

Abstract
The promising clinical results obtained with engineered T cells, including chimeric antigen receptor (CAR) therapy, call for further advancements to facilitate and broaden their applicability. One potentially beneficial innovation is to exploit new T cell sources that reduce the need for autologous cell manufacturing and enable cell transfer across histocompatibility barriers. Here we review emerging T cell engineering approaches that utilize alternative T cell sources, which include virus-specific or T cell receptor-less allogeneic T cells, expanded lymphoid progenitors, and induced pluripotent stem cell (iPSC)-derived T lymphocytes. The latter offer the prospect for true off-the-shelf, genetically enhanced, histocompatible cell therapy products.
T cells are essential mediators of immune defense against infectious pathogens and cancer. Their insufficiency, which occurs in hereditary or acquired immune deficiencies, results in life threatening infections, increased cancer incidence, and disrupted immunoregulation. T cells can also be harmful and cause normal tissue destruction, as seen in autoimmune disorders, graft rejection, and graft-versus-host disease (GVHD). T cells develop from precursors that rearrange germline antigen receptor VDJ genes in the thymus, thereby generating clonotypic T cell receptors (TCRs) that undergo positive and negative thymic selection (Figure 1). The resulting T cells are self-restricted and tolerant of self tissues. The newly generated T cell clones, known as naive T cells, initially circulate throughout the body at low frequency. Upon encountering antigen, T cells expand and acquire effector and/or memory functions. This T cell priming requires TCR engagement by Human Leucocyte Antigen (HLA)-peptide complexes on the surface of antigen presenting cells (APCs) and concomitant ligation of costimulatory receptors by ligands borne by the APCs (Chen and Flies, 2013; Krogsgaard and Davis, 2005).
Figure 1. Human T Lymphocyte Development.
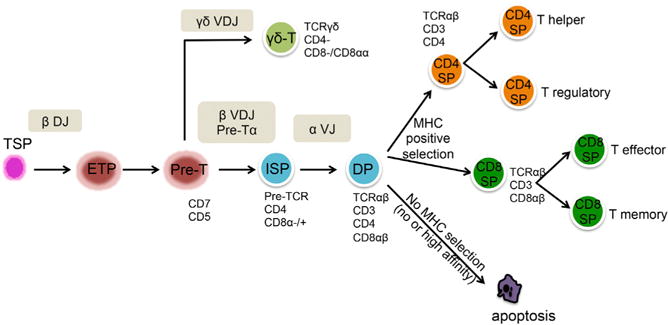
Hematopoietic stem cell-derived thymus-seeding progenitors (TSPs) migrate into the thymus and differentiate into an Early Thymic Progenitor (ETP) upon rearrangement of the diversity (D) and joining (J) regions of the TCR β locus. ETPs progress to a pre-T cell state expressing CD1a and CD5. At this stage, recombination of the variable (V) region of the TCR β locus to form a complete rearranged VDJ TCR β locus occurs almost simultaneously with the rearrangement of the gene segments encoding the γδ TCR. Depending on the outcome of the TCR segment rearrangements, the cells can then follow an αβ or a γδ differentiation path. A successful TCR β rearrangement leads to the process of β-selection and emergence of a CD4+ immature single positive (ISP) T cell. The CD4 ISP cell then develops into a double-positive (DP) cell that expresses both CD4 and CD8 and has begun to rearrange the V and J regions of the TCRα locus. The life span of DP thymocytes is limited as they quickly proceed to apoptosis if they do not receive a TCR-mediated survival signal provided by the self-HLA molecules of the thymic epithelium before maturing into CD4+CD8− and CD4− CD8+ single-positive (SP) T cells.
Pathogen-specific T cells can be effectively expanded through vaccination, a medical intervention that allows prevention of a number of infectious diseases. In this instance, immunization proceeds in vivo within secondary lymphoid organs where T cells engage their TCRs on professional APCs that initiate productive T cell activation and clonal expansion. Active immunization has, however, proven far less effective when infection or cancer is already established and progressing. In such circumstances, T cells, whether they are naturally activated or elicited through immunization, often fail to eradicate disease owing to their inadequate number or suboptimal function.
The infusion of T cells, or adoptive transfer, has proven to overcome the limitations of active immunization in some pathologies. The therapeutic use of isolated T cells began somewhat inadvertently with allogeneic bone marrow transplantation (BMT). The use of whole marrow grafts containing donor T cells revealed the beneficial (graft-versus-tumor responses) and deleterious (GVHD) effects of adoptive T cell transfer (Ferrara and Deeg, 1991). Several forms of T cell therapy subsequently developed, including donor leukocyte infusion (Kolb et al., 2005) and virus-specific T cell therapy (Riddell and Greenberg, 1995). These therapies utilize “donor-derived T cells,” which tap into the alloreactive potential of T cells harvested from a healthy donor but expose the recipient to the risk of normal tissue destruction by graft versus host (GVH) responses. In contrast, autologous T cells, harvested from the intended recipient (Rosenberg et al., 1986), are devoid of such toxic potential. However, autologous T cells with therapeutic potential may be lacking or functionally impaired in patients with refractory infections or progressing cancer. Allogeneic and autologous T cells thus have their respective advantages and disadvantages.
For some cancers, T cells may be isolated from surgically removed tumors, which are enriched in tumor-reactive T cells relative to peripheral blood. Tumor infiltrating lymphocytes (TILs) can be isolated at quite a high frequency from melanoma specimens, but this technique is not feasible or effective in many other tumor types (Rosenberg et al., 2008; Wu et al., 2012). Thus, we and others have sought to generate tumor-targeted T cells through genetic engineering (Ho et al., 2003; Sadelain et al., 2003). The rationale for T cell engineering is to rapidly generate populations of T cells specific for any antigen and, furthermore, to enhance their therapeutic (e.g., anti-tumor) functions. Peripheral blood T cells are easily accessible and are a perfectly suitable cell source for this purpose. Most current therapies utilizing engineered T cells process autologous peripheral blood T cells that are targeted to tumor antigens following retroviral transduction of a TCR or a chimeric antigen receptor (CAR). In recent years, a few clinical trials have resulted in encouraging and sometimes dramatic clinical responses (Couzin-Frankel, 2013). This Perspective article focuses on the sources of T cells for adoptive cell therapy, starting from blood, hematopoietic stem cell-derived lymphoid progenitor cells, embryonic stem cell (ESC), or induced pluripotent stem cell (iPSC)-derived T cells.
T Cell Engineering with TCRs and CARs
The general premise for engineering T cells for cancer immunotherapy is to rapidly generate tumor-targeted T cells, bypassing the obstacles that preclude the induction and execution of effective immune responses in vivo. Two categories of antigen receptors are used to retarget T cell specificity: physiological TCRs and synthetic receptors referred to as CARs (Figure 2). The design of TCRs and CARs has steadily improved over the past 2 decades (Cohen et al., 2006, 2007; Robbins et al., 2008; Sadelain et al., 2003, 2009, 2013; Voss et al., 2008). TCRs are typically cloned from patient tumor-reactive T cell clones (Johnson et al., 2006), from humanized murine models (Cohen et al., 2005; Parkhurst et al., 2009), or through the use of phage display technology (Li et al., 2005; Varela-Rohena et al., 2008). In CARs, tumor recognition is mediated by a single chain variable fragment (scFv) derived from a monoclonal antibody or an antigen-binding region isolated from an immunoglobulin (Ig) heavy and light chain library. Unlike TCR-mediated antigen recognition, CARs function independently of HLA and can therefore be used in any genetic background. Second-generation CARs (Maher et al., 2002, Figure 2) not only mediate antigen recognition and initiate T cell activation but also harness costimulation to enhance T cell function and prolong T cell persistence (Sadelain et al., 2009). Over a decade ago, we selected the CD19 antigen as a potential CAR target for B cell malignancies (Brentjens et al., 2003) and made it the focus of our CAR therapy program. Impressive results were obtained in patients with relapsed, chemorefractory B cell malignancies. A number of patients with chemorefractory B cell malignancies developed complete responses after a single infusion of CAR T cells, as first reported by the National Cancer Institute for B cell lymphoma (Kochenderfer et al., 2010, 2012), the University of Pennsylvania for chronic lymphocytic leukemia (Kalos et al., 2011; Porter et al., 2011; Brentjens et al., 2011), and Memorial Sloan Kettering Cancer Center for acute lymphoblastic leukemia (ALL) (Brentjens et al., 2013b; Davila et al., 2014a; Grupp et al., 2013; Lee et al., 2015; Maude et al., 2014). These patients were treated with autologous T lymphocytes that were retrovirally transduced with second-generation, CD19-specific CARs (Davila et al., 2012). To date, the most dramatic results have been obtained in adult and pediatric patients with ALL (Brentjens et al., 2013b; Davila et al., 2014a; Grupp et al., 2013; Lee et al., 2015; Maude et al., 2014). Encouraging results have also been obtained in patients with CD19+ lymphomas, reviewed in Ramos et al. (2014) and Kochenderfer and Rosenberg (2013). Third-generation CARs, which contain two costimulatory domains along with an activation domain, may provide superior T cell function (Carpenito et al., 2009; Pule et al., 2005; Till et al., 2012; Zhong et al., 2010), although their effectiveness remains to be evaluated in clinical trials.
Figure 2. Antigen Receptors Used for T Cell Engineering.
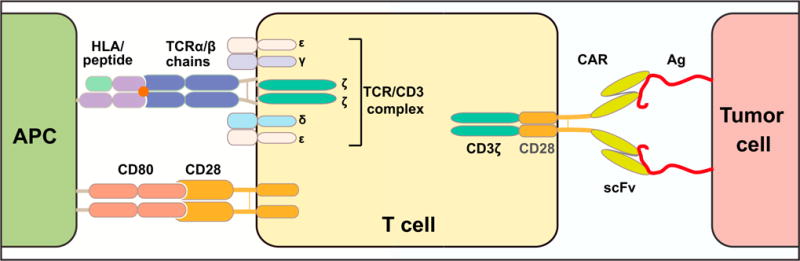
Left: Structure of the αβ heterodimeric T cell receptor (TCR) associated with the multi-chain CD3 complex (γ, δ, ε, and ζ) and flanked by CD28, a costimulatory receptor constitutively expressed in most αβ-T cells. Right: Structure of a prototypical second-generation chimeric antigen receptor (CAR), comprising an scFv for antigen recognition, the CD3ζ cytoplasmic domain for T cell activation, and the CD28 cytoplasmic domain to enhance T cell function and persistence (Maher et al., 2002).
Autologous T Cell Manufacture
The genetic modification of autologous peripheral blood T lymphocytes to generate tumor-targeted T cells is now a well-established approach that was developed in a handful of academic centers. The power and promise of TCR and CAR therapies utilizing these manufacturing processes are best illustrated by the exciting clinical results obtained with NY-ESO-1 TCR (Robbins et al., 2011) and CD19 CAR T cells (Brentjens et al., 2013b; Davila et al., 2014a; Grupp et al., 2013; Kochenderfer et al., 2012, 2014).
These cell manufacturing processes combine T cell activation and transduction steps to generate expanded, genetically targeted T cell products. For example, T cells engineered to express specific CARs or TCRs may be initiated from Ficoll-purified PBMCs, which are next activated with anti-CD3 monoclonal antibody (mAb) in the presence of irradiated allogeneic feeder cells and transduced with a vector encoding either the CAR or TCR α and β chains (Till et al., 2012; Morgan et al., 2006). We and others have established cGMP-compliant large-scale transduction and expansion processes, which are applicable to CARs or TCRs, utilizing either γ-retroviral or lentiviral T cell manufacturing (Figure 3). These processes begin with the selection and activation of T cells from patient apheresis products using materials coated with anti-CD3 and anti-CD28 mAbs. In the case of iron beads, CD3+CD28+ T cells are enriched using a magnetic particle concentrator and subsequently cultured. Activated T cells are retrovirally transduced in RetroNectin-coated cell bags and inoculated in a WAVE bioreactor where they are expanded with a continuous perfusion regimen, reaching cell densities of 10 million T cells/ml or more (Hollyman et al., 2009). At the end of the production, the beads are removed and the cells are formulated for immediate infusion or frozen for deferred use. The entire process typically takes 10–14 days, depending on the disease and the targeted T cell dose. This semi-closed large-scale manufacturing platform can be easily adapted for various vectors and for the expansion of either patient (autologous) or donor (allogeneic) T cells. It successfully supports several ongoing clinical trials in which therapeutic efficacy has been demonstrated (Brentjens et al., 2011, 2013a; Davila et al., 2014b). This process starts from bulk T cells harvested from each individual subject. Several groups are currently evaluating what T cell phenotype and T cell subset or subsets account for the anti-tumor activity of these cells and what will be optimal tools to activate and expand T cells for different T cell therapies. Various means to enhance the activation and expansion of T cells for adoptive cell therapy have been reviewed elsewhere (Vacchelli et al., 2013).
Figure 3. General Schema of Autologous T Cell Manufacturing.
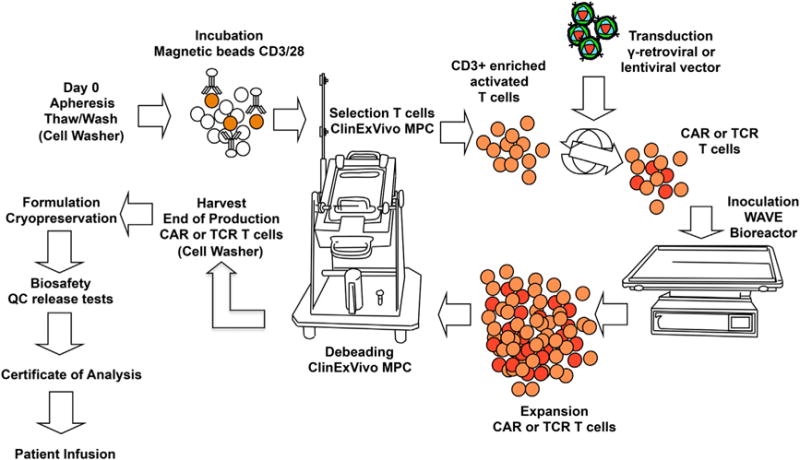
The semi-closed system relies on the use of a cell washer to wash the apheresis product before freezing and after thawing, the capture of CD3+CD28+ T cells with magnetic beads or microbeads. For magnetic beads subsequent selection is performed on the ClinExVivo magnetic particle concentrator (MPC). Thereafter, T cells are transduced with a viral vector expressing a TCR or a CAR, expanded in a bioreactor (e.g., Wave Bioreactor), debeaded on the ClinExVivo MPC, and formulated. Adapted from Hollyman et al. (2009).
The functional, proliferative, and persistence potential of adoptively transferred T lymphocytes is determined by multiple factors. These include the TCR or CAR design, the manufacturing platform, the selected T cell subsets, and the differentiation stage of the harvested T cells. Peripheral blood T cells comprise naive (TN), stem cell memory (TSCM), central memory (TCM), effector memory (TEM), and terminal effector (TE) cells (Klebanoff et al., 2012). Several groups have investigated which of these T cell subsets are best suited for use in different adoptive therapy settings (Klebanoff et al., 2012; Riddell et al., 2014). In non-human primates and murine NSG models, T cell transfer studies have shown that virus-specific and CAR-redirected anti-tumor CD8 TEM rapidly mature to terminal effector T cells and do not persist beyond 7–14 days, while a subset of transferred CD8+ TE/CM can acquire memory cell features and persist for months and even years (Wang et al., 2012). Polyclonal CD8+ TCM isolation from leukopheresis products, followed by CD3/CD28 activation without exogenous feeder cells and cell expansion in IL-2/IL-15, has thus been developed on a clinical scale and is currently in use for the generation of autologous CAR-redirected CD19-specific CD8+ TE/CM for adoptive transfer after autologous hematopoietic stem cell transplantation (HSCT) for high-risk CD19+ non-Hodgkin lymphomas (Wang et al., 2012). Additional variations on the manufacturing schemas exemplified here have been reported or are under development (DiGiusto and Cooper, 2007; Laport et al., 2003; Savoldo et al., 2011; Somerville et al., 2012; Wang and Rivière, 2015).
It remains to be determined how the cell attributes imparted by in vivo persistence of antigen-specific T cells correlate with those conferring increased anti-tumor efficacy (Biasco et al., 2015; Flynn and Gorry, 2014; Xu et al., 2014). Defining optimal, potent T cell products of specified composition for adoptive cell therapy will require careful phenotypic and biological characterization, taking in account manufacturing and economic practicalities (Heathman et al., 2015).
Allogeneic T Cells as a Substrate for T Cell Engineering
The promising clinical results of engineered T cell therapy could be further amplified and broadened if potent and histocompatible T cells were readily available. Autologous approaches have a proven track record, but personalized manufacture may be challenging in some instances, for example in patients with chemotherapy or HIV-induced immune deficiency or in small infants. While T cells can be easily harvested from donors, their use is compromised by the high alloreactive potential. Owing to their ontogeny, TCRs are naturally prone to react against non-autologous tissues, recognizing either allogeneic HLA molecules or other polymorphic gene products, referred to as minor antigens (Afzali et al., 2007). This propensity underlies the high risk of graft rejection in transplant recipients and of GVHD in recipients of donor-derived T cells. Thus, bulk, unselected donor T cells are prone to cause normal tissue destruction and may be lethal on occasion. To provide an acceptable risk-benefit ratio, allogeneic T cells must be devoid of alloreactive potential. Two strategies designed to overcome the risk of GVH reactions have been proposed, based on the selection of virus-specific TCRs devoid of GVH reactivity or the ablation of TCR expression.
Virus-Specific T Cells
Donor-derived virus-specific T cells can be administered to virus-infected, HLA-matched recipients with a reduced risk of GVHD. For this purpose, donor T cells from a seropositive donor are stimulated in vitro with APCs that present the relevant viral antigens. The APCs may consist of syngeneic Epstein-Barr virus (EBV)-transformed B cells (Heslop et al., 1994), peptide pulsed dendritic cells, or artificial APCs (Latouche and Sadelain, 2000; Papanicolaou et al., 2003). Repeated stimulation with viral antigen gradually increases viral specificity and concomitantly depletes alloreactivity. Although alloreactivity and unanticipated TCR cross-reactivity cannot be prospectively eliminated with full certainty (Cameron et al., 2013; Morgan et al., 2013), virus-specific T cell lines generated in this manner have shown dramatic responses in EBV, cytomegalovirus (CMV) and adenovirus-infected recipients without causing severe GVHD (Heslop et al., 1996; Papadopoulos et al., 1994). Recent studies have suggested that virus-specific T cells can be administered to multiple recipients with limited risk of GVHD (Doubrovina et al., 2012; Haque et al., 2007). Virus-specific T cells may thus serve as cellular vehicles for TCR or CAR therapy. A first trial testing this approach showed that T cells expanded in vivo in response to viral reactivation although anti-tumor activity was modest (Cruz et al., 2013). While the relatively limited expansion potential of virus-specific T cells and the sometimes unpredictable cross-reactivity of TCR-mediated antigen recognition are valid concerns, this approach to treat viral infections represents a first step toward multi-recipient T cell product manufacturing (Wang and Rivière, 2015).
