

Abstract
Because the backbone of most organic molecules is composed primarily of carbon–carbon bonds, the development of efficient methods for their construction is one of the central challenges of organic synthesis. Transition-metal-catalyzed cross-coupling reactions between organic electrophiles and nucleophiles serve as particularly powerful tools for achieving carbon–carbon bond formation. Until recently, the vast majority of cross-coupling processes had employed either aryl or alkenyl electrophiles as one of the coupling partners. In the past 15 years, versatile new methods have been developed that effect cross-couplings of an array of alkyl electrophiles, thereby greatly expanding the diversity of target molecules that are readily accessible. The ability to couple alkyl electrophiles opens the door to a stereochemical dimension that significantly enhances the already remarkable utility of cross-coupling processes, specifically, enantioconvergent couplings of racemic electrophiles.
Introduction
The construction of carbon–carbon bonds is central to organic chemistry, which is the chemistry of molecules that contain carbon. During the past several decades, a wide array of powerful new methods for carbon–carbon bond formation have been developed, including two transition-metal-catalyzed processes that have recently been recognized with Nobel Prizes in Chemistry (olefin metathesis in 2005 (1) and cross-coupling in 2010 (2)). The discovery of efficient approaches to the creation of carbon–carbon bonds has an impact not only on synthetic organic chemistry, but also on the many other disciplines that employ organic compounds, including biology and materials science.
Metal-catalyzed cross-coupling can provide a particularly straightforward, modular approach to carbon–carbon bond formation through the union of two coupling partners, an organic electrophile and an organometallic nucleophile, which may be either commercially available or readily synthesized (Fig. 1A) (2). Early studies of such processes were dominated by the use of palladium catalysts to accomplish couplings that generate a bond between two sp2-hybridized carbons, and these methods have found application in industry (Fig. 1; R and R1 = aryl or alkenyl).
Fig. 1.

Transition-metal-catalyzed cross-coupling to form carbon–carbon bonds. A: General scheme. B: Application of a Suzuki cross-coupling to form a Csp2–Csp2 bond in an industrial synthesis of Boscalid.
Although methods to construct carbon–carbon bonds between sp2-hybridized carbons (e.g., “aryl–aryl” bonds) are exceptionally powerful tools in organic synthesis, bonds between sp3-hybridized carbons (“alkyl–alkyl” bonds) are much more common than are aryl–aryl bonds. Fig. 2 provides illustrative examples of bioactive compounds that include a variety of alkyl–alkyl bonds. The development of effective cross-coupling catalysts that could generate such bonds at will would dramatically impact the retrosynthetic analysis (3) and, in turn, the synthesis of a broad array of organic molecules. As described herein, until recent years, progress in addressing this challenge had been limited; thus, it was observed in 2004 that, despite the pervasiveness of alkyl–alkyl bonds, “Alkyl–alkyl cross-coupling reactions have historically been the most difficult to realize” (4).
Fig. 2.
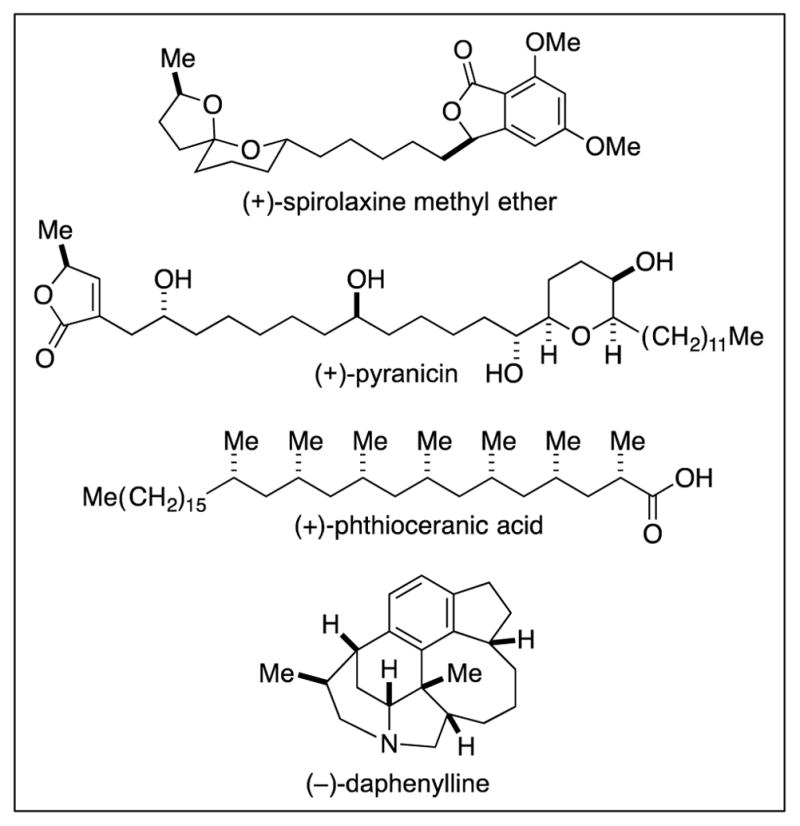
Bioactive compounds that include an array of alkyl–alkyl bonds.
Examination of the bioactive molecules depicted in Fig. 2 serves as a reminder that, in addition to constructing the carbon–carbon bond itself, another challenge can arise when an alkyl, rather than an aryl, electrophile is employed as the cross-coupling partner, specifically, controlling the stereochemistry at the carbon derived from the electrophile (Fig. 3A) (5). Stereochemistry can of course play a key role in determining properties such as structure and biological activity (6).
Fig. 3.

Stereochemistry as an added dimension in cross-coupling reactions of alkyl electrophiles. A: Aryl electrophiles versus alkyl electrophiles. B: Use of a chiral catalyst to control stereochemistry: enantioconvergent cross-couplings of racemic alkyl electrophiles.
Thus, for many cross-coupling reactions of alkyl electrophiles, the challenge may in fact be two-fold: forming the carbon–carbon bond as well as controlling the stereochemistry of the product, independent of the stereochemistry of the starting material (Fig. 3B). In this review, we describe recent progress in addressing these objectives, with a particular focus on initial breakthroughs (new families of coupling partners and new transition-metal catalysts) in alkyl–alkyl cross-couplings of unactivated alkyl electrophiles (7,8,9,10).
An Impediment to Alkyl–Alkyl Cross-Coupling: β-Hydride Elimination
As mentioned above, most early studies of cross-couplings employed palladium catalysts and focused on reactions of aryl electrophiles (2). An outline of a mechanism for such processes (Fig. 5A) involves a sequence of oxidative addition of the organic electrophile (R–X) to a Pd(0) complex (1) to generate an organopalladium(II) complex (2), transmetalation by the nucleophilic coupling partner (M–R1) to furnish a diorganopalladium(II) complex (3), and reductive elimination to form the carbon–carbon bond (R–R1) and regenerate a Pd(0) complex (1) (2).
Fig. 5.

Mechanistic aspects of metal-catalyzed cross-couplings, illustrated for palladium. A: An outline of a catalytic cycle. B: β-Hydride elimination as a side reaction.
Organopalladium(II) complex 2 is a key intermediate in this catalytic cycle (Fig. 5A). Organometallic compounds that bear a hydrogen in the β position have the potential to undergo β-hydride elimination, an intramolecular process that generates a metal–hydride (Fig. 5B). In the case of arylpalladium(II) complexes, there is no precedent for β-hydride elimination to form a palladium–aryne, and, correspondingly, cross-couplings of aryl electrophiles are not diverted by this undesired side reaction. In contrast, β-hydride elimination of an alkylpalladium(II) complex to generate a palladium–alkene is a common pathway, the efficiency of which is critical for important palladium-catalyzed processes such as the Wacker (11) and Heck reactions (12). The unwanted, but often facile, β-hydride elimination of alkylmetal complexes presents a key impediment to efficient cross-coupling of alkyl electrophiles.
Primary Alkyl Electrophiles
In early studies, primary alkyl electrophiles were coupled with alkylmagnesium reagents (Grignard reagents) in the presence of transition-metal catalysts such as copper (Fig. 6A) (13,14). While such methods have found application in the total synthesis of natural products (Fig. 6B) (15), Grignard reagents can be incompatible with many functional groups, such as carbonyl compounds, that are commonly encountered in organic chemistry (16). More recently, it has been shown that palladium and nickel catalysts can achieve cross-couplings of primary alkyl halides with alkylboron and alkylzinc reagents (Fig. 6C) (17), both of which have good functional-group compatibility (2).
Fig. 6.

Cross-couplings of primary alkyl electrophiles: Early (pre-2000) methods. A: Examples. B: An application in the total synthesis of a natural product. C: Use of nucleophiles that have improved functional-group compatibility.
Since 2000, the scope of methods for the cross-coupling of primary alkyl electrophiles with mild organometallic nucleophiles has increased considerably. For example, palladium complexes that bear a bulky, electron-rich phosphine have proved to be versatile catalysts (Fig. 7A) (18), enabling the coupling of an array of alkyl electrophiles with alkylboron reagents; in contrast to the earlier palladium/PPh3-based method (Fig. 6C), which was only applied to primary alkyl iodides, palladium/trialkylphosphine catalysts are effective for cross-couplings of alkyl bromides, chlorides, and tosylates. More recently, copper and iron catalysts have also been shown to be useful for cross-couplings of primary alkyl electrophiles with alkylboron reagents (Fig. 7A). The palladium-based method has been applied to late-stage fragment couplings in the total synthesis of natural products such as (+)-spirolaxine methyl ether and (+)-pyranicin (Fig. 7B) (19,20).
Fig. 7.

Cross-couplings of primary alkyl electrophiles: Recent (post-2000) methods that use alkylboron reagents as nucleophiles. A: Examples. B: Applications in the total synthesis of natural products.
The use of a bulky, electron-rich ligand has enabled palladium-catalyzed cross-couplings not only of alkylboron reagents, but also of alkylzinc reagents, opening the door to carbon–carbon bond formation with a wide range of primary alkyl electrophiles, including iodides, bromides, chlorides, and tosylates (Fig. 8A) (21). Subsequently, a copper catalyst has also been shown to achieve alkyl–alkyl cross-couplings of primary alkyl bromides with alkylzinc reagents (Fig. 8A). These new methods have found application in the synthesis of bioactive compounds such as MaR1n–3 DPA (Fig. 8B) (22).
Fig. 8.

Cross-couplings of primary alkyl electrophiles: Recent (post-2000) methods that use alkylzinc reagents as nucleophiles. A: Examples. B: An application in the total synthesis of a natural product.
Stereochemical studies of a palladium/trialkylphosphine-catalyzed alkyl–alkyl coupling were consistent with an SN2 pathway for oxidative addition under these conditions (23). This mechanism can account for the inability of this catalyst to accomplish alkyl–alkyl cross-couplings of secondary alkyl electrophiles.
Secondary Alkyl Electrophiles
As in the case of metal-catalyzed alkyl–alkyl cross-coupling reactions of primary alkyl electrophiles, early studies provided proof-of-principle that couplings of secondary alkyl electrophiles are indeed possible (Fig. 9A) (24). As with primary electrophiles (Fig. 6A), these early methods employed reactive Grignard reagents as the nucleophilic coupling partner. Despite this limitation, such cross-couplings have found application in the total synthesis of natural products (Fig. 9B) (25).
Fig. 9.

Cross-couplings of secondary alkyl electrophiles: Early (pre-2000) methods. A: Examples. B: An application in the total synthesis of a natural product.
More recently, the first methods for coupling secondary alkyl electrophiles with mild organometallic nucleophiles (alkylboron and alkylzinc reagents) have been described (Fig. 10A and 10B) (26,27). To date, nickel-based complexes have proved to be the most versatile, enabling alkyl–alkyl couplings of a range of secondary alkyl electrophiles (iodides, bromides, and chlorides), although copper- (limited to allylboron reagents) and iron-catalyzed methods have also been reported. Nickel-catalyzed cross-couplings have been applied, for example, to the diastereoselective synthesis of C-alkyl glycosides, an important family of bioactive molecules (28).
Fig. 10.

