

SUMMARY
The development and function of epithelia depend on the establishment and maintenance of cell-cell adhesion and intercellular junctions, which operate as mechanosensor hubs for the transduction of biochemical signals regulating cell proliferation, differentiation, survival, and regeneration. Here, we show that αE-catenin, a key component of adherens junctions, functions as a positive regulator of pancreatic islet cell lineage differentiation by repressing the sonic hedgehog pathway (SHH). Thus, deletion of αE-catenin in multipotent pancreatic progenitors resulted in (1) loss of adherens junctions, (2) constitutive activation of SHH, (3) decrease in islet cell lineage differentiation, and (4) accumulation of immature Sox9+ progenitors. Pharmacological blockade of SHH signaling in pancreatic organ cultures and in vivo rescued this defect, allowing αE-catenin-null Sox9+ pancreatic progenitors to differentiate into endocrine cells. The results uncover crucial functions of αE-catenin in pancreatic islet development and harbor significant implications for the design of β cell replacement and regeneration therapies in diabetes.
Graphical abstract
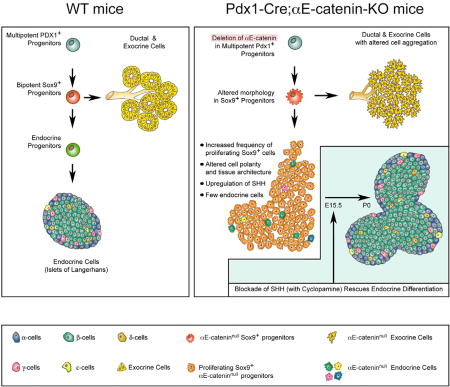
Jimenez-Caliani et al. examine a regulatory function for αE-catenin in the endocrine differentiation of pancreatic progenitors. Ablation of αE-catenin in multipotent Pdx1+ progenitors disrupts cell-cell adhesion and leads to constitutive activation of SHH signaling that precludes endocrine differentiation and leads to the accumulation of proliferating Sox9+ cells. Pharmacological blockade of SHH rescues the competency of αE-cateninnullSox9+ progenitors to acquire an endocrine phenotype.
INTRODUCTION
Epithelial tissues are rich in different types of intercellular junctions, including desmosomes, tight junctions, adherens junctions, and gap junctions, which collectively ensure the adhesion of cells to each other, and modulate a number of intercellular signaling pathways that are crucial for the establishment and maintenance of cell polarity, cell differentiation, proliferation, survival, and function, during both embryonic and postnatal life (Kobielak and Fuchs, 2004; Perez-Moreno and Fuchs, 2006; Pokutta and Weis, 2007; Rimm et al., 1995). In epithelial systems, adherens junctions provide for a mechanical docking between the cytoskeleton of adjacent cells through the stabilizing function of β-catenin and αE-catenin (Perez-Moreno et al., 2003). While β-catenin has been extensively studied for its contribution to the homeostasis of junctional complexes and the regulation of the Wnt pathway in tissues derived from all three germ layers, the function of αE-catenin has been primarily studied in ectoderm derivatives, in which it negatively regulates the activity of the MAPK/ERK, SHH, and Hippo pathways (Flores and Halder, 2011; Lien et al., 2006a, 2006b; Vasioukhin et al., 2001). Irrespective of the cell context, significant evidence indicates that αE-catenin can also inhibit β-catenin signaling through a mechanism of transcriptional repression of Wnt target genes (Choi et al., 2013; Daugherty et al., 2014; Giannini et al., 2000).
The pancreatic epithelium provides an interesting model to investigate the function of αE-catenin, as this tissue is composed of distinct cell lineages (i.e., ductal, acinar, and endocrine) arising from common Pdx1+ multipotent progenitors (Pan and Wright, 2011), engaging both the Wnt and the SHH pathways early during development, and at later stages of cell lineages differentiation (Cervantes et al., 2010; Hebrok et al., 1998, 2000; Heiser et al., 2006; Murtaugh et al., 2005). Alterations of such a complex differentiation program are thought to be causal to severe clinical conditions including diabetes, pancreatitis, and cancer (Puri and Hebrok, 2010).
In this study, we report that αE-catenin functions as a selective positive regulator of the pancreatic islet cell lineage differentiation through the repression of the SHH pathway. Thus, we show that the genetic ablation of αE-catenin in Pdx1+ multipotent pancreatic progenitors results in altered cell-cell aggregation, constitutive activation of SHH signaling, dramatic reduction of endocrine cell differentiation, and accumulation of Sox9+ pancreatic progenitors. Furthermore, chemical blockade of SHH signaling rescues this defect in Pdx1-Cre;αE-catenin-KO embryos. These results uncover hitherto unknown functions of αE-catenin in the development of the endocrine pancreatic cell lineage and harbor significant implications for the design of replacement and regeneration therapies to treat diabetes.
RESULTS
Targeting the Deletion of αE-Catenin to Pdx1+ Progenitors Disrupts the Architecture of the Pancreatic Epithelium
We used a Cre-mediated strategy to ablate a floxed αE-catenin allele in Pdx1+ pancreatic progenitors, by breeding Pdx1-CreEarly mice, henceforth referred to as Pdx1-Cre, with αE-cateninflox/flox mice (Figure S1A) to generate Pdx1-Cre;αE-cateninflox/− heterozygous mice. These animals were then crossed back to α-cateninflox/flox animals to obtain Pdx1-Cre;αE-catenin−/− homozygous recombinant mice (Figure S1B), henceforth referred to as Pdx1-Cre;αE-catenin-KO.
At birth (P0), the pancreas of heterozygous Pdx1-Cre;α-cateninflox/− mice did not reveal notable abnormalities in tissue architecture and cellular composition (data not shown). In contrast, the pancreas of homozygous Pdx1-Cre;αE-catenin-KO offsprings exhibited altered cell-cell aggregation (Figure 1B). Thus, compared to wild-type (WT) littermates, exhibiting compact acinar and islet tissues (Figure 1A), the pancreatic epithelium of Pdx1-Cre;αE-catenin-KO mice had a fragmented appearance, with loosely associated epithelial cells (Figure 1B). Immunostaining detected αE-catenin in the exocrine and endocrine compartments of the pancreas of WT mice (Figure 1C), but not in those of Pdx1-Cre;αE-catenin-KO mice (Figure 1D), demonstrating the successful Cre-mediated deletion of αE-catenin under control of the Pdx1 promoter.