TRC-less Allogeneic Peripheral Blood T Cells
If the endogenous TCR cannot be tamed, one may abrogate its expression, making the engineered TCR or CAR the sole driver of T cell activation and clonal expansion. With the advent of gene disruption technologies, this approach is now within reach. Four technologies based on the use of targeted nucleases, including meganucleases, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALEN), and CRISPR/Cas9, enable gene disruption in human cells (Kim and Kim, 2014; Sander and Joung, 2014). ZFNs and CRISPR/Cas9 are presently the most developed of these tools and have been shown to efficiently target the HIV co-receptor CCR5 (Holt et al., 2010; Mandal et al., 2014; Tebas et al., 2014). The ablation of endogenous TCR expression has been achieved using targeted ZFNs or TALENs that disrupt the constant regions of TCRA and TCRB genes (Berdien et al., 2014; Provasi et al., 2012; Torikai et al., 2012). Unlike unedited, allogeneic T cells, TCR-deleted lymphocytes retargeted by CAR or TCR gene transfer do not mediate GVH reactivity. Their long-term persistence could potentially be compromised, since homeostatic proliferation is partially dependent on TCR-major histocompatibility complex (MHC) interactions. However, T lymphocytes that have acquired a central memory phenotype are less dependent on the TCR for homeostatic proliferation (Surh and Sprent, 2008) and proliferate in response to cytokines (Provasi et al., 2012). Their long-term persistence, relative to that of unedited T lymphocytes, has not yet been fully characterized. Furthermore, gene disruption technologies are still in early stages of development and require optimization to afford high frequency bi-allelic gene targeting without causing off-target mutations, which could potentially alter T cell function or predispose to cell transformation. Prevention of GVHD would require that virtually all T cells bear a disrupted TCR gene, requiring high-efficiency targeting and robust purging of unmodified cells to ensure T cell safety. Similar to the virus-specific T cell paradigm, it is unknown to what extent mature T cells undergoing extensive manipulation, including antigen-specific restimulation, TCR or CAR transduction, gene editing, and cell selection, will yield sufficiently large batches of functional T cells that meet the needs for multiple recipient infusions. Thus, allogeneic T cell approaches are still labor intensive and constrained by the limited replicative potential of mature T cells (Gattinoni et al., 2012).
Interestingly, allogeneic T cells may not cause GVHD in some particular circumstances. Thus, patients infused with T cells collected after allogeneic transplantation have not developed any GVHD-like syndrome (Davila et al., 2014a; Lee et al., 2015; Maude et al., 2014). More strikingly, CD19 CAR-targeted donor T cells show reduced GVHD potential in allogeneic recipient mice (reported at the American Society of Hematology annual meeting in 2012) and in human patients infused with CD19 CAR-modified donor leukocytes (Kochenderfer et al., 2013). A mechanistic explanation for these intriguing observations is still lacking.
Lymphoid Progenitor Therapy
While T cells can cause GVHD, their precursors do not, as they undergo positive and negative selection in the recipient’s thymus. Taking advantage of this requires the ability to expand T cell precursors in culture, which is now possible due to advances in understanding T cell development (Awong et al., 2007; Rothenberg, 2011; Shah and Zúñiga-Pflücker, 2014). T cell precursors lack the ability to initiate GVH reactions because they complete their differentiation in the recipient’s thymus wherein they become restricted to host MHC and yield T lymphocytes that are host tolerant (Zakrzewski et al., 2006). When transduced with a CAR, allogeneic lymphoid progenitors yield tumor-targeted T cells without causing GVHD (Zakrzewski et al., 2008). The main advantage of using T cell precursors for immunotherapy is that this approach does not require strict histocompatibility between donors and recipients. In mice, this therapy works with unrelated fully mismatched cells just as well as with autologous cells. T cell precursor immunotherapy may therefore allow for a true “off-the-shelf” therapy, if lymphoid progenitor cell manufacturing can be scaled up.
Pluripotent Stem Cells as a Source of Therapeutic T Lymphocytes
The development of cellular therapeutics relying on functionally validated, banked, broadly histocompatible cell types would have a major impact on the applicability and cost of adoptive T cell therapies. This prospect raises the challenge of artificially generating ideal T cells rather than modifying those naturally formed. Pluripotent stem cells can give rise to a variety of somatic cells (Inoue et al., 2014; Murry and Keller, 2008; Takahashi et al., 2007) and thus have in principle the potential to serve as an endless supply of therapeutic T lymphocytes. A few reports support the feasibility of generating T lymphocytes from human ESCs and iPSCs in vitro (Kennedy et al., 2012; Nishimura et al., 2013; Themeli et al., 2013; Timmermans et al., 2009; Vizcardo et al., 2013).
Antigen Specificity
The first requirement for therapeutic function is specific antigen recognition, which is physiologically mediated by the TCR. ESCs and most iPSCs bear TCR α and β loci in the germline configuration. These undergo random rearrangements during lymphoid differentiation, thus generating polyclonal T cells of undetermined specificity and HLA restriction. This unpredictable repertoire severely limits the usefulness and potential for expansion and functional characterization of T cells derived from ESCs/iPSCs. Two approaches to dictate the specificity of iPSC-derived T cells have been hitherto reported. One utilizes iPSCs that bear rearranged TCR genes, providing a known antigen specificity (Nishimura et al., 2013; Vizcardo et al., 2013; Wakao et al., 2013). Re-differentiation of iPSCs derived from mucosal-associated invariant T (MAIT) cells expressing the invariant T cell receptor Vα7.2 or established viral- and tumor-specific T cell clones gives rise to T lymphocytes bearing the same TCR as the parental T cell from which the iPSC clone was established, although re-rearrangement of a remaining germline TCR α locus may result in multiple TCRs. This approach to afford antigen-specificity for cancer immunotherapy requires laborious cloning of antigen-specific T cells and the availability of the desired antigen-specific T cells for every prospective recipient. Another approach is to genetically transfer a receptor for antigen of known specificity. We previously demonstrated that T cell-derived iPSCs (TiPSCs) expressing a CAR (CAR-TiPSC) provide an effective means to concomitantly exploit the unlimited proliferative potential of iPSCs and direct the antigen specificity of iPSC-derived T cells (Themeli et al., 2013). In contrast to TCR transfer, CAR engineering yields T cells with unrestricted antigen recognition and enhanced potency owing to the costimulatory signals provided through the CAR.
Determining T Lineage Commitment and T Cell Fate
The functional properties of T lymphocytes depend not only on their differentiation stage and engineered features, as discussed above, but also on their lineage subtype (γδ or αβ T cells, effector or regulatory subsets) (Vantourout and Hayday, 2013). It is therefore essential to generate T cells of the desired functional subset. Natural human T lymphoid development is outlined in Figure 1. Lymphopoietic progenitors seem to follow these steps overall throughout ESC/iPSC in vitro differentiation. TiPSCs generated from an αβ-TCR-bearing T cell indeed give rise to αβ-TCR+ cells. However, their phenotype, whether pre or post antigen-expansion, may not be that of a typical αβ-T cell. Expanded TiPSC-derived T cells are CD3+CD7+CD5loTCRαβ+CD56+ and either double negative for CD4 and CD8 or CD8α+CD8β− (Nishimura et al., 2013; Themeli et al., 2013). Antigen-activated and expanded CAR-TiPSC-T cells display an effector memory phenotype (CD45RA+CD27−CD28−CCR7−) (Themeli et al., 2013). Microarray gene expression analyses and detailed immunophenotypic profiling provided some clarification for these unexpected findings, establishing that the in vitro generated CAR-TiPSC-T cells possess an innate γδ T cell-like profile, even though they express their endogenous αβ-TCR (Themeli et al., 2013). Significantly, their in vivo anti-tumor function was comparable to natural, peripheral blood-derived γδ T cells collected from the same donor and transduced with the same CAR (Themeli et al., 2013). Furthermore, Nishimura et al. (2013) observed the emergence of a few central memory TiPSC-derived T cells (CCR7+CD27+CD28+), although these cells had very low CCR7 and CD28 expression. In aggregate, these findings indicate that although the TiPSC-T cells express their rearranged endogenous αβ-TCR on their surface, they acquire a phenotype and functional properties that do not correspond to that of natural naive or memory CD8αβ+ T lymphocytes. One study showed efficient generation of CD4+CD8α+TCR+ cells, which almost exclusively differentiated into CD8αα+ cells upon stimulation with antigen (Vizcardo et al., 2013). Other studies showed absent or minor generation of double positive CD4+CD8α+ cells, but no detection of CD8β has yet been reported. A better understanding of the requirements for inducing CD4+CD8αβ+ cells will pave the way for the generation of CD8αβ + and CD4+ T cells, including effector and regulatory T cells.