Cross-couplings of secondary alkyl electrophiles: Recent (post-2000) methods that use nucleophiles that have improved functional-group compatibility. A: Alkylboron reagents. B: Alkylzinc reagents. C: An application in synthesis.
Catalytic Asymmetric Carbon–Carbon Bond Formation
The ability to employ secondary electrophiles as partners opened the door to an additional dimension in cross-coupling chemistry, catalytic enantioselective carbon–carbon bond formation starting with racemic alkyl electrophiles (Fig. 3B) (29). Preliminary mechanistic data indicated that the nickel-catalyzed cross-coupling methods described in the previous section proceed via the formation of a radical intermediate from the electrophile, which is ideal for an enantioconvergent process (Fig. 11A). Thus, both enantiomers of the electrophile could generate the same secondary radical upon homolytic cleavage of the C–X bond, thereby ablating the original stereochemistry and enabling a chiral catalyst to react with the alkyl radical and transform both enantiomers of the electrophile into a single enantiomer of the product. Because catalytic asymmetric alkyl–alkyl cross-couplings are still in a relatively early stage of development, in this section we describe couplings not only of unactivated alkyl electrophiles, but also of several activated alkyl electrophiles (30).
Fig. 11.

Catalytic asymmetric carbon–carbon bond formation. A: Enantioconvergent cross-coupling via a radical intermediate. B: Methods for activated electrophiles. C: An application in the total synthesis of a natural product.
Activated racemic alkyl halides, specifically, α-bromoamides and benzylic halides, served as the electrophilic partner in early examples of catalytic asymmetric alkyl–alkyl cross-coupling (Fig. 11B) (31). In the presence of a chiral nickel catalyst, an array of alkylzinc reagents can be employed as the nucleophilic partner. These methods have found application in the total synthesis of natural products such as (−)-daphenylline (Fig. 11C) (32).
Unactivated electrophiles can also serve as useful partners in enantioconvergent alkyl–alkyl cross-couplings. In this case, alkylboron reagents have proved to be the nucleophiles of choice, coupling with an array of racemic alkyl halides in good ee and yield with the aid of chiral nickel/diamine catalysts (Fig. 12) (33). For these methods, the presence of a directing group, which likely interacts with the chiral catalyst in the stereochemistry-determining step of the cross-coupling, is essential for high enantioselectivity.
Fig. 12.

Enantioconvergent cross-couplings of unactivated alkyl electrophiles, directed by the indicated functional groups.
Enantioconvergent reactions of racemic electrophiles are not the only opportunity in the field of catalytic asymmetric alkyl–alkyl cross-coupling. The umpolung (polarity inverted) (34) process, i.e., enantioconvergent couplings of racemic nucleophiles, can also be envisioned (Fig. 13A) (35). An example of such a process has recently been described, wherein a racemic alkylzinc reagent is coupled with an array of alkyl halides (Fig. 13B) (36). This report sets the stage for addressing an interesting new challenge, the doubly stereoconvergent cross-coupling of two racemic partners (electrophile and nucleophile) to generate each of the four possible stereoisomeric products simply through the appropriate choice of catalyst (Fig. 13C).
Fig. 13.

Enantioconvergent alkyl–alkyl cross-couplings. A: Use of a racemic electrophile or a racemic nucleophile. B: Enantioconvergent nickel-catalyzed coupling of a racemic alkylzinc reagent. C: Doubly stereoconvergent coupling of a racemic electrophile and a racemic nucleophile.
Tertiary Alkyl Electrophiles
Although quaternary carbons are less common than tertiary carbons, the development of alkyl–alkyl cross-couplings to generate such fully substituted centers is nevertheless an important objective in organic synthesis. As in the case of primary and secondary alkyl electrophiles, the initial advances in employing tertiary electrophiles as coupling partners involved the use of Grignard reagents as nucleophiles. For example, cobalt, silver, and copper complexes serve as effective catalysts for such cross-couplings, but only in the case of allylmagnesium and benzylmagnesium reagents (Fig. 14A) (37). More recently, silver-catalyzed couplings of tertiary alkyl bromides with organozinc reagents have been described (Fig. 14B) (38), although these methods are also limited to allyl and benzyl nucleophiles, and the mechanism of these processes has not been elucidated. In contrast to alkyl–alkyl cross-couplings of secondary electrophiles, general methods that employ organozinc or organoboron nucleophiles have not yet been developed for couplings of tertiary electrophiles, nor have highly enantioselective variants.
Fig. 14.

Cross-couplings of tertiary alkyl electrophiles. A: Early methods. B: Recent method that uses a nucleophile that has improved functional-group compatibility.
Prospective and Conclusions
Because alkyl–alkyl bonds are commonplace in organic molecules, the development of increasingly powerful methods for their construction (and for the control of any associated stereochemistry) from readily available coupling partners will have a significant impact on the many disciplines that employ organic compounds. In the case of a synthesis of a particular target compound, the availability of tools to reliably achieve alkyl–alkyl bond formation will provide new options for retrosynthetic analysis and, in turn, the synthesis of the desired molecule.
The development of versatile new methods for alkyl–alkyl cross-coupling, which represents a powerful fragment-coupling process, can be expected to have an impact beyond the target-oriented synthesis of a particular compound. In recent years, diversity-oriented library synthesis, which is focused on the efficient generation of families of molecules rather than a particular molecule, has become an increasingly important tool in science, especially in drug discovery (39,40). This strategy depends upon reliable reactions that provide ready access to diverse collections of molecules for compound libraries and for lead optimization. Recently, medicinal chemists have observed that many current efforts in drug development may be biased toward compounds that have aromatic subunits, as a consequence of the dependability of (and therefore the reliance upon) cross-coupling reactions of readily available aryl electrophiles and nucleophiles (41). On the other hand, an analysis has suggested that a higher percentage of sp3-hybridized (rather than sp2-hybridized) carbons, as well as a larger number of stereogenic centers, can increase the probability of clinical success for a compound (41,42). Thus, the development of increasingly versatile tools for alkyl–alkyl bond formation, including stereoselective processes, may facilitate an “escape from flatland” (41,42).
In closing, substantial progress has been described in recent years in the development of new methods for the construction of alkyl–alkyl bonds through the metal-catalyzed cross-coupling of readily available electrophiles and nucleophiles (Fig. 15) (43,44). In particular, through the use of nickel catalysts, carbon–carbon bond formation can be achieved under mild conditions with an array of coupling partners, and, in certain instances, a chiral catalyst can accomplish stereoconvergent reactions of racemic starting materials with high enantioselectivity. A number of challenges remain, including further expanding the scope of coupling partners and of enantioselective processes.
Fig. 15.

Alkyl–alkyl cross-coupling.
Fig. 4.