Figure 1. Deletion of αE-Catenin in Pdx1+ Pancreatic Progenitors Disrupts Tissue Architecture and Endocrine Differentiation.

(A and B) H&E staining of P0 pancreatic sections from WT (A) and Pdx1-Cre;αE-catenin mutants (B) reveals that in the absence of αE-catenin the pancreatic epithelium looses its ability to develop a compact architectural organization.
(C–H) Immunolocalization of exocrine and endocrine markers in the pancreas of WT (C, E, and G) and Pdx1-Cre;αE-catenin-KO mice (D, F, and H) shows that ablation of αE-catenin in Pdx1+ pancreatic progenitors severely impairs the differentiation of the islet cell lineage and causes the association of hormone-negative cells into islet-like clusters (D and F, arrowheads).
(I and J) Morphometric assessment of islet cell-type representation (I) and frequency of islet hormone+ cell clusters distribution (J). Reference bar, 50 µm. Data in (I) and (J) are presented as mean ± SEM from n = 8 WT and n = 8 Pdx1-Cre;αE-catenin-KO pancreata. **p < 0.001.
Ablation of αE-Catenin Selectively Impairs the Ability of Pdx1+ Progenitors to Produce Islet Tissue, Resulting in the Accumulation of Putative Sox9+ Islet Progenitors
We found that the ablation of αE-catenin did not impair the differentiation of the exocrine cell lineage, as measured by positive immunostaining for amylase (Figure 1F). In contrast, the differentiation of the endocrine cell lineage appeared impaired, inasmuch as only few cells positive for either insulin, glucagon, somatostatin, pancreatic polypeptide (PP), or ghrelin were detected in amylase-negative islet-like cell clusters (Figures 1D and 1F, arrowheads). Morphometric analysis confirmed that the number of the endocrine cells producing insulin, glucagon, somatostatin, PP, or ghrelin, was reduced in the pancreas of Pdx1-Cre;αE-catenin-KO mice (Figure 1I). The size of hormone-positive cell clusters was also significantly lower in the pancreata of Pdx1-Cre;αE-catenin-KO as compared to that detected in WT mice (Figure 1J).
Immunostaining for transcription factors known to define developing islet progenitors revealed that most of the hormone-negative cells identified in islet-like clusters expressed Sox9+ and that the cells expressing this transcription factor were about four times more numerous in the pancreas of Pdx1-Cre;αE-catenin-KO than of WT mice (Figures 2A, 2B, and 2I). In contrast, the transcription factors Pdx1 and Nkx6.1 were detected only in the very few insulin-positive cells still identifiable in the mutant pancreas (Figures 2D and 2F). Immunoreactivity for Ngn3 was virtually undetectable in the mutant tissue (Figure 2H). Gene expression profiling by qPCR revealed that the transcripts coding for these three transcription factors were significantly lower in the pancreas Pdx1-Cre;αE-catenin-KO as compared to WT mice (Figure 2J).
Figure 2. Accumulation of Sox9+ Putative Progenitors in the Pancreas of Pdx1-Cre;αE-Catenin Mutants.

(A, C, E, and G) At P0, the expression of Sox9 in WT pancreata is restricted to ductal domains (A), whereas Pdx1 (C) and Nkx6.1 (E) are found only in islet β cells, some of them expressing low levels of Ngn3 (G).
(B, D, F, and H) This pattern is disrupted in the Pdx1-Cre;αE-catenin mutant pancreas, which displays an accumulation of Sox9+ cells, mostly arranged as loosely aggregated clusters (B, D, F, and H) and a minimal immunoreactivity for Pdx1 (D), Nkx6.1 (F), and Ngn3 (H).
(I and J) Morphometry reveals 4-fold increase of Sox9+ cells in Pdx1-Cre;αE-catenin mutants (I). The expression of Pdx1, Nkx6.1, and Ngn3 is also decreased at the transcriptional level (J). Data in (I) and (J) are presented as mean ± SEM from n = 6 WT and n = 6 Pdx1-Cre;αE-catenin mutant pancreata; **p < 0.001.
(K–N) Analysis of the cellular localization of E-cadherin and β-catenin in WT pancreata reveals a typical cell-surface pattern, restricted to cell-cell contacts (K and M, insets), whereas mutant Pdx1-Cre;αE-catenin pancreata also show significant intracellular staining (L and N, insets), suggesting reduced engagement in adhesion complexes. Reference bar, 50 µm.
Collectively, these results suggest that, in the absence of αE-catenin, clusters of pancreatic epithelial cells exhibiting an islet-like organization are populated by a large pool of Sox9+ progenitors that are unable to further develop into endocrine cells.
αE-Catenin Is Required for the Establishment of Adhesion Complexes and the Architectural Organization of the Pancreatic Epithelium
Epithelial cell-cell interactions play crucial functions in the establishment and maintenance of cell polarity, which, in turn, positively regulates cellular differentiation. To further characterize possible downstream effects of αE-catenin ablation in Pdx1+ pancreatic progenitors, we investigated the expression pattern of E-cadherin, a primary regulator of direct cell-cell aggregation and formation of adherens junctions in epithelia, and its intracellular partner β-catenin, which acts as a mechano-signaling coupler and stabilizer of adherens junctions in polarized epithelia. These experiments revealed that in the WT pancreatic epithelium, E-cadherin-specific immunoreactivity was restricted to cell-cell boundaries (Figure 2K, inset), whereas, in the pancreas of Pdx1-Cre;αE-catenin-KO mice, the protein was detected both at the cell surface and in the cytoplasm, suggesting that, in the absence of αE-catenin, E-cadherin may be weakly associated to β-catenin complexes (Figure 2L, inset). Accordingly, we noted a similar expression pattern in WT versus Pdx1-Cre;αE-catenin-KO for β-catenin (Figures 2M, and 2N, insets).