Interestingly, both CAR-TiPSC-T cells and regenerated MAIT cells (Wakao et al., 2013) express CD56 and CD161, suggesting that the innate nature of the TiPSC-T cells is independent of the identity of the initially reprogrammed T cell subtype. It is noteworthy that lineage diversion has been previously observed in transgenic TCRαβ mice (Baldwin et al., 2005; Egawa et al., 2008; Terrence et al., 2000), wherein T cells distinct from wild-type natural killer (NK), NK-T, or CD4 or CD8 single-positive T cells displayed γδ T cell features, including expression of CD8α and low levels of CD5 (Terrence et al., 2000). Furthermore, in vitro differentiated T cells derived from TCR-engineered human CD34+ hematopoietic progenitors display an NK cell-like phenotype (Zhao et al., 2007). These observations suggest that the presence of rearranged TCR genes influences T cell fate, similar to reports in TCR transgenic mice (Baldwin et al., 2005). Accordingly, mature CD4+CD8+ and single-positive T cells developed from TCR-engineered CD34+ hematopoietic progenitors when the TCR cDNAs were introduced in the pre-T cell stage of differentiation (Snauwaert et al., 2014), consistent with time-dependent TCR expression causing lineage diversion. Alternatively, considering that some features of TiPSC-derived T cells, such as their CD8α+CD8β− phenotype, expression of CD161 and low expression of CD5, are also found in innate-like T cells generated in fetal development (Cupedo et al., 2009; Spits and Cupedo, 2012), it may be that their innate character is imparted by a fetal-like hematopoietic stem cell intermediate committed to innate lymphopoiesis (Kennedy et al., 2012; Mold et al., 2010; Yuan et al., 2012) and intrinsically skewed toward embryonic characteristics (Murry and Keller, 2008). Further investigation of the mechanisms underlying in vitro T lymphoid differentiation of TiPSCs is needed to better direct T cell subset differentiation and further shape the functional attributes of induced T cells.
T Cell Functionality
Beyond antigen specificity, two critical features that will determine the therapeutic relevance of pluripotent cell-derived T cells are their potential for in vivo persistence and sustained functionality. Few studies have comprehensively assessed the functional profile of ESC or iPSC-derived T cells in vitro or upon adoptive transfer in vivo. As previously mentioned, the random TCR rearrangements occurring in ESC/iPSC-derived T cells limit the feasibility of studying antigen-specific expansion and function. Therefore, ESC/iPSC-derived T cell function has been assessed only in in vitro assays showing IFNγ secretion after unspecific stimulation (Timmermans et al., 2009). Expanded tumor- and viral-specific TiPSC-derived T cells (100- to 1,000-fold) secrete IFN-γ after unspecific stimulation and lyse target cells in an antigen-specific manner in vitro (Nishimura et al., 2013). Re-differentiated MAIT cells were shown to successfully function in vivo against mycobacterium infection, although in a non-antigen-specific manner (Wakao et al., 2013). CAR-TiPSC T cells generated in culture expanded robustly upon CD19 engagement by the CAR (up to 1,000-fold over 3 weeks) and showed anti-tumor efficacy against a CD19+ lymphoma in a xenogeneic murine model, comparable to their natural counterparts harvested from peripheral blood (from the same donor) and transduced with the same CAR (Themeli et al., 2013). The latter study provided the proof of principle that human iPSC-derived T lymphocytes generated in vitro possessed anti-tumor function in vivo. Further studies are needed to investigate the in vitro expansion potential and the in vivo capabilities of iPSC-derived T lymphocytes.
Striking a Balance between Preventing Anti-Host Reactivity and Escaping Immune Rejection Is Key
Whereas anti-host reactivity may cause unacceptable toxicity, immune rejection of non-autologous T cells will curtail their efficacy. Therefore, escaping immune rejection, ensuring sufficient persistence and possibly long term engraftment, are further critical requirements to enable off-the-shelf adoptive T cell therapy. One immediate approach to solve this problem is to bank cells with common HLA haplotypes, as proposed for EBV-reactive T cells (Gallot et al., 2014; Leen et al., 2013) or iPSC/ESCs (Gourraud et al., 2012; Nakatsuji et al., 2008; Stacey et al., 2013; Turner et al., 2013). Although this approach is certainly a valuable first step toward broader applicability of adoptive T cell therapy, it is still constrained by HLA matching and by donor availability. Furthermore, the establishment of iPSC/ESC banks requires the generation of multiple iPSC lines from multiple donors, compounded by the eventual need to identify T cells of an appropriate specificity and HLA restriction, followed by extensive safety studies and validation of the individual clones. The alternative is to genetically target HLA genes to generate histocompatible cell products (Riolobos et al., 2013; Torikai et al., 2012). Targeting multiple HLA loci in primary T lymphocytes may be feasible but poses technical challenges owing to the substantial safety validation required for each cell product. HLA engineering and biosafety testing may be easier to perform in pluripotent stem cells (Riolobos et al., 2013). In contrast to primary T cell manipulations, the genetic engineering of iPSCs results in fully modified clonal lines, which can be extensively evaluated (Papapetrou et al., 2011). However, disruption of HLA loci would expose cells to NK cell-mediated rejection. Further cell engineering including overexpression of HLA-E or HLA-G has been proposed as a solution to confer NK resistance (Riolobos et al., 2013; Torikai et al., 2012).
Perspectives for “Synthetic T Cells”
Stem cell reprogramming not only offers potential access to an unlimited source of therapeutic T lymphocytes, but it also provides an excellent platform for performing additional engineering intended to enhance the therapeutic value of induced T cells. The genetic engineering of TiPSCs with CARs is the first example of an efficient strategy to concomitantly harness the unlimited availability of iPSCs and direct the specificity and functional potential of iPSC-derived T cells (Themeli et al., 2013). The use of iPSCs further opens up new perspectives for the generation of histocompatible, off-the-shelf T cells that could eventually be administered to multiple recipients. The combination of iPSC technology and immune engineering may thus provide an opportunity to generate T cells that uniquely combine favorable attributes including antigen specificity, lack of alloreactivity, enhanced functional properties and histocompatibility (Figure 4). Several challenges remain, including the ability to control T lineage specification (to αβ- or γδ-T cells, NK-T, CD8, CD4, or regulatory T cells), differentiation to an optimal maturation stage (e.g., naive or stem central memory T cells) (Gattinoni et al., 2012), and acquisition of an optimal functional and proliferative potential (Sadelain et al., 2003). Natural, autologous T cells represent the best-defined cell source for adoptive cell therapy today, and they are the cornerstone of present cell-based cancer immunotherapy. Induced, engineered T cells derived from allogeneic pluripotent stem cell sources may play an important role in the future.
Figure 4. Perspectives for Synthetic TiPSC-Derived T Cell Generation.
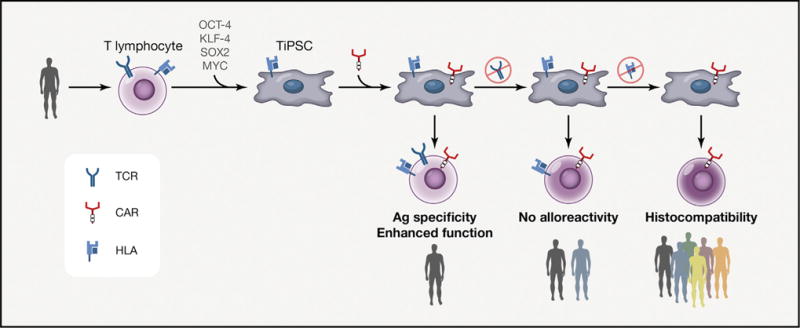
TiPSCs can be generated from one donor and utilized for the production of synthetic T lymphocytes engineered to possess optimized properties such as (1) antigen specificity through a CAR or TCR, (2) enhanced function (e.g., introducing costimulatory molecules), (3) elimination of alloreactivity and partial broadening of applicability (e.g., strategies to knock out the TCR expression without perturbing the T lymphoid differentiation process), and (4) broad histocompatibility by selection or genetic modification of the HLA loci.
Acknowledgments
The authors thank Dr. M. Hamieh for help with figures. Michel Sadelain and Isabelle Riviere are scientific co-founders of Juno Thearapeutics.
References
- Afzali B, Lechler RI, Hernandez-Fuentes MP. Allorecognition and the alloresponse: clinical implications. Tissue Antigens. 2007;69:545–556. doi: 10.1111/j.1399-0039.2007.00834.x. [DOI] [PubMed] [Google Scholar]
- Awong G, La Motte-Mohs RN, Zúñiga-Pflücker JC. Generation of pro-T cells in vitro: potential for immune reconstitution. Semin Immunol. 2007;19:341–349. doi: 10.1016/j.smim.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Baldwin TA, Sandau MM, Jameson SC, Hogquist KA. The timing of TCR alpha expression critically influences T cell development and selection. J Exp Med. 2005;202:111–121. doi: 10.1084/jem.20050359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdien B, Mock U, Atanackovic D, Fehse B. TALEN-mediated editing of endogenous T-cell receptors facilitates efficient reprogramming of T lymphocytes by lentiviral gene transfer. Gene Ther. 2014;21:539–548. doi: 10.1038/gt.2014.26. [DOI] [PubMed] [Google Scholar]
- Biasco L, Scala S, Basso Ricci L, Dionisio F, Baricordi C, Calabria A, Giannelli S, Cieri N, Barzaghi F, Pajno R, et al. In vivo tracking of T cells in humans unveils decade-long survival and activity of genetically modified T memory stem cells. Sci Transl Med. 2015;7:273ra213. doi: 10.1126/scitranslmed.3010314. [DOI] [PubMed] [Google Scholar]
- Brentjens RJ, Latouche JB, Santos E, Marti F, Gong MC, Lyddane C, King PD, Larson S, Weiss M, Rivière I, Sadelain M. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat Med. 2003;9:279–286. doi: 10.1038/nm827. [DOI] [PubMed] [Google Scholar]
- Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013a;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013b;5:177ra138. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5:197ra103. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA. 2009;106:3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Zheng Z, Bray R, Zhao Y, Sherman LA, Rosenberg SA, Morgan RA. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006;66:8878–8886. doi: 10.1158/0008-5472.CAN-06-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA. Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res. 2007;67:3898–3903. doi: 10.1158/0008-5472.CAN-06-3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, Diouf O, Liu E, Barrett AJ, Ito S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood. 2013;122:2965–2973. doi: 10.1182/blood-2013-06-506741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupedo T, Crellin NK, Papazian N, Rombouts EJ, Weijer K, Grogan JL, Fibbe WE, Cornelissen JJ, Spits H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat Immunol. 2009;10:66–74. doi: 10.1038/ni.1668. [DOI] [PubMed] [Google Scholar]
- Davila ML, Brentjens R, Wang X, Rivière I, Sadelain M. How do CARs work?: Early insights from recent clinical studies targeting CD19. OncoImmunology. 2012;1:1577–1583. doi: 10.4161/onci.22524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014a;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014b;6:224ra225. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiusto D, Cooper L. Preparing clinical grade Ag-specific T cells for adoptive immunotherapy trials. Cytotherapy. 2007;9:613–629. doi: 10.1080/14653240701650320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doubrovina E, Oflaz-Sozmen B, Prockop SE, Kernan NA, Abramson S, Teruya-Feldstein J, Hedvat C, Chou JF, Heller G, Barker JN, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119:2644–2656. doi: 10.1182/blood-2011-08-371971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa T, Kreslavsky T, Littman DR, von Boehmer H. Lineage diversion of T cell receptor transgenic thymocytes revealed by lineage fate mapping. PloS one. 2008;3:e1512. doi: 10.1371/journal.pone.0001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara JL, Deeg HJ. Graft-versus-host disease. N Engl J Med. 1991;324:667–674. doi: 10.1056/NEJM199103073241005. [DOI] [PubMed] [Google Scholar]
- Flynn JK, Gorry PR. Stem memory T cells (TSCM)-their role in cancer and HIV immunotherapies. Clin Transl Immunol. 2014;3:e20. doi: 10.1038/cti.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallot G, Vollant S, Saïagh S, Clémenceau B, Vivien R, Cerato E, Bignon JD, Ferrand C, Jaccard A, Vigouroux S, et al. T-cell therapy using a bank of EBV-specific cytotoxic T cells: lessons from a phase I/II feasibility and safety study. J Immunother. 2014;37:170–179. doi: 10.1097/CJI.0000000000000031. [DOI] [PubMed] [Google Scholar]
- Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12:671–684. doi: 10.1038/nrc3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourraud PA, Gilson L, Girard M, Peschanski M. The role of human leukocyte antigen matching in the development of multiethnic “haplobank” of induced pluripotent stem cell lines. Stem Cells. 2012;30:180–186. doi: 10.1002/stem.772. [DOI] [PubMed] [Google Scholar]
- Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P, Burns D, McAulay K, Turner M, Bellamy C, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. 2007;110:1123–1131. doi: 10.1182/blood-2006-12-063008. [DOI] [PubMed] [Google Scholar]
- Heathman TR, Nienow AW, McCall MJ, Coopman K, Kara B, Hewitt CJ. The translation of cell-based therapies: clinical landscape and manufacturing challenges. Regen Med. 2015;10:49–64. doi: 10.2217/rme.14.73. [DOI] [PubMed] [Google Scholar]
- Heslop HE, Brenner MK, Rooney CM. Donor T cells to treat EBV-associated lymphoma. N Engl J Med. 1994;331:679–680. doi: 10.1056/NEJM199409083311017. [DOI] [PubMed] [Google Scholar]
- Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA, Brenner MK, Rooney CM. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2:551–555. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- Ho WY, Blattman JN, Dossett ML, Yee C, Greenberg PD. Adoptive immunotherapy: engineering T cell responses as biologic weapons for tumor mass destruction. Cancer Cell. 2003;3:431–437. doi: 10.1016/s1535-6108(03)00113-2. [DOI] [PubMed] [Google Scholar]
- Hollyman D, Stefanski J, Przybylowski M, Bartido S, Borquez-Ojeda O, Taylor C, Yeh R, Capacio V, Olszewska M, Hosey J, et al. Manufacturing validation of biologically functional T cells targeted to CD19 antigen for autologous adoptive cell therapy. J Immunother. 2009;32:169–180. doi: 10.1097/CJI.0b013e318194a6e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nagata N, Kurokawa H, Yamanaka S. iPS cells: a game changer for future medicine. EMBO J. 2014;33:409–417. doi: 10.1002/embj.201387098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Heemskerk B, Powell DJ, Jr, Cohen CJ, Morgan RA, Dudley ME, Robbins PF, Rosenberg SA. Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J Immunol. 2006;177:6548–6559. doi: 10.4049/jimmunol.177.9.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Science translational medicine. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M, Awong G, Sturgeon CM, Ditadi A, LaMotte-Mohs R, Zúñiga-Pflücker JC, Keller G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012;35:651–660. doi: 10.1097/CJI.0b013e31827806e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. 2013;10:267–276. doi: 10.1038/nrclinonc.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood. 2012;119:2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, Hakim FT, Halverson DC, Fowler DH, Hardy NM, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122:4129–4139. doi: 10.1182/blood-2013-08-519413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM, et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J Clinic Oncol. 2014 doi: 10.1200/JCO.2014.56.2025. in press. Published online August 25, 2014. http://dx.doi.org/10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed]
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb HJ, Schmid C, Buhmann R, Tischer J, Ledderose G. DLI: where are we know? Hematology. 2005;10(1):115–116. doi: 10.1080/10245330512331390122. [DOI] [PubMed] [Google Scholar]
- Krogsgaard M, Davis MM. How T cells ‘see’ antigen. Nat Immunol. 2005;6:239–245. doi: 10.1038/ni1173. [DOI] [PubMed] [Google Scholar]
- Laport GG, Levine BL, Stadtmauer EA, Schuster SJ, Luger SM, Grupp S, Bunin N, Strobl FJ, Cotte J, Zheng Z, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003;102:2004–2013. doi: 10.1182/blood-2003-01-0095. [DOI] [PubMed] [Google Scholar]
- Latouche JB, Sadelain M. Induction of human cytotoxic T lymphocytes by artificial antigen-presenting cells. Nat Biotechnol. 2000;18:405–409. doi: 10.1038/74455. [DOI] [PubMed] [Google Scholar]
- Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, Kapoor N, Pai SY, Rowley SD, Kebriaei P, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–5123. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Moysey R, Molloy PE, Vuidepot AL, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat Biotechnol. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. Nature biotechnology. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, Vrbanac V, Garrison BS, Stortchevoi A, Bryder D, et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mold JE, Venkatasubrahmanyam S, Burt TD, Michaëlsson J, Rivera JM, Galkina SA, Weinberg K, Stoddart CA, McCune JM. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010;330:1695–1699. doi: 10.1126/science.1196509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, Royal RE, Topalian SL, Kammula US, Restifo NP, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Nakatsuji N, Nakajima F, Tokunaga K. HLA-haplotype banking and iPS cells. Nat Biotechnol. 2008;26:739–740. doi: 10.1038/nbt0708-739. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Kaneko S, Kawana-Tachikawa A, Tajima Y, Goto H, Zhu D, Nakayama-Hosoya K, Iriguchi S, Uemura Y, Shimizu T, et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:114–126. doi: 10.1016/j.stem.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Papadopoulos EB, Ladanyi M, Emanuel D, Mackinnon S, Boulad F, Carabasi MH, Castro-Malaspina H, Childs BH, Gillio AP, Small TN, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330:1185–1191. doi: 10.1056/NEJM199404283301703. [DOI] [PubMed] [Google Scholar]
- Papanicolaou GA, Latouche JB, Tan C, Dupont J, Stiles J, Pamer EG, Sadelain M. Rapid expansion of cytomegalovirus-specific cytotoxic T lymphocytes by artificial antigen-presenting cells expressing a single HLA allele. Blood. 2003;102:2498–2505. doi: 10.1182/blood-2003-02-0345. [DOI] [PubMed] [Google Scholar]
- Papapetrou EP, Lee G, Malani N, Setty M, Riviere I, Tirunagari LM, Kadota K, Roth SL, Giardina P, Viale A, et al. Genomic safe harbors permit high β-globin transgene expression in thalassemia induced pluripotent stem cells. Nat Biotechnol. 2011;29:73–78. doi: 10.1038/nbt.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst MR, Joo J, Riley JP, Yu Z, Li Y, Robbins PF, Rosen-berg SA. Characterization of genetically modified T-cell receptors that recognize the CEA:691–699 peptide in the context of HLA-A2.1 on human colorectal cancer cells. J Clin Cancer Res. 2009;15:169–180. doi: 10.1158/1078-0432.CCR-08-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. The New England journal of medicine. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provasi E, Genovese P, Lombardo A, Magnani Z, Liu PQ, Reik A, Chu V, Paschon DE, Zhang L, Kuball J, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–815. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Therapy. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Ramos CA, Savoldo B, Dotti G. CD19-CAR trials. Cancer J. 2014;20:112–118. doi: 10.1097/PPO.0000000000000031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell SR, Greenberg PD. Principles for adoptive T cell therapy of human viral diseases. Annu Rev Immunol. 1995;13:545–586. doi: 10.1146/annurev.iy.13.040195.002553. [DOI] [PubMed] [Google Scholar]
- Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, Hudecek M, Maloney DG, Turtle CJ. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J. 2014;20:141–144. doi: 10.1097/PPO.0000000000000036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riolobos L, Hirata RK, Turtle CJ, Wang PR, Gornalusse GG, Zavajlevski M, Riddell SR, Russell DW. HLA Engineering of Human Pluripotent Stem Cells. Mol Therapy. 2013;21:1232–1241. doi: 10.1038/mt.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, Xu H, Morgan RA, Feldman SA, Johnson LA, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol. 2008;180:6116–6131. doi: 10.4049/jimmunol.180.9.6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, Nahvi AV, Helman LJ, Mackall CL, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;233:1318–1321. doi: 10.1126/science.3489291. [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV. T cell lineage commitment: identity and renunciation. J Immunol. 2011;186:6649–6655. doi: 10.4049/jimmunol.1003703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Rivière I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21:215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Disc. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah DK, Zúñiga-Pflücker JC. An overview of the intrathymic intricacies of T cell development. J Immunol. 2014;192:4017–4023. doi: 10.4049/jimmunol.1302259. [DOI] [PubMed] [Google Scholar]
- Snauwaert S, Verstichel G, Bonte S, Goetgeluk G, Vanhee S, Van Caeneghem Y, De Mulder K, Heirman C, Stauss H, Heemskerk MH, et al. In vitro generation of mature, naive antigen-specific CD8(+) T cells with a single T-cell receptor by agonist selection. Leukemia. 2014;28:830–841. doi: 10.1038/leu.2013.285. [DOI] [PubMed] [Google Scholar]
- Somerville RP, Devillier L, Parkhurst MR, Rosenberg SA, Dudley ME. Clinical scale rapid expansion of lymphocytes for adoptive cell transfer therapy in the WAVE bioreactor. J Transl Med. 2012;10:69. doi: 10.1186/1479-5876-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H, Cupedo T. Innate lymphoid cells: emerging insights in development, lineage relationships, and function. Annu Rev Immunol. 2012;30:647–675. doi: 10.1146/annurev-immunol-020711-075053. [DOI] [PubMed] [Google Scholar]
- Stacey GN, Crook JM, Hei D, Ludwig T. Banking human induced pluripotent stem cells: lessons learned from embryonic stem cells? Cell Stem Cell. 2013;13:385–388. doi: 10.1016/j.stem.2013.09.007. [DOI] [PubMed] [Google Scholar]
- Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrence K, Pavlovich CP, Matechak EO, Fowlkes BJ. Premature expression of T cell receptor (TCR)alphabeta suppresses TCRgammadelta gene rearrangement but permits development of gammadelta lineage T cells. J Exp Med. 2000;192:537–548. doi: 10.1084/jem.192.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, Sadelain M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol. 2013;31:928–933. doi: 10.1038/nbt.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans F, Velghe I, Vanwalleghem L, De Smedt M, Van Coppernolle S, Taghon T, Moore HD, Leclercq G, Langerak AW, Kerre T, et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J Immunol. 2009;182:6879–6888. doi: 10.4049/jimmunol.0803670. [DOI] [PubMed] [Google Scholar]
- Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovitch B, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–5705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner M, Leslie S, Martin NG, Peschanski M, Rao M, Taylor CJ, Trounson A, Turner D, Yamanaka S, Wilmut I. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell. 2013;13:382–384. doi: 10.1016/j.stem.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Vacchelli E, Eggermont A, Fridman WH, Galon J, Tartour E, Zitvogel L, Kroemer G, Galluzzi L. Trial Watch: Adoptive cell transfer for anticancer immunotherapy. Oncoimmunology. 2013;2:e24238. doi: 10.4161/onci.24238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantourout P, Hayday A. Six-of-the-best: unique contributions of gd T cells to immunology. Nat Rev Immunol. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varela-Rohena A, Molloy PE, Dunn SM, Li Y, Suhoski MM, Carroll RG, Milicic A, Mahon T, Sutton DH, Laugel B, et al. Control of HIV-1 immune escape by CD8 T cells expressing enhanced T-cell receptor. Nat Med. 2008;14:1390–1395. doi: 10.1038/nm.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcardo R, Masuda K, Yamada D, Ikawa T, Shimizu K, Fujii S, Koseki H, Kawamoto H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8(+) T cells. Cell Stem Cell. 2013;12:31–36. doi: 10.1016/j.stem.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Voss RH, Willemsen RA, Kuball J, Grabowski M, Engel R, Intan RS, Guillaume P, Romero P, Huber C, Theobald M. Molecular design of the Calphabeta interface favors specific pairing of introduced TCRalphabeta in human T cells. J Immunol. 2008;180:391–401. doi: 10.4049/jimmunol.180.1.391. [DOI] [PubMed] [Google Scholar]
- Wakao H, Yoshikiyo K, Koshimizu U, Furukawa T, Enomoto K, Matsunaga T, Tanaka T, Yasutomi Y, Yamada T, Minakami H, et al. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell. 2013;12:546–558. doi: 10.1016/j.stem.2013.03.001. [DOI] [PubMed] [Google Scholar]
- Wang X, Rivière I. Manufacture of tumor- and virus-specific T lymphocytes for adoptive cell therapies. Cancer Gene Ther. 2015;22:85–94. doi: 10.1038/cgt.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang WC, Aguilar B, Ostberg JR, Riddell SR, Forman SJ, Jensen MC. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother. 2012;35:689–701. doi: 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu R, Forget MA, Chacon J, Bernatchez C, Haymaker C, Chen JQ, Hwu P, Radvanyi LG. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: current status and future outlook. Cancer J. 2012;18:160–175. doi: 10.1097/PPO.0b013e31824d4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, Liu H, Creighton CJ, Gee AP, Heslop HE, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR. CD19-T cells and are preserved by IL-7 and IL-15 Blood. 2014;123:3750–3759. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science. 2012;335:1195–1200. doi: 10.1126/science.1216557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewski JL, Kochman AA, Lu SX, Terwey TH, Kim TD, Hubbard VM, Muriglan SJ, Suh D, Smith OM, Grubin J, et al. Adoptive transfer of T-cell precursors enhances T-cell reconstitution after allogeneic hematopoietic stem cell transplantation. Nat Med. 2006;12:1039–1047. doi: 10.1038/nm1463. [DOI] [PubMed] [Google Scholar]
- Zakrzewski JL, Suh D, Markley JC, Smith OM, King C, Goldberg GL, Jenq R, Holland AM, Grubin J, Cabrera-Perez J, et al. Tumor immunotherapy across MHC barriers using allogeneic T-cell precursors. Nat Biotechnol. 2008;26:453–461. doi: 10.1038/nbt1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Parkhurst MR, Zheng Z, Cohen CJ, Riley JP, Gattinoni L, Restifo NP, Rosenberg SA, Morgan RA. Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoietic stem cells via Notch signaling. Cancer Res. 2007;67:2425–2429. doi: 10.1158/0008-5472.CAN-06-3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]