structure of this initial hit and elegant fine-tuning of the interactions in the induced pocket, a highly potent inhibitor of DOT1L, 6 (Figure 22), was discovered, displaying an IC50 of 14 nM in a biochemical assay. These inhibitors were identified as SAM-competitive because, upon binding, they engage the lid loop of the SAM binding pocket and form a conformation incompatible with SAM binding, based on the cocrystal structures of DOT1L in complexes with the compounds developed in this study. Compound 6 constitutes the first DOT1L inhibitor with a chemotype that is distinctly different from SAM derivatives and interacts at a site different than the SAM binding site. In a subsequent report, the same research group described another structurally novel DOT1L inhibitor series that targets the same induced pocket.285 HTS, followed by hit optimization guided by structure-based design, resulted in the discovery of potent DOT1L inhibitors 7 and 8 (Figure 22), which displayed very high potencies (IC50 values of 1.4 and 0.4 nM and Ki values of 0.36 and 0.08 nM, respectively) in biochemical assays. Compounds 7 and 8 displayed selectivity against a panel of 22 PKMTs and PRMTs, showing no inhibitory activity at up to 50 μM. Both compounds also exhibited long residence times, as assessed by surface plasmon resonance experiments (τ = 43 min for 7 and >240 min for 8 (the detection limit of the SPR assay used). Importantly, 7 and 8 potently decreased H3K79me2 levels (IC50 = 23 and 16 nM, respectively) and reduced the activity of the HoxA9 promoter (IC50 = 384 and 340 nM, respectively) in cellular assays. Moreover, both compounds efficiently inhibited the proliferation of MV4–11 cells carrying the oncogenic MLL-AF4 fusion with IC50 values of 85 and 128 nM, respectively. In addition, 7 was evaluated in PK experiments in rats and showed a high total blood clearance, a high volume of distribution, a moderate half-life, and oral bioavailability. Overall, these two novel series (compounds 6 and 7/8) of potent, selective, SAM-competitive DOT1L inhibitors are exciting. Compounds 7 and 8 are useful chemical tools for cellular and in vivo studies. In addition, a recent report described the identification of another non-SAM scaffold with IC50 values in the micromolar range for DOT1L by virtual screening.286
Figure 22.

Structures of recently reported DOT1L inhibitors.
2.2. Protein Arginine Methyltransferases
Protein arginine methylation is another significant and widely observed PTM in eukaryotic cells.287−289 Arginine is able to form five potential hydrogen bonds with surrounding hydrogen bond acceptors via its guanidinium group. Therefore, every methylation of arginine prevents a potential hydrogen bond, creating steric bulkiness and increasing hydrophobicity. Furthermore, methylation does not neutralize the cationic charge of arginine residues and could enhance their interactions toward aromatic rings via cation-pi interactions.290 The methylation of arginine residues is very important since protein recognition and in turn its physiological functions can be altered.291
Nine PRMTs have been identified to date, and they are responsible for mono- and/or dimethylation of the guanidino group of arginine residues.287 The two possible ways for dimethylation after monomethylation of arginine (MMA) are shown in Figure 2. Methylation of the same nitrogen yields asymmetrically dimethylated arginine (aDMA) or methylation of another nitrogen gives symmetrically dimethylated arginine (sDMA). On the basis of their methylation functions, PRMTs are divided into 3 subcategories: type I, type II, and type III PRMTs.292 Type I PRMTs, which include PRMT1, PRMT2, PRMT3, CARM1 (coactivator-associated arginine methyltransferase 1, also known as PRMT4), PRMT6, and PRMT8, catalyze monomethylation and asymmetric dimethylation of arginine residues. PRMT5 and PRMT9 are type II PRMTs, which catalyze monomethylation and symmetrical dimethylation of arginine residues.293 PRMT7 is the only known type III PRMT as it catalyzes arginine monomethylation only. PRMT 10 and PRMT11 have been identified as putative PRMT genes, but no methylation activity has been shown yet.294,295
All PRMTs contain a conserved core region of about 310 amino acids.296 They typically have additions to the N-terminal. CARM1, on the other hand, also has C-terminal additions.28 The monomeric structure of the PRMT core comprises a methyltransferase domain, a β-barrel296 that is unique to PRMTs, and a dimerization arm. Type I PRMTs adopt a head-to-tail homodimeric structure. In the homodimer, the dimerization arm that extends out of the β-barrel of one monomeric subunit interacts with the Rossman fold of another subunit.296,297 Moreover, a dynamic α-helix (consisting of two segments: α-X and α-Y) at the N-terminus of the Rossman fold bends on the bound SAM like a lid and also participates in proper positioning of the peptide substrate.296,298 PRMTs generally methylate glycine and arginine-rich (GAR) motifs in their substrates289 with the exception of CARM1, which specifically methylates proline-, glycine-, and methionine-rich (PGM) motifs.299,300 PRMT5, on the other hand, can symmetrically dimethylate both of these motifs.301 PRMTs methylate nonhistone proteins as well as histones.287,291,302,303 Dysregulation of PRMTs and arginine methylation have been implicated in cancer and other diseases.287,292
2.2.1. Inhibitors of PRMT1
PRMT1 was the first mammalian protein arginine methyltransferase identified304 and is responsible for most of the type I arginine methyltransferase activity in mammalian cells.305 PRMT1 catalyzes asymmetric dimethylation of H4R3 (H4R3me2a), which is associated with transcriptional activation.306,307 PRMT1 also methylates nonhistone substrates, including DNA repair proteins MRE11,308 p53 binding protein 1 (53BP1),309 ASH2L,310 and the tumor suppressor BRCA1.311 Therefore, PRMT1 has been implicated in numerous cellular processes, including transcription, RNA processing, and signal transduction.292 In addition, PRMT1 has been linked to human telomeres312 and shown to directly regulate the AKT signaling pathway.313,314 The alternative splicing of PRMT1 has been well-studied, and seven PRMT1 isoforms (PRMT1v1– v7) have been found by alternative splicing in the 5′ region of its pre-mRNA. Of these isoforms, PRMT1v1 is the most abundant variant.315 Overexpression and aberrant splicing of PRMT1 have been implicated in diseases, including breast,316,317 prostate,318 colon,319,320 lung, and bladder cancers.321 The overexpression of PRMT1v1 has been observed in colon cancer,319 whereas PRMT1v2 has been upregulated in breast cancer and, in turn, could promote the survival and invasiveness of breast cancer cells.316 Furthermore, PRMT1 is crucial for the development of AML322 and is part of an MLL transcriptional complex.323
In 2016, inspired by the recent discoveries of potent and selective PRMT6 and CARM1 inhibitors (see below), Eram and co-workers developed MS023 (Figure 23), a selective inhibitor of type I PRMTs, based on the hypothesis that the ethylenediamino group of MS023 is an excellent arginine mimetic and a critical moiety for targeting type I PRMTs.324 In biochemical assays, MS023 was highly potent for PRMT1 (IC50 = 30 ± 9 nM), PRMT3 (IC50 = 119 ± 14 nM), PRMT4 (IC50 = 83 ± 10 nM), PRMT6 (IC50 = 4 ± 0.5 nM), and PRMT8 (IC50 = 5 ± 0.1 nM). Importantly, it was inactive against all type II and type III PRMTs, 25 PKMTs, and DNMTs, and other epigenetic modifiers including histone lysine demethylases and various methyllysine and methylarginine reader proteins. Direct binding of MS023 to PRMT6 was confirmed by ITC with high affinity (Kd = 6 nM) and DSF (ΔTm = 20 °C). In addition, an X-ray cocrystal structure of MS023 in complex with PRMT6 was obtained and revealed that the inhibitor occupies the substrate-binding site and the ethylenediamino group of MS023 indeed serves as an arginine mimetic (PDB ID: 5E8R). Importantly, MS023 potently inhibited PRMT1-mediated asymmetric dimethylation of H4R3 in cells. It also reduced global levels of arginine asymmetric dimethylation and concurrently increased arginine monomethylation and symmetric dimethylation in cells. A close analog of MS023 that was inactive in biochemical and cellular assays was also developed as a negative control for future chemical biology studies. Overall, MS023 and its negative control are valuable chemical tools for the biomedical community to investigate the role of type I PRMTs in health and disease.
Figure 23.

Structures of type I PRMT inhibitor MS023 and reported PRMT1 inhibitors.
In 2004, Cheng and co-workers reported the discovery of AMIs (arginine methyltransferase inhibitors) by HTS of a 9000-compound library.325 Among the nine hits identified, only AMI-1, a symmetric sulfonated urea salt, and AMI-6 showed activity for PRMTs (Figure 23). These compounds were selective for PRMT3, CARM1, and PRMT6 over PRMT5 and a series of PKMTs.325 AMI-1 inhibited PRMT1 with an IC50 of 8.8 μM and was shown to be noncompetitive with the cofactor SAM. Therefore, it was proposed to bind in the substrate-binding pocket in a substrate-competitive manner. It was also reported that AMI-1 inhibited the methylation of Npl3p in HeLA cells in a concentration-dependent manner. In 2007, Ragno and co-workers disclosed structure- and ligand-based modeling studies that focused on AMIs and their analogs confirming AMI-5 (Figure 23) as a PRMT1 inhibitor (IC50 = 1.4 μM). The selectivity of these inhibitors, however, was not reported.326 In 2010, Feng and co-workers reported the discovery of NS-1 (naphthalene-sulfo derivative 1) via virtual screening of 400000 compounds (Figure 23).327 NS-1 inhibited PRMT1 with an IC50 of 13 ± 0.1 μM and was substrate-competitive with a Ki of 1.7 ± 0.54 μM. While NS-1 did not inhibit CARM1, it did inhibit PRMT3 and PRMT6 with similar potencies. Interestingly, Feng and co-workers determined that AMI-1, NS-1, and related naphthalenesulfonate derivatives directly targeted the peptide substrates (i.e., H4 and GAR peptides) instead of PRMT1. This mechanism of action was believed to be largely responsible for the observed PRMT1 inhibition by way of preventing the recognition of substrates by the enzyme.327
Spannhoff and co-workers applied a target-based approach328 to discover new PRMT inhibitors. As a result, Stilbamidine and Allantodapsone were reported as PRMT1 inhibitors, with IC50 values of 57 ± 6.2 and 1.7 ± 3 μM, respectively.329 After virtual screening, the same research group reported another PRMT1 inhibitor, RM65 (Figure 23), which possessed an IC50 of 55 ± 3.4 μM.330 A similar virtual screening approach was also reported by Heinke and co-workers in search of a different chemotype.331 Hit optimization resulted in the generation of inhibitors with low micromolar potency against PRMT1, which were not fully characterized.332 A recent virtual screening study also resulted in the discovery of PRMT1 inhibitors with IC50 values around 20 μM.333 It is important to note, however, that all of these PRMT1 inhibitors lack extensive selectivity and characterization data, thereby limiting their use as selective PRMT1 inhibitors.
In 2010, Bonham and co-workers discovered compound 9 (Figure 23), which inhibited PRMT1 with an IC50 of 4.2 ± 1.6 μM and CARM1 with an IC50 = 2.6 ± 0.6 μM.334 Although it did not inhibit SETD7, this compound was not selective over PRMT5 and PRMT6. In 2011, Dowden and co-workers reported a SAM derivative, 10 (Figure 23), as a PRMT1 inhibitor with an IC50 of 3.9 ± 1.8 μM. This compound was inactive against CARM1 and SETD7.335
In 2008, Osborne and co-workers, in search of PRMT1 inhibitors, incubated a SAM derivative, 5′-(diaminobutyric acid)-N-iodoethyl-5′-deoxyadenosine ammonium hydrochloride (AAI)336 (Figure 23) and acetylated H4–21 peptide with PRMT1 and showed that the peptide substrate was covalently modified with comparable kinetics to H4.337 Control experiments in the absence of PRMT1 did not result in any modification, indicating that PRMT1 promotes the transfer of this SAM-like moiety to the peptide substrate. When the H4–21R3K mutant peptide is used instead of the H4–21 peptide, no modification was observed, indicating that R3 is the targeted residue and the PRMT1-directed modification is regiospecific. Extended incubation time did not improve the yield of the modified peptide. The lack of time dependence is therefore attributed to the generation of a bisubstrate inhibitor. Further experiments established that PRMT1 activity was inhibited when preincubated with AAI with similar potency to SAH. The observed IC50 (18.5 ± 4.2 μM) was ∼10-fold lower than that of AMI-1. Further studies indicated that the AcH4–21-AAI bisubstrate inhibitor preferentially inhibits PRMT1 over CARM1 (4.4-fold). The ability of PRMT1 to catalyze the transfer of AAI to a peptide substrate and in the process generate a bisubstrate inhibitor provided a proof of concept for the chemoenzymatic generation of PRMT-targeted bisubstrate analogues. It was proposed that AAI would undergo an intramolecular cyclization to form a reactive aziridinium intermediate, which would then react with the guanidinium group of R3 of AcH4–21 to covalently link to peptide. It should be noted that the same inhibition mechanism was later suggested for DOT1L inhibitor compound 4 (Figure 20) by Yao and co-workers in 2011.278
In 2010, inspired by the earlier work on chloroacetamidine-based protein arginine deiminase (PAD) inactivators,338 Obianyo and co-workers reported C21, a 21-residue peptide featuring a chloroacetamidine warhead (Figure 23) as a covalent PRMT1 inhibitor.339 The peptide sequence of C21 is based on the N terminus of histone H4. Further examination revealed that C21 inhibits PRMT1 (IC50 = 1.8 ± 0.1 μM) in a time- and concentration-dependent manner with the kinact/KI value of 4.6 × 106 min–1 M–1 (kinact = 3.1 ± 0.2 min–1, KI = 0.8 ± 0.4 μM) (please see section 3.1.2 for more detailed discussion on these constants). Although the specific residue modified by this compound was not clearly identified in this study, it was later speculated that the active residue was C101.340 Inhibitor C21 was selective for PRMT1 over CARM1 (>250-fold), PRMT3 (>250-fold), and PRMT6 (4.9-fold), based on IC50 values. Given that haloacetamidine-containing compounds also inhibit PADs, the kinact/KI value for PAD4 was measured. C21 inactivates PAD4 with kinact/KI of 300 min–1 M–1, showing >15000-fold selectivity. In addition, it was demonstrated that C21 inhibits PRMT1 in cellular assays using 293T cells. Following up this study, the same research group developed activity-based proteomic probes (ABPs) that selectively modify PRMT1. Fluorescein-conjugated C21 and biotin-conjugated C21 were used as PRMT1-specific ABPs and provided the first evidence that PRMT1 activity is negatively regulated in a spatial and temporal fashion.341
In 2012, Dillon and co-workers reported the discovery of covalent inhibitors CID 5380390 and CID 2818500 (Figure 23), which inhibited PRMT1 and PRMT8.340 The inhibition of PRMT1 and PRMT8 was thought to be related to the presence of a cysteine residue (C101) in the SAM binding site of PRMT1 and PRMT8, which is absent in the SAM binding site of other PRMTs.340 These inhibitors were inactive against CARM1 and SETD7. In the same year, Wang and co-workers employed pharmacophore-based virtual screening methods and discovered A36 (Figure 23), a substrate-competitive inhibitor of PRMT1, which displayed an IC50 of 12 ± 0.2 μM.342 A36 was found to be 7-fold selective for PRMT1 over CARM1 but only 2-fold selective for PRMT1 over PRMT5.
In 2014, Yan and co-workers reported compound DB75 (Figure 23), a diamidine-containing PRMT1 inhibitor with an IC50 of 9.4 ± 1.1 μM.343 It displayed selectivity for PRMT1 over CARM1 (>42-fold), PRMT5 (around 18-fold), and PRMT6 (around 30-fold). It was also found to inhibit the proliferation of several leukemia cell lines. Interestingly, cell lines derived from Down syndrome patients and leukemia patients (CMY, CHRF-288-1, and MOLM-13 cells) appeared to display enhanced sensitivity to this compound compared to the other cell lines tested (HEL, Jurkat, and HL-60). More recently, this same research group investigated the SAR of cyanine structures. This study led to the identification of a pentamethine compound, E-84 (Figure 23), as a PRMT1 inhibitor with an IC50 of 3.4 μM.344,345 Furthermore, E-84 displayed a 6-fold selectivity over CARM1, a 10-fold selectivity over PRMT5, and a 25-fold selectivity over PRMT8.344,345 Fluorescence intensity was measured, and a 6-fold increase was observed upon E-84 binding to PRMT1, with a calculated Kd of 2.3 μM, suggesting a direct interaction between the inhibitor and PRMT1. In cell-based studies, E-84 was found to inhibit leukemia cell growth at 100 nM in Meg01 and MOLM13 cells and at 200 nM in HEL cells.
Recently, a series of nitropyrimidine-containing compounds were reported, and follow-up optimization led to the discovery of a PRMT1 inhibitor with an IC50 of 2.0 μM.346 This compound was inactive against PRMT5 and PRMT6 (IC50 > 100 μM) but inhibited CARM1 with an IC50 of 10 μM. However, biophysical assays indicated that the inhibitor did not display characteristics of direct binding. Finally, in cell-based studies, this compound exhibited antiproliferative activity against three tumor cell lines (DLD-1, T24, and SH-SY-5Y) with IC50 values in the micromolar range.
It is important to note that, despite a growing interest in the discovery of PRMT1 selective inhibitors, most of the PRMT1 inhibitors described above lack thorough characterization in biochemical and biophysical assays regarding their selectivity, thus limiting their potential use in functional studies.
2.2.2. Inhibitors of PRMT3
PRMT3 (protein arginine methyltransferase 3) is a type I PRMT that catalyzes the mono- and asymmetric dimethylation of arginine residues. PRMT3 contains a zinc finger domain at its N-terminus and was first reported in 1998.347 It was shown to be a cytosolic protein, primarily methylating the 40S ribosomal protein S2 (rpS2).348,349 Asymmetric dimethylation of rpS2 by PRMT3 results in stabilization of rpS2 and impacts ribosomal biosynthesis.348−351 Recently, it was shown that in cells treated with palmitic acid or T0901317 [a liver X receptor α (LXRα) agonist], PRMT3 colocalizes with LXRα in the cell nucleus, regulating hepatic lipogenesis. However, this effect is considered to be independent of PRMT3’s methyltransferase activity. PRMT3, as well as PRMT1, have also been described to methylate the recombinant mammalian nuclear poly(A)-binding protein (PABPN1) and have been connected to oculopharyngeal muscular dystrophy, which is a consequence of polyalanine expansion in PABPN1.352−354 PRMT3 also methylates a histone peptide (H4 1–24) in vitro.355 The protein complex that includes PRMT3, the von Hippel–Lindau (VHL) tumor suppressor, and ARF (alternative reading frame) has been shown to methylate the tumor suppressor p53.356 Furthermore, the interaction of PRMT3 with the tumor suppressor DAL-1 (differentially expressed in adenocarcinoma of the lung-1) resulted in the inhibition of its methyltransferase activity. Consequently, it was proposed that DAL-1, the downregulation of which has been associated with a number of cancers,357−359 might be affecting tumor growth by regulating PRMT3 function.360 PRMT3 function has also been reported to be crucial for dendritic spine maturation in rats.361 Moreover, PRMT3 overexpression was observed in myocardial tissues from patients with atherosclerosis.362
In 2012, Siarheyeva and co-workers reported the discovery of the first selective PRMT3 inhibitor, compound 11 (Figure 24A), via screening of a diverse library of 16000 compounds.298 Compound 11 inhibited PRMT3 with an IC50 of 1.6 ± 0.3 μM and was selective for PRMT3 over a number of other methyltransferases. One of the most interesting findings in this report was that this inhibitor was noncompetitive with both the cofactor SAM and the peptide substrate, thereby suggesting an allosteric mechanism of inhibition. The cocrystal structure of 11 in complex with PRMT3 confirmed that 11 is an allosteric inhibitor (PDB ID: 3SMQ). It occupies a novel allosteric binding site located at the interface of the two subunits of the PRMT3 homodimer (Figure 24B). This was the first example of an allosteric inhibition of a protein methyltransferase by a small molecule. The cocrystal structure revealed that the cyclohexenyl moiety of the inhibitor interacts with the α-Y segment of the activation helix of the opposite subunit. This interaction most likely leads to the α-X segment becoming disordered. Since it has been shown that the proper folding of the α-X segment is essential for binding of both cofactor and substrate, it is likely that the binding of 11 to the allosteric site prevents the proper positioning or folding of the α-X segment, thus resulting in the inhibition of PRMT3 enzymatic activity.
Figure 24.

(A) Structures of PRMT3 inhibitors 11, 12, and 13. (B) Key interactions of 11 with PRMT3 allosteric pocket (3SMQ). (C) Structure of SGC707. (D) Co-crystal structure of PRMT3 in complex with SGC707 (PDB ID: 4RYL).
Other key ligand–protein interactions revealed by the cocrystal structure include: (1) a hydrogen bond between the middle nitrogen of the tightly fit benzothiadiazole moiety with the hydroxyl group of T466, (2) two hydrogen bonds between the two nitrogens of the central urea moiety and the carboxylate group of E422, and (3) a hydrogen bond between the oxygen of the urea moiety with the guanidinium group of R396 (Figure 24B). The key hydrogen bond interactions were confirmed by SAR studies as well as site-directed mutagenesis studies. Subsequent SAR studies by Liu and co-workers resulted in the discovery of more potent inhibitors such as 12 and 13 (IC50 = 0. 48 and 0.23 μM, respectively).363 Furthermore, a scaffold hopping exercise was employed to replace the left-hand side benzothiadiazole moiety and resulted in the identification of the isoquinoline moiety as a preferred benzothiadiazole replacement.364 Various amides, ketones, and bicyclic heterocycles were extensively investigated as the right-hand side moiety. These modifications resulted in the discovery of SGC707 (Figure 24C), the most potent PRMT3 inhibitor to date with an IC50 of 31 ± 2 nM in a biochemical assay.364 Binding of SGC707 to PRMT3 was confirmed by ITC and SPR, with Kd values of 53 ± 2 nM and 85 ± 1 nM, respectively. Importantly, SGC707 was selective for PRMT3 over 31 other methyltransferases, as well as a broad range of nonepigenetic targets including more than 250 kinases, GPCRs, ion channels, and transporters. As expected, in MOA studies, SGC707 was noncompetitive with both the cofactor and peptide substrate, consistent with an allosteric inhibition mechanism. This MOA was confirmed by the cocrystal structure of the PRMT3-SGC707 complex (PDB ID: 4RYL), which clearly showed binding of SGC707 to the same allosteric site of PRMT3 mentioned earlier (Figure 24D). Similar to the benzothiadiazole group, the isoquinoline group forms a hydrogen bond with T466. The urea group of SGC707 also forms three hydrogen bonds with E422 and R396 side chains, and the pyrrolidine amide of SGC707 is buttressed against the α-helix of the other PRMT3 subunit. In addition, XY1, a naphthyl analog of SGC707 that lacks the key hydrogen bond with T466, was developed as an inactive control (IC50 > 100 μM) for chemical biology studies.
The cellular target engagement of SGC707 was assessed using an InCELL Hunter assay, which measures the intracellular binding of SGC707 to the methyltransferase domain of PRMT3 in cell lines expressing the methyltransferase domain of PRMT3 tagged with a short fragment of β-galactosidase. Indeed, SGC707 stabilized PRMT3 in both HEK293 and A549 cells with EC50 values of 1.3 and 1.6 μM, respectively. The effect of SGC707 on H4R3 asymmetric dimethylation in cells was investigated. Due to the relatively slow turnover of methylated arginine residues, the overexpressed human Flag-tagged PRMT3 was used and the methylation of both endogenous H4 and exogenously introduced GFP-tagged H4 was examined. Overexpressed PRMT3 increased the endogenous levels H4R3me2a from the baseline, and treatment with SGC707 was able to mitigate this increase with an IC50 of 225 nM. The asymmetric dimethylation of exogenous H4R3 was also inhibited (IC50 = 91 nM), indicating a potent cellular effect of SGC707. These results clearly indicate that SGC707 can engage PRMT3 and effectively inhibit its catalytic activity in cells. In addition, SGC707 displayed no toxicity in cell growth assays. In mouse PK studies, intraperitoneal injection of SGC707 at 30 mg/kg resulted in good plasma exposure over 6 h. This dose was well-tolerated. Thus, SGC707 was bioavailable and can be used for animal studies in addition to cellular studies. In summary, SGC707 is a potent, selective, cell-active allosteric inhibitor of PRMT3. SGC707 and its negative control XY1 comprise a pair of valuable chemical tools for elucidating biological functions and disease associations of PRMT3.
2.2.3. Inhibitors of CARM1
Co-activator-associated arginine methyltransferase 1 (CARM1, also known as PRMT4) was first identified as a steroid receptor coactivator and was the first member of the PRMTs to be associated with transcriptional regulation.365,366 It is responsible for the asymmetric dimethylation of H3R17 and H3R26, though it prefers the former to the latter.367,368 In addition to histones, CARM1 methylates a variety of nonhistone proteins, such as CBP/p300, PABP1, HuR, HuD, SRC-3, NCOA2, CA150, SAP49, SmB, and U1C.369−374 CARM1 has been shown to play a role in mRNA splicing,369 RNA processing and stability,375 cell cycle progression,376 and DNA damage response.377 Furthermore, the loss of CARM1 results in neonatal lethality evidenced by the fact that newborn knockout mice die shortly after birth.375 CARM1 functions as a coactivator for various proteins that have been linked to cancer, including p53, NF-κB, β-catenin, E2F1, and steroid hormone receptor ERα.378−380 In addition, CARM1 levels have been shown to be elevated in castration-resistant prostate cancer,381,382 aggressive breast tumors,376 and lung cancer.383 Given its association in a wide variety of biological processes and diseases,292 CARM1 has been pursued as a potential therapeutic target.
Several HTS campaigns and SAR studies resulted in the identification of pyrrazole-amide- and benzo[d]imidazole-containing scaffolds as CARM1 inhibitors.384−388 Further optimization of these hits led to the discovery of potent CARM1 inhibitors 14 and 15 (Figure 25) with IC50 values of 27 nM and 30 nM, respectively.389 These inhibitors displayed selectivity for CARM1 over PRMT1 and PRMT3. The selectivity over other PRMTs, however, was not reported. In addition, the inhibitors were either not tested in cells or lack significant cellular activity. Co-crystal structures of the CARM1 catalytic domain in complexes with inhibitors 14 and 15 were obtained (PDB ID: 2Y1W and 2Y1X, respectively) (Figure 26A).389 A wealth of structural information revealed by the cocrystal structures has enabled structure-based design of PRMT4 selective inhibitors.
Figure 25.

Structures of CARM1 inhibitors.
Figure 26.
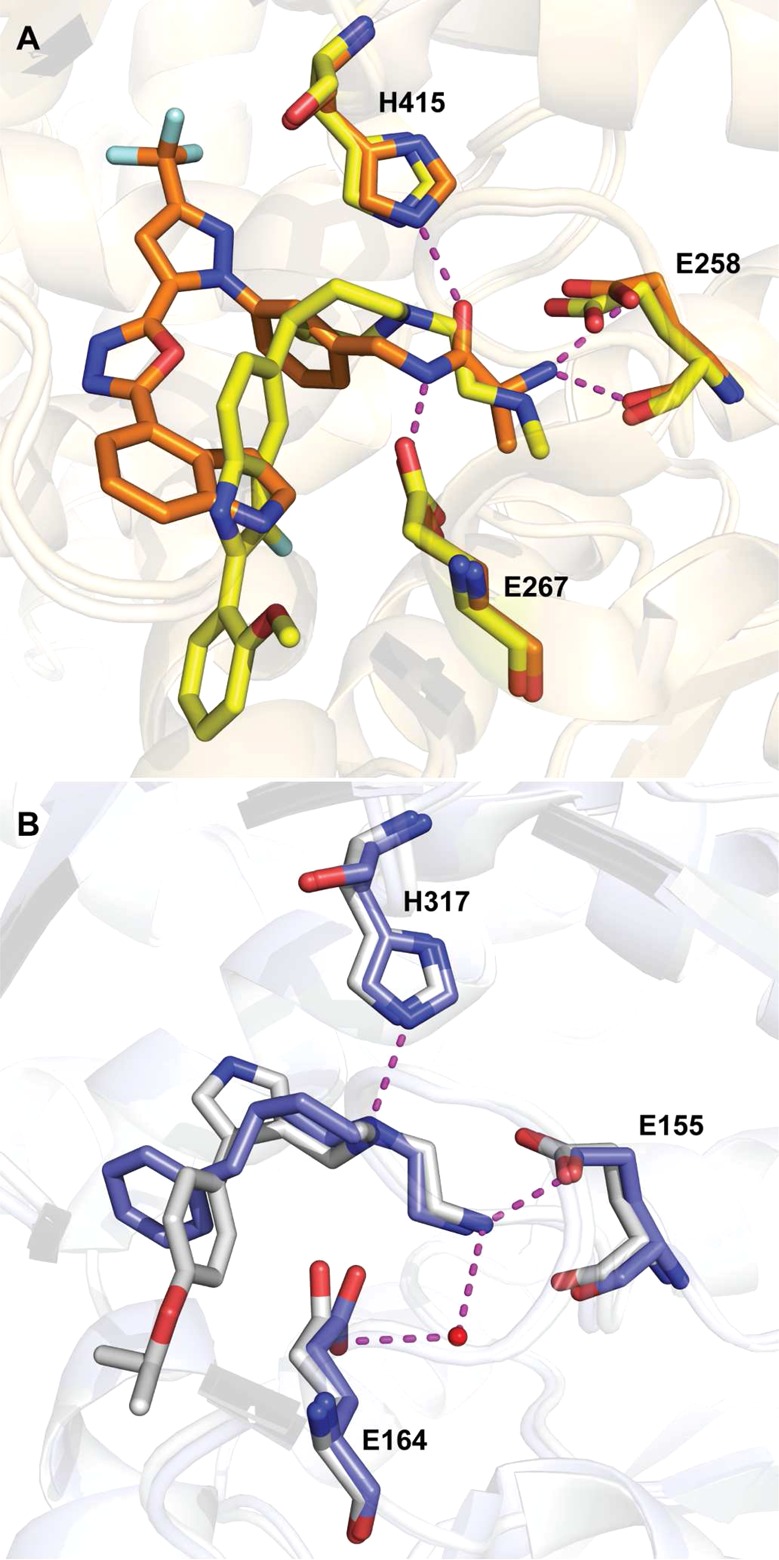
(A) Crystal structures of 14 (orange) and 15 (yellow) bound to CARM1 are superimposed (PDB ID: 2Y1W and 2Y1X). Key interactions of 14 in arginine binding channel are indicated. (B) Crystal structures of MS023 (gray) and 16 (blue) bound to PRMT6 are superimposed (PDB ID: 5E8R and 5EGS), and key interactions for 16 are shown. Hydrogen bonds are represented as magenta dashed lines and water molecule as red sphere.
In a study aiming to develop PRMT inhibitors via a fragment-based approach, a common feature of type I PRMTs (MS023), CARM1 (compounds 14 and 15), and PRMT6 inhibitors (EPZ020411, see section 2.2.5) was recognized. All of these inhibitors were anchored in the PRMT arginine-binding channel through a basic alkyl-diamino or alanine-amide tail (Figure26, panels A and B).390 Therefore, a commercially available, diverse fragment library of compounds mimicking these basic amino tails was tested against PRMT6, resulting in the discovery of compound 16 (Figure 25), an inhibitor of PRMT6 (IC50 = 300 ± 40 nM), CARM1 (IC50 = 1000 ± 40 nM), and PRMT8 (IC50 = 2100 ± 200 nM). It was selective for PRMT6 over PRMT1 (40-fold), PRMT3 (>60-fold), PRMT5 (inactive), and PRMT7 (inactive). The cocrystal structure of PRMT6 in complex with compound 16 (PDB ID: 5EGS) was solved, and the ethylenediamino group was indeed deeply buried in the arginine–binding pocket where the amino groups made direct and water-mediated hydrogen bonds (Figure 26B). This fragment was also able to inhibit asymmetric dimethylation of H3R2 in HEK293 cells transfected with PRMT6 (IC50 of 21 ± 3 μM) without cell cytotoxicity.
Recently, a dual CARM1 and PRMT6 inhibitor, MS049 (Figure 25), was discovered via SAR studies based on the fragment hit, compound 16. MS049 is a potent, selective, and cell-active dual inhibitor of CARM1 and PRMT6, displaying high potency (IC50 = 34 ± 10 and 43 ± 7 nM, respectively) in biochemical assays.391 It was selective for CARM1 and PRMT6 over other PRMTs (>300-fold over PRMT1 and PRMT3; > 30-fold over PRMT8; and no inhibition against PRMT5 and 7) and a broad range of epigenetic modifiers (including PKMTs, DNMTs, KDMs, and methyllysine/methylarginine reader proteins) and nonepigenetic targets (including GPCRs, ion channels, transporters, and kinases). The direct binding of this inhibitor to both CARM1 and PRMT6 was confirmed by ITC and DSF experiments. MOA studies showed that increasing the peptide substrate or SAM concentrations had no effect on the IC50 values of MS049 against CARM1 and PRMT6, suggesting that this inhibitor is noncompetitive with both the cofactor SAM and the peptide substrate. As we described earlier, active site-binding inhibitors can display noncompetitive behavior in MOA studies. It was postulated that the substrate binds outside the catalytic pocket of CARM1 with significant affinity, and it is not completely displaced by the inhibitor in competition assays.182,324,390,392,393 Another possible explanation is that the binding of MS049 induces major protein conformational changes, and traditional enzyme kinetics may not apply.324 In cellular assays, MS049 potently inhibited the methyltransferase activity of CARM1 and PRMT6 and reduced the levels of Med12me2a and H3R2me2a in HEK293 cells. MS049N, a close analog of MS049, was also developed as a negative control for chemical biology studies. It was inactive in biochemical and cellular assays. In addition, neither MS049 nor MS049N displayed toxicity in HEK293 cells. Overall, MS049 and its negative control MS049N are valuable chemical tools for the research community to investigate biological functions and disease associations of CARM1 and PRMT6.
The same research group also reported a potent and selective inhibitor of CARM1, compound 17 (Figure 25), which was developed based on the fragment hit 16.394 In biochemical assays, compound 17, which contains a (piperidinyl)ethyl-1-amine core, displayed high potency (IC50 = 94 ± 23 nM) for CARM1. It was also around 20-fold selective for CARM1 over PRMT6 and highly selective over PRMT1, PRMT3, PRMT5, PRMT7, and PRMT8. In MOA studies, this inhibitor was noncompetitive with the cofactor SAM and peptide substrate. On the basis of the structural similarity of this inhibitor to MS023 and MS049, it was believed that this inhibitor also binds to the substrate-binding pocket. No cellular studies were conducted with this CARM1 selective inhibitor.
Concurrent with the two studies described above, another potent and selective CARM1 inhibitor was discovered via virtual screening.395 This study also focused on amino-containing fragments such as the ethylenediamino group in compound 15 and alanine-amide group in compound 14. As described earlier, these amino-containing fragments were hypothesized to serve as arginine mimetics targeting PRMTs. Therefore, a focused virtual library composed of commercially available compounds where diverse scaffolds were attached to these two basic amino tails was created and docked into a PRMT6 crystal structure. The selected compounds at the end of this virtual screening study were then tested in biochemical assays. The hits were then optimized to yield SGC2085 (Figure 25) as a potent CARM1 inhibitor with an IC50 of 50 ± 20 nM. This inhibitor was inactive against other PRMTs, with the exception of PRMT6 (IC50 = 5.2 μM, around 100-fold selective for CARM1). SGC2085 also showed complete selectivity against a panel of 21 other methyltransferases. Although this compound is believed to bind to the CARM1 substrate-binding site, MOA studies showed that it was noncompetitive with the substrate, similar to the inhibitors depicted above. However, this inhibitor was inactive in cell-based assays at concentrations up to 10 μM. The lack of cellular activity is likely due to its poor cell membrane permeability.
Recently, the first potent, selective, and cell-active inhibitor of CARM1, TP-064, was discovered (Figure 25).396 TP-064 potently inhibited CARM1 with an IC50 < 10 nM and displayed a > 100-fold selectivity for CARM1 over other methyltransferases and nonepigenetic targets. In cellular assays, TP-064 inhibited the methylation of MED12 with an IC50 of 43 nM. A closely related compound was developed as a negative control. It exhibited no activity in the same biochemical and cellular assays.
Several other CARM1 inhibitors have also been reported. Recently, a series of PRMT inhibitors were designed to simultaneously occupy both the SAM and substrate binding sites.397 A relatively potent CARM1 inhibitor (IC50 = 0.12 ± 0.02 μM) was synthesized, which contains a two-carbon linker connecting adenosine and guanidine moieties. Aside from CARM1, this inhibitor was only tested against PRMT1 (IC50 = 11.1 ± 2.8 μM), PRMT6 (IC50 = 20.2 ± 8.7 μM), and G9a (IC50 > 50 μM). A set of curcumin derivatives were also found to be potent CARM1 inhibitors. A lead compound from this set showed inhibition against CARM1 with an IC50 of 8.6 μM and was >80-fold selective for CARM1 over PRMT1 and SETD7.398 In addition, this compound was tested against a panel of PRMTs and PKMTs. At 100 μM, this compound inhibited PRMT3, PRMT5, PRMT6, DOT1L, SUV39H1, and G9a to various extents; however, the inhibition was weaker than the observed inhibition of CARM1. This inhibitory effect against CARM1 was investigated by treating LNCaP cells that had been transfected with prostate-specific antigen promoter. Inhibition of the level of transcription was observed starting at 4 μM. In 2010, Selvi and co-workers identified TBBD (ellagic acid), which was isolated from pomegranate crude extract, as a CARM1 inhibitor.399 TBBD inhibited CARM1 but did not inhibit G9a or histone acetyltransferase CBP/p300. It was also determined to be uncompetitive with both H3 and SAM. ITC experiments showed minimal interaction between TBBD and CARM1 without SAM. Thus, it was postulated that the partial inhibition of CARM1 by TBBD could be mediated via its interaction with the enzyme-cofactor complex. At 5 μM, TBBD reduced more than 50% of H3R17 methylation levels.
2.2.4. Inhibitors of PRMT5
PRMT5 is the major type II PRMT that is responsible for the symmetric dimethylation of arginine residues.301 PRMT5 symmetrically dimethylates H2AR3, H4R3, H3R2, and H3R8 in vivo.301,400−402 Importantly, these marks are associated with a variety of transcriptional regulatory processes.400,403−406 PRMT5 has also been found to regulate transcription and many downstream events through methylation of transcription factors, including NF-κB, p53, and E2F-1.407−409 PRMT5 modulates the RAS to ERK signaling pathway through the methylation of RAF proteins410 and regulates ribosome biogenesis through the methylation of ribosomal protein S10 (RPS10).411 PRMT5 interacts with a variety of binding partners, including Blimp1, RioK1, pICLn, MBD/NuRD, and MEP50.412−416 MEP50, a member of the WD40 family of proteins, is the most common partner of PRMT5. It is required for PRMT5 enzymatic activity and is likely present in every PRMT5-containing complex in vivo.417,418 The human PRMT5 contains a triosephosphate isomerase (TIM) barrel on its N-terminus, a Rossmann-fold, and a C-terminal β-barrel enclosing a dimerization domain. As we mentioned before, the head-to-tail ring-shaped homodimeric structure is conserved in all Type I PRMT structures; however, human PRMT5, in particular, forms a heterooctomeric complex composed of four PRMT5 proteins and four MEP50 proteins.417,418 In this complex, however, PRMT5 molecules form two dimers in the head-to-tail arrangement, which is typical of PRMTs. The PRMT5–MEP50 complex displays a higher level of methyltransferase activity than that of PRMT5 alone.418 The overexpression of PRMT5 has been reported in several human malignancies, including lymphomas,401,404,419 melanoma,420 lung cancer,421 breast cancer,422 and colorectal cancer.423 In addition, in epithelial ovarian cancer, elevated PRMT5 correlates with decreased patient survival.424 Elevated PRMT5 and MEP50 expression are also highly correlated with nonsmall cell lung cancer (NSCLC) incidence.425 PRMT5 is reported to have a role in MCL, and its upregulation has been observed in patient samples.401,404,419
In April 2015, a potent, selective, cell-active, and orally bioavailable inhibitor of PRMT5 with antiproliferative effects in both in vitro and in vivo models of MCL was disclosed.426,427 HTS of a diversity library containing 370000 small molecules and subsequent optimization of hits identified led to the discovery of EPZ015666 (also known as GSK3235025, Figure 27) with an IC50 of 22 ± 14 nM in a PRMT5 biochemical assay. It was competitive with the peptide substrate (Ki = 5 ± 0.3 nM) and uncompetitive with the cofactor SAM. In addition, while the inhibitor exhibited some modest affinity for the free enzyme, its affinity for the enzyme was greatly enhanced by SAM binding. These findings were consistent with crystallographic data for the PRMT5:MEP50-SAM-EPZ015666 complex (PDB ID: 4X61) (Figure 28). The cocrystal structure revealed a unique binding mode within the substrate channel of PRMT5, indicating a key cation-π interaction between EPZ015666 and the cofactor SAM. It is believed that this interaction may be contributing to the high selectivity of EPZ015666 against other PMTs. EPZ015666 was inactive against a panel of 20 other PRMTs and PKMTs at concentrations up to 50 μM. However, it was not evaluated against PRMT9. The inhibitor had a favorable PK profile in mice, with a plasma clearance of 30 mL/min/kg and oral bioavailability of 69% following oral administration at a dose of 10 mg/kg. Thus, EPZ015666 is an appropriate tool for both cellular and in vivo studies. As such, cellular methylation and proliferation effects of this inhibitor were assessed in a panel of five MCL cell lines (Z-138, Maver-1, Mino, Granta-519, and Jeko-1). EPZ015666 treatment led to a concentration-dependent decrease in SmD3, a previously reported substrate that colocalizes with PRMT5. The same decrease in SmD3 was also observed in PRMT5 knockdown cells. Concentration-dependent decreases in SmD3me2s were also observed when whole-cell lysates from the panel of MCL cell lines treated with EPZ015666 were harvested after 4 days and evaluated via Western blotting. Target engagement was further confirmed through the use of cellular thermal shift assays (CETSA). The presence of EPZ015666 resulted in a 5.9 °C shift in the melting curve of PRMT5 in A375 cells, while no shift was seen in cells treated with a structurally similar negative control. However, cellular levels of H4R3me2s and H3R8me2s were not significantly decreased in Z-138 leukemia cells treated with EPZ015666 for 4 days. The lack of reduction in global histone methylation was attributed to the absence of existing antibodies that were sensitive and specific for those marks.
Figure 27.

Structures of PRMT5 inhibitors.
Figure 28.
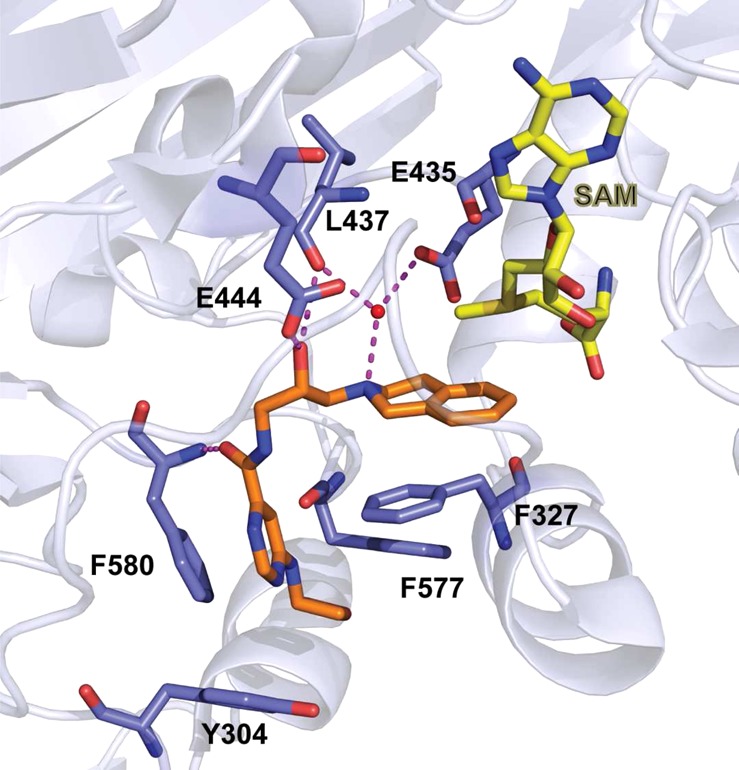
Crystal structure of EPZ015666 (orange) bound to PRMT5:MEP50 complex in the presence of SAM (yellow) (PDB ID: 4x61). Hydrogen bonds are represented as magenta dashed lines and water molecule as red spheres.
EPZ015666 demonstrated potent, concentration-dependent antiproliferative effects with IC50 values of 96 nM and 450 nM in Z-138 and Maver-1 cells, respectively. Similar effects were also observed in additional MCL cell lines, with IC50 values ranging from 61 to 904 nM. Importantly, oral dosing of EPZ015666 resulted in dose-dependent antitumor activity in multiple MCL xenograft models, with near 95% tumor-growth inhibition after 21 days of dosing (at 200 mg/kg). EPZ015666 was well-tolerated in all the models used with minimal weight loss observed. Observation of the diminished levels of symmetrically dimethylated PRMT5 substrates in the excised tumors strongly suggested that the antiproliferative effects were a direct consequence of PRMT5 inhibition. Overall, EPZ015666 is the first potent, selective, cell-active, and orally bioavailable inhibitor of PRMT5 that has been well-characterized. This inhibitor is an excellent chemical tool to decipher biological functions of PRMT5 and test therapeutic hypotheses.
A very close analog of EPZ015666, GSK591 (Figure 27), was recently disclosed as a chemical probe of PRMT5.428 It potently inhibited the methylation of H4 by the PRMT5/MEP50 complex, with an IC50 of 11 nM in a biochemical assay. It also inhibited symmetric arginine methylation of SmD3 with an EC50 of 56 nM in Z-138 cells. Furthermore, GSK591 was selective for PRMT5 over a panel of other methyltransferases at concentrations up to 50 μM.
Most notably, GSK3326595 (formerly known as EPZ015938, structure not disclosed), a PRMT5 inhibitor that potently inhibited tumor growth in cellular and animal models, has entered phase 1 clinical trials.429 It is being evaluated in subjects with advanced or recurrent solid tumors and non-Hodgkin’s lymphoma. In March 2015, Alinari and co-workers investigated the role of PRMT5 in the regulation of epigenetic repressive marks during lymphomagenesis in the context of Epstein–Barr virus (EBV)-induced B-cell transformation.430 The EBV is a human B-lymphotropic γ-herpesvirus associated with the development of B-cell lymphomas and EBV lymphomas. Transformed cell lines are known to display abundant expression of PRMT5. Since PRMT5 expression was limited to EBV-transformed cells, and not resting or activated B lymphocytes, targeting PRMT5 could be an interesting therapeutic approach for treating EBV lymphomas and B-cell lymphomas. This study disclosed a small-molecule PRMT5 inhibitor that was capable of blocking EBV-driven B-lymphocyte transformation and survival without affecting normal B cells. Virtual screening of a 10000-compound library using a human PRMT5 catalytic site model identified potential hits, which were tested in JeKo cells (a mantle cell lymphoma cell line) to assess their effects on inhibition of symmetric methylation of H4R3. Compound 5 (CMP5 or BLL-1, Figure 27) was discovered as the best candidate. CMP5 selectively blocked symmetric dimethylation of H4R3, while it was inactive against PRMT1, PRMT4, and PRMT7 in biochemical assays (IC50’s not reported). Direct binding of the inhibitor to PRMT5 was not reported, nor was a cocrystal structure of this inhibitor in complex with PRMT5 included in the paper. Cytotoxicity studies with increasing concentrations of CMP5 showed that it was toxic to lymphoma cells, while demonstrating limited toxicity to normal resting B lymphocytes even after an extended incubation period. Furthermore, while CMP5 treatment of fully transformed lympho-blastoid cell lines (LCLs) resulted in the loss of H4R3me2s and H3R8me2s marks, the asymmetric methylation of H4R3 was not affected, suggesting that this inhibitor may be selective for PRMT5 over type I PRMTs. In 2016, a second generation of CMP5, termed HLCL-61, was developed by replacing the pyridine ring with an ortho-methoxyphenyl group (Figure 27).431 It was postulated that this modification resulted in a new hydrogen-bond interaction with the protein. HLCL-61, again, did not display activity against PRMT1, PRMT4, and PRMT7. However, it inhibited H4R3me2s and H3R8me2s in AML samples. Treatment of AML cell lines (MV4–11 and THP-1) and primary AML blasts with this inhibitor resulted in a decrease in cell viability with IC50 of 7.2–21.5 μM for cell lines and 4.0–8.7 μM for patient blasts.
LLY-283, the first potent and selective SAM-competitive chemical probe for PRMT5 was recently discovered.432 LLY-283 potently inhibited PRMT5 with an IC50 of 20 nM in a biochemical assay. It was >100-fold selective for PRMT5 over other methyltransferases and nonepigenetic targets. In cellular assays, it inhibited the methylation of SmBB′ with an IC50 of 25 nM in MCF7 cells and also affected MDM4 splicing with an IC50 of 40 nM in A375 cells.
Very recently, structure-based virtual screening and subsequent SAR studies led to the discovery of a new PRMT5 inhibitor, which displayed an IC50 of 0.57 μM and selectivity for PRMT5 against other PRMTs tested in biochemical assays.433 However, direct binding of this compound to PRMT5 was not shown by biophysical assays.
Similar to PRMT5, PRMT7 plays a role in the methylation of H3R2 and Sm proteins.402,434 While PRMT7 is the sole type III PRMT and only monomethylates arginine side-chains,435 it also interacts with PRMT5, suggesting that these two enzymes may function in combination to symmetrically dimethylate protein substrates.287 It has been shown that genetic silencing of PRMT7 reduces H4R3me2s, derepresses E-cadherin expression, and diminishes cell migration and invasion in breast cancer cells.436 Using available PRMT5 structural information, DS-437 was designed and found to inhibit both PRMT5 and PRMT7 with an IC50 of 6 μM.437 It did not inhibit 29 other human protein-, DNA-, and RNA-methyltransferases. The compound also reduced symmetric dimethylation of PRMT5 substrates in cells. DS-437 is a SAM derivative and was shown to be a cofactor-competitive inhibitor. It could be a useful tool to interrogate the potential of the PRMT5–PRMT7 axis as a therapeutic target.
2.2.5. Inhibitors of PRMT6
PRMT6 is a nuclear-localized protein that catalyzes the methylation of H4R3 and H3R2.438,439 PRMT6 is the sole methyltransferase modifying the H3R2 mark, which acts as a repressive mark that antagonizes the trimethylation of H3K4 by MLL.439−442 Asymmetric dimethylation of H3R2 weakens its binding to WDR5 and prevents the recruitment of WDR5 to euchromatic regions.402 WDR5 is a mutual component of the SET1/MLL family of HMTs and has been shown to bind different peptides containing arginine, as well as H3R2.443−446 Interestingly, symmetric dimethylation of H3R2 by PRMT5 enhances its binding to WDR5 and keeps the target genes poised for transcriptional activation.402
PRMT6 has been implicated in a variety of cellular processes, such as regulation of cell cycle,447 hormone receptor-mediated transcription,448 maintenance of stem cell pluripotency,449 and DNA repair.450 PRMT6 also acts as a limiting factor for viral replication in HIV pathogenesis by methylating TAT and other HIV proteins.451 PRMT6 overexpression has been reported in several cancer types, including melanoma452 and prostate carcinoma.179 Knockdown of PRMT6 significantly suppresses growth of bladder and lung cancer cells.321 Therefore, PRMT6 is a potential therapeutic target, and development of small-molecule inhibitors as tool compounds for in vitro and in vivo studies is befitting.
The discovery of the first PRMT6 selective inhibitor was described in 2015. HTS of a corporate proprietary compound library yielded an aryl pyrazole bearing a diamino side-chain, which displayed nanomolar potency against PRMT1, PRMT6, and PRMT8.392 A cocrystal structure of this hit in complex with PRMT6 and SAH (PDB ID: 4Y2H) was obtained. This cocrystal structure revealed an extensive set of interactions, as well as the fact that there is available space in the binding pocket for expansion of the ligand at the para-position of the aryl moiety. SAR studies focused on optimizing the aryl moiety led to the discovery of EPZ020411 (Figure 29), which had an IC50 of 10 nM in a PRMT6 biochemical assay. This inhibitor was selective for PRMT6 over PRMT1 (12-fold) and PRMT8 (22-fold) and was over 100-fold selective for PRMT6/1/8 over other methyltransferases, including PRMT3, PRMT4, PRMT5, and PRMT7. A cocrystal structure of EPZ020411 in complex with PRMT6 and SAH was also solved (PDB ID: 4Y30), showing many similar interactions compared to those observed for the initial hit.
Figure 29.

