

Abstract
Cutaneous lymphomas (CL) are a heterogeneous group of neoplasms characterized with clinical and histopathological variation, as well as overlap with benign dermatoses. Diagnosis and treatment of CLs is challenging and often requires a multi-disciplinary approach. However, prognostic knowledge of these conditions and awareness of treatment options can help optimize appropriate use of available regimens, thereby improving care for patients. Here we review the most recent literature and outline treatment themes for managing patients with cutaneous B-cell and T-cell lymphomas other than mycosis fungoides.
Keywords: Primary cutaneous B- cell lymphoma, cutaneous T- cell lymphoma, non-mycosis fungoides, systemic treatment, skin-directed treatment, multi-disciplinary approach
INTRODUCTION
Cutaneous lymphomas (CL) are a heterogeneous group of neoplasms characterized by an accumulation of mononuclear, mostly lymphocytic cells in the skin.1 They are the second most prevalent extranodal non-Hodgkin lymphomas (after gastrointestinal), representing approximately 19% of extranodal non-Hodgkin lymphomas1 with mycosis fungoides (MF) and Sézary syndrome (SS) accounting for over 50% of all cutaneous T-cell lymphomas (CTCL). Non-mycosis fungoides (non-MF) CTCL and cutaneous B-cell lymphomas (CBCL) include several other subtypes and variants, whose clinical and histopathological presentations vary significantly.
While this review will focus mainly on treatment of such non-MF CTCL and CBCL, accurate diagnosis is a crucial, and often challenging, initial step. Given the rarity of these cutaneous lymphomas and the variability in presentations, clinical-pathological correlation is paramount to making a sound diagnosis. Furthermore, there can be significant overlap with reactive benign inflammatory conditions. Application of modern PCR-based methods for determining monoclonality in formalin-fixed tissue has greatly increased our ability to diagnose cutaneous lymphomas. At least 85% of cases demonstrate monoclonality.2 However, since monoclonality can be seen rarely in reactive processes, interpretation should be performed in clinical, histopathologic and immunophenotypic context2–4. At times, long term clinical follow up with multiple biopsies is needed to confirm the final diagnosis.
Once the diagnosis is established, there are some common treatment themes. Our overall approach is to best understand the patient’s presenting condition within the realm of the existing World Health Organization-European Organization for Research and Treatment of Cancer (WHO-EORTC) classification, thereby understanding the disease prognosis as indolent or aggressive. For the indolent lymphomas, our approach is to balance efficacy with safety of the available regimens. Given that several indolent conditions have a high rate of relapse with a retained excellent overall prognosis; treatments with low potential of immediate and long-term risks are preferable for early lines of therapy. At times, observation is a reasonable option. In contrast, early diagnosis of the aggressive cutaneous lymphomas, followed by combination chemotherapy and often a strong consideration of hematopoietic stem cell transplant (HSCT) is a mainstay of treatment.
CUTANEOUS T-CELL LYMPHOMA
Primary Cutaneous CD30+ lymphoproliferative disorders (CD 30+ LPD)
Primary cutaneous CD30-positive lymphoproliferative disorders are the second most common form of cutaneous T-cell lymphomas representing about 30% of all CTCLs. They include lymphomatoid papulosis (LyP), primary cutaneous anaplastic large cell lymphoma (PCALCL) and borderline/overlapping cases that may include features of both.1 Clinical follow up is occasionally the only way to establish the final diagnosis.
Lymphomatoid papulosis (LyP)
Overview
Overview: chronic, recurrent self resolving papular necrotic/papular nodular skin disease, with variable histology, and CD3+, CD4+, typically CD8− immunophenotype, with type A and type C revealing large CD30+ cells.5 Given the spontaneous resolution of these lesions the prognosis is excellent and treatment is often not needed. One series with 118 patients with LyP, reported a 2% mortality over a follow up of 77 months.6 There is a reported incidence of second lymphomas (most commonly cutaneous lymphomas such as MF and cALCL) in this subgroup of patient as high as 20–40% but a very low risk of systemic lymphomas.6,7,8 The risk factors to predict a secondary lymphoma are not well delineated. Number of lesions, severity of symptoms, ethnicity, and lactate dehydrogenase levels did not affect development of lymphoma in one series. Moreover, treatment of LyP provided symptomatic relief but did not prevent progression of lymphoma.8
Treatment guidelines
The natural course of LyP is chronic and relapsing, without a curative treatment option. Hence, we support a conservative treatment approach, balancing long- term safety profile and adverse effects of the available treatment regimens. For patients with few or infrequent, non-scarring lesions, active observation should be considered. Occasionally lesions may ulcerate or become infected, in which case topical or oral antibiotics maybe considered. Several anecdotal reports have demonstrated responses to phototherapy modalities such as Psolaren and UVA (PUVA), narrowband ultraviolet B (nbUVB), excimer laser, photodynamic treatment (PDT), however, relapses are common after discontinuation of treatment.6 In addition, LyP lesions have been reported to appear de novo in a PUVA treated field in MF patients.9 Other reported effected skin directed treatments include topical steroids, intralesional steroids, nitrogen mustard and topical imiquimod with variable treatment responses.8 Low-dose oral methotrexate (MTX, 5–20 mg/wk) is effective in suppressing the development of new skin lesions and can be considered in patients with widespread disease.10 Other novel options include oral and topical bexarotene11, as well as brentuximab vedotin, a CD30−directed antibody-toxin conjugate. In a phase II clinical trial, of brentuximab vedotin in patients with CD30+ lymphoproliferative disorders (n=9 LyP patients), a 100% overall response rate (ORR) was achieved; time to response was 3 weeks (range, 3 to 9 weeks), and median duration of response was 26 weeks (range, 6 to 44 weeks). Grade 1 to 2 peripheral neuropathy was the most common (54%) and ongoing side effect (45%).12
Primary cutaneous anaplastic large cell lymphoma (pcALCL)
Overview
Primary cutaneous anaplastic large cell lymphoma is an indolent cutaneous T-cell lymphoma, characterized by large cells characterized by the expression of CD30 antigen in more than 75% of the tumor cells, without history of concomitant MF.13 Clinically, lesions present as solitary or grouped, rapidly growing and ulcerating tumors or thick plaques, and may involve regional lymph nodes. Histopathology demonstrates non-epidermotropic sheets of large CD30+ cells with anaplastic, pleomorphic, or immunoblastic cytomorphology. Immunophenotype is CD4+ (but can be CD8+), with variable loss of CD3, CD2, and CD5. Cells show expression of cutaneous lymphocyte antigen (CLA), but are negative for epithelial membrane antigen (EMA) and anaplastic lymphoma kinase (ALK). CD 15 staining is negative, distinguishing this entity from Hodgkin’s lymphoma.1
Clinical course is indolent with 5-year disease-related survival ranging from 96% (skin only) to 91% (skin and regional lymph node involvement). Spontaneous regression (complete or partial) maybe observed in skin lesions in up to 42% of the patients.6 However, relapse rate after therapy can be as high as 42%.14 In a small study, extensive limb disease (ELD) and extracutaneous spread were found to be negative prognostic factors for disease specific survival in a multivariate analysis, with an aggressive course and poorer outcomes.15 A newly described DUSP22-IRF4 locus on 6p25.3 (DUSP22 rearrangement) in systemic ALK-neg ALCL is associated with an improved prognosis.16 Although it is also reported in 28% of pcALCL.17 as well as in LyP18, the prognostic significance of this remains unclear.
Treatment guidelines
Given that CD30+LPDs demonstrate an overall indolent course. For localized disease, surgical excision for small lesions and/or radiation are considered to be the first line treatment, with 95% complete response rate (CRR). Relapses after local therapy may occur in up to 40% of patients, with no treatment more superior in controlling relapses.19 Multiagent chemotherapy with CRR of 80–92%, but 62–80% relapse rate in skin and/or draining lymph nodes, has fallen out of favor as first line treatment, due to lack of durable responses. Single agent chemotherapies including MTX (intralesional/systemic), etoposide, gemcitabine in addition to anecdotal reports of response to interferons, imiquimod, thalidomide, bexarotene (alone or in combination with Interferon α), as well as HSCT have been summarized elsewhere.19
In the relapsed or refractory pcALCL, Pralatrexate, an anti-folate approved for use in peripheral T cell lymphoma, has shown activity. It is an anti-folate with an acceptable safety profile for continuous long-term dosing. In a dose-de-escalation strategy, Pralatrexate 15mg/m(2)/wk for 3 of 4 weeks showed high activity with acceptable toxicity in patients with relapsed/refractory CTCL.20 Duvic et al12 showed that CD30+ LPDs, including primary cutaneous anaplastic T-cell lymphomas (n = 2) showed ORR of 100% to Brentuximab vedotin (BV); time to response was approximately 3 weeks, and median duration of response was 26weeks for LyP/pcALCL patients. Onset of response was faster than MF patients but the duration of response was also shorter.12 An international, randomized, prospective study of Brentuximab vedotin+methotrexate vs Brentuximab alone has also recently completed accrual with anticipated results on the horizon.
Subacute Panniculitis-like T-cell lymphoma
Overview
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is a rare primary cutaneous T-cell lymphoma that makes up <1% of T-cell lymphomas.21 Clinically, lesions presents as non-specific subcutaneous nodules that may regress and remit spontaneously.22–24 Histopathology shows atypical lymphoid cells rimming individual adipocytes with associated reactive histiocytes often associated with coagulation necrosis. Atypical lymphocytes show CD8+, CD56+, alpha/beta+ immunophenotype. Involvement of lymph nodes, spleen or bone marrow is uncommon. Hemophagocytic syndrome (HPS)/hemophagocytic lymphohistiocytosis (HLH) can be observed and carries a poor prognosis. The gamma/delta expressing phenotype of this disease is now classified as cutaneous gamma/delta T-cell lymphoma, under primary cutaneous peripheral T-cell lymphoma and tends to have a more aggressive clinical course.24,25
Treatment guidelines
Therapy for SPTCL remains controversial. Since SPTCL usually follows an indolent course and carries a favorable prognosis similar to MF (particularly when hemophagocytic lymphohistiocytosis is not present), approaches more commonly used for other indolent CTCLs have proven effective. Historically, responses to combination chemotherapy are usually of short duration and CRs are rare.26,27 There are reports of successful allogeneic stem cell transplantation28–30 and prolonged remissions with combination chemotherapy followed by an autologous stem cell transplant.31 However, the rarity of this disease and the infrequent need for aggressive therapy hampers our understanding of the utility of such approaches. Importantly, as with other indolent lymphomas, relapses often have not correlated with shortened survival. Single agent bexarotene has significant clinical activity in SPTCL with an ORR of 82% including CRs.32 Other systemic agents as used for CTCL, such as oral methotrexate and histone deacetylase inhibitors also have activity. Anecdotal responses to glucocorticoids, interferon-α, zidovudine, and cyclosporine also have been observed.33–37 The use of denileukin diftitox in two patients has been reported with evidence of activity.38
It has been our approach to treat those with SPTCL or the rare clinically indolent cutaneous gamma-delta T-cell lymphoma with systemic therapies that may be less aggressive but more tolerable for long periods of time. This strategy mimics what we often do for CTCL, in contrast to aggressive systemic PTCL. Therefore, we prefer initial therapy with either bexarotene or oral methotrexate. At times, observation alone may be appropriate. We consider combination chemotherapy, often adding HST consolidation with curative intent, in physically fit patients who continue to progress on these and other milder therapies, show a more aggressive disease course, and/or develop HLH.
Primary Cutaneous Peripheral T-cell lymphoma
Cutaneous gamma/delta T-cell lymphoma
Overview
As mentioned above, the term SPTCL now restricted to the a/b phenotype and the gamma/delta expressing phenotype of this disease is now classified as cutaneous gamma/delta T-cell lymphoma under the classification of PTCL. It often carries a highly aggressive clinical course.24,25 Clinically, lesions appear as generalized plaques and/or ulceronecrotic nodules or tumors involving the extremities, mucosal sites, other extranodal sites. Immunophenotype of the PTCL, gamma/delta type is represented by a proliferation of mature, activated gamma/delta T-cells w/a cytotoxic phenotype with CD2+, CD3+, bF1−, g/d+, CD5−, CD56+, cytotoxic proteins. It is generally CD4−, CD8−, EBV−.39 Involvement of lymph nodes, spleen or bone marrow is uncommon.
Treatment guidelines
In contrast to STPCL alpha/beta, cutaneous gamma/delta T-cell lymphoma is more frequently associated with hemophagocytic lymphohistiocytosis (HLH). In the largest multicenter retrospective series of this disorder, the median survival was 31 months but others have cited medium survivals of approximately 1 year.39,40 Given the poor prognosis, aggressiveness, and poor responsiveness to conventional-dose treatments, we and others prefer to manage most patients with aggressive cutaneous gamma/delta lymphomas patients similarly to other cytotoxic T-cell lymphomas (e.g. HSTCL) with non-CHOP based induction chemotherapy followed by SCT with a preference fir allogeneic over autologous when feasible. More recently, as our ability to perform gamma staining by immunohistochemistry the has become more reliable, it has been observed that there may be a less aggressive form of cutaneous gamma/delta T-cell lymphoma that has histologic features and clinical behavior skin to SPTCL.41–43
Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (provisional)
Overview
Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma is a rare subtype representing<1% of cutaneous T-cell lymphoma (CTCL) with about 45 cases44 reported in the literature, and a more recently, additional 18 cases reported through a EORTC Cutaneous Lymphoma Task Force Workshop45. It is characterized by widely distributed ulcerated papules and nodules, with occasional mucosal involvement, with an aggressive and fatal course.44,46 Histology reveals epidermotropic infiltrates of CD8+ (CD2−/CD4−/CD45RA−/CD45RO−/CD5−/CCD56−) cytotoxic T cells, expressing cytotoxic markers (beta F1+, TIA-1+, granzyme B+, perforin+); EBV is generally negative.1,45 Clinical course is rapidly progressive, (median survival 12–32 months45,46) and usually unresponsive to conventional CTCL treatments. There is a high tendency for spread to extranodal sites, mucosa and central nervous system, generally sparing lymph nodes and bone marrow.45,46 Of note, there are reported cases of indolent CD8+ lymphoid proliferation of acral sites (CD8+ LP), which are thought to be low grade lymphomas with clinical presentation similar to CD4+ small/medium pleomorphic T-cell lymphoma (see below), comparable disease course, and infrequent need for systemic therapy, which should not be confused with aggressive CD8+ epidermotropic TCL.47,48
Treatment guidelines
Optimal treatment for this condition is not defined, due to rarity of this disease and its aggressive course, systemic spread and frequent relapse. In general, skin directed therapies are ineffective, including radiotherapy, unless used in combination with systemic therapy.44 Partial responses may result with oral bexarotene plus total skin electron beam treatment (TSEBT).49 Multi-agent CHOP like regimens are used most commonly but rates relapse are high, potentially improved with auto- or allogeneic HSCT.45 When the clinical aggressiveness is clear is, our approach is to treat them with our same approach for cutaneous gamma/delta T-cell lymphoma.
CD4+ small- medium pleomorphic T-cell lymphoma (CD4+ SMPTCL)/CD8+ lymphoid proliferations of acral sites (CD8+ LP) (provisional)
Overview
Primary cutaneous CD4+ small- medium pleomorphic T- cell lymphoma (CD4+ SMPTCL) is a low grade indolent lymphoma (provisional entity) presenting as solitary papule or nodule, usually located on the head and neck.47 Trunk and extremities can be involved, and a multifocal presentation has been very rarely associated with a more aggressive course with systemic involvement. No surface markers are predictive of an aggressive course, including Ki-67 index.50 Histologically, the lesions are characterized by dense dermal infiltrate of small to medium sized atypical lymphocytes (CD4+/CD8+) without significant epidermotropism.51 Immunohistochemistry reveals expression of T- follicular helper cell markers (PD-1, CXCL-13 and BCL-6) by the lymphocytic infiltrate in CD4+ SMPTCL, which can aide in diagnosis in cases with a polymorphous infiltrate along with a dominant T-cell clone.51 While CD4+SMPTCL is a provisional entity in WHO-EORTC classification of cutaneous lymphomas, and its true malignant potential is questioned, a discussion of this entity is included here to emphasize the favorable prognosis and indolent nature of these conditions.50, 52
Treatment guidelines
Data suggests excellent prognosis in these patients and durable response to skin directed treatment modalities.48,50,52–57 Treatment options include local excision, topical/intra-lesional steroids and local radiation. The rates of remission are high irrespective of the treatment modality used, and the relapse rates are low. Patients with multifocal progressive disease and/or symptoms suggestive of systemic involvement should be evaluated with staging studies such as computed tomography (CT), or positron emission tomography (PET) scan to exclude PTCL, NOS with subsequent dissemination to the skin as opposed to CD4+ SMPTCL.50
CUTANEOUS B-CELL LYMPHOMAS (CBCL)
Primary cutaneous B-cell lymphomas represent less than one third of cutaneous lymphomas1,58. The WHO-EORTC has categorized primary cutaneous B-cell lymphomas to 3 main subtypes (Table 1). The relevance to distinguish these subtypes is in the different treatment options as well as different prognosis59. Primary cutaneous marginal zone lymphoma (PCMZL) and primary cutaneous follicle center lymphoma (PCFCL) are characterized as indolent diseases with 99%–95% 5 year survival in comparison to DLBCL, leg type that has a more aggressive disease course and less than a 50% 5year survival60. The diagnosis of primary cutaneous B-cell lymphoma is only established when the staging is negative after the initial clinical and histopathological diagnosis. Of note, a bone marrow biopsy is often not required as routine staging in indolent CBCL, such as PCMZL, but is required in clinically intermediate to aggressive forms of cutaneous B-cell lymphomas.61
Table 1.
The WHO/EORTC classification for cutaneous lymphomas1
| Mature T-cell and NK-cell neoplasms |
|---|
| Mycosis fungoides (MF) |
| Variants of MF |
| Pagetoid reticulosis (localized disease) |
| Folliculotropic |
| Subtype of MF |
| Granulomatous slack skin |
| Sezary syndrome |
| CD30+ T-cell lymphoproliferative disorders of the skin |
| Lymphomatoid papulosis |
| Primary cutaneous anaplastic large cell lymphoma |
| Subcutaneous panniculitis-like T-cell lymphoma – alpha/beta origin |
| Primary cutaneous peripheral T-Cell lymphoma (PTL), unspecified |
| Subtypes of PTL |
| Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma (provisional) |
| Cutaneous gamma/delta-positive T-cell lymphoma (provisional) |
| Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma (provisional) |
| Extranodal NK/T-cell lymphoma, nasal type* |
| Hydroa vacciniforme-like lymphoma (variant)* |
| Adult T-cell leukemia/lymphoma* |
| Angioimmunoblastic T-cell lymphoma* |
|
Mature B-cell neoplasms |
| Cutaneous marginal zone B-cell lymphoma |
| Primary cutaneous follicle center lymphoma |
| Growth patterns |
| Follicular |
| Follicular and diffuse |
| Diffuse |
| Cutaneous diffuse large B-cell lymphoma, leg type |
| Cutaneous diffuse large B-cell lymphoma, others |
| Subtype of DLBCL, other |
| Intravascular large B-cell lymphoma* |
|
Immature hematopoietic malignancies |
| Blastic NK-cell lymphoma CD4+/CD56+ hematodermic neoplasm (Blastic NK lymphoma)* |
This table also contains entities of extracutaneous lymphomas frequently involving the skin as a secondary site, thereby excluded from discussion in this review.
Primary cutaneous marginal zone lymphomas (PCMZL)
Primary cutaneous marginal zone lymphomas of MALT-type (PCMZL), previously known as primary cutaneous immunocytomas, comprise 24% of all primary cutaneous B-cell lymphomas60. Clinically, they present as single or multiple, red to violaceous infiltrated cutaneous or subcutaneous papules, plaques or multi-focal nodules with 2 cm or less in diameter (70%), with multifocal lesions being most common (72%).62 The lesions are slow growing and usually do not ulcerate. Biopsies (i.e. punch biopsy or surgical excision) should be adequately deep in order to evaluate the extent of the infiltrate. Some authors have suggested an association of MZL is associated with chronic inflammatory processes or infections.63,64 In Europe there have been cases of PCMZL associated with Borrelia burgdorferi infection however, recent reports are contradictory62,65, however, recent reports are contradictory.66–69
Immunohistochemistry reveals CD19+, CD20+, CD5−, CD10− with monotypic immunoglobulin light chains often demonstrable in paraffin sections. The cells are often class-switched (IgG, IgA, IgE), unlike other types of extranodal marginal zone B-cell lymphomas. BCL2 is expressed in most cases70–72. The characteristic presence of reactive follicles can be highlighted by a CD21 stain that marks follicular dendritic cells.
Primary cutaneous follicle center lymphoma (PCFCL)
PCFCL is the most common type of primary cutaneous B-cell lymphomas, making up 57% of cases in a recent large study. The median age at diagnosis is 58 years with a male:female ratio of 1.8.60 It presents as an erythematous papule, plaque, or nodule most commonly located on the trunk or head/neck. Lesions may be single or multiple but are localized when multiple. It is only rarely seen on the upper (2.3%) or lower (6.4%) extremities. The latter is at times difficult to differentiate from diffuse large B-cell lymphoma, leg type (DLBCL – LT). Immunohistochemistry reveals pan-B-cell markers CD19 and CD20, with coexpression of BCL6. Other germinal center B-cell markers are also often expressed including CD10 and human germinal center –associated lymphoma (HGAL)73 Unlike nodal follicular lymphoma, in which BCL2 expression is a hallmark that reflects a t(14;18)(q32;q21) translocation, PCFCL is characteristically negative for BCL2. However, in examples that are follicular, especially when predominantly small cleaved cells, BCL2 is expressed in approximately 40% of cases.73,74
Primary cutaneous diffuse large B-cell lymphoma, leg type (PCDLBL-LT)
PCDLBCL-LT comprises approximately 20% of primary cutaneous B-cell lymphomas and has several distinctive clinical features. Compared to the above two types, this lymphoma occurs in an older population (median age 78 years) and has a striking female predominance (M:F ratio of 0.5). As the name implies, it presents most commonly (88% of patients) on the lower extremity. However, it can occur at other sites including the head/neck, trunk, and upper extremities in 5–12% of patients1,60. Clinically it presents as a nodule, either singly or as multiple regional lesions. Multifocal disease is seen in 20% of cases. Uncharacteristic to other cutaneous lymphomas, PCDLBCL-LT will often disseminate to nodal and visceral sites, which likely portends a transition in its already aggressive behavior75,76. Immunohistochemistry reveals expression of CD19 and CD20. Unlike PCFCL, expression of BCL2 is the rule and the post-germinal center B-cell maker MUM1 is usually expressed. BCL6 is expressed by most cases but CD10 is not.73,77–80
Molecular genetics
t(11;18)(q21;q21) translocation involving API2-MALT1 and t(3;14)(p14;q32) involving FOXP1 and IGH@ are seen in less than 10% of cases. The t(14;18)(q32;q21) also involving IGH@ and MALT1 is present in less than 15% of cases.72,81 The IGH@-BCL2 translocation typically seen in nodal follicular lymphoma can be seen in 0–40% of cases PCFCL with a follicular growth pattern.74,82,83 Variation may be related to technique.82 Gene expression profiling studies have shown that the profile resembles germinal center-like diffuse large B-cell lymphomas.84
PCDLBL-LT lacks translocations seen in MALT-type lymphomas or follicular lymphoma. However, translocations of BCL6, MYC, and IGH@ and amplification of BCL2 are commonly seen. Deletion in the region of cell cycle inhibitors CDKN2A and CDK2NB (chromosome 9p21.3) or promoter methylation is frequent and associated with poor outcome.85,86 Gene expression profiling shows a distinct profile from PCFCL and similarity to activated B-cell type of diffuse large B-cell lymphoma.84
Treatment guidelines
The standard treatment for indolent cutaneous B-cell lymphoma (MZL and FCL) depends on number and size of the lesions. Although there is no strong support in the literature for “watch and wait”, it is recommended by the National Comprehensive Cancer Network (NCCN) guidelines87 and practiced by some experts for multifocal lesions or extensive disease.