Recent progress in transition-metal-catalyzed alkyl–alkyl coupling.
Acknowledgments
Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences: R01–GM62871).
References
- 1.Grela K, editor. Olefin Metathesis: Theory and Practice. John Wiley & Sons; Hoboken, NJ: 2014. [Google Scholar]
- 2.de Meijere A, Bräse S, Oestreich M, editors. Metal-Catalyzed Cross-Coupling Reactions and More. Wiley–VCH; Weinheim, Germany: 2014. [Google Scholar]
- 3.Corey EJ, Cheng X-M. The Logic of Chemical Synthesis. John Wiley & Sons; New York, NY: 1995. [Google Scholar]
- 4.Denmark SE, Sweis RF. In: Metal-Catalyzed Cross-Coupling Reactions. de Meijere A, Diederich F, editors. Wiley–VCH; Weinheim, Germany: 2004. p. 191. [Google Scholar]
- 5.In the case of cross-couplings that generate biaryl compounds, the opportunities for enantioselective catalysis are limited. For a review of methods that control axial chirality, see: Loxq P, Manoury E, Poli R, Deydier E, Labande A. Coord Chem Rev. 2016;308:131–190.
- 6.Lin G-Q, You Q-D, Cheng J-F, editors. Chiral Drugs: Chemistry and Biological Action. John Wiley & Sons; Hoboken, NJ: 2011. [Google Scholar]
- 7.Except as noted, this review focuses on cross-couplings of unactivated alkyl electrophiles and therefore does not include reactions of electrophiles that bear a π system adjacent to the leaving group (specifically, allylic, benzylic, and propargylic electrophiles, as well as α-halocarbonyl compounds).
- 8.Iwasaki T, Kambe N. In: Comprehensive Organic Synthesis. Knochel P, Molander GA, editors. Vol. 3. Elsevier; Amsterdam, Netherlands: 2004. pp. 337–391. [Google Scholar]
- 9.Geist E, Kirschning A, Schmidt T. Nat Prod Rep. 2014;31:441–448. doi: 10.1039/c3np70108e. [DOI] [PubMed] [Google Scholar]
- 10.Glasspoole BW, Crudden CM. Nat Chem. 2011;3:912–913. doi: 10.1038/nchem.1210. [DOI] [PubMed] [Google Scholar]
- 11.Michel BW, Steffens LD, Sigman MS. Org React. 2014;84:75–413. [Google Scholar]
- 12.Oestreich M, editor. The Mizoroki–Heck Reaction. John Wiley & Sons; Chichester, UK: 2009. [Google Scholar]
- 13.Fig. 6A: Tamura M, Kochi J. Synthesis. 1971:303–305.Donkervoort JG, Vicario JL, Jastrzebski JTBH, Cahiez G, van Koten G. Rec Trav Chim Pays-Bas. 1996;115:547–548.
- 14.Castle PL, Widdowson DA. Tetrahedron Lett. 1986;27:6013–6016.See also Yuan K, Scott WJ. Tetrahedron Lett. 1989;30:4779–4782.
- 15.Fig. 6B: Herber C, Breit B. Angew Chem Int Ed. 2005;44:5267–5269. doi: 10.1002/anie.200501612.
- 16.Rappoport Z, Marek I, editors. The Chemistry of Organomagnesium Compounds. John Wiley & Sons; Chichester, UK: 2008. [Google Scholar]
- 17.Fig. 6C: Ishiyama T, Abe S, Miyaura N, Suzuki A. Chem Lett. 1992:691–694.Devasagayaraj A, Stüdemann T, Knochel P. Angew Chem Int Ed Engl. 1995;34:2723–2725.
- 18.Fig. 7A: Netherton MR, Dai C, Neuschütz K, Fu GC. J Am Chem Soc. 2001;123:10099–10100. doi: 10.1021/ja011306o.Yang CT, Zhang ZQ, Liu YC, Liu L. Angew Chem Int Ed. 2011;50:3904–3907. doi: 10.1002/anie.201008007.Hatakeyama T, Hashimoto T, Kathriarachchi KADS, Zenmyo T, Seike H, Nakamura M. Angew Chem Int Ed. 2012;51:8834–8837. doi: 10.1002/anie.201202797.
- 19.Fig. 7B: Keaton KA, Phillips AJ. Org Lett. 2007;9:2717–2719. doi: 10.1021/ol0710111.Griggs ND, Phillips AJ. Org Lett. 2008;10:4955–4957. doi: 10.1021/ol802041c.
- 20.For another application, see: Das S, Abraham S, Sinha SC. Org Lett. 2007;9:2273–2276. doi: 10.1021/ol070517g.
- 21.Fig. 8A: Zhou J, Fu GC. J Am Chem Soc. 2003;125:12527–12530. doi: 10.1021/ja0363258.Organ MG, Avola S, Dubovyk I, Hadei N, Kantchev EAB, O’Brien CJ, Valente C. Chem Eur J. 2006;12:4749–4755. doi: 10.1002/chem.200600206.Erdik E, Serdar EZ. J Organomet Chem. 2012;703:1–8.
- 22.Fig. 8B: Tungen JE, Aursnes M, Dalli J, Arnardottir H, Serhan CN, Hansen TV. Chem Eur J. 2014;20:14575–14578. doi: 10.1002/chem.201404721.
- 23.Netherton MR, Fu GC. Angew Chem Int Ed. 2002;41:3910–3912. doi: 10.1002/1521-3773(20021018)41:20<3910::AID-ANIE3910>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 24.Fig. 9A: Burns DH, Miller JD, Chan HK, Delaney MO. J Am Chem Soc. 1997;119:2125–2123.Donkervoort JG, Vicario JL, Jastrzebski JTBH, Gossage RA, Cahiez G, van Koten G. J Organomet Chem. 1998;558:61–69.
- 25.Fig. 9B: Xu S, Oda A, Bobinski T, Li H, Matsueda Y, Negishi E-i. Angew Chem Int Ed. 2015;54:9319–9322. doi: 10.1002/anie.201503818.
- 26.Fig. 10A: Saito B, Fu GC. J Am Chem Soc. 2007;129:9602–9603. doi: 10.1021/ja074008l.Wang GZ, Jiang J, Bu XS, Dai JJ, Xu J, Fu Y, Xu HJ. Org Lett. 2015;17:3682–3685. doi: 10.1021/acs.orglett.5b01612.(c) Reference 18c
- 27.Fig. 10B: Zhou J, Fu GC. J Am Chem Soc. 2003;125:14726–14727. doi: 10.1021/ja0389366.
- 28.Fig. 10C: Gong H, Sinisi R, Gagné MR. J Am Chem Soc. 2007;129:1908–1909. doi: 10.1021/ja068950t.
- 29.Glorius F. Angew Chem Int Ed. 2008;47:8347–8349. doi: 10.1002/anie.200803509. [DOI] [PubMed] [Google Scholar]
- 30.Because there are numerous reports of transition-metal-catalyzed enantioselective coupling reactions that employ allylic electrophiles, they are not discussed herein. For leading references, see: Helmchen G, Kazmaier U, Förster S. In: Catalytic Asymmetric Synthesis. Ojima I, editor. Chapter 8B John Wiley & Sons; Hoboken, NJ: 2010.
- 31.Fig. 11B: Fischer C, Fu GC. J Am Chem Soc. 2005;127:4594–4595. doi: 10.1021/ja0506509.Arp FO, Fu GC. J Am Chem Soc. 2005;127:10482–10483. doi: 10.1021/ja053751f.Binder JT, Cordier CJ, Fu GC. J Am Chem Soc. 2012;134:17003–17006. doi: 10.1021/ja308460z.
- 32.Fig. 11C: Yamada R, Adachi Y, Yokoshima S, Fukuyama T. Angew Chem Int Ed. 2016;55:6067–6070. doi: 10.1002/anie.201601958.
- 33.Fig. 12: Saito B, Fu GC. J Am Chem Soc. 2008;130:6694–6695. doi: 10.1021/ja8013677.Jiang X, Gandelman M. J Am Chem Soc. 2015;137:2542–2547. doi: 10.1021/jacs.5b00473.Owston NA, Fu GC. J Am Chem Soc. 2010;132:11908–11909. doi: 10.1021/ja105924f.Lu Z, Wilsily A, Fu GC. J Am Chem Soc. 2011;133:8154–8157. doi: 10.1021/ja203560q.Wilsily A, Tramutola F, Owston NA, Fu GC. J Am Chem Soc. 2012;134:5794–5797. doi: 10.1021/ja301612y.
- 34.Seebach D. Angew Chem Int Ed Engl. 1979;18:239–258. [Google Scholar]
- 35.Hayashi T, Tajika M, Tamao K, Kumada M. J Am Chem Soc. 1976;98:3718–3719. [Google Scholar]
- 36.Fig. 13B: Cordier CJ, Lundgren RJ, Fu GC. J Am Chem Soc. 2013;135:10946–10949. doi: 10.1021/ja4054114.
- 37.Fig. 14A: Tsuji T, Yorimitsu H, Oshima K. Angew Chem Int Ed. 2002;41:4137–4139. doi: 10.1002/1521-3773(20021104)41:21<4137::AID-ANIE4137>3.0.CO;2-0.Someya H, Ohmiya H, Yorimitsu H, Oshima K. Org Lett. 2008;10:969–971. doi: 10.1021/ol800038a.Sai M, Yorimitsu H, Oshima K. Bull Chem Soc Jpn. 2009;82:1194–1196.
- 38.Fig. 14B: Mitamura Y, Asada Y, Murakami K, Someya H, Yorimitsu H, Oshima K. Chem Asian J. 2010;5:1487–1493. doi: 10.1002/asia.201000068.
- 39.Schreiber SL. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 40.Bienstock RJ, editor. Library Design, Search Methods, and Applications of Fragment-Based Drug Design. American Chemical Society; Washington, D. C: 2011. [Google Scholar]
- 41.Lovering F, Bikker J, Humblet C. J Med Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 42.Lovering F. Med Chem Commun. 2013;4:515–519. [Google Scholar]
- 43.Very recently, nickel-catalyzed decarboxylative alkyl–alkyl cross-couplings have been reported: Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS. Science. 2016;352:801–805. doi: 10.1126/science.aaf6123.Johnston CP, Smith RT, Allmendinger S, MacMillan DWC. Nature. 2016;536:322–325. doi: 10.1038/nature19056.
- 44.Reductive alkyl–alkyl cross-couplings of alkyl electrophiles have been described: Yu X, Yang T, Wang S, Xu H, Gong H. Org Lett. 2011;13:2138–2141. doi: 10.1021/ol200617f.Liu JH, Yang CT, Lu XY, Zhang ZQ, Xu L, Cui M, Lu X, Xiao B, Fu Y, Liu L. Chem Eur J. 2014;20:15334–15338. doi: 10.1002/chem.201405223.