Structures of PRMT6 inhibitors.
The cellular activity of EPZ020411 was examined in an engineered model where PRMT6 was transiently expressed in A375 cells, a melanoma cell line. In this model, selective methylation of the PRMT6 substrate H3R2 was strongly induced after 48 h of PRMT6 expression. Upon treatment with EPZ020411, a concentration-dependent decrease in H3R2 methylation (IC50 = 0.64 ± 0.24 μM) was seen. Treatment with an inactive analog of EPZ020411 did not have an effect at concentrations up to 20 μM. The cellular activity of EPZ020411 against PRMT1 was determined by measuring monomethyl R*GG levels, which has previously been demonstrated to be selectively modulated by PRMT1, rather than PRMT6.453 EPZ020411 displayed less potency (>10-fold) on this PRMT1-specific mark than that of the H3R2 methyl mark, agreeing with the biochemical potencies of EPZ020411 on these two enzymes. In in vivo PK studies, EPZ020411 demonstrated good bioavailability following subcutaneous dosing in rats. Thus, it is suitable for potential in vivo studies.
Very recently, a study investigating the structural basis of PRMT6-mediated asymmetric dimethylation disclosed a bisubstrate inhibitor, 6′-methyleneamine sinefungin (GMS, Figure 29), as well as its cocrystal structure in complex with PRMT6.454 GMS showed an IC50 of 90 nM, which is significantly more potent than SAH and SNF for PRMT6. GMS, however, also inhibited several other type I PRMTs, such as PRMT8 (11 nM) and CARM1 (<15 nM).
3. Histone Demethylases
Until the early 2000’s, histone methylation was largely accepted to be a stable modification. The turnover of methyl groups on histones was suggested to be the result of the replacement of methylated histones with histone variants or clipping of histone tails in the cell or during DNA replication.455,456 Another proposal involved the existence of histone demethylases that catalyze the removal of methyl groups from lysine or arginine residues, which in turn, regulate the dynamic methylation process.457 In 2004, the latter proposal was verified by Shi and co-workers by the discovery of lysine specific demethylase (LSD1, also known as KDM1A or AOF2, BHC110, and KIAA0601) as a demethylase of histone H3 lysine 4 (H3K4).5 In 2005, shortly after this discovery, Tsukada and co-workers identified and functionally characterized lysine demethylase 2A (KDM2A, also known as JHDM1A or FBXL11) as a Jumonji C (JmjC) domain containing H3K36 demethylase.458 Since the initial discoveries of LSD1 and KDM2A, an extended family of related histone demethylases have been identified and their substrate specificities have also been characterized. Today, histone demethylases (KDMs) are divided into two classes depending upon their sequence homology and catalytic mechanism: (1) LSDs (also known as KDM1s), which are members of a flavin adenine dinucleotide (FAD)-dependent amine oxidases superfamily and function to remove the methyl groups from mono- and dimethylated H3K4 (H3K4me1 and H3K4me2) via enzymatic oxidation and (2) the JmjC domain containing KDMs (KDM2 through KDM7), Fe(II), and 2-oxoglutarate (α-ketoglutarate)-dependent enzymes, which catalyze the demethylation of mono-, di-, and trimethylated lysine residues at various histone sites. LSDs are comprised of two members, LSD1 and LSD2459 (also known as KDM1B and AOF1), while the JmjC KDMs are structurally similar to nucleic acid oxygenase and contain over 20 members. In the upcoming sections, we discuss these two classes of demethylases in greater detail, including their mechanisms of action, substrate specificities, and biological functions, as well as their associations with human diseases.20,460 Finally, we provide a comprehensive review of the selective inhibitors of histone demethylases that have been published in academic literature to date.16,461,462
3.1. LSD Family of Demethylases
There are two identified members in the LSD (or KDM1) family of demethylases, LSD1 and LSD2. After the identification of the founding member LSD1,5 another FAD-dependent demethylase, LSD2, was discovered by Karytinos and co-workers in 2009.459 Similar to LSD1, LSD2 selectively demethylates H3K4me1 and H3K4me2 over H3K9, 27, 36, and 79.459 It should be noted, however, that demethylation of H3K9me2 and H3K9me1 by LSD1 in the presence of androgen receptor (AR) is reported in a cell-based assay.463
LSD1 features three major domains: an N-terminal SWIRM (small α-helical domain), a C-terminal AOL (amine oxidase like) domain, and a Tower domain in the center (Figure 30).464 The sequence identity of LSD1 and LSD2 is largely similar between their SWIRM and AOL domains, but their N-terminal domains differ.465 LSD2 contains an N-terminal CW-type zinc finger domain,465,466 while lacking the Tower domain that inserts into the AOL domain in LSD1. The Tower domain has been shown to be responsible for the binding to the corepressor for RE1-silencing transcription factor (CoREST, also known as RCOR1), which forms a heterodimeric complex with LSD1 and increases both the stability and the catalytic activity of LSD1.467 While LSD1 alone can demethylate H3K4me2/me1 in peptides or bulk histones, only the LSD1-CoREST complex can efficiently demethylate H3K4me2/me1 in nucleosomes.467,468 In addition to CoREST, LSD1 has been shown to be part of various transcriptional corepressor complexes, such as histone deacetylases (HDACs),469,470 nucleosome remodeling and deacetylation complexes (NuRDs),471 and C-terminal binding proteins (CtBPs).472 LSD1 has also been found in complexes such as AR463 and estrogen receptor (ER),473 which are associated with transcriptional activation.
Figure 30.
Domain architecture of LSD1 and LSD2.
LSDs execute their demethylase activity by oxidatively removing the methyl group, using FAD as the cofactor.5,474,475 The reaction starts with a hydride transfer from the Nε-methyl group of lysine onto FAD, thereby forming an iminium ion (Figure 31).474,475 Subsequent hydrolysis of the iminium ion results in the demethylated amine and formaldehyde as products. As shown in the first step of the demethylation mechanism, the lone pair on the starting N-methylated lysine is required for the hydride transfer. Consequently, quaternary amines (Kme3) cannot be the substrate for the LSD family of demethylases; rather, only Kme2 and Kme1 can be demethylated by LSDs. The FAD cofactor undergoes a two-electron reduction while the lysine substrate is oxidized. The intermediate FADH– then gets converted to FADH2, which is then reoxidized by molecular oxygen to yield FAD and hydrogen peroxide to complete the catalytic cycle.
Figure 31.

FAD-dependent enzymatic oxidation mechanism of LSDs.
Depending on the various protein complexes that LSD1 forms, it plays a role either in transcriptional repression (H3K4 demethylation) or activation (H3K9 demethylation). In addition, LSD1 targets nonhistone proteins such as p53 (K370me2 demethylation),476 the retinoblastoma protein 1 (RB1) regulator myosin phosphatase target subunit 1 (MYPT1, K442 demethylation),477 DNA methyltransferase 1 (DNMT1, K1096 demethylation),478 and signal transducer and activator of transcription 3 (STAT3, K140 demethylation).161 Overexpression of LSD1 has been reported in many human cancers,479 including ER-negative breast cancer,480 prostate cancer,481 nonsmall cell lung cancer,482 and bladder cancer,479 as well as neuroblastoma483 and some subtypes of acute myeloid leukemia (AML).484−486 As a result, LSD1 has emerged as a promising therapeutic target for various cancers, and interest in discovering selective LSD1 inhibitors has dramatically increased over the past decade.16,18,487,488
3.1.1. Background of LSD Inhibitors
As members of a FAD-dependent amine oxidases superfamily, LSDs share sequence and structure similarity with monoamine oxidases (MAOs) A and B. Therefore, shortly after the identification of LSD1 as the first lysine demethylase, several well-known MAO inhibitors, which were approved by US Food and Drug Administration (FDA) as antidepressants, including (±)-tranylcypromine ((±)-2-phenylcyclopropan-1-amine (also known as TCP, PCPA)), phenelzine (phenethylhydrazine), and pargyline (N-benzyl-N-methylprop-2-yn-1-amine), were tested as potential LSD1 inhibitors (Figure 32).489 As reported by Lee and co-workers in 2006, among the cohort of tested MAO inhibitors, tranylcypromine was the most promising inhibitor with an IC50 of 2 μM for LSD1 and a 10-fold selectivity for LSD1 against MAO A and B (IC50 = 20 μM).489 The closely related LSD2 had not yet been identified at the time of this study. While phenelzine showed some inhibitory activity, as measured by an increased H3K4me2 mark at 1 mM, pargyline was virtually inactive even at 5 mM concentration. Interestingly, pargyline was previously reported to inhibit LSD1 and demethylation of H3K9me2 and H3K9me1 marks when in complex with AR.463 Lee and co-workers also demonstrated an increase in the H3K4me2 mark at the OCT4 promoter after treating P19 cells with a 2 μM concentration of (±)-tranylcypromine. Similarly, after treatment with (±)-tranylcypromine, H3K4me2 levels on the EGR1 promoter were significantly enhanced, resulting in derepression of the EGR1 gene expression in the same cell line. Following this report, studies toward the discovery of more potent and selective inhibitors of LSDs mainly focused on the scaffolds represented by (±)-tranylcypromine, phenelzine, and pargyline. In the following sections, we detail the discovery, selectivity, mechanism of action, as well as functional studies of the potent inhibitors based on these three “mechanism-based” MAO inhibitors (also referred to as suicide inhibitors) that assert their activity via covalent modification of the cofactor FAD.
Figure 32.

Structures of mechanism-based MAO inhibitors.
In 2005, Forneris and co-workers published a study aiming to understand the capacity of LSD1 in recognizing and acting on various epigenetic marks on H3.490 This study demonstrated the inhibition of LSD1 by a dimethylated peptide (21 amino acid N-terminal H3 peptide) with an IC50 of 1.8 μM. In addition to mechanism-based LSD inhibitors, this work and subsequent studies on the discovery of peptide-based LSD inhibitors will be discussed. Moreover, we will discuss discovery of reversible inhibitors of the LSD family that have gained momentum in recent years.
3.1.2. Tranylcypromine-Based LSD1 Inhibitors
The AOL domain of LSD1 is homologous to amine oxidase domains found in polyamine oxidase (PAO, 22.4% identity), MAO A (17.6% identity), and MAO B (17.6% identity).491 As mentioned earlier, based on the similarities between the catalytic sites of MAO A and B and LSD1, Lee and co-workers previously tested the ability of irreversible monoamine oxidase inhibitors to inhibit the function of LSD1.489 Among the MAO inhibitors tested, (±)-tranylcypromine displayed the best inhibitory potency (IC50 = 2 μM) and was also effective at inhibiting histone demethylation in vivo.489 Following this first report of the inhibitory activity of (±)-tranylcypromine on LSD1, the same group published their work investigating this inhibition in a more quantitative fashion via kinetic analysis and pursuing a better understanding of the mechanism of inhibition.491 Using a different assay that utilized a defined peptide substrate representing the N-terminal tail of histone H3K4me2 and detected hydrogen peroxide formation by LSD1, Schmidt and co-workers reported IC50 values of 20.7 ± 2.1 μM for LSD1, 2.3 ± 0.2 μM for MAO A, and 0.95 ± 0.07 μM for MAO B with (±)-tranylcypromine. These measurements were inconsistent and appeared to disagree with their initial findings (IC50 = 2 μM for LSD1 and 20 μM for MAO A and MAO B). The large differences between IC50 values were attributed to the Western blot assays using bulk histones and nucleosomes as substrates, which therefore caused much lower effective concentrations of the substrate in their initial work. It should be noted here that (±)-tranylcypromine is an irreversible inhibitor of LSD1, and therefore IC50 is not a very good measure of its potency. A more accurate measure of the potencies of irreversible inhibitors is the ratio of kinact over KI inhibitory constants, where kinact is the first-order rate constant for inactivation of the enzyme at saturating inhibitor concentration and KI (Ki(inact)) is equivalent to the apparent affinity of the inhibitor for the enzyme in the initial encounter complex, prior to enzyme processing.492−494 However, given the complex nature of these measurements, potencies of irreversible inhibitors are still being reported as IC50 values, which are greatly dependent on the assay conditions used and, consequently, make it difficult to judge the potency and form a comparison between the inhibitors tested in different studies.
After demonstrating the irreversible inhibition of LSD1 by (±)-tranylcypromine, Schmidt and co-workers also reported kinetic parameters for the inhibition against LSD1 (kinact/KI (M–1 s–1) = 44 ± 6.0), MAO A (kinact/KI (M–1 s–1) = 107 ± 43.4), and MAO B (kinact/KI (M–1 s–1) = 706 ± 368.9).491 These results clearly showed that (±)-tranylcypromine most effectively inhibited MAO B and showed around 16- and 7-fold selectivity over LSD1 and MAO A, respectively. It is worth noting that the kinact values for LSD1 (0.0106 ± 0.0006 s–1), MAO A (0.0109 ± 0.0013 s–1), and MAO B (0.0113 ± 0.0023 s–1) are very similar, indicating that the differences in kinact/KI for these enzymes results from the value of KI. Another known inhibitor of MAOs, pargyline (Figure 32), was also investigated for LSD1 inhibition in this study, and the kinact/KI was found to be 0.364 M–1 s–1, showing that this inhibitor is 120-fold less potent against LSD1 than (±)-tranylcypromine.
It has been demonstrated that characteristic UV absorbance of LSD1-bound FAD (two peaks at approximately 382 and 456 nm) resolves into a single absorbance peak (approximately 416 nm) in a time-dependent manner upon incubation with (±)-tranylcypromine (free-FAD UV spectrum remains unchanged when treated with inhibitor), indicating LSD1-mediated deactivation of FAD.491,495,496 It has also been shown that covalent inhibitors of FAD-dependent amine oxidases can form a covalent bond at either the N(5) or C(4a) atoms of FAD,495 very similar to modification of FAD in MAO B497 and monomeric sarcosine oxidase (MSOX).498 A single electron transfer mechanism was proposed for the modification of FAD by (±)-tranylcypromine.491,495,499 According to this mechanism (Figure 33), an electron is transferred from the primary amine nitrogen of (±)-tranylcypromine to FAD, forming a cation radical. After the initial SET, there are two possible pathways for the modification of FAD. In pathway 1, ring opening of the radical cation of (±)-tranylcypromine yields the formation of a stable benzylic radical which then forms a bond at C(4a) of FAD to result in an iminium ion (18), which when hydrolyzed by water gives the aldehyde (19). This aldehyde then intramolecularly reacts with N(5) of FAD to yield a hemiaminal (20) that, following water elimination and tautomerization, results in the cyclic adduct 21. An alternative ring opening to give a carbon radical is also possible (pathway 2), but because of the energetically unfavorable nature of this radical, a concerted ring opening and bond formation reaction at C(4a) was proposed to give iminium 22. Again, this iminum ion can be easily hydrolyzed by water to yield the corresponding aldehyde 23.
Figure 33.

Proposed structures and mechanisms of formation of FAD-(±)-tranylcypromine adduct(s).
Even though this pathway seems unfavorable, it has been shown that the major product formed during the covalent inhibition of FAD in MAO B by (±)-tranylcypromine was indeed the aldehyde 23.497 Since FAD is not covalently bound to LSD1, FAD-inhibitor covalent adducts could be isolated and analyzed by mass spectroscopy, and formation of adducts 19 and 20 (Figure 33, pathway 1) can be detected by mass spectroscopy. It should be noted that expected mass for compounds 19 and 20 are the same, but the observation of the mass of 21 strongly suggested that the mechanism of the reaction was following pathway 1 for the covalent modification of FAD in LSD1. Yang and co-workers have also obtained the crystal structure of (±)-tranylcypromine-modified FDA in the LSD1-CoREST complex at a 2.75 Å resolution (PDB ID: 2UXX).495 While in this resolution the chemical structure of (±)-tranylcypromine-FDA adduct could not be assigned unambiguously; the electron density was a very good fit for one of the diastereomers of the compound 20, but it did not fit for 23 at all. The cocrystal structure provided information about stereochemistry of the C(4a), C(11), and C(13) of compound 20 (Figure 33). The C(4a) stereochemistry was explained by the fact that FAD in LSD1 has only one accessible solvent-exposed face to allow for the approach of the inhibitor, thus leading to the shown stereochemistry. The alternative stereochemistry at the C(11) would cause a steric clash between the phenyl group and a tyrosine residue of the LSD1, and stereochemistry at the C(13) could be rationalized by the intramolecular hydrogen bonding interaction of the hydroxyl hydrogen with the C(4) carbonyl oxygen of FAD. The cocrystal structure also revealed that the C(11) phenyl substituent is located in a hydrophobic pocket but that these hydrophobic interactions could be improved, indicating that derivatives of (±)-tranylcypromine could be more potent and selective inhibitors of LSD1. Due to the key differences of residues in their active sites, FAD in LSD1 forms the covalent adduct 20 with (±)-tranylcypromine, while MAO B forms adduct 23 preferentially. These critical studies on the structure and mechanism of LSD1 inhibition together with MAO B inhibition laid the groundwork for the discovery of more potent and selective inhibitors of LSD1. In addition, another crystal structure of LSD1 in complex with (±)-tranylcypromine at 2.25 Å (PDB ID: 2EJR) suggested the possibility of another adduct via a different mechanism. In this work, Mimasu and co-workers observed a new N(5)-modified FAD adduct 24 (N(5) adduct) rather than compound 20 (Figure 33)).500 This new adduct formation was explained by a different mechanism that was suggested to involve a nucleophilic addition of the heterolytically opened cyclopropane ring to FAD, which will not be discussed here.
(±)-Tranylcypromine used as a LSD1 inhibitor is a racemic mixture of trans-2-phenylcyclopropan-1-amine. To investigate whether or not the enantiomers of this compound have different inhibitory activities, (+)-tranylcypromine and (−)-tranylcypromine were obtained.501,502 Binda and co-workers reported that two enantiomers inhibited LSD1 and LSD2 with similar potencies with respect to each other and the racemic form [Ki(±) = 271 μM, Ki(+) = 284 μM, Ki(−) = 168 μM for LSD1; Ki(±) = 186 μM, Ki(+) = 137 μM, Ki(−) = 127 μM for LSD2] by using a peroxide-coupled assay. On the other hand, the difference was significant for MAO B [Ki(±) = 16 μM, Ki(+) = 4.4 μM, and Ki(−) = 89 μM].501 Through the use of a fluorescence-based assay with full length LSD1, Benelkebir and co-workers also found that the enantiomers had very similar potencies [Ki(±) = 25.0 ± 9.5 μM, Ki(+) = 26.6 ± 12.2 μM, Ki(−) = 28.1 ± 12.9 μM].502 Interestingly, the cocrystal structures of these enantiomers in complex with LSD1-CoREST revealed that the binding orientations and the resulting adducts were different for the enantiomers (PDB ID: 2XAH and 2XAJ). The FAD adduct corresponding to the N(5) adduct (Figure 33) was observed for para-bromo-(−)-tranylcypromine, while for (+)-tranylcypromine, the covalent bond formed between the benzylic carbon of the (+)-enantiomer and N(5) of the flavin ring (adduct not shown).501 More recently, Vianello and co-workers described 1-substituted (α-amino substituted) tranylcypromines as LSD1 inhibitors and based on the observed covalent FAD-inhibitor products, an alternative mechanism, which will not be discussed here, was also proposed.503
In 2008, the first series of (+)-tranylcypromine derivatives substituted at the phenyl ring (e.g., para-CF3, -Br, and -Me) were synthesized and evaluated against LSD1 as well as MAOs by Gooden and co-workers.504 While these derivatives were more potent than (+)-tranylcypromine for inhibiting LSD1, none of them were selective inhibitors of LSD1 over MAOs. In 2009, Ueda and co-workers published a series of LSD1 inhibitors,505 which were designed based on the crystal structures of (±)-tranylcypromine-FAD adduct (PDB ID: 2UXX)495 and N-propargyl lysine peptide-FAD adduct (PDB ID: 2UXN).506 The peptide-based inhibitors will be discussed in section 3.1.4. This study disclosed NCL-1 as an inhibitor, featuring an amino acid with its side chain linked to the meta position of (±)-tranylcypromine phenyl ring via an ether bond (Figure 34). In this structure, the benzoyl and benzylamino groups cap the amino and acid moieties of the amino acid chain, providing additional hydrophobicity to the inhibitor (Figure 34). NCL-1 is a potent, time-dependent, therefore irreversible inhibitor of LSD1 (kinact/KI of 2000 ± 670 M–1 s–1 (kinact = 0.011 ± 0.0024 s–1, KI = 5.7 ± 2.4 μM)], MAO A (kinact/KI of 79 ± 1.6 M–1 s–1) and MAO B (kinact/KI of 2.6 ± 0.025 M–1 s–1). These results showed that NCL-1 inhibited LSD1 selectively over MAO A (25-fold) and MAO B (770-fold). NCL-1 was also around 50-fold more potent than (±)-tranylcypromine (kinact/KI = 38 ± 7.0 M–1 s–1) for LSD1. A para-ether-linked derivative of NCL-1 was also synthesized and tested. It was about 3-fold less potent for LSD1 than NCL-1. A concentration-dependent increase in H3K4me2 levels after treatment with NCL-1 for 8 h was shown by Western blot analysis in HEK293 cells. Tumor growth inhibition by NCL-1 was also evaluated against various cancer cell lines and growth inhibition with a half maximal growth inhibitory concentration (GI50) values in the range of 6.0–67 μM were reported.
Figure 34.

(+)-Tranylcypromine-based inhibitors with substitution at the phenyl ring. *Absolute configuration.
In 2010, Mimasu and co-workers reported LSD1 inhibitors generated on the basis of (±)-tranylcypromine bound crystal structures of LSD1 and MAO B (PDB ID: 2EJR and 2XFU).507 Following iterative synthesis and structural evaluation of the inhibitors, their studies resulted in the discovery of compound S2101 (Figure 34) as their most potent LSD1 inhibitor. The strong inhibitory effect of this compound was attributed to the increased number of hydrophobic interactions of the ortho-substituent of the phenyl group as well as the two fluorine atoms at meta positions. S2101 inhibited LSD1 with a kinact/KI of 4560 M–1 s–1 (kinact = 0.0028 ± 0.000101 s–1, KI = 0.61 ± 0.13 μM), while kinact/KI values were reported to be 18 and 60 M–1 s–1 for MAO B and MAO A, respectively. Therefore, S2101 was around 250- and 75-fold selective for LSD1 over MAO B and MAO A, respectively. As a comparison, the kinact/KI values for (±)-tranylcypromine were 58 (LSD1), 271 (MAO B), and 1050 (MAO A) M–1 s–1, indicating that S2101 was >75-fold more potent than (±)-tranylcypromine for LSD1. The treatment of HEK293T cells with S2101 resulted in a concentration-dependent increase of H3K4me2 levels by Western blotting.
On the basis of the structural and mechanistic insights gained from their earlier studies, Binda and co-workers designed and synthesized (±)-tranylcypromine derivatives to exploit the differences between the binding sites of LSD1/LSD2 and MAOs to achieve more selective inhibitors of LSDs.501 The (±)-tranylcypromine derivatives containing the phenyl ring with larger substituents comprised of various hydrophobic and hydrophilic groups were investigated. Compound 25 (Figure 34) emerged as the best inhibitor with a Ki of 1.3 μM for LSD1, 38 μM for LSD2, 1.2 μM for MAO A, and no inhibition for MAO B. While this compound showed some selectivity over LSD2 (29-fold) and MAO B, it was not selective for LSD1 over MAO A. The potency difference between MAOs was attributed to a larger active site cavity of MAO A with greater flexibility to accommodate larger compounds such as 25 compared with MAO B. This compound was evaluated in a cellular model of acute promyeloctic leukemia (APL) due to the fact that the development of this disease is associated with chromatin modifications such as histone deacetylation and histone methylation. Treatment of APL-derived NB4 cells with 25 for 6 h showed a concentration-dependent increase in H3K4me2 levels. After 12 h, a concentration-dependent increase was observed to be less apparent, suggesting irreversible inhibition by the compound. While a small increase in acetylation of H4 was recorded, no substantial changes were observed in H3K9me2 levels. Even though the compound did not affect the growth of NB4 cells for 7 days at 2 μM, it strongly enhanced the efficacy of retinoic acid on growth and differentiation of APL cells.
All the (±)-tranylcypromine derivatives we have discussed so far featured various changes to the phenyl ring of the compound. In 2012, Neelamegam and co-workers reported derivatives with modifications to both the phenyl ring and amino group.508 These inhibitors were tested against LSD1 by three orthogonal biochemical assays: Horseradish peroxide (HRP)-coupled assay, a time-resolved fluorescence energy transfer (TR-FRET) assay, and a label-free, direct mass spectroscopy (MS) assay. A commercial MAO-Glo assay was used for assessing their inhibitory activities against MAO A and B. The most potent and selective inhibitor, RN-1 (Figure 35), was determined based on IC50 values [0.07 μM (HRP); 0.01 μM (TR-FRET); and 0.02 μM (MS)] from these biochemical assays, and kinact/KI values were not reported, thus making a comparison of this inhibitor to the previous ones difficult. RN-1 showed some selectivity against MAO A and B (IC50 = 0.51 and 2.78 μM, respectively). In mouse PK studies, plasma and brain concentrations were still detectable (Tmax = 0.08 and 2.0 h, respectively) 24 h after the IP administration of RN-1 at 10 mg/kg. The effect of RN-1 on long-term memory was evaluated using a novel object recognition (NOR) test. After 24 h, RN-1 treated mice displayed significant impairment in long-term memory for the familiar object compared to vehicle-treated mice. In a similar short-term memory for the familiar object test (90 min), vehicle- and RN-1-treated mice did not show a significant difference. It was thus concluded that the LSD-1 inhibitor RN-1 significantly impaired long-term memory. However, the effect of RN-1 on histone methylation in cells was not reported.
Figure 35.

(+)-Tranylcypromine-based inhibitors with substitution at both the phenyl ring and amino group. *Absolute configuration.
Ogasawara and co-workers designed peptide and small-molecule inhibitors of LSD1 based on a strategy that they termed “protein targeted drug delivery mechanism”.509 They mimicked a substrate peptide (H3-21mer) that contains tranylcypromine functionalized K4, which will be discussed in section 3.1.4. Their study also resulted in the discovery of a series of small-molecule inhibitors, among which compound 26 appeared to be the best inhibitor (Figure 35). Kinetic properties of this irreversible inhibitor was measured as follows: kinact = 0.0037 ± 0.00026 s–1, KI = 0.89 ± 0.20 μM, and thus kinact/KI = 4100 ± 980 M –1 s–1. It was about 100-fold more potent than (±)-tranylcypromine (kinact/KI = 44 ± 12 μM –1 s–1) and was also >180- and >200-fold selective for LSD1 over MAO A and B, respectively (calculated using IC50 values). This compound and its derivatives also showed growth inhibition of cancer cell lines such as SH-SY5Y neuroblastoma cells with GI50 values in the micromolar range.
ORY-1001 (Figure 35) was the first irreversible LSD1 inhibitor that entered a Phase I clinical trial for the treatment of relapsed or refractory acute leukemia (AL).510 ORY-1001 was reported to be highly potent and selective for LSD1 over the MAO enzymes and LSD2. ORY-1001 reduced AML tumor growth in mouse and rat xenograft models and increased survival in a disseminated model of T-ALL.488 We cannot provide any details of potency, selectivity, or functional activity of this compound since this data has not been published in academic literature.
Screening of a collection of 2.5 million compounds and subsequent optimization resulted in the discovery of LSD1 inhibitors GSK2879552 (Figure 35) and GSK-LSD1 (the latter is available as a chemical probe through Structural Genomics Consortium.496,511 GSK2879552 is a (±)-tranylcypromine derivative that is functionalized in the primary amine group (Figure 35). Kinetic parameters of this inhibitor were measured as kinact = 0.11 ± 0.01 min–1, KI = 1.7 ± 0.5 μM, and kinact/KI = 6.47 × 10–2 ± 3.07 × 10–3 μM–1 min–1 (1078.4 ± 51.2 M–1 s–1). Observation of very weak inhibition of MAO B and A by GSK2879552 was reported, but kinact and KI could not be calculated and therefore were not reported. This inhibitor was shown to be selective against a panel of GPCRs, ion channels, nuclear receptors, kinases, and transporters. The treatment of a panel of tumor cell lines with GSK2879552 showed that small cell lung carcinoma (SCLC) and acute myeloid leukemia (AML) cells were sensitive to pharmacological inhibition of LSD1. In particular, the treatment of SCLC cells resulted in growth inhibition in vivo by using SCLC xenograft bearing mice. GSK2879552 was orally bioavailable and well-tolerated by the animals. The antitumor activity was mainly cytostatic rather than cytotoxic. Interestingly, little effect was observed until at least 4 days of exposure in vitro, suggesting that extended inhibition of LSD1 is likely required for maximal efficacy. Since SCLC is a neuroendocrine tumor, related marker genes were surveyed, and while many hallmarks of these cells were altered in a panel of SCLC lines, gastrin releasing peptide (GRP) was found to be consistently lower upon LSD1 inhibition. Such a change in neuroendocrine marker expression in SCLC was attributed to alterations in cell state, showing similarities to the pro-differentiation effect observed in leukemia upon loss of LSD1. LSD1 inhibition in SCLC cell lines resulted in altered gene expression in vitro and in vivo in a time- and dose-dependent manner. While global effects on H3K4 methylation or genomic distribution of LSD1 were not seen, increased local H3K4 methylation levels were observed after the treatment of SCLC cells with GSK2879552. Gene ontology (GO) analysis of LSD1 ChIP-Seq data showed that genes important for neuron differentiation and cell development were the most strongly bound by LSD1, further emphasizing a possible role of LSD1 in differentiation of SCLC. Despite the presence of gene expression changes, only a limited number of SCLC cell lines tested (9/28) were sensitive to the GSK2879552 treatment. DNA methylation analyses of SCLC cell lines revealed a differentially methylated gene signature in sensitive versus resistant cell lines, and the DNA hypomethylation signature identified in SCLC cell lines was also found in primary SCLC samples. This biomarker was then assessed in three patien- derived xenograft (PDX) models to predict sensitivity to LSD1 inhibition. Tumor growth inhibition was only observed in models with the sensitivity-associated DNA methylation signature. In 2013, GSK2879552 entered phase 1 clinical trial for patients with relapsed/refractory SCLC. Recently, this compound entered into clinical trials for AML and Myelodysplastic Syndromes (MDS).512−514
A Phase I/II trial of (±)-tranylcypromine in combination with ATRA (Tretinoin) has also been initiated for relapsed or refractory AML patients, for whom no intensive treatment is currently available.515−517
In 2015, Kahn and co-workers published an N-alkylated analog of NCL-1 (27) by incorporating a piperazine acyl moiety on NCL-1 (Figure 35).518 This hybrid compound, which contains modifications to both the amino and phenyl groups of (±)-tranylcypromine, was 6-fold more potent than NCL-1 and did not inhibit MAO A or B. Not surprisingly, it inhibited LSD1 in a time-dependent manner with kinact = 0.0035 s–1 and KI = 2.4 μM (kinact/KI = 1458 M –1 s–1). This compound also showed some growth inhibitory activity in SH-SY5Y neuroblastoma cells at high concentrations, but no further cellular activities were reported for this compound.
In summary, significant advancements on discovering irreversible or covalent/suicide inhibitors of LSD1 have been made by exploiting known MAO inhibitors as likely LSD1 inhibitors and subsequent optimization based on structural insights revealed by a number of critical cocrystal structures (see Table 1). Most notably, ORY-1001 and GSK2879552 have been advanced to clinical trials. These compounds, along with other potent, selective, well-characterized inhibitors discussed above, are valuable chemical tools to investigate the role of LSD1 in cancer and other human diseases and validate therapeutic hypotheses.
Table 1. Summary Table of Kinact/KI Data of LSD1 Inhibitorsa.
| inhibitor | Kinact/KI (M–1 s–1) | Ki (μM) |
|---|---|---|
| NCL-1 | 2000 ± 670 | 5.7 ± 2.4 |
| S2101 | 4560 | 0.61 |
| 25 | N.R. | 1.3 |
| RN-1 | N.R. | N.R. |
| 26 | 4100 ± 980 | 0.75 ± 0.5 |
| GSK2879552 | 1078 | N.R. |
| 27 | 1458 | 2.4 |
N.R. = not reported.
3.1.3. Phenelzine- and Pargyline-Based LSD1 Inhibitors
The initial report by Lee and co-workers investigating MAOs inhibitors as potential LSD1 inhibitors indicated that phenelzine could inhibit LSD1 at 200 μM, while (±)-tranylcypromine displayed inhibition at 100 nM.489 In 2010, Culhane and co-workers reinvestigated the inhibitory activity of phenelzine (Figure 36) against LSD1 and reported kinact = 0.955 ± 0.085 min–1, KI = 17.6 ± 2.8 μM, and thus kinact/KI = 900 M –1 s–1, which was about 35-fold more potent than (±)-tranylcypromine (kinact/KI = 25.3 M –1 s–1).519 This finding concerning the potency of phenelzine against LSD1 contradicted the initial report by Lee and co-workers. It should also be noted, however, that the assays used in the two studies are different. The inhibition was confirmed by an orthogonal mass spectroscopy assay showing that in the presence of phenelzine, LSD1 was not able to demethylate a H3-21-K4me2 substrate peptide. Culhane and co-workers further investigated the effects of phenelzine on a thyroid hormone (T3)-inhibited TSHα luciferase reporter transfected in cells and determined the methylation levels of H3K4 by ChIP experiments.519 They reported that mono- and dimethylation of the TSHα reporter region were enhanced by phenelzine treatment, while the trimethylation level was unaltered. Therefore, it was postulated that mono- and dimethylation of H3K4 might enhance basal transcription of the TSHα promoter in the absence or presence of T3. On the basis of these results, the same research group explored a series of phenelzine analogs with modifications to the hydrazine moiety, alkyl chain length and rigidity, and phenyl ring to improve LSD1 potency and selectivity.520 These SAR studies resulted in the discovery of a compound, which was named as bizine (Figure 36) and contains a propylphenyl group tethered to the phenelzine para position via an amide spacer. Bizine was the most potent LSD1 inhibitor in this study with kinact = 0.15 ± 0.017 min–1, KI (inact) = 0.059 ± 0.021 μM, and kinact/KI = 2.5 ± 0.96 μM –1 min–1 (41,666 M–1 s–1). As a reference, the kinetic parameters for phenelzine were measured in parallel as kinact = 0.35 ± 0.056 min–1, KI = 5.6 ± 1.3 μM, and thus kinact/KI = 0.063 ± 0.018 μM –1 min–1 (previously measured as 0.054 μM –1 min–1) using the same colorimetric peroxide assay.521 In addition, selectivity of bizine for LSD1 versus MAO A, MAO B, and LSD2 was assessed. On the basis of kinact/KI values, it was found that bizine was 23-, 63-, and >100-fold selective for LSD1 over MAO A, MAO B, and LSD2, respectively. On the other hand, the parent compound, phenelzine, preferentially inhibited MAO A and was equipotent at inhibiting MAO B and LSD1.
Figure 36.

Structures of LSD1 inhibitors phenelzine and bizine.
Culhane and co-workers found a concentration-dependent increase in H3K4me2 levels (EC50 ∼ 2 μM) in LNCaP cells, a prostate cancer cell line, treated with bizine for 48 h.519 On the other hand, no significant alterations in H3K4me1, H3K4me3, unmethylated H3K4, and other histone H3 marks were observed. LSD1 protein levels were also unchanged. Phenelzine, however, which is ∼40-fold less potent than bizine, did not alter H3K4me2 levels at concentrations up to 40 μM in LNCaP cells. Bizine was also tested in additional cancer cell lines, such as H460 and A549 lung cancer cells and MDA-MB-231 breast cancer cells. While similar effects of bizine on H3K4me2 levels were observed in H460 cells, increases in H3K4me2 levels were detected only at a higher concentration (20 μM) in A549 and MDA-MB-231 cells. The effects of bizine on cell proliferation were also investigated in H460 cells, and the studies suggested that LSD1 inhibition by bizine might be contributing to cancer cell growth inhibition; however, concentrations well above the EC50 values in the cellular demethylation assay were necessary for inhibition of the proliferation. The examination of additive or synergistic effects of bizine in combination with several HDAC inhibitors and the DNMT inhibitor azacytidine revealed some moderate additive or synergistic effects on growth inhibition in H460 cells. Only two out of the five HDAC inhibitors examined exhibited these effects at the highest concentrations tested. Furthermore, in this study, bizine was also proposed as a possible neuroprotective agent against oxidative stress.
In 2013, Schmitt and co-workers reported a study pursuing potent LSD1 inhibitors based on the parygline structure.522 Their similarity based virtual screening and subsequent optimization resulted in a nonpeptidic pargyline derivative, which inhibited LSD1 with an IC50 of 44 ± 2.2 μM. This inhibitor, however, was much more potent against MAO A (IC50 = 0.55 ± 0.06 μM) and especially MAO B (IC50 = 0.06 ± 0.003 μM) in a biochemical assay.
It is worth noting that most of the LSD1 inhibitors described here were investigated against MAOs for selectivity, but they lack selectivity data against LSD2. In addition, a report describing tranylcypromine-containing small molecules as potent and selective dopamine D3 receptor antagonists underlines the importance of thoroughly investigating the selectivity profile. In fact, selectivity of the LSD1 inhibitors described here against a broad range of other epigenetic and nonepigenetic targets including GPCRs, kinases, ion channels, and transported was not reported. Therefore, one should be cautious to attribute the observed phenotypic effects to pharmacological inhibition of LSD1.523
Similar to (±)-tranylcypromine, LSD1 inhibitors derived from phenelzine and pargyline exert their inhibitory effects via covalent inactivation of FAD. Their inhibition mechanism will be discussed in the peptide-based LSD1 inhibitors section below.
3.1.4. Peptide-Based LSD1 Inhibitors
The peptide substrates having less than 16 amino acid residues have been shown to be inactive against LSD1, while optimal binding requires 21 amino acid residues.490 In 2007, Forneris and co-workers studied the structural basis for LSD1-CoREST selectivity in H3 recognition by examining the X-ray crystal structure of its complex with a 21-amino acid H3 peptide, in which K4 is mutated to methionine (H3-21-K4M) (PDB ID: 2 V1D).524 This H3K4M mutation led to a 30-fold increase in binding affinity (Ki = 0.05 ± 0.02 μM) compared with the wild-type peptide (Ki = 1.8 ± 0.02 μM) and was one of the first examples of peptide-based LSD1 inhibitors.
In 2006, based on the premise that pargyline is a LSD1 inhibitor,463 Culhane and co-workers reported a propargyl-lysine derivatized peptide (28) as a potent and irreversible inhibitor of LSD1 (Figure 37).525 This initial publication on the peptide inhibitor was followed by mechanistic and kinetic studies. A kinact = 0.29 ± 0.02 min–1, KI = 0.69 ± 0.10 μM, and kinact/KI = 0.42 μM –1 min–1 (2590 M–1 s–1) were measured for this peptide inhibitor.526 Interestingly, a 13-fold increase for the kinact/KI value was observed for bacterially (E. coli) produced GST-LSD1 over that of the insect cell-derived enzyme in this study. The formation of an FDA-inhibitor covalent adduct was demonstrated by UV and mass spectroscopy, while the structure of the adduct (Figure 37) was elucidated by NMR analysis. The proposed mechanism starts with a hydride transfer from propargyl-lysine derivatized peptide (28) to FAD, resulting in FADH– and a propargylic iminium ion (29), which then undergoes Michael addition with N(5) of the FADH– (Figure 37, top). The resulting iminium adduct (30) could be in different stereochemical and tautomeric forms which cannot be distinguished by NMR analysis. A biotinylated derivative of the peptide inhibitor was also synthesized and used successfully in pull down studies for LSD1-CoREST.
Figure 37.

Proposed structures and mechanism of formation of FAD-(±)-propargyl-lysine (30) and FAD-(±)-hydrazine-lysine derivatized peptide (32) adduct(s).
The same research group further investigated incorporation of other known MAO inhibitor motives and related derivatives into the H3-21 peptide, including cyclopropyl amine, propargyl amine, 3-chloroallyl amine, as well as a hydrazine functionality.519 Among these peptides, the one featuring the hydrazine group on K4 (H3-21-K4-hydrazino, 31) was discovered to be most potent with a kinact = 0.247 ± 0.018 min–1, KI = 0.00435 ± 0.00086 μM, and kinact/KI = 56.8 ± 0.82 μM –1 min–1 (Figure 37). The phenelzine was also tested in the same conditions as a reference with a kinact/KI = 0.0543 ± 0.00077 μM –1 min–1, therefore making this new peptide inhibitor 31 1000-fold more potent than phenelzine and 25-fold more potent than the N-methyl-derivative (Figure 38) (kinact = 0.208 ± 0.068 min–1, KI = 0.107 ± 0.057 μM, and kinact/KI = 1.94 ± 0. 34 μM –1 min–1). The MS analysis revealed a product with a mass that is consistent with the formation of the peptide-FAD adduct with the loss of N2. It was proposed that (Figure 37, bottom) a two-electron oxidation to form the corresponding diazene, followed by another two-electron oxidation of the diazene (with reoxidized FAD by molecular oxygen) yields the diazonium moiety. The subsequent attack from the N(5) of the reduced flavin displaces the diazonium group, which is an excellent leaving group, and produces the inhibitor-FAD covalent adduct 32 with loss of N2. Two possible peptide degradation pathways were also proposed, supported by the MS data of the byproducts. This study showed that while the chlorovinyl- and alkynyl-containing peptide inhibitors are comparable in potency, the hydrazine-containing peptide inhibitor was the most potent inhibitor for LSD1. As mentioned in the previous section, phenelzine is a more potent LSD1 inhibitor (∼35-fold) than (±)-tranylcypromine.
Figure 38.

Structures of peptide-based LSD1 inhibitors.
Ogasawara and co-workers designed H3-21-K4-tranylcypromine (33, Figure 38), which bears a (±)-tranylcypromine moiety at K4 of the 21-amino-acid H3 peptide, and evaluated its LSD1 inhibitory activity.509 This peptide inhibited LSD1 (IC50 = 0.16 ± 0.036 μM) in a time- and concentration-dependent manner but did not inhibit MAO A or MAO B (IC50 > 100 μM) in a horseradish peroxidase coupled assay. Compound 33 is an irreversible LSD1 inhibitor with a kinact/KI value of 5900 ± 3000 M –1 s–1, showing greater binding affinity and potency for LSD1 than that of (±)-tranylcypromine (kinact/KI = 22 ± 8.2 M –1 s–1). However, cellular potency of 33 was weak against cervical HeLa and neuroblastoma SH-SY5Y cancer cell lines, in which LSD1 is overexpressed (GI50 = 27 ± 17 and >160 μM, respectively). This was likely caused by poor cell-membrane permeability. The same research group later explored replacing the tranylcypromine moiety with 2,5-dihydro-1H-pyrrole (DHP) and 1,2,3,6-tetrahydropyridine (THP), as well as H3K9-based tranylcypromine-bearing peptides and truncated H3K4-tranylcypromine peptide inhibitors. None of these inhibitors, however, showed better potency than that of 33.527
In 2013, various cyclic peptides based on H3-21-K4M (in which K4 is mutated to methionine) were synthesized and evaluated as LSD1 inhibitors.528 These cyclic peptides were constructed by substituting selected positions with one lysine residue and one glutamic acid residue and cyclizing these residues to form a lactam bridge. Among the cyclic peptides constructed, H3-21-K4M in which the lactam bridge was between lysine 5 and glutamic acid 10, showed the greatest inhibitory activity in a biochemical assay with an IC50 of 2.1 μM. This cyclic peptide was shown to be a competitive inhibitor with a Ki of 385 nM. It also exhibited modest antitumor activity in MCF7 breast and Calu-6 lung tumor cell lines.
Recently, short peptides that inhibited LSD1 in a reversible manner were reported by Tortorici and co-workers.529 SNAIL1 is a member of the SNAIL family of transcription factors, and it has been shown that the N-terminal 21 amino acids of the SNAG domain of SNAIL1 binds to the LSD1 active site in a conformation similar to that of H3. As a result, SNAIL1 is a competitive inhibitor of LSD1, with respect to the H3 substrate. As mentioned earlier, while LSD1 binding to the H3 substrate requires at least the first 16 N-terminal residues, in this study, analysis of the crystal structure of LSD1/CoREST in complex with the SNAIL1 N-terminal peptide indicated that only the first 9 amino acids had well-defined electron densities (PDB ID: 3ZMT). Indeed, it has been demonstrated that the SNAIL1 1–9 peptide inhibited the LSD1/CoREST complex with a similar affinity (Ki = 0.14 ± 0.06 μM) to that of the SNAIL1 1–20 peptide (Ki = 0.21 ± 0.07 μM) and that the first 6 amino acid residues were required to achieve low micromolar affinity (Ki = 28.4 ± 4.8 μM). Variations in the sequence of this minimal peptide were also found to modulate binding affinities.
3.1.5. Reversible LSD1 Inhibitors
As described in previous sections, outstanding progress has been made in the development of irreversible LSD1 inhibitors. Although there is a growing interest in developing reversible, noncovalent inhibitors, the large binding site of LSD1 presents a major challenge. Despite that, significant progress has been made in the discovery of reversible LSD1 inhibitors.530 We covered peptide-based reversible LSD1 inhibitors in the section above. In this section, we discuss small-molecule noncovalent inhibitors of LSD1.
Biguanide and bisguanidine polyamine analogues were one of the earliest reported LSD1 inhibitors with >50% inhibition at 1 μM.531 Two of the most potent compounds, 34 and 35 (Figure 39), have been shown to be noncompetitive inhibitors with respect to the substrate; however, no further IC50 measurements, biophysical binding, or selectivity data were reported. These compounds affected re-expression of multiple, aberrantly silenced genes in HCT116 human colon carcinoma cells. The re-expression coincided with increased H3K4me2/me1 and H3K9ac marks and decreased H3K9me2/me1 repressive marks. In 2012, a study was reported using the same or new polyamine analogs [e.g., PG11144 (Figure 39)] in MDA-MB-231, a triple negative human breast cancer cell line.532 Another series of polyamines containing (bis)thiourea and a 3–5–3 or 3–6–3 backbone architecture was also reported by the same research group as inhibitors of recombinant LSD1.533,534 Among these compounds, 36 (Figure 39) was the most potent with IC50 value of 4.8 μM in a biochemical assay and was shown to be competitive with the peptide substrate (Ki of 2.2 μM). While it was selective against MAO A (IC50 > 100 μM), it was only about 4-fold selective over MAO B (IC50 = 19 μM). These polyamine derivatives were relatively large inhibitors with limited potency and selectivity.
Figure 39.

Biguanide and bisguanidine polyamine analogues that were reported as LSD1 inhibitors.
In 2012, Hazeldine and co-workers reported the identification of a series of amidoximes as potent, reversible LSD1 inhibitors using a virtual screening strategy.535 The most potent compound from this study exhibited an IC50 of 16.8 μM in a biochemical assay. In 2013, Sorna and co-workers identified a novel series of LSD1 inhibitors with much improved potencies, again utilizing a structured-based virtual screening strategy.536 The subsequent SAR studies resulted in the discovery of compound 37, featuring a benzohydrazide moiety (Figure 40). This compound inhibited LSD1 with an IC50 of 13 nM and showed selectivity for LSD1 over MAO A and B (no activity up to 300 μM). Compound 37 was further screened against d-lactate dehydrogenase and glucose oxidase, exhibiting IC50 values around or above 10 μM. In DSF experiments, compound 37 shifted the Tm in a subtle but statistically significant manner, suggesting that it binds LSD1. It also has been shown that compound 37 is a noncompetitive, reversible inhibitor with the peptide substrate with a Ki of 34 ± 1.9 nM. The experiments showing direct binding and reversibility are important since hydroxyl phenyl hydrazones are common PAIN structures that are observed in many assays.537,538 The effect of compound 37 on cell growth was evaluated in a panel of cancer cell lines. Nine of the 17 cell lines tested, including endometrial, breast, colorectal, and pancreatic cancers cells, were sensitive to compound 37, with an EC50 < 1 μM. Furthermore, the effects of compound 37 on histone methylation in VCaP cells (an androgen-sensitive prostate cancer cell line) were also investigated. H3K9me2 was reported as a target for LSD1 when in complex with AR.463 An increase in H3K9me2 levels were observed at 24 h with both 1 and 10 μM of treatment with 37, suggesting that effects of the compound were mediated through LSD1 inhibition.
Figure 40.

Structures of reversible LSD1 inhibitors.
In 2016, Zhou and co-workers aimed to improve potency and synthesized constrained derivatives of compound 37.539 Among these derivatives, compound 38 (Figure 40) was the most potent with an IC50 of 1.4 ± 0.3 nM in a biochemical assay, which was about 10-fold more potent than compound 37. Compound 38 inhibited the proliferation of cancer cell lines such as A549, H1299, A2780, HCT116, MCF-7, MDA-MB-231, and DU145 with an IC50 < 1 μM. Similar to the previous study, the effect of this compound on the H3K4 and H3K9 methylation levels were assessed in A2780 cells (human ovarian carcinoma cells). It was found that compounds 38 concentration-dependently increased H3K4me2 and H3K9me2 levels in A2780 cells.
In 2016, Wu and co-workers reported the discovery of a series of 3-(piperidin-4-ylmethoxy)pyridine-containing compounds as LSD1 inhibitors, inspired by earlier SAR studies of tranylcypromine-containing inhibitors.540 Structure-guided molecular modeling and ligand optimization resulted in potent LSD1 inhibitors. Among them, compound 39 (Figure 40) emerged as the most potent inhibitor with a Ki value of 29 nM. Enzyme kinetics studies were performed on a close derivative of 39 and showed that the inhibitor was competitive with the H3K4me2 peptide substrate. Compound 39 was highly selective for LSD1 over MAO A (Ki > 50 μM) and MAO B (Ki = 18.7 ± 6.9 μM). Since structurally similar compounds were reported to be potent Akt inhibitors, selectivity of compound 39 against human Akt was also evaluated. It was found that the inhibitor displayed high selectivity for LSD1 over Akt (IC50 = 87.6 ± 21.6 μM). Compound 39 at 1 μM increased H3K4me2 levels in cells. The inhibitor also exhibited good potency at blocking the proliferation of MV4–11 and Molm-13 cells (leukemia cell lines with an MLL gene translocation), as well as MCF-7 (estrogen receptor positive) and MDA-MB-231 (estrogen receptor negative) breast cancer cells with EC50 values ranging from 0.28 to 8.6 μM. On the other hand, little to no effect on the growth of normal fibroblast cells was detected. It should be noted that, in 2013, a study aiming to develop reversible LSD1 inhibitors used a very close analog of compound 39, GSK354, as part of their research. In this study, GSK354 displayed an IC50 of 90 ± 10 nM for LSD1 in biochemical assays, as well as a cellular IC50 of 1.4 ± 0.3 μM in a CD86 cell-based assay.541 The compound also displayed selectivity over MAO A (IC50 > 200 μM) with no apparent cytotoxicity at 20 μM against the THP-1 cell line.
Another report on the design and synthesis of reversible LSD1 inhibitors came from Zheng and co-workers in 2013.542 This new class of triazole–dithiocarbamate-containing inhibitors was based on their early work on the chemotype as potential anticancer agents.543 In this study, compound 40 (Figure 40) was reported to inhibit LSD1 with an IC50 of 2.1 ± 0.7 μM. The reversibility of the compound for LSD1 was demonstrated by a dilution assay. The binding affinity of compound 40 was determined using microscale thermophoresis (MST) (Kd = 0.35 μM). Importantly, compound 40 was selective for LSD1 over LSD2 (IC50 > 36.6 ± 4.5 μM, about 17-fold selective) and MAO A and B (IC50 > 1250 μM). In MOA studies, compound 40 was characterized as a noncompetitive inhibitor with the substrate H3K4me2 but as a competitive inhibitor with the cofactor FAD. This finding was interesting because it suggested that compound 40 could potentially displace FAD, which had a measured Kd of 0.182 μM and showed tight binding to LSD1. An increase in H3K4me2/me1 and H3K9me2 levels was observed via treatment of MGC-803 cells (human gastric cancer cell line) with compound 40. Compound 40 exhibited strong cytotoxicity against human gastric cancer cell lines MGC-803 and HGC-27 (IC50 = 0.89 μM and 1.13 μM, respectively), which comprise higher LSD1 expression, as evaluated by MTT assays. On the other hand, it did not show any discernible effects on normal gastric epithelial cell line GES-1 and gastric cancer cell line SGC-7901 (IC50 > 45 μM), which are known to contain a lower LSD1 expression. The effect of compound 40 on tumor growth was also investigated in vivo in a xenograft model where tumors were generated by subcutaneous implantation of MGC-803 cells into nude mice. A significant inhibition of tumor growth and a 68% reduction in tumor weight were observed over time without any clear body weight loss during the 21-day treatment (dosed at 20 mg/kg), suggesting that compound 40 was efficacious in vivo with no apparent toxicity.
The same research group designed and synthesized coumarin–1, 2, 3-triazole–dithiocarbamate hybrids by introducing a coumarin moiety to the compound 40 scaffold to create more potent LSD1 inhibitors.544 Compound 41 was disclosed as the most potent inhibitor from this series, with an IC50 of 0.39 ± 0.15 μM (Figure 40). Compound 41 was about 5-fold more potent than compound 40 and showed high selectivity for LSD1 over MAO A and B, similar to compound 40. It concentration-dependently increased H3K4me2/me1 and H3K9me2 levels in MGC-803 cells.
In 2015, Ma and co-workers reported a new series of small-molecules as potential LSD1 inhibitors.545 These compounds contain aminothiourea and propargyl pharmacophores that were linked together by a pyrimidine moiety. The most potent compound, 42 (Figure 40), inhibited LSD1 with an IC50 of 0.65 ± 0.12 μM and was selective for LSD1 over MAO A and B (IC50 > 1250 μM). The direct interaction between the compound and LSD1 was demonstrated by biolayer interferometry (BLI), which produced a Kd = 3.7 μM. Similar to compounds 37 and 41, this inhibitor increased H3K4me2/me1 and H3K9me2 levels in a concentration-dependent manner in MGC-803 cells. Levels of H3K4me3, LSD1, and H4 were unaffected. Compound 42 inhibited the growth of MGC-803 and HGC-27 cells with IC50 values of 4.01 ± 0.21 μM and 8.92 ± 0.52 μM, respectively. In addition, this inhibitor was reported to be orally active in vivo.
Other reversible LSD1 inhibitors with less potency (IC50 values in micromolar range) and/or selectivity have been reported. The 3,5-diaminotriazole scaffold was discovered as a potential LSD1 inhibitor by virtual screening. The most potent compound identified from this study was inhibitor 43 (Figure 41), which has an IC50 of 1.19 μM and good selectivity for LSD1 over MAOs (IC50 > 100 μM).546 Direct binding of this reversible inhibitor was shown by ITC with a Ki of 2.2 μM. It was also shown to be competitive with the substrate by kinetics experiments. In another study using the pharmacophore-based virtual screening, compound XZ09 (Figure 41) was identified as a potent and selective LSD1 inhibitor (IC50 = 2.41 μM and IC50 for MAO A = 685 μM and for MAO B = 27.5 μM).547 A series of functionalized phenyl oxazole derivatives was screened in vitro for their activities against LSD1 and effects on viability of cervical and breast cancer cells.548 Among the compounds tested, 44 (Figure 41) showed modest inhibitory activity against LSD1 with IC50 around 10–16 μM and blocked the growth of cancer cells (IC50 = 1.2–1.4 nM). However, direct binding of this compound to LSD1 was not experimentally tested and no selectivity data were reported in this study. Wang and co-workers developed CBB-1007 (Figure 41) as a reversible LSD1 inhibitor (IC50 = 5.27 μM), which exhibited selectivity for LSD1 over the closely related LSD2 and JARID1A.549 However, its selectivity against MAOs was not reported. Compounds 45 (Figure 41) and its derivatives were also reported as reversible LSD1 inhibitors. Compound 45 has modest potency (IC50 = 9.5 ± 3.4 μM) and was selective for LSD1 over MAO A (IC50 > 500 μM).541 Lastly, namoline (Figure 41) was also reported as a selective and reversible inhibitor of LSD1 with very modest potency in biochemical assays (IC50 = 51 μM).550 It was reported to affect global histone methylation levels at >20 μM.
Figure 41.