Excision and local radiotherapy is commonly considered as first line therapy especially for solitary lesions. Recent studies have shown treatment with radiation could result in 99% complete response rate but the relapse rate in these studies widely varied.88 Neelis et al used low dose (2×4Gy) radiation in 18 indolent CBCL patients with 72% complete response rate.89 Low-dose local radiation may have fewer side effects and moreover, it provides the possibility of repeating radiation when there is evidence of relapse.
In small studies, intralesional interferon α90, intralesional adenovirus-interferon γ91, intralesional steroids92–94, intralesional rituximab95,96 have been administered successfully with an acceptable relapse rate. Systemic rituximab monotherapy is often administered when there is multifocal disease or other therapies are contraindicated or unwanted97–99. Topical imiquimod, an immune response modulator, is an option in certain cases100,101.
In cases with high suspicion for an infectious trigger such as Borrelia or H.pylori, appropriate antibiotic therapy can be attempted as first line therapy with anecdotal responses reported.102–105 Systemic mono- or multi-agent chemotherapy such as chlorambucil106 or CHOP-like regimens have been used in the past when the true indolent nature of this lymphoma was less well understood. These are now only considered in cases of extensive disease, failure prior therapies, and extracutaneous spread, in which case their management is similar to the more common systemic indolent B-cell lymphomas.88
The treatment of PCDLBCL-LT is extrapolated from the diffuse large B-cell lymphoma, the most common systemic non-Hodgkin lymphoma. Therefore, if manageable, immunotherapy with rituximab plus multiagent anthracycline based chemotherapy is recommended.75,107 Localized radiation therapy to a solitary lesion or grouped lesions is an option as an adjuvant treatment to systemic chemoimunotherapy or alone in cases where comorbidities preclude chemotherapy. The most common regimens used in the up-front management of PCDLBCL-LT are R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) or the infusional regimen, dose adjusted R-EPOCH (cyclophosphamide, doxorubicin vincristine, etoposide, and prednisone). Commonly, full course therapy with 6 cycles is used as there is a lack of evidence for “short-course” combined modality therapy in PCDLBCL-LT despite its use extensively in localized DLBCL.108 Rituximab monotherapy for PCDLBCL-LT is thought to be inferior therapy, however, remains an option for patients who are unable to tolerate multi-agent chemotherapy.109 To date, there are no randomized studies to provide guidance.
CONCLUSION
Given the rarity of these lymphomas, prospective data comparing treatments is lacking. Randomized trials are rare and our management decisions are often extrapolated from very imperfect data. In addition, clinical trials evaluating novel therapies frequently accrue CTCL patients across several subtypes. However, despite lack of high-level evidence, patients need treatment. While a strict algorithm cannot be written, we attempt to highlight themes and principles that have emerged through the existing literature, coupled with clinical experience, allowing reasonable and logical decisions.
Figure 1.
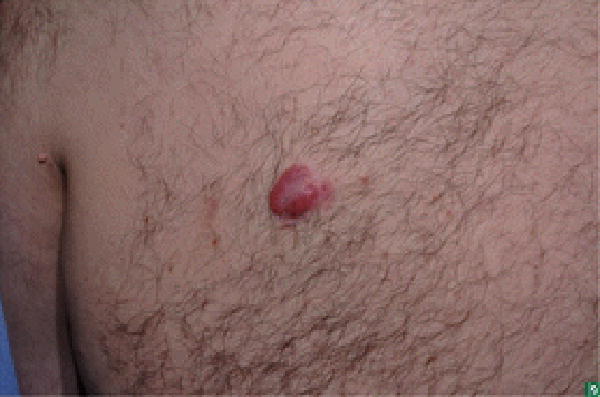
Lymphomatoid papulosis: lesions in different stages of healing, one active lesion on R calf (arrow).
Figure 2.
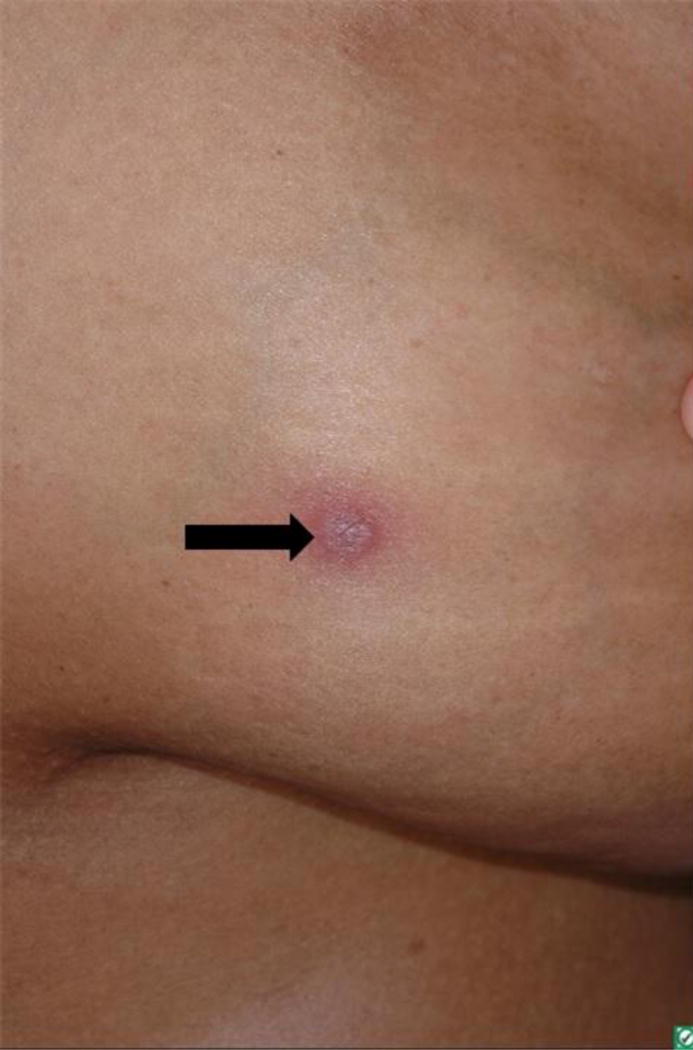
Primary cutaneous marginal zone lymphoma: solitary non-ulcerated, erythematous nodule.
Acknowledgments
Alison Moskowitz reports grants from Seattle Genetics.
Steven M. Horwitz reports grants and personal fees from Celgene, Seattle Genetics, Takeda Millennium, and Spectrum.
Footnotes
Conflict of Interest
Meenal Kheterpal, Neha Mehta-Shah, Pooja Virmani, and Patricia L. Myskowski each declare no potential conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105(10):3768–3785. doi: 10.1182/blood-2004-09-3502. [DOI] [PubMed] [Google Scholar]
- 2.Morales AV, Arber DA, Seo K, Kohler S, Kim YH, Sundram UN. Evaluation of B-cell clonality using the BIOMED-2 PCR method effectively distinguishes cutaneous B-cell lymphoma from benign lymphoid infiltrates. The American Journal of dermatopathology. 2008;30(5):425–430. doi: 10.1097/DAD.0b013e31818118f7. [DOI] [PubMed] [Google Scholar]
- 3.Fujiwara M, Morales AV, Seo K, Kim YH, Arber DA, Sundram UN. Clonal Identity and Differences in Primary Cutaneous B-Cell Lymphoma Occurring at Different Sites or Time Points in the Same Patient. The American Journal of dermatopathology. 2012 doi: 10.1097/DAD.0b013e318255dbae. [DOI] [PubMed] [Google Scholar]
- 4.Nihal M, Mikkola D, Wood GS. Detection of clonally restricted immunoglobulin heavy chain gene rearrangements in normal and lesional skin: analysis of the B cell component of the skin-associated lymphoid tissue and implications for the molecular diagnosis of cutaneous B cell lymphomas. J Mol Diagn. 2000;2(1):5–10. doi: 10.1016/S1525-1578(10)60609-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kadin MNK, Sako D, Said J, Vonderheid E. Lymphomatoid papulosis. A cutaneous proliferation of activated helper T cells expressing Hodgkin’s disease-associated antigens. Am J Pathol. 1985;119(2):315–25. (2):315–325. [PMC free article] [PubMed] [Google Scholar]
- 6.Bekkenk MW1GF, van Voorst Vader PC, Heule F, Geerts ML, van Vloten WA, Meijer CJ, Willemze R. Primary and secondary cutaneous CD30(+) lymphoproliferative disorders: a report from the Dutch Cutaneous Lymphoma Group on the long-term follow-up data of 219 patients and guidelines for diagnosis and treatment. Blood. 2000 Jun;95(12):3653–3661. [PubMed] [Google Scholar]
- 7.Kunishige JH1MH, Alvarez G, Johnson M, Prieto V, Duvic M. Lymphomatoid papulosis and associated lymphomas: a retrospective case series of 84 patients. Clin Exp Dermatol. 2009 Jul;34(5):576–581. doi: 10.1111/j.1365-2230.2008.03024.x. [DOI] [PubMed] [Google Scholar]
- 8.Wieser IOC, Talpur R, Duvic M. Lymphomatoid papulosis: Treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016 Jan;74(1):59–67. doi: 10.1016/j.jaad.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Wolf P1CL, Smolle J, Kerl H. PUVA-induced lymphomatoid papulosis in a patient with mycosis fungoides. J Am Acad Dermatol. 1991 Aug;25(2):422–426. doi: 10.1016/0190-9622(91)70220-v. [DOI] [PubMed] [Google Scholar]
- 10.Vonderheid ECSA, Kadin ME. Methotrexate is effective therapy for lymphomatoid papulosis and other primary cutaneous CD30-positive lymphoproliferative disorders. J Am Acad Dermatol. 1996;34(3):470–481. doi: 10.1016/s0190-9622(96)90442-9. [DOI] [PubMed] [Google Scholar]
- 11.Krathen RAWS, Duvic M. Bexarotene is a new treatment option for lymphomatoid papulosis. Dermatology. 2003;206(2):142–147. doi: 10.1159/000068451. [DOI] [PubMed] [Google Scholar]
- 12.Duvic MTM, Gangar P, et al. Results of a Phase II Trial of Brentuximab Vedotin for CD30+ Cutaneous T-Cell Lymphoma and Lymphomatoid Papulosis. J Clin Oncol. 2015 Nov;33(32):3759–3765. doi: 10.1200/JCO.2014.60.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willemze R1BR. Spectrum of primary cutaneous CD30 (Ki-1)-positive lymphoproliferative disorders. A proposal for classification and guidelines for management and treatment. J Am Acad Dermatol. 1993 Jun;28(6):973–980. doi: 10.1016/0190-9622(93)70140-o. [DOI] [PubMed] [Google Scholar]
- 14.Liu HLHR, Kohler S, Harvell JD, Reddy S, Kim YH. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003 Dec;49(6):1049–1058. doi: 10.1016/s0190-9622(03)02484-8. [DOI] [PubMed] [Google Scholar]
- 15.Woo DKJC, Vanoli-Storz MN, Kohler S, Reddy S, Advani R, Hoppe RT, Kim YH. Prognostic factors in primary cutaneous anaplastic large cell lymphoma: characterization of clinical subset with worse outcome. Arch Dermatol. 2009 Jun;145(6):667–674. doi: 10.1001/archdermatol.2009.74. [DOI] [PubMed] [Google Scholar]
- 16.Parrilla Castillar ERJE, Said J, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014;124:1473–1480. doi: 10.1182/blood-2014-04-571091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feldman ALLM, Remstein ED, et al. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009;23:574–580. doi: 10.1038/leu.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karai LJKM, Hsi ED, et al. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am J Surg Pathol. 2013;37:1173–1181. doi: 10.1097/PAS.0b013e318282d01e. [DOI] [PubMed] [Google Scholar]
- 19.Kempf W1PK, Vermeer MH, et al. EORTC, ISCL, and USCLC consensus recommendations for the treatment of primary cutaneous CD30-positive lymphoproliferative disorders: lymphomatoid papulosis and primary cutaneous anaplastic large-cell lymphoma. Blood. 2011 Oct 13;118(15):4024–4035. doi: 10.1182/blood-2011-05-351346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horwitz SMKY, Foss F, Zain JM, Myskowski PL, Lechowicz MJ, Fisher DC, Shustov AR, Bartlett NL, Delioukina ML, Koutsoukos T, Saunders ME, O’Connor OA, Duvic M. Identification of an active, well-tolerated dose of pralatrexate in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood. 2012 May;119(18):4115–4122. doi: 10.1182/blood-2011-11-390211. [DOI] [PubMed] [Google Scholar]
- 21.Gallardo F, Pujol RM. Subcutaneous panniculitic-like T-cell lymphoma and other primary cutaneous lymphomas with prominent subcutaneous tissue involvement. Dermatol Clin. 2008;26(4):529–540. viii. doi: 10.1016/j.det.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 22.Takeshita M, Okamura S, Oshiro Y, et al. Clinicopathologic differences between 22 cases of CD56-negative and CD56-positive subcutaneous panniculitis-like lymphoma in Japan. Human pathology. 2004;35(2):231–239. doi: 10.1016/j.humpath.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 23.Paulli M, Berti E. Cutaneous T-cell lymphomas (including rare subtypes). Current concepts. II. Haematologica. 2004;89(11):1372–1388. [PubMed] [Google Scholar]
- 24.Willemze R, Hodak E, Zinzani PL, Specht L, Ladetto M, Group EGW Primary cutaneous lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology: official journal of the European Society for Medical Oncology/ESMO. 2013;24(Suppl 6):vi149–154. doi: 10.1093/annonc/mdt242. [DOI] [PubMed] [Google Scholar]
- 25.WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon, France: International Agency for Research on Cancer; 2008. [Google Scholar]
- 26.Go RS, Wester SM. Immunophenotypic and molecular features, clinical outcomes, treatments, and prognostic factors associated with subcutaneous panniculitis-like T-cell lymphoma: a systematic analysis of 156 patients reported in the literature. Cancer. 2004;101(6):1404–1413. doi: 10.1002/cncr.20502. [DOI] [PubMed] [Google Scholar]
- 27.Matsue K, Itoh M, Tsukuda K, Miyazaki K, Kokubo T. Successful treatment of cytophagic histiocytic panniculitis with modified CHOP-E. Cyclophosphamide, adriamycin, vincristine, predonisone, and etoposide. American journal of clinical oncology. 1994;17(6):470–474. doi: 10.1097/00000421-199412000-00003. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Persona E, Mateos-Mazon JJ, Lopez-Villar O, et al. Complete remission of subcutaneous panniculitic T-cell lymphoma after allogeneic transplantation. Bone marrow transplantation. 2006;38(12):821–822. doi: 10.1038/sj.bmt.1705527. [DOI] [PubMed] [Google Scholar]
- 29.Ichii M, Hatanaka K, Imakita M, Ueda Y, Kishino B, Tamaki T. Successful treatment of refractory subcutaneous panniculitis-like T-cell lymphoma with allogeneic peripheral blood stem cell transplantation from HLA-mismatched sibling donor. Leukemia & lymphoma. 2006;47(10):2250–2252. doi: 10.1080/10428190600783619. [DOI] [PubMed] [Google Scholar]
- 30.Yuan L, Sun L, Bo J, et al. Durable remission in a patient with refractory subcutaneous panniculitis-like T-cell lymphoma relapse after allogeneic hematopoietic stem cell transplantation through withdrawal of cyclosporine. Annals of transplantation: quarterly of the Polish Transplantation Society. 2011;16(3):135–138. doi: 10.12659/aot.882007. [DOI] [PubMed] [Google Scholar]
- 31.Mukai HY, Okoshi Y, Shimizu S, et al. Successful treatment of a patient with subcutaneous panniculitis-like T-cell lymphoma with high-dose chemotherapy and total body irradiation. European journal of haematology. 2003;70(6):413–416. doi: 10.1034/j.1600-0609.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 32.Mehta N, Wayne AS, Kim YH, et al. Bexarotene is active against subcutaneous panniculitis-like T-cell lymphoma in adult and pediatric populations. Clinical lymphoma, myeloma & leukemia. 2012;12(1):20–25. doi: 10.1016/j.clml.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang CY, Su WP, Kurtin PJ. Subcutaneous panniculitic T-cell lymphoma. International journal of dermatology. 1996;35(1):1–8. doi: 10.1111/j.1365-4362.1996.tb01606.x. [DOI] [PubMed] [Google Scholar]
- 34.Papenfuss JS, Aoun P, Bierman PJ, Armitage JO. Subcutaneous panniculitis-like T-cell lymphoma: presentation of 2 cases and observations. Clinical lymphoma. 2002;3(3):175–180. doi: 10.3816/clm.2002.n.024. [DOI] [PubMed] [Google Scholar]
- 35.Springinsfeld G, Guillaume JC, Boeckler P, Tortel MC, Cribier B. Two cases of subcutaneous panniculitis-like T-cell lymphoma (CD4− CD8+ CD56−) Annales de dermatologie et de venereologie. 2009;136(3):264–268. doi: 10.1016/j.annder.2008.07.061. [DOI] [PubMed] [Google Scholar]
- 36.Jung HR, Yun SY, Choi JH, Bae SH, Ryoo HM, Kum YS. Cyclosporine in Relapsed Subcutaneous Panniculitis-like T-Cell Lymphoma after Autologous Hematopoietic Stem Cell Transplantation. Cancer research and treatment: official journal of Korean Cancer Association. 2011;43(4):255–259. doi: 10.4143/crt.2011.43.4.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee WS, Hwang JH, Kim MJ, et al. Cyclosporine A as a Primary Treatment for Panniculitis-like T Cell Lymphoma: A Case with a Long-Term Remission. Cancer research and treatment: official journal of Korean Cancer Association. 2014;46(3):312–316. doi: 10.4143/crt.2014.46.3.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hathaway T, Subtil A, Kuo P, Foss F. Efficacy of denileukin diftitox in subcutaneous panniculitis-like T-cell lymphoma. Clinical lymphoma & myeloma. 2007;7(8):541–545. doi: 10.3816/clm.2007.n.040. [DOI] [PubMed] [Google Scholar]
- 39.Guitart J, Weisenburger DD, Subtil A, et al. Cutaneous gammadelta T-cell lymphomas: a spectrum of presentations with overlap with other cytotoxic lymphomas. The American journal of surgical pathology. 2012;36(11):1656–1665. doi: 10.1097/PAS.0b013e31826a5038. [DOI] [PubMed] [Google Scholar]
- 40.Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101(9):3407–3412. doi: 10.1182/blood-2002-05-1597. [DOI] [PubMed] [Google Scholar]
- 41.Takahashi Y, Takata K, Kato S, et al. Clinicopathological analysis of 17 primary cutaneous T-cell lymphoma of the gammadelta phenotype from Japan. Cancer science. 2014;105(7):912–923. doi: 10.1111/cas.12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Herrera A, Song JY, Chuang SS, et al. Nonhepatosplenic gammadelta T-cell lymphomas represent a spectrum of aggressive cytotoxic T-cell lymphomas with a mainly extranodal presentation. The American journal of surgical pathology. 2011;35(8):1214–1225. doi: 10.1097/PAS.0b013e31822067d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magro CM, Wang X. Indolent primary cutaneous gamma/delta T-cell lymphoma localized to the subcutaneous panniculus and its association with atypical lymphocytic lobular panniculitis. American journal of clinical pathology. 2012;138(1):50–56. doi: 10.1309/AJCPQGVLTZQ77VFF. [DOI] [PubMed] [Google Scholar]
- 44.Nofal A1A-MM, Assaf M, Salah E. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma: proposed diagnostic criteria and therapeutic evaluation. J Am Acad Dermatol. 2012 Oct;67(4):748–759. doi: 10.1016/j.jaad.2011.07.043. [DOI] [PubMed] [Google Scholar]
- 45.Robson AAC, Bagot M, et al. Aggressive epidermotropic cutaneous CD8+ lymphoma: a cutaneous lymphoma with distinct clinical and pathological features. Report of an EORTC Cutaneous Lymphoma Task Force Workshop. Histopathology. 2015 Oct;67(4):425–441. doi: 10.1111/his.12371. [DOI] [PubMed] [Google Scholar]
- 46.Berti DT E, Vermeer MH, Meijer CJ, Alessi E, Willemze R. Primary cutaneous CD8-positive epidermotropic cytotoxic T-cell lymphomas: a distinct clinicopathological entity with an aggressive clinical behavior. Am J Pathol. 1999;155:483–492. doi: 10.1016/S0002-9440(10)65144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beltraminelli HMR, Cerroni L. Indolent CD8+ lymphoid proliferation of the ear: a phenotypic variant of the small-medium pleomorphic cutaneous T-cell lymphoma? Journal of cutaneous pathology. 2010 Jan;37(1):81–84. doi: 10.1111/j.1600-0560.2009.01278.x. [DOI] [PubMed] [Google Scholar]
- 48.Swick BLBC, Venkat AP, Liu V. Indolent CD8+ lymphoid proliferation of the ear: report of two cases and review of the literature. Journal of cutaneous pathology. 2011 Feb;38(2):209–215. doi: 10.1111/j.1600-0560.2010.01647.x. [DOI] [PubMed] [Google Scholar]
- 49.Gormley RH1HS, Anand D, Junkins-Hopkins J, Rook AH, Kim EJ. Primary cutaneous aggressive epidermotropic CD8+ T-cell lymphoma. J Am Acad Dermatol. 2010 Feb;62(2):300–307. doi: 10.1016/j.jaad.2009.02.035. [DOI] [PubMed] [Google Scholar]
- 50.James ESJ, Gibson JF, et al. CD4 + primary cutaneous small/medium-sized pleomorphic T-cell lymphoma: a retrospective case series and review of literature. Leukemia & lymphoma. 2015 Apr;56(4):951–957. doi: 10.3109/10428194.2014.938331. [DOI] [PubMed] [Google Scholar]
- 51.Rodriguez Pinilla SMRG, Rodriguez-Peralto JL, et al. Primary cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma expresses follicular T-cell markers. The American journal of surgical pathology. 2009 Jan;33(1):81–90. doi: 10.1097/PAS.0b013e31818e52fe. [DOI] [PubMed] [Google Scholar]
- 52.Beltraminelli HLB, Kerl H, Cerroni L. Primary cutaneous CD4+ small-/medium-sized pleomorphic T-cell lymphoma: a cutaneous nodular proliferation of pleomorphic T lymphocytes of undetermined significance? A study of 136 cases. The American Journal of Dermatopathol. 2009 Jun;31(4):317–322. doi: 10.1097/DAD.0b013e31819f19bb. [DOI] [PubMed] [Google Scholar]
- 53.Bekkenk MWVM, Jansen PM, et al. Peripheral T-cell lymphomas unspecified presenting in the skin: analysis of prognostic factors in a group of 82 patients. Blood. 2003 Sep 15;102(6):2213–2219. doi: 10.1182/blood-2002-07-1960. [DOI] [PubMed] [Google Scholar]
- 54.Grogg KLJS, Erickson LA, McClure RF, Dogan A. Primary cutaneous CD4-positive small/medium-sized pleomorphic T-cell lymphoma: a clonal T-cell lymphoproliferative disorder with indolent behavior. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2008 Jun;21(6):708–715. doi: 10.1038/modpathol.2008.40. [DOI] [PubMed] [Google Scholar]
- 55.Williams VLT-CC, Duvic M. Primary cutaneous small- to medium-sized CD4+ pleomorphic T-cell lymphoma: a retrospective case series and review of the provisional cutaneous lymphoma category. American journal of clinical dermatology. 2011 Dec 1;12(6):389–401. doi: 10.2165/11590390-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 56.Baum CLLB, Neppalli VT, Swick BL, Liu V. Reappraisal of the provisional entity primary cutaneous CD4+ small/medium pleomorphic T-cell lymphoma: a series of 10 adult and pediatric patients and review of the literature. Journal of the American Academy of Dermatology. 2011 Oct;65(4):739–748. doi: 10.1016/j.jaad.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 57.Suchak ROCS, McNamara C, Robson A. Indolent CD8-positive lymphoid proliferation on the face: part of the spectrum of primary cutaneous small-/medium-sized pleomorphic T-cell lymphoma or a distinct entity? Journal of cutaneous pathology. 2010 Sep;37(9):977–981. doi: 10.1111/j.1600-0560.2009.01448.x. [DOI] [PubMed] [Google Scholar]
- 58.Bradford PT, Devesa SS, Anderson WF, Toro JR. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113(21):5064–5073. doi: 10.1182/blood-2008-10-184168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dummer R, Asagoe K, Cozzio A, et al. Recent advances in cutaneous lymphomas. J Dermatol Sci. 2007;48(3):157–167. doi: 10.1016/j.jdermsci.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 60.Senff NJ, Hoefnagel JJ, Jansen PM, et al. Reclassification of 300 primary cutaneous B-Cell lymphomas according to the new WHO-EORTC classification for cutaneous lymphomas: comparison with previous classifications and identification of prognostic markers. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25(12):1581–1587. doi: 10.1200/JCO.2006.09.6396. [DOI] [PubMed] [Google Scholar]
- 61.Kim YH, Willemze R, Pimpinelli N, et al. TNM classification system for primary cutaneous lymphomas other than mycosis fungoides and Sezary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC) Blood. 2007;110(2):479–484. doi: 10.1182/blood-2006-10-054601. [DOI] [PubMed] [Google Scholar]
- 62.Hoefnagel JJ, Vermeer MH, Jansen PM, et al. Primary cutaneous marginal zone B-cell lymphoma: clinical and therapeutic features in 50 cases. Arch Dermatol. 2005;141(9):1139–1145. doi: 10.1001/archderm.141.9.1139. [DOI] [PubMed] [Google Scholar]
- 63.Zendri E, Venturi C, Ricci R, Giordano G, De Panfilis G. Primary cutaneous plasmacytoma: a role for a triggering stimulus? Clin Exp Dermatol. 2005;30(3):229–231. doi: 10.1111/j.1365-2230.2004.01692.x. [DOI] [PubMed] [Google Scholar]
- 64.May SA, Netto G, Domiati-Saad R, Kasper C. Cutaneous lymphoid hyperplasia and marginal zone B-cell lymphoma following vaccination. J Am Acad Dermatol. 2005;53(3):512–516. doi: 10.1016/j.jaad.2005.04.036. [DOI] [PubMed] [Google Scholar]
- 65.Aberer E, Fingerle V, Wutte N, Fink-Puches R, Cerroni L. Within European margins. Lancet. 2011;377(9760):178. doi: 10.1016/S0140-6736(10)62241-6. [DOI] [PubMed] [Google Scholar]
- 66.Cerroni L, Zochling N, Putz B, Kerl H. Infection by Borrelia burgdorferi and cutaneous B-cell lymphoma. J Cutan Pathol. 1997;24(8):457–461. doi: 10.1111/j.1600-0560.1997.tb01318.x. [DOI] [PubMed] [Google Scholar]
- 67.Goodlad JR, Davidson MM, Hollowood K, et al. Primary cutaneous B-cell lymphoma and Borrelia burgdorferi infection in patients from the Highlands of Scotland. Am J Surg Pathol. 2000;24(9):1279–1285. doi: 10.1097/00000478-200009000-00012. [DOI] [PubMed] [Google Scholar]
- 68.Ponzoni M, Ferreri AJ, Mappa S, et al. Prevalence of Borrelia burgdorferi infection in a series of 98 primary cutaneous lymphomas. Oncologist. 2011;16(11):1582–1588. doi: 10.1634/theoncologist.2011-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wood GS, Kamath NV, Guitart J, et al. Absence of Borrelia burgdorferi DNA in cutaneous B-cell lymphomas from the United States. J Cutan Pathol. 2001;28(10):502–507. doi: 10.1034/j.1600-0560.2001.281002.x. [DOI] [PubMed] [Google Scholar]
- 70.Servitje O, Gallardo F, Estrach T, et al. Primary cutaneous marginal zone B-cell lymphoma: a clinical, histopathological, immunophenotypic and molecular genetic study of 22 cases. Br J Dermatol. 2002;147(6):1147–1158. doi: 10.1046/j.1365-2133.2002.04961.x. [DOI] [PubMed] [Google Scholar]
- 71.Edinger JT, Kant JA, Swerdlow SH. Cutaneous marginal zone lymphomas have distinctive features and include 2 subsets. The American journal of surgical pathology. 2010;34(12):1830–1841. doi: 10.1097/PAS.0b013e3181f72835. [DOI] [PubMed] [Google Scholar]
- 72.Cho-Vega JH, Vega F, Rassidakis G, Medeiros LJ. Primary cutaneous marginal zone B-cell lymphoma. American journal of clinical pathology. 2006;125(Suppl):S38–49. doi: 10.1309/CVFYBQNMX1PKNAA7. [DOI] [PubMed] [Google Scholar]
- 73.Xie X, Sundram U, Natkunam Y, et al. Expression of HGAL in primary cutaneous large B-cell lymphomas: evidence for germinal center derivation of primary cutaneous follicular lymphoma. Mod Pathol. 2008 doi: 10.1038/modpathol.2008.30. [DOI] [PubMed] [Google Scholar]
- 74.Mirza I, Macpherson N, Paproski S, et al. Primary cutaneous follicular lymphoma: an assessment of clinical, histopathologic, immunophenotypic, and molecular features. J Clin Oncol. 2002;20(3):647–655. doi: 10.1200/JCO.2002.20.3.647. [DOI] [PubMed] [Google Scholar]
- 75.Grange F, Beylot-Barry M, Courville P, et al. Primary cutaneous diffuse large B-cell lymphoma, leg type: clinicopathologic features and prognostic analysis in 60 cases. Arch Dermatol. 2007;143(9):1144–1150. doi: 10.1001/archderm.143.9.1144. [DOI] [PubMed] [Google Scholar]
- 76.Vermeer MH, Geelen FA, van Haselen CW, et al. Primary cutaneous large B-cell lymphomas of the legs. A distinct type of cutaneous B-cell lymphoma with an intermediate prognosis. Dutch Cutaneous Lymphoma Working Group [see comments] Arch Dermatol. 1996;132(11):1304–1308. [PubMed] [Google Scholar]
- 77.Geelen FA, Vermeer MH, Meijer CJ, et al. bcl-2 protein expression in primary cutaneous large B-cell lymphoma is site-related. J Clin Oncol. 1998;16(6):2080–2085. doi: 10.1200/JCO.1998.16.6.2080. [DOI] [PubMed] [Google Scholar]
- 78.Grange F, Petrella T, Beylot-Barry M, et al. Bcl-2 protein expression is the strongest independent prognostic factor of survival in primary cutaneous large B-cell lymphomas. Blood. 2004;103(10):3662–3668. doi: 10.1182/blood-2003-08-2726. [DOI] [PubMed] [Google Scholar]
- 79.Hoefnagel JJ, Vermeer MH, Jansen PM, Fleuren GJ, Meijer CJ, Willemze R. Bcl-2, Bcl-6 and CD10 expression in cutaneous B-cell lymphoma: further support for a follicle centre cell origin and differential diagnostic significance. Br J Dermatol. 2003;149(6):1183–1191. doi: 10.1111/j.1365-2133.2003.05649.x. [DOI] [PubMed] [Google Scholar]
- 80.Sundram U, Kim Y, Mraz-Gernhard S, Hoppe R, Natkunam Y, Kohler S. Expression of the bcl-6 and MUM1/IRF4 proteins correlate with overall and disease-specific survival in patients with primary cutaneous large B-cell lymphoma: a tissue microarray study. J Cutan Pathol. 2005;32(3):227–234. doi: 10.1111/j.0303-6987.2005.00298.x. [DOI] [PubMed] [Google Scholar]
- 81.Streubel B, Simonitsch-Klupp I, Mullauer L, et al. Variable frequencies of MALT lymphoma-associated genetic aberrations in MALT lymphomas of different sites. Leukemia. 2004;18(10):1722–1726. doi: 10.1038/sj.leu.2403501. [DOI] [PubMed] [Google Scholar]
- 82.Streubel B, Scheucher B, Valencak J, et al. Molecular cytogenetic evidence of t(14;18)(IGH;BCL2) in a substantial proportion of primary cutaneous follicle center lymphomas. Am J Surg Pathol. 2006;30(4):529–536. doi: 10.1097/00000478-200604000-00015. [DOI] [PubMed] [Google Scholar]
- 83.Cerroni L, Arzberger E, Putz B, et al. Primary cutaneous follicle center cell lymphoma with follicular growth pattern. Blood. 2000;95(12):3922–3928. [PubMed] [Google Scholar]
- 84.Hoefnagel JJ, Dijkman R, Basso K, et al. Distinct types of primary cutaneous large B-cell lymphoma identified by gene expression profiling. Blood. 2004 doi: 10.1182/blood-2004-04-1594. [DOI] [PubMed] [Google Scholar]
- 85.Dijkman R, Tensen CP, Jordanova ES, et al. Array-based comparative genomic hybridization analysis reveals recurrent chromosomal alterations and prognostic parameters in primary cutaneous large B-cell lymphoma. J Clin Oncol. 2006;24(2):296–305. doi: 10.1200/JCO.2005.02.0842. [DOI] [PubMed] [Google Scholar]
- 86.Hallermann C, Kaune KM, Gesk S, et al. Molecular cytogenetic analysis of chromosomal breakpoints in the IGH, MYC, BCL6, and MALT1 gene loci in primary cutaneous B-cell lymphomas. J Invest Dermatol. 2004;123(1):213–219. doi: 10.1111/j.0022-202X.2004.22720.x. [DOI] [PubMed] [Google Scholar]
- 87.NCCN guidelines. http://www.nccn.org.
- 88.Senff NJ, Noordijk EM, Kim YH, et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood. 2008;112(5):1600–1609. doi: 10.1182/blood-2008-04-152850. [DOI] [PubMed] [Google Scholar]
- 89.Neelis KJ, Schimmel EC, Vermeer MH, Senff NJ, Willemze R, Noordijk EM. Low-dose palliative radiotherapy for cutaneous B- and T-cell lymphomas. Int J Radiat Oncol Biol Phys. 2009;74(1):154–158. doi: 10.1016/j.ijrobp.2008.06.1918. [DOI] [PubMed] [Google Scholar]
- 90.Cozzio A, Kempf W, Schmid-Meyer R, et al. Intra-lesional low-dose interferon alpha2a therapy for primary cutaneous marginal zone B-cell lymphoma. Leukemia & lymphoma. 2006;47(5):865–869. doi: 10.1080/10428190500399698. [DOI] [PubMed] [Google Scholar]
- 91.Audigé A, Urosevic M, Schlaepfer E, et al. Anti-HIV state but not apoptosis depends on IFN signature in CD4+ T cells. J Immunol. 2006;177(9):6227–6237. doi: 10.4049/jimmunol.177.9.6227. [DOI] [PubMed] [Google Scholar]
- 92.Perry A, Vincent BJ, Parker SR. Intralesional corticosteroid therapy for primary cutaneous B-cell lymphoma. Br J Dermatol. 2010;163(1):223–225. doi: 10.1111/j.1365-2133.2010.09798.x. [DOI] [PubMed] [Google Scholar]
- 93.Burg G, Dummer R, Kerl H. Classification of cutaneous lymphomas. Dermatol Clin. 1994;12(2):213–217. [PubMed] [Google Scholar]
- 94.Wong KC, Weller PA. Primary cutaneous B cell lymphoma: outcomes and treatment. Australas J Dermatol. 1998;39(4):261–264. doi: 10.1111/j.1440-0960.1998.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 95.Heinzerling L, Dummer R, Kempf W, Schmid MH, Burg G. Intralesional therapy with anti-CD20 monoclonal antibody rituximab in primary cutaneous B-cell lymphoma. Arch Dermatol. 2000;136(3):374–378. doi: 10.1001/archderm.136.3.374. [DOI] [PubMed] [Google Scholar]
- 96.Kyrtsonis MC, Siakantaris MP, Kalpadakis C, et al. Favorable outcome of primary cutaneous marginal zone lymphoma treated with intralesional rituximab. European journal of haematology. 2006;77(4):300–303. doi: 10.1111/j.1600-0609.2006.00720.x. [DOI] [PubMed] [Google Scholar]
- 97.Fink-Puches R, Wolf IH, Zalaudek I, Kerl H, Cerroni L. Treatment of primary cutaneous B-cell lymphoma with rituximab. J Am Acad Dermatol. 2005;52(5):847–853. doi: 10.1016/j.jaad.2005.01.093. [DOI] [PubMed] [Google Scholar]
- 98.Gitelson E, Al-Saleem T, Millenson M, Lessin S, Smith MR. Cutaneous B-cell lymphoma responds to rituximab: a report of five cases and a review of the literature. Leukemia & lymphoma. 2006;47(9):1902–1907. doi: 10.1080/10428190600688099. [DOI] [PubMed] [Google Scholar]
- 99.Heinzerling LM, Urbanek M, Funk JO, et al. Reduction of tumor burden and stabilization of disease by systemic therapy with anti-CD20 antibody (rituximab) in patients with primary cutaneous B-cell lymphoma. Cancer. 2000;89(8):1835–1844. [PubMed] [Google Scholar]
- 100.Farkas A, Kemeny L, French LE, Dummer R. New and experimental skin-directed therapies for cutaneous lymphomas. Skin Pharmacol Physiol. 2009;22(6):322–334. doi: 10.1159/000241302. [DOI] [PubMed] [Google Scholar]
- 101.Coors EA, Schuler G, Von Den Driesch P. Topical imiquimod as treatment for different kinds of cutaneous lymphoma. European journal of dermatology: EJD. 2006;16(4):391–393. [PubMed] [Google Scholar]
- 102.Bogle MA, Riddle CC, Triana EM, Jones D, Duvic M. Primary cutaneous B-cell lymphoma. J Am Acad Dermatol. 2005;53(3):479–484. doi: 10.1016/j.jaad.2005.04.043. [DOI] [PubMed] [Google Scholar]
- 103.Grange F, Wechsler J, Guillaume JC, et al. Borrelia burgdorferi-associated lymphocytoma cutis simulating a primary cutaneous large B-cell lymphoma. J Am Acad Dermatol. 2002;47(4):530–534. doi: 10.1067/mjd.2002.120475. [DOI] [PubMed] [Google Scholar]
- 104.Hofbauer GF, Kessler B, Kempf W, Nestle FO, Burg G, Dummer R. Multilesional primary cutaneous diffuse large B-cell lymphoma responsive to antibiotic treatment. Dermatology. 2001;203(2):168–170. doi: 10.1159/000051735. [DOI] [PubMed] [Google Scholar]
- 105.Kutting B, Bonsmann G, Metze D, Luger TA, Cerroni L. Borrelia burgdorferi-associated primary cutaneous B cell lymphoma: complete clearing of skin lesions after antibiotic pulse therapy or intralesional injection of interferon alfa-2a. J Am Acad Dermatol. 1997;36(2):311–314. doi: 10.1016/s0190-9622(97)80405-7. [DOI] [PubMed] [Google Scholar]
- 106.Hoefnagel JJ, Vermeer MH, Jansen PM, et al. Primary cutaneous marginal zone B-cell lymphoma: clinical and therapeutic features in 50 cases. Arch Dermatol. 2005;141(9):1139–1145. doi: 10.1001/archderm.141.9.1139. [DOI] [PubMed] [Google Scholar]
- 107.Senff NJ, Noordijk EM, Kim YH, et al. European Organization for Research and Treatment of Cancer and International Society for Cutaneous Lymphoma consensus recommendations for the management of cutaneous B-cell lymphomas. Blood. 2008;112(5):1600–1609. doi: 10.1182/blood-2008-04-152850. [DOI] [PubMed] [Google Scholar]
- 108.Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26(14):2258–2263. doi: 10.1200/JCO.2007.13.6929. [DOI] [PubMed] [Google Scholar]
- 109.Fenot M, Quereux G, Brocard A, Renaut JJ, Dreno B. Rituximab for primary cutaneous diffuse large B-cell lymphoma-leg type. European journal of dermatology: EJD. 2010;20(6):753–757. doi: 10.1684/ejd.2010.1102. [DOI] [PubMed] [Google Scholar]