Immunostaining for other components of junctional complexes revealed that in WT pancreata Claudin-3 and Claudin-7 were mostly expressed in ductal cells and at lower levels in endocrine and acinar cells (Figures 3B and 3E, respectively). In contrast, the immunoreactivity ϕoρ both proteins was increased in all cell types of the pancreatic epithelium in Pdx1-Cre;α-catenin-KO mice (Figures 3H and 3K). We also assessed the expression pattern of other proteins associated with, and/or regulating the stability of, adhesion complexes such as Plakophilin-3, Desmoplakin, and Desmoglein (Chun and Hanahan, 2010). The results of these experiments revealed a high Phakophilin-3-specific immunoreactivity in the cytoplasm of the pancreatic epithelium of mutant Pdx1-Cre;α-catenin-KO mice, compared to the membrane-restricted pattern observed in the WT pancreas (Figure S2). Upregulation and cytoplasmic mis-localization of Desmoplakin (Figure S3) and Desmoglein (Figure S4) was also noted, as previously reported in hyperproliferating epithelia (Getsios et al., 2004).
Figure 3. Cell-Cell Aggregation and Tissue Architectural Organization Are Altered in the Pancreatic Epithelium of Pdx1-Cre;αE-Catenin-KO Mice.

(A–L) Pancreatic sections from P0 WT (A–F) and Pdx1-Cre;αE-catenin-KO mice (G–L) immunostained for markers of cell polarity Claudin-3 (B and H) and Claudin-7 (E and K) and E-cadherin (A, D, G, J). Co-staining with insulin (blue) is shown in (C), (I), (F), and (L). Note the strong immunoreactivity for both Claudin-3 and Cladin-7 throughout the pancreatic epithelium of Pdx1-Cre;αE-catenin-KO; (G–L), with cells arranged into disorganized cord-like clusters lacking the typical ductal or islet architecture.
(M–O) Ultrastructural analysis by TEM identifies close cell-cell aggregation in islet (M, arrowheads), duct (N), and acinar cells (O) in the pancreas of WT mice, with notable electron-dense plasma membrane domains characteristic of junctional complexes sealing the basolateral from the apical domains of ductal and intra-acinar lumens (circles in N and O).
(P–R) In contrast, all epithelial cell types identified in Pdx1-Cre;αE-catenin-KO pancreata exhibit profound disruption of cell-cell interactions, with rare β- (P), ductal (Q), and acinar cells (R) barely contacting with each other through a dense network of lamillipodia-like projections, across enlarged intercellular spaces (*), and lacking recognizable junctional complexes. Reference bar in (A)–(L), 50 µm, and in (M)–(R), 1 µm.
Ultrastructural studies to assess the state the cell-cell aggregation by transmitted electron microscopy (TEM) of WT pancreata showed that endocrine (Figure 3M), ductal (Figure 3N), and acinar cells (Figure 3O) closely adhered to companion cells, with narrow intercellular spaces between adjacent cells (Figure 3M, arrowheads). Most notable, ductal and acinar cells exhibited numerous junctional complexes, highlighted by the electron-dense concentration of cytoskeletal filaments binding to junctional components (red circles in Figures 3N and 3O). In sharp contrast, the ultrastructural analysis of Pdx1-Cre;αE-catenin-KO pancreata disclosed a significantly altered cell-cell adhesion of endocrine (Figure 3P), ductal (Figure 3Q), and acinar cells (Figure 3R). Thus, intercellular spaces were markedly enlarged (Figures 3P, 3Q, and 3R, stars) and were populated by numerous filopodia-like projections, whereas cell membranes lacked recognizable junctional complexes (Figures 3P–3R). Quantitative analysis revealed close to a 15-fold increase of the intercellular space in Pdx1-Cre;αE-catenin-KO compared to WT pancreata, further supporting the notion that in the absence of αE-catenin the pancreatic epithelium is unable to establish intimate cell-cell adhesion complexes (Figures S5, S6, and S7).
αE-Catenin Is Required for Sox9+ Pancreatic Progenitors to Adopt an Endocrine Fate at Early Stages of Islet Neogenesis
To determine whether the pancreatic phenotype observed at P0 in Pdx1-Cre;αE-catenin-KO mice could be traced back to embryonic stages of islet cell neogenesis, we examined the phenotype of the pancreas at E15.5, a stage when significant endocrine differentiation is occurring and islet cells start to cluster. While the pancreas of WT mice contained a normal endocrine compartment (Figures 4A, 4G, and 4I), Pdx1-Cre;αE-catenin-KO pancreata exhibited a significant reduction in the frequency of insulin and glucagon-containing cells (Figures 4B, 4H, and 4I) and in the number of Ngn3+, Pdx1+, and Nk6.1+ progenitors (Figures 4D, 4F, 4H, and 4I). Concomitantly, we observed a significant increase in the number of Sox9+ cells, which appeared loosely clustered and did not form the ductal structures typical of WT pancreata (Figures 4B, 4D, 4F, and 4H). qPCR confirmed a dramatic reduction of Ngn3, Pdx1, and Nk6.1 transcripts in the pancreatic epithelium of Pdx1-Cre;αE-catenin-KO mice (Figure 4J). These results point to a positive regulatory function of αE-catenin in the endocrine commitment and differentiation of Sox9+ progenitors.
Figure 4. αE-Catenin Is Required for Islet Cell Lineage Commitment and Differentiation during Development.

(A–I) Pancreatic sections from E15.5 WT (A, C, E, and G) and Pdx1-Cre;αE-catenin-KO embryos (B, D, F, and H) immunostained for Ngn3 (C and D), Pdx1 (E and F), and Nkx6.1 (G and H) reveals a significant reduction in numbers of endocrine/progenitor cells in Pdx1-Cre;αE-catenin-KO embryos (B, D, F, and H), whereas the frequency of multipotent pancreatic progenitors marked by Sox9 is increased at this early stage of islet cell development (I).
(J) Expression of transcription factors Ngn3, Pdx1, and Nkx6.1 is also reduced at the transcriptional level (J) in mutant Pdx1-Cre;αE-catenin-KO embryos.
Data in (I) and (J) are presented as mean ± SEM from n = 6 WT and n = 6 Pdx1-Cre;αE-catenin-KO pancreata; *p < 0.01; **p < 0.001. Reference bar in (A)–(H), 50 µm.
Ablation of αE-Catenin in Pancreatic Progenitors Causes the Activation of the MAPK/ERK and Sonic Hedgehog Pathways, Leading to a Sustained Proliferation of Sox9+ Cells
We investigated the state of activation of the MAPK/ERK and SHH pathways, two signaling mechanisms that enhance cell growth and can be repressed by αE-catenin (Lien et al., 2006a; Vasioukhin et al., 2001). We found that, at the P0 stage, the pancreas of Pdx1-Cre;αE-catenin-KO mice exhibited an increased frequency of the overall number of PCNA+ cells (Figure 5B), and more proliferating Sox9+ cells (Figure 5C), when compared to WT pancreata (Figures 5A and 5C). In parallel experiments, western blotting analysis revealed that the pancreatic epithelium of Pdx1-Cre;αE-catenin-KO mice exhibited a significant upregulation of Sox9, and increased levels of p-Erk1/2, when compared to WT samples (Figure 5D). Together, these results suggest that both the upregulation of Sox9 and the activation of the MAPK/ERK, known positive regulators of cell-cycle progression (Bastide et al., 2007) (Rovida and Stecca, 2015), may contribute to the increased cell proliferation observed in the pancreatic epithelium of Pdx1-Cre;αE-catenin-KO mice.
Figure 5. Sustained Proliferation of Sox9+ Cells and Activation of the MAPK/ERK in Pdx1-Cre;αE-Catenin-KO Pancreas.

(A–C) At P0, the WT pancreas (A) exhibits a compartmentalization of Sox9+ cells within the ductal epithelium. In contrast, in Pdx1-Cre;αE-catenin-KO embryos (B) Sox9+ cells are found disorderly distributed throughout the pancreatic epithelium. Immunostaining (A and B; green) shows that PCNA+ cells are 4 times more frequent in the Pdx1-Cre;αE-catenin-KO than in the WT pancreas (C). Data in (C) are presented as mean ± SEM of 8 pancreata in each group; **p < 0.001; Reference bar, 50 µm.
(D) Western blotting analysis revealed a significant upregulation of Sox9 protein and levels of phosphorylated Erk1/2.
Data shown are representative of 3 independent experiments.
Next, based on the notion that MAPK ERK1/2 can activate Hedgehog signaling in a number of cell types (Ji et al., 2007; Stecca et al., 2007) and that SHH signaling antagonizes pancreatic development (Hebrok et al., 1998, 2000), we sought to determine the expression levels of key genes required for SHH signaling. These studies revealed that all components of the SHH pathway were significantly upregulated, during both fetal (E15.5) and perinatal life (P0) (Figures 6A and 6B). Together, these results provide evidence for a positive regulatory role of αE-catenin in pancreatic development and specifically in the endocrine differentiation of the islet cell lineage, through the repression of MAPK/ERK and SHH pathways. Hence, the impaired endocrine cell development observed in Pdx1-Cre;αE-catenin-KO pancreata may stem from an untamed activation of the SHH pathway.
Figure 6. Constitutive Activation of the Sonic Hedgehog Pathways in Pdx1-Cre;αE-Catenin-KO Pancreas Prevents the Acquisition of an Endocrine Cell Phenotype.

The expression of genes positively regulating the activity of the SHH pathways is significantly upregulated in the pancreatic epithelium of Pdx1-Cre;αE-catenin-KO pancreas, both during development at E15.5 (A) and at birth day P0 (B).
(A and B) Data are shown as mean ± SEM from n = 4 independent experiments; *p < 0.05; **p < 0.001.
(C–M) Organ cultures of P0 pancreata from WT (C–E) and Pdx1-Cre;αE-catenin-KO mice (F–H) under control conditions. Addition of cyclopamine to organ cultures from Pdx1-Cre;αE-catenin-KO pancreata (I–K) rescued endocrine cell differentiation. Cyclopamine treatment normalizes the overall endocrine cell area (L) and reduces the frequency of proliferating Sox9+ cells to levels close to those measured in WT pancreata (M). qPCR data presented in (A) and (B) are from 4 independent experiments. Morphometric analysis on organ cultures (C–M) was performed on samples from n = 8 independent experiments. Reference bar, 50 µm.
Inhibition of the Sonic Hedgehog Pathway Rescues the Differentiation and Development of Endocrine Cells in Pdx1-Cre;αE-Catenin-KO Pancreata
To functionally validate the involvement of SHH signaling in the observed phenotype of Pdx1-Cre;αE-catenin-KO mice and determine whether the mutant pancreatic epithelium could regain the ability to acquire an endocrine fate under conditions that inhibit SHH activation, we conducted organ culture experiments in which P0 pancreata were treated with cyclopamine, a steroid alkaloid that blocks SHH signaling (Cooper et al., 1998). The results of these studies revealed that after 3 days in culture in the presence of cyclopamine, the pancreatic epithelium isolated from P0 Pdx1-Cre;αE-catenin-KO mice had fully recovered the ability to produce all endocrine cell types (Figures 6C–6K). These conditions also decreased the frequency of Sox9+ cells, as well as the rate of proliferation, as determined by the enumeration of PCNA+ cells (Figures 6E, 6H, 6K, and 6M). Interestingly, Pdx1-Cre;αE-catenin-KO pancreata cultured in the presence of cyclopamine consistently produced higher numbers of insulin+, glucagon+, somatostatin+, and ghrelin+ cells than WT explants (Figure 6L). This suggests that Sox9+ cells accumulating in Pdx1-Cre;αE-catenin-KO pancreata represent a large pool of endocrine committed precursors that are unable to proceed further in their differentiation program when SHH is activated.
Given that the in vitro conditions do not fully recapitulate the native in vivo pancreatic microenvironment, we next assessed the effects of cyclopamine in vivo. Pregnant dams from Pdx1-Cre;α-cateninflox/−;α-cateninflox/flox mating were injected intraperitoneally with cyclopamin for 6 consecutive days, starting at day E15.5 (Figure 7A). In P0 pups, we observed a complete rescue of the pancreatic phenotype in Pdx1-Cre;αE-catenin-KO mice, with normal-looking islet clusters (Figure 7B), and normalized insulin+ and glucagon+ areas (Figure 7C, upper panels). In addition, Sox9+ cells regained a duct restricted distribution (Figure 7B, red), as observed in WT pancreata, and were no longer found as loosely aggregated islet-like clusters (Figure 2B). Finally, the frequency of Sox9+PCNA+ cells was also reduced to levels similar to those recorded in WT mice (Figure 7C, lower panel). Collectively, these in vivo experiments demonstrate that blockade of SHH rescues the ability of Sox9+ αE-cateninnull cells to acquire an endocrine phenotype, even in the absence of αE-catenin.
Figure 7. In Vivo Blockade of Sonic Hedgehog in Pdx1-Cre;αE-Catenin-KO Pancreas Rescues Endocrine Cell Differentiation.

(A) Daily injection of cyclopamine in pregnant mice, starting at E15.5, rescues the ability of putative pancreatic progenitors contained in the pancreas of Pdx1-Cre;αE-catenin-KO mice to acquire an endocrine phenotype, as shown by immunostaining for insulin, (green).
(B) Two adjacent microscopic fields from the same pancreas, identifying numerous insulin+ cell clusters (green). A normal-looking compartmentalization of Sox9+ cells to ductal domains (red) can also be observed.
(C) Morphometric analysis performed on pancreata immunostained for insulin and glucagon shows normalized values of insulin+ area, and slight increase in glucagon+ areas (C). The frequency of Sox9+PCNA+ cells is also reduced to levels close to those measured in WT pancreata (C).
Data are presented as mean ± SEM from 6 independent experiments, each comprising three to five Pdx1-Cre;αE-catenin-KO and five to eight WT animals. Reference bar, 50 µm.
DISCUSSION
The establishment and maintenance of specialized cellular phenotypes that dictate tissue organization depend on cell-autonomous gene activation programs, as well as on the ability of individual cells to communicate with one another. In this respect, adherens junctions play crucial functions as they not only mediate the mechanical adhesion of contacting cells but also regulate a number signaling pathways, which collectively contribute to the acquisition and maintenance of differentiated cellular functions (Kobielak and Fuchs, 2004; Perez-Moreno and Fuchs, 2006; Pokutta and Weis, 2007; Rimm et al., 1995). In this study, we demonstrate that αE-catenin, a critical component of adherens junctions, is required for the development of a proper tissue architecture of the pancreatic epithelium and for the differentiation of the endocrine cell lineage, through a negative regulation of the SHH signaling pathway.
Our results demonstrate that the genetic deletion of αE-catenin in early Pdx1+ pancreatic progenitors prevents them from establishing stable adhesive contacts, thus impairing their development into a normal, compact epithelium, in both the exocrine and endocrine compartments of the mouse pancreas. In spite of this global disorganization, the differentiation of the exocrine cell lineage proceeded normally, as judged by the expression of amylase, whereas that of endocrine cells was significantly prevented, as assessed by the decreased expression of islet hormones. These results point to distinct requirements of signaling pathways downstream of αE-catenin for the specification of the exocrine and endocrine pancreatic cell lineages, thus uncovering a previously unappreciated functional dichotomy with respect to the requirement of αE-catenin in the acquisition of specific cell phenotypes.
Within putative regions of islet formation, the abnormal increase in the frequency of Sox9+ cells, many of which proliferated, points to the activation of pro-growth pathways that negatively impact on the ability of these precursors to differentiate. Hence, we observed that the loss of αE-catenin sustains the activation of the MAPK and SHH pathways, as measured by increased levels of phosphorylated Erk1/2, and elevated expression of GL1, GL2, SMO, and PTCH1, respectively. Furthermore, and at variance with WT pancreata in which Sox9+ only populated the ductal epithelium, in the pancreas of Pdx1-Cre;αE-catenin-KO mutants Sox9+ cells lost their ductal restricted distribution and formed islet-like clusters that failed to acquire markers of endocrine determination such as Pdx1, Ngn3, or Nkx6.1 and islet hormones.
During embryonic life, with the onset of the “secondary transition” at ~E13.5, large numbers of pancreatic progenitors are recruited from trunk regions of the ductal epithelium to enter an endocrine specification fate. This event, referred to as islet cell neogenesis, is marked by the downregulation of Sox9, and the transient expression of the transcription factor Ngn3 (Gradwohl et al., 2000; Gu et al., 2002; Jacquemin et al., 2000; Schwitzgebel et al., 2000), followed by the induction of the islet-specific hormones insulin, glucagon, somatostatin, pancreatic polypeptide, and ghrelin (Pan and Wright, 2011). Our studies show that these events are largely compromised in the absence of αE-catenin, resulting in a significant accumulation of Sox9+ progenitors. The finding that these alterations are associated with an increased proliferation of Sox9+ cells and a concomitant upregulation of Sox9 protein levels, are consistent with previous work demonstrating that this transcription factor can also function as a downstream target of MAPK (Murakami et al., 2000; Zhang et al., 2006) and promote intestinal and pancreatic progenitor cell proliferation (Bastide et al., 2007; Seymour et al., 2007).
Collectively, these observations support a plausible scenario in which loss of αE-catenin in early pancreatic progenitors prevents their ability to establish functional junctional complexes and cell polarity (Bryant and Mostov, 2008; Drees et al., 2005; Perez-Moreno and Fuchs, 2006; Vasioukhin et al., 2000; Yamada et al., 2005), as supported by our ultrastructural observations, which in turn leads to a sustained activation of the MAPK. In addition, the observed upregulation of genes required for SHH activity, known to enhance the growth of pancreatic epithelial cells (Reichert and Rustgi, 2011; Thayer et al., 2003), further interferes with the endocrine differentiation of Sox9+ putative progenitors (Hebrok et al., 1998, 2000). In agreement with this scenario, we observed that the pharmacological blockade of SHH signaling by cyclopamine restores the competency for the endocrine differentiation of Sox9+ αE-cateninnull cells, both in vitro and in vivo. Interestingly, the in utero blockade of SHH in Pdx1-Cre;αE-catenin-KO mice also restored the spatial positioning of Sox9+ cells within ductal structures, the organization of endocrine cells into islet clusters, and decreases the frequency of proliferating Sox9+ cells. Our results show that αE-catenin is a critical upstream regulator of the SHH activity that controls multiple steps of pancreatic development, spanning from the establishment and maintenance of adhesion complexes, acquisition of cell polarity, lineage specification, expansion, and differentiation.
EXPERIMENTAL PROCEDURES
Ethics Statement
All animal experiments were performed according to the Institutional Animal Care and Use Committee (IACUCC) procedures approved by the University of Washington. Experimental procedures and analysis described in this study were performed using both males and females mice, between embryonic age E13.5 and postnatal age P10, with no significant difference in results between the two sexes.
Generation of Pdx1-Cre;αE-Catenin-KO Mice and Genotyping
Mice containing a floxed allele of αE-catenin (i.e. αE-cateninfl/fl) were previously described (Vasioukhin et al., 2001). To generate conditional deletion of αE-catenin in pancreatic progenitors, we utilized a Pdx1-cre transgenic strain, a generous gift of Dr. Dough Melton (Harvard University, Boston, MA), previously reported to mediate efficient recombination in the developing pancreatic epithelium (Gu et al., 2002). Genomic DNA was extracted from mouse tail using DNeasy Blood & Tissue kit (QIAGEN) according to the manufacturer’s instructions. To identify Cre, the following primers were used to amplify a 475-bp product: forward 5′-AGATGTTCGCGATTATCTTC-3′; reverse 5′-AGCTACACCAGAGACGG-3′. PCR conditions were 94°C for 3 min and then 30 cycles at 94°C for 30 s, 58°C for 45 s, and 72°C for 45 s, followed by 72°C for 5 min. To identify αE-cateninfl/fl animals, we used the following primers: forward 5′-ACTGCCTTTGTTCTCTTCCCTTCTG-3′; reverse 5′-CAGCCAAGGAGAGCAGGTGAGG-3′, amplifying a 150-bp product for WT αE-catenin, and a 200-bp product for αE-cateninfl/fl mice. PCR conditions were 94°C for 3 min and then 30 cycles at 94°C for 30 s, 63°C for 45 s, and 72°C for 45 s, followed by 72°C for 5 min.
Histology and Morphometric Analysis
Pancreatic tissue from embryos and newborn mice was fixed in 4% formaldehyde, from freshly prepared paraformaldehyde in PBS overnight, dehydrated, embedded in paraffin, and sectioned at 5 µm, as previously described (Diaferia et al., 2013). Antigen retrieval was performed in citrate buffer (pH 6.0). For frozen sections, tissues were fixed for 4 hr at 4°C in 4%formaldehyde (from paraformaldehyde), washed in cold PBS, and placed overnight in 30% sucrose (w/v) in PBS, prior to embedding and freezing in OCT, and sections were cut at 5 µm. Sections were then permeabilized and incubated in blocking buffer containing 1% normal donkey serum in 0.1% Triton × 100 in PBS for 30 min at room temperature. Apoptosis was detected by transferase-mediated dUTP nick-end labeling (TUNEL) and a digoxygenin-labeling kit (Chemicon). The primary antibodies used in this study are summarized in the Table S1. Fluorophore-conjugated secondary antibodies used (FITC, Rhodamine, Cy5) were all from Jackson ImmunoResearch. Morphometric analysis for the quantitative assessment of cell-type representation was performed on every fifth section throughout the entire newborn pancreas or on every third section of embryonic pancreata. Image analysis was conducted using NIS-Elements AR 3.2 (Nikon).
Western Blotting
Total protein extracts from freshly dissected newborn pancreata were prepared in lysis buffer (10 mM Tris-HCl [pH 7.8]; 150 mM NaCl; 1 mM EDTA; protease inhibitor cocktail tablet-IP 1×, MALT, 1 mM DTT, 1 mM PMSF), separated on an SDS-Page gel (30 µg/lane) and transferred onto a nitrocellulose membrane as previously described (Diaferia et al., 2013; Yebra et al., 2003). Membranes were then washed in PBST (1 × PBS, 0.1% Tween [pH 7.4]), blocked with 3% non-fat milk for 1 hr, incubated in primary antibody diluted in 1% BSA in PBST overnight at 4°C and then washed with PBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hr at room temperature. After washing, chemiluminescence was detected using the ECL Western Blotting Analysis System (Amersham/GE Healthcare).
RNA Extraction and qRT-PCR
RNA was isolated from pancreas using Aurum Total RNA Mini Kit (Bio-Rad) according to the manufacturer’s instructions and retrotranscribed into cDNA using the Superscript III (Invitrogen), and the primers are listed in Table S2. qPCRs were performed in triplicate using the Sybr Green kit (Bio-Rad), and data were analyzed using Bio-Rad CFX Manager software. Data are shown as ±SEM.
Organ Culture
Pancreas of newborn mice were split into four pieces and laid on Millicell culture plates inserts (Millipore) in 24-well plates containing 2 mL RPMI 1640 medium (Invitrogen) supplemented with penicillin (100 units/mL), streptomycin (100 µg/mL), HEPES (10 mmol/L), L-glutamine (2 mmol/L), nonessential amino acids (1×; GIBCO), 10% heat-inactive calf serum (HyClone), recombinant human FGF10 (50 ng/mL; R&D Systems), and heparin (50 µg/m; Sigma). Two of the pancreatic tissue pieces were cultured in the presence of cyclopamine (10 µM) (Thayer et al., 2003) and the other two in the absence of cyclopamine. Under these culture conditions, the explants grew at the air/medium interface (Attali et al., 2007). Cultures were maintained at 37°C in humidified 95% air/5% CO2 for 3 days, and media was changed daily. After three days, the pancreas were harvested and fixed in 4% formaldehyde, from freshly prepared paraformaldehyde, and later processed for paraffin embedding and morphometric studies.
In Vivo Administration of Cyclopamine
Pregnancies were timed by the detection of vaginal plugs, corresponding to E0.5. Pregnant mice were given daily i.p. injections of 2 mg/kg cyclopamine (Stemgent) complexed with 2-hydroxypropyl-β-cyclodextrin (HBC; Sigma) from day E15.5 to day 20 (van den Brink et al., 2001). A cyclopamine-HBC stock solution was produced by suspending 1 mg cyclopamine in 1 mL 45% HBC in sterile PBS and stirring for 60 min at 65°C. The cyclopamine-HBC stock was stored at −20°C until use. Mice were injected i.p. daily with 10 mg/kg of body weight, for six days. Six independent experiments were run, each comprising three to five Pdx1-Cre;αE-catenin-KO and five to eight WT animals.
Transmission Electron Microscopy
Ultrastructural analysis by transmission electron microscopy (TEM) was performed as previously described (Diaferia et al., 2013). Briefly, freshly microdissected pancreata were fixed for 1 hr in ice-cold 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer and then postfixed with 1% osmium tetroxide in 0.15 M cacodylate buffer at room temperature for 1 hr, dehydrated, and immersed in propylene oxide. After incubation in a mix of propylene oxide and EPON at room temperature, blocks were embedded in EPON and polymerized at 60°C for 48 hr. Ultrathin sections mounted on gold grids were then contrasted with 2% uranyl acetate and counterstained with Sato lead before analysis at a JEOL/JEM-1200 (120 kV) microscope.
Statistics
Were applicable, statistical significance of differences was validated by analysis of variance (ANOVA), followed by Bonferroni’s multiple comparison Test, or by two tailed Student’s t test, using the Prism-6 statistical package (Graph-Pad, San Diego, CA), with significance limit set at p < 0.05.
Supplementary Material
Highlights.
αE-catenin controls cell-to-cell architectural organization of pancreatic progenitors
Loss of αE-catenin in Pdx1+ cells leads to an increased pool of Sox9+ progenitors
Activation of SHH in αE-cateninnull/Sox9+ cells precludes endocrine specification
Blockade of SHH in αE-cateninnull/Sox9+ cells rescues their endocrine differentiation
Acknowledgments
We are grateful to Dr. Robert Florkiewicz (University of Washington, Department of Medicine, Seattle, WA), Dr. Valeri Vasioukhin (Fred Hutchinson Cancer Research Center, Seattle, WA), and members of the Cirulli and Crisa laboratories for their insightful advice and comments on the manuscript. We would like to thank Dr. Matthias Hebrok (University of California, San Francisco, San Francisco, CA) for his insightful advice on mechanisms of SHH inhibition for the in vivo experiments conducted in Pdx1-Cre;αE-catenin-KO mice. V.C. was supported by JDRF grants #1-2005-1084 and #1-2004-13, by the WA State Life Sciences Discovery Fund Program grant #4553677, and by R01 DK103711-01; L.C. was supported by RO1 HL075270 and WA State Life Sciences Discovery Fund Program grant #4553677; W.Y. was the recipient of a Pharm. Sci. Training grant #5T32GM007750-34, a pilot project from the Diabetes Research Connection, and a scholarship from the Howard Hughes Medical Institute (Med-Into-Grad program #56006778) to the University of Washington. G.R.D. was the recipient of a postdoctoral fellowship from the Juvenile Diabetes Research Foundation (JDRF). We also acknowledge the expert assistance of Thomas Deerinck and Mason Mackey at the National Center for Microscopy and Imaging Research, University of California, San Diego (La Jolla, CA) and Edward Parker at the Vision Science Center, University of Washington (Seattle, WA) for ultrastructural studies by transmission electron microscopy. The National Center for Microscopy and Imaging Research (University of California, San Diego) is supported by NIH grant #P41GM103412 to M.H. Ellisman and the Vision Science Center (University of Washington, Seattle) by NIH NEI Center Core Grant #P30 EY001730.
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.07.035.
AUTHOR CONTRIBUTIONS
A.J.J-C., R.P., W.Y., and G.R.D. performed experiments and analyzed data; P.M. analyzed TEM data; L.C. designed and performed experiments, analyzed data, and edited the manuscript; and V.C. designed experiments, performed TEM experiments, analyzed data, prepared the figures, and wrote the manuscript.
References
- Attali M, Stetsyuk V, Basmaciogullari A, Aiello V, Zanta-Boussif MA, Duvillie B, Scharfmann R. Control of beta-cell differentiation by the pancreatic mesenchyme. Diabetes. 2007;56:1248–1258. doi: 10.2337/db06-1307. [DOI] [PubMed] [Google Scholar]
- Bastide P, Darido C, Pannequin J, Kist R, Robine S, Marty-Double C, Bibeau F, Scherer G, Joubert D, Hollande F, et al. Sox9 regulates cell proliferation and is required for Paneth cell differentiation in the intestinal epithelium. J. Cell Biol. 2007;178:635–648. doi: 10.1083/jcb.200704152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, Mostov KE. From cells to organs: Building polarized tissue. Nat. Rev. Mol. Cell Biol. 2008;9:887–901. doi: 10.1038/nrm2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes S, Lau J, Cano DA, Borromeo-Austin C, Hebrok M. Primary cilia regulate Gli/Hedgehog activation in pancreas. Proc. Natl. Acad. Sci. USA. 2010;107:10109–10114. doi: 10.1073/pnas.0909900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Estarás C, Moresco JJ, Yates JR, 3rd, Jones KA. α-Catenin interacts with APC to regulate β-catenin proteolysis and transcriptional repression of Wnt target genes. Genes Dev. 2013;27:2473–2488. doi: 10.1101/gad.229062.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun MG, Hanahan D. Genetic deletion of the desmosomal component desmoplakin promotes tumor microinvasion in a mouse model of pancreatic neuroendocrine carcinogenesis. PLoS Genet. 2010;6:e1001120. doi: 10.1371/journal.pgen.1001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- Daugherty RL, Serebryannyy L, Yemelyanov A, Flozak AS, Yu HJ, Kosak ST, deLanerolle P, Gottardi CJ. α-Catenin is an inhibitor of transcription. Proc. Natl. Acad. Sci. USA. 2014;111:5260–5265. doi: 10.1073/pnas.1308663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaferia GR, Jimenez-Caliani AJ, Ranjitkar P, Yang W, Hardiman G, Rhodes CJ, Crisa L, Cirulli V. β1 integrin is a crucial regulator of pancreatic β-cell expansion. Development. 2013;140:3360–3372. doi: 10.1242/dev.098533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Halder G. Stem cell proliferation in the skin: alpha-catenin takes over the hippo pathway. Sci. Signal. 2011;4:pe34. doi: 10.1126/scisignal.2002311. [DOI] [PubMed] [Google Scholar]
- Getsios S, Huen AC, Green KJ. Working out the strength and flexibility of desmosomes. Nat. Rev. Mol. Cell Biol. 2004;5:271–281. doi: 10.1038/nrm1356. [DOI] [PubMed] [Google Scholar]
- Giannini AL, Vivanco Md, Kypta RM. alpha-catenin inhibits beta-catenin signaling by preventing formation of a beta-catenin*T-cell factor*DNA complex. J. Biol. Chem. 2000;275:21883–21888. doi: 10.1074/jbc.M001929200. [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Hebrok M, Kim SK, Melton DA. Notochord repression of endodermal Sonic hedgehog permits pancreas development. Genes Dev. 1998;12:1705–1713. doi: 10.1101/gad.12.11.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebrok M, Kim SK, St Jacques B, McMahon AP, Melton DA. Regulation of pancreas development by hedgehog signaling. Development. 2000;127:4905–4913. doi: 10.1242/dev.127.22.4905. [DOI] [PubMed] [Google Scholar]
- Heiser PW, Lau J, Taketo MM, Herrera PL, Hebrok M. Stabilization of beta-catenin impacts pancreas growth. Development. 2006;133:2023–2032. doi: 10.1242/dev.02366. [DOI] [PubMed] [Google Scholar]
- Jacquemin P, Durviaux SM, Jensen J, Godfraind C, Gradwohl G, Guillemot F, Madsen OD, Carmeliet P, Dewerchin M, Collen D, et al. Transcription factor hepatocyte nuclear factor 6 regulates pancreatic endocrine cell differentiation and controls expression of the proendocrine gene ngn3. Mol. Cell. Biol. 2000;20:4445–4454. doi: 10.1128/mcb.20.12.4445-4454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007;282:14048–14055. doi: 10.1074/jbc.M611089200. [DOI] [PubMed] [Google Scholar]
- Kobielak A, Fuchs E. Alpha-catenin: At the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 2004;5:614–625. doi: 10.1038/nrm1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Klezovitch O, Fernandez TE, Delrow J, Vasioukhin V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science. 2006a;311:1609–1612. doi: 10.1126/science.1121449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien WH, Klezovitch O, Vasioukhin V. Cadherin-catenin proteins in vertebrate development. Curr. Opin. Cell Biol. 2006b;18:499–506. doi: 10.1016/j.ceb.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Murakami S, Kan M, McKeehan WL, de Crombrugghe B. Upregulation of the chondrogenic Sox9 gene by fibroblast growth factors is mediated by the mitogen-activated protein kinase pathway. Proc. Natl. Acad. Sci. USA. 2000;97:1113–1118. doi: 10.1073/pnas.97.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh LC, Law AC, Dor Y, Melton DA. Beta-catenin is essential for pancreatic acinar but not islet development. Development. 2005;132:4663–4674. doi: 10.1242/dev.02063. [DOI] [PubMed] [Google Scholar]
- Pan FC, Wright C. Pancreas organogenesis: From bud to plexus to gland. Dev. Dyn. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- Perez-Moreno M, Fuchs E. Catenins: keeping cells from getting their signals crossed. Dev. Cell. 2006;11:601–612. doi: 10.1016/j.devcel.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Moreno M, Jamora C, Fuchs E. Sticky business: Orchestrating cellular signals at adherens junctions. Cell. 2003;112:535–548. doi: 10.1016/s0092-8674(03)00108-9. [DOI] [PubMed] [Google Scholar]
- Pokutta S, Weis WI. Structure and mechanism of cadherins and catenins in cell-cell contacts. Annu. Rev. Cell Dev. Biol. 2007;23:237–261. doi: 10.1146/annurev.cellbio.22.010305.104241. [DOI] [PubMed] [Google Scholar]
- Puri S, Hebrok M. Cellular plasticity within the pancreas—lessons learned from development. Dev. Cell. 2010;18:342–356. doi: 10.1016/j.devcel.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichert M, Rustgi AK. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Invest. 2011;121:4572–4578. doi: 10.1172/JCI57131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc. Natl. Acad. Sci. USA. 1995;92:8813–8817. doi: 10.1073/pnas.92.19.8813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovida E, Stecca B. Mitogen-activated protein kinases and Hedgehog-GLI signaling in cancer: A crosstalk providing therapeutic opportunities? Semin. Cancer Biol. 2015;35:154–167. doi: 10.1016/j.semcancer.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Schwitzgebel VM, Scheel DW, Conners JR, Kalamaras J, Lee JE, Anderson DJ, Sussel L, Johnson JD, German MS. Expression of neurogenin3 reveals an islet cell precursor population in the pancreas. Development. 2000;127:3533–3542. doi: 10.1242/dev.127.16.3533. [DOI] [PubMed] [Google Scholar]
- Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G, Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. USA. 2007;104:1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, Beermann F, Ruiz I Altaba A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA. 2007;104:5895–5900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, Qi YP, Gysin S, Fernández-del Castillo C, Yajnik V, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink GR, Hardwick JC, Tytgat GN, Brink MA, Ten Kate FJ, Van Deventer SJ, Peppelenbosch MP. Sonic hedgehog regulates gastric gland morphogenesis in man and mouse. Gastroenterology. 2001;121:317–328. doi: 10.1053/gast.2001.26261. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Yin M, Fuchs E. Directed actin polymerization is the driving force for epithelial cell-cell adhesion. Cell. 2000;100:209–219. doi: 10.1016/s0092-8674(00)81559-7. [DOI] [PubMed] [Google Scholar]
- Vasioukhin V, Bauer C, Degenstein L, Wise B, Fuchs E. Hyperproliferation and defects in epithelial polarity upon conditional ablation of alpha-catenin in skin. Cell. 2001;104:605–617. doi: 10.1016/s0092-8674(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yebra M, Montgomery AM, Diaferia GR, Kaido T, Silletti S, Perez B, Just ML, Hildbrand S, Hurford R, Florkiewicz E, et al. Recognition of the neural chemoattractant Netrin-1 by integrins alpha6beta4 and alpha3-beta1 regulates epithelial cell adhesion and migration. Dev. Cell. 2003;5:695–707. doi: 10.1016/s1534-5807(03)00330-7. [DOI] [PubMed] [Google Scholar]
- Zhang R, Murakami S, Coustry F, Wang Y, de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc. Natl. Acad. Sci. USA. 2006;103:365–370. doi: 10.1073/pnas.0507979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.