Structures of additional reversible LSD1 inhibitors.
3.2. JmjC Containing Lysine Demethylases
The second and largest class of histone demethylases contains a Jumonji C (JmjC) domain, named after the “Jumonji” protein in which this domain was first identified.551 JmjC domain-containing lysine demethylases [JmjC KDMs, also known as the JmjC domain-containing histone demethylases (JHDMs)] can demethylate all three lysine-methylation states, in contrast to LSDs, which can only demethylate mono- and dimethylated lysine residues. In 2006, Tsukada and co-workers identified the first JmjC domain-containing protein showing histone demethylase activity specifically at H3K36me2 and recognized the JmjC domain as a signature motif for demethylation.458 The same year, JmjC KDMs were reported to be able to remove the H3K9me3 mark. Since then, the discovery of JmjC domain-containing demethylases was expanded significantly.552−556 Today, members of JmjC KDMs have been shown to modify all three methylated states of H3K4, H3K9, H3K27, and H3K36 residues.557
The JmjC KDMs are part of a larger JmjC subfamily of Fe(II)- and 2-oxoglutarate- (2-OG) dependent oxygenases, which also catalyze protein hydroxylation reactions that do not involve demethylation.19,558 In 2006, Klose and co-workers investigated JmjC KDMs in the context of JmjC domain evolution and domain architecture via analysis of public protein-domain databases. They defined seven groups of evolutionarily conserved proteins, including JHDM1, PHF2/PHF8, JARID1/JARID2, JHDM3/JMJD2, UTX/UTY and JHDM2 families, which have at least one additional protein domain besides the JmjC domain, and a seventh group containing only the JmjC domain.557 Recently, an alternative classification contains six main subfamilies: KDM2, KDM3, KDM4, KDM5, KDM6, and KDM7 (see the phylogenetic tree in Figure 42), based on sequence analysis of their catalytic domains.19 In some reports, KDM2 and KDM7 are considered as a combined subfamily because of their close sequence identity in their catalytic domains.559 We choose to use this subfamily classification in this review. The names of the members of JmjC KDMs subfamilies, including their alternative names and reported substrate specificities, are summarized in Table 2. Most of the JmjC KDMs have additional noncatalytic domains, including histone binding and nucleic acid binding domains, which allow them to target specific histone modifications. For example, KDM7A and PHF8 utilize their plant homeobox domains (PHD) to bind H3K4me3 and direct their KDM domains to H3K27 and H3K9, respectively.560 KDM4A binds H3K4me3 and H4K20me3 sites via its tudor domains and catalyzes demethylation of H3K9me3/2, H3K36me3/2, and H1.4K26me3/2.561,562 The domain architectures of the subfamilies will be discussed in the context of inhibitors below (Figure 43).
Figure 42.
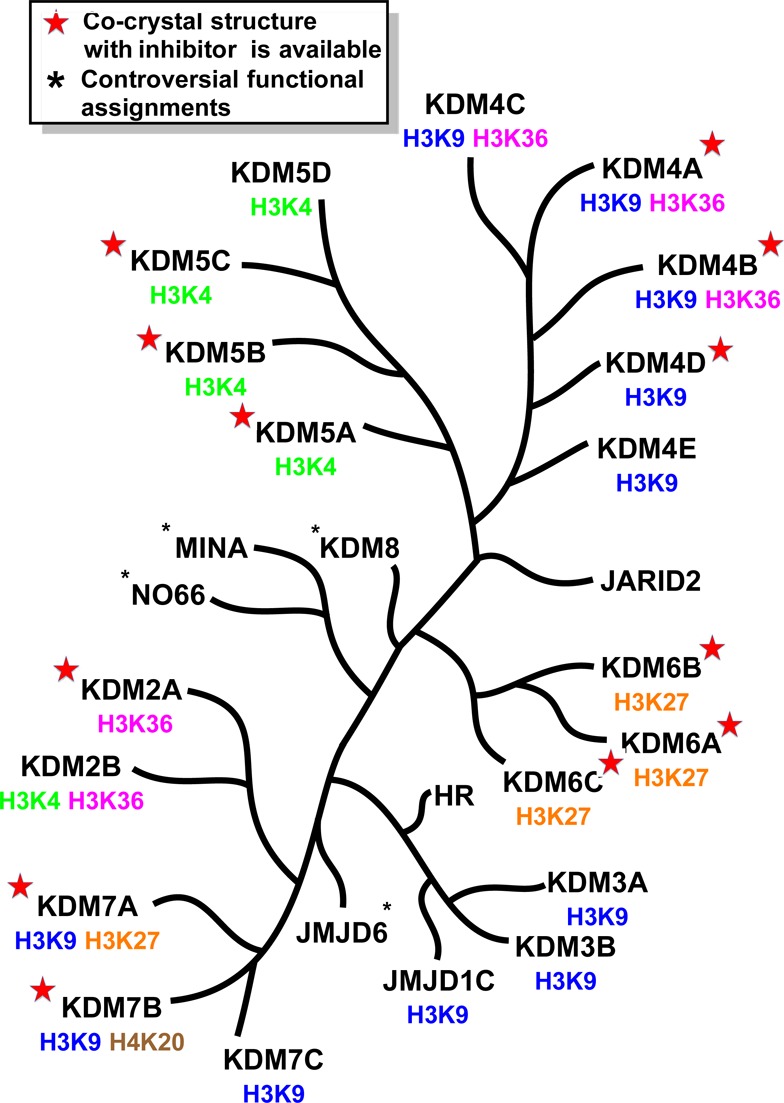
Phylogenetic tree of JmjC KDMs.
Table 2. KDM Subfamilies, Their Alternative Names, and Substrates.
| subfamily | members | other names | histone substrate |
|---|---|---|---|
| KDM2/7 | KDM2A | FBXL11, JHDM1A | H3K36me2/me1 |
| KDM2B | FBXL10, JHDM1B | H3K36me2/me1, H3K4me3 | |
| KDM7A | KIA1718, JHDM1D | H3K9me2/me1, H3K27me2/me1 | |
| KDM7B | PHF8, JHDM1F | H3K9me2/me1, H4K20me1 | |
| KDM7C | PHF2, JHDM1E | H3K9me2/me1 | |
| KDM3 | KDM3A | JMJD1A, JHDM2A | H3K9me2/me1 |
| KDM3B | JMJD1B, JHDM2B | H3K9me2/me1 | |
| JMJD1C | – | H3K9me2/me1 | |
| KDM4 | KDM4A | JMJD2A, JHDM3A | H3K9me2/me1, H3K36me2/me1 |
| KDM4B | JMJD2B, JHDM3B | H3K9me2/me1, H3K36me2/me1 | |
| KDM4C | GASC1, JMJD2C, JHDM3C | H3K9me2/me1, H3K36me2/me1 | |
| KDM4D | JMJD2D, JHDM3D | H3K9me2/me1 | |
| KDM4E | JMJD2E | H3K9me2/me1 | |
| KDM5 | KDM5A | JARID1A, RBP2 | H3K4me3/me2/me1 |
| KDM5B | JARID1B, PLU1 | H3K4me3/me2/me1 | |
| KDM5C | JARID1C, SMCX | H3K4me3/me2/me1 | |
| KDM5D | JARID1D, SMCY | H3K4me3/me2/me1 | |
| KDM6 | KDM6A | UTX | H3K27me3/me2/me1 |
| KDM6B | JMJD3 | H3K27me3/me2/me1 | |
| KDM6C | UTY | H3K27me3/me2/me1 |
Figure 43.

Domain architectures of some JmjC KDMs.
Some of the JmjC-containing oxygenases, including MYC-induced nuclear antigen 53 (MINA53) and nucleolar protein 66 (NO66), were initially assigned as JmjC KDMs;563−565 however, later studies demonstrated that they also have hydroxylase activity.566,567 Earlier studies, as well as recent work from Williams and co-workers using isolated proteins, provided NMR assignments of products. Qualitative cellular studies by these groups support the idea that MINA53 and NO66 are hydroxylases, rather than KDMs.566,568 In addition, the JmjC oxygenase, JMJD6, was reported as the first methylarginine demethylase of histone H3/H4 residues.569 However, this finding is controversial, as several following studies have been unable to reproduce the results.570 Furthermore, JMJD6 was also identified as a C-5 lysine-hydroxylase that acted on mRNA splicing-regulatory proteins and potentially even histone proteins.570,571 Very recently, Walport and co-workers reported that a subset of JmjC KDMs, namely KDM3A, KDM4A, KDM5C, and KDM6B, catalyze arginine demethylation of histone and nonhistone fragments.572 While this reported arginine demethylation activity is very exciting, more studies are required to conclusively establish arginine demethylation activity in cells. UTY (KDM6C), a JmjC enzyme that was previously reported not to display KDM activity,573 was recently discovered to catalyze the demethylation of H3K27, albeit with reduced activity compared to UTX (KDM6A) and KDM6B.574 In addition, there are some reports on KDM activity of PHF2 (KDM7C)575 and Hairless (HR),576 as well as JMJD5 (KDM8).577 Some of these activities, however, have not been reproduced clearly by others.578−580
Similar to all identified 2-OG oxygenases, JmjC KDMs have a barrel-like double-stranded β-helix (DSBH) fold, comprising eight antiparallel strands that form the conserved active site which bind to 2-OG and Fe(II).581 The iron binding motif is highly conserved and is comprised of one aspartic/glutamic acid and two histidine residues, which form part of the metal binding site, with an octahedral metal binding geometry where the rest of the sites are occupied by water molecules.582,583 The demethylation mechanism is proposed to begin with the binding of ferrous iron (Fe(II)) in the active site, followed by 2-OG coordination to the metal in a bidentate manner via its 1-carboxylate and 2-oxo groups, which displaces two water molecules (Figure 44).558,584,585 The observed coordination position of the 2-oxo group is always positioned trans to the metal-coordinating carboxylate of aspartic/glutamic acid. Following the 2-OG binding, the methylated lysine substrate then binds to the active site and weakens binding of the remaining water to the metal, thus activating oxygen binding. These proposals are supported by both crystallographic and spectroscopic studies in the literature.558,584−586 The oxygen then binds to the Fe(II) and reacts with the iron-bound 2-OG in an oxidative decarboxylation process to generate an active Fe(IV) intermediate and CO2. This reactive Fe(IV) species then reacts with the methylated lysine to give a hemiaminal intermediate, which fragments to give formaldehyde and a demethylated lysine substrate. Release of the succinate and coordination of water molecules regenerates the Fe(II) to complete the catalytic cycle.
Figure 44.

Ferrous iron (Fe(II)) and 2-OG-based oxidation mechanism of JmjC KDMs.
The JmjC KDMs, including but not limited to KDM2B, KDM3A, the KDM4 subfamily, and KDM5B, are implicated in various cancers, such as prostate cancer,587 breast cancer,588 bladder cancer,589 esophageal squamous cancer,553 primary mediastinal B cell lymphoma, and Hodgkin lymphoma.590 They have also been associated with neural development and/or function and conditions such as X-linked mental retardation.591,592 In addition, hypoxic conditions that activate the hypoxia-inducible factor (HIF) transcription factor are common in tumor tissues. KDM3A, KDM4B, and KDM5B are reported to be direct targets of HIF and regulate the transcriptional response to the hypoxic tumor environment, enhancing cancer progression.593−595 Therefore, selective inhibitors of JmjC KDMs are valuable chemical tools for studying the functions of these enzymes and testing therapeutic hypotheses concerning these enzymes. Therefore, they have been the focus of an increasing number of studies in recent years.20,596,597 In the following sections, we provide more details about each KDM subfamily, including their substrate specificity, domain architecture, and disease relevance, and importantly, we cover JmjC KDM inhibitors in the academic literature and discuss characterization of these inhibitors in biochemical, cellular, and/or in vivo assays.
3.2.1. JmjC KDMs Inhibitors
There has been a growing interest in discovering JmjC KDM inhibitors in recent years. The majority of these inhibitors reported to date are active site iron chelators and compete with the cofactor 2-OG in the active site. Although some potent inhibitors have been identified, achieving selectivity for different KDM subfamilies, within subfamilies, and against other Fe(II)-/2-OG-dependent enzymes is still the biggest challenge. In 2008, Rose and co-workers reported their studies on pursuing KDM4 inhibitors.598 The 2-OG analog, NOG (Figure 45), was one of the first identified JmjC KDM inhibitors and showed inhibitory activity against KDM4E (IC50 = 24 μM). However, it was not selective over the human HIF prolyl hydroxylase PHD2 or the asparaginyl hydroxylase factor-inhibiting-HIF (FIH). In this report, pyridine-2,4-dicarboxylic acid (2,4-PDCA) was also identified as a KDM4A and KDM4E inhibitor (IC50 = 0.7 and 1.4 μM, respectively).598 The bipyridine compound (46) also inhibited KDM4E with an IC50 of 6.6 μM. An additional scaffold that was examined in this study was hydroxamic acid, which is a well-known motif of HDAC inhibitors. 2-OG analogs, pyridyl carboxylates, bipyridyl and pyrimidine carboxylate-based scaffolds, as well as hyroxamic acid bearing compounds have been very common KDM inhibitor scaffolds that act as Fe(II) chelating agents. Therefore, the report by Rose and co-workers provided starting points for the development of more potent and selective KDM inhibitors.598
Figure 45.

Structures of JmjC KDMs inhibitors that contain common motifs.
Another common scaffold of KDM inhibitors is 8-hydroxyquinoline (8-HQ), which was discovered via an HTS campaign.599 5-Carboxy-8-hydroxyquinoline (IOX1, Figure 45) emerged as the most potent inhibitor of KDM4A and KDM4E (IC50 = 1.7 and 2.4 μM, respectively) in a biochemical assay from subsequent SAR studies of the 8-HQ scaffold.599 While IOX1 was equipotent for the KDM3, KDM4, and KDM6 subfamilies, it displayed slight selectivity over the KDM2 and KDM5 subfamilies as well as PHD2 and FIH.600 It should be noted that both NOG and IOX1 suffer from low cell permeability and need to be used in their methyl ester forms as prodrugs for cellular studies.601
On the basis of the crystal structure of KDM4A in complex with NOG and trimethylated lysine peptide (PDB ID: 2OQ6),602 Hamada and co-workers designed and synthesized hydroxamic acid derivatives as potential KDM4 inhibitors.603 Compound 47 (Figure 45) showed inhibitory activity for KDM4C and KDM4A, with IC50 values of 1 and 3 μM, respectively. This inhibitor showed better potency for KDM4C and KDM4A than NOG (IC50 values of 9.4 and 4.2 μM, respectively) and 2,4-PDCA (IC50 values of 500 and 250 μM, respectively) under the same assay conditions. While it was shown to be selective over PHD1 and PHD2, its selectivity over other KDM subfamilies was not reported. In addition, neither this compound nor its methyl ester derivative showed any activity in cell-based assays. Another hydroxamic acid derivative, methylstat (Figure 45), was designed based on an HDAC inhibitor, MS275 (Entinostat).604 The carboxylic acid derivative of methylstat (OH instead of OMe, 48) was equipotent against KDM4A, KDM4C, and KDM4E (IC50 = 4.3, 3.4, and 5.9 μM, respectively), displaying moderate selectivity against KDM6B, PHF8, and PHD1–3 (IC50 = 43, 10, and 31–83 μM, respectively). This compound showed virtually no inhibition against LSD1 and HDACs. The cellular activity of methylstat in KYSE150 (a JMJD2C-sensitive esophageal carcinoma cell line) in a growth inhibition assay was investigated and methylstat inhibited KYSE150 cell growth with GI50 at approximately 5.1 μM. The carboxylic acid derivative (48) did not show any significant growth inhibition at up to 100 μM, due to its poor cell permeability. Methylstat increased H3K4, H3K9, H3K27, and H3K36 methylation at almost all methylation states in a concentration-dependent manner in KYSE150 cells. Similar results were observed in MCF7 cells, albeit to a different degree for different marks. Quantification of the activity of metylstat in KYSE150 cells provided EC50 values of 10.3 and 8.6 μM for H3K4me3 and H3K9me3, respectively. On the other hand, in MCF7 cells, the EC50 values for H3K4me3 and H3K9me3 were 6.7 and 6.3 μM, respectively. Interestingly, increased cellular levels of H3K79me3 and H4K20me3 were also observed upon treatment with methylstat. The same research group reported a fluorescent probe based on methylstat and developed a fluorescence polarization (FP) binding assay for KDM2A.605 The same group also generated a peptidic affinity probe derived from methylstat for histone demethylases. This probe was then utilized to purify KDM2A from a mixture of purified enzymes and histone proteins and to enrich other H3K36 targeting JmjC-KDMs from HeLa cell extracts.606
JIB-04, a pyridine hydrazone containing compound, was identified as a pan JmjC KDM inhibitor (Figure 45).607,608 In biochemical assays, JIB-04 was most potent for KDM5A with an IC50 of 230 ± 40 nM and KDM4D with an IC50 of 290 ± 18 nM. It also inhibited KDM4A, 4B, and 4E with similar potencies (IC50 = 445 ± 30, 435 ± 70, and 340 ± 50 nM, respectively) and was slightly less potent for KDM4C (IC50 = 1100 ± 200 nM) and KDM6B (IC50 = 855 ± 40 nM). JIB-04 did not inhibit LSD1 or other epigenetic enzymes including sirtuins, PRMTs, and PKMTs. Compared to 2,4-PDCA, it was about 5-fold more potent at inhibiting KDM4D activity in vitro. Competition assays with JIB-04 showed that, unlike known JmjC KDM inhibitors, it was not competitive with 2-OG. It was postulated that JIB-04 might be competitive with iron, and potentially, with the histone substrate. Altogether, JIB-04 is a pan JmjC KDM inhibitor in biochemical assays. However, its MOA is still not very clear and requires further work.
JIB-04 showed anticancer activity in cell cultures for several tumor types and in vivo in mouse xenograft models without causing general toxicity. Importantly, this inhibitor increased survival in an aggressive breast cancer model. Antiproliferative effects of JIB-04 are suggested to be mediated, at least in part, through direct cancer-specific transcriptional changes in genes that control cell growth, thereby resulting in tumor cell death.
The compounds shown in Figure 45 represent the most common inhibitor scaffolds and are broad-spectrum inhibitors of KDMs. In the following sections, we discuss the studies toward achieving selectivity between and within subfamilies of KDMs.
3.2.2. Inhibitors of KDM2/7 Subfamily
KDM2 and Plant Homeodomain Finger (PHF) proteins are closely related to KDM7, and therefore, they can be considered all together as a KDM2/7 subfamily of KDMs that is comprised of KDM2A (F-box and leucine rich repeat protein 11 (FBXL11), also known as JHDM1A), KDM2B (FBXL10, also known as JHDM1B), KDM7A (KIAA1718, also known as JHDM1D), PHF8 (KDM7B, also known as JHDM1F), and KDM7C (PHF2, also known as JHDM1E).559 KDM2A is the first JmjC KDM identified, and it demethylates H3K36me2/me1.458 KDM2B demethylates H3K4me3609 as well as H3K36me2,610 while in contrasting reports it was suggested to be an H3K4me3 demethylase rather than an H3K36me2 demethylase.611,612 The KDM2 family contains a CxxC zinc finger domain, a PHD domain, and an Fbox domain along with the JmjC domain.
KDM2 demethylases are implicated in both tumor promotion and suppression, depending upon cellular context.613,614 KDM2A has a role in cell differentiation,615 proliferation,616 and regulation of NF-κB.617 It has been associated with various cancers, including lung618 and gastric cancers.619 Overexpression of KDM2B is observed in leukemias,620 pancreatic cancer,621 as well as triple-negative breast cancer.622
The catalytic domains of the PHF proteins are highly similar to that of the KDM2 proteins, however, the PHF proteins recognize H3K4me3 through their PHD domain and demethylate H3K9me2/me1, H3K27me2/me1, and nucleosomal H4K20me1.560,623−625 PHF8 was initially characterized as a H3K9me2/me1 demethylase,623 while KDM7A was reported as a dual specificity histone demethylase targeting both H3K27me2/me1 and H3K9me2/me1.624,626 Substrate specificity contradicting the initial reports has also been reported and attributed to different distances between the JmjC and PHD finger domains in these two proteins.560 It was suggested that, in vivo, both enzymes recognize or “read” the H3K4me3 mark via their PHD domains, but in PHF8, the shorter and possibly more flexible linker between the two domains enables the active site of the JmjC domain to reach the target H3K9me2/me1.560 On the other hand, in KDM7A, the linker is longer but more ordered, resulting in a conformation that renders the enzyme inactive toward H3K9me2/me1 marks and selectively active toward H3K27me2/me1 marks.560 However, it should be noted that studies on the C. elegans and zebrafish homologues of KDM7A suggest that the presence of H3K4me3 does not necessarily lead to preferential demethylation of H3K27me2 over H3K9me2/me1 in vivo.624,627 Another proposal is that in a trans-histone peptide-binding mechanism, the enzyme recognizes H3K4me3 on one H3 tail and targets H3K9me2/me1 or H3K27me2/me1 on another H3 tail. PHF2 has been suggested to demethylate H3K9me1 in vivo;575 however, the catalytic activity of this enzyme has not been reproduced in vitro.580 It was suggested that there must be other regulatory factors necessary for the observed enzymatic activity in vivo for PHF2, which shares significant similarity in its catalytic domain with KDM7A and PHF8, or that it perhaps acts on nonhistone substrates.580
KDM7A is required for brain development,626 PHF8 is vital for normal development, and the mutations in the gene cause X-linked mental retardation.628 Overexpression of PHF8 has also been shown in several cancers.629,630 PHF2 has been described as a potential tumor suppressor in association with p53.631
Given their association with important biological functions and diseases, there has been a growing interest in developing inhibitors of the KDM2/7 subfamily of demethylases. In 2012, Rose and co-workers identified the plant growth regulator daminozide (Figure 46) as a selective inhibitor of KDM2A (IC50 = 1.5 μM) in an AlphaSecreen assay.559 The potency of daminozide against KDM2A (IC50 = 1.5 μM) was confirmed by an orthogonal, formaldehyde dehydrogenase (FDH) coupled assay, which monitored formaldehyde production. Daminozide was also active against two other members of the KDM2/7 subfamily, PHF8 and KDM7A, with IC50 values of 0.55 μM and 2.1 μM, respectively. Daminozide was selective for the KDM2/7 subfamily over the other representative demethylase subfamily members tested (KDM3A, KDM4E, KDM5C, KDM6B: IC50 > 100 μM). In addition, no inhibition was observed at 1 mM against the other 2-OG oxygenases (PHD2, FIH, and BBOX1) that catalyze hydroxylation. Kinetic experiments showed that daminozide was a competitive inhibitor of KDM2A with respect to 2-OG (Ki = 1.97 μM). On the other hand, mixed inhibition with respect to the peptide substrate was observed, such that inhibitor binding was primarily to the enzyme-peptide complex (Ki = 85 μM). Given its structure similarity to NOG, the selectivity achieved is quite interesting. On the basis of the cocrystal structures of the inhibitor in complex with KDM4A, FIH, and PHF8 (PDB IDs: 4AI9, 4AI8, 4DO0), it was proposed that the selectivity of daminozide for the KDM2/7 subfamily could be, at least in part, a result of a tight fit. This tight fit arises from the binding of the dimethylamino group trans to His247, where its two methyl groups are accommodated in a hydrophobic pocket that is conserved in the KDM2/7 subfamily and is observed (by crystallography) or predicted (by sequence alignments) to be less tight in other demethylases/oxygenases tested. Supporting this proposal, the removal of two methyl groups resulted in a slightly more potent (IC50 = 0.25 and 0.48 μM for KDM2A and PHF8, respectively) but less selective inhibitor (IC50 = 0.20 and 0.48 μM for KDM4E and KDM5C, respectively). The biological effects of this inhibitor in cell-based studies were not reported.
Figure 46.

Structures of KDM2/7 subfamily inhibitors.
While daminozide was once widely used as a plant growth retardant, it was later withdrawn because of genotoxicity that was believed to arise from its 1,1-dimethylhydrazine group. Therefore, in 2013, Suzuki and co-workers aimed to identify novel KDM2/7 inhibitors without the 1,1-dimethylhydrazine moiety and used hydroxymate derivatives they previously prepared (e.g., compound 47, Figure 45 in section 3.2.1) as starting points.614 Compound 49 (Figure 46) emerged as the best KDM2/7 inhibitor, showing the highest potency for KDM7A (KDM7A: IC50 = 0.2 μM; KDM2A: IC50 = 6.8 μM; and KDM7B: IC50 = 1.2 μM). It also exhibited selectivity for KDM2/7 over KDM4A (IC50 > 120 μM), KDM4C (IC50 = 83 μM), KDM5A (IC50 = 55 μM), and KDM6A (IC50 > 100 μM). It should be noted, however, that different assay conditions were used for different subfamilies, making the direct comparison rather difficult. The H3K27me2 mark was concentration-dependently raised in the presence of 49, displaying inhibition of KDM7A and KDM7B in N2a cells in which KDM7 was expressed. In addition, growth inhibition in HeLa and KYSE150 cells was observed following treatment with compound 49. On the basis of the study which reported that KDM7B stimulates the transcription of the E2F1 transcription factor in HeLa cells and promotes cell cycle progression,625 downregulation of the expression of E2F1 in HeLa cells was investigated and compound 49 significantly decreased the mRNA level of E2F1 at 80 μM.
A new scaffold based on the bipyridine-containing inhibitors was designed by replacing one of the pyridine rings with a triazole ring.632 It was postulated that the triazole nitrogen atom would be able to coordinate the Fe(II) similar as a pyridine nitrogen, and that the construction of the triazole ring via copper-catalyzed click reactions would allow for rapid SAR exploration of this new scaffold. The enantiomerically pure (R)-50 (Figure 46) potently inhibited KDM2A (IC50 = 63 nM), while the (S)-enantiomer was about 50-fold less potent. This change in potency showed that the stereochemistry of the substituted piperidine played an important role in binding, and that the (R)-enantiomer positioned the amide substituent in a favored position for binding to KDM2A. Compound 50 was shown to be selective against several other KDMs including: KDM5C (30-fold), KDM4A, KDM4C (>200-fold) and KDM3A, KDM4E, and KDM6B (>1,500-fold). In addition, another derivative from this scaffold, which replaced the phenyl group with an ethyl group, possessed lower potency but showed selectivity for KDM5C over other KDMs. While (R)-50 showed high in vitro potency and selectivity for KDM2A over other KDM subfamilies, it was not reported whether or not it possessed selectivity against other members of the KDM2/7 subfamily. In addition, this study did not report activity of this inhibitor in cell-based assays.
3.2.3. Inhibitors of KDM4 Subfamily
The KDM4 subfamily is comprised of five members: KDM4A (also known as JMJD2A, JHDM3A, and JMJD2), KDM4B (also known as JMJD2B), KDM4C (also known as GASC1, JMJD2C, and JHDM3C), KDM4D (also known as JMJD2D), and KDM4E (also known as JMJD2E and KDM4DL). KDM4F is identified as a pseudogene. KDM4A-C catalyzes the demethylation of H3K9me3/me2 and H3K36me3/me2.552−555 KDM4D can only demethylate H3K9me3/me2, while KDM4E catalyzes the removal of methyl groups from H3K9me3 and H3K56me3.554,633 It has also been shown that the KDM4 family can demethylate H1.4K26me3/me2.634 KDM4A, 4B, and 4C share more than 50% sequence identity and include JmjN, JmjC, two PHD, and two Tudor domains. On the other hand, KDM4D and KDM4E are significantly shorter proteins missing the C-terminal region, which includes the PHD and Tudor domains. The JmjN domain interacts extensively with JmjC and provides structural integrity.
KDM4 subfamily members are coactivators of AR and are key in androgen signaling.587,635−637 They are overexpressed in a variety of human cancers as well as cardiovascular diseases and mental retardation. Overexpression of KDM4A is highly dominant in squamous cell carcinoma, breast, and colon cancers.638−640 KDM4B is highly expressed in ER+ breast cancer and also associated with prostate cancer.637,641 KDM4C is also overexpressed in numerous hematological and solid cancers, including squamous cell carcinoma and AML.588,642,643 KDM4D and 4E interact with AR as well. KDM4D was shown to be involved in the TNF-α response and, therefore, might influence tumorigenesis in cancer cells.644 Thus, the KDM4 subfamily of demethylases is one of the most well-studied, and there is a considerable interest in pursuing them as potential therapeutic targets.
A series of 8HQ-containing KDM4 inhibitors645 were tested in LNCaP cells. A few of them inhibited LNCaP cell growth with IC50 values in the micromolar range.646 Among these compounds, B3 (Figure 47) was the most potent, inhibiting KDM4B with an IC50 of ∼10 nM in biochemical assays. B3 is a close analogue of ML324 (with a dimethylamino group instead of the phenyl group of B3), which was identified as a KDM4A and KDM4E (IC50 = 0.92 μM) inhibitor by Rai and co-workers.645 No selectivity data was reported against other members of the subfamily for ML324. B3 showed some selectivity (not quantified) against KDM4A, KDM4C, KDM4D, and KDM5A, while selectivity for other KDMs were not reported. The physical binding of B3 to KDM4B was shown by transverse relaxation enhanced spectroscopy (TROSY)-based heteronuclear single quantum correlation (HSQC) spectra via observed shifts in specific cross-peaks of the 1H–15N TROSY-HSQC spectrum of the catalytic domain of KDM4B. B3 exhibited high sensitivity to the fast-growing AR-negative PC3 cells (IC50 = 40 nM) and also inhibited AR-positive cell lines, including LNCaP and VCaP with IC50 values in the submicromolar concentrations. B3 also abrogated androgen-stimulated LNCaP cell growth. In addition, it was effective (with micromolar IC50 values) in inhibiting the growth of other cancer cell lines, such as MDA-MB2 and MCF-7 breast cancer cell lines. Furthermore, treatment with B3 resulted in notable inhibition of tumor growth in an in vivo PC3 xenograft tumor model. On the basis of these results, it was suggested that KDM4B promotes prostate tumorigenesis by activating the transcription of Myb-related protein B (BMYB)-targeted genes, such as polo-like kinase 1 (PLK1), which are critical for cell-cycle progression via an AR-independent mechanism. Overall, B3 showed good in vitro and in vivo potency. However, further characterization and optimization are needed to establish or achieve better selectivity against other KDMs.
Figure 47.

Structures of 8HQ-containing KDM4 inhibitors.
Two other studies exploring the 8HQ chemotype as KDM4 inhibitors were published in 2015. Thinnes and co-workers utilized a three-component Betti reaction for the construction of 7-substituted 8HQ derivatives and identified CCT1 (Figure 47) as their most potent inhibitor (IC50 = 5 μM) against KDM4C and 4E.647 In biochemical assays, CCT1 displayed 7- to 20-fold selectivity against other KDMs (KDM2A, KDM3A, KDM5C, and KDM6B) as well as PHD2 and FIH. CCT1 increased H3K9me3 levels in MCF7 cells (EC50 = 12 μM) and in HeLa cells (EC50 = 9 μM). However, no or little inhibition of isolated PHD2 and FIH by this inhibitor in vitro (IC50 = 96 and >100 μM, respectively) did not correlate with blockage of HIF hydroxylation observed in cell-based assays, suggesting that the effects of CCT1 may not be exclusively due to direct JmjC KDM inhibition. Modifications at the 2-position of 5-carboxy-8HQ led to the identification of compound 51 (Figure 47) with micromolar range IC50 values in biochemical assays.648 While some antiproliferative activity in several cancer cell lines was reported, the selectivity of this compound over other KDMs and related enzymes was not reported, with the exception of the hydroxylase PHD2.
Very recently, Westaway and co-workers identified pyridine-4-carboxylic acids 52 and 53 as KDM4 subfamily inhibitors with IC50 ≤ 100 nM and with ≥50-fold selectivity over KDM6B in biochemical assays (Figure 48).649 However, compounds 52 and 53 were also found to be potent inhibitors of KDM5C with IC50 = 100–125 nM. These two inhibitors showed activity in a cellular high content imaging assay against overexpressed KDM4C with IC50 values of <10 μM, while activity was also observed in U2OS cells overexpressing KDM5C, albeit with lower potency. Additional work is required to achieve selectivity for the KDM4 subfamily over KDM5C.
Figure 48.

Pyridine-4-carboxylic acid containing KDM4 inhibitors.
In 2016, Roatsch and co-workers described the identification of KDM4A inhibitors via virtual screening and follow-up optimization. The study yielded compound 54 (Figure 48) with a Ki of 186 nM (IC50 = 0.37 ± 0.028 μM).650 However, testing of closely related analogs of this inhibitor against two other demethylases revealed that while the compounds were selective for KDM4A over KDM6B, they were slightly more potent (2-fold) for KDM5A. A cocrystal structure (PDB ID: 5ANQ) and kinetic experiments revealed that these inhibitors were competitive with 2-OG. KYSE-150 cells, which have been shown to possess high levels of KDM4C, were used to test cellular activity of the inhibitors. However, cell permeability and solubility were problematic issues for these inhibitors, and for some of them, a reduction of KYSE-150 cell proliferation was seen at high concentrations (e.g., 50 μM). The conversion of the carboxylic acid to its ester as a prodrug was also attempted, but no clear improvement in cellular activity was observed.
Docking a library of 600000 fragments into a model of the KDM4A active site resulted in the identification of 5-aminosalicylate fragments with docking poses in two distinct but overlapping orientations.651 The fragments were then covalently linked and derivatized further based on the docking model. The resulting compounds contained a pyridyl-4-carboxylate moiety, which in turn, resembled 2,4-PDCA or pyridine-containing KDM inhibitors. This approach resulted in the discovery of KDM4C inhibitors 55 and 56 (Ki = 43 and 680 nM, IC50 = 12 and 3.8 μM, respectively) (Figure 48). While 56 was selective (>11–26-fold) for KDM4C over FIH, KDM2A, and KDM6B, it lacked selectivity against KDM3A and KDM5B. Both inhibitors were equipotent for KDM4C and KDM4D. Although 55 was the most potent, it did not show any selectivity except against FIH. The docking predictions were supported by cocrystal structures (PDB ID: 5A7O and 5A7W). These inhibitors are 2-OG competitive. No cellular studies were reported with these inhibitors. Limited selectivity and lack of cellular studies limit the potential utility of these compounds. Additional optimization is likely needed to achieve better selectivity.
Woon and co-workers analyzed KDM4 structures and recognized that the 2-OG and substrate binding sites are adjacent, creating a large pocket.652 As described earlier, KDM4A–C accepts both H3K9me3/me2 and H3K36me3/me2 as their substrates, while KDM4D–E only accepts H3K9me3/me2 as their substrate. Therefore, it was postulated that inhibitors combining 2-OG and substrate mimics might achieve selectivity within the KDM4 subfamily, as well as across other KDMs. N-oxalyl-d-cysteine (DNOC, Figure 49), which is a 2-OG mimic, and a H3 substrate fragment (residues 7–14) containing H3K9me3, were used to develop bisubstrate inhibitors (Figure 49). A suitable cross-linking with DNOC between residues 10–13 was obtained via the addition of a thinyl radical to a corresponding allylglycine-containing peptide, resulting in the discovery of compound 57 (Figure 49), which potently inhibited KDM4A (IC50 = 0.27 μM, Tm = +7.1 °C) and KDM4E (IC50 = 0.09 μM, Tm = +12.1 °C) with no significant inhibition against other KDMs (>300-fold) including KDM2A, PHF8, PHF2, KDM3A and KDM6B, as well as PHD2 and FIH. It is important to note that selectivity for KDM4A/E over KDM3A (IC50 > 100 μM, Tm = −0.3 °C) was achieved despite that KDM3A targets the same substrate, H3K9me3/me2. It was also found that the K9me3 form of the bisubstrate inhibitors, such as compound 57, exhibited better inhibitory activities. It was also proposed that an H3K36me3-mimicking inhibitor could be selective for KDM4A over KDM4E. Indeed, compound 58 (Figure 49), obtained by cross-linking DNOC with P38 of another H3 fragment (residues 31–41) containing H3K36me3, exhibited 60-fold selectivity for KDM4A (IC50 = 1.5 μM, Tm = 5.8 °C) over KDM4E (IC50 = 91 μM, Tm < 1 °C) with no inhibitory activity or Tm shift against any of the other aforementioned KDMs (around 40-fold selective over KDM2A and KDM3A and at least 130-fold for others). This study has demonstrated that by exploiting the inherent substrate selectivity of JmjC KDMs, it is possible to achieve a high degree of selectivity within a KDM subfamily and between KDM subfamilies. Selectivity over PHD2 and FIH also points to possible discrimination against 2-OG oxygenases. However, cell membrane permeability of these inhibitors was not reported. It is likely that these inhibitors lack sufficient cell permeability, thus limiting their use in cell-based studies.
Figure 49.

Structures of peptide-based KDM4 inhibitors.
Wang and co-workers conducted a HTS campaign, which resulted in two pyridine-containing compounds 59 and 60 (Figure 50) with inhibitory activities against KDM4A and KDM2A (Ki = 0.52 ± 0.06 and 0.42 ± 0.005 μM and IC50 = 2.58 ± 0.25 and 3.17 ± 0.37 μM, respectively).653 However, selectivity against other KDMs was not reported.
Figure 50.

Structures of other KDM4 inhibitors.
Rüger and co-workers used an approach651,654 that is reminiscent of a method involving the covalent binding of docked fragments.651 They proposed to replace the α-keto acid of 2-OG with an acylhydrazide moiety, inspired by the success of this functional group in daminozide.654 Due to the ease of synthesis, however, they used the hydrazone as a Fe(II)-binding moiety instead. The second carboxylic acid of 2-OG was replaced by a tetrazole moiety. Inhibitors combining these two moieties attached by a linker of different length were tested to find the optimum connection length. As a result, an inhibitor (61, with an IC50 of 2.38 ± 0.37 μM and 46.6 ± 0.94 μM in two orthogonal KDM4A biochemical assays) was obtained as the most potent compound (Figure 50). This compound showed some selectivity for KDM4A over KDM5A (4-fold) and KDM6B (40-fold). On the other hand, selectivity over other members of the KDM4 subfamily and additional KDMs was not reported. Since this inhibitor is a metal chelator by nature, kinetic experiments were performed to show that the effects were not a result of promiscuous metal chelation or ejection. This inhibitor was found to be competitive with 2-OG, while other known metal chelators as controls did not display similar inhibitory profiles.
Several other KDM4 inhibitors (Figure 50), which utilized different scaffolds than those mentioned up to this point, have been reported. For example, an iridium(III) complex (62) was synthesized and found to inhibit KDM4D with an IC50 of around 15 μM in a fluorescence-based assay.655 It has been shown that this complex was inert to Fe(II); however, no further explanation or MOA for this complex was reported.
Leurs and co-workers screened DNA-encoded peptide libraries against KDM4 by phage display. Through this effort, two cyclic peptides targeting KDM4C were identified.656 These initial peptides were optimized via amino acid replacement, truncation, and other modifications, leading to the discovery of an inhibitor (63, Figure 50) with an IC50 of 0.6 ± 0.02 μM. This peptide-based inhibitor targets KDM4C independent of the substrate and cofactor via interactions located on the surface, remote from the active site, within less conserved regions of KDM4C. This inhibitor was not active in cell-based assays, most likely due to its poor cell permeability. The interaction sites identified in this study may provide new opportunities for targeting KDM4C to develop potent, selective inhibitors and biological probes for the KDM4 subfamily. In a similar study, a lysine analog containing a geometrically constrained side chain (64, Figure 50) was shown to be recognized as a substrate by KDM4E and KDM7B.657
Derivatives of curcumin were also identified as potential KDM4 inhibitors in vitro.658 A curcuminoid (65) that was identified via virtual screening demonstrated competitive inhibition against KDM4A (IC50 = 6.4 μM and Ki (H3K9me3) = 5.5 ± 1.6 μM) (Figure 50). While KDM4B was also inhibited with equal potency, weaker inhibitory effects were observed toward KDM4D and KDM4E.659 The selectivity over other KDMs was not reported.
3.2.4. Inhibitors of KDM5 Subfamily
The KDM5 (also know as JARID) subfamily of demethylases catalyzes the demethylation of H3K4me3/me2, a transcription-activating mark.660 This subfamily is comprised of four members: KDM5A-D, which show a high degree of homology in sequence and domain organization. The KDM5 subfamily differentiates itself from other histone demethylases in that each member contains an atypical split catalytic Jumonji domain with insertion of a DNA-binding ARID and histone-interacting PHD1 domain, which separates it into two sections, JmjN and JmjC.661 It has been shown that the ARID and PHD1 domains are expendable for in vitro enzymatic activity, though the Zn-binding domain, which is immediately next to C-terminal to JmjC, is essential.662 The constructed linked JmjN-JmjC domain from KDM5A retains full structural integrity, as well as the metal ion- and 2-OG-binding features of the other structurally characterized Jumonji domain demethylases.662
KDM5A (also known as JARID1A and RBP2) was first identified as Retinoblastoma-Binding Protein 2 (RBP2) and is ubiquitously expressed.663 It is implicated in cellular differentiation and cell cycle regulation due to its interaction with retinoblastoma protein (pRB).663,664 KDM5A is overexpressed in gastric cancer,665 lung cancer,666 and hepatocellular carcinoma.667 The amplification of the KDM5A locus in breast cancer contributes to drug resistance in breast cancer.668 KDM5A is found in association with the PRC2 complex,96 as well as other repressive chromatin modulators, such as G9a and the HDAC1/2/REST complex.591,669 Overexpression of KDM5B (also known as PLU-1 and JARID1B) has been identified in bladder cancer,670 prostate cancer,671 colorectal cancer,670,672 lung cancers,670 and malignant melonoma.673 KDM5B-expressing cells form a distinct subpopulation that is slow-cycling and chemo-resistant in melanoma.673 KDM5C (also known as JARID1C and SMCX) appears to be involved in the development of renal carcinoma via regulation of the von Hippel-Lindau tumor suppressor protein.674 It has also been associated with cervical cancer as an oncogenic target of human papillomavirus.675 KDM5D (also known as JARID1D and SMCY) is 85% identical to KDM5C, and it has been implicated in prostate cancer.676
Very recently, Vinogradova and co-workers reported the discovery of a KDM5 subfamily selective inhibitor, CPI-455 (Figure 51), which displayed high potency for KDM5A (IC50 of 10 ± 1 nM) and similar potencies for KDM5B and KDM5C.677,678 Importantly, it showed significantly weaker potency toward KDM4C and KDM7B (∼200- and 770-fold, respectively), and no considerable inhibition of KDM2B, KDM3B, and KDM6A. A closely related compound that was 25-fold less potent for KDM5A was also developed as an inactive (or less active) control in this study. In MOA studies, CPI-455 was competitive with 2-OG. Furthermore, the cocrystal structure of the intact amino-terminal half of the KDM5A enzyme, including its JmjN, ARID, PHD1, catalytic JmjC, and α-helical domains, in complex with CPI-455 was obtained (PDB ID: 5CEH). The cocrystal structure showed that the inhibitor binds at the demethylase active site, and the nitrile group of the compound makes a single interaction with the active site metal ion. The pocket occupied by the inhibitor fully overlapped with the 2-OG binding site, which indicated a competitive MOA, thereby confirming the biochemical assay results. KDM5 inhibition by CPI-455 resulted in a concentration-dependent increase in global H3K4me3 levels that were detected only after 2 or more days of treatment in HeLa cells. The less active compound did not affect H3K4me3 levels at the same concentrations. Removal of CPI-455 led to a rapid reversal of H3K4me3 increases in HeLa cells. H3K4me3 and H3K4me2 levels were also concentration-dependently increased in melanoma (M14), breast cancer (SKBR3), and NSCLC (PC9) cells treated with CPI-455 for 5 days. Again, the less active compound did not affect the levels of these marks in the same cell lines. Altogether, these data indicate that CPI-455 is a KDM5 subfamily selective inhibitor and a valuable chemical tool for investigating biological functions of the KDM5 subfamily.
Figure 51.

Structures of KDM5 inhibitors.
It has been previously described that a drug-tolerant state (DTP) gives rise to drug-tolerant expanded persister cancer cells (DTEPs); DTEPs display increased expression of KDM5A, and the emergence of these populations is dependent on KDM5A. CPI-455 significantly reduced the number of DTPs in cell culture models, suggesting that the demethylase activity was required to establish drug tolerance. In addition, CPI-455 showed low plasma clearance (CL = 4.4 mL/min/kg) and excellent oral exposure (F = 100%) in mice. However, it had relatively high plasma protein binding (PPB) in mice (98.8%). Therefore, further optimization of this lead was conducted.679 On the basis of the cocrystal structure of KDM5A in complex with CPI-455 (PDB ID: 5CEH), SAR around the isopropyl and phenyl moieties were investigated, leading to the identification of compound 66 (Figure 51) with an IC50 of 15 nM in a KDM5A biochemical assay and an EC50 of 340 nM in a cellular assay (PC9 H3K4me3). Compound 66 also inhibited KDM5B and KDM5C with high potency (IC50 of 4.7 and 65.5 nM, respectively). It was selective for the KDM5 subfamily over KDM1A, 2B, 3B, 4C (IC50 = 1.9 μM, > 100-fold selective for KDM5A), KDM6A, and KDM7B. It was also selective for the KDM5 subfamily over a broad panel of nonepigenetic targets including >300 kinases and GPCRs (<50% inhibition at 10 μM against all proteins tested). When dosed in mice at 50 mg/kg twice daily, 66 showed an unbound total maximum concentration (Cmax) > 15-fold of its cell EC50 value. Therefore, inhibitor 66 is a promising KDM5 subfamily selective inhibitor that is suitable for studying biological functions of KDM5 in vivo. The same research group also reported hybrid molecules of CPI-455 with an HTS hit.680 The resulting compounds, 1,7-naphthyridones, displayed high potency and selectivity for KDM5A over KDM4C and KDM2B. However, these compounds lacked cellular activity.
Concurrently, Johansson and co-workers reported crystal structures of the catalytic core of the human KDM5B enzyme in apo state and in complex with different inhibitor chemotypes.681 In this study, KDM5B structures were superimposed on KDM6A (PDB ID: 3AVR) and KDM4A (PDB ID: 2P5B) structures and it was observed that while the domain architecture and the overall fold of KDM5B were similar to those of KDM6A, the loop structures and the JmjC domain differed significantly. On the other hand, the KDM5B JmjC domain was shown to be more closely related to that of KDM4A. Comparison of the 2-OG-binding pockets between KDM5B (PDB ID: 5A1F) and KDM6A revealed considerable differences, while comparison with KDM4A (PDB ID: 2P5B) showed a similar shape of the 2-OG-binding pocket. It was also found that inhibitor KDM5-C49 (Figure 51) displayed high potency for the KDM5 subfamily (IC50 = 7, 4, 13, 15 nM for KDM5A, 5B, 5C, and 5D, respectively) in biochemical assays. Importantly, this inhibitor was selective for the KDM5 subfamily over KDM4C (IC50 = 210 nM), KDM6B (IC50 = 1.4 μM), KDM3A (IC50 = 780 nM), and KDM2A (IC50 = 2.2 μM). KDM5-C70 (Figure 51) was developed as a cell-permeable prodrug, which can be hydrolyzed by an esterase within the cell to generate KDM5-C49. The crystal structure of KDM5-C49 in complex with KDM5B revealed that the inhibitor occupies the 2-OG-binding site, with the pyridine nitrogen and the aminomethyl nitrogen forming a bidentate interaction with the catalytic metal (Figure 52A). Comparison of Ki values calculated from the IC50 values above showed a 25–150-fold selectivity for the KDM5 subfamily over the KDM6 subfamily, whereas the higher structural similarity to the KDM4 subfamily was reflected by 12–76-fold differences in Ki for the KDM5 subfamily over KDM4C. In addition, cocrystal structures of KDM5-C49 in complexes with KDM6C (PDB ID: 4UF0) and KDM4A (PDB ID: 5FPV) revealed numerous critical similarities and differences between these subfamilies. The inhibitor occupied the cofactor site and coordinated the metal in a similar manner in KDM5B, KDM4A, and KDM6C structures. In KDM4A, the interactions with KDM5-C49 were almost identical to that in KDM5B, therefore supporting the observed low selectivity (7–8-fold for KDM5B over KDM4C). The cell-permeable derivative KDM5-C70 had an antiproliferative effect in myeloma cells (7 days, 50% reduction at 20 μM) and also resulted in a genome-wide increase in H3K4me3 levels, as determined through ChIP-Seq experiments. While this compound represents one of the most selective inhibitors for the KDM5 subfamily, the relatively weak cellular potency decreases its potential as a useful tool. Thus, further optimization to improve cellular potency is required.
Figure 52.

(A) KDM5-C49 (orange) binding interactions with KDM5B (PDB ID: 5A3T) and (B) GSK467 binding interactions with KDM5B (PDB ID: 5FUN). Side chains are displayed in gray, water molecules as red spheres, and the metal centers as green spheres.
Shortly after the publication by Johansson and co-workers, Horton and co-workers published their work investigating the structural basis of KDM5A inhibition by using a set of inhibitors, including a close analog of CPI-455 and KDM5-C49, in complex with KDM5A in the presence of Mn(II) (PDB ID: 5IVE and 5ISL).682 The in vitro potencies and binding affinities of these inhibitors with all four KDM5 family members were also reported. Most of these inhibitors contain an iso-nicotinic acid core, similar to KDM5-C49. Here, we focus on KDM5-C49 and KDM5-C70, for which Horton and co-workers explored selectivity and cellular activities alongside structural analysis of inhibitor binding. KDM5-C49 inhibited KDM5A, KDM5B, and KDM5C with similar potencies with IC50 values in the nanomolar range in two independent assays. The results are in agreement with the ones generated by Johansson and co-workers. KDM5-C49 did not inhibit KDM6A and KDM6B by more than 50% even at 50 μM. It showed 10-fold weaker potency against KDM4A than KDM5B. To demonstrate cellular activity, KDM5-C70 was tested in breast cancer cells (MCF7, MDA-MB-231, BT474, and ZR-75–1) since KDM5A and KDM5B have been shown to be overexpressed in human breast cancers.683,684 Treatment of MCF7 and MDA-MB-231 breast cancer cells with KDM5-C70 significantly increased global levels of H3K4me3, while this inhibitor had only slight effects on H3K4me2/me1 and H3K27me3 marks (KDM6 subfamily) and H3K9me3/H3K36me3 marks (KDM4 subfamily). KDM5-C70 at 5 μM inhibited the growth of MCF7, BT474, and ZR-75–1 cells by 85%, 97%, and 70%, respectively. In MDA-MB-231, PC9 lung cancer, and MCF10A immortalized, nontransformed mammary epithelial cells that have been shown to be relatively resistant to KDM5A or KDM5B knockdown,684 KDM5-C70 did not display significant effects. It is interesting to note that KDM5-C70 at the same concentrations led to the accumulation of global H3K4me3 levels both in sensitive (MCF7) and resistant (MDA-MB-231) cell lines, while effects on cell growth differed. Treatment of MCF7 cells with KDM5-C70 led to an increase in the expression of some, but not all, of KDM5B target genes while knockdown of KDM5B affected most of them, suggesting that while the demethylase activities of KDM5 enzymes are important to maintain the repression of KDM5 target genes, KDM5 proteins might have additional functions in gene regulation beyond their catalytic activities. Importantly, KDM5A crystal structures in complex with several inhibitors revealed important interactions and provided insights and strategies that can be utilized for the design of selective and potent KDM5 inhibitors. For example, it was suggested that the additional space near Cys-481 (a residue unique to the KDM5 subfamily) could be potentially exploited. In addition, existing inhibitors could be modified so that they can extend into a nearby water-filled channel lined with several unique residues of the KDM5 subfamily.
Westaway and co-workers reported GSK467 (Figure 51, a dual KDM5 and KDM4 inhibitor generated from their optimization study for the KDM4 family inhibitors discussed in the previous section.649,685 GSK467 displayed a Ki of 10 nM for KDM5A and KDM5B (calculated from an IC50 of ∼25 nM in biochemical assays) and was about 10-fold less potent for KDM5C and KDM5D. However, it was highly potent for KDM4C (Ki = 1.86 nM). GSK467 exhibited no significant inhibitory activity against KDM6 and other KDMs tested.649 The cocrystal structure of GSK467 in complex with KDM5B (PDB ID: 5FUN) revealed that this inhibitor bound in the 2-OG binding pocket, where the inhibitor was engaged in a monodentate interaction with the catalytic metal via its pyrido-nitrogen and the two remaining coordination sites were occupied by water molecules (Figure 52B). This unique binding mode is interesting. However, GSK467 did not display activities in cellular assays. The lack of cellular potency unfortunately hampers its use as a chemical tool. Nevertheless, GSK467 presents a good starting point for developing more selective and cell-active KDM inhibitors.
In a continuation of their earlier work,685 Bavetsias and co-workers reported bicyclic pyrido[3,4-d]pyrimidin-4(3H)-one containing, cell-permeable KDM inhibitors with good potency (Figure 53).686 Extensive structure-based optimization resulted in the discovery of KDM inhibitors 67 and 68 (Figure 53). Compound 68 exhibited equal potencies for KDM4 (IC50 = 0.080 and 0.017 μM for KDM4A and KDM4B, respectively) and KDM5 (IC50 = 0.014 and 0.051 μM for KDM5B and KDM5C, respectively) but were selective over other KDM subfamilies, such as KDM3A (IC50 = 6.1 μM), KDM6B (4% inhibition at 100 μM), and KDM2A (IC50 = 2.4 μM). A crystal structure of 68 in complex with KDM4A (PDB ID: 5F3I) revealed key ligand–protein interactions, including the bidentate coordination of the metal ion by the inhibitor. 67 inhibited H3K9me3 and H3K4me3 demethylation in a cell-based assay. As we discussed in an earlier section, pyridine-4-carboxylic acids 52 and 53, which were identified as KDM4 subfamily inhibitors with IC50 ≤ 100 nM, were also potent inhibitors of KDM5C with IC50 = 100–125 nM.649,685
Figure 53.

Structures of other KDM5 inhibitors.
In 2014, Mannironi and co-workers established a screening system to test the effects of small-molecule inhibitors on an S. cerevisiae strain, which requires KDM5 demethylase activity to efficiently grow in the presence of rapamycin.687 Series of structurally different compounds selected by a computer-aided drug design approach were screened, leading to the identification of compound 69 (Figure 53), which inhibited KDM5 in vitro and in vivo. Compound 69 also inhibited human KDM5B (IC50 = 1–2.5 μM) and KDM5D (IC50 ∼ 2.0 μM) in biochemical assays and significantly increased H3K4me3 levels in HeLa cell nuclear extracts at 30 μM. When added to HeLa cells, the compound led to an increase of H3K4me3 but did not affect H3K9me3 levels.
In another study geared toward identification of KDM5A inhibitors, a hydroxamic acid derivative 70 (Figure 53) was identified.688 It selectively inhibited KDM5A (IC50 = 3.3 μM) over KDM3A, 4C, and 7B (>10–100-fold). However, its selectivity over other KDMs was not reported. While the methyl ester of this compound, as a prodrug, slightly increased H3K4me3 levels at relatively high concentrations, it did not affect the proliferation of A549 lung cancer cells even at concentrations up to 300 μM.
3.2.5. Inhibitors of KDM6 Subfamily
The KDM6 subfamily members UTX (ubiquitously transcribed tetratricopeptide repeat X-chromosome protein, also known as KDM6A), KDM6B (also known as jumonji domain-containing protein D3 or JMJD3), and UTY (ubiquitously transcribed tetratricopeptide repeat Y-chromosome protein, also known as KDM6C) specifically demethylate H3K27me3/me2.573,574,689 UTX and KDM6B have been shown to have critical roles in development and differentiation both in vitro and in vivo.690−692 UTX has been linked to the regulation of HOX transcriptional network.693 Loss of function mutations in UTX have been observed in a variety of cancers, including renal cell carcinoma, multiple myeloma, esophageal carcinoma, myeloid leukemias, breast and colorectal cancers, and glioblastoma.694 KDM6 family members, including UTX, are also components of multiprotein complexes such as MLL2, MLL3, and MLL4 that regulate the activity of polycomb family proteins.693,695 KDM6B has been implicated in colon cancer696 and lymphoma697 and has been shown to be associated with different oncogenes and tumor suppression genes.698,699 As we mentioned earlier, UTY was initially reported not to have KDM activity; however, it was recently shown to be a male-specific demethylase.574 While it displays reduced activity in vitro due to a point substitution (isoleucine to proline) in the substrate binding site compared to UTX and KDM6B, it contains all three of the predicted Fe-binding residues, as well as those predicted to be important in 2-OG binding.574 Sequence alignments predict that UYX and UTY share similar domain organizations, with N-terminal tetratricopeptide repeat (TPR) domains, C-terminal JmjC, and zinc binding domains. It was suggested that KDM6B might also contain TPR-like domains. While UTX and UTY share >96% similarity in their catalytic JmjC domains,574 KDM6B shares only 84% similarity.573 Given the known associations to important biological functions and cancers, there is a growing interest in developing KDM6 selective inhibitors as tools for further studying the roles of the KDM6 subfamily in diseases.
By screening of a 2 million-compound collection, GSK-J1 (Figure 54) was identified as a lead compound with an IC50 of 60 nM in a KDM6B biochemical assay.700 It was about 3-fold selective for KDM6B over UTX. GSK-J1 was inactive against a panel of demethylases, including KDM4A, KDM4C-E, KDM3A, and KDM5C, as measured by a combination of thermal shift, mass spectrometry, and antibody-based assays. GSK-J1 also did not significantly inhibit 100 protein kinases, even at a concentration of 30 μM in a competition-binding assay and had negligible activities against a panel of 60 unrelated proteins, including other chromatin-modifying enzymes such as histone deacetylases. In MOA studies, GSK-J1 was found to be competitive with 2-OG but noncompetitive with the peptide substrate. A closely related compound was developed as a negative control by moving the pyridine substitution from the 2- to the 3-position. GSK-J4 (Figure 54), the ethyl ester of GSK-J1, was also developed as a pro-drug for cellular studies. The cocrystal structure of GSK-J1 in complex with human KDM6B (Co2+ was used to mimic the Fe2+) revealed that the propionic acid of GSK-J1 mimics 2-OG binding by maintaining key interactions, while the aromatic ring of the tetrahydrobenzazepine of GSK-J1 sits in a narrow cleft in between, mimicking P30 of the histone peptide (PDB ID: 4ASK) (Figure 55). The pyridyl-pyrimidine biaryl moiety of GSK-J1 forms a bidentate interaction with the catalytic metal and induces a shift in the Co2+ ion away from the three conserved Fe-binding residues (HHE triad). Interestingly, this shift results in the metal cation exchanging positions with a previously apical water molecule, causing H1470 to make an indirect water-bridged interaction with the metal (Figure 55). It was suggested that this dynamic metal shift in KDM6B could create an opportunity to design selective compounds. Specific precipitation of flag-tagged, full-length KDM6B and UTX from transiently transfected HEK-293 cells through the use of an immobilized derivative of GSK-J1 suggested target engagement since addition of free GSK-J1 inhibited the binding of immobilized probe to the target proteins. In quantitative MS experiments, KDB6B was the sole protein captured, suggesting selectivity in a cellular context. The ethyl ester prodrug GSK-J4 increased total nuclear H3K27me3 levels in cells. Studies were also conducted to investigate the efficacy of GSK-J4 in inhibiting lipopolysaccharide (LPS)-induced response of human primary macrophages derived from healthy volunteers. Treatment with GSK-J4 significantly reduced the expression of 16 out of the 34 LPS-driven cytokines, including tumor-necrosis factor-α (TNF-α). Inhibition of TNF-α, an important cytokine in inflammatory disorders, was demonstrated in a concentration-dependent manner (IC50 = 9 μM) without any apparent cellular toxicity after treatment with GSK-J4. Altogether, GSK-J1 was identified to be a selective H3K27 demethylase (KDM6B and UTX) inhibitor that modulates proinflammatory macrophage responses.
Figure 54.

Structures of KDM6 inhibitor GSK-J1 and its prodrug GSK-J4.
Figure 55.
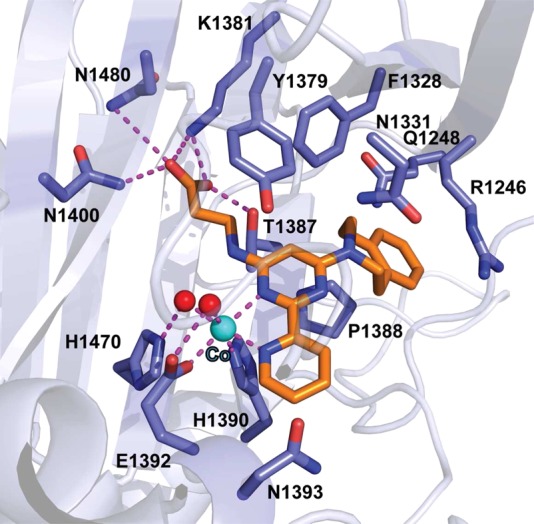
Co-crystal structure of KDM6B in complex with GSK-J1 (orange). The key residues that formed the catalytic pocked are indicated in blue. Hydrogen bonds are represented as magenta dashed lines and water molecules as red spheres.
In 2014, Heinemann and co-workers revisited the selectivity of GSK-J1, expanding the panel of JmjC KDMs tested.701 Their findings primarily supported the selectivity of GSK-J1 in inhibiting KDM6B and UTX over other KDMs (KDM2B, KDM3A-B, and KDM4A-C). However, in contrast to the previous study, in which KDM5C was the only KDM5 subfamily member tested, KDM5A and KDM5B were also investigated by Heinemann and co-workers. It was found that, in addition to its high potency for KDM6B and UTX (IC50 = 28 and 53 nM, respectively), GSK-J1 was quite potent for KDM5B and KDM5C (IC50 = 170 and 550 nM, respectively), exhibiting limited selectivity, especially over KDM5B (around 6- and 3-fold for KDM6B and UTX, respectively) in biochemical assays. In cells transfected with KDM6B, KDM5B, and KDM4C, the potency of GSK-J4, the prodrug of GSK-J1, was found to be very close with IC50 values of 3.1, 3.1, and 7.3 μM, respectively. The selectivity profile of GSK-J1 has recently been updated by Johansson and co-workers (KDM5B: Ki = 0.14 μM; KDM5C: Ki = 0.71 μM; KDM5D: Ki = 0.34 μM KDM6B: Ki = 0.02 μM).681
A study that explored the SAR by replacing the pyridine ring of GSK-J1 with other potential chelating heterocycles was also published.702 Several compounds featuring a pyrazole or triazole ring instead of the pyridine ring with IC50 values of 0.15–0.27 μM were identified. The ethyl esters of these compounds showed activity against TNF-production in Raw 264.7 cells, a LPS-stimulated murine macrophage cell line. However, no selectivity data against other KDMs were reported.
3.3. Dual LSD1 and JmjC KDM Inhibitors
Rotili and co-workers reported hybrid LSD1/JmjC KDMs inhibitors as “pan-KDM” inhibitors by coupling the chemical features of tranylcypromine (LSD1 inhibitor) with the 2,2′-bipyridine-dicarboxylate or 5-carboxy-8HQ scaffolds (2-OG-competitive JmjC inhibition scaffolds) (Figure 56).703 Compound 70 inhibited LSD1 (IC50 = 2.2 μM) as well as JmjC enzymes (IC50 values for KDM2A = 0.22 μM; KDM3A = 0.14 μM; KDM4C = 0.42 μM; and KDM5C = 0.19 μM). It was selective over MAO A (IC50 = 35 μM), MAO B (IC50 = 47 μM), FIH (IC50 > 100 μM), and PHD2 (IC50 = 278 μM). Compounds 71 and 72 (Figure 56) were also active for LSD1 (IC50 < 1 μM) and for JmjC KDMs, albeit with lower potencies, and displayed some intersubfamily selectivity (IC50 in range of 1.2–76 μM, where highest potencies were observed for KDM4C).
Figure 56.

Structures of dual LSD1-JmjC KDM inhibitors.
In human prostate cancers, it was shown that LSD1 and JmjC KDM4A/C are coexpressed and colocalized with AR.587 In addition, as mentioned earlier, KDM4A/C inhibitors were not found to inhibit prostate (LNCaP and PC3) or colon (HCT116) cancer cell growth in isolation, but they did display antiproliferative effects when combined with NCL-1 derivatives (LSD1 inhibitors as discussed in Figure 34), suggesting that a potential synergistic or additive effect may exist for dual inhibition of LSDs and JmjC KDMs.603 Compounds 71 and 72 resulted in simultaneous increases in H3K4me2/3 and H3K9me2/3 levels, as well as high incidences of growth arrest and apoptosis in LNCaP prostate and HCT116 colon cancer cells. On the other hand, the corresponding LSD1 inhibitor tranylcypromine and KDM inhibitor 4-carboxy-4′-carbomethoxy-2,2′-bipyridine were inactive both separately and in combination. However, although little toxicities were observed in MePR noncancer cells, careful examination of broader toxic effects of these hybrid compounds is warranted given that the compounds inhibited a wide range of demethylases including LSDs and KDM1–7 and likely 2-OG dependent hydroxylases.
4. Concluding Remarks and Future Directions
A growing body of evidence supports the fact that protein methyltransferases and histone demethylases play key roles in the regulation of transcriptional activity and are implicated in cancer and many other human diseases. Not surprisingly, given these key functions, there has been a steadily increasing interest toward assessing the potential of these enzymes as therapeutic targets. Consequently, discovering selective inhibitors of these enzymes has become a very active and fast-paced area of research over the past decade. There has been remarkable progress in the PMT and KDM inhibitor field as a result of collective advances made in assay development, high-throughput screening, structural biology, medicinal chemistry, and cellular and animal models. The first PKMT and PRMT inhibitors were discovered in 2005 and 2004, respectively, followed by the first LSD1 inhibitor in 2006. In 2012, less than a decade after initial inhibitors were discovered, the first human clinical trials for the DOT1L inhibitor EPZ005676 were initiated. Since then, numerous PKMT, PRMT, and KDM inhibitors have entered clinical trials. EZH2 inhibitors [EPZ-6438 (2013), GSK126 (2014), and CPI-1205 (2015)], PRMT5 inhibitor [GSK3326595 (2016)], and LSD1 inhibitors [ORY-1001 (2013) and GSK2879552 (2014)] are all currently in human clinical trials, underscoring the rapid progress made in the field.
The discovery and development of several highly potent, selective, and well-characterized small-molecule inhibitors of methyltransferases with robust on-target activities in cells have been achieved during the past decade. For example, PKMT inhibitors, including UNC0638, UNC0642, EPZ005687, GSK126, EI1, UNC1999, EPZ-6438, CPI-1205, EPZ004777, SGC0946, and EPZ-5676, are valuable chemical tools for further investigating biological functions of the targeted enzymes and have already been extensively used in assessing the therapeutic potential of these proteins. As we already described, three EZH2 inhibitors (EPZ-6438, GSK126, and CPI-1205) have already entered clinical trials. The discoveries of substrate-competitive inhibitors of G9a/GLP (e.g., BIX-01294, UNC0638, UNC0642, and A-366), SMYD2 (e.g., AZ-505, A-893, LLY-507, and BAY-598), SMYD3 (e.g., EPZ0316867 and EPZ030456), SETD8 (e.g., MS2177), and SETD7 [e.g., (R)-PFI-2] suggest that the substrate-binding grooves of PKMTs can be targeted to yield potent and selective inhibitors. Similarly, the discoveries of highly potent, selective, and SAM-competitive inhibitors of DOT1L (e.g., EPZ004777, SGC0946, and EPZ-5676) and EZH2 (e.g., EPZ005687, GSK126, EI1, UNC1999, EPZ-6438, and CPI-1205) provided experimental evidence that extremely high selectivity can be achieved by targeting the SAM-binding site of PKMTs, which is analogous to targeting the ATP-binding site of protein kinases. In addition, highly potent, selective, substrate-competitive PRMT inhibitors including MS023 (type I PRMTs), EPZ015666 (PRMT5), MS049 (CARM1 and PRMT6) and EPZ020411 (PRMT6) have been accomplished, suggesting that the substrate-binding grooves of PRMTs can also be successfully targeted. The identification of a novel allosteric binding site of PRMT3, which is the first reported allosteric binding site of any PKMTs and PRMTs, was another major advancement in the PMT inhibitor field. The discovery of the first allosteric PRMT3 inhibitor and the development of the PRMT3 chemical probe SGC707 have demonstrated that the allosteric binding site of PRMT3 can be exploited to yield potent, selective, and cell-active inhibitors, opening the door for discovering allosteric inhibitors of other PRMTs. Furthermore, the discovery of the covalent SETD8 inhibitor MS453 has demonstrated that cysteine residues in active sites of PKMTs and PRMTs can be selectively targeted. We expect that this success will pave the way for developing additional selective, covalent PKMT and PRMT inhibitors that target an active site cysteine residue.
While there has been significant progress, there is still much to be achieved in the PMT inhibitor field. For example, a systematic coverage of PMTs as a protein family with potent and selective inhibitors is needed since, currently, many individual targets and subgroups of targets on the PMT phylogenetic tree lack selective inhibitors, including MLL family, MMSET (NSD-2), and PRDMs (Figure 3). There is a limited understanding of biological functions and potential disease implications for many of these targets, partly due to that selective inhibitors of these PMTs have not been generated. Potent, selective, and cell-active inhibitors of these methyltransferases would be invaluable chemical tools to better understand their biological functions and test therapeutic hypotheses concerning these proteins.
Similar to PMTs, significant progress has been made on the development of histone demethylase inhibitors as chemical tools and potential therapeutic agents. Two LSD1 inhibitors have recently entered clinical trials. GSK2879552 was advanced into clinical trials in the U.S. for the treatment of small cell lung carcinoma in 2013, AML in 2014, and myelodysplastic syndrome in 2016. ORY-1001 entered phase I clinical study in the European Union for the treatment of relapsed or refractory acute leukemia (AL) in 2013. Although significant work has been done in understanding the mechanism and selectivity of LSD1, there are still opportunities for further work. For example, although it is now evident that potent and selective inhibitors of LSD1 can be achieved, little or no progress has been made on generating selective inhibitors for LSD1 over LSD2. The vast majority of the reported LSD1 inhibitors are “mechanism-based” or “suicide” ligands that covalently modify FAD, yet promising reversible small-molecule inhibitors of LSD1 (e.g., 37, 39) have been recently discovered. We believe that more progress can be made in this area. In the case of JmjC KDMs, it is imperative to focus future work on improving selectivity between and within the subfamilies so that resulting inhibitors have sufficient potency, selectivity, and cellular activity and can be confidently utilized to study biological functions of the targeted KDMs and test relevant therapeutic hypotheses. While most of the JmjC KDM inhibitors reported to date lack sufficient selectivity, the recent discovery of KDM5 subfamily selective inhibitors (e.g., CPI-455, 66) has demonstrated that high selectivity between JmjC KDM subfamilies can be achieved.
As we discussed, most LSD1 and JmjC KDM inhibitor discovery efforts have focused on the extension or continuation of previously known types of inhibitors. For example, for LSD1, efforts focused mostly on mechanism-based inhibition of LSD1 and for the JmjC KDMs, on the active site iron chelators. Drawing from the advances in the methyltransferase inhibitor field, one can expect that as time progresses, it is likely that new types of inhibitors will emerge, such as those targeting the substrate-binding site or potential allosteric binding site(s). Moreover, KDMs contain noncatalytic binding domains, and the interactions of these domains with chromatin might be considered as protein–protein/protein–nucleic acid interactions. Selective disruption of these interactions could offer a potential approach for developing selective KDM inhibitors.
As we emphasized throughout this review, thorough characterization in biochemical, biophysical, and cellular assays is very important and needed for many of the reported inhibitors. Without sufficient characterization, caution should be taken when attributing the observed phenotypic effects to pharmacological inhibition of the intended target(s). For an inhibitor to be useful in in vitro studies, direct binding between the protein target and the inhibitor should be demonstrated by a biophysical method (e.g., ITC and SPR) or an NMR solution or X-ray crystal structure of the protein–ligand complex, in addition to activities in biochemical assays. One of the most common issues is that the selectivity of reported inhibitors was not sufficiently characterized. It is critical to assess the selectivity of key inhibitors for the targeted PMT(s) or KDM(s) over a broad panel of other methyltransferases and demethylases and other relevant biological targets. For the inhibitors to be used in cellular studies, sufficient cell permeability and target engagement in cells should be demonstrated in addition to the in vitro target engagement and selectivity assessment described above.
Understanding the structural basis of high subtype selectivity is important to advance this field. Significant progress has been made in obtaining high-resolution crystal structures of the targeted enzymes in complex with inhibitors, which provide structural basis and insights for designing more selective inhibitors. We expect that efforts in this area will continue and yield more high-resolution X-ray or NMR structures. In addition, better understanding the dynamics of the protein–inhibitor interactions is urgently needed, which we believe will offer critical insights for achieving selectivity between highly homologous PMTs and KMDs. Furthermore, there are opportunities to generate new chemical tools such as biotinylated compounds as affinity ligands based on the highly potent and selective inhibitors developed recently. These tools are useful in chemical biology studies such as chemoproteomics, Chem-ChIP, and Chem-Seq.
In this review, we thoroughly covered the discovery, characterization, and application of selective PMT and KDM inhibitors for investigating the physiological functions and disease implications of the target proteins. We highlighted key advances and discussed challenges, future directions, and opportunities in the PMT and KDM inhibitor fields. Over the last 15 years, we have witnessed astonishing growth and progress in these emerging research fields, culminated by DOT1L, EZH2, PRMT5, and LSD1 inhibitors entering clinical trials in 2012–2016 for diseases such as leukemia, lymphoma, and SCLC. We expect that amazing progress and successes will continue in the very active research fields. We hope that this review will be a useful resource and inspire new and original discoveries.
Acknowledgments
This work was supported by Grant R01GM103893 (J.J.) and R01GM122749 (J.J.) from the National Institute of General Medical Sciences of the U.S. National Institutes of Health.
Glossary
Abbreviations
- PTMs
post translational modifications
- DNMTs
DNA methyltransferases
- HDACs
histone deacetylases
- FDA
Food and Drug Administration
- PMTs
protein methyltransferases
- HMTs
histone methyltransferases
- SAM
S-5′-adenosyl-L-methionine
- SAH
S-5′-adenosyl-L-homocysteine
- PKMTs
protein lysine methyltransferases
- PRMTs
protein arginine methyltransferases
- H3
Histone 3
- H4
Histone 4
- MMA
monomethylation of arginine
- sDMA
symmetrical monomethylation of arginine
- aDMA
asymmetrical monomethylation of arginine
- H3K4
histone 3, lysine 4
- SUV39H1
suppressor of variegation 3–9 homologue 1
- EHMT2
euchromatic histone-lysine N-methyltransferase 2
- GLP
G9a-like protein 1
- SETDB1
SET domain, bifurcated 1
- PRDM2
PR domain containing 2, with ZNF domain
- ETP
epidithiodiketopiperazine
- DTT
dithiothreitol
- mES
mouse embryonic stem
- ES cell
embryonic stem cell
- HSPCs
hematopoietic stem and progenitor cells
- AML
acute myeloid leukemia
- HTS
high-throughput screening
- PRMT1
protein arginine methyltransferase 1
- MEFs
mouse embryonic fibroblasts
- ChIP
chromatin immunoprecipitation
- wt
wild-type
- ITC
isothermal titration calorimetry
- DSF
differential scanning fluorimetry
- GPCRs
G-protein coupled receptors
- SPR
surface plasmon resonance
- PRC2
Polycomb repressive complex 2
- EZH1
enhancer of zeste homologue 1
- EZH2
enhancer of zeste 2 polycomb repressive complex 2 subunit
- SUZ12
suppressor of zeste 12
- EED
embryonic ectoderm development
- DLBCLs
diffuse large B-cell lymphomas
- SWI/SNF
switch/sucrose nonfermentable
- MLL
mixed lineage leukemia
- ALL
acute lymphoblastic leukemia
- SETD7
SET domain containing (lysine methyltransferase) 7
- SMYD
SET and MYND domain containing
- MYND
myeloid translocation protein-8, Nervy, and DEAF-1
- SMYD2
SET and MYND domain containing 2
- SETD2
SET domain containing 2
- HGGs
hemispheric high-grade gliomas
- SETD8
SET domain containing (lysine methyltransferase) 8
- H4K20me
H4K20 monomethylation
- PCNA
proliferating cell nuclear antigen
- DOT1L
disruptor of telomeric silencing 1-like
- CARM1
coactivator-associated arginine methyltransferase 1
- GAR
glycine and arginine rich
- PGM
proline-, glycine-, and methionine-rich
- AMIs
arginine methyltransferase inhibitors
- PRMT3
protein arginine methyltransferase 3
- PABPN1
mammalian nuclear poly(A)-binding protein
- VHL
von Hippel–Lindau
- ARF
alternative reading frame
- DAL-1
differentially expressed in adenocarcinoma of the lung-1
- ELISA
enzyme-linked immunosorbent assay
- MS
mass spectrometry
- mESC
mouse embryonic stem cells
- SAHH
SAH hydrolase
- SPA
scintillation proximity assay
- HP1
heterochromatin protein 1
- GSEA
gene set enrichment analysis
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- ChIP-chip
(chromatin immunoprecipitation–DNA microarray)
- LTR
long terminal repeat
- PK
pharmacokinetic
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia and Rad3-related
- SC
subcutaneous
- IV
intravenous
- WDR5
WD repeat-containing protein 5
- CCR2
CC chemokine receptor 2
- CCL2
CC chemokine ligand 2
- CCR5
CC chemokine receptor 5
- TLC
thin layer chromatography
- KDMs
histone demethylases
- LSD1
lysine specific demethylase
- KDM2A
lysine demethylase 2A
- FAD
flavin adenine dinucleotide
- JmjC
Jumonji C
- AR
androgen receptor
- SWIRM
small α-helical domain
- AOL
amine oxidase like
- CoREST
corepressor for RE1-silencing transcription factor
- NuRDs
nucleosome remodeling and deacetylation complexes
- CtBPs
C-terminal binding proteins
- ER
estrogen receptor
- STAT3
signal transducer and activator of transcription 3
- MAOs
monoamine oxidases
- PAO
polyamine oxidase
- MSOX
monomeric sarcosine oxidase
- APL
acute promyeloctic leukemia
- HRP
horseradish peroxide
- TR-FRET
time-resolved fluorescence energy transfer
- NOR
novel object recognition
- GRP
gastrin releasing peptide
- GO
gene ontology
- PDX
patient-derived xenograft
- MDS
myelodysplastic syndromes
- DHP
2,5-dihydro-1H-pyrrole
- THP
1,2,3,6-tetrahydropyridine
- PHD
plant homeobox domains
- MINA53
MYC-induced nuclear antigen 53
- NO66
nucleolar protein 66
- HR
hairless
- DSBH
barrel-like double-stranded β-helix
- 2-OG
2-oxoglutarate
- HIF
hypoxia-inducible factor
- FIH
factor-inhibiting-HIF
- 2,4-PDCA
pyridine-2,4-dicarboxylic acid
- PHF
plant homeodomain finger
- FBXL11
F-box and leucine rich repeat protein 11
- HSQC
heteronuclear single quantum correlation
- TROSY
transverse relaxation enhanced spectroscopy
- BMYB
Myb-related protein B
- PLK1
polo-like kinase 1
- DNOC
N-oxalyl-d-cysteine
- DTP
drug-tolerant state
- DTEPs
drug-tolerant expanded persister cancer cells
- TPR
tetratricopeptide repeat
- PPB
plasma protein binding
- UTX
ubiquitously transcribed tetratricopeptide repeat X-chromosome protein
- UTY
ubiquitously transcribed tetratricopeptide repeat Y-chromosome protein
Biographies
H. Ümit Kaniskan earned his Ph.D. in organic chemistry at Case Western Reserve University under the supervision of Dr. Philip Garner. During his doctoral study, he completed the formal total synthesis of Bioxalomycin β2 and Cyanocycline A. He then pursued his postdoctoral studies in Dr. Movassaghi’s group at Massachusetts Institute of Technology, while working on the synthesis of Myrmicarin alkaloids. In January 2013, Dr. Kaniskan joined Dr. Jin’s laboratory at University of North Carolina at Chapel Hill and later at the Icahn School of Medicine at Mount Sinai as a postdoctoral research associate in the Department of Pharmacological Sciences. He is currently an Assistant Professor in the Department of Pharmacological Sciences at the Icahn School of Medicine at Mount Sinai. His research interests include small-molecule organic synthesis, structure-based drug discovery, and development of small molecule inhibitors of protein methyltransferases.
Michael L. Martini graduated summa cum laude with a bachelor’s degree in Molecular Biology and Biochemistry from Middlebury College in 2015. His undergraduate thesis, conducted under the tutelage of Dr. Richard Bunt, focused on investigating bite angle as a ligand parameter influencing reversibility in Pd-catalyzed allylic aminations. Michael spent his summers conducting chemical biology research on serine hydrolases in the laboratory of Dr. Benjamin Cravatt at The Scripps Research Institute. He later returned to Scripps and worked as a laboratory technician in Dr. Dale Boger’s medicinal chemistry group. During his undergraduate career, Michael received an Amgen Scholarship in 2013, a Barry Goldwater Scholarship for Excellence in Science in 2014, and upon graduation, was inducted into the Phi Beta Kappa honor society and named Salutatorian of the Class of 2015. Michael then moved to New York City, where he is now an M.D./Ph.D. candidate in the Medical Scientist Training Program at the Icahn School of Medicine at Mount Sinai. He is currently doing graduate research with Dr. Jian Jin in the Department of Pharmacological Sciences.
Jian Jin received a Ph.D. degree in synthetic organic chemistry from Pennsylvania State University and completed postdoctoral training at Ohio State University. He joined GlaxoSmithKline as a medicinal chemist in 1998 and had been a manager of medicinal chemistry from 2003 to 2008. In 2008, Dr. Jin joined University of North Carolina at Chapel Hill (UNC–CH) as a faculty member. He had also served as an associate director of medicinal chemistry in the Center for Integrative Chemical Biology and Drug Discovery at UNC–CH from 2008 to 2014. In 2014, Dr. Jin was recruited to Icahn School of Medicine at Mount Sinai (Mount Sinai) as a Professor with Tenure. He is currently a professor in the Departments of Pharmacological Sciences and Oncological Sciences at Mount Sinai. Since 2008, Dr. Jin’s laboratory has focused on discovering selective inhibitors of protein methyltransferases and functionally selective ligands of G protein-coupled receptors. To date, Dr. Jin has published more than 100 research papers and delivered more than 70 invited talks. He is also an inventor of 10 issued U.S. patents and 38 published PCT patent applications.
The authors declare no competing financial interest.
References
- Bernstein B. E.; Meissner A.; Lander E. S. The Mammalian Epigenome. Cell 2007, 128, 669–681. 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Allis C. D.; Jenuwein T.; Reinberg D.. Epigenetics; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, N.Y., 2007; p. 502. [Google Scholar]
- Gelato K. A.; Fischle W. Role of Histone Modifications in Defining Chromatin Structure and Function. Biol. Chem. 2008, 389, 353–363. 10.1515/BC.2008.048. [DOI] [PubMed] [Google Scholar]
- Rea S.; Eisenhaber F.; O’Carroll D.; Strahl B. D.; Sun Z. W.; Schmid M.; Opravil S.; Mechtler K.; Ponting C. P.; Allis C. D.; et al. Regulation of Chromatin Structure by Site-Specific Histone H3Methyltransferases. Nature 2000, 406, 593–599. 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic Protein Families: A New Frontier for Drug Discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Solomon M. E.; Richon V. M. Protein Methyltransferases as a Target Class for Drug Discovery. Nat. Rev. Drug Discovery 2009, 8, 724–732. 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Konze K. D.; Jin J. Selective Inhibitors of Protein Methyltransferases. J. Med. Chem. 2015, 58, 1596–1629. 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K.; Dhanak D. Chromatin Proteins and Modifications as Drug Targets. Nature 2013, 502, 480–488. 10.1038/nature12751. [DOI] [PubMed] [Google Scholar]
- Zagni C.; Chiacchio U.; Rescifina A. Histone Methyltransferase Inhibitors: Novel Epigenetic Agents for Cancer Treatment. Curr. Med. Chem. 2013, 20, 167–185. 10.2174/092986713804806667. [DOI] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Jin J. Chemical Probes Of Histone Lysine Methyltransferases. ACS Chem. Biol. 2015, 10, 40–50. 10.1021/cb500785t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann J.; Clancy K. W.; Thompson P. R. Chemical Biology of Protein Arginine Modifications in Epigenetic Regulation. Chem. Rev. 2015, 115, 5413–5461. 10.1021/acs.chemrev.5b00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M. Chemical Inhibition of Protein Methyltransferases. Cell Chem. Biol. 2016, 23, 1067–1076. 10.1016/j.chembiol.2016.07.014. [DOI] [PubMed] [Google Scholar]
- Hu H.; Qian K.; Ho M. C.; Zheng Y. G. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin. Invest. Drugs 2016, 25, 335–358. 10.1517/13543784.2016.1144747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann J.; Thompson P. R. Protein Arginine Methylation and Citrullination in Epigenetic Regulation. ACS Chem. Biol. 2016, 11, 654–668. 10.1021/acschembio.5b00942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister T. E.; England K. S.; Hopkinson R. J.; Brennan P. E.; Kawamura A.; Schofield C. J. Recent Progress in Histone Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1308–1329. 10.1021/acs.jmedchem.5b01758. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Patel D. J. Small Molecule Epigenetic Inhibitors Targeted to Histone Lysine Methyltransferases and Demethylases. Q. Rev. Biophys. 2013, 46, 349–373. 10.1017/S0033583513000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y. C.; Ma J.; Wang Z.; Li J.; Jiang B.; Zhou W.; Shi X.; Wang X.; Zhao W.; Liu H. M. A Systematic Review of Histone Lysine-Specific Demethylase 1 and Its Inhibitors. Med. Res. Rev. 2015, 35, 1032–1071. 10.1002/med.21350. [DOI] [PubMed] [Google Scholar]
- Markolovic S.; Leissing T. M.; Chowdhury R.; Wilkins S. E.; Lu X.; Schofield C. J. Structure-Function Relationships of Human JmjC Oxygenases-Demethylases versus Hydroxylases. Curr. Opin. Struct. Biol. 2016, 41, 62–72. 10.1016/j.sbi.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Hojfeldt J. W.; Agger K.; Helin K. Histone Lysine Demethylases as Targets for Anticancer Therapy. Nat. Rev. Drug Discovery 2013, 12, 917–930. 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Audia J. E.; Austin C.; Baell J.; Bennett J.; Blagg J.; Bountra C.; Brennan P. E.; Brown P. J.; Bunnage M. E.; et al. The Promise and Peril of Chemical Probes. Nat. Chem. Biol. 2015, 11, 536–541. 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C.; Zhang Y. The Diverse Functions of Histone Lysine Methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- Shi X.; Kachirskaia I.; Yamaguchi H.; West L. E.; Wen H.; Wang E. W.; Dutta S.; Appella E.; Gozani O. Modulation of p53 Function by SET8-Mediated Methylation at Lysine 382. Mol. Cell 2007, 27, 636–646. 10.1016/j.molcel.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S.; Kurash J. K.; Wilson J. R.; Xiao B.; Justin N.; Ivanov G. S.; McKinney K.; Tempst P.; Prives C.; Gamblin S. J.; et al. Regulation of p53 Activity Through Lysine Methylation. Nature 2004, 432, 353–360. 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell 2007, 128, 693–705. 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Jenuwein T.; Laible G.; Dorn R.; Reuter G. SET Domain Proteins Modulate Chromatin Domains in Eu- and Heterochromatin. Cell. Mol. Life Sci. 1998, 54, 80–93. 10.1007/s000180050127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbusch L. O.; Thorstensen T.; Krauss V.; Fischer A.; Naumann K.; Assalkhou R.; Schulz I.; Reuter G.; Aalen R. B. The Arabidopsis Thaliana Genome Contains at least 29 Active Genes Encoding SET Domain Proteins that can be Assigned to Four Evolutionarily Conserved Classes. Nucleic Acids Res. 2001, 29, 4319–4333. 10.1093/nar/29.21.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Collins R. E.; Zhang X. Structural and Sequence Motifs of Protein (Histone) Methylation Enzymes. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 267–294. 10.1146/annurev.biophys.34.040204.144452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S. A.; Harp J. M.; Devarakonda S.; Kim Y.; Rastinejad F.; Khorasanizadeh S. The Active Site of the SET Domain is Constructed on a Knot. Nat. Struct. Biol. 2002, 9, 833–838. 10.1038/nsb861. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Histone Methylation in Transcriptional Control. Curr. Opin. Genet. Dev. 2002, 12, 198–209. 10.1016/S0959-437X(02)00287-3. [DOI] [PubMed] [Google Scholar]
- Allis C. D.; Berger S. L.; Cote J.; Dent S.; Jenuwien T.; Kouzarides T.; Pillus L.; Reinberg D.; Shi Y.; Shiekhattar R.; et al. New Nomenclature for Chromatin-Modifying Enzymes. Cell 2007, 131, 633–636. 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Tamaru H.; Khan S. I.; Horton J. R.; Keefe L. J.; Selker E. U.; Cheng X. Structure of the Neurospora SET Domain Protein DIM-5, A Histone H3 Lysine Methyltransferase. Cell 2002, 111, 117–127. 10.1016/S0092-8674(02)00999-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S. D.; Li H.; Ruthenburg A. J.; Allis C. D.; Patel D. J. How Chromatin-Binding Modules Interpret Histone Modifications: Lessons from Professional Pocket Pickers. Nat. Struct. Mol. Biol. 2007, 14, 1025–1040. 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister A. J.; Kouzarides T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A.; Cuddapah S.; Cui K.; Roh T. Y.; Schones D. E.; Wang Z.; Wei G.; Chepelev I.; Zhao K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Pugh B. F. High-Resolution Genome-Wide Mapping of the Primary Structure of Chromatin. Cell 2011, 144, 175–186. 10.1016/j.cell.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauss V. Glimpses of Evolution: Heterochromatic Histone H3K9Methyltransferases Left its Marks Behind. Genetica 2008, 133, 93–106. 10.1007/s10709-007-9184-z. [DOI] [PubMed] [Google Scholar]
- Bannister A. J.; Zegerman P.; Partridge J. F.; Miska E. A.; Thomas J. O.; Allshire R. C.; Kouzarides T. Selective Recognition of Methylated Lysine 9 on Histone H3 by the HP1 Chromo Domain. Nature 2001, 410, 120–124. 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Chen M. W.; Hua K. T.; Kao H. J.; Chi C. C.; Wei L. H.; Johansson G.; Shiah S. G.; Chen P. S.; Jeng Y. M.; Cheng T. Y.; et al. H3K9 Histone Methyltransferase G9a Promotes Lung Cancer Invasion and Metastasis by Silencing the Cell Adhesion Molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. 10.1158/0008-5472.CAN-10-0833. [DOI] [PubMed] [Google Scholar]
- Wen B.; Wu H.; Shinkai Y.; Irizarry R. A.; Feinberg A. P. Large Histone H3 Lysine 9 Dimethylated Chromatin Blocks Distinguish Differentiated from Embryonic Stem Cells. Nat. Genet. 2009, 41, 246–250. 10.1038/ng.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y.; Shen L.; Ahmed S.; Boumber Y.; Sekido Y.; Haddad B. R.; Issa J. P. Downregulation of Histone H3 lysine 9 Methyltransferase G9a Induces Centrosome Disruption and Chromosome Instability in Cancer Cells. PLoS One 2008, 3, e2037. 10.1371/journal.pone.0002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K.; Togami H.; Okamoto T. Involvement of Histone H3 Lysine 9 (H3K9) Methyl Transferase G9a in the Maintenance of HIV-1 Latency and its Reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. 10.1074/jbc.M110.103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maze I.; Covington H. E. 3rd; Dietz D. M.; LaPlant Q.; Renthal W.; Russo S. J.; Mechanic M.; Mouzon E.; Neve R. L.; Haggarty S. J.; et al. Essential Role of the Histone Methyltransferase G9a in Cocaine-Induced Plasticity. Science 2010, 327, 213–216. 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aagaard L.; Laible G.; Selenko P.; Schmid M.; Dorn R.; Schotta G.; Kuhfittig S.; Wolf A.; Lebersorger A.; Singh P. B.; et al. Functional Mammalian Homologues of the Drosophila PEV-Modifier Su(var)3–9 Encode Centromere-Associated Proteins which Complex with the Heterochromatin Component M31. EMBO journal 1999, 18, 1923–1938. 10.1093/emboj/18.7.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A. H.; O’Carroll D.; Scherthan H.; Mechtler K.; Sauer S.; Schofer C.; Weipoltshammer K.; Pagani M.; Lachner M.; Kohlmaier A.; et al. Loss of the Suv39h Histone Methyltransferases Impairs Mammalian Heterochromatin and Genome Stability. Cell 2001, 107, 323–337. 10.1016/S0092-8674(01)00542-6. [DOI] [PubMed] [Google Scholar]
- Muramatsu D.; Singh P. B.; Kimura H.; Tachibana M.; Shinkai Y. Pericentric Heterochromatin Generated by HP1 Protein Interaction-Defective Histone Methyltransferase Suv39h1. J. Biol. Chem. 2013, 288, 25285–25296. 10.1074/jbc.M113.470724. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nielsen S. J.; Schneider R.; Bauer U. M.; Bannister A. J.; Morrison A.; O’Carroll D.; Firestein R.; Cleary M.; Jenuwein T.; Herrera R. E.; et al. Rb Targets Histone H3Methylation and HP1 to Promoters. Nature 2001, 412, 561–565. 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Greiner D.; Bonaldi T.; Eskeland R.; Roemer E.; Imhof A. Identification of A Specific Inhibitor of the Histone Methyltransferase SU(VAR)3–9. Nat. Chem. Biol. 2005, 1, 143–145. 10.1038/nchembio721. [DOI] [PubMed] [Google Scholar]
- Cherblanc F. L.; Chapman K. L.; Brown R.; Fuchter M. J. Chaetocin is A Nonspecific Inhibitor of Histone Lysine Methyltransferases. Nat. Chem. Biol. 2013, 9, 136–137. 10.1038/nchembio.1187. [DOI] [PubMed] [Google Scholar]
- Cherblanc F. L.; Chapman K. L.; Reid J.; Borg A. J.; Sundriyal S.; Alcazar-Fuoli L.; Bignell E.; Demetriades M.; Schofield C. J.; DiMaggio P. A. Jr.; et al. On the histone Lysine Methyltransferase Activity of Fungal Metabolite Chaetocin. J. Med. Chem. 2013, 56, 8616–8625. 10.1021/jm401063r. [DOI] [PubMed] [Google Scholar]
- Greiner D.; Bonaldi T.; Eskeland R.; Roemer E.; Imhof A. Reply to ″Chaetocin is A Nonspecific Inhibitor of Histone Lysine Methyltransferases. Nat. Chem. Biol. 2013, 9, 137–137. 10.1038/nchembio.1188. [DOI] [PubMed] [Google Scholar]
- Tachibana M.; Sugimoto K.; Nozaki M.; Ueda J.; Ohta T.; Ohki M.; Fukuda M.; Takeda N.; Niida H.; Kato H.; et al. G9a Histone Methyltransferase Plays A Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and is Essential for Early Embryogenesis. Genes Dev. 2002, 16, 1779–1791. 10.1101/gad.989402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M.; Ueda J.; Fukuda M.; Takeda N.; Ohta T.; Iwanari H.; Sakihama T.; Kodama T.; Hamakubo T.; Shinkai Y. Histone Methyltransferases G9a and GLP form Heteromeric Complexes and are both Crucial for Methylation of Euchromatin at H3-K9. Genes Dev. 2005, 19, 815–826. 10.1101/gad.1284005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Dorsey J.; Chuikov S.; Zhang X.; Jenuwein T.; Reinberg D.; Berger S. L. G9a and GLP Methylate Lysine 373 in the Tumor Suppressor p53. J. Biol. Chem. 2010, 285, 9636–9641. 10.1074/jbc.M109.062588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H.; Soejima K.; Yasuda H.; Kawada I.; Nakachi I.; Yoda S.; Naoki K.; Ishizaka A. Deregulation of Histone Lysine Methyltransferases Contributes to Oncogenic Transformation of Human Bronchoepithelial Cells. Cancer Cell Int. 2008, 8, 15–26. 10.1186/1475-2867-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyama S.; Nitta E.; Yoshino T.; Kako S.; Watanabe-Okochi N.; Shimabe M.; Imai Y.; Takahashi K.; Kurokawa M. EVI-1 Interacts with Histone Methyltransferases SUV39H1 and G9a for Transcriptional Repression and Bone Marrow Immortalization. Leukemia 2010, 24, 81–88. 10.1038/leu.2009.202. [DOI] [PubMed] [Google Scholar]
- Lehnertz B.; Pabst C.; Su L.; Miller M.; Liu F.; Yi L.; Zhang R.; Krosl J.; Yung E.; Kirschner J.; et al. The Methyltransferase G9a Regulates HoxA9-Dependent Transcription in AML. Genes Dev. 2014, 28, 317–327. 10.1101/gad.236794.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A.; Sampath S. C.; Intrator A.; Min A.; Gertler T. S.; Surmeier D. J.; Tarakhovsky A.; Greengard P. Control of Cognition and Adaptive Behavior by the GLP/G9a Epigenetic Suppressor Complex. Neuron 2009, 64, 678–691. 10.1016/j.neuron.2009.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington H. E. 3rd; Maze I.; Sun H.; Bomze H. M.; DeMaio K. D.; Wu E. Y.; Dietz D. M.; Lobo M. K.; Ghose S.; Mouzon E.; et al. A Role for Repressive Histone Methylation in Cocaine-Induced Vulnerability to Stress. Neuron 2011, 71, 656–670. 10.1016/j.neuron.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antignano F.; Burrows K.; Hughes M. R.; Han J. M.; Kron K. J.; Penrod N. M.; Oudhoff M. J.; Wang S. K.; Min P. H.; Gold M. J.; et al. Methyltransferase G9a Regulates T Cell Differentiation During Murine Intestinal Inflammation. J. Clin. Invest. 2014, 124, 1945–1955. 10.1172/JCI69592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. T.; Kim K. B.; Chae Y. C.; Kang J. Y.; Hahn Y.; Seo S. B. H3K9 Histone Methyltransferase G9a-Mediated Transcriptional Activation of p21. FEBS Lett. 2014, 588, 685–691. 10.1016/j.febslet.2014.01.039. [DOI] [PubMed] [Google Scholar]
- Link P. A.; Gangisetty O.; James S. R.; Woloszynska-Read A.; Tachibana M.; Shinkai Y.; Karpf A. R. Distinct Roles for Histone Methyltransferases G9a and GLP in Cancer Germ-line Antigen Gene Regulation in Human Cancer Cells and Murine Embryonic Stem Cells. Mol. Cancer Res. 2009, 7, 851–862. 10.1158/1541-7786.MCR-08-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M.; Matsumura Y.; Fukuda M.; Kimura H.; Shinkai Y. G9a/GLP Complexes Independently Mediate H3K9 and DNA Methylation to Silence Transcription. EMBO J. 2008, 27, 2681–2690. 10.1038/emboj.2008.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong K. B.; Maksakova I. A.; Mohn F.; Leung D.; Appanah R.; Lee S.; Yang H. W.; Lam L. L.; Mager D. L.; Schubeler D.; et al. DNA Methylation in ES Cells Requires the Lysine Methyltransferase G9a but not its Catalytic Activity. EMBO J. 2008, 27, 2691–2701. 10.1038/emboj.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Skutt-Kakaria K.; Davison J.; Ou Y. L.; Choi E.; Malik P.; Loeb K.; Wood B.; Georges G.; Torok-Storb B.; et al. G9a/GLP-Dependent Histone H3K9me2 Patterning During human Hematopoietic Stem Cell Lineage Commitment. Genes Dev. 2012, 26, 2499–2511. 10.1101/gad.200329.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T.; Brunner H. G.; Amiel J.; Oudakker A. R.; Nillesen W. M.; Magee A.; Genevieve D.; Cormier-Daire V.; van Esch H.; Fryns J. P.; et al. Loss-of-Function Mutations in Euchromatin Histone Methyl Transferase 1 (EHMT1) Cause the 9q34 Subtelomeric Deletion Syndrome. Am. J. Hum. Genet. 2006, 79, 370–377. 10.1086/505693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T.; van Zelst-Stams W. A.; Nillesen W. M.; Cormier-Daire V.; Houge G.; Foulds N.; van Dooren M.; Willemsen M. H.; Pfundt R.; Turner A.; et al. Further Clinical and Molecular Delineation of the 9q Subtelomeric Deletion Syndrome Supports A Major Contribution of EHMT1 Haploinsufficiency to the Core Phenotype. J. Med. Genet. 2009, 46, 598–606. 10.1136/jmg.2008.062950. [DOI] [PubMed] [Google Scholar]
- Ohno H.; Shinoda K.; Ohyama K.; Sharp L. Z.; Kajimura S. EHMT1 Controls Brown Adipose Cell Fate and Thermogenesis Through the PRDM16 Complex. Nature 2013, 504, 163–167. 10.1038/nature12652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek S.; O’Sullivan R. J.; August E. M.; Hickey E. R.; Zhang Q.; Teodoro M. L.; Rea S.; Mechtler K.; Kowalski J. A.; Homon C. A.; et al. Reversal of H3K9me2 by A Small-Molecule Inhibitor for the G9a Histone Methyltransferase. Mol. Cell 2007, 25, 473–481. 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Chang Y.; Zhang X.; Horton J. R.; Upadhyay A. K.; Spannhoff A.; Liu J.; Snyder J. P.; Bedford M. T.; Cheng X. Structural Basis for G9a-Like Protein Lysine Methyltransferase Inhibition by BIX-01294. Nat. Struct. Mol. Biol. 2009, 16, 312–317. 10.1038/nsmb.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Chen X.; Allali-Hassani A.; Quinn A. M.; Wigle T. J.; Wasney G. A.; Dong A.; Senisterra G.; Chau I.; Siarheyeva A.; et al. Protein Lysine Methyltransferase G9a Inhibitors: Design, Synthesis, and Structure Activity Relationships of 2,4-Diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 2010, 53, 5844–5857. 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.; Ganesh T.; Horton J. R.; Spannhoff A.; Liu J.; Sun A.; Zhang X.; Bedford M. T.; Shinkai Y.; Snyder J. P.; et al. Adding A Lysine Mimic in the Design of Potent Inhibitors of HistoneLysine Methyltransferases. J. Mol. Biol. 2010, 400, 1–7. 10.1016/j.jmb.2010.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Barsyte-Lovejoy D.; Allali-Hassani A.; He Y.; Herold J. M.; Chen X.; Yates C. M.; Frye S. V.; Brown P. J.; Huang J.; et al. Optimization of Cellular Activity of G9a Inhibitors 7-Aminoalkoxy-quinazolines. J. Med. Chem. 2011, 54, 6139–6150. 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedadi M.; Barsyte-Lovejoy D.; Liu F.; Rival-Gervier S.; Allali-Hassani A.; Labrie V.; Wigle T. J.; DiMaggio P. A.; Wasney G. A.; Siarheyeva A.; et al. A Chemical Probe Selectively Inhibits G9a and GLP Methyltransferase Activity in Cells. Nat. Chem. Biol. 2011, 7, 566–574. 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Barsyte-Lovejoy D.; Li F.; Xiong Y.; Korboukh V.; Huang X. P.; Allali-Hassani A.; Janzen W. P.; Roth B. L.; Frye S. V.; et al. Discovery of an in vivo Chemical Probe of the Lysine Methyltransferases G9a and GLP. J. Med. Chem. 2013, 56, 8931–8942. 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Lee H.-M.; Xiong Y.; Sciaky N.; Hulbert S. W.; Cao X.; Everitt J. I.; Jin J.; Roth B. L.; Jiang Y.-h. Targeting the Histone Methyltransferase G9a Activates Imprinted Genes and Improves Survival of a Mouse Model of Prader-Willi Syndrome. Nat. Med. 2017, 23, 213–222. 10.1038/nm.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D.; Pattenden S. G.; Liu F.; Barsyte-Lovejoy D.; Li F.; Simon J. M.; Davis I. J.; Vedadi M.; Jin J. A Chemical Tool for in vitro and in vivo Precipitation of Lysine Methyltransferase G9a. ChemMedChem 2014, 9, 549–553. 10.1002/cmdc.201300450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Wang Q.; Paulk J.; Kubicek S.; Kemp M. M.; Adams D. J.; Shamji A. F.; Wagner B. K.; Schreiber S. L. A Small-Molecule Probe of the Histone Methyltransferase G9a Induces Cellular Senescence in Pancreatic Adenocarcinoma. ACS Chem. Biol. 2012, 7, 1152–1157. 10.1021/cb300139y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweis R. F.; Pliushchev M.; Brown P. J.; Guo J.; Li F.; Maag D.; Petros A. M.; Soni N. B.; Tse C.; Vedadi M.; et al. Discovery and Development of Potent and Selective Inhibitors of Histone Methyltransferase G9a. ACS Med. Chem. Lett. 2014, 5, 205–209. 10.1021/ml400496h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devkota K.; Lohse B.; Liu Q.; Wang M. W.; Staerk D.; Berthelsen J.; Clausen R. P. Analogues of the Natural Product Sinefungin as Inhibitors of EHMT1 and EHMT2. ACS Med. Chem. Lett. 2014, 5, 293–297. 10.1021/ml4002503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondengaden S. M.; Luo L. F.; Huang K.; Zhu M. Y.; Zang L. L.; Bataba E.; Wang R. L.; Luo C.; Wang B. H.; Li K. K.; et al. Discovery of Novel Small Molecule Inhibitors of Lysine Methyltransferase G9a and their Mechanism in Leukemia Cell Lines. Eur. J. Med. Chem. 2016, 122, 382–393. 10.1016/j.ejmech.2016.06.028. [DOI] [PubMed] [Google Scholar]
- Xiong Y.; Li F.; Babault N.; Dong A.; Zeng H.; Wu H.; Chen X.; Arrowsmith C. H.; Brown P. J.; Liu J., et al. Discovery of Potent and Selective Inhibitors for G9a-Like Protein (GLP) Lysine Methyltransferase. J. Med. Chem. 2017, Article ASAP, 60, 1876. 10.1021/acs.jmedchem.6b01645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Meara M. M.; Simon J. A. Inner Workings and Regulatory Inputs that Control Polycomb Repressive Complex 2. Chromosoma 2012, 121, 221–234. 10.1007/s00412-012-0361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R.; Wang L.; Wang H.; Xia L.; Erdjument-Bromage H.; Tempst P.; Jones R. S.; Zhang Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- Kuzmichev A.; Nishioka K.; Erdjument-Bromage H.; Tempst P.; Reinberg D. Histone Methyltransferase Activity Associated with A HumanMultiprotein Complex Containing the Enhancer of Zeste Protein. Genes Dev. 2002, 16, 2893–2905. 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R.; Reinberg D. The Polycomb Complex PRC2 and its Mark in Life. Nature 2011, 469, 343–349. 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czermin B.; Melfi R.; McCabe D.; Seitz V.; Imhof A.; Pirrotta V. Drosophila Enhancer of Zeste/ESC Complexes have A Histone H3Methyltransferase Activity that Marks Chromosomal Polycomb Sites. Cell 2002, 111, 185–196. 10.1016/S0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- Muller J.; Hart C. M.; Francis N. J.; Vargas M. L.; Sengupta A.; Wild B.; Miller E. L.; O’Connor M. B.; Kingston R. E.; Simon J. A. Histone Methyltransferase Activity of A Drosophila Polycomb Group Repressor Complex. Cell 2002, 111, 197–208. 10.1016/S0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- Joshi P.; Carrington E. A.; Wang L.; Ketel C. S.; Miller E. L.; Jones R. S.; Simon J. A. Dominant Alleles Identify SET Domain Residues Required for Histone Methyltransferase of Polycomb Repressive Complex 2. J. Biol. Chem. 2008, 283, 27757–27766. 10.1074/jbc.M804442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S. K.; Tian X.; LaFrance L. V.; Duquenne C.; Suarez D. P.; Newlander K. A.; Romeril S. P.; Burgess J. L.; Grant S. W.; Brackley J. A.; et al. Identification of Potent, Selective, Cell-Active Inhibitors of the Histone Lysine Methyltransferase EZH2. ACS Med. Chem. Lett. 2012, 3, 1091–1096. 10.1021/ml3003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R.; Li G.; Sarma K.; Blais A.; Zavadil J.; Woodcock C. L.; Dynlacht B. D.; Reinberg D. Ezh1 and Ezh2Maintain Repressive Chromatin through Different Mechanisms. Mol. Cell 2008, 32, 503–518. 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X.; Liu Y.; Hsu Y.-J.; Fujiwara Y.; Kim J.; Mao X.; Yuan G.-C.; Orkin S. H. EZH1Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 2008, 32, 491–502. 10.1016/j.molcel.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zee B. M.; Levin R. S.; Xu B.; LeRoy G.; Wingreen N. S.; Garcia B. A. In Vivo Residue-Specific Histone Methylation Dynamics. J. Biol. Chem. 2010, 285, 3341–3350. 10.1074/jbc.M109.063784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R.; Zhang Y. SUZ12 is Required for both the Histone Methyltransferase Activity and the Silencing Function of the EED-EZH2 Complex. Mol. Cell 2004, 15, 57–67. 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Ketel C. S.; Andersen E. F.; Vargas M. L.; Suh J.; Strome S.; Simon J. A. Subunit Contributions to Histone Methyltransferase Activities of Fly and Worm Polycomb Group Complexes. Mol. Cell. Biol. 2005, 25, 6857–6868. 10.1128/MCB.25.16.6857-6868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasini D.; Hansen K. H.; Christensen J.; Agger K.; Cloos P. A.; Helin K. Coordinated Regulation of Transcriptional Repression by the RBP2 H3K4 Demethylase and Polycomb-Repressive Complex 2. Genes Dev. 2008, 22, 1345–1355. 10.1101/gad.470008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Kang K.; Kim J. AEBP2 as A Potential Targeting Protein for Polycomb Repression Complex PRC2. Nucleic Acids Res. 2009, 37, 2940–2950. 10.1093/nar/gkp149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasov M.; Klymenko T.; Fraterman S.; Papp B.; Oktaba K.; Kocher T.; Cohen A.; Stunnenberg H. G.; Wilm M.; Muller J. Pcl-PRC2 is Needed to Generate High Levels of H3-K27 Trimethylation at Polycomb Target Genes. EMBO J. 2007, 26, 4078–4088. 10.1038/sj.emboj.7601837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker E.; Chang W. Y.; Hunkapiller J.; Cagney G.; Garcha K.; Torchia J.; Krogan N. J.; Reiter J. F.; Stanford W. L. Polycomb-like 2 Associates with PRC2 and Regulates Transcriptional Networks during Mouse Embryonic Stem Cell Self-Renewal and Differentiation. Cell Stem Cell 2010, 6, 153–166. 10.1016/j.stem.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Margueron R.; Ku M. C.; Chambon P.; Bernstein B. E.; Reinberg D. Jarid2 and PRC2, Partners in Regulating Gene Expression. Genes Dev. 2010, 24, 368–380. 10.1101/gad.1886410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin R. D.; Johnson N. A.; Severson T. M.; Mungall A. J.; An J.; Goya R.; Paul J. E.; Boyle M.; Woolcock B. W.; Kuchenbauer F.; et al. Somatic Mutations Altering EZH2 (Tyr641) in Follicular and Diffuse Large B-Cell Lymphomas of Germinal-Center Origin. Nat. Genet. 2010, 42, 181–185. 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap D. B.; Chu J.; Berg T.; Schapira M.; Cheng S. W.; Moradian A.; Morin R. D.; Mungall A. J.; Meissner B.; Boyle M.; et al. Somatic Mutations at EZH2 Y641 Act Dominantly through A Mechanism of Selectively Altered PRC2 Catalytic Activity, to Increase H3K27 Trimethylation. Blood 2011, 117, 2451–2459. 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneeringer C. J.; Scott M. P.; Kuntz K. W.; Knutson S. K.; Pollock R. M.; Richon V. M.; Copeland R. A. Coordinated Activities of Wild-Type Plus Mutant EZH2 Drive Tumor-Associated Hypertrimethylation of Lysine 27 on Histone H3 (H3K27) in Human B-Cell Lymphomas. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 20980–20985. 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe M. T.; Graves A. P.; Ganji G.; Diaz E.; Halsey W. S.; Jiang Y.; Smitheman K. N.; Ott H. M.; Pappalardi M. B.; Allen K. E.; et al. Mutation of A677 in Histone Methyltransferase EZH2 in Human B-Cell Lymphoma Promotes Hypertrimethylation of Histone H3 on Lysine 27 (H3K27). Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2989–2994. 10.1073/pnas.1116418109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken A. P.; Pasini D.; Capra M.; Prosperini E.; Colli E.; Helin K. EZH2 is Downstream of the pRB-E2F Pathway, Essential for Proliferation and Amplified in Cancer. EMBO J. 2003, 22, 5323–5335. 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J. A.; Lange C. A. Roles of the EZH2 histone Methyltransferase in Cancer Epigenetics. Mutat. Res., Fundam. Mol. Mech. Mutagen. 2008, 647, 21–29. 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Ding L.; Erdmann C.; Chinnaiyan A. M.; Merajver S. D.; Kleer C. G. Identification of EZH2 as A Molecular Marker for A Precancerous sState in Morphologically Normal Breast Tissues. Cancer Res. 2006, 66, 4095–4099. 10.1158/0008-5472.CAN-05-4300. [DOI] [PubMed] [Google Scholar]
- Kleer C. G.; Cao Q.; Varambally S.; Shen R.; Ota I.; Tomlins S. A.; Ghosh D.; Sewalt R. G.; Otte A. P.; Hayes D. F.; et al. EZH2 is A Marker of Aggressive Breast Cancer and Promotes Neoplastic Transformation of Breast Epithelial Cells. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 11606–11611. 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S.; Dhanasekaran S. M.; Zhou M.; Barrette T. R.; Kumar-Sinha C.; Sanda M. G.; Ghosh D.; Pienta K. J.; Sewalt R. G.; Otte A. P.; et al. The Polycomb Group Protein EZH2 is Involved in Progression of Prostate Cancer. Nature 2002, 419, 624–629. 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- Velichutina I.; Shaknovich R.; Geng H.; Johnson N. A.; Gascoyne R. D.; Melnick A. M.; Elemento O. EZH2-Mediated Epigenetic Silencing in GerminalCenter B Cells Contributes to Proliferation and Lymphomagenesis. Blood 2010, 116, 5247–5255. 10.1182/blood-2010-04-280149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando H.; Gelato K. A.; Lesche R.; Beckmann G.; Koehr S.; Otto S.; Steigemann P.; Stresemann C. EZH2 Inhibition Blocks Multiple Myeloma Cell Growth through Upregulation of Epithelial Tumor Suppressor Genes. Mol. Cancer Ther. 2016, 15, 287–298. 10.1158/1535-7163.MCT-15-0486. [DOI] [PubMed] [Google Scholar]
- Shih A. H.; Abdel-Wahab O.; Patel J. P.; Levine R. L. The Role of Mutations in Epigenetic Regulators in Myeloid Malignancies. Nat. Rev. Cancer 2012, 12, 599–612. 10.1038/nrc3343. [DOI] [PubMed] [Google Scholar]
- Neff T.; Sinha A. U.; Kluk M. J.; Zhu N.; Khattab M. H.; Stein L.; Xie H.; Orkin S. H.; Armstrong S. A. Polycomb Repressive Complex 2 is Required for MLL-AF9 Leukemia. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5028–5033. 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Wang E.; Zuber J.; Rappaport A.; Taylor M.; Johns C.; Lowe S. W.; Vakoc C. R. The Polycomb Complex PRC2 Supports Aberrant Self-Renewal in A Mouse Model of MLL-AF9;Nras(G12D) Acute Myeloid Leukemia. Oncogene 2013, 32, 930–938. 10.1038/onc.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B. W.; On D. M.; Ma A. Q.; Parton T.; Konze K. D.; Pattenden S. G.; Allison D. F.; Cai L.; Rockowitz S.; Liu S. C.; et al. Selective Inhibition of EZH2 and EZH1 Enzymatic Activity by A Small Molecule Suppresses MLL-Rearranged Leukemia. Blood 2015, 125, 346–357. 10.1182/blood-2014-06-581082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Wigle T. J.; Warholic N. M.; Sneeringer C. J.; Allain C. J.; Klaus C. R.; Sacks J. D.; Raimondi A.; Majer C. R.; Song J.; et al. A Selective Inhibitor of EZH2 Blocks H3K27 Methylation and Kills Mutant Lymphoma Cells. Nat. Chem. Biol. 2012, 8, 890–896. 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- McCabe M. T.; Ott H. M.; Ganji G.; Korenchuk S.; Thompson C.; Van Aller G. S.; Liu Y.; Graves A. P.; Iii A. D.; Diaz E.; et al. EZH2 Inhibition as A Therapeutic Strategy for Lymphoma with EZH2-Activating Mutations. Nature 2012, 492, 108–112. 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- Beguelin W.; Popovic R.; Teater M.; Jiang Y.; Bunting K. L.; Rosen M.; Shen H.; Yang S. N.; Wang L.; Ezponda T.; et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2Mutations Promote Lymphoid Transformation. Cancer Cell 2013, 23, 677–692. 10.1016/j.ccr.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT02082977. A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK2816126 in Subjects With Relapsed/Refractory Diffuse Large B Cell and Transformed Follicular Lymphoma, Transformed Follicular Lymphoma, Other Non-Hodgkin’s Lymphomas, Solid Tumors and Multiple Myeloma (accessed Nov 2016).
- Qi W.; Chan H.; Teng L.; Li L.; Chuai S.; Zhang R.; Zeng J.; Li M.; Fan H.; Lin Y.; et al. Selective Inhibition of Ezh2 by A Small Molecule Inhibitor Blocks Tumor Cells Proliferation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 21360–21365. 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konze K. D.; Ma A.; Li F.; Barsyte-Lovejoy D.; Parton T.; MacNevin C. J.; Liu F.; Gao C.; Huang X. P.; Kuznetsova E.; et al. An Orally Bioavailable Chemical Probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem. Biol. 2013, 8, 1324–1334. 10.1021/cb400133j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. B.; Li F. L.; Konze K. D.; Meslamani J.; Ma A. Q.; Brown P. J.; Zhou M. M.; Arrowsmith C. H.; Kaniskan H. Ü.; Vedadi M.; et al. Structure-Activity Relationship Studies for Enhancer of Zeste Homologue 2 (EZH2) and Enhancer of Zeste Homologue 1 (EZH1) Inhibitors. J. Med. Chem. 2016, 59, 7617–7633. 10.1021/acs.jmedchem.6b00855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson S. K.; Warholic N. M.; Wigle T. J.; Klaus C. R.; Allain C. J.; Raimondi A.; Porter Scott M.; Chesworth R.; Moyer M. P.; Copeland R. A.; et al. Durable Tumor Regression in Genetically Altered Malignant Rhabdoid Tumors by Inhibition of Methyltransferase EZH2. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7922–7927. 10.1073/pnas.1303800110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson B. G.; Roberts C. W. SWI/SNF Nucleosome Remodellers and Cancer. Nat. Rev. Cancer 2011, 11, 481–492. 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- Wilson B. G.; Wang X.; Shen X.; McKenna E. S.; Lemieux M. E.; Cho Y. J.; Koellhoffer E. C.; Pomeroy S. L.; Orkin S. H.; Roberts C. W. Epigenetic Antagonism between Polycomb and SWI/SNF Complexes During Oncogenic Transformation. Cancer Cell 2010, 18, 316–328. 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT01897571. An Open-Label, Multicenter, Phase 1/2 Study of E7438 (EZH2 Histone Methyl Transferase [HMT] Inhibitor) as a Single Agent in Subjects With Advanced Solid Tumors or With B-cell Lymphomas (accessed Nov 2016).
- ClinicalTrials.gov Identifier: NCT02889523. A Phase Ib-II Study of Tazemetostat (EPZ-6438) in Newly Diagnosed Diffuse Large B Cell Lymphoma (DLBCL) Patients With Poor Prognosis Treated by Rituximab-Cyclophosphamide-Vincristine-Doxorubicin-Prednisolone Chemiotherapy (R-CHOP) (accessed Nov 2016).
- ClinicalTrials.gov Identifier: NCT02601950. A Phase II, Multicenter Study of the EZH2 Inhibitor Tazemetostat in Adult Subjects With INI1-Negative Tumors or Relapsed/Refractory Synovial Sarcoma (accessed Nov 2016).
- ClinicalTrials.gov Identifier: NCT02860286. A Multicenter Study of the EZH2 Inhibitor Tazemetostat in Adult Subjects With Relapsed or Refractory Malignant Mesothelioma With BAP1 Loss of Function (accessed Nov 2016).
- ClinicalTrials.gov Identifier: NCT02601937. A Phase 1 Study of the EZH2 Inhibitor Tazemetostat in Pediatric Subjects With Relapsed or Refractory INI1-Negative Tumors or Synovial Sarcoma (accessed Nov 2016).
- Garapaty-Rao S.; Nasveschuk C.; Gagnon A.; Chan E. Y.; Sandy P.; Busby J.; Balasubramanian S.; Campbell R.; Zhao F.; Bergeron L.; et al. Identification of EZH2 and EZH1 Small Molecule Inhibitors with Selective Impact on Diffuse Large B Cell Lymphoma Cell Growth. Chem. Biol. 2013, 20, 1329–1339. 10.1016/j.chembiol.2013.09.013. [DOI] [PubMed] [Google Scholar]
- Nasveschuk C. G.; Gagnon A.; Garapaty-Rao S.; Balasubramanian S.; Campbell R.; Lee C.; Zhao F.; Bergeron L.; Cummings R.; Trojer P.; et al. Discovery and Optimization of Tetramethylpiperidinyl Benzamides as Inhibitors of EZH2. ACS Med. Chem. Lett. 2014, 5, 378–383. 10.1021/ml400494b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley W. D.; Arora S.; Busby J.; Balasubramanian S.; Gehling V. S.; Nasveschuk C. G.; Vaswani R. G.; Yuan C. C.; Hatton C.; Zhao F.; et al. EZH2 Inhibitor Efficacy in Non-Hodgkin’s Lymphoma Does Not Require Suppression of H3K27 Monomethylation. Chem. Biol. 2014, 21, 1463–1475. 10.1016/j.chembiol.2014.09.017. [DOI] [PubMed] [Google Scholar]
- Vaswani R. G.; Gehling V. S.; Dakin L. A.; Cook A. S.; Nasveschuk C. G.; Duplessis M.; Iyer P.; Balasubramanian S.; Zhao F.; Good A. C.; et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), A Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J. Med. Chem. 2016, 59, 9928–9941. 10.1021/acs.jmedchem.6b01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J. E.; Kuntz K. W.; Knutson S. K.; Warholic N. M.; Keilhack H.; Wigle T. J.; Raimondi A.; Klaus C. R.; Rioux N.; Yokoi A.; et al. EPZ011989, A Potent, Orally-Available EZH2 Inhibitor with Robust In Vivo Activity. ACS Med. Chem. Lett. 2015, 6, 491–495. 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X.; Gao T.; Wang N.; Feng Q.; You X.; Ye T.; Lei Q.; Zhu Y.; Xiong M.; Xia Y.; et al. Selective Inhibition of EZH2 by ZLD1039 Blocks H3K27 Methylation and Leads to Potent Anti-Tumor Activity in Breast Cancer. Sci. Rep. 2016, 6, 20864. 10.1038/srep20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T. T.; Zhang L. D.; Zhu Y. X.; Song X. J.; Feng Q.; Lei Q.; Shi S. X.; Deng H. X.; Xiong M. H.; You X. Y.; et al. ZLD1122, A Novel EZH2 and EZH1 Small Molecular Inhibitor, Blocks H3K27 Methylation and Diffuse Large B Cell Lymphoma Cell Growth. RSC Adv. 2016, 6, 28512–28521. 10.1039/C6RA00618C. [DOI] [Google Scholar]
- Jiao L. Y.; Liu X. Structural Basis of Histone H3K27 Trimethylation by an Active Polycomb Repressive Complex 2. Science 2015, 350, 1–8. 10.1126/science.aac4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justin N.; Zhang Y.; Tarricone C.; Martin S. R.; Chen S.; Underwood E.; De Marco V.; Haire L. F.; Walker P. A.; Reinberg D.; et al. Structural Basis of Oncogenic Histone H3K27M Inhibition of Human Polycomb Repressive Complex 2. Nat. Commun. 2016, 7, 11316. 10.1038/ncomms11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooun A.; Gajiwala K. S.; Deng Y. L.; Liu W.; Bolanos B.; Bingham P.; He Y. A.; Diehl W.; Grable N.; Kung P. P.; et al. Polycomb Repressive Complex 2 Structure with Inhibitor Reveals A Mechanism of Activation and Drug Resistance. Nat. Commun. 2016, 7, 11384–11395. 10.1038/ncomms11384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker T.; Nerle S.; Pritchard J.; Zhao B. Y.; Rivera V. M.; Garner A.; Gonzalvez F. Acquisition of A Single EZH2 D1 Domain Mutation Confers Acquired Resistance to EZH2-Targeted Inhibitors. Oncotarget 2015, 6, 32646–32655. 10.18632/oncotarget.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibaja V.; Shen F.; Harari J.; Korn J.; Ruddy D.; Saenz-Vash V.; Zhai H.; Rejtar T.; Paris C. G.; Yu Z.; et al. Development of Secondary Mutations in Wild-Type and Mutant EZH2 Alleles Cooperates to Confer Resistance to EZH2 Inhibitors. Oncogene 2016, 35, 558–566. 10.1038/onc.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung P. P.; Rui E.; Bergqvist S.; Bingham P.; Braganza J.; Collins M.; Cui M.; Diehl W.; Dinh D.; Fan C.; et al. Design and Synthesis of Pyridone-Containing 3,4-Dihydroisoquinoline-1(2H)-ones as a Novel Class of Enhancer of Zeste Homolog 2 (EZH2) Inhibitors. J. Med. Chem. 2016, 59, 8306–8325. 10.1021/acs.jmedchem.6b00515. [DOI] [PubMed] [Google Scholar]
- Souroullas G. P.; Jeck W. R.; Parker J. S.; Simon J. M.; Liu J. Y.; Paulk J.; Xiong J.; Clark K. S.; Fedoriw Y.; Qi J.; et al. An Oncogenic Ezh2Mutation Induces Tumors Through Global Redistribution of Histone 3 Lysine 27 Trimethylation. Nat. Med. 2016, 22, 632–640. 10.1038/nm.4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X. Q.; Chen L. M.; Jiao L. Y.; Jiang X. R.; Lian F. L.; Lu J. Y.; Zhu K. K.; Du D. H.; Liu J. Q.; Ding H.; et al. Astemizole Arrests the Proliferation of Cancer Cells by Disrupting the EZH2-EED Interaction of Polycomb Repressive Complex 2. J. Med. Chem. 2014, 57, 9512–9521. 10.1021/jm501230c. [DOI] [PubMed] [Google Scholar]
- He Y.; Selvaraju S.; Curtin M. L.; Jakob C. G.; Zhu H.; Comess K. M.; Shaw B.; The J.; Lima-Fernandes E.; Szewczyk M. M.; et al. The EED Protein-Protein Interaction Inhibitor A-395 Inactivates the PRC2 Complex. Nat. Chem. Biol. 2017, 10.1038/nchembio.2306. [DOI] [PubMed] [Google Scholar]
- Qi W.; Zhao K.; Gu J.; Huang Y.; Wang Y.; Zhang H.; Zhang M.; Zhang J.; Yu Z.; Li L.; et al. An Allosteric PRC2 Inhibitor Targeting the H3K27me3 Binding Pocket of EED. Nat. Chem. Biol. 2017, 10.1038/nchembio.2304. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Zhang J.; Yu Z.; Zhang H.; Wang Y.; Lingel A.; Qi W.; Gu J.; Zhao K.; Shultz M. D.; et al. Discovery of First-in-Class, Potent, and Orally Bioavailable Embryonic Ectoderm Development (EED) Inhibitor with Robust Anticancer Efficacy. J. Med. Chem. 2017, 10.1021/acs.jmedchem.6b01576. [DOI] [PubMed] [Google Scholar]
- Lingel A.; Sendzik M.; Huang Y.; Shultz M. D.; Cantwell J.; Dillon M. P.; Fu X.; Fuller J.; Gabriel T.; Gu J.; et al. Structure-Guided Design of EED Binders Allosterically Inhibiting the Epigenetic Polycomb Repressive Complex 2 (PRC2) Methyltransferase. J. Med. Chem. 2017, 60, 415–427. 10.1021/acs.jmedchem.6b01473. [DOI] [PubMed] [Google Scholar]
- Kim W.; Bird G. H.; Neff T.; Guo G. J.; Kerenyi M. A.; Walensky L. D.; Orkin S. H. Targeted Disruption of the EZH2-EED Complex Inhibits EZH2-Dependent Cancer. Nat. Chem. Biol. 2013, 9, 643–650. 10.1038/nchembio.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Allis C. D.; Wysocka J. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading A Single Epigenetic Mark. Mol. Cell 2007, 25, 15–30. 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Brown M. A.; Sims R. J. 3rd; Gottlieb P. D.; Tucker P. W. Identification and Characterization of Smyd2: A Split SET/MYND Domain-Containing Histone H3 Lysine 36-Specific Methyltransferase that Interacts with the Sin3 Histone Deacetylase Complex. Mol. Cancer 2006, 5, 26. 10.1186/1476-4598-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Farha M.; Lambert J. P.; Al-Madhoun A. S.; Elisma F.; Skerjanc I. S.; Figeys D. The Tale of Two Domains: Proteomics and Genomics Analysis of SMYD2, A New Histone Methyltransferase. Mol. Cell. Proteomics 2007, 7, 560–572. 10.1074/mcp.M700271-MCP200. [DOI] [PubMed] [Google Scholar]
- Zhou P. P.; Wang Z. L.; Yuan X. J.; Zhou C. H.; Liu L. L.; Wan X. L.; Zhang F.; Ding X. D.; Wang C. G.; Xiong S. D.; et al. Mixed Lineage Leukemia 5 (MLL5) Protein Regulates Cell Cycle Progression and E2F1-responsive Gene Expression via Association with Host Cell Factor-1 (HCF-1). J. Biol. Chem. 2013, 288, 17532–17543. 10.1074/jbc.M112.439729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer L. L.; Lima-Fernandes E.; Gibson E.; Eram M. S.; Loppnau P.; Arrowsmith C. H.; Schapira M.; Vedadi M. PR Domain-Containing Protein 7 (PRDM7) Is A Histone 3 Lysine 4 Trimethyltransferase. J. Biol. Chem. 2016, 291, 13509–13519. 10.1074/jbc.M116.721472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Mathioudakis N.; Diagouraga B.; Dong A. P.; Dombrovski L.; Baudat F.; Cusack S.; de Massy B.; Kadlec J. Molecular Basis for the Regulation of the H3K4Methyltransferase Activity of PRDM9. Cell Rep. 2013, 5, 13–20. 10.1016/j.celrep.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Nishioka K.; Chuikov S.; Sarma K.; Erdjument-Bromage H.; Allis C. D.; Tempst P.; Reinberg D. Set9, a Novel Histone H3Methyltransferase that Facilitates Transcription by Precluding Histone Tail Modifications Required for Heterochromatin Formation. Genes Dev. 2002, 16, 479–489. 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Cao R.; Xia L.; Erdjument-Bromage H.; Borchers C.; Tempst P.; Zhang Y. Purification and Functional Characterization of A Histone H3-Lysine 4-Specific Methyltransferase. Mol. Cell 2001, 8, 1207–1217. 10.1016/S1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- Pradhan S.; Chin H. G.; Esteve P. O.; Jacobsen S. E. SET7/9 Mediated Methylation of Non-Histone Proteins in Mammalian Cells. Epigenetics 2009, 4, 383–387. 10.4161/epi.4.6.9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K.; Jia D.; Kapoor-Vazirani P.; Powell D. R.; Collins R. E.; Sharma D.; Peng J.; Cheng X.; Vertino P. M. Regulation of Estrogen Receptor Alpha by the SET7 Lysine Methyltransferase. Mol. Cell 2008, 30, 336–347. 10.1016/j.molcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr S. M.; Munro S.; Kessler B.; Oppermann U.; La Thangue N. B. Interplay between Lysine Methylation and CDK Phosphorylation in Growth Control by the Retinoblastoma Protein. EMBO J. 2011, 30, 317–327. 10.1038/emboj.2010.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Huang J.; Dasgupta M.; Sears N.; Miyagi M.; Wang B.; Chance M. R.; Chen X.; Du Y.; Wang Y.; et al. Reversible Methylation of Promoter-Bound STAT3 by Histone-Modifying Enzymes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 21499–21504. 10.1073/pnas.1016147107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Chen Z.; Xu C.; Leng X.; Cao H.; Ouyang G.; Xiao W. Repression of Hypoxia-Inducible Factor Alpha Signaling by Set7-Mediated Methylation. Nucleic Acids Res. 2015, 43, 5081–5098. 10.1093/nar/gkv379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calnan D. R.; Webb A. E.; White J. L.; Stowe T. R.; Goswami T.; Shi X.; Espejo A.; Bedford M. T.; Gozani O.; Gygi S. P.; et al. Methylation by Set9Modulates FoxO3 Stability and Transcriptional Activity. Aging 2012, 4, 462–479. 10.18632/aging.100471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteve P. O.; Chin H. G.; Benner J.; Feehery G. R.; Samaranayake M.; Horwitz G. A.; Jacobsen S. E.; Pradhan S. Regulation of DNMT1 Stability through SET7-Mediated lysine Methylation in Mammalian Cells. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5076–5081. 10.1073/pnas.0810362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea C. K.; Baltimore D. Regulation of NF-kappaB Activity through Lysine Monomethylation of p65. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18972–18977. 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Reddy M. A.; Miao F.; Shanmugam N.; Yee J.-K.; Hawkins D.; Ren B.; Natarajan R. Role of the Histone H3 Lysine 4 Methyltransferase, SET7/9, in the Regulation of NF-{kappa}B-dependent Inflammatory Genes: Relevance to Diabetes and Inflammation. J. Biol. Chem. 2008, 283, 26771–26781. 10.1074/jbc.M802800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebel A. L.; Fernandez A. Z.; El-Osta A. Glycemic Memory Associated Epigenetic Changes. Biochem. Pharmacol. 2010, 80, 1853–1859. 10.1016/j.bcp.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Barsyte-Lovejoy D.; Li F.; Oudhoff M. J.; Tatlock J. H.; Dong A.; Zeng H.; Wu H.; Freeman S. A.; Schapira M.; Senisterra G. A.; et al. (R)-PFI-2 is a Potent and Selective Inhibitor of SETD7Methyltransferase Activity in Cells. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 12853–12858. 10.1073/pnas.1407358111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M. Structural Chemistry of Human SET Domain Protein Methyltransferases. Curr. Chem. Genomics 2011, 5, 85–94. 10.2174/1875397301005010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez G.; McBean J. L.; Astudillo Y. M.; Luo M. An Enzyme-Coupled Ultrasensitive Luminescence Assay for Protein Methyltransferases. Anal. Biochem. 2010, 401, 203–210. 10.1016/j.ab.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Mori S.; Iwase K.; Iwanami N.; Tanaka Y.; Kagechika H.; Hirano T. Development of Novel Bisubstrate-Type Inhibitors of Histone Methyltransferase SET7/9. Bioorg. Med. Chem. 2010, 18, 8158–8166. 10.1016/j.bmc.2010.10.022. [DOI] [PubMed] [Google Scholar]
- Francis N. J.; Rowlands M.; Workman P.; Jones K.; Aherne W. Small-Molecule Inhibitors of the Protein Methyltransferase SET7/9 Identified in A High-Throughput Screen. J. Biomol. Screening 2012, 17, 1102–1109. 10.1177/1087057112452137. [DOI] [PubMed] [Google Scholar]
- Meng F.; Cheng S.; Ding H.; Liu S.; Liu Y.; Zhu K.; Chen S.; Lu J.; Xie Y.; Li L.; et al. Discovery and Optimization of Novel, Selective Histone Methyltransferase SET7 Inhibitors by Pharmacophore- and Docking-Based Virtual Screening. J. Med. Chem. 2015, 58, 8166–8181. 10.1021/acs.jmedchem.5b01154. [DOI] [PubMed] [Google Scholar]
- Takemoto Y.; Ito A.; Niwa H.; Okamura M.; Fujiwara T.; Hirano T.; Handa N.; Umehara T.; Sonoda T.; Ogawa K.; et al. Identification of Cyproheptadine as an Inhibitor of SET Domain Containing Lysine Methyltransferase 7/9 (Set7/9) That Regulates Estrogen-Dependent Transcription. J. Med. Chem. 2016, 59, 3650–3660. 10.1021/acs.jmedchem.5b01732. [DOI] [PubMed] [Google Scholar]
- Gottlieb P. D.; Pierce S. A.; Sims R. J.; Yamagishi H.; Weihe E. K.; Harriss J. V.; Maika S. D.; Kuziel W. A.; King H. L.; Olson E. N.; et al. Bop Encodes A Muscle-Restricted Protein Containing MYND and SET Domains and is Essential for Cardiac Differentiation and Morphogenesis. Nat. Genet. 2002, 31, 25–32. 10.1038/ng866. [DOI] [PubMed] [Google Scholar]
- Hamamoto R.; Furukawa Y.; Morita M.; Iimura Y.; Silva F. P.; Li M.; Yagyu R.; Nakamura Y. SMYD3 Encodes A Histone Methyltransferase Involved in the Proliferation of Cancer Cells. Nat. Cell Biol. 2004, 6, 731–740. 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- Mazur P. K.; Reynoird N.; Khatri P.; Jansen P. W. T. C.; Wilkinson A. W.; Liu S. C.; Barbash O.; Van Aller G. S.; Huddleston M.; Dhanak D.; et al. SMYD3 Links Lysine Methylation of MAP3K2 to Ras-Driven Cancer. Nature 2014, 510, 283–287. 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Aller G. S.; Reynoird N.; Barbash O.; Huddleston M.; Liu S.; Zmoos A. F.; McDevitt P.; Sinnamon R.; Le B.; Mas G.; et al. SMYD3 Regulates Cancer Cell Phenotypes and Catalyzes Histone H4 Lysine 5 Methylation. Epigenetics 2012, 7, 340–343. 10.4161/epi.19506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira F. Q.; Costa-Pinheiro P.; Ramalho-Carvalho J.; Pereira A.; Menezes F. D.; Antunes L.; Carneiro I.; Oliveira J.; Henrique R.; Jeronimo C. Deregulated Expression of Selected Histone Methylases and Demethylases in Prostate Carcinoma. Endocr.-Relat. Cancer 2014, 21, 51–61. 10.1530/ERC-13-0375. [DOI] [PubMed] [Google Scholar]
- Hamamoto R.; Silva F. P.; Tsuge M.; Nishidate T.; Katagiri T.; Nakamura Y.; Furukawa Y. Enhanced SMYD3 Expression is Essential for the Growth of Breast Cancer Cells. Cancer Sci. 2006, 97, 113–118. 10.1111/j.1349-7006.2006.00146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarris M. E.; Moulos P.; Haroniti A.; Giakountis A.; Talianidis I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366. 10.1016/j.ccell.2016.01.013. [DOI] [PubMed] [Google Scholar]
- Mitchell L. H.; Boriack-Sjodin P. A.; Smith S.; Thomenius M.; Rioux N.; Munchhof M.; Mills J. E.; Klaus C.; Totman J.; Riera T. V.; et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 134–138. 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A.Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, 2nd ed.; 2013, pp 1–538 10.1002/9781118540398. [DOI] [PubMed] [Google Scholar]
- Van Aller G. S.; Graves A. P.; Elkins P. A.; Bonnette W. G.; McDevitt P. J.; Zappacosta F.; Annan R. S.; Dean T. W.; Su D. S.; Carpenter C. L.; et al. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure 2016, 24, 774–781. 10.1016/j.str.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Peserico A.; Germani A.; Sanese P.; Barbosa A. J.; Di Virgilio V.; Fittipaldi R.; Fabini E.; Bertucci C.; Varchi G.; Moyer M. P.; et al. A SMYD3 Small-Molecule Inhibitor Impairing Cancer Cell Growth. J. Cell. Physiol. 2015, 230, 2447–2460. 10.1002/jcp.24975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon S. C.; Zhang X.; Trievel R. C.; Cheng X. The SET-Domain Protein Superfamily: Protein Lysine Methyltransferases. Genome Biol. 2005, 6, 227. 10.1186/gb-2005-6-8-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdic V.; Zhang P.; Lanouette S.; Groulx A.; Tremblay V.; Brunzelle J.; Couture J. F. Structural and Biochemical Insights into MLL1 Core Complex Assembly. Structure 2011, 19, 101–108. 10.1016/j.str.2010.09.022. [DOI] [PubMed] [Google Scholar]
- Krivtsov A. V.; Armstrong S. A. MLL Translocations, Histone Modifications and Leukaemia Stem-Cell Development. Nat. Rev. Cancer 2007, 7, 823–833. 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- Albert M.; Helin K. Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 2010, 21, 209–220. 10.1016/j.semcdb.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Kohlmann A.; Schoch C.; Dugas M.; Schnittger S.; Hiddemann W.; Kern W.; Haferlach T. New Insights into MLL Gene Rearranged Acute Leukemias using Gene Expression Profiling: Shared Pathways, Lineage Commitment, and Partner Genes. Leukemia 2005, 19, 953–964. 10.1038/sj.leu.2403746. [DOI] [PubMed] [Google Scholar]
- Hess J. L. MLL: a Histone Methyltransferase Disrupted in Leukemia. Trends Mol. Med. 2004, 10, 500–507. 10.1016/j.molmed.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Yu B. D.; Hess J. L.; Horning S. E.; Brown G. A.; Korsmeyer S. J. Altered Hox Expression and Segmental Identity in MLL-Mutant Mice. Nature 1995, 378, 505–508. 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- Li Y.; Han J.; Zhang Y.; Cao F.; Liu Z.; Li S.; Wu J.; Hu C.; Wang Y.; Shuai J.; et al. Structural Basis for Activity Regulation of MLL Family Methyltransferases. Nature 2016, 530, 447–452. 10.1038/nature16952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southall S. M.; Wong P. S.; Odho Z.; Roe S. M.; Wilson J. R. Structural Basis for the Requirement of Additional Factors for MLL1 SET Domain Activity and Recognition of Epigenetic Marks. Mol. Cell 2009, 33, 181–191. 10.1016/j.molcel.2008.12.029. [DOI] [PubMed] [Google Scholar]
- Karatas H.; Townsend E. C.; Cao F.; Chen Y.; Bernard D.; Liu L.; Lei M.; Dou Y.; Wang S. High-Affinity, Small-Molecule Peptidomimetic Inhibitors of MLL1/WDR5 Protein-Protein Interaction. J. Am. Chem. Soc. 2013, 135, 669–682. 10.1021/ja306028q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senisterra G.; Wu H.; Allali-Hassani A.; Wasney G. A.; Barsyte-Lovejoy D.; Dombrovski L.; Dong A.; Nguyen K. T.; Smil D.; Bolshan Y.; et al. Small-Molecule Inhibition of MLL aActivity by Disruption of its Interaction with WDR5. Biochem. J. 2013, 449, 151–159. 10.1042/BJ20121280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F.; Townsend E. C.; Karatas H.; Xu J.; Li L.; Lee S.; Liu L.; Chen Y.; Ouillette P.; Zhu J. D.; et al. Targeting MLL1 H3K4Methyltransferase Activity in Mixed-Lineage Leukemia. Mol. Cell 2014, 53, 247–261. 10.1016/j.molcel.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getlik M.; Smil D.; Zepeda-Velazquez C.; Bolshan Y.; Poda G.; Wu H.; Dong A. P.; Kuznetsova E.; Marcellus R.; Senisterra G.; et al. Structure-Based Optimization of a Small Molecule Antagonist of the Interaction Between WD Repeat-Containing Protein 5 (WDR5) and Mixed-Lineage Leukemia 1 (MLL1). J. Med. Chem. 2016, 59, 2478–2496. 10.1021/acs.jmedchem.5b01630. [DOI] [PubMed] [Google Scholar]
- Borkin D.; He S.; Miao H.; Kempinska K.; Pollock J.; Chase J.; Purohit T.; Malik B.; Zhao T.; Wang J.; et al. Pharmacologic Inhibition of the Menin-MLL Interaction Blocks Progression of MLL Leukemia in vivo. Cancer Cell 2015, 27, 589–602. 10.1016/j.ccell.2015.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkin D.; Pollock J.; Kempinska K.; Purohit T.; Li X. Q.; Wen B.; Zhao T.; Miao H. Z.; Shukla S.; He M.; et al. Property Focused Structure-Based Optimization of Small Molecule Inhibitors of the Protein-Protein Interaction between Menin and Mixed Lineage Leukemia (MLL). J. Med. Chem. 2016, 59, 892–913. 10.1021/acs.jmedchem.5b01305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka J.; He S. H.; Shi A. B.; Purohit T.; Muntean A. G.; Sorenson R. J.; Showalter H. D.; Murai M. J.; Belcher A. M.; Hartley T.; et al. Menin-MLL Inhibitors Reverse Oncogenic Activity of MLL Fusion Proteins in Leukemia. Nat. Chem. Biol. 2012, 8, 277–284. 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S. H.; Senter T. J.; Pollock J.; Han C. H.; Upadhyay S. K.; Purohit T.; Gogliotti R. D.; Lindsley C. W.; Cierpicki T.; Stauffer S. R.; et al. High-Affinity Small-Molecule Inhibitors of the Menin-Mixed Lineage Leukemia (MLL) Interaction Closely Mimic a Natural Protein-Protein Interaction. J. Med. Chem. 2014, 57, 1543–1556. 10.1021/jm401868d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski D. S.; Grembecka J.; Cierpicki T. H3K36 Methyltransferases as Cancer Drug Targets: Rationale and Perspectives for Inhibitor Development. Future Med. Chem. 2016, 8, 1589–1607. 10.4155/fmc-2016-0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G. L.; Liu G. L.; Xiong S. D.; Liu H. Y.; Chen X.; Zheng B. The Histone Methyltransferase Smyd2 Is a Negative Regulator of Macrophage Activation by Suppressing Interleukin 6 (IL-6) and Tumor Necrosis Factor alpha (TNF-alpha) Production. J. Biol. Chem. 2015, 290, 5414–5423. 10.1074/jbc.M114.610345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H.; Allali-Hassani A.; Antonysamy S.; Chang S.; Chen L. H.; Curtis C.; Emtage S.; Fan L.; Gheyi T.; Li F.; et al. LLY-507, a Cell-active, Potent, and Selective Inhibitor of Protein-lysine Methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. 10.1074/jbc.M114.626861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Perez-Burgos L.; Placek B. J.; Sengupta R.; Richter M.; Dorsey J. A.; Kubicek S.; Opravil S.; Jenuwein T.; Berger S. L. Repression of p53 Activity by SMYD2-Mediated Methylation. Nature 2006, 444, 629–632. 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- Saddic L. A.; West L. E.; Aslanian A.; Yates J. R. 3rd; Rubin S. M.; Gozani O.; Sage J. Methylation of the Retinoblastoma Tumor Suppressor by SMYD2. J. Biol. Chem. 2010, 285, 37733–37740. 10.1074/jbc.M110.137612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Farha M.; Lanouette S.; Elisma F.; Tremblay V.; Butson J.; Figeys D.; Couture J. F. Proteomic Analyses of the SMYD Family Interactomes Identify HSP90 as A Novel Target for SMYD2. J. Mol. Cell Biol. 2011, 3, 301–308. 10.1093/jmcb/mjr025. [DOI] [PubMed] [Google Scholar]
- Piao L.; Kang D.; Suzuki T.; Masuda A.; Dohmae N.; Nakamura Y.; Hamamoto R. The Histone Methyltransferase SMYD2Methylates PARP1 and Promotes Poly(ADP-ribosyl)ation Activity in Cancer Cells. Neoplasia 2014, 16, 257–264. 10.1016/j.neo.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Tanaka K.; Yan J.; Li J.; Peng D.; Jiang Y.; Yang Z.; Barton M. C.; Wen H.; Shi X. Regulation of Estrogen Receptor alpha by Histone Methyltransferase SMYD2-Mediated Protein Methylation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17284–17289. 10.1073/pnas.1307959110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto L. H.; Andrade R. V.; Felipe M. S.; Motoyama A. B.; Pittella Silva F. SMYD2 is highly Expressed in Pediatric Acute LymphoblasticLeukemia and Constitutes A Bad Prognostic Factor. Leuk. Res. 2014, 38, 496–502. 10.1016/j.leukres.2014.01.013. [DOI] [PubMed] [Google Scholar]
- Komatsu S.; Imoto I.; Tsuda H.; Kozaki K. I.; Muramatsu T.; Shimada Y.; Aiko S.; Yoshizumi Y.; Ichikawa D.; Otsuji E.; et al. Overexpression of SMYD2 Relates to Tumor Cell Proliferation and Malignant Outcome of Esophageal Squamous Cell Carcinoma. Carcinogenesis 2009, 30, 1139–1146. 10.1093/carcin/bgp116. [DOI] [PubMed] [Google Scholar]
- Ferguson A. D.; Larsen N. A.; Howard T.; Pollard H.; Green I.; Grande C.; Cheung T.; Garcia-Arenas R.; Cowen S.; Wu J.; et al. Structural Basis of Substrate Methylation and Inhibition of SMYD2. Structure 2011, 19, 1262–1273. 10.1016/j.str.2011.06.011. [DOI] [PubMed] [Google Scholar]
- Cowen S. D.; Russell D.; Dakin L. A.; Chen H. R.; Larsen N. A.; Godin R.; Throner S.; Zheng X.; Molina A.; Wu J.; Cheung T.; Howard T.; Garcia-Arenas R.; Keen N.; Pendleton C. S.; Pietenpol J. A.; Ferguson A. D. Design, Synthesis and Biological Activity of Substrate Competitive SMYD2 Inhibitors. J. Med. Chem. 2016, 59, 11079–11097. 10.1021/acs.jmedchem.6b01303. [DOI] [PubMed] [Google Scholar]
- Sweis R. F.; Wang Z.; Algire M.; Arrowsmith C. H.; Brown P. J.; Chiang G. G.; Guo J.; Jakob C. G.; Kennedy S.; Li F.; et al. Discovery of A-893, A New Cell-Active Benzoxazinone Inhibitor of Lysine Methyltransferase SMYD2. ACS Med. Chem. Lett. 2015, 6, 695–700. 10.1021/acsmedchemlett.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LLY-507: A chemical probe for SMYD2 protein lysine methyltransferase. http://www.thesgc.org/chemical-probes/LLY-507 (accessed November 2016).
- Eggert E.; Hillig R. C.; Koehr S.; Stockigt D.; Weiske J.; Barak N.; Mowat J.; Brumby T.; Christ C. D.; Ter Laak A.; et al. Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem. 2016, 59, 4578–4600. 10.1021/acs.jmedchem.5b01890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieda Y.; Tsukita S.; Tsukita S. A New High Molecular Mass Protein Showing Unique Localization in Desmosomal Plaque. J. Cell Biol. 1989, 109, 1511–1518. 10.1083/jcb.109.4.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. B.; Cao X. J.; Han B.; Chen L. H.; Horvath A.; Richardson T. I.; Campbell R. M.; Garcia B. A.; Nguyen H. Quantitative Profiling of the Activity of Protein Lysine Methyltransferase SMYD2 Using SILAC-Based Proteomics. Mol. Cell. Proteomics 2016, 15, 892–905. 10.1074/mcp.M115.053280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida S. F.; Grosso A. R.; Koch F.; Fenouil R.; Carvalho S.; Andrade J.; Levezinho H.; Gut M.; Eick D.; Gut I.; et al. Splicing Enhances Recruitment of Methyltransferase HYPB/Setd2 and Methylation of Histone H3 Lys36. Nat. Struct. Mol. Biol. 2011, 18, 977–U1501. 10.1038/nsmb.2123. [DOI] [PubMed] [Google Scholar]
- Newbold R. F.; Mokbel K. Evidence for A Tumour Suppressor Function of SETD2 in Human Breast Cancer: A New Hypothesis. Anticancer Res. 2010, 30, 3309–3311. [PubMed] [Google Scholar]
- Hu M.; Sun X. J.; Zhang Y. L.; Kuang Y.; Hu C. Q.; Wu W. L.; Shen S. H.; Du T. T.; Li H.; He F.; et al. Histone H3 Lysine 36 Methyltransferase Hypb/Setd2 is Required for Embryonic Vascular Remodeling. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2956–2961. 10.1073/pnas.0915033107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duns G.; van den Berg E.; van Duivenbode I.; Osinga J.; Hollema H.; Hofstra R. M.; Kok K. Histone Methyltransferase Gene SETD2 is A Novel Tumor Suppressor Gene in Clear Cell Renal Cell Carcinoma. Cancer Res. 2010, 70, 4287–4291. 10.1158/0008-5472.CAN-10-0120. [DOI] [PubMed] [Google Scholar]
- Gossage L.; Murtaza M.; Slatter A. F.; Lichtenstein C. P.; Warren A.; Haynes B.; Marass F.; Roberts I.; Shanahan S. J.; Claas A.; et al. Clinical and Pathological Impact of VHL, PBRM1, BAP1, SETD2, KDM6A, and JARID1c in Clear Cell Renal Cell Carcinoma. Genes, Chromosomes Cancer 2014, 53, 38–51. 10.1002/gcc.22116. [DOI] [PubMed] [Google Scholar]
- Hao C.; Wang L.; Peng S.; Cao M.; Li H.; Hu J.; Huang X.; Liu W.; Zhang H.; Wu S.; et al. Gene Mutations in Primary Tumors and Corresponding Patient-Derived Xenografts Derived From Non-Small Cell Lung Cancer. Cancer Lett. 2015, 357, 179–185. 10.1016/j.canlet.2014.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso A. M.; Schwartzentruber J.; Khuong-Quang D. A.; Liu X. Y.; Sturm D.; Korshunov A.; Jones D. T.; Witt H.; Kool M.; Albrecht S.; et al. Mutations in SETD2 and Genes Affecting Histone H3k36 Methylation Target Hemispheric High-Grade Gliomas. Acta Neuropathol. 2013, 125, 659–669. 10.1007/s00401-013-1095-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; He F.; Zeng H.; Ling S.; Chen A.; Wang Y.; Yan X.; Wei W.; Pang Y.; Cheng H.; et al. Identification of Functional Cooperative Mutations of Setd2 in Human Acute Leukemia. Nat. Genet. 2014, 46, 287–293. 10.1038/ng.2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Ibáñez G.; Wu H.; Blum G.; Zeng H.; Dong A.; Li F.; Hajian T.; Allali-Hassani A.; Amaya M. F.; et al. Sinefungin Derivatives as Inhibitors and Structure Probes of Protein Lysine Methyltransferase SETD2. J. Am. Chem. Soc. 2012, 134, 18004–18014. 10.1021/ja307060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisi D.; Chiarparin E.; Tamanini E.; Pathuri P.; Coyle J. E.; Hold A.; Holding F. P.; Amin N.; Martin A. C.; Rich S. J.; et al. Structure of the Epigenetic Oncogene MMSET and Inhibition by N-Alkyl Sinefungin Derivatives. ACS Chem. Biol. 2016, 11, 3093–3105. 10.1021/acschembio.6b00308. [DOI] [PubMed] [Google Scholar]
- Qiao Q.; Li Y.; Chen Z.; Wang M.; Reinberg D.; Xu R. M. The Structure of NSD1 Reveals an Autoregulatory Mechanism Underlying Histone H3K36 Methylation. J. Biol. Chem. 2011, 286, 8361–8368. 10.1074/jbc.M110.204115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E. J.; Carpenter P. B. Understanding the Language of Lys36 Methylation at Histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucio-Eterovic A. K.; Singh M. M.; Gardner J. E.; Veerappan C. S.; Rice J. C.; Carpenter P. B. Role for the Nuclear Receptor-Binding SET Domain Protein 1 (NSD1) Methyltransferase in Coordinating Lysine 36 Methylation at Histone 3 with RNA Polymerase II Function. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16952–16957. 10.1073/pnas.1002653107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabella F.; Wu P.; Wardell C. P.; Kaiser M. F.; Walker B. A.; Johnson D. C.; Morgan G. J. MMSET is the Key Molecular Target in t(4;14) Myeloma. Blood Cancer J. 2013, 3, e114. 10.1038/bcj.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Garcia E.; Popovic R.; Min D. J.; Sweet S. M.; Thomas P. M.; Zamdborg L.; Heffner A.; Will C.; Lamy L.; Staudt L. M.; et al. The MMSET Histone Methyl Transferase Switches Global Histone Methylation and Alters Gene Expression in t(4;14) Multiple Myeloma Cells. Blood 2011, 117, 211–220. 10.1182/blood-2010-07-298349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyer J. A.; Huang X.; Zheng Y.; Shim J.; Ezponda T.; Carpenter Z.; Allegretta M.; Okot-Kotber C. I.; Patel J. P.; Melnick A.; et al. Point mutation E1099K in MMSET/NSD2 Enhances its Methyltranferase Activity and Leads to Altered Global Chromatin Methylation in Lymphoid Malignancies. Leukemia 2014, 28, 198–201. 10.1038/leu.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudlebusch H. R.; Santoni-Rugiu E.; Simon R.; Ralfkiaer E.; Rossing H. H.; Johansen J. V.; Jorgensen M.; Sauter G.; Helin K. The Histone Methyltransferase and Putative Oncoprotein MMSET is Overexpressed in A Large Variety of Human Tumors. Clin. Cancer Res. 2011, 17, 2919–2933. 10.1158/1078-0432.CCR-10-1302. [DOI] [PubMed] [Google Scholar]
- Hudlebusch H. R.; Skotte J.; Santoni-Rugiu E.; Zimling Z. G.; Lees M. J.; Simon R.; Sauter G.; Rota R.; De Ioris M. A.; Quarto M.; et al. MMSET is Highly Expressed and Associated with Aggressiveness in Neuroblastoma. Cancer Res. 2011, 71, 4226–4235. 10.1158/0008-5472.CAN-10-3810. [DOI] [PubMed] [Google Scholar]
- Beck D. B.; Oda H.; Shen S. S.; Reinberg D. PR-Set7 and H4K20me1: At the Crossroads of Genome Integrity, Cell Cycle, Chromosome Condensation, and Transcription. Genes Dev. 2012, 26, 325–337. 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K.; Rice J. C.; Sarma K.; Erdjument-Bromage H.; Werner J.; Wang Y.; Chuikov S.; Valenzuela P.; Tempst P.; Steward R.; et al. PR-Set7 is A Nucleosome-Specific Methyltransferase that Modifies Lysine 20 of Histone H4 and is Associated with Silent Chromatin. Mol. Cell 2002, 9, 1201–1213. 10.1016/S1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- Fang J.; Feng Q.; Ketel C. S.; Wang H.; Cao R.; Xia L.; Erdjument-Bromage H.; Tempst P.; Simon J. A.; Zhang Y. Purification and Functional Characterization of SET8, A Nucleosomal Histone H4-Lysine 20-Specific Methyltransferase. Curr. Biol. 2002, 12, 1086–1099. 10.1016/S0960-9822(02)00924-7. [DOI] [PubMed] [Google Scholar]
- Jorgensen S.; Schotta G.; Sorensen C. S. Histone H4 Lysine 20 Methylation: Key Player in Epigenetic Regulation of Genomic Integrity. Nucleic Acids Res. 2013, 41, 2797–2806. 10.1093/nar/gkt012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustel J.; Tardat M.; Kirsh O.; Grimaud C.; Julien E. Coupling Mitosis to DNA Replication: the Emerging Role of the Histone H4-Lysine 20 Methyltransferase PR-Set7. Trends Cell Biol. 2011, 21, 452–460. 10.1016/j.tcb.2011.04.006. [DOI] [PubMed] [Google Scholar]
- Takawa M.; Cho H. S.; Hayami S.; Toyokawa G.; Kogure M.; Yamane Y.; Iwai Y.; Maejima K.; Ueda K.; Masuda A.; et al. Histone Lysine Methyltransferase SETD8 Promotes Carcinogenesis by Deregulating PCNA Expression. Cancer Res. 2012, 72, 3217–3227. 10.1158/0008-5472.CAN-11-3701. [DOI] [PubMed] [Google Scholar]
- Williams D. E.; Dalisay D. S.; Li F.; Amphlett J.; Maneerat W.; Chavez M. A. G.; Wang Y. A.; Matainaho T.; Yu W.; Brown P. J.; et al. Nahuoic Acid A Produced by a Streptomyces sp. Isolated From a Marine Sediment Is a Selective SAM-Competitive Inhibitor of the Histone Methyltransferase SETD8. Org. Lett. 2013, 15, 414–417. 10.1021/ol303416k. [DOI] [PubMed] [Google Scholar]
- Williams D. E.; Izard F.; Arnould S.; Dalisay D. S.; Tantapakul C.; Maneerat W.; Matainaho T.; Julien E.; Andersen R. J. Structures of Nahuoic Acids B-E Produced in Culture by a Streptomyces sp Isolated from a Marine Sediment and Evidence for the Inhibition of the Histone Methyl Transferase SETD8 in Human Cancer Cells by Nahuoic Acid A. J. Org. Chem. 2016, 81, 1324–1332. 10.1021/acs.joc.5b02569. [DOI] [PubMed] [Google Scholar]
- Ma A.; Yu W.; Li F.; Bleich R. M.; Herold J. M.; Butler K. V.; Norris J. L.; Korboukh V.; Tripathy A.; Janzen W. P.; et al. Discovery of A Selective, Substrate-Competitive Inhibitor of the Lysine Methyltransferase SETD8. J. Med. Chem. 2014, 57, 6822–6833. 10.1021/jm500871s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A.; Yu W.; Xiong Y.; Butler K. V.; Brown P. J.; Jin J. Structure-Activity Relationship Studies of SETD8 Inhibitors. MedChemComm 2014, 5, 1892–1898. 10.1039/C4MD00317A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veschi V.; Liu Z.; Voss T. C.; Ozbun L.; Gryder B.; Yan C.; Hu Y.; Ma A.; Jin J.; Mazur S. J.; Lam N.; Souza B. K.; Giannini G.; Hager G. L.; Arrowsmith C. H.; Khan J.; Appella A.; Thiele C. J. Epigenetic siRNA and Chemical Screens Identify SETD8 Inhibition as a Therapeutic Strategy for p53 Activation in High-risk Neuroblastoma. Cancer Cell 2017, 31, 50–63. 10.1016/j.ccell.2016.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler K. V.; Ma A.; Yu W.; Li F.; Tempel W.; Babault N.; Pittella-Silva F.; Shao J.; Wang J.; Luo M.; et al. Structure-Based Design of a Covalent Inhibitor of the SET Domain-Containing Protein 8 (SETD8) Lysine Methyltransferase. J. Med. Chem. 2016, 59, 9881–9889. 10.1021/acs.jmedchem.6b01244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G.; Ibanez G.; Rao X.; Shum D.; Radu C.; Djaballah H.; Rice J. C.; Luo M. Small-Molecule Inhibitors of SETD8 with Cellular Activity. ACS Chem. Biol. 2014, 9, 2471–2478. 10.1021/cb500515r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge R. A.; Zhu H.; Upadhyay A. K.; Bodelle P. M.; Hutchins C. W.; Torrent M.; Marin V. L.; Yu W.; Vedadi M.; Li F.; Brown P. J.; Pappano W. N.; Sun C.; Petros A. M.; Turning A. Substrate Peptide into a Potent Inhibitor for the Histone Methyltransferase SETD8. ACS Med. Chem. Lett. 2016, 7, 1102–1106. 10.1021/acsmedchemlett.6b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga M. F.; Ballestar E.; Villar-Garea A.; Boix-Chornet M.; Espada J.; Schotta G.; Bonaldi T.; Haydon C.; Ropero S.; Petrie K.; et al. Loss of Acetylation at Lys16 and Trimethylation at Lys20 of Histone H4 is A Common Hallmark of Human Cancer. Nat. Genet. 2005, 37, 391–400. 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- Bromberg K. D.; Mitchell T. R.; Upadhyay A. K.; Jakob C. G.; Jhala M. A.; Comess K. M.; Lasko L. M.; Li C.; Tuzon C. T.; Dai Y.; et al. The SUV4–20 Inhibitor A-196 Verifies A Role for Epigenetics in Genomic Integrity. Nat. Chem. Biol. 2017, 13, 317. 10.1038/nchembio.2282. [DOI] [PubMed] [Google Scholar]
- Feng Q.; Wang H.; Ng H. H.; Erdjument-Bromage H.; Tempst P.; Struhl K.; Zhang Y. Methylation of H3-Lysine 79 is Mediated by A New Family of HMTases without A SET Domain. Curr. Biol. 2002, 12, 1052–1058. 10.1016/S0960-9822(02)00901-6. [DOI] [PubMed] [Google Scholar]
- Min J.; Feng Q.; Li Z.; Zhang Y.; Xu R. M. Structure of the Catalytic Domain of Human DOT1L, A Non-SET Domain Nucleosomal Histone Methyltransferase. Cell 2003, 112, 711–723. 10.1016/S0092-8674(03)00114-4. [DOI] [PubMed] [Google Scholar]
- Schubert H. L.; Blumenthal R. M.; Cheng X. D. Many Paths to Methyltransfer: A Chronicle of Convergence. Trends Biochem. Sci. 2003, 28, 329–335. 10.1016/S0968-0004(03)00090-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederiks F.; Tzouros M.; Oudgenoeg G.; van Welsem T.; Fornerod M.; Krijgsveld J.; van Leeuwen F. Nonprocessive Methylation by DOT1 Leads to Functional Redundancy of Histone H3K79 Methylation States. Nat. Struct. Mol. Biol. 2008, 15, 550–557. 10.1038/nsmb.1432. [DOI] [PubMed] [Google Scholar]
- Park J. W.; Kim K. B.; Kim J. Y.; Chae Y. C.; Jeong O. S.; Seo S. B. RE-IIBP Methylates H3K79 and Induces MEIS1-mediated Apoptosis via H2BK120 Ubiquitination by RNF20. Sci. Rep. 2015, 5 (12485), 1–12. 10.1038/srep12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; Zhang Y. The Diverse Functions of DOT1 and H3K79 Methylation. Genes Dev. 2011, 25, 1345–1358. 10.1101/gad.2057811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; He J.; Taranova O.; Zhang Y. Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res. 2011, 21, 1370–1373. 10.1038/cr.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; Xiao B.; Neppl R. L.; Kallin E. M.; Li J. A.; Chen T. P.; Wang D. Z.; Xiao X. A.; Zhang Y. DOT1L Regulates Dystrophin Expression and is Critical for Cardiac Function. Genes Dev. 2011, 25, 263–274. 10.1101/gad.2018511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; Taranova O.; He J.; Zhang Y. DOT1L, the H3K79 Methyltransferase, is Required for MLL-AF9-Mediated Leukemogenesis. Blood 2011, 117, 6912–6922. 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitoun E.; Oliver P. L.; Davies K. E. The Mixed-Lineage Leukemia Fusion Partner AF4 Stimulates RNA Polymerase Ii Transcriptional Elongation And Mediates Coordinated Chromatin Remodeling. Hum. Mol. Genet. 2006, 16, 92–106. 10.1093/hmg/ddl444. [DOI] [PubMed] [Google Scholar]
- Mueller D.; Bach C.; Zeisig D.; Garcia-Cuellar M. P.; Monroe S.; Sreekumar A.; Zhou R.; Nesvizhskii A.; Chinnaiyan A.; Hess J. L.; et al. A Role for the MLL Fusion Partner ENL in Transcriptional Elongation and Chromatin Modification. Blood 2007, 110, 4445–4454. 10.1182/blood-2007-05-090514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A.; Lin M.; Naresh A.; Kitabayashi I.; Cleary M. L. A Higher-Order Complex Containing AF4 and ENL Family Proteins with P-TEFb Facilitates Oncogenic and Physiologic MLL-Dependent Transcription. Cancer Cell 2010, 17, 198–212. 10.1016/j.ccr.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas D.; Milne T. A.; Basrur V.; Kim J.; Elenitoba-Johnson K. S. J.; Allis C. D.; Roeder R. G. Function of Leukemogenic Mixed Lineage Leukemia 1 (MLL) Fusion Proteins through Distinct Partner Protein Complexes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 15751–15756. 10.1073/pnas.1111498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin J. L.; Song Y. A Medicinal Chemistry Perspective for Targeting Histone H3 Lysine-79 Methyltransferase DOT1L. J. Med. Chem. 2013, 56, 8972–8983. 10.1021/jm4007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Majer C. R.; Sneeringer C. J.; Song J.; Johnston L. D.; Scott M. P.; Smith J. J.; Xiao Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by A Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavapathruni A.; Jin L.; Daigle S. R.; Majer C. R.; Therkelsen C. A.; Wigle T. J.; Kuntz K. W.; Chesworth R.; Pollock R. M.; Scott M. P.; et al. Conformational Adaptation Drives Potent, Selective and Durable Inhibition of the Human Protein Methyltransferase DOT1L. Chem. Biol. Drug Des. 2012, 80, 971–980. 10.1111/cbdd.12050. [DOI] [PubMed] [Google Scholar]
- Yu W.; Chory E. J.; Wernimont A. K.; Tempel W.; Scopton A.; Federation A.; Marineau J. J.; Qi J.; Barsyte-Lovejoy D.; Yi J.; et al. Catalytic Site Remodelling of the DOT1L Methyltransferase by Selective Inhibitors. Nat. Commun. 2012, 3, 1288. 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- Bernt K. M.; Zhu N.; Sinha A. U.; Vempati S.; Faber J.; Krivtsov A. V.; Feng Z. H.; Punt N.; Daigle A.; Bullinger L.; et al. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 2011, 20, 66–78. 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle S. R.; Olhava E. J.; Therkelsen C. A.; Basavapathruni A.; Jin L.; Boriack-Sjodin P. A.; Allain C. J.; Klaus C. R.; Raimondi A.; Scott M. P.; et al. Potent Inhibition of DOT1L as Treatment of MLL-Fusion Leukemia. Blood 2013, 122, 1017–1025. 10.1182/blood-2013-04-497644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT02141828. A Phase 1 Dose Escalation and Expanded Cohort Study of EPZ-5676 in the Treatment of Pediatric Patients With Relapsed/Refractory Leukemias Bearing a Rearrangement of the MLL Gene (accessed Aug 2016).
- ClinicalTrials.gov Identifier: NCT01684150. A Phase 1, Open-Label, Dose-Escalation & Expanded Cohort, Continuous IV Infusion, Multi-center Study of the Safety, Tolerability, PK & PD of EPZ-5676 in Treatment Relapsed/Refractory Patients With Leukemias Involving Translocation of the MLL Gene at 11q23 or Advanced Hematologic Malignancies (accessed Aug 2016).
- Chen L.; Deshpande A. J.; Banka D.; Bernt K. M.; Dias S.; Buske C.; Olhava E. J.; Daigle S. R.; Richon V. M.; Pollock R. M.; et al. Abrogation of MLL-AF10 and CALM-AF10-Mediated Transformation through Genetic Inactivation or Pharmacological Inhibition of The H3K79 Methyltransferase DOT1L. Leukemia 2013, 27, 813–822. 10.1038/leu.2012.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande A. J.; Chen L.; Fazio M.; Sinha A. U.; Bernt K. M.; Banka D.; Dias S.; Chang J.; Olhava E. J.; Daigle S. R.; et al. Leukemic Transformation by the MLL-AF6 Fusion Oncogene Requires the H3K79 Methyltransferase DOT1L. Blood 2013, 121, 2533–2541. 10.1182/blood-2012-11-465120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters N. J.; Smith S. A.; Olhava E. J.; Duncan K. W.; Burton R. D.; O’Neill J.; Rodrigue M. E.; Pollock R. M.; Moyer M. P.; Chesworth R. Metabolism and Disposition of the DOT1L Inhibitor, Pinometostat (EPZ-5676), in Rat, Dog and Human. Cancer Chemother. Pharmacol. 2016, 77, 43–62. 10.1007/s00280-015-2929-y. [DOI] [PubMed] [Google Scholar]
- Yao Y.; Chen P.; Diao J.; Cheng G.; Deng L.; Anglin J. L.; Prasad B. V. V; Song Y. Selective Inhibitors of Histone Methyltransferase DOT1L: Design, Synthesis and Crystallographic Studies. J. Am. Chem. Soc. 2011, 133, 16746–16749. 10.1021/ja206312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglin J. L.; Deng L.; Yao Y.; Cai G.; Liu Z.; Jiang H.; Cheng G.; Chen P.; Dong S.; Song Y. Synthesis and Structure-Activity Relationship Investigation of Adenosine-Containing Inhibitors of Histone Methyltransferase DOT1L. J. Med. Chem. 2012, 55, 8066–8074. 10.1021/jm300917h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurr S. S.; Bayle E. D.; Yu W.; Li F.; Tempel W.; Vedadi M.; Schapira M.; Fish P. V. New Small Molecule Inhibitors of Histone Methyl Transferase DOT1L with A Nitrile as A Non-Traditional Replacement for Heavy Halogen Atoms. Bioorg. Med. Chem. Lett. 2016, 26, 4518–4522. 10.1016/j.bmcl.2016.07.041. [DOI] [PubMed] [Google Scholar]
- Yu W.; Smil D.; Li F.; Tempel W.; Fedorov O.; Nguyen K. T.; Bolshan Y.; Al-Awar R.; Knapp S.; Arrowsmith C. H.; et al. Bromo-deaza-SAH: A Potent and Selective DOT1L Inhibitor. Bioorg. Med. Chem. 2013, 21, 1787–1794. 10.1016/j.bmc.2013.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J. S.; Federation A. J.; Qi J.; Dhe-Paganon S.; Hadler M.; Xu X. St; St. Pierre R.; Varca A. C.; Wu L.; Marineau J. J.; et al. Structure-Guided DOT1L Probe Optimization by Label-Free Ligand Displacement. ACS Chem. Biol. 2015, 10, 667–674. 10.1021/cb500796d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B.; Zhang H.; Pan S.; Wang C.; Ge J.; Lee J. S.; Yao S. Q. In Situ Proteome Profiling and Bioimaging Applications of Small-Molecule Affinity-Based Probes Derived From DOT1L Inhibitors. Chem. - Eur. J. 2016, 22, 7824–7836. 10.1002/chem.201600259. [DOI] [PubMed] [Google Scholar]
- Scheufler C.; Mobitz H.; Gaul C.; Ragot C.; Be C.; Fernandez C.; Beyer K. S.; Tiedt R.; Stauffer F. Optimization of a Fragment-Based Screening Hit toward Potent DOT1L Inhibitors Interacting in an Induced Binding Pocket. ACS Med. Chem. Lett. 2016, 7, 730–734. 10.1021/acsmedchemlett.6b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Zhu H.; Stauffer F.; Caravatti G.; Vollmer S.; Machauer R.; Holzer P.; Mobitz H.; Scheufler C.; Klumpp M.; et al. Discovery of Novel DOT1L Inhibitors through A Structure-Based Fragmentation Approach. ACS Med. Chem. Lett. 2016, 7, 735–740. 10.1021/acsmedchemlett.6b00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. J.; Li L. J.; Chen Y. T.; Hu J. C.; Liu J. Q.; Liu Y. C.; Liu R. F.; Zhang Y. Y.; Meng F. W.; Zhu K. K.; et al. Identification of Novel Disruptor of Telomeric Silencing 1-like (DOT1L) Inhibitors through Structure-Based Virtual Screening and Biological Assays. J. Chem. Inf. Model. 2016, 56, 527–534. 10.1021/acs.jcim.5b00738. [DOI] [PubMed] [Google Scholar]
- Bedford M. T.; Clarke S. G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najbauer J.; Johnson B. A.; Young A. L.; Aswad D. W. Peptides with Sequences Similar to Glycine, Arginine-Rich Motifs in Proteins Interacting with Rna Are Efficiently Recognized by Methyltransferase(S) Modifying Arginine in Numerous Proteins. J. Biol. Chem. 1993, 268, 10501–10509. [PubMed] [Google Scholar]
- Boffa L. C.; Karn J.; Vidali G.; Allfrey V. G. Distribution of NG, NG,-Dimethylarginine in Nuclear Protein Fractions. Biochem. Biophys. Res. Commun. 1977, 74, 969–976. 10.1016/0006-291X(77)91613-8. [DOI] [PubMed] [Google Scholar]
- Hughes R. M.; Waters M. L. Arginine Methylation in A Beta-Hairpin Peptide: Implications for Arg-Pi Interactions, DeltaCp(O), and the Cold Denatured State. J. Am. Chem. Soc. 2006, 128, 12735–12742. 10.1021/ja061656g. [DOI] [PubMed] [Google Scholar]
- Bedford M. T.; Richard S. Arginine Methylation An Emerging Regulator of Protein Function. Mol. Cell 2005, 18, 263–272. 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Bedford M. T. Protein Arginine Methyltransferases and Cancer. Nat. Rev. Cancer 2012, 13, 37–50. 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Hadjikyriacou A.; Xia Z.; Gayatri S.; Kim D.; Zurita-Lopez C.; Kelly R.; Guo A.; Li W.; Clarke S. G.; et al. PRMT9 is A type II Methyltransferase that Methylates the Splicing Factor SAP145. Nat. Commun. 2015, 6, 6428. 10.1038/ncomms7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu L. F.; Lu F. L.; Pei Y. X.; Liu C. Y.; Cao X. F. Regulation of Flowering Time by The Protein Arginine Methyltransferase AtPRMT10. EMBO Rep. 2007, 8, 1190–1195. 10.1038/sj.embor.7401111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scebba F.; De Bastiani M.; Bernacchia G.; Andreucci A.; Galli A.; Pitto L. PRMT11: A New Arabidopsis MBD7 Protein Partner with Arginine Methyltransferase Activity. Plant J. 2007, 52, 210–222. 10.1111/j.1365-313X.2007.03238.x. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Zhou L.; Cheng X. Crystal Structure of The Conserved Core of Protein Arginine Methyltransferase PRMT3. EMBO J. 2000, 19, 3509–3519. 10.1093/emboj/19.14.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Cheng X. Structure of the Predominant Protein Arginine Methyltransferase PRMT1 and Analysis of its Binding to Substrate Peptides. Structure 2003, 11, 509–520. 10.1016/S0969-2126(03)00071-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarheyeva A.; Senisterra G.; Allali-Hassani A.; Dong A.; Dobrovetsky E.; Wasney; Gregory A.; Chau I.; Marcellus R.; Hajian T.; Liu F.; et al. An Allosteric Inhibitor of Protein Arginine Methyltransferase 3. Structure 2012, 20, 1425–1435. 10.1016/j.str.2012.06.001. [DOI] [PubMed] [Google Scholar]
- Lee J.; Bedford M. T. PABP1 Identified as An Arginine Methyltransferase Substrate Using High-Density Protein Arrays. EMBO Rep. 2002, 3, 268–273. 10.1093/embo-reports/kvf052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D.; Cote J.; Shaaban S.; Bedford M. T. The Arginine Methyltransferase CARM1 Regulates the Coupling of Transcription and mRNA Processing. Mol. Cell 2007, 25, 71–83. 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Branscombe T. L.; Frankel A.; Lee J. H.; Cook J. R.; Yang Z.; Pestka S.; Clarke S. PRMT5 (Janus Kinase-Binding protein 1) Catalyzes the Formation of Symmetric Dimethylarginine Residues in Proteins. J. Biol. Chem. 2001, 276, 32971–32976. 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- Jansson M.; Durant S. T.; Cho E. C.; Sheahan S.; Edelmann M.; Kessler B.; La Thangue N. B. Arginine Methylation Regulates the p53 Response. Nat. Cell Biol. 2008, 10, 1431–1439. 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- Wei H.; Mundade R.; Lange K. C.; Lu T. Protein Arginine Methylation of Non-Histone Proteins and Its Role in Diseases. Cell Cycle 2014, 13, 32–41. 10.4161/cc.27353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W. J.; Gary J. D.; Yang M. C.; Clarke S.; Herschman H. R. The Mammalian Immediate-Early TIS21 Protein and the Leukemia-Associated BTG1 Protein Interact with A Protein-Arginine N-Methyltransferase. J. Biol. Chem. 1996, 271, 15034–15044. 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- Tang J.; Frankel A.; Cook R. J.; Kim S.; Paik W. K.; Williams K. R.; Clarke S.; Herschman H. R. PRMT1 is the Predominant Type I Protein Arginine Methyltransferase in Mammalian Cells. J. Biol. Chem. 2000, 275, 7723–7730. 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- Wang H.; Huang Z. Q.; Xia L.; Feng Q.; Erdjument-Bromage H.; Strahl B. D.; Briggs S. D.; Allis C. D.; Wong J.; Tempst P.; et al. Methylation of Histone H4 at Arginine 3 Facilitating Transcriptional Activation by Nuclear Hormone Receptor. Science 2001, 293, 853–857. 10.1126/science.1060781. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Briggs S. D.; Brame C. J.; Caldwell J. A.; Koh S. S.; Ma H.; Cook R. G.; Shabanowitz J.; Hunt D. F.; Stallcup M. R.; et al. Methylation of Histone H4 at Arginine 3 Occurs In Vivo and is Mediated by the Nuclear Receptor Coactivator PRMT1. Curr. Biol. 2001, 11, 996–1000. 10.1016/S0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- Yu Z.; Vogel G.; Coulombe Y.; Dubeau D.; Spehalski E.; Hebert J.; Ferguson D. O.; Masson J. Y.; Richard S. The MRE11 GAR Motif Regulates DNA Double-Strand Break Processing and ATR Activation. Cell Res. 2012, 22, 305–320. 10.1038/cr.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert F. M.; Rhie A.; Richard S.; Doherty A. J. The GAR Motif of 53BP1 is Arginine Methylated by PRMT1 and is Necessary for 53BP1 DNA Binding Activity. Cell Cycle 2005, 4, 1834–1841. 10.4161/cc.4.12.2250. [DOI] [PubMed] [Google Scholar]
- Butler J. S.; Zurita-Lopez C. I.; Clarke S. G.; Bedford M. T.; Dent S. Y. Protein-Arginine Methyltransferase 1 (PRMT1) Methylates ASH2L, A Shared Component of Mammalian Histone H3K4Methyltransferase Complexes. J. Biol. Chem. 2011, 286, 12234–12244. 10.1074/jbc.M110.202416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guendel I.; Carpio L.; Pedati C.; Schwartz A.; Teal C.; Kashanchi F.; Kehn-Hall K. Methylation of the tumor suppressor Protein, BRCA1, Influences its Transcriptional Cofactor Function. PLoS One 2010, 5, e11379. 10.1371/journal.pone.0011379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell T. R.; Glenfield K.; Jeyanthan K.; Zhu X. D. Arginine Methylation Regulates Telomere Length and Stability. Mol. Cell. Biol. 2009, 29, 4918–4934. 10.1128/MCB.00009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamaki J.; Daitoku H.; Ueno K.; Hagiwara A.; Yamagata K.; Fukamizu A. Arginine Methylation of BCL-2 Antagonist of Cell Death (BAD) Counteracts its Phosphorylation and Inactivation by AKT. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6085–6090. 10.1073/pnas.1015328108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K.; Daitoku H.; Takahashi Y.; Namiki K.; Hisatake K.; Kako K.; Mukai H.; Kasuya Y.; Fukamizu A. Arginine Methylation of FOXO Transcription Factors Inhibits their Phosphorylation by AKT. Mol. Cell 2008, 32, 221–231. 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Goulet I.; Gauvin G.; Boisvenue S.; Cote J. Alternative Splicing Yields Protein Arginine Methyltransferase 1 Isoforms with Distinct Activity, Substrate Specificity, and Subcellular Localization. J. Biol. Chem. 2007, 282, 33009–33021. 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- Baldwin R. M.; Morettin A.; Paris G.; Goulet I.; Cote J. Alternatively Spliced Protein Arginine Methyltransferase 1 Isoform PRMT1v2 Promotes the Survival and Invasiveness of Breast Cancer Cells. Cell Cycle 2012, 11, 4597–4612. 10.4161/cc.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathioudaki K.; Scorilas A.; Ardavanis A.; Lymberi P.; Tsiambas E.; Devetzi M.; Apostolaki A.; Talieri M. Clinical Evaluation of PRMT1 Gene Expression in Breast Cancer. Tumor Biol. 2011, 32, 575–582. 10.1007/s13277-010-0153-2. [DOI] [PubMed] [Google Scholar]
- Seligson D. B.; Horvath S.; Shi T.; Yu H.; Tze S.; Grunstein M.; Kurdistani S. K. Global Histone Modification Patterns Predict Risk of Prostate Cancer Recurrence. Nature 2005, 435, 1262–1266. 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- Mathioudaki K.; Papadokostopoulou A.; Scorilas A.; Xynopoulos D.; Agnanti N.; Talieri M. The PRMT1 Gene Expression Pattern in Colon Cancer. Br. J. Cancer 2008, 99, 2094–2099. 10.1038/sj.bjc.6604807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadokostopoulou A.; Mathioudaki K.; Scorilas A.; Xynopoulos D.; Ardavanis A.; Kouroumalis E.; Talieri M. Colon Cancer and Protein Arginine Methyltransferase 1 Gene Expression. Anticancer Res. 2009, 29, 1361–1366. [PubMed] [Google Scholar]
- Yoshimatsu M.; Toyokawa G.; Hayami S.; Unoki M.; Tsunoda T.; Field H. I.; Kelly J. D.; Neal D. E.; Maehara Y.; Ponder B. A.; et al. Dysregulation of PRMT1 and PRMT6, Type I arginine Methyltransferases, is Involved in Various Types of Human Cancers. Int. J. Cancer 2011, 128, 562–573. 10.1002/ijc.25366. [DOI] [PubMed] [Google Scholar]
- Shia W. J.; Okumura A. J.; Yan M.; Sarkeshik A.; Lo M. C.; Matsuura S.; Komeno Y.; Zhao X.; Nimer S. D.; Yates J. R. 3rd; et al. PRMT1 Interacts with AML1-ETO to Promote its Transcriptional Activation and Progenitor Cell Proliferative Potential. Blood 2012, 119, 4953–4962. 10.1182/blood-2011-04-347476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N.; Chan L. C.; Thompson A.; Cleary M. L.; So C. W. Protein Arginine-Methyltransferase-Dependent Oncogenesis. Nat. Cell Biol. 2007, 9, 1208–1215. 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- Eram M. S.; Shen Y.; Szewczyk M. M.; Wu H.; Senisterra G.; Li F.; Butler K. V.; Kaniskan H. Ü.; Speed B. A.; Dela Sena C.; et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D.; Yadav N.; King R. W.; Swanson M. S.; Weinstein E. J.; Bedford M. T. Small Molecule Regulators of Protein Arginine Methyltransferases. J. Biol. Chem. 2004, 279, 23892–23899. 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- Ragno R.; Simeoni S.; Castellano S.; Vicidomini C.; Mai A.; Caroli A.; Tramontano A.; Bonaccini C.; Trojer P.; Bauer I.; et al. Small Molecule Inhibitors of Histone Arginine Methyltransferases: Homology Modeling, Molecular Docking, Binding Mode Analysis, and Biological Evaluations. J. Med. Chem. 2007, 50, 1241–1253. 10.1021/jm061213n. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Li M.; Wang B.; Zheng Y. G. Discovery and Mechanistic Study of A Class of Protein Arginine Methylation Inhibitors. J. Med. Chem. 2010, 53, 6028–6039. 10.1021/jm100416n. [DOI] [PubMed] [Google Scholar]
- Sams-Dodd F. Target-Based Drug Discovery: Is Something Wrong?. Drug Discovery Today 2005, 10, 139–147. 10.1016/S1359-6446(04)03316-1. [DOI] [PubMed] [Google Scholar]
- Spannhoff A.; Heinke R.; Bauer I.; Trojer P.; Metzger E.; Gust R.; Schule R.; Brosch G.; Sippl W.; Jung M. Target-based approach to inhibitors of histone arginine methyltransferases. J. Med. Chem. 2007, 50, 2319–2325. 10.1021/jm061250e. [DOI] [PubMed] [Google Scholar]
- Spannhoff A.; Machmur R.; Heinke R.; Trojer P.; Bauer I.; Brosch G.; Schule R.; Hanefeld W.; Sippl W.; Jung M. A Novel Arginine Methyltransferase Inhibitor With Cellular Activity. Bioorg. Med. Chem. Lett. 2007, 17, 4150–4153. 10.1016/j.bmcl.2007.05.088. [DOI] [PubMed] [Google Scholar]
- Heinke R.; Spannhoff A.; Meier R.; Trojer P.; Bauer I.; Jung M.; Sippl W. Virtual Screening and Biological Characterization of Novel Histone Arginine Methyltransferase PRMT1 Inhibitors. ChemMedChem 2009, 4, 69–77. 10.1002/cmdc.200800301. [DOI] [PubMed] [Google Scholar]
- Bissinger E. M.; Heinke R.; Spannhoff A.; Eberlin A.; Metzger E.; Cura V.; Hassenboehler P.; Cavarelli J.; Schule R.; Bedford M. T.; et al. Acyl Derivatives of p-Aminosulfonamides and Dapsone as New Inhibitors of the Arginine Methyltransferase hPRMT1. Bioorg. Med. Chem. 2011, 19, 3717–3731. 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- Xie Y. Q.; Zhou R.; Lian F. L.; Liu Y.; Chen L. M.; Shi Z.; Zhang N. X.; Zheng M. Y.; Shen B. R.; Jiang H. L.; et al. Virtual Screening and Biological Evaluation of Novel Small Molecular Inhibitors Against Protein Arginine Methyltransferase 1 (PRMT1). Org. Biomol. Chem. 2014, 12, 9665–9673. 10.1039/C4OB01591F. [DOI] [PubMed] [Google Scholar]
- Bonham K.; Hemmers S.; Lim Y. H.; Hill D. M.; Finn M. G.; Mowen K. A. Effects of A Novel Arginine Methyltransferase Inhibitor on T-Helper Cell Cytokine Production. FEBS J. 2010, 277, 2096–2108. 10.1111/j.1742-4658.2010.07623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowden J.; Pike R. A.; Parry R. V.; Hong W.; Muhsen U. A.; Ward S. G. Small Molecule Inhibitors that Discriminate between Protein Arginine N-Methyltransferases PRMT1 and CARM1. Org. Biomol. Chem. 2011, 9, 7814–7821. 10.1039/c1ob06100c. [DOI] [PubMed] [Google Scholar]
- Weller R. L.; Rajski S. R. Design, Synthesis, and Preliminary Biological Evaluation of A DNA Methyltransferase-Directed Alkylating Agent. ChemBioChem 2006, 7, 243–245. 10.1002/cbic.200500362. [DOI] [PubMed] [Google Scholar]
- Osborne T.; Roska R. L. W.; Rajski S. R.; Thompson P. R. In Situ Generation of A Bisubstrate Analogue for Protein Arginine Methyltransferase 1. J. Am. Chem. Soc. 2008, 130, 4574–4575. 10.1021/ja077104v. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Arita K.; Bhatia M.; Knuckley B.; Lee Y. H.; Stallcup M. R.; Sato M.; Thompson P. R. Inhibitors and Inactivators of Protein Arginine Deiminase 4: Functional and Structural Characterization. Biochemistry 2006, 45, 11727–11736. 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obianyo O.; Causey C. P.; Osborne T. C.; Jones J. E.; Lee Y. H.; Stallcup M. R.; Thompson P. R. A Chloroacetamidine-Based Inactivator of Protein Arginine Methyltransferase 1: Design, Synthesis, and In Vitro and In Vivo Evaluation. ChemBioChem 2010, 11, 1219–1223. 10.1002/cbic.201000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon M. B. C.; Bachovchin D. A.; Brown S. J.; Finn M. G.; Rosen H.; Cravatt B. F.; Mowen K. A. Novel Inhibitors for PRMT1 Discovered by High-Throughput Screening Using Activity-Based Fluorescence Polarization. ACS Chem. Biol. 2012, 7, 1198–1204. 10.1021/cb300024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obianyo O.; Causey C. P.; Jones J. E.; Thompson P. R. Activity-based protein profiling of protein arginine methyltransferase 1. ACS Chem. Biol. 2011, 6, 1127–1135. 10.1021/cb2001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Chen L.; Sinha S. H.; Liang Z.; Chai H.; Muniyan S.; Chou Y. W.; Yang C.; Yan L.; Feng Y.; et al. Pharmacophore-Based Virtual Screening and Biological Evaluation of Small Molecule Inhibitors for Protein Arginine Methylation. J. Med. Chem. 2012, 55, 7978–7987. 10.1021/jm300521m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L.; Yan C.; Qian K.; Su H.; Kofsky-Wofford S. A.; Lee W. C.; Zhao X.; Ho M. C.; Ivanov I.; Zheng Y. G. Diamidine Compounds for Selective Inhibition of Protein Arginine Methyltransferase 1. J. Med. Chem. 2014, 57, 2611–2622. 10.1021/jm401884z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H.; Owens E. A.; Su H. R.; Yan L. L.; Levitz A.; Zhao X. Y.; Henary M.; Zheng Y. G. Exploration of Cyanine Compounds as Selective Inhibitors of Protein Arginine Methyltransferases: Synthesis and Biological Evaluation. J. Med. Chem. 2015, 58, 1228–1243. 10.1021/jm501452j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. H.; Owens E. A.; Feng Y.; Yang Y. T.; Xie Y.; Tu Y. P.; Henary M.; Zheng Y. G. Synthesis and Evaluation of Carbocyanine Dyes as PRMT Inhibitors and Imaging Agents. Eur. J. Med. Chem. 2012, 54, 647–659. 10.1016/j.ejmech.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X. R.; Tang Y.; Wang W. J.; Ji S.; Ma S.; Zhong L.; Zhang C. H.; Yang J.; Wu X. A.; Fu Z. Y.; et al. Discovery and Structure-Activity Analysis of 4-((5-Nitropyrimidin-4-Yl)Amino)Benzimidamide Derivatives as Novel Protein Arginine Methyltransferase 1 (PRMT1) Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5449–5453. 10.1016/j.bmcl.2015.06.095. [DOI] [PubMed] [Google Scholar]
- Tang J.; Gary J. D.; Clarke S.; Herschman H. R. Prmt 3, A Type I Protein Arginine N-Methyltransferase that Differs From PRMT1 in its Oligomerization, Subcellular Localization, Substrate Specificity, and Regulation. J. Biol. Chem. 1998, 273, 16935–16945. 10.1074/jbc.273.27.16935. [DOI] [PubMed] [Google Scholar]
- Bachand F.; Silver P. A. PRMT3 is A Ribosomal Protein Methyltransferase that Affects the Cellular Levels of Ribosomal Subunits. EMBO J. 2004, 23, 2641–2650. 10.1038/sj.emboj.7600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiercz R.; Person M. D.; Bedford M. T. Ribosomal Protein S2 is A Substrate for Mammalian PRMT3 (Protein Arginine Methyltransferase 3). Biochem. J. 2005, 386, 85–91. 10.1042/BJ20041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A.; Bedford M. T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiercz R.; Cheng D.; Kim D.; Bedford M. T. Ribosomal Protein rpS2 is Hypomethylated in PRMT3-Deficient Mice. J. Biol. Chem. 2007, 282, 16917–16923. 10.1074/jbc.M609778200. [DOI] [PubMed] [Google Scholar]
- Smith J. J.; Rucknagel K. P.; Schierhorn A.; Tang J.; Nemeth A.; Linder M.; Herschman H. R.; Wahle E. Unusual Sites of Arginine Methylation in Poly(A)-Binding Protein II and In Vitro Methylation by Protein Arginine Methyltransferases PRMT1 and PRMT3. J. Biol. Chem. 1999, 274, 13229–13234. 10.1074/jbc.274.19.13229. [DOI] [PubMed] [Google Scholar]
- Fronz K.; Otto S.; Kolbel K.; Kuhn U.; Friedrich H.; Schierhorn A.; Beck-Sickinger A. G.; Ostareck-Lederer A.; Wahle E. Promiscuous Modification of the Nuclear Poly(A)-Binding Protein by Multiple Protein-Arginine Methyltransferases Does Not Affect The Aggregation Behavior. J. Biol. Chem. 2008, 283, 20408–20420. 10.1074/jbc.M802329200. [DOI] [PubMed] [Google Scholar]
- Tavanez J. P.; Bengoechea R.; Berciano M. T.; Lafarga M.; Carmo-Fonseca M.; Enguita F. J. Hsp70 Chaperones and Type I PRMTs are Sequestered at Intranuclear Inclusions Caused by Polyalanine Expansions in PABPN1. PLoS One 2009, 4, e6418. 10.1371/journal.pone.0006418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allali-Hassani A.; Wasney G. A.; Siarheyeva A.; Hajian T.; Arrowsmith C. H.; Vedadi M. Fluorescence-Based Methods For Screening Writers And Readers Of Histone Methyl Marks. J. Biomol. Screening 2012, 17, 71–84. 10.1177/1087057111422256. [DOI] [PubMed] [Google Scholar]
- Lai Y.; Song M.; Hakala K.; Weintraub S. T.; Shiio Y. Proteomic Dissection of the von Hippel-Lindau (VHL) Interactome. J. Proteome Res. 2011, 10, 5175–5182. 10.1021/pr200642c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y.; Iwai M.; Kawai T.; Arakawa A.; Ito T.; Sakurai-Yageta M.; Ito A.; Goto A.; Saito M.; Kasumi F.; et al. Aberrant Expression of Tumor Suppressors CADM1 and 4.1B in Invasive Lesions of Primary Breast Cancer. Breast Cancer 2012, 19, 242–252. 10.1007/s12282-011-0272-7. [DOI] [PubMed] [Google Scholar]
- Alexiou G. A.; Markoula S.; Gogou P.; Kyritsis A. P. Genetic and Molecular Alterations in Meningiomas. Clin. Neurol. Neurosurgery 2011, 113, 261–267. 10.1016/j.clineuro.2010.12.007. [DOI] [PubMed] [Google Scholar]
- Heller G.; Geradts J.; Ziegler B.; Newsham I.; Filipits M.; Markis-Ritzinger E. M.; Kandioler D.; Berger W.; Stiglbauer W.; Depisch D.; et al. Downregulation of TSLC1 and DAL-1 Expression Occurs Frequently in Breast Cancer. Breast Cancer Res. Treat. 2007, 103, 283–291. 10.1007/s10549-006-9377-7. [DOI] [PubMed] [Google Scholar]
- Singh V.; Miranda T. B.; Jiang W.; Frankel A.; Roemer M. E.; Robb V. A.; Gutmann D. H.; Herschman H. R.; Clarke S.; Newsham I. F. DAL-1/4.1B Tumor Suppressor Interacts with Protein Arginine N-Methyltransferase 3 (PRMT3) and Inhibits its Ability to Methylate Substrates In Vitro and In Vivo. Oncogene 2004, 23, 7761–7771. 10.1038/sj.onc.1208057. [DOI] [PubMed] [Google Scholar]
- Miyata S.; Mori Y.; Tohyama M. PRMT3 is Essential for Dendritic Spine Maturation in Rat Hippocampal Neurons. Brain Res. 2010, 1352, 11–20. 10.1016/j.brainres.2010.07.033. [DOI] [PubMed] [Google Scholar]
- Chen X.; Niroomand F.; Liu Z.; Zankl A.; Katus H. A.; Jahn L.; Tiefenbacher C. P. Expression of Nitric Oxide Related Enzymes in Coronary Heart Disease. Basic Res. Cardiol. 2006, 101, 346–353. 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- Liu F.; Li F.; Ma A.; Dobrovetsky E.; Dong A.; Gao C.; Korboukh I.; Liu J.; Smil D.; Brown P. J.; et al. Exploiting An Allosteric Binding Site of PRMT3 Yields Potent and Selective Inhibitors. J. Med. Chem. 2013, 56, 2110–2124. 10.1021/jm3018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Szewczyk M. M.; Yu Z.; Eram M. S.; Yang X.; Schmidt K.; Luo X.; Dai M.; He F.; Zang I.; et al. A Potent, Selective and Cell-Active Allosteric Inhibitor of Protein Arginine Methyltransferase 3 (PRMT3). Angew. Chem., Int. Ed. 2015, 54, 5166–5170. 10.1002/anie.201412154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Ma H.; Hong H.; Koh S. S.; Huang S. M.; Schurter B. T.; Aswad D. W.; Stallcup M. R. Regulation of Transcription by A Protein Methyltransferase. Science 1999, 284, 2174–2177. [DOI] [PubMed] [Google Scholar]
- Bauer U. M.; Daujat S.; Nielsen S. J.; Nightingale K.; Kouzarides T. Methylation at Arginine 17 of Histone H3 is Linked to Gene Activation. EMBO Rep. 2002, 3, 39–44. 10.1093/embo-reports/kvf013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurter B. T.; Koh S. S.; Chen D.; Bunick G. J.; Harp J. M.; Hanson B. L.; Henschen-Edman A.; Mackay D. R.; Stallcup M. R.; Aswad D. W. Methylation of Histone H3 by Coactivator-Associated Arginine Methyltransferase 1. Biochemistry 2001, 40, 5747–5756. 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- Jacques S. L.; Aquino K. P.; Gureasko J.; Boriack-Sjodin P. A.; Porter Scott M.; Copeland R. A.; Riera T. V. CARM1 Preferentially Methylates H3R17 over H3R26 through a Random Kinetic Mechanism. Biochemistry 2016, 55, 1635–1644. 10.1021/acs.biochem.5b01071. [DOI] [PubMed] [Google Scholar]
- Cheng D. H.; Cote J.; Shaaban S.; Bedford M. T. The Arginine Methyltransferase CARM1 Regulates the Coupling of Transcription and mRNA Processing. Mol. Cell 2007, 25, 71–83. 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Fujiwara T.; Mori Y.; Chu D. L.; Koyama Y.; Miyata S.; Tanaka H.; Yachi K.; Kubo T.; Yoshikawa H.; Tohyama M. CARM1Regulates Proliferation of PC12 Cells by Methylating HuD. Mol. Cell. Biol. 2006, 26, 2273–2285. 10.1128/MCB.26.6.2273-2285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. W.; Park S. M.; Kilburn B.; Jelinek M. A.; Henschen-Edman A.; Aswad D. W.; Stallcup M. R.; Laird-Offringa I. A. Lipopolysaccharide-Induced Methylation of HuR, An mRNA-Stabilizing Protein, by CARM1. J. Biol. Chem. 2002, 277, 44623–44630. 10.1074/jbc.M206187200. [DOI] [PubMed] [Google Scholar]
- Xu W.; Chen H. W.; Du K. Y.; Asahara H.; Tini M.; Emerson B. M.; Montminy M.; Evans R. M. A Transcriptional Switch Mediated by Cofactor Methylation. Science 2001, 294, 2507–2511. 10.1126/science.1065961. [DOI] [PubMed] [Google Scholar]
- Chevillard-Briet M.; Trouche D.; Vandel L. Control of CBP Co-Activating Activity by Arginine Methylation. EMBO J. 2002, 21, 5457–5466. 10.1093/emboj/cdf548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q.; Yi P.; Wong J.; O’Malley B. W. Signaling within A Coactivator Complex: Methylation of SRC-3/AIB1 is A Molecular Switch for Complex Disassembly. Mol. Cell. Biol. 2006, 26, 7846–7857. 10.1128/MCB.00568-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav N.; Lee J.; Kim J.; Shen J. J.; Hu M. C. T.; Aldaz C. M.; Bedford M. T. Specific Protein Methylation Defects and Gene Expression Perturbations in Coactivator-Associated Arginine Methyltransferase 1-Deficient Mice. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 6464–6468. 10.1073/pnas.1232272100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Messaoudi S.; Fabbrizio E.; Rodriguez C.; Chuchana P.; Fauquier L.; Cheng D. H.; Theillet C.; Vandel L.; Bedford M. T.; Sardet C. Coactivator-Associated Arginine Methyltransferase 1 (CARM1) is A Positive Regulator of the Cyclin E1 Gene. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13351–13356. 10.1073/pnas.0605692103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. H.; Stallcup M. R. Roles of Protein Arginine Methylation in DNA Damage Signaling Pathways is CARM1 A Life-or-Death Decision Point?. Cell Cycle 2011, 10, 1343–1344. 10.4161/cc.10.9.15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S. S.; Li H.; Lee Y. H.; Widelitz R. B.; Chuong C. M.; Stallcup M. R. Synergistic Coactivator Function by Coactivator-Associated Arginine Methyltransferase (CARM) 1 and Beta-Catenin with Two Different Classes of DNA-Binding Transcriptional Activators. J. Biol. Chem. 2002, 277, 26031–26035. 10.1074/jbc.M110865200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covic M.; Hassa P. O.; Saccani S.; Buerki C.; Meier N. I.; Lombardi C.; Imhof R.; Bedford M. T.; Natoli G.; Hottiger M. O. Arginine Methyltransferase CARM1 is A Promoter-Specific Regulator of NF-kappaB-Dependent Gene Expression. EMBO J. 2005, 24, 85–96. 10.1038/sj.emboj.7600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Dhaheri M.; Wu J.; Skliris G. P.; Li J.; Higashimato K.; Wang Y.; White K. P.; Lambert P.; Zhu Y.; Murphy L.; et al. CARM1 is An Important Determinant of ERalpha-Dependent Breast Cancer Cell Differentiation and Proliferation in Breast Cancer Cells. Cancer Res. 2011, 71, 2118–2128. 10.1158/0008-5472.CAN-10-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong H.; Kao C.; Jeng M. H.; Eble J. N.; Koch M. O.; Gardner T. A.; Zhang S.; Li L.; Pan C. X.; Hu Z.; et al. Aberrant Expression of CARM1, A Transcriptional Coactivator of Androgen Receptor, in the Development of Prostate Carcinoma and Androgen-Independent Status. Cancer 2004, 101, 83–89. 10.1002/cncr.20327. [DOI] [PubMed] [Google Scholar]
- Majumder S.; Liu Y.; Ford O. H. 3rd; Mohler J. L.; Whang Y. E. Involvement of Arginine Methyltransferase CARM1 In Androgen Receptor Function and Prostate Cancer Cell Viability. Prostate 2006, 66, 1292–1301. 10.1002/pros.20438. [DOI] [PubMed] [Google Scholar]
- Elakoum R.; Gauchotte G.; Oussalah A.; Wissler M. P.; Clement-Duchene C.; Vignaud J. M.; Gueant J. L.; Namour F. CARM1 and PRMT1 are Dysregulated in Lung Cancer without Hierarchical Features. Biochimie 2014, 97, 210–218. 10.1016/j.biochi.2013.10.021. [DOI] [PubMed] [Google Scholar]
- Huynh T.; Chen Z.; Pang S. H.; Geng J. P.; Bandiera T.; Bindi S.; Vianello P.; Roletto F.; Thieffine S.; Galvani A.; et al. Optimization of Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 2924–2927. 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- Purandare A. V.; Chen Z.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Oconnell J.; Nowak K.; Jayaraman L. Pyrazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2008, 18, 4438–4441. 10.1016/j.bmcl.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Wan H. H.; Huynh T.; Pang S. H.; Geng J. P.; Vaccaro W.; Poss M. A.; Trainor G. L.; Lorenzi M. V.; Gottardis M.; Jayaraman L.; et al. Benzo[d]imidazole Inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1)-Hit to Lead Studies. Bioorg. Med. Chem. Lett. 2009, 19, 5063–5066. 10.1016/j.bmcl.2009.07.040. [DOI] [PubMed] [Google Scholar]
- Allan M.; Manku S.; Therrien E.; Nguyen N.; Styhler S.; Robert M. F.; Goulet A. C.; Petschner A. J.; Rahil G.; MacLeod A. R.; et al. N-Benzyl-1-Heteroaryl-3-(Trifluoromethyl)-1h-Pyrazole-5-Carboxamides as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 1218–1223. 10.1016/j.bmcl.2008.12.075. [DOI] [PubMed] [Google Scholar]
- Therrien E.; Larouche G.; Manku S.; Allan M.; Nguyen N.; Styhler S.; Robert M. F.; Goulet A. C.; Besterman J. M.; Nguyen H.; et al. 1,2-Diamines as Inhibitors of Co-Activator Associated Arginine Methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2009, 19, 6725–6732. 10.1016/j.bmcl.2009.09.110. [DOI] [PubMed] [Google Scholar]
- Sack J. S.; Thieffine S.; Bandiera T.; Fasolini M.; Duke G. J.; Jayaraman L.; Kish K. F.; Klei H. E.; Purandare A. V.; Rosettani P.; et al. Structural Basis for CARM1 Inhibition by Indole and Pyrazole Inhibitors. Biochem. J. 2011, 436, 331–339. 10.1042/BJ20102161. [DOI] [PubMed] [Google Scholar]
- Ferreira de Freitas R.; Eram M. S.; Szewczyk M. M.; Steuber H.; Smil D.; Wu H.; Li F.; Senisterra G.; Dong A.; Brown P. J.; et al. Discovery of a Potent Class I Protein Arginine Methyltransferase Fragment Inhibitor. J. Med. Chem. 2016, 59, 1176–1183. 10.1021/acs.jmedchem.5b01772. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Szewczyk M. M.; Eram M. S.; Smil D.; Kaniskan H. Ü.; Ferreira de Freitas R.; Senisterra G.; Li F.; Schapira M.; Brown P. J.; et al. Discovery of A Potent, Selective, and Cell-Active Dual Inhibitor of Protein Arginine Methyltransferase 4 and Protein Arginine Methyltransferase 6. J. Med. Chem. 2016, 59, 9124–9139. 10.1021/acs.jmedchem.6b01033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell L. H.; Drew A. E.; Ribich S. A.; Rioux N.; Swinger K. K.; Jacques S. L.; Lingaraj T.; Boriack-Sjodin P. A.; Waters N. J.; Wigle T. J.; et al. Aryl Pyrazoles as Potent Inhibitors of Arginine Methyltransferases: Identification of the First PRMT6 Tool Compound. ACS Med. Chem. Lett. 2015, 6, 655–659. 10.1021/acsmedchemlett.5b00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blat Y. Non-Competitive Inhibition by Active Site Binders. Chem. Biol. Drug Des. 2010, 75, 535–540. 10.1111/j.1747-0285.2010.00972.x. [DOI] [PubMed] [Google Scholar]
- Kaniskan H. Ü.; Eram M. S.; Liu J.; Smil D.; Martini M. L.; Shen Y.; Santhakumar V.; Brown P. J.; Arrowsmith C. H.; Vedadi M.; et al. Design and Synthesis of Selective, Small Molecule Inhibitors of Coactivator-Associated Arginine Methyltransferase 1 (CARM1). MedChemComm 2016, 7, 1793–1796. 10.1039/C6MD00342G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira de Freitas R.; Eram M. S.; Smil D.; Szewczyk M. M.; Kennedy S.; Brown P. J.; Santhakumar V.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Vedadi M.; et al. Discovery of a Potent and Selective Coactivator Associated Arginine Methyltransferase 1 (CARM1) Inhibitor by Virtual Screening. J. Med. Chem. 2016, 59, 6838–6847. 10.1021/acs.jmedchem.6b00668. [DOI] [PubMed] [Google Scholar]
- TP-064: A Chemical Probe For PRMT4. http://www.thesgc.org/chemical-probes/TP-064 (accessed January 2017).
- van Haren M.; van Ufford L. Q.; Moret E. E.; Martin N. I. Synthesis and Evaluation of Protein Arginine N-Methyltransferase Inhibitors Designed to Simultaneously Occupy both Substrate Binding Sites. Org. Biomol. Chem. 2015, 13, 549–560. 10.1039/C4OB01734J. [DOI] [PubMed] [Google Scholar]
- Cheng D.; Valente S.; Castellano S.; Sbardella G.; Di Santo R.; Costi R.; Bedford M. T.; Mai A. Novel 3,5-Bis(Bromohydroxybenzylidene)Piperidin-4-Ones as Coactivator-Associated Arginine Methyltransferase 1 Inhibitors: Enzyme Selectivity and Cellular Activity. J. Med. Chem. 2011, 54, 4928–4932. 10.1021/jm200453n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvi B. R.; Batta K.; Kishore A. H.; Mantelingu K.; Varier R. A.; Balasubramanyam K.; Pradhan S. K.; Dasgupta D.; Sriram S.; Agrawal S.; et al. Identification of a Novel Inhibitor of Coactivator-associated Arginine Methyltransferase 1 (CARM1)-mediated Methylation of Histone H3 Arg-17. J. Biol. Chem. 2010, 285, 7143–7152. 10.1074/jbc.M109.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Vishwanath S. N.; Erdjument-Bromage H.; Tempst P.; Sif S. Human SWI/SNF-Associated PRMT5Methylates Histone H3 Arginine 8 and Negatively Regulates Expression of ST7 and NM23 Tumor Suppressor Genes. Mol. Cell. Biol. 2004, 24, 9630–9645. 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Baiocchi R. A.; Byrd J. C.; Grever M. R.; Jacob S. T.; Sif S. Low Levels of miR-92b/96 Induce PRMT5 Translation and H3R8/H4R3Methylation in Mantle Cell Lymphoma. EMBO J. 2007, 26, 3558–3569. 10.1038/sj.emboj.7601794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori V.; Muller J.; Phalke S.; Low D.; Bezzi M.; Mok W. C.; Sahu S. K.; Gunaratne J.; Capasso P.; Bassi C.; et al. Symmetric Dimethylation of H3R2 is A Newly Identified Histone Mark that Supports Euchromatin Maintenance. Nat. Struct. Mol. Biol. 2012, 19, 136–144. 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- Xu X.; Hoang S.; Mayo M. W.; Bekiranov S. Application of Machine Learning Methods to Histone Methylation ChIP-Seq Data Reveals H4R3me2 Globally Represses Gene Expression. BMC Bioinf. 2010, 11, 396. 10.1186/1471-2105-11-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Pal S.; Sif S. Protein Arginine Methyltransferase 5 Suppresses the Transcription of the RB Family of Tumor Suppressors in Leukemia and Lymphoma Cells. Mol. Cell. Biol. 2008, 28, 6262–6277. 10.1128/MCB.00923-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbrizio E.; El Messaoudi S.; Polanowska J.; Paul C.; Cook J. R.; Lee J. H.; Negre V.; Rousset M.; Pestka S.; Le Cam A.; et al. Negative Regulation of Transcription by the Type II Arginine Methyltransferase PRMT5. EMBO Rep. 2002, 3, 641–645. 10.1093/embo-reports/kvf136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth-Vollenweider T.; Joshi S.; Dhawan P.; Sif S.; Christakos S. Novel mechanism of negative regulation of 1,25-dihydroxyvitamin D3-induced 25-hydroxyvitamin D3 24-hydroxylase (Cyp24a1) Transcription: epigenetic modification involving cross-talk between protein-arginine methyltransferase 5 and the SWI/SNF complex. J. Biol. Chem. 2014, 289, 33958–33970. 10.1074/jbc.M114.583302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Wang B.; Miyagi M.; She Y.; Gopalan B.; Huang D. B.; Ghosh G.; Stark G. R.; Lu T. PRMT5 Dimethylates R30 of the p65 Subunit to Activate NF-kappaB. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 13516–13521. 10.1073/pnas.1311784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson M.; Durant S. T.; Cho E. C.; Sheahan S.; Edelmann M.; Kessler B.; La Thangue N. B. Arginine Methylation Regulates the p53 Response. Nat. Cell Biol. 2008, 10, 1431–1439. 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- Zheng S.; Moehlenbrink J.; Lu Y. C.; Zalmas L. P.; Sagum C. A.; Carr S.; McGouran J. F.; Alexander L.; Fedorov O.; Munro S.; et al. Arginine Methylation-Dependent Reader-Writer Interplay Governs Growth Control by E2F-1. Mol. Cell 2013, 52, 37–51. 10.1016/j.molcel.2013.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu-Perez P.; Esteve-Puig R.; de Torre-Minguela C.; Lopez-Fauqued M.; Bech-Serra J. J.; Tenbaum S.; Garcia-Trevijano E. R.; Canals F.; Merlino G.; Avila M. A.; et al. Protein Arginine Methyltransferase 5 Regulates ERK1/2 Signal Transduction Amplitude and Cell Fate through CRAF. Sci. Signaling 2011, 4, ra58. 10.1126/scisignal.2001936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Wang Y.; Liang Y.; Zhang Y.; Bao S.; Xu Z. Methylation of Ribosomal Protein S10 by Protein-Arginine Methyltransferase 5 Regulates Ribosome Biogenesis. J. Biol. Chem. 2010, 285, 12695–12705. 10.1074/jbc.M110.103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancelin K.; Lange U. C.; Hajkova P.; Schneider R.; Bannister A. J.; Kouzarides T.; Surani M. A. Blimp1 Associates with PRMT5 and Directs Histone Arginine Methylation in Mouse Germ Cells. Nat. Cell Biol. 2006, 8, 623–630. 10.1038/ncb1413. [DOI] [PubMed] [Google Scholar]
- Friesen W. J.; Paushkin S.; Wyce A.; Massenet S.; Pesiridis G. S.; Van Duyne G.; Rappsilber J.; Mann M.; Dreyfuss G. The Methylosome, A 20S Complex Containing JBP1 and pICln, Produces Dimethylarginine-Modified Sm Proteins. Mol. Cell. Biol. 2001, 21, 8289–8300. 10.1128/MCB.21.24.8289-8300.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen W. J.; Wyce A.; Paushkin S.; Abel L.; Rappsilber J.; Mann M.; Dreyfuss G. A Novel WD Repeat Protein Component of the Methylosome Binds Sm Proteins. J. Biol. Chem. 2002, 277, 8243–8247. 10.1074/jbc.M109984200. [DOI] [PubMed] [Google Scholar]
- Guderian G.; Peter C.; Wiesner J.; Sickmann A.; Schulze-Osthoff K.; Fischer U.; Grimmler M. RioK1, a New Interactor of Protein Arginine Methyltransferase 5 (PRMT5), Competes with pICln for Binding and Modulates PRMT5 Complex Composition and Substrate Specificity. J. Biol. Chem. 2011, 286, 1976–1986. 10.1074/jbc.M110.148486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guezennec X.; Vermeulen M.; Brinkman A. B.; Hoeijmakers W. A.; Cohen A.; Lasonder E.; Stunnenberg H. G. MBD2/NuRD and MBD3/NuRD, Two Distinct Complexes with Different Biochemical and Functional Properties. Mol. Cell. Biol. 2006, 26, 843–851. 10.1128/MCB.26.3.843-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M. C.; Wilczek C.; Bonanno J. B.; Xing L.; Seznec J.; Matsui T.; Carter L. G.; Onikubo T.; Kumar P. R.; Chan M. K.; et al. Structure of the Arginine Methyltransferase PRMT5-MEP50 Reveals A Mechanism for Substrate Specificity. PLoS One 2013, 8, e57008. 10.1371/journal.pone.0057008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y.; Rauch C.; et al. Crystal Structure of the Human PRMT5:MEP50 Complex. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 17960–17965. 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J. H.; Karkhanis V.; Tae S.; Yan F. T.; Smith P.; Ayers L. W.; Agostinelli C.; Pileri S.; Denis G. V.; Baiocchi R. A.; et al. Protein Arginine Methyltransferase 5 (PRMT5) Inhibition Induces Lymphoma Cell Death through Reactivation of the Retinoblastoma Tumor Suppressor Pathway and Polycomb Repressor Complex 2 (PRC2) Silencing. J. Biol. Chem. 2013, 288, 35534–35547. 10.1074/jbc.M113.510669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas C.; Yang J.; Peters S. B.; Bill M. A.; Baiocchi R. A.; Yan F. T.; Sif S.; Tae S.; Gaudio E.; Wu X.; et al. PRMT5 Is Upregulated in Malignant and Metastatic Melanoma and Regulates Expression of MITF and p27(Kip1). PLoS One 2013, 8, e74710. 10.1371/journal.pone.0074710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T. Y. W.; Juan C. C.; Hisa J. Y.; Su L. J.; Lee Y. C. G.; Chou H. Y.; Chen J. M. M.; Wu Y. C.; Chiu S. C.; Hsu C. P.; et al. Protein Arginine Methyltransferase 5 is A Potential Oncoprotein that Upregulates G1 Cyclins/Cyclin-Dependent Kinases and the Phosphoinositide 3-Kinase/AKT Signaling Cascade. Cancer Sci. 2012, 103, 1640–1650. 10.1111/j.1349-7006.2012.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers M. A.; Fay M. M.; Factor R. E.; Welm A. L.; Ullman K. S. Protein Arginine Methyltransferase 5 Accelerates Tumor Growth by Arginine Methylation of the Tumor Suppressor Programmed Cell Death 4. Cancer Res. 2011, 71, 5579–5587. 10.1158/0008-5472.CAN-11-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho E. C.; Zheng S. S.; Munro S.; Liu G.; Carr S. M.; Moehlenbrink J.; Lu Y. C.; Stimson L.; Khan O.; Konietzny R.; et al. Arginine Methylation Controls Growth Regulation by E2F-1. EMBO J. 2012, 31, 1785–1797. 10.1038/emboj.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X. X.; Zhao S.; Liu T.; Liu Y.; Liu Y. Y.; Yang X. S. Overexpression of PRMT5 Promotes Tumor Cell Growth and is Associated with Poor Disease Prognosis in Epithelial Ovarian Cancer. J. Histochem. Cytochem. 2013, 61, 206–217. 10.1369/0022155413475452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy B.; Surowiak P.; Budczies J.; Lanczky A. Online Survival Analysis Software to Assess the Prognostic Value of Biomarkers Using Transcriptomic Data in Non-Small-Cell Lung Cancer. PLoS One 2013, 8, e82241. 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan-Penebre E.; Kuplast K. G.; Majer C. R.; Boriack-Sjodin P. A.; Wigle T. J.; Johnston L. D.; Rioux N.; Munchhof M. J.; Jin L.; Jacques S. L.; et al. A Selective Inhibitor of PRMT5 with In Vivo and In Vitro Potency in MCL Models. Nat. Chem. Biol. 2015, 11, 432–437. 10.1038/nchembio.1810. [DOI] [PubMed] [Google Scholar]
- Duncan K. W.; Rioux N.; Boriack-Sjodin P. A.; Munchhof M. J.; Reiter L. A.; Majer C. R.; Jin L.; Johnston L. D.; Chan-Penebre E.; Kuplast K. G.; et al. Structure and Property Guided Design in the Identification of PRMT5 Tool Compound EPZ015666. ACS Med. Chem. Lett. 2016, 7, 162–166. 10.1021/acsmedchemlett.5b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GSK591: A chemical probe for PRMT5. http://www.thesgc.org/chemical-probes/GSK591 (accessed November 2016).
- ClinicalTrials.gov Identifier: NCT02783300. A Phase I, Open-label, Dose Escalation Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK3326595 in Subjects With Solid Tumors and Non-Hodgkin’s Lymphoma (accessed September 2016).
- Alinari L.; Mahasenan K. V.; Yan F. T.; Karkhanis V.; Chung J. H.; Smith E. M.; Quinion C.; Smith P. L.; Kim L.; Patton J. T.; et al. Selective Inhibition of Protein Arginine Methyltransferase 5 Blocks Initiation and Maintenance of B-Cell Transformation. Blood 2015, 125, 2530–2543. 10.1182/blood-2014-12-619783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarighat S. S.; Santhanam R.; Frankhouser D.; Radomska H. S.; Lai H.; Anghelina M.; Wang H.; Huang X.; Alinari L.; Walker A.; et al. The Dual Epigenetic Role of PRMT5 in Acute Myeloid Leukemia: Gene Activation and Repression via Histone Arginine Methylation. Leukemia 2016, 30, 789–799. 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LLY-283: A Chemical Probe For PRMT5. http://www.thesgc.org/chemical-probes/LLY-283 (accessed November 2016).
- Ji S.; Ma S.; Wang W. J.; Huang S. Z.; Wang T. Q.; Xiang R.; Hu Y. G.; Chen Q.; Li L. L.; Yang S. Y.. Discovery of Selective Protein Arginine Methyltransferase 5 (PRMT5) Inhibitors and Biological Evaluations. Chem. Biol. Drug Des. 2016, 10.1111/cbdd.12881. [DOI] [PubMed] [Google Scholar]
- Gonsalvez G. B.; Tian L.; Ospina J. K.; Boisvert F. M.; Lamond A. I.; Matera A. G. Two Distinct Arginine Methyltransferases are Required for Biogenesis of Sm-Class Ribonucleoproteins. J. Cell Biol. 2007, 178, 733–740. 10.1083/jcb.200702147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurita-Lopez C. I.; Sandberg T.; Kelly R.; Clarke S. G. Human Protein Arginine Methyltransferase 7 (PRMT7) Is a Type III Enzyme Forming omega-N-G-Monomethylated Arginine Residues. J. Biol. Chem. 2012, 287, 7859–7870. 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao R. S.; Jiang H.; Ma Y. H.; Wang L. P.; Wang L.; Du J.; Hou P. F.; Gao Y. Y.; Zhao L.; Wang G. N.; et al. PRMT7 Induces Epithelial-to-Mesenchymal Transition and Promotes Metastasis in Breast Cancer. Cancer Res. 2014, 74, 5656–5667. 10.1158/0008-5472.CAN-14-0800. [DOI] [PubMed] [Google Scholar]
- Smil D.; Eram M. S.; Li F.; Kennedy S.; Szewczyk M. M.; Brown P. J.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Vedadi M.; Schapira M. Discovery of A Dual PRMT5-PRMT7 Inhibitor. ACS Med. Chem. Lett. 2015, 6, 408–412. 10.1021/ml500467h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A.; Yadav N.; Lee J.; Branscombe T. L.; Clarke S.; Bedford M. T. The Novel Human Protein Arginine N-Methyltransferase PRMT6 is A Nuclear Enzyme Displaying Unique Substrate Specificity. J. Biol. Chem. 2002, 277, 3537–3543. 10.1074/jbc.M108786200. [DOI] [PubMed] [Google Scholar]
- Hyllus D.; Stein C.; Schnabel K.; Schiltz E.; Imhof A.; Dou Y. L.; Hsieh J.; Bauer U. M. Prmt6-Mediated Methylation of R2 in Histone H3 Antagonizes H3K4 Trimethylation. Genes Dev. 2007, 21, 3369–3380. 10.1101/gad.447007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guccione E.; Bassi C.; Casadio F.; Martinato F.; Cesaroni M.; Schuchlautz H.; Luscher B.; Amati B. Methylation of Histone H3R2 by PRMT6 and H3K4 by An MLL Complex are Mutually Exclusive. Nature 2007, 449, 933–U918. 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- Kirmizis A.; Santos-Rosa H.; Penkett C. J.; Singer M. A.; Vermeulen M.; Mann M.; Bahler J.; Green R. D.; Kouzarides T. Arginine Methylation at Histone H3R2 Controls Deposition of H3K4 Trimethylation. Nature 2007, 449, 928–U917. 10.1038/nature06160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iberg A. N.; Espejo A.; Cheng D.; Kim D.; Michaud-Levesque J.; Richard S.; Bedford M. T. Arginine Methylation of the Histone H3 Tail Impedes Effector Binding. J. Biol. Chem. 2008, 283, 3006–3010. 10.1074/jbc.C700192200. [DOI] [PubMed] [Google Scholar]
- Schuetz A.; Allali-Hassani A.; Martin F.; Loppnau P.; Vedadi M.; Bochkarev A.; Plotnikov A. N.; Arrowsmith C. H.; Min J. R. Structural Basis for Molecular Recognition and Presentation of Histone H3 by WDR5. EMBO J. 2006, 25, 4245–4252. 10.1038/sj.emboj.7601316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmarajan V.; Lee J. H.; Patel A.; Skalnik D. G.; Cosgrove M. S. Structural Basis for WDR5 Interaction (WIN) Motif Recognition in Human SET1 Family Histone Methyltransferases. J. Biol. Chem. 2012, 287, 27275–27289. 10.1074/jbc.M112.364125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A.; Dharmarajan V.; Cosgrove M. S. Structure of WDR5 Bound to Mixed Lineage Leukemia Protein-1 Peptide. J. Biol. Chem. 2008, 283, 32158–32161. 10.1074/jbc.C800164200. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Lee H.; Brunzelle J. S.; Couture J. F. The Plasticity of WDR5 Peptide-Binding Cleft Enables the Binding of the SET1 Family of Histone Methyltransferases. Nucleic Acids Res. 2012, 40, 4237–4246. 10.1093/nar/gkr1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinschmidt M. A.; de Graaf P.; van Teeffelen H. A. A. M.; Timmers H. T. M. Cell Cycle Regulation by the PRMT6 Arginine Methyltransferase through Repression of Cyclin-Dependent Kinase Inhibitors. PLoS One 2012, 7, e41446. 10.1371/journal.pone.0041446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison M. J.; Tang Y. H.; Dowhan D. H. Protein Arginine Methyltransferase 6 Regulates Multiple Aspects Of Gene Expression. Nucleic Acids Res. 2010, 38, 2201–2216. 10.1093/nar/gkp1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. H.; Ma H.; Tan T. Z.; Ng S. S.; Soong R.; Mori S.; Fu X. Y.; Zernicka-Goetz M.; Wu Q. Protein Arginine Methyltransferase 6 Regulates Embryonic Stem Cell Identity. Stem Cells Dev. 2012, 21, 2613–2622. 10.1089/scd.2011.0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Andaloussi N.; Valovka T.; Toueille M.; Steinacher R.; Focke F.; Gehrig P.; Covic M.; Hassa P. O.; Schar P.; Hubscher U.; et al. Arginine Methylation Regulates DNA Polymerase Beta. Mol. Cell 2006, 22, 51–62. 10.1016/j.molcel.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Boulanger M. C.; Liang C.; Russell R. S.; Lin R. T.; Bedford M. T.; Wainberg M. A.; Richard S. Methylation of Tat by PRMT6 Regulates Human Immunodeficiency Virus Type 1 Gene Expression. J. Virol. 2005, 79, 124–131. 10.1128/JVI.79.1.124-131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limm K.; Ott C.; Wallner S.; Mueller D. W.; Oefner P.; Hellerbrand C.; Bosserhoff A. K. Deregulation of Protein Methylation in Melanoma. Eur. J. Cancer 2013, 49, 1305–1313. 10.1016/j.ejca.2012.11.026. [DOI] [PubMed] [Google Scholar]
- Dhar S.; Vemulapalli V.; Patananan A. N.; Huang G. L.; Di Lorenzo A.; Richard S.; Comb M. J.; Guo A.; Clarke S. G.; Bedford M. T.. Loss of the Major Type I Arginine Methyltransferase PRMT1 Causes Substrate Scavenging by Other PRMTs. Sci. Rep. 2013, 3, 10.1038/srep01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Zheng W.; Eram M. S.; Vhuiyan M.; Dong A.; Zeng H.; He H.; Brown P.; Frankel A.; Vedadi M.; et al. Structural Basis of Arginine Asymmetrical Dimethylation by PRMT6. Biochem. J. 2016, 473, 3049–3063. 10.1042/BCJ20160537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis C. D.; Bowen J. K.; Abraham G. N.; Glover C. V.; Gorovsky M. A. Proteolytic Processing of Histone H3 in Chromatin: A Physiologically Regulated Event in Tetrahymena Micronuclei. Cell 1980, 20, 55–64. 10.1016/0092-8674(80)90234-2. [DOI] [PubMed] [Google Scholar]
- Ahmad K.; Henikoff S. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 2002, 9, 1191–1200. 10.1016/S1097-2765(02)00542-7. [DOI] [PubMed] [Google Scholar]
- Paik W. K.; Kim S. Enzymatic Demethylation of Calf Thymus Histones. Biochem. Biophys. Res. Commun. 1973, 51, 781–788. 10.1016/0006-291X(73)91383-1. [DOI] [PubMed] [Google Scholar]
- Tsukada Y.; Fang J.; Erdjument-Bromage H.; Warren M. E.; Borchers C. H.; Tempst P.; Zhang Y. Histone Demethylation by A Family of JmjC Domain-Containing Proteins. Nature 2005, 439, 811–816. 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Karytinos A.; Forneris F.; Profumo A.; Ciossani G.; Battaglioli E.; Binda C.; Mattevi A. A Novel Mammalian Flavin-Dependent Histone Demethylase. J. Biol. Chem. 2009, 284, 17775–17782. 10.1074/jbc.M109.003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova E.; Turberfield A. H.; Klose R. J. Histone Demethylases in Chromatin Biology and Beyond. EMBO Rep. 2015, 16, 1620–1639. 10.15252/embr.201541113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinnes C. C.; England K. S.; Kawamura A.; Chowdhury R.; Schofield C. J.; Hopkinson R. J. Targeting Histone Lysine Demethylases - Progress, Challenges, and the Future. Biochim. Biophys. Acta, Gene Regul. Mech. 2014, 1839, 1416–1432. 10.1016/j.bbagrm.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Miyata N. Lysine Demethylases Inhibitors. J. Med. Chem. 2011, 54, 8236–8250. 10.1021/jm201048w. [DOI] [PubMed] [Google Scholar]
- Metzger E.; Wissmann M.; Yin N.; Muller J. M.; Schneider R.; Peters A. H.; Gunther T.; Buettner R.; Schule R. LSD1 Demethylates Repressive Histone Marks to Promote Androgen-Receptor-Dependent Transcription. Nature 2005, 437, 436–439. 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Yang Y.; Wang F.; Wan K.; Yamane K.; Zhang Y.; Lei M. Crystal Structure of Human Histone Lysine-Specific Demethylase 1 (LSD1). Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13956–13961. 10.1073/pnas.0606381103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Ma H. Evolutionary History Ootone Demethylase Families: Distinct Evolutionary Patterns Suggest Functional Divergence. BMC Evol. Biol. 2008, 8, 294. 10.1186/1471-2148-8-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry J.; Zhao Y. The CW Domain, A Structural Module Shared Amongst Vertebrates, Vertebrate-Infecting Parasites and Higher Plants. Trends Biochem. Sci. 2003, 28, 576–580. 10.1016/j.tibs.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Lee M. G.; Wynder C.; Cooch N.; Shiekhattar R. An Essential Role for CoREST in Nucleosomal Histone 3 Lysine 4 Demethylation. Nature 2005, 437, 432–435. 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Shi Y. J.; Matson C.; Lan F.; Iwase S.; Baba T.; Shi Y. Regulation of LSD1 Histone Demethylase Activity by its Associated Factors. Mol. Cell 2005, 19, 857–864. 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Hakimi M. A.; Bochar D. A.; Chenoweth J.; Lane W. S.; Mandel G.; Shiekhattar R. A Core-BRAF35 Complex Containing Histone Deacetylase Mediates Repression of Neuronal-Specific Genes. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 7420–7425. 10.1073/pnas.112008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi M. A.; Dong Y.; Lane W. S.; Speicher D. W.; Shiekhattar R. A Candidate X-Linked Mental Retardation Gene is A Component of A New Family of Histone Deacetylase-Containing Complexes. J. Biol. Chem. 2003, 278, 7234–7239. 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang H.; Chen Y.; Sun Y.; Yang F.; Yu W.; Liang J.; Sun L.; Yang X.; Shi L.; et al. LSD1 is A Subunit of the NuRD Complex and Targets the Metastasis Programs in Breast Cancer. Cell 2009, 138, 660–672. 10.1016/j.cell.2009.05.050. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Sawada J.; Sui G.; El Bachir A.; Whetstine J. R.; Lan F.; Ogawa H.; Po-Shan Luke M.; Nakatani Y.; Shi Y. Coordinated Histone Modifications Mediated by A CtBP Co-Repressor Complex. Nature 2003, 422, 735–738. 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Perillo B.; Ombra M. N.; Bertoni A.; Cuozzo C.; Sacchetti S.; Sasso A.; Chiariotti L.; Malorni A.; Abbondanza C.; Avvedimento E. V. DNA Oxidation as Triggered by H3K9me2 Demethylation Drives Estrogen-Induced Gene Expression. Science 2008, 319, 202–206. 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- Culhane J. C.; Cole P. A. LSD1 and the Chemistry of Histone Demethylation. Curr. Opin. Chem. Biol. 2007, 11, 561–568. 10.1016/j.cbpa.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X.; Ouyang S.; Liang Z.; Lu J.; Chen L.; Shen B.; Li D.; Zheng M.; Li K. K.; Luo C.; et al. Catalytic Mechanism Investigation of Lysine-Specific Demethylase 1 (LSD1): A Computational Study. PLoS One 2011, 6, e25444. 10.1371/journal.pone.0025444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Sengupta R.; Espejo A. B.; Lee M. G.; Dorsey J. A.; Richter M.; Opravil S.; Shiekhattar R.; Bedford M. T.; Jenuwein T.; et al. p53 is Regulated by the Lysine Demethylase LSD1. Nature 2007, 449, 105–108. 10.1038/nature06092. [DOI] [PubMed] [Google Scholar]
- Cho H. S.; Suzuki T.; Dohmae N.; Hayami S.; Unoki M.; Yoshimatsu M.; Toyokawa G.; Takawa M.; Chen T.; Kurash J. K.; et al. Demethylation of RB Regulator MYPT1 by Histone Demethylase LSD1 Promotes Cell Cycle Progression in Cancer Cells. Cancer Res. 2011, 71, 655–660. 10.1158/0008-5472.CAN-10-2446. [DOI] [PubMed] [Google Scholar]
- Wang J.; Hevi S.; Kurash J. K.; Lei H.; Gay F.; Bajko J.; Su H.; Sun W.; Chang H.; Xu G.; et al. The Lysine Demethylase LSD1 (KDM1) is Required for Maintenance of Global DNA Methylation. Nat. Genet. 2009, 41, 125–129. 10.1038/ng.268. [DOI] [PubMed] [Google Scholar]
- Hayami S.; Kelly J. D.; Cho H. S.; Yoshimatsu M.; Unoki M.; Tsunoda T.; Field H. I.; Neal D. E.; Yamaue H.; Ponder B. A.; et al. Overexpression of LSD1 Contributes to Human Carcinogenesis through Chromatin Regulation in Various Cancers. Int. J. Cancer 2011, 128, 574–586. 10.1002/ijc.25349. [DOI] [PubMed] [Google Scholar]
- Serce N.; Gnatzy A.; Steiner S.; Lorenzen H.; Kirfel J.; Buettner R. Elevated Expression of LSD1 (Lysine-Specific Demethylase 1) During Tumour Progression from Pre-Invasiveto Invasive Ductal Carcinoma of the Breast. BMC Clin. Pathol. 2012, 12, 13. 10.1186/1472-6890-12-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahl P.; Gullotti L.; Heukamp L. C.; Wolf S.; Friedrichs N.; Vorreuther R.; Solleder G.; Bastian P. J.; Ellinger J.; Metzger E.; et al. Androgen Receptor Coactivators Lysine-Specific Histone Demethylase 1 and Four and A Half LIM Domain Protein 2 Predict Risk of Prostate Cancer Recurrence. Cancer Res. 2006, 66, 11341–11347. 10.1158/0008-5472.CAN-06-1570. [DOI] [PubMed] [Google Scholar]
- Lv T.; Yuan D.; Miao X.; Lv Y.; Zhan P.; Shen X.; Song Y. Over-Expression of LSD1 Promotes Proliferation, Migration and Invasion in Non-Small Cell Lung Cancer. PLoS One 2012, 7, e35065. 10.1371/journal.pone.0035065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte J. H.; Lim S.; Schramm A.; Friedrichs N.; Koster J.; Versteeg R.; Ora I.; Pajtler K.; Klein-Hitpass L.; Kuhfittig-Kulle S.; et al. Lysine-Specific Demethylase 1 is Strongly Expressed in Poorly Differentiated Neuroblastoma: Implications for Therapy. Cancer Res. 2009, 69, 2065–2071. 10.1158/0008-5472.CAN-08-1735. [DOI] [PubMed] [Google Scholar]
- Rhodes D. R.; Kalyana-Sundaram S.; Mahavisno V.; Varambally R.; Yu J.; Briggs B. B.; Barrette T. R.; Anstet M. J.; Kincead-Beal C.; Kulkarni P.; et al. Oncomine 3.0: Genes, Pathways, and Networks in A Collection of 18,000 Cancer Gene Expression Profiles. Neoplasia 2007, 9, 166–180. 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radich J. P.; Dai H.; Mao M.; Oehler V.; Schelter J.; Druker B.; Sawyers C.; Shah N.; Stock W.; Willman C. L.; et al. Gene Expression Changes Associated with Progression and Response in Chronic Myeloid Leukemia. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 2794–2799. 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters B. J.; Lowenberg B.; Erpelinck-Verschueren C. A.; van Putten W. L.; Valk P. J.; Delwel R. Double CEBPA Mutations, but not Single CEBPA Mutations, Define A Subgroup of Acute Myeloid Leukemia with A Distinctive Gene Expression Profile that is Uniquely Associated with A Favorable Outcome. Blood 2009, 113, 3088–3091. 10.1182/blood-2008-09-179895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Huang B.; Suzuki T.; Liu X.; Zhan P. Medicinal Chemistry Insights in the Discovery of Novel LSD1 Inhibitors. Epigenomics 2015, 7, 1379–1396. 10.2217/epi.15.86. [DOI] [PubMed] [Google Scholar]
- Maes T.; Mascaro C.; Ortega A.; Lunardi S.; Ciceri F.; Somervaille T. C.; Buesa C. KDM1 Histone Lysine Demethylases as Targets for Treatments of Oncological and Neurodegenerative Disease. Epigenomics 2015, 7, 609–626. 10.2217/epi.15.9. [DOI] [PubMed] [Google Scholar]
- Lee M. G.; Wynder C.; Schmidt D. M.; McCafferty D. G.; Shiekhattar R. Histone H3 Lysine 4 Demethylation is A Target of Nonselective Antidepressive Medications. Chem. Biol. 2006, 13, 563–567. 10.1016/j.chembiol.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Forneris F.; Binda C.; Vanoni M. A.; Battaglioli E.; Mattevi A. Human Histone Demethylase LSD1 Reads the Histone Code. J. Biol. Chem. 2005, 280, 41360–41365. 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- Schmidt D. M.; McCafferty D. G. Trans-2-Phenylcyclopropylamine is A Mechanism-Based Inactivator of the Histone Demethylase LSD1. Biochemistry 2007, 46, 4408–4416. 10.1021/bi0618621. [DOI] [PubMed] [Google Scholar]
- Kitz R.; Wilson I. B. Esters of Methanesulfonic Acid as Irreversible Inhibitors of Acetylcholinesterase. J. Biol. Chem. 1962, 237, 3245–3249. [PubMed] [Google Scholar]
- Silverman R. B. Mechanism-Based Enzyme Inactivators. Methods Enzymol. 1995, 249, 240–283. 10.1016/0076-6879(95)49038-8. [DOI] [PubMed] [Google Scholar]
- Hayward D.; Cole P. A. LSD1 Histone Demethylase Assays and Inhibition. Methods Enzymol. 2016, 573, 261–278. 10.1016/bs.mie.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Culhane J. C.; Szewczuk L. M.; Jalili P.; Ball H. L.; Machius M.; Cole P. A.; Yu H. Structural Basis for the Inhibition of the LSD1 Histone Demethylase by the Antidepressant Trans-2-Phenylcyclopropylamine. Biochemistry 2007, 46, 8058–8065. 10.1021/bi700664y. [DOI] [PubMed] [Google Scholar]
- Mohammad H. P.; Smitheman K. N.; Kamat C. D.; Soong D.; Federowicz K. E.; Van Aller G. S.; Schneck J. L.; Carson J. D.; Liu Y.; Butticello M.; et al. A DNA Hypomethylation Signature Predicts Antitumor Activity of LSD1 Inhibitors in SCLC. Cancer Cell 2015, 28, 57–69. 10.1016/j.ccell.2015.06.002. [DOI] [PubMed] [Google Scholar]
- Binda C.; Li M.; Hubalek F.; Restelli N.; Edmondson D. E.; Mattevi A. Insights into the Mode of Inhibition of Human Mitochondrial Monoamine Oxidase B from High-Resolution Crystal Structures. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 9750–9755. 10.1073/pnas.1633804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. W.; Zhao G.; Martinovic S.; Jorns M. S.; Mathews F. S. Structure of The Sodium Borohydride-Reduced N-(Cyclopropyl)Glycine Adduct of the Flavoenzyme Monomeric Sarcosine Oxidase. Biochemistry 2005, 44, 15444–15450. 10.1021/bi0515422. [DOI] [PubMed] [Google Scholar]
- Silverman R. B. Radical Ideas About Monoamine-Oxidase. Acc. Chem. Res. 1995, 28, 335–342. 10.1021/ar00056a003. [DOI] [Google Scholar]
- Mimasu S.; Sengoku T.; Fukuzawa S.; Umehara T.; Yokoyama S. Crystal Structure of Histone Demethylase LSD1 and Tranylcypromine at 2.25 Angstrom. Biochem. Biophys. Res. Commun. 2008, 366, 15–22. 10.1016/j.bbrc.2007.11.066. [DOI] [PubMed] [Google Scholar]
- Binda C.; Valente S.; Romanenghi M.; Pilotto S.; Cirilli R.; Karytinos A.; Ciossani G.; Botrugno O. A.; Forneris F.; Tardugno M.; et al. Biochemical, Structural, and Biological Evaluation of Tranylcypromine Derivatives as Inhibitors of Histone Demethylases LSD1 and LSD2. J. Am. Chem. Soc. 2010, 132, 6827–6833. 10.1021/ja101557k. [DOI] [PubMed] [Google Scholar]
- Benelkebir H.; Hodgkinson C.; Duriez P. J.; Hayden A. L.; Bulleid R. A.; Crabb S. J.; Packham G.; Ganesan A. Enantioselective Synthesis of Tranylcypromine Analogues as Lysine Demethylase (LSD1) Inhibitors. Bioorg. Med. Chem. 2011, 19, 3709–3716. 10.1016/j.bmc.2011.02.017. [DOI] [PubMed] [Google Scholar]
- Vianello P.; Botrugno O. A.; Cappa A.; Ciossani G.; Dessanti P.; Mai A.; Mattevi A.; Meroni G.; Minucci S.; Thaler F.; et al. Synthesis, Biological Activity and Mechanistic Insights of 1-Substituted Cyclopropylamine Derivatives: A Novel Class of Irreversible Inhibitors of Histone Demethylase KDM1A. Eur. J. Med. Chem. 2014, 86, 352–363. 10.1016/j.ejmech.2014.08.068. [DOI] [PubMed] [Google Scholar]
- Gooden D. M.; Schmidt D. M.; Pollock J. A.; Kabadi A. M.; McCafferty D. G. Facile Synthesis of Substituted trans-2-Arylcyclopropylamine Inhibitors of the Human Histone Demethylase LSD1 and Monoamine Oxidases A and B. Bioorg. Med. Chem. Lett. 2008, 18, 3047–3051. 10.1016/j.bmcl.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda R.; Suzuki T.; Mino K.; Tsumoto H.; Nakagawa H.; Hasegawa M.; Sasaki R.; Mizukami T.; Miyata N. Identification of Cell-Active Lysine Specific Demethylase 1-Selective Inhibitors. J. Am. Chem. Soc. 2009, 131, 17536–17537. 10.1021/ja907055q. [DOI] [PubMed] [Google Scholar]
- Yang M. J.; Culhane J. C.; Szewczuk L. M.; Gocke C. B.; Brautigam C. A.; Tomchick D. R.; Machius M.; Cole P. A.; Yu H. T. Structural Basis of Histone Demethylation by LSD1 Revealed by Suicide Inactivation. Nat. Struct. Mol. Biol. 2007, 14, 535–539. 10.1038/nsmb1255. [DOI] [PubMed] [Google Scholar]
- Mimasu S.; Umezawa N.; Sato S.; Higuchi T.; Umehara T.; Yokoyama S. Structurally Designed trans-2-Phenylcyclopropylamine Derivatives Potently Inhibit Histone Demethylase LSDI/KDM1. Biochemistry 2010, 49, 6494–6503. 10.1021/bi100299r. [DOI] [PubMed] [Google Scholar]
- Neelamegam R.; Ricq E. L.; Malvaez M.; Patnaik D.; Norton S.; Carlin S. M.; Hill I. T.; Wood M. A.; Haggarty S. J.; Hooker J. M. Brain-Penetrant LSD1 Inhibitors Can Block Memory Consolidation. ACS Chem. Neurosci. 2012, 3, 120–128. 10.1021/cn200104y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara D.; Itoh Y.; Tsumoto H.; Kakizawa T.; Mino K.; Fukuhara K.; Nakagawa H.; Hasegawa M.; Sasaki R.; Mizukami T.; et al. Lysine-Specific Demethylase 1-Selective Inactivators: Protein-Targeted Drug Delivery Mechanism. Angew. Chem., Int. Ed. 2013, 52, 8620–8624. 10.1002/anie.201303999. [DOI] [PubMed] [Google Scholar]
- EudraCT Number: 2013-002447-29. A phase I study of Human Pharmacokinetics and Safety of ORY-1001, and LSD1 inhibitor, in relapsed or refractory acute leukaemia (AL). https://www.clinicaltrialsregister.eu (accessed October 1, 2016).
- GSK-LSD1: A chemical probe for LSD1. http://www.thesgc.org/chemical-probes/GSK-LSD1 (accessed August 26, 2016).
- ClinicalTrials.gov Identifier: NCT02034123. A Phase I Open-label, Dose Escalation Study to Investigate The Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK2879552 Given Orally in Subjects With Relapsed/Refractory Small Cell Lung Carcinoma (accessed November 2016).
- ClinicalTrials.gov Identifier: NCT02177812. A Phase I Open-label, Dose Escalation Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics and Clinical Activity of GSK2879552 Given Orally in Subjects With Relapsed/Refractory Acute Myeloid Leukemia (accessed October 2016).
- ClinicalTrials.gov Identifier: NCT02929498. A Phase I/II, Open-label, 2 Arm Study to Investigate the Safety, Clinical Activity, Pharmacokinetics and Pharmacodynamics of GSK2879552 Administered Alone or in Combination With Azacitidine, in Adult Subjects With IPSS-R High and Very High Risk Myelodysplastic Syndromes (MDS) Previously Treated With Hypomethylating Agents (HMA) (accessed November 2016).
- ClinicalTrials.gov Identifier: NCT02261779. Phase I/II Pilot Trial of ATRA (Tretinoin) and TCP (Tranylcypromine) in Patients With Relapsed or Refractory Acute Myeloid Leukemia (AML) and no Intensive Treatment is Possible (accessed July 2015).
- ClinicalTrials.gov Identifier: NCT02273102. An Open-Label, Dose Escalation, Phase 1 Study of Tranylcypromine (TCP) in Combination With ATRA (Tretinoin) for Adult Patients With Acute Myelogenous Leukemia (AML) and Myelodysplastic Syndromes (MDS) (accessed November 2016).
- ClinicalTrials.gov Identifier: NCT02717884. Phase I/II Study of Sensitization of Non-M3 Acute Myeloid Leukemia (AML) Blasts to All-trans Retinoic Acid (ATRA) by Epigenetic Treatment With Tranylcypromine (TCP), an Inhibitor of the Histone Lysine Demethylase 1 (LSD1) (accessed June 2016).
- Khan M. N. A.; Tsumoto H.; Itoh Y.; Ota Y.; Suzuki M.; Ogasawara D.; Nakagawa H.; Mizukami T.; Miyata N.; Suzuki T. Design, Synthesis, and Biological Activity of N-Alkylated Analogue of NCL1, A Selective Inhibitor of Lysine-Specific Demethylase 1. MedChemComm 2015, 6, 407–412. 10.1039/C4MD00330F. [DOI] [Google Scholar]
- Culhane J. C.; Wang D. Q.; Yen P. M.; Cole P. A. Comparative Analysis of Small Molecules and Histone Substrate Analogues as LSD1 Lysine Demethylase Inhibitors. J. Am. Chem. Soc. 2010, 132, 3164–3176. 10.1021/ja909996p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusevich P.; Kalin J. H.; Ming S. A.; Basso M.; Givens J.; Li X.; Hu J. F.; Taylor M. S.; Cieniewicz A. M.; Hsiao P. Y.; et al. A Selective Phenelzine Analogue Inhibitor of Histone Demethylase LSD1. ACS Chem. Biol. 2014, 9, 1284–1293. 10.1021/cb500018s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt A.; Sharman D. F.; Baker G. B.; Palcic M. M. A Continuous Spectrophotometric Assay for Monoamine Oxidase and Related Enzymes in Tissue Homogenates. Anal. Biochem. 1997, 244, 384–392. 10.1006/abio.1996.9911. [DOI] [PubMed] [Google Scholar]
- Schmitt M. L.; Hauser A. T.; Carlino L.; Pippel M.; Schulz-Fincke J.; Metzger E.; Willmann D.; Yiu T.; Barton M.; Schule R.; et al. Nonpeptidic Propargylamines as Inhibitors of Lysine Specific Demethylase 1 (LSD1) with Cellular Activity. J. Med. Chem. 2013, 56, 7334–7342. 10.1021/jm400792m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Levant B.; Jiang C.; Keck T. M.; Newman A. H.; Wang S. Tranylcypromine Substituted cis-Hydroxycyclobutylnaphthamides as Potent and Selective Dopamine D(3) Receptor Antagonists. J. Med. Chem. 2014, 57, 4962–4968. 10.1021/jm401798r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forneris F.; Binda C.; Adamo A.; Battaglioli E.; Mattevi A. Structural Basis of LSD1-CoREST Selectivity in Histone H3Recognition. J. Biol. Chem. 2007, 282, 20070–20074. 10.1074/jbc.C700100200. [DOI] [PubMed] [Google Scholar]
- Culhane J. C.; Szewczuk L. M.; Liu X.; Da G.; Marmorstein R.; Cole P. A. A Mechanism-Based Inactivator for Histone Demethylase LSD1. J. Am. Chem. Soc. 2006, 128, 4536–4537. 10.1021/ja0602748. [DOI] [PubMed] [Google Scholar]
- Szewczuk L. M.; Culhane J. C.; Yang M.; Majumdar A.; Yu H.; Cole P. A. Mechanistic Analysis of A Suicide Inactivator of Histone Demethylase LSD1. Biochemistry 2007, 46, 6892–6902. 10.1021/bi700414b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakizawa T.; Ota Y.; Itoh Y.; Tsumoto H.; Suzuki T. Histone H3 Peptide Based LSD1-Selective Inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 1925–1928. 10.1016/j.bmcl.2015.03.030. [DOI] [PubMed] [Google Scholar]
- Kumarasinghe I. R.; Woster P. M. Synthesis and Evaluation of Novel Cyclic Peptide Inhibitors of Lysine-Specific Demethylase 1. ACS Med. Chem. Lett. 2014, 5, 29–33. 10.1021/ml4002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M.; Borrello M. T.; Tardugno M.; Chiarelli L. R.; Pilotto S.; Ciossani G.; Vellore N. A.; Bailey S. G.; Cowan J.; O’Connell M.; et al. Protein Recognition by Short Peptide Reversible Inhibitors of the Chromatin-Modifying LSD1/CoREST Lysine Demethylase. ACS Chem. Biol. 2013, 8, 1677–1682. 10.1021/cb4001926. [DOI] [PubMed] [Google Scholar]
- Mould D. P.; McGonagle A. E.; Wiseman D. H.; Williams E. L.; Jordan A. M. Reversible Inhibitors of LSD1 as Therapeutic Agents in Acute Myeloid Leukemia: Clinical Significance and Progress to Date. Med. Res. Rev. 2015, 35, 586–618. 10.1002/med.21334. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Greene E.; Murray Stewart T.; Goodwin A. C.; Baylin S. B.; Woster P. M.; Casero R. A. Jr. Inhibition of Lysine-Specific Demethylase 1 by Polyamine Analogues Results in Reexpression of Aberrantly Silenced Genes. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 8023–8028. 10.1073/pnas.0700720104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Huang Y.; Marton L. J.; Woster P. M.; Davidson N. E.; Casero R. A. Jr. Polyamine Analogs Modulate Gene Expression by Inhibiting Lysine-Specific Demethylase 1 (LSD1) and Altering Chromatin Structure in Human Breast Cancer Cells. Amino Acids 2012, 42, 887–898. 10.1007/s00726-011-1004-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowotarski S. L.; Pachaiyappan B.; Holshouser S. L.; Kutz C. J.; Li Y.; Huang Y.; Sharma S. K.; Casero R. A. Jr.; Woster P. M. Structure-Activity Study for (Bis)Ureidopropyl- and (Bis)Thioureidopropyldiamine LSD1 Inhibitors with 3–5-3 and 3–6-3 Carbon Backbone Architectures. Bioorg. Med. Chem. 2015, 23, 1601–1612. 10.1016/j.bmc.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. K.; Wu Y.; Steinbergs N.; Crowley M. L.; Hanson A. S.; Casero R. A.; Woster P. M. Bis)Urea and (Bis)Thiourea Inhibitors of Lysine-Specific Demethylase 1 as Epigenetic Modulators. J. Med. Chem. 2010, 53, 5197–5212. 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazeldine S.; Pachaiyappan B.; Steinbergs N.; Nowotarski S.; Hanson A. S.; Casero R. A. Jr.; Woster P. M. Low Molecular Weight Amidoximes that Act as Potent Inhibitors of Lysine-Specific Demethylase 1. J. Med. Chem. 2012, 55, 7378–7391. 10.1021/jm3002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorna V.; Theisen E. R.; Stephens B.; Warner S. L.; Bearss D. J.; Vankayalapati H.; Sharma S. High-Throughput Virtual Screening Identifies Novel N’-(1-Phenylethylidene)-Benzohydrazides as Potent, Specific, and Reversible LSD1 Inhibitors. J. Med. Chem. 2013, 56, 9496–9508. 10.1021/jm400870h. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Simeonov A.; Jadhav A.; Babaoglu K.; Inglese J.; Shoichet B. K.; Austin C. P. A High-Throughput Screen for Aggregation-Based Inhibition in A Large Compound Library. J. Med. Chem. 2007, 50, 2385–2390. 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Caselli E.; Grigorieff N.; Shoichet B. K. A Common Mechanism Underlying Promiscuous Inhibitors from Virtual and High-Throughput Screening. J. Med. Chem. 2002, 45, 1712–1722. 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Li Y.; Wang W. J.; Xiang P.; Luo X. M.; Yang L.; Yang S. Y.; Zhao Y. L. Synthesis and Biological Evaluation of Novel (E)-N’-(2,3-Dihydro-1H-Inden-1-Ylidene) Benzohydrazides as Potent LSD1 Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4552–4557. 10.1016/j.bmcl.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Wu F.; Zhou C.; Yao Y.; Wei L.; Feng Z.; Deng L.; Song Y. 3-(Piperidin-4-ylmethoxy)pyridine Containing Compounds are Potent Inhibitors of Lysine Specific Demethylase 1. J. Med. Chem. 2016, 59, 253–263. 10.1021/acs.jmedchem.5b01361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitchin J. R.; Blagg J.; Burke R.; Burns S.; Cockerill M. J.; Fairweather E. E.; Hutton C.; Jordan A. M.; McAndrew C.; Mirza A.; et al. Development and Evaluation of Selective, Reversible LSD1 Inhibitors Derived from Fragments. MedChemComm 2013, 4, 1513–1522. 10.1039/c3md00226h. [DOI] [Google Scholar]
- Zheng Y. C.; Duan Y. C.; Ma J. L.; Xu R. M.; Zi X.; Lv W. L.; Wang M. M.; Ye X. W.; Zhu S.; Mobley D.; et al. Triazole-Dithiocarbamate Based Selective Lysine Specific Demethylase 1 (LSD1) Inactivators Inhibit Gastric Cancer Cell Growth, Invasion, and Migration. J. Med. Chem. 2013, 56, 8543–8560. 10.1021/jm401002r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y. C.; Ma Y. C.; Zhang E.; Shi X. J.; Wang M. M.; Ye X. W.; Liu H. M. Design and Synthesis of Novel 1,2,3-Triazole-Dithiocarbamate Hybrids as Potential Anticancer Agents. Eur. J. Med. Chem. 2013, 62, 11–19. 10.1016/j.ejmech.2012.12.046. [DOI] [PubMed] [Google Scholar]
- Ye X. W.; Zheng Y. C.; Duan Y. C.; Wang M. M.; Yu B.; Ren J. L.; Ma J. L.; Zhang E.; Liu H. M. Synthesis and Biological Evaluation of Coumarin-1,2,3-Triazole-Dithiocarbamate Hybrids as Potent LSD1 Inhibitors. MedChemComm 2014, 5, 650–654. 10.1039/c4md00031e. [DOI] [Google Scholar]
- Ma L. Y.; Zheng Y. C.; Wang S. Q.; Wang B.; Wang Z. R.; Pang L. P.; Zhang M.; Wang J. W.; Ding L.; Li J.; et al. Design, Synthesis, and Structure-Activity Relationship of Novel LSD1 Inhibitors Based on Pyrimidine-Thiourea Hybrids as Potent, Orally Active Antitumor Agents. J. Med. Chem. 2015, 58, 1705–1716. 10.1021/acs.jmedchem.5b00037. [DOI] [PubMed] [Google Scholar]
- Kutz C. J.; Holshouser S. L.; Marrow E. A.; Woster P. M. 3,5-Diamino-1,2,4-Triazoles as A Novel Scaffold for Potent, Reversible LSD1 (KDM1A) Inhibitors. MedChemComm 2014, 5, 1863–1870. 10.1039/C4MD00283K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C.; Kang D.; Xu Y.; Zhang L.; Zha X. Identification of Novel Selective Lysine-Specific Demethylase 1 (LSD1) Inhibitors Using a Pharmacophore-Based Virtual Screening Combined with Docking. Chem. Biol. Drug Des. 2015, 85, 659–671. 10.1111/cbdd.12461. [DOI] [PubMed] [Google Scholar]
- Dulla B.; Kirla K. T.; Rathore V.; Deora G. S.; Kavela S.; Maddika S.; Chatti K.; Reiser O.; Iqbal J.; Pal M. Synthesis and Evaluation of 3-Amino/Guanidine Substituted Phenyl Oxazoles as A Novel Class of LSD1 Inhibitors with Anti-Proliferative Properties. Org. Biomol. Chem. 2013, 11, 3103–3107. 10.1039/c3ob40217g. [DOI] [PubMed] [Google Scholar]
- Wang J.; Lu F.; Ren Q.; Sun H.; Xu Z. S.; Lan R. F.; Liu Y. Q.; Ward D.; Quan J. M.; Ye T.; et al. Novel Histone Demethylase LSD1 Inhibitors Selectively Target Cancer Cells with Pluripotent Stem Cell Properties. Cancer Res. 2011, 71, 7238–7249. 10.1158/0008-5472.CAN-11-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann D.; Lim S.; Wetzel S.; Metzger E.; Jandausch A.; Wilk W.; Jung M.; Forne I.; Imhof A.; Janzer A.; et al. Impairment of Prostate Cancer Cell Growth by A Selective and Reversible Lysine-Specific Demethylase 1 Inhibitor. Int. J. Cancer 2012, 131, 2704–2709. 10.1002/ijc.27555. [DOI] [PubMed] [Google Scholar]
- Takeuchi T.; Yamazaki Y.; Katoh-Fukui Y.; Tsuchiya R.; Kondo S.; Motoyama J.; Higashinakagawa T. Gene Trap Capture of A Novel Mouse Gene, Jumonji, Required for Neural Tube Formation. Genes Dev. 1995, 9, 1211–1222. 10.1101/gad.9.10.1211. [DOI] [PubMed] [Google Scholar]
- Klose R. J.; Yamane K.; Bae Y.; Zhang D.; Erdjument-Bromage H.; Tempst P.; Wong J.; Zhang Y. The Transcriptional Repressor JHDM3A Demethylates Trimethyl Histone H3 Lysine 9 and Lysine 36. Nature 2006, 442, 312–316. 10.1038/nature04853. [DOI] [PubMed] [Google Scholar]
- Cloos P. A.; Christensen J.; Agger K.; Maiolica A.; Rappsilber J.; Antal T.; Hansen K. H.; Helin K. The Putative Oncogene GASC1 Demethylates tri- and dimethylated Lysine 9 on Histone H3. Nature 2006, 442, 307–311. 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]
- Whetstine J. R.; Nottke A.; Lan F.; Huarte M.; Smolikov S.; Chen Z.; Spooner E.; Li E.; Zhang G.; Colaiacovo M.; et al. Reversal of Histone Lysine Trimethylation by the JMJD2 Family of Histone Demethylases. Cell 2006, 125, 467–481. 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Fodor B. D.; Kubicek S.; Yonezawa M.; O’Sullivan R. J.; Sengupta R.; Perez-Burgos L.; Opravil S.; Mechtler K.; Schotta G.; Jenuwein T. JMJD2B Antagonizes H3K9 trimethylation at Pericentric Heterochromatin in Mammalian Cells. Genes Dev. 2006, 20, 1557–1562. 10.1101/gad.388206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K.; Toumazou C.; Tsukada Y.; Erdjument-Bromage H.; Tempst P.; Wong J.; Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell 2006, 125, 483–495. 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Klose R. J.; Kallin E. M.; Zhang Y. JmjC-Domain-Containing Proteins and Histone Demethylation. Nat. Rev. Genet. 2006, 7, 715–727. 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]
- Rose N. R.; McDonough M. A.; King O. N. F.; Kawamura A.; Schofield C. J. Inhibition of 2-Oxoglutarate Dependent Oxygenases. Chem. Soc. Rev. 2011, 40, 4364–4397. 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- Rose N. R.; Woon E. C.; Tumber A.; Walport L. J.; Chowdhury R.; Li X. S.; King O. N.; Lejeune C.; Ng S. S.; Krojer T.; et al. Plant Growth Regulator Daminozide is A Selective Inhibitor of Human KDM2/7 Histone Demethylases. J. Med. Chem. 2012, 55, 6639–6643. 10.1021/jm300677j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; Upadhyay A. K.; Qi H. H.; Zhang X.; Shi Y.; Cheng X. Enzymatic and Structural Insights for Substrate Specificity of A Family of Jumonji Histone Lysine Demethylases. Nat. Struct. Mol. Biol. 2010, 17, 38–43. 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Fang J.; Bedford M. T.; Zhang Y.; Xu R. M. Recognition of Histone H3 lysine-4 Methylation by the Double Tudor Domain of JMJD2A. Science 2006, 312, 748–751. 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- Lee J.; Thompson J. R.; Botuyan M. V.; Mer G. Distinct Binding Modes Specify the Recognition of Methylated Histones H3K4 and H4K20 by JMJD2A-Tudor. Nat. Struct. Mol. Biol. 2008, 15, 109–111. 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Chang Q.; Zhang Y.; Beezhold K.; Rojanasakul Y.; Zhao H.; Castranova V.; Shi X.; Chen F. Lung Cancer-Associated JmjC Domain Protein Mdig Suppresses Formation of Tri-Methyl Lysine 9 Of Histone H3. Cell Cycle 2009, 8, 2101–2109. 10.4161/cc.8.13.8927. [DOI] [PubMed] [Google Scholar]
- Sinha K. M.; Yasuda H.; Coombes M. M.; Dent S. Y.; de Crombrugghe B. Regulation of theOsteoblast-Specific Transcription Factor Osterix by NO66, A Jumonji Family Histone Demethylase. EMBO J. 2010, 29, 68–79. 10.1038/emboj.2009.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Yu M.; Chang Q.; Lu Y.; Thakur C.; Ma D.; Yi Z.; Chen F. Mdig De-represses H19 Large Intergenic Non-Coding RNA (lincRNA) by Down-Regulating H3K9me3 and Heterochromatin. Oncotarget 2013, 4, 1427–1437. 10.18632/oncotarget.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge W.; Wolf A.; Feng T.; Ho C. H.; Sekirnik R.; Zayer A.; Granatino N.; Cockman M. E.; Loenarz C.; Loik N. D.; et al. Oxygenase-Catalyzed Ribosome Hydroxylation Occurs in Prokaryotes and Humans. Nat. Chem. Biol. 2012, 8, 960–962. 10.1038/nchembio.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Sekirnik R.; Brissett N. C.; Krojer T.; Ho C. H.; Ng S. S.; Clifton I. J.; Ge W.; Kershaw N. J.; Fox G. C.; et al. Ribosomal Oxygenases are Structurally Conserved from Prokaryotes to Humans. Nature 2014, 510, 422–426. 10.1038/nature13263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. T.; Walport L. J.; Hopkinson R. J.; Madden S. K.; Chowdhury R.; Schofield C. J.; Kawamura A. Studies on the Catalytic Domains of Multiple JmjC Oxygenases Using Peptide Substrates. Epigenetics 2014, 9, 1596–1603. 10.4161/15592294.2014.983381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B.; Chen Y.; Zhao Y.; Bruick R. K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. 10.1126/science.1145801. [DOI] [PubMed] [Google Scholar]
- Bottger A.; Islam M. S.; Chowdhury R.; Schofield C. J.; Wolf A. The Oxygenase JmjD6--A Case Study in Conflicting Assignments. Biochem. J. 2015, 468, 191–202. 10.1042/BJ20150278. [DOI] [PubMed] [Google Scholar]
- Webby C. J.; Wolf A.; Gromak N.; Dreger M.; Kramer H.; Kessler B.; Nielsen M. L.; Schmitz C.; Butler D. S.; Yates J. R. 3rd; et al. JmjD6 Catalyses Lysyl-Hydroxylation of U2AF65, A Protein Associated with RNA Splicing. Science 2009, 325, 90–93. 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- Walport L. J.; Hopkinson R. J.; Chowdhury R.; Schiller R.; Ge W.; Kawamura A.; Schofield C. J. Arginine Demethylation is Catalysed by A Subset of JmjC Histone Lysine Demethylases. Nat. Commun. 2016, 7, 11974. 10.1038/ncomms11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.; Cho Y. W.; Yu L. R.; Yu H.; Veenstra T. D.; Ge K. Identification of JmjC Domain-Containing UTX and JMJD3 as Histone H3 Lysine 27 Demethylases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18439–18444. 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walport L. J.; Hopkinson R. J.; Vollmar M.; Madden S. K.; Gileadi C.; Oppermann U.; Schofield C. J.; Johansson C. Human UTY(KDM6C) is A Male-Specific N-Methyl Lysyl Demethylase. J. Biol. Chem. 2014, 289, 18302–18313. 10.1074/jbc.M114.555052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H.; Li J.; Song T.; Lu M.; Kan P. Y.; Lee M. G.; Sha B.; Shi X. Recognition of Histone H3K4 Trimethylation by the Plant Homeodomain of PHF2Modulates Histone Demethylation. J. Biol. Chem. 2010, 285, 9322–9326. 10.1074/jbc.C109.097667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Kim H.; Casta A.; Kobayashi Y.; Shapiro L. S.; Christiano A. M. Hairless is A Histone H3K9 Demethylase. FASEB J. 2014, 28, 1534–1542. 10.1096/fj.13-237677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia D. A.; Tepper C. G.; Pochampalli M. R.; Hsia E. Y.; Izumiya C.; Huerta S. B.; Wright M. E.; Chen H. W.; Kung H. J.; Izumiya Y. KDM8, a H3K36me2 Histone Demethylase that Acts in the Cyclin A1 Coding Region to Regulate Cancer Cell Proliferation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 9671–9676. 10.1073/pnas.1000401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn M. Y.; Yokoyama A.; Fujiyama-Nakamura S.; Ohtake F.; Minehata K.; Yasuda H.; Suzuki T.; Kato S.; Imai Y. JMJD5, a Jumonji C (JmjC) Domain-Containing Protein, Negatively Regulates Osteoclastogenesis by Facilitating NFATc1 Protein Degradation. J. Biol. Chem. 2012, 287, 12994–13004. 10.1074/jbc.M111.323105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rizzo P. A.; Krishnan S.; Trievel R. C. Crystal Structure and Functional Analysis of JMJD5 Indicate An Alternate Specificity and Function. Mol. Cell. Biol. 2012, 32, 4044–4052. 10.1128/MCB.00513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. R.; Upadhyay A. K.; Hashimoto H.; Zhang X.; Cheng X. Structural Basis for Human PHF2 Jumonji Domain Interaction with Metal Ions. J. Mol. Biol. 2011, 406, 1–8. 10.1016/j.jmb.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton I. J.; McDonough M. A.; Ehrismann D.; Kershaw N. J.; Granatino N.; Schofield C. J. Structural Studies on 2-Oxoglutarate Oxygenases and Related Double-Stranded beta-Helix Fold Proteins. J. Inorg. Biochem. 2006, 100, 644–669. 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- Hegg E. L.; Que L. Jr. The 2-His-1-Carboxylate Facial Triad--An Emerging Structural Motif in Mononuclear Non-heme Iron(II) Enzymes. Eur. J. Biochem. 1997, 250, 625–629. 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- Hausinger R. P. FeII/Alpha-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 21–68. 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Ren J.; Stammers D. K.; Baldwin J. E.; Harlos K.; Schofield C. J. Structural Origins of the Selectivity of the Trifunctional Oxygenase Clavaminic Acid Synthase. Nat. Struct. Biol. 2000, 7, 127–133. 10.1038/72398. [DOI] [PubMed] [Google Scholar]
- Martinez S.; Hausinger R. P. Catalytic Mechanisms of Fe(II)-and 2-Oxoglutarate-Dependent Oxygenases. J. Biol. Chem. 2015, 290, 20702–20711. 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissmann M.; Yin N.; Muller J. M.; Greschik H.; Fodor B. D.; Jenuwein T.; Vogler C.; Schneider R.; Gunther T.; Buettner R.; et al. Cooperative Demethylation by JMJD2C and LSD1 Promotes Androgen Receptor-Dependent Gene Expression. Nat. Cell Biol. 2007, 9, 347–353. 10.1038/ncb1546. [DOI] [PubMed] [Google Scholar]
- Liu G.; Bollig-Fischer A.; Kreike B.; van de Vijver M. J.; Abrams J.; Ethier S. P.; Yang Z. Q. Genomic Amplification and Oncogenic Properties of the GASC1 Histone Demethylase Gene in Breast Cancer. Oncogene 2009, 28, 4491–4500. 10.1038/onc.2009.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman E. C.; Robinson B. D.; Downes M. J.; Powell L. G.; Lee M. M.; Scherr D. S.; Gudas L. J.; Mongan N. P. Role of Androgen Receptor and Associated Lysine-Demethylase Coregulators, LSD1 and JMJD2A, in Localized and Advanced Human Bladder Cancer. Mol. Carcinog. 2011, 50, 931–944. 10.1002/mc.20758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L.; Emre N. C.; Kruhlak M. J.; Chung H. J.; Steidl C.; Slack G.; Wright G. W.; Lenz G.; Ngo V. N.; Shaffer A. L.; et al. Cooperative Epigenetic Modulation by Cancer Amplicon Genes. Cancer Cell 2010, 18, 590–605. 10.1016/j.ccr.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M.; Mei P.; Fang R.; Leonor T.; Rutenberg M.; Shimizu F.; Li J.; Rao A.; Shi Y. The Histone H3K4 Demethylase SMCX Links REST Target Genes to X-Linked Mental Retardation. Nature 2007, 447, 601–605. 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Laumonnier F.; Holbert S.; Ronce N.; Faravelli F.; Lenzner S.; Schwartz C. E.; Lespinasse J.; Van Esch H.; Lacombe D.; Goizet C.; et al. Mutations in PHF8 are Associated with X Linked Mental Retardation And Cleft Lip/Cleft Palate. J. Med. Genet. 2005, 42, 780–786. 10.1136/jmg.2004.029439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Ledaki I.; Turley H.; Gatter K. C.; Montero J. C.; Li J. L.; Harris A. L. Role of Hypoxia-Inducible Factors in Epigenetic Regulation via Histone Demethylases. Ann. N. Y. Acad. Sci. 2009, 1177, 185–197. 10.1111/j.1749-6632.2009.05027.x. [DOI] [PubMed] [Google Scholar]
- Beyer S.; Kristensen M. M.; Jensen K. S.; Johansen J. V.; Staller P. The Histone Demethylases JMJD1A and JMJD2B are Transcriptional Targets of Hypoxia-Inducible Factor HIF. J. Biol. Chem. 2008, 283, 36542–36552. 10.1074/jbc.M804578200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg A. J.; Rankin E. B.; Chan D.; Razorenova O.; Fernandez S.; Giaccia A. J. Regulation of the Histone Demethylase JMJD1A by Hypoxia-Inducible Factor 1 Alpha Enhances Hypoxic Gene Expression and Tumor Growth. Mol. Cell. Biol. 2010, 30, 344–353. 10.1128/MCB.00444-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J.; Trojer P. Targeting Histone Lysine Methylation in Cancer. Pharmacol. Ther. 2015, 150, 1–22. 10.1016/j.pharmthera.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Johansson C.; Tumber A.; Che K.; Cain P.; Nowak R.; Gileadi C.; Oppermann U. The Roles of Jumonji-Type Oxygenases in Human Disease. Epigenomics 2014, 6, 89–120. 10.2217/epi.13.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose N. R.; Ng S. S.; Mecinovic J.; Lienard B. M.; Bello S. H.; Sun Z.; McDonough M. A.; Oppermann U.; Schofield C. J. Inhibitor Scaffolds for 2-Oxoglutarate-Dependent Histone Lysine Demethylases. J. Med. Chem. 2008, 51, 7053–7056. 10.1021/jm800936s. [DOI] [PubMed] [Google Scholar]
- King O. N.; Li X. S.; Sakurai M.; Kawamura A.; Rose N. R.; Ng S. S.; Quinn A. M.; Rai G.; Mott B. T.; Beswick P.; et al. Quantitative High-Throughput Screening Identifies 8-Hydroxyquinolines as Cell-Active Histone Demethylase Inhibitors. PLoS One 2010, 5, e15535. 10.1371/journal.pone.0015535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkinson R. J.; Tumber A.; Yapp C.; Chowdhury R.; Aik W.; Che K. H.; Li X. S.; Kristensen J. B.; King O. N.; Chan M. C.; et al. 5-Carboxy-8-hydroxyquinoline is A Broad Spectrum 2-Oxoglutarate Oxygenase Inhibitor which Causes Iron Translocation. Chem. Sci. 2013, 4, 3110–3117. 10.1039/c3sc51122g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller R.; Scozzafava G.; Tumber A.; Wickens J. R.; Bush J. T.; Rai G.; Lejeune C.; Choi H.; Yeh T. L.; Chan M. C.; et al. A Cell-Permeable Ester Derivative of the JmjC Histone Demethylase Inhibitor IOX1. ChemMedChem 2014, 9, 566–571. 10.1002/cmdc.201300428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S. S.; Kavanagh K. L.; McDonough M. A.; Butler D.; Pilka E. S.; Lienard B. M.; Bray J. E.; Savitsky P.; Gileadi O.; von Delft F.; et al. Crystal Structures of Histone Demethylase JMJD2A Reveal Basis for Substrate Specificity. Nature 2007, 448, 87–91. 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]
- Hamada S.; Suzuki T.; Mino K.; Koseki K.; Oehme F.; Flamme I.; Ozasa H.; Itoh Y.; Ogasawara D.; Komaarashi H.; et al. Design, Synthesis, Enzyme-Inhibitory Activity, and Effect on Human Cancer Cells of A Novel Series of Jumonji Domain-Containing Protein 2 Histone Demethylase Inhibitors. J. Med. Chem. 2010, 53, 5629–5638. 10.1021/jm1003655. [DOI] [PubMed] [Google Scholar]
- Luo X.; Liu Y.; Kubicek S.; Myllyharju J.; Tumber A.; Ng S.; Che K. H.; Podoll J.; Heightman T. D.; Oppermann U.; et al. A Selective Inhibitor and Probe of the Cellular Functions of Jumonji C Domain-Containing Histone Demethylases. J. Am. Chem. Soc. 2011, 133, 9451–9456. 10.1021/ja201597b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Podoll J. D.; Dong X.; Tumber A.; Oppermann U.; Wang X. Quantitative Analysis of Histone Demethylase Probes Using Fluorescence Polarization. J. Med. Chem. 2013, 56, 5198–5202. 10.1021/jm3018628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marholz L. J.; Chang L.; Old W. M.; Wang X. Development of Substrate-Selective Probes for Affinity Pulldown of Histone Demethylases. ACS Chem. Biol. 2015, 10, 129–137. 10.1021/cb5006867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easmon J.; Heinisch G.; Purstinger G.; Langer T.; Osterreicher J. K.; Grunicke H. H.; Hofmann J. Azinyl and Diazinyl Hydrazones Derived from Aryl N-Heteroaryl Ketones: Synthesis and Antiproliferative Activity. J. Med. Chem. 1997, 40, 4420–4425. 10.1021/jm970255w. [DOI] [PubMed] [Google Scholar]
- Wang L.; Chang J.; Varghese D.; Dellinger M.; Kumar S.; Best A. M.; Ruiz J.; Bruick R.; Pena-Llopis S.; Xu J.; et al. A Small Molecule Modulates Jumonji Histone Demethylase Activity And Selectively Inhibits Cancer Growth. Nat. Commun. 2013, 4, 2035. 10.1038/ncomms3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzatsos A.; Pfau R.; Kampranis S. C.; Tsichlis P. N. Ndy1/KDM2B Immortalizes Mouse Embryonic Fibroblasts by Repressing the Ink4a/Arf Locus. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 2641–2646. 10.1073/pnas.0813139106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Kallin E. M.; Tsukada Y.; Zhang Y. The H3K36 Demethylase Jhdm1b/Kdm2b Regulates Cell Proliferation and Senescence through p15(Ink4b). Nat. Struct. Mol. Biol. 2008, 15, 1169–1175. 10.1038/nsmb.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frescas D.; Guardavaccaro D.; Bassermann F.; Koyama-Nasu R.; Pagano M. JHDM1B/FBXL10 is A Nucleolar Protein That Represses Transcription of Ribosomal RNA Genes. Nature 2007, 450, 309–313. 10.1038/nature06255. [DOI] [PubMed] [Google Scholar]
- Janzer A.; Stamm K.; Becker A.; Zimmer A.; Buettner R.; Kirfel J. The H3K4me3 Histone Demethylase Fbxl10 is A Regulator of Chemokine Expression, Cellular Morphology, and the Metabolome of Fibroblasts. J. Biol. Chem. 2012, 287, 30984–30992. 10.1074/jbc.M112.341040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar S. S.; Alam H.; Li N.; Wagner K. W.; Chung J.; Ahn Y. W.; Lee M. G. Transcriptional Repression of Histone Deacetylase 3 By The Histone Demethylase KDM2A is Coupled to Tumorigenicity of Lung Cancer Cells. J. Biol. Chem. 2014, 289, 7483–7496. 10.1074/jbc.M113.521625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T.; Ozasa H.; Itoh Y.; Zhan P.; Sawada H.; Mino K.; Walport L.; Ohkubo R.; Kawamura A.; Yonezawa M.; et al. Identification of the KDM2/7 Histone Lysine Demethylase Subfamily Inhibitor and its Antiproliferative Activity. J. Med. Chem. 2013, 56, 7222–7231. 10.1021/jm400624b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J.; Ma Y.; Ma P.; Wang S.; Fan Z. Demethylation of Epiregulin Gene by Histone Demethylase FBXL11 and BCL6 Corepressor Inhibits Osteo/Dentinogenic Differentiation. Stem Cells 2013, 31, 126–136. 10.1002/stem.1255. [DOI] [PubMed] [Google Scholar]
- Gao R.; Dong R.; Du J.; Ma P.; Wang S.; Fan Z. Depletion of Histone Demethylase KDM2A Inhibited Cell Proliferation of Stem Cells from Apical Papilla by De-repression of p15INK4B and p27Kip1. Mol. Cell. Biochem. 2013, 379, 115–122. 10.1007/s11010-013-1633-7. [DOI] [PubMed] [Google Scholar]
- Lu T.; Jackson M. W.; Wang B.; Yang M.; Chance M. R.; Miyagi M.; Gudkov A. V.; Stark G. R. Regulation of NF-kappaB by NSD1/FBXL11-Dependent Reversible Lysine Methylation of p65. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 46–51. 10.1073/pnas.0912493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner K. W.; Alam H.; Dhar S. S.; Giri U.; Li N.; Wei Y.; Giri D.; Cascone T.; Kim J. H.; Ye Y.; et al. KDM2A Promotes Lung Tumorigenesis by Epigenetically Enhancing ERK1/2 Signaling. J. Clin. Invest. 2013, 123, 5231–5246. 10.1172/JCI68642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Liu Y.; Yu L.; Chen J.; Hou J.; Cui L.; Ma D.; Lu W. Histone Demethylase KDM2A Promotes Tumor Cell Growth and Migration in Gastric Cancer. Tumor Biol. 2015, 36, 271–278. 10.1007/s13277-014-2630-5. [DOI] [PubMed] [Google Scholar]
- He J.; Nguyen A. T.; Zhang Y. KDM2b/JHDM1b, An H3K36me2-Specific Demethylase, is Required for Initiation and Maintenance of Acute Myeloid Leukemia. Blood 2011, 117, 3869–3880. 10.1182/blood-2010-10-312736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzatsos A.; Paskaleva P.; Ferrari F.; Deshpande V.; Stoykova S.; Contino G.; Wong K. K.; Lan F.; Trojer P.; Park P. J.; et al. KDM2B Promotes Pancreatic Cancer via Polycomb-Dependent and -Independent Transcriptional Programs. J. Clin. Invest. 2013, 123, 727–739. 10.1172/JCI64535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottakis F.; Foltopoulou P.; Sanidas I.; Keller P.; Wronski A.; Dake B. T.; Ezell S. A.; Shen Z.; Naber S. P.; Hinds P. W.; et al. NDY1/KDM2B Functions as A Master Regulator of Polycomb Complexes and Controls Self-Renewal of Breast Cancer Stem Cells. Cancer Res. 2014, 74, 3935–3946. 10.1158/0008-5472.CAN-13-2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L.; Wang Y.; Huang S.; Wang J.; Deng Z.; Zhang Q.; Wu W.; Zhang X.; Liu Z.; Gong W.; et al. Structural Insights into A Novel Histone Demethylase PHF8. Cell Res. 2010, 20, 166–173. 10.1038/cr.2010.8. [DOI] [PubMed] [Google Scholar]
- Tsukada Y.; Ishitani T.; Nakayama K. I. KDM7 is A Dual Demethylase for Histone H3 Lys 9 and Lys 27 and Functions in Brain Development. Genes Dev. 2010, 24, 432–437. 10.1101/gad.1864410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Tanasa B.; Tyurina O. V.; Zhou T. Y.; Gassmann R.; Liu W. T.; Ohgi K. A.; Benner C.; Garcia-Bassets I.; Aggarwal A. K.; et al. PHF8Mediates Histone H4 Lysine 20 Demethylation Events Involved in Cell Cycle Progression. Nature 2010, 466, 508–512. 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Hu L.; Wang P.; Hou H.; Lin Y.; Liu Y.; Li Z.; Gong R.; Feng X.; Zhou L.; et al. Structural Insights into A Dual-Specificity Histone Demethylase ceKDM7A from Caenorhabditis Elegans. Cell Res. 2010, 20, 886–898. 10.1038/cr.2010.86. [DOI] [PubMed] [Google Scholar]
- Lin H.; Wang Y.; Wang Y.; Tian F.; Pu P.; Yu Y.; Mao H.; Yang Y.; Wang P.; Hu L.; et al. Coordinated Regulation of Active and Repressive Histone Methylations by A Dual-Specificity Histone Demethylase ceKDM7A from Caenorhabditis Elegans. Cell Res. 2010, 20, 899–907. 10.1038/cr.2010.84. [DOI] [PubMed] [Google Scholar]
- Abidi F.; Miano M.; Murray J.; Schwartz C. A Novel Mutation in the PHF8 Gene is Associated with X-Linked Mental Retardation with Cleft Lip/Cleft Palate. Clin. Genet. 2007, 72, 19–22. 10.1111/j.1399-0004.2007.00817.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Qiu J. J.; Zhu S.; Cao B.; Sun L.; Li S.; Li P.; Zhang S.; Dong S. Oncogenic Features of PHF8 Histone Demethylase in Esophageal Squamous Cell Carcinoma. PLoS One 2013, 8, e77353. 10.1371/journal.pone.0077353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Pan X.; Zhao H. The Histone Demethylase PHF8 is An Oncogenic Protein in Human Non-Small Cell Lung Cancer. Biochem. Biophys. Res. Commun. 2014, 451, 119–125. 10.1016/j.bbrc.2014.07.076. [DOI] [PubMed] [Google Scholar]
- Lee K. H.; Park J. W.; Sung H. S.; Choi Y. J.; Kim W. H.; Lee H. S.; Chung H. J.; Shin H. W.; Cho C. H.; Kim T. Y.; et al. PHF2 Histone Demethylase Acts as A Tumor Suppressor in Association with p53 in Cancer. Oncogene 2015, 34, 2897–2909. 10.1038/onc.2014.219. [DOI] [PubMed] [Google Scholar]
- England K. S.; Tumber A.; Krojer T.; Scozzafava G.; Ng S. S.; Daniel M.; Szykowska A.; Che K.; von Delft F.; Burgess-Brown N. A.; et al. Optimisation of A Triazolopyridine Based Histone Demethylase Inhibitor Yields A Potent and Selective KDM2A (FBXL11) Inhibitor. MedChemComm 2014, 5, 1879–1886. 10.1039/C4MD00291A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack A. P.; Bussemer S.; Hahn M.; Punzeler S.; Snyder M.; Wells M.; Csankovszki G.; Solovei I.; Schotta G.; Hake S. B. H3K56me3 is A Novel, Conserved Heterochromatic Mark that Largely but not Completely Overlaps with H3K9me3 in both Regulation and Localization. PLoS One 2013, 8, e51765. 10.1371/journal.pone.0051765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojer P.; Zhang J.; Yonezawa M.; Schmidt A.; Zheng H.; Jenuwein T.; Reinberg D. Dynamic Histone H1 Isotype 4 Methylation and Demethylation by Histone Lysine Methyltransferase G9a/KMT1C and the Jumonji Domain-Containing JMJD2/KDM4 Proteins. J. Biol. Chem. 2009, 284, 8395–8405. 10.1074/jbc.M807818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.; Alumkal J. Epigenetic Regulation of Androgen Receptor Signaling in Prostate Cancer. Epigenetics 2010, 5, 100–104. 10.4161/epi.5.2.10778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S.; Janknecht R. Activation of Androgen Receptor by Histone Demethylases JMJD2A and JMJD2D. Biochem. Biophys. Res. Commun. 2007, 359, 742–746. 10.1016/j.bbrc.2007.05.179. [DOI] [PubMed] [Google Scholar]
- Coffey K.; Rogerson L.; Ryan-Munden C.; Alkharaif D.; Stockley J.; Heer R.; Sahadevan K.; O’Neill D.; Jones D.; Darby S.; et al. The Lysine Demethylase, KDM4B, is A Key Molecule in Androgen Receptor Signalling and Turnover. Nucleic Acids Res. 2013, 41, 4433–4446. 10.1093/nar/gkt106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J. C.; Manning A. L.; Van Rechem C.; Kim J.; Ladd B.; Cho J.; Pineda C. M.; Murphy N.; Daniels D. L.; Montagna C.; et al. KDM4A Lysine Demethylase Induces Site-Specific Copy Gain and Rereplication of Regions Amplified in Tumors. Cell 2013, 154, 541–555. 10.1016/j.cell.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry W. L.; Shin S.; Lightfoot S. A.; Janknecht R. Oncogenic Features of the JMJD2A Histone Demethylase in Breast Cancer. Int. J. Oncol. 2012, 41, 1701–1706. 10.3892/ijo.2012.1618. [DOI] [PubMed] [Google Scholar]
- Kim T. D.; Shin S.; Berry W. L.; Oh S.; Janknecht R. The JMJD2A Demethylase Regulates Apoptosis and Proliferation in Colon Cancer Cells. J. Cell. Biochem. 2012, 113, 1368–1376. 10.1002/jcb.24009. [DOI] [PubMed] [Google Scholar]
- Yang J.; Jubb A. M.; Pike L.; Buffa F. M.; Turley H.; Baban D.; Leek R.; Gatter K. C.; Ragoussis J.; Harris A. L. The Histone Demethylase JMJD2B is Regulated by Estrogen Receptor Alpha and Hypoxia, and is A Key Mediator of Estrogen Induced Growth. Cancer Res. 2010, 70, 6456–6466. 10.1158/0008-5472.CAN-10-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Q.; Imoto I.; Fukuda Y.; Pimkhaokham A.; Shimada Y.; Imamura M.; Sugano S.; Nakamura Y.; Inazawa J. Identification of A Novel Gene, GASC1, within An Amplicon at 9p23–24 Frequently Detected in Esophageal Cancer Cell Lines. Cancer Res. 2000, 60, 4735–4739. [PubMed] [Google Scholar]
- Helias C.; Struski S.; Gervais C.; Leymarie V.; Mauvieux L.; Herbrecht R.; Lessard M. Polycythemia Vera Transforming to Acute Myeloid Leukemia and Complex Abnormalities Including 9p Homogeneously Staining Region with Amplification of MLLT3, JMJD2C, JAK2, and SMARCA2. Cancer Genet. Cytogenet. 2008, 180, 51–55. 10.1016/j.cancergencyto.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; van Essen D.; Saccani S. Cell-Type-Specific Control of Enhancer Activity by H3K9 Trimethylation. Mol. Cell 2012, 46, 408–423. 10.1016/j.molcel.2012.05.011. [DOI] [PubMed] [Google Scholar]
- Rai G.; Kawamura A.; Tumber A.; Liang Y.; Vogel J. L.; Arbuckle J. H.; Rose N. R.; Dexheimer T. S.; Foley T. L.; King O. N., et al. Discovery of ML324, A JMJD2 Demethylase Inhibitor with Demonstrated Antiviral Activity. In Probe Reports from the NIH Molecular Libraries Program; National Institute of Health: Bethesda, MD, 2010. [PubMed] [Google Scholar]
- Duan L.; Rai G.; Roggero C.; Zhang Q. J.; Wei Q.; Ma S. H.; Zhou Y.; Santoyo J.; Martinez E. D.; Xiao G.; et al. KDM4/JMJD2 Histone Demethylase Inhibitors Block Prostate Tumor Growth by Suppressing the Expression of AR and BMYB-Regulated Genes. Chem. Biol. 2015, 22, 1185–1196. 10.1016/j.chembiol.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinnes C. C.; Tumber A.; Yapp C.; Scozzafava G.; Yeh T.; Chan M. C.; Tran T. A.; Hsu K.; Tarhonskaya H.; Walport L. J.; et al. Betti Reaction Enables Efficient Synthesis of 8-Hydroxyquinoline Inhibitors of 2-Oxoglutarate Oxygenases. Chem. Commun. 2015, 51, 15458–15461. 10.1039/C5CC06095H. [DOI] [PubMed] [Google Scholar]
- Feng T.; Li D.; Wang H.; Zhuang J.; Liu F.; Bao Q.; Lei Y.; Chen W.; Zhang X.; Xu X.; et al. Novel 5-Carboxy-8-HQ Based Histone Demethylase JMJD2A Inhibitors: Introduction of An Additional Carboxyl Group at The C-2 Position of Quinoline. Eur. J. Med. Chem. 2015, 105, 145–155. 10.1016/j.ejmech.2015.09.013. [DOI] [PubMed] [Google Scholar]
- Westaway S. M.; Preston A. G.; Barker M. D.; Brown F.; Brown J. A.; Campbell M.; Chung C. W.; Diallo H.; Douault C.; Drewes G.; et al. Cell Penetrant Inhibitors of the KDM4 and KDM5 Families of Histone Lysine Demethylases. 1. 3-Amino-4-pyridine Carboxylate Derivatives. J. Med. Chem. 2016, 59, 1357–1369. 10.1021/acs.jmedchem.5b01537. [DOI] [PubMed] [Google Scholar]
- Roatsch M.; Robaa D.; Pippel M.; Nettleship J. E.; Reddivari Y.; Bird L. E.; Hoffmann I.; Franz H.; Owens R. J.; Schule R.; et al. Substituted 2-(2-Aminopyrimidin-4-Yl)Pyridine-4-Carboxylates as Potent Inhibitors of JumonjiC Domain-Containing Histone Demethylases. Future Med. Chem. 2016, 8, 1553–1571. 10.4155/fmc.15.188. [DOI] [PubMed] [Google Scholar]
- Korczynska M.; Le D. D.; Younger N.; Gregori-Puigjane E.; Tumber A.; Krojer T.; Velupillai S.; Gileadi C.; Nowak R. P.; Iwasa E.; et al. Docking and Linking of Fragments To Discover Jumonji Histone Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1580–1598. 10.1021/acs.jmedchem.5b01527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woon E. C.; Tumber A.; Kawamura A.; Hillringhaus L.; Ge W.; Rose N. R.; Ma J. H.; Chan M. C.; Walport L. J.; Che K. H.; et al. Linking of 2-Oxoglutarate and Substrate Binding Sites Enables Potent and Highly Selective Inhibition of JmjC Histone Demethylases. Angew. Chem., Int. Ed. 2012, 51, 1631–1634. 10.1002/anie.201107833. [DOI] [PubMed] [Google Scholar]
- Wang W.; Marholz L. J.; Wang X. Novel Scaffolds of Cell-Active Histone Demethylase Inhibitors Identified from High-Throughput Screening. J. Biomol. Screening 2015, 20, 821–827. 10.1177/1087057115579637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüger N.; Roatsch M.; Emmrich T.; Franz H.; Schule R.; Jung M.; Link A. Tetrazolylhydrazides as Selective Fragment-Like Inhibitors of the JumonjiC-Domain-Containing Histone Demethylase KDM4A. ChemMedChem 2015, 10, 1875–1883. 10.1002/cmdc.201500335. [DOI] [PubMed] [Google Scholar]
- Liu L. J.; Lu L.; Zhong H. J.; He B.; Kwong D. W.; Ma D. L.; Leung C. H. An Iridium(III) Complex Inhibits JMJD2 Activities and Acts as A Potential Epigenetic Modulator. J. Med. Chem. 2015, 58, 6697–6703. 10.1021/acs.jmedchem.5b00375. [DOI] [PubMed] [Google Scholar]
- Leurs U.; Lohse B.; Rand K. D.; Ming S.; Riise E. S.; Cole P. A.; Kristensen J. L.; Clausen R. P. Substrate- and Cofactor-Independent Inhibition of Histone Demethylase KDM4C. ACS Chem. Biol. 2014, 9, 2131–2138. 10.1021/cb500374f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley G. W.; Brinko A.; Munzel M.; Walport L. J.; Schofield C. J.; Hopkinson R. J. Analysis of JmjC Demethylase-Catalyzed Demethylation Using Geometrically-Constrained Lysine Analogues. ACS Chem. Biol. 2016, 11, 755–762. 10.1021/acschembio.5b00738. [DOI] [PubMed] [Google Scholar]
- Kim T. D.; Fuchs J. R.; Schwartz E.; Abdelhamid D.; Etter J.; Berry W. L.; Li C.; Ihnat M. A.; Li P. K.; Janknecht R. Pro-Growth Role of the JMJD2C Histone Demethylase In HCT-116 Colon Cancer Cells And Identification Of Curcuminoids as JMJD2 Inhibitors. Am. J. Transl. Res. 2014, 6, 236–247. [PMC free article] [PubMed] [Google Scholar]
- Chu C. H.; Wang L. Y.; Hsu K. C.; Chen C. C.; Cheng H. H.; Wang S. M.; Wu C. M.; Chen T. J.; Li L. T.; Liu R.; et al. KDM4B as A Target for Prostate Cancer: Structural Analysis and Selective Inhibition by A Novel Inhibitor. J. Med. Chem. 2014, 57, 5975–5985. 10.1021/jm500249n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen J.; Agger K.; Cloos P. A.; Pasini D.; Rose S.; Sennels L.; Rappsilber J.; Hansen K. H.; Salcini A. E.; Helin K. RBP2 Belongs to A Family of Demethylases, Specific for Tri-and Dimethylated Lysine 4 on Histone 3. Cell 2007, 128, 1063–1076. 10.1016/j.cell.2007.02.003. [DOI] [PubMed] [Google Scholar]
- Pilka E. S.; James T.; Lisztwan J. H. Structural Definitions of Jumonji Family Demethylase Selectivity. Drug Discovery Today 2015, 20, 743–749. 10.1016/j.drudis.2014.12.013. [DOI] [PubMed] [Google Scholar]
- Horton J. R.; Engstrom A.; Zoeller E. L.; Liu X.; Shanks J. R.; Zhang X.; Johns M. A.; Vertino P. M.; Fu H.; Cheng X. Characterization of A Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J. Biol. Chem. 2016, 291, 2631–2646. 10.1074/jbc.M115.698449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defeo-Jones D.; Huang P. S.; Jones R. E.; Haskell K. M.; Vuocolo G. A.; Hanobik M. G.; Huber H. E.; Oliff A. Cloning of cDNAs for Cellular Proteins that Bind to the Retinoblastoma Gene Product. Nature 1991, 352, 251–254. 10.1038/352251a0. [DOI] [PubMed] [Google Scholar]
- Benevolenskaya E. V.; Murray H. L.; Branton P.; Young R. A.; Kaelin W. G. Jr. Binding of pRB to the PHD Protein RBP2 Promotes Cellular Differentiation. Mol. Cell 2005, 18, 623–635. 10.1016/j.molcel.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Zeng J.; Ge Z.; Wang L.; Li Q.; Wang N.; Bjorkholm M.; Jia J.; Xu D. The Histone Demethylase RBP2 is Overexpressed in Gastric Cancer and its Inhibition Triggers Senescence of Cancer Cells. Gastroenterology 2010, 138, 981–992. 10.1053/j.gastro.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Wang S.; Wang Y.; Wu H.; Hu L. RBP2 Induces Epithelial-Mesenchymal Transition in Non-Small Cell Lung Cancer. PLoS One 2013, 8, e84735. 10.1371/journal.pone.0084735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X.; Zeng J.; Wang L.; Fang M.; Wang Q.; Zhao M.; Xu X.; Liu Z.; Li W.; Liu S.; et al. Histone Demethylase Retinoblastoma Binding Protein 2 is Overexpressed in Hepatocellular Carcinoma and Negatively Regulated by hsa-miR-212. PLoS One 2013, 8, e69784. 10.1371/journal.pone.0069784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J.; Wu J.; Dombkowski A.; Zhang K.; Holowatyj A.; Boerner J. L.; Yang Z. Q. Genomic Amplification and A Role in Drug-Resistance for the KDM5A Histone Demethylase in Breast Cancer. Am. J. Transl. Res. 2012, 4, 247–256. [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi C. P.; Somasundaram B.; Singh K.; Carpenedo R. L.; Stanford W. L.; Dilworth F. J.; Brand M. Maintenance of Gene Silencing by the Coordinate Action of the H3K9Methyltransferase G9a/KMT1C and the H3K4 Demethylase Jarid1a/KDM5A. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 18845–18850. 10.1073/pnas.1213951109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayami S.; Yoshimatsu M.; Veerakumarasivam A.; Unoki M.; Iwai Y.; Tsunoda T.; Field H. I.; Kelly J. D.; Neal D. E.; Yamaue H.; et al. Overexpression of the JmjC Histone Demethylase KDM5B in Human Carcinogenesis: Involvement in the Proliferation of Cancer Cells through the E2F/RB pathway. Mol. Cancer 2010, 9, 59. 10.1186/1476-4598-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y.; Zhu Z.; Han G.; Ye X.; Xu B.; Peng Z.; Ma Y.; Yu Y.; Lin H.; Chen A. P.; et al. JARID1B is A Histone H3 Lysine 4 Demethylase Up-Regulated in Prostate Cancer. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19226–19231. 10.1073/pnas.0700735104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta K.; Haraguchi N.; Kano Y.; Kagawa Y.; Konno M.; Nishikawa S.; Hamabe A.; Hasegawa S.; Ogawa H.; Fukusumi T.; et al. Depletion of JARID1B Induces Cellular Senescence in Human Colorectal Cancer. Int. J. Oncol. 2013, 42, 1212–1218. 10.3892/ijo.2013.1799. [DOI] [PubMed] [Google Scholar]
- Roesch A.; Vultur A.; Bogeski I.; Wang H.; Zimmermann K. M.; Speicher D.; Korbel C.; Laschke M. W.; Gimotty P. A.; Philipp S. E.; et al. Overcoming Intrinsic Multidrug Resistance In Melanoma by Blocking the Mitochondrial Respiratory Chain of Slow-Cycling JARID1B(high) Cells. Cancer Cell 2013, 23, 811–825. 10.1016/j.ccr.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu X.; Zhang T.; Liao L.; Zhou L.; Lindner D. J.; Zhou M.; Rini B.; Yan Q.; Yang H. The von Hippel-Lindau Tumor Suppressor Protein Regulates Gene Expression and Tumor Growth Through Histone Demethylase JARID1C. Oncogene 2012, 31, 776–786. 10.1038/onc.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. A.; White E. A.; Sowa M. E.; Powell M. L.; Ottinger M.; Harper J. W.; Howley P. M. Genome-Wide siRNA Screen Identifies SMCX, EP400, and Brd4 as E2-Dependent Regulators of Human Papillomavirus Oncogene Expression. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3752–3757. 10.1073/pnas.0914818107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perinchery G.; Sasaki M.; Angan A.; Kumar V.; Carroll P.; Dahiya R. Deletion of Y-Chromosome Specific Genes in Human Prostate Cancer. J. Urol. 2000, 163, 1339–1342. 10.1016/S0022-5347(05)67774-9. [DOI] [PubMed] [Google Scholar]
- Vinogradova M.; Gehling V. S.; Gustafson A.; Arora S.; Tindell C. A.; Wilson C.; Williamson K. E.; Guler G. D.; Gangurde P.; Manieri W.; et al. An Inhibitor of KDM5 Demethylases Reduces Survival o Drug-Tolerant Cancer Cells. Nat. Chem. Biol. 2016, 12, 531–538. 10.1038/nchembio.2085. [DOI] [PubMed] [Google Scholar]
- Gehling V. S.; Bellon S. F.; Harmange J. C.; LeBlanc Y.; Poy F.; Odate S.; Buker S.; Lan F.; Arora S.; Williamson K. E.; et al. Identification of Potent, Selective KDM5 Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4350–4354. 10.1016/j.bmcl.2016.07.026. [DOI] [PubMed] [Google Scholar]
- Liang J.; Zhang B.; Labadie S.; Ortwine D. F.; Vinogradova M.; Kiefer J. R.; Gehling V. S.; Harmange J. C.; Cummings R.; Lai T.; et al. Lead Optimization of A Pyrazolo[1,5-A]Pyrimidin-7(4h)-One Scaffold to Identify Potent, Selective and Orally Bioavailable KdDM5 Inhibitors Suitable for In Vivo Biological Studies. Bioorg. Med. Chem. Lett. 2016, 26, 4036–4041. 10.1016/j.bmcl.2016.06.078. [DOI] [PubMed] [Google Scholar]
- Labadie S. S.; Dragovich P. S.; Cummings R. T.; Deshmukh G.; Gustafson A.; Han N.; Harmange J. C.; Kiefer J. R.; Li Y.; Liang J.; et al. Design and Evaluation of 1,7-Naphthyridones as Novel KDM5 Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 4492–4496. 10.1016/j.bmcl.2016.07.070. [DOI] [PubMed] [Google Scholar]
- Johansson C.; Velupillai S.; Tumber A.; Szykowska A.; Hookway E. S.; Nowak R. P.; Strain-Damerell C.; Gileadi C.; Philpott M.; Burgess-Brown N.; et al. Structural Analysis of Human KDM5B Guides Histone Demethylase Inhibitor Development. Nat. Chem. Biol. 2016, 12, 539–545. 10.1038/nchembio.2087. [DOI] [PubMed] [Google Scholar]
- Horton J. R.; Liu X.; Gale M.; Wu L.; Shanks J. R.; Zhang X.; Webber P. J.; Bell J. S.; Kales S. C.; Mott B. T.; et al. Structural Basis for KDM5A Histone Lysine Demethylase Inhibition by Diverse Compounds. Cell Chem. Biol. 2016, 23, 769–781. 10.1016/j.chembiol.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J.; Liu Z.; Cheung W. K.; Zhao M.; Chen S. Y.; Chan S. W.; Booth C. J.; Nguyen D. X.; Yan Q. Histone Demethylase RBP2 is Critical for Breast Cancer Progression and Metastasis. Cell Rep. 2014, 6, 868–877. 10.1016/j.celrep.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S.; Wu Z.; Russnes H. G.; Takagi S.; Peluffo G.; Vaske C.; Zhao X.; Moen Vollan H. K.; Maruyama R.; Ekram M. B.; et al. JARID1B is A Luminal Lineage-Driving Oncogene in Breast Cancer. Cancer Cell 2014, 25, 762–777. 10.1016/j.ccr.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway S. M.; Preston A. G.; Barker M. D.; Brown F.; Brown J. A.; Campbell M.; Chung C. W.; Drewes G.; Eagle R.; Garton N.; et al. Cell Penetrant Inhibitors of the KDM4 and KDM5 Families of Histone Lysine Demethylases. 2. Pyrido[3,4-d]pyrimidin-4(3H)-one Derivatives. J. Med. Chem. 2016, 59, 1370–1387. 10.1021/acs.jmedchem.5b01538. [DOI] [PubMed] [Google Scholar]
- Bavetsias V.; Lanigan R. M.; Ruda G. F.; Atrash B.; McLaughlin M. G.; Tumber A.; Mok N. Y.; Le Bihan Y. V.; Dempster S.; Boxall K. J.; et al. 8-Substituted Pyrido[3,4-d]pyrimidin-4(3H)-one Derivatives As Potent, Cell Permeable, KDM4 (JMJD2) and KDM5 (JARID1) Histone Lysine Demethylase Inhibitors. J. Med. Chem. 2016, 59, 1388–1409. 10.1021/acs.jmedchem.5b01635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannironi C.; Proietto M.; Bufalieri F.; Cundari E.; Alagia A.; Danovska S.; Rinaldi T.; Famiglini V.; Coluccia A.; La Regina G.; et al. An High-Throughput In Vivo Screening System to Select H3K4-Specific Histone Demethylase Inhibitors. PLoS One 2014, 9, e86002. 10.1371/journal.pone.0086002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y.; Sawada H.; Suzuki M.; Tojo T.; Sasaki R.; Hasegawa M.; Mizukami T.; Suzuki T. Identification of Jumonji AT-Rich Interactive Domain 1A Inhibitors and Their Effect on Cancer Cells. ACS Med. Chem. Lett. 2015, 6, 665–670. 10.1021/acsmedchemlett.5b00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K.; Cloos P. A.; Christensen J.; Pasini D.; Rose S.; Rappsilber J.; Issaeva I.; Canaani E.; Salcini A. E.; Helin K. UTX and JMJD3 are Histone H3K27 Demethylases Involved in HOX Gene Regulation and Development. Nature 2007, 449, 731–734. 10.1038/nature06145. [DOI] [PubMed] [Google Scholar]
- Thieme S.; Gyarfas T.; Richter C.; Ozhan G.; Fu J.; Alexopoulou D.; Muders M. H.; Michalk I.; Jakob C.; Dahl A.; et al. The Histone Demethylase UTX Regulates Stem Cell Migration and Hematopoiesis. Blood 2013, 121, 2462–2473. 10.1182/blood-2012-08-452003. [DOI] [PubMed] [Google Scholar]
- Mansour A. A.; Gafni O.; Weinberger L.; Zviran A.; Ayyash M.; Rais Y.; Krupalnik V.; Zerbib M.; Amann-Zalcenstein D.; Maza I.; et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 2012, 488, 409–413. 10.1038/nature11272. [DOI] [PubMed] [Google Scholar]
- Zhao W.; Li Q.; Ayers S.; Gu Y.; Shi Z.; Zhu Q.; Chen Y.; Wang H. Y.; Wang R. F. Jmjd3 Inhibits Reprogramming by Upregulating Expression of INK4a/Arf and Targeting PHF20 for Ubiquitination. Cell 2013, 152, 1037–1050. 10.1016/j.cell.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. G.; Villa R.; Trojer P.; Norman J.; Yan K. P.; Reinberg D.; Di Croce L.; Shiekhattar R. Demethylation of H3K27 Regulates Polycomb Recruitment and H2A Ubiquitination. Science 2007, 318, 447–450. 10.1126/science.1149042. [DOI] [PubMed] [Google Scholar]
- van Haaften G.; Dalgliesh G. L.; Davies H.; Chen L.; Bignell G.; Greenman C.; Edkins S.; Hardy C.; O’Meara S.; Teague J.; et al. Somatic Mutations of the Histone H3K27 Demethylase Gene UTX in Human Cancer. Nat. Genet. 2009, 41, 521–523. 10.1038/ng.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y. W.; Hong T.; Hong S.; Guo H.; Yu H.; Kim D.; Guszczynski T.; Dressler G. R.; Copeland T. D.; Kalkum M.; et al. PTIP Associates with MLL3- and MLL4-Containing Histone H3 Lysine 4 Methyltransferase Complex. J. Biol. Chem. 2007, 282, 20395–20406. 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira F.; Barbachano A.; Silva J.; Bonilla F.; Campbell M. J.; Munoz A.; Larriba M. J. KDM6B/JMJD3 Histone Demethylase is Induced by Vitamin D and Modulates its Effects in Colon Cancer Cells. Hum. Mol. Genet. 2011, 20, 4655–4665. 10.1093/hmg/ddr399. [DOI] [PubMed] [Google Scholar]
- Anderton J. A.; Bose S.; Vockerodt M.; Vrzalikova K.; Wei W.; Kuo M.; Helin K.; Christensen J.; Rowe M.; Murray P. G.; et al. The H3K27me3 Demethylase, KDM6B, is Induced by Epstein-Barr Virus and Over-Expressed in Hodgkin’s Lymphoma. Oncogene 2011, 30, 2037–2043. 10.1038/onc.2010.579. [DOI] [PubMed] [Google Scholar]
- Svotelis A.; Bianco S.; Madore J.; Huppe G.; Nordell-Markovits A.; Mes-Masson A. M.; Gevry N. H3K27 Demethylation by JMJD3 at A Poised Enhancer of Anti-Apoptotic Gene BCL2 Determines ERalpha Ligand Dependency. EMBO J. 2011, 30, 3947–3961. 10.1038/emboj.2011.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K.; Cloos P. A.; Rudkjaer L.; Williams K.; Andersen G.; Christensen J.; Helin K. The H3K27me3 Demethylase JMJD3 Contributes to the Activation of the INK4A-ARF Locus in Response to Oncogene- and Stress-Induced Senescence. Genes Dev. 2009, 23, 1171–1176. 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidenier L.; Chung C. W.; Cheng Z.; Liddle J.; Che K.; Joberty G.; Bantscheff M.; Bountra C.; Bridges A.; Diallo H.; et al. A Selective Jumonji H3K27 Demethylase Inhibitor Modulates the Proinflammatory Macrophage Response. Nature 2012, 488, 404–408. 10.1038/nature11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann B.; Nielsen J. M.; Hudlebusch H. R.; Lees M. J.; Larsen D. V.; Boesen T.; Labelle M.; Gerlach L. O.; Birk P.; Helin K. Inhibition of Demethylases by GSK-J1/J4. Nature 2014, 514, E1–2. 10.1038/nature13688. [DOI] [PubMed] [Google Scholar]
- Hu J.; Wang X.; Chen L.; Huang M.; Tang W.; Zuo J.; Liu Y. C.; Shi Z.; Liu R.; Shen J.; et al. Design and Discovery of New Pyrimidine Coupled Nitrogen Aromatic Rings as Chelating Groups of JMJD3 Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 721–725. 10.1016/j.bmcl.2016.01.006. [DOI] [PubMed] [Google Scholar]
- Rotili D.; Tomassi S.; Conte M.; Benedetti R.; Tortorici M.; Ciossani G.; Valente S.; Marrocco B.; Labella D.; Novellino E.; et al. Pan-Histone Demethylase Inhibitors Simultaneously Targeting Jumonji C and Lysine-Specific Demethylases Display High Anticancer Activities. J. Med. Chem. 2014, 57, 42–55. 10.1021/jm4012802. [DOI] [PubMed] [Google Scholar]