

Abstract
The aims of this study were to test the hypothesis that mice expressing mitochondrially targeted human glutathione reductase (GR) driven by a surfactant protein C promoter (spc–mthGR) are functionally riboflavin deficient and that this deficiency exacerbates hyperoxic lung injury. The authors further hypothesized that dietary supplementation with riboflavin (FADH) will improve the bioactivity of GR, thus enhancing resistance to hyperoxic lung injury. Transgenic mt–spchGR mice and their nontransgenic littermates were fed control or riboflavin-supplemented diets upon weaning. At 6 weeks of age the mice were exposed to either room air (RA) or >95% O2 for up to 84 hours. GR activities (with and without exogenous FADH) and GR protein levels were measured in lung tissue homogenates. Glutathione (GSH) and glutathione disulfide (GSSG) concentrations were assayed to identify changes in GR activity in vivo. Lung injury was assessed by right lung to body weight ratios and bronchoalveolar lavage protein concentrations. The data showed that enhanced GR activity in the mitochondria of lung type II cells does not protect adult mice from hyperoxic lung injury. Furthermore, the addition of riboflavin to the diets of spc–mthGR mice neither enhances GR activities nor offers protection from hyperoxic lung injury. The results indicated that modulation of mitochondrial GR activity in lung type II cells is not an effective therapy to minimize hyperoxic lung injury.
Keywords: glutathione reductase, hyperoxic lung injury, riboflavin
Glutathione reductase (GR) is an essential component of the glutathione (GSH) antioxidant system. GSH is a tripeptide thiol found in high concentrations in lung tissues and alveolar lining fluid [1-4] and is an important first line of defense against reactive oxygen species generated by normal respiration or by inhalation of elevated oxygen concentrations. In reducing cellular H2O2, GSH is oxidized to glutathione disulfide (GSSG) by the activity of glutathione peroxidases (GPx) [5]. Once oxidized, GSSG can be exported from cells, react with free protein thiols, or be reduced by the activity of glutathione reductase (GR), thus replenishing the cellular pool and reducing capacity of GSH. Alterations in the ratios of GSH and GSSG in serum and tracheal aspirates are associated with lung injury in preterm infants and in chronic obstructive pulmonary disease (COPD) patients [6-8]. Pharmacologic inhibition of GR is associated with increased toxicity in response to oxidative stress in both rodents and cultured lung cells [9, 10]. We have previously reported that mice lacking GR compensated for this deficiency by thioredoxin-dependent mechanisms and inhibition of the thioredoxin system increased the susceptibility of these mice to hyperoxic lung injury [11]. Other studies have demonstrated that increased expression of GR protects against oxidant stress in lung cells in vitro [12, 13], suggesting that GR is a reasonable target for interventions in vivo against oxidant-mediated injury.
Our original studies revealed that transgenic mice expressing mitochondrially targeted human GR in type II alveolar epithelial cells driven by the surfactant protein C (SPC) promoter (spc-mthGR) had more severe lung injury than did the wild-type controls when exposed to hyperoxia [14]. These surprising results led to investigations designed to define the mechanisms responsible for the deleterious effects of increased GR expression during hyperoxia exposure.
GR protein forms a “head to tail” homodimer. Once dimerized, GR enzymatic activity requires association with flavin adenine dinucleotide (FAD), a coenzyme derived from riboflavin, as a cofactor. In our previous studies, we detected human GR mRNA and protein expression in whole-lung homogenates from spc–mthGR mice; however, increases in GR activities were not detected without the addition of riboflavin ex vivo to the assay [14]. Lower GSSG levels were observed in lungs from spc–mthGR mice than in lungs from wild-type mice, suggesting that GR activities were likely to be higher in vivo than the results obtained from the ex vivo analysis. Thus, we hypothesized that GR activities in vivo in the targeted GR overexpressing mice are restricted by limited quantities of the essential cofactor, riboflavin.
In the present studies, we tested the hypothesis that spc–mthGR mice are functionally riboflavin-deficient and that this deficiency exacerbates hyperoxic lung injury. We further hypothesized that dietary supplementation with riboflavin would improve the bioactivity of GR and enhance resistance to hyperoxic lung injury.
METHODS
Mice
Two distinct lines of GR transgenic mice were generated as previously described [14]. For the purpose of these studies, we have focused on Line 2, which exhibited greater GR expression than Line 1. Line 2 will be referred to as spc–mthGR. The wild-type (WT) mice were nontransgenic littermates. Beginning at weaning, spc–mthGR and WT littermates were fed ad libitum with either a control diet containing 7 mg/kg riboflavin (TD.06706; Harlan Teklad, Madison, WI) or with a custom high-riboflavin diet containing 70 mg/kg riboflavin (TD.07398; Harlan Teklad). At 6 weeks of age, mice were exposed to either >95% O2 (10 L per minute) or room air (RA) for 84 hours. Hyperoxia exposures were performed in a Plexiglas chamber with soda lime (Fisher Scientific) to absorb excess CO2 and oxygen levels were monitored daily. After 84 hours of exposure, mice were weighed and sacrificed using lethal doses of sodium pentobarbital (200 mg/kg). Right lungs were removed, weighed, snap-frozen, and placed in liquid nitrogen. Tracheas were cannulated with PE-50 tubing (Becton Dickinson, Franklin Lakes, NJ), and bronchoalveolar lavage (BAL) contents were collected from the left lungs by flushing 3 times with 0.3 mL saline. Left lungs were then snap-frozen. Right lung/body weight ratios were assessed as an indicator of lung injury.
Biochemical assays
Ten percent lung homogenates were prepared in 0.1 M KHPO4, 6.4 mM EDTA buffer, pH 7.4, using dounce homogenizers. GR activities, GSH concentrations, and GSSG concentrations were measured as previously described [15, 16]. GR activities were measured with and without the addition of ex vivo riboflavin (FAD) [14]. Clinically, riboflavin deficiency is assessed by GR activation coefficient, which represents the ratio of GR activity with ex vivo FAD (stimulated) divided by GR activities without ex vivo FAD (unstimulated) [17].
Protein quantitation
Protein contents in lung homogenates and BAL (only samples in which >70% of the instilled volume was recovered were included for analyses) were measured using the Bio-Rad protein assay as previously described [14].
Western blots
Lung hGR and mouse GR (mGR) immunoreactivities were assessed by Western blot as previously described [14]. Briefly, 30 μg protein from 10% lung tissue homogenates was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to nitrocellulose membranes, and were blocked with 10% nonfat dry milk in 20mM Tris-HCl (pH 7.6), 0.15 M NaCl, 0.2% Tween-20 (TBS-T) for 1.5 hours at room temperature. Membranes were incubated with primary antibody, either polyclonal rabbit anti-hGR or anti-mGR (both developed in our laboratory) in 5% milk in TBS-T overnight at 4°C, followed by secondary goat anti-rabbit-HRP in (Bio-Rad, Hercules, CA) in 5% milk in TBS-T for 1 hour at room temperature. Immunoreactivity was assessed using enhanced chemiluminescence (GE Healthcare, Buckinghamshire, UK). Membranes were stripped and reprobed with monoclonal mouse anti-β-actin antibody (Abcam, Cambridge, UK) to normalize for protein load.
Statistics
Data were analyzed by 3-way analysis of variance (ANOVA) with transgene, diet, and oxygen as factors. Secondarily, the data were analyzed by 2-way ANOVA with oxygen or RA exposure as a fixed factor followed by modified t tests post hoc to identify individual differences. Non-normal data were modified by Box-Cox power transformations to approach normality (as assessed by q-q plot fit) prior to analyses, with the transformations used described in legends for each figure. Data are expressed as means ± standard error and P ≤.05 was chosen to indicate statistical significance.
RESULTS
Control and spc–mthGR mice were fed purified control or high-riboflavin diets beginning at weaning. No effects of diet on body weights were detected in WT or spc–mthGR mice maintained in RA for 6 weeks (data not shown), indicating that riboflavin supplementation did not alter normal body growth.
GR activities were measured in right lung homogenates of WT and spc–mthGR mice to assess the effect of dietary riboflavin supplementation. GR activities in lung tissues obtained from the spc–mthGR control diet–fed mice were higher than in WT mice exposed to either RA and O2 (Figure 1). However, GR activities in lung tissues obtained from the spc–mthGR riboflavin diet–fed mice were not different than WT in RA. In hyperoxia, however, GR activities in the lungs of riboflavin-supplemented mice spc–mthGR were modestly elevated (Figure 1). Statistical analyses indicated individual effects of transgene and diet, and interactions between diet and transgene, transgene and exposure, and diet and exposure.
FIGURE 1.
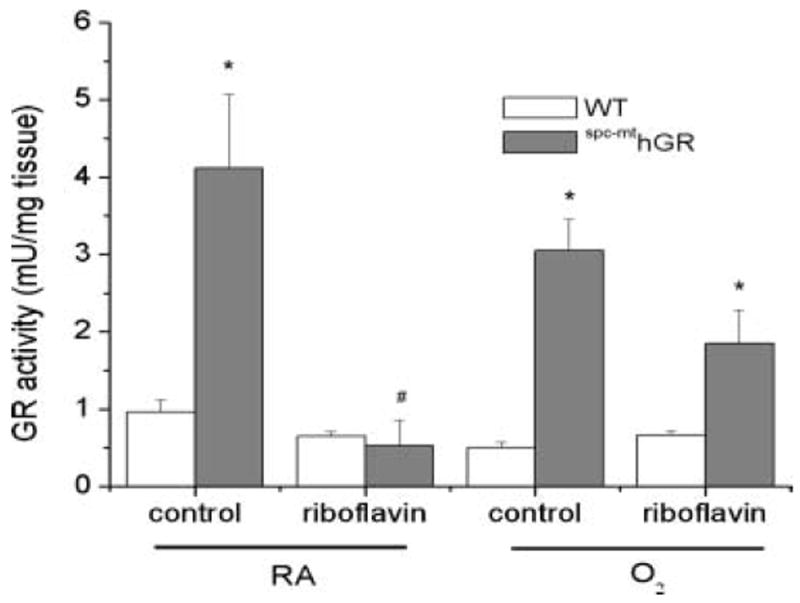
GR enzyme activities. Transgenic spc–mthGR mice and WT littermates were fed either a control or high-riboflavin diet beginning at 3 weeks of age. At 6 weeks of age the mice were exposed to RA or >95% O2 for 84 hours. Mice were sacrificed and tissues snap-frozen for analyses. GR activities were assessed in 10% lung homogenates by enzymatic assay. Data were natural-log transformed, and 3-way ANOVA indicated individual effects of transgene, diet, and interactions between transgene and diet, transgene and exposure, and diet and exposure. Two-way ANOVA with exposure as a fixed factor and modified t test post hoc were performed to identify individual differences. *Difference compared to WT, same diet; #difference between diets, same genotype; n = 5–22; P ≤ .05.
GR activities in lung tissue homogenates were also expressed as an activation coefficent by determining the ratio of the measured GR activities in the presence of exogenous FAD (1.25 μM) divided by the GR activities measured in the absence of exogenous FAD (Figure 2). These analyses identified any potential GR activity not measured in the original assay due to insufficient quantities of cofactor; a higher ratio correlates with increased cofactor deficiency. The data indicate that the GR activity in the WT strain is maximal and cannot be further enhanced. The addition of exogenous riboflavin to the assay increased GR activities in the lung homogenates from spc–mthGR mice, and the most significant activation occurred in riboflavin-supplemented, RA-exposed mice. Statistical analyses revealed individual effects of transgene, diet, and exposure, and interactions between transgene and diet, transgene and exposure, and diet and exposure, and a 3-way interaction between transgene, diet, and exposure.
FIGURE 2.
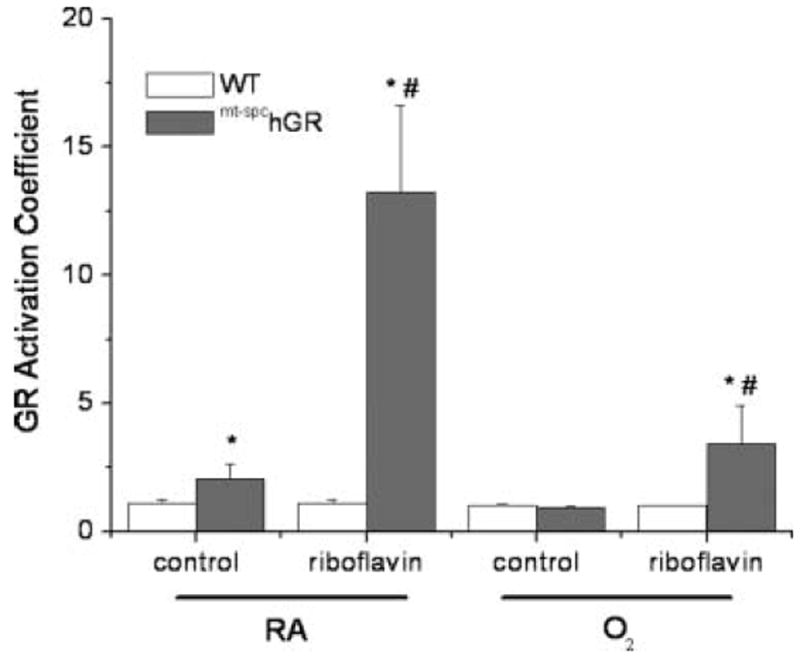
GR activation coefficient. GR activities were also measured in the presence of exogenous FADH (1.25 μM) and the ratios of GR activities with and without ex vivo riboflavin addition were calculated as activation coefficient. Data were analyzed by 3-way ANOVA after natural-log transformation and individual effects of transgene, diet, and exposure, and interactions between transgene and diet, transgene and exposure, and diet and exposure were detected. Two-way ANOVA with exposure as a fixed factor and modified t test post hoc were performed to identify individual differences between groups. *Difference compared to WT, same diet; #difference between diets, same genotype; n = 5–22; P ≤ .05.
To determine the effects of riboflavin supplementation on GR protein expression, Western blots were performed using lung tissue homogenates from spc–mthGR fed control and riboflavin-supplemented diets using specific anti-hGR antibodies that do not cross-react with mGR. We have previously shown that WT mice do not express hGR [14]. Western blots confirmed that riboflavin supplementation had no effect on hGR protein levels (Figure 3). Endogenous GR protein levels were also assessed using antibodies specific for mGR. No effects of transgene or oxygen exposure were detected (data not shown).
FIGURE 3.
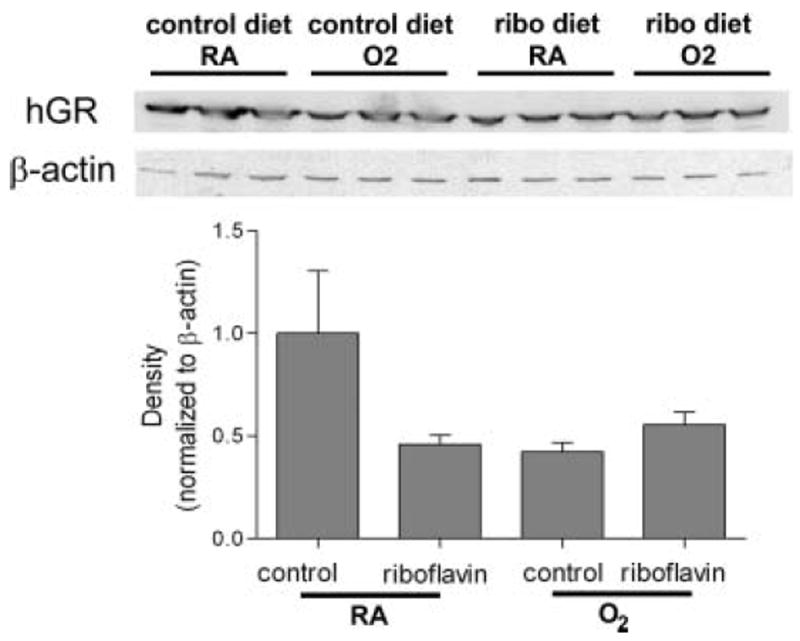
Human GR protein immunoreactivities. hGR protein levels were assessed by Western blot. Ten percent tissue homogenates were prepared from the lungs of mice treated as described in Methods and Figure 1. Protein were separated by SDS-PAGE and probed with anti-human GR or β-actin antibody. Band density was observed by enhanced chemiluminescence and evaluated by densitometry. No differences in band densities due to riboflavin supplementation were detected.
At tissue harvest, right lungs were weighed and right lung/body weight ratios were calculated as an assessment of lung edema and injury (Figure 4A). The data indicated an effect of hyperoxia on right lung/body weight ratios in all animals but no effect of diet. The greater right lung/body weight ratios in hyperoxia-exposed spc–mthGR mice are consistent with our previous report [14]. Riboflavin supplementation did not attenuate the effects of hyperoxia on right lung/body weight ratios. Individual effects of transgene and exposure, and an interaction between transgene and exposure were detected.
FIGURE 4.
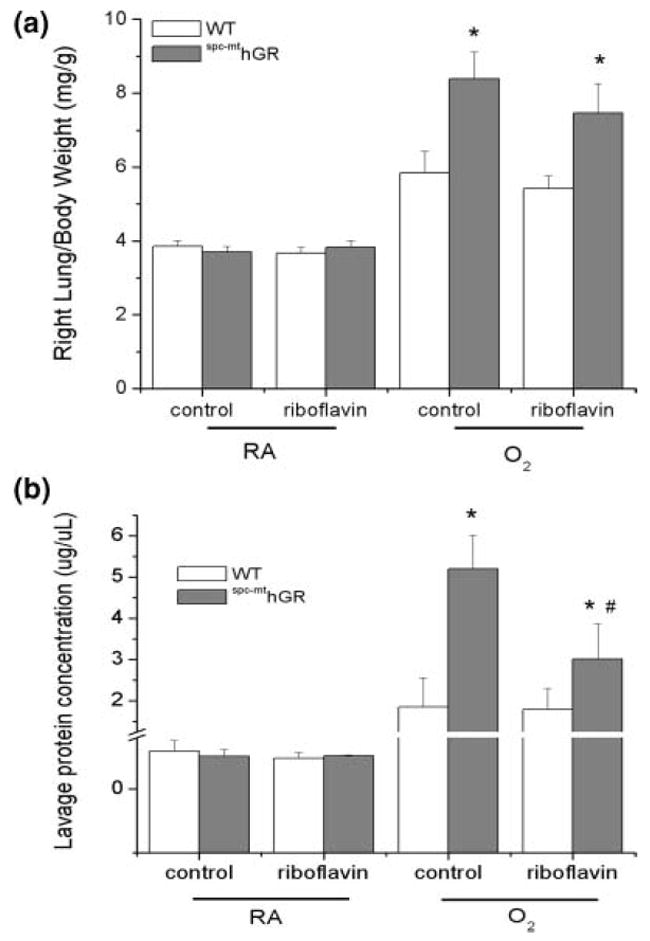
Indices of lung injury due to hyperoxia exposure. Mice were treated as described in Figure 1. (A) Total body weights and right lungs weights were measured at the time of sacrifice. Right lung/body weight ratios were calculated as indices of lung injury. Three-way ANOVA indicated individual effects of transgene and exposure and an interaction between transgene and exposure. (B) Bronchoalveolar lavage was collected from the left lung at the time of sacrifice. Protein concentrations in lavage fluids were measured by the Bio-Rad protein assay. Data were natural-log transformed and were analyzed by 3-way ANOVA. Individual effects of diet and exposure were detected. Two-way ANOVA with exposure as a fixed factor and modified t tests post hoc were performed to identify individual differences. *Difference compared to WT, same diet; #difference between diets, same genotype; n = 5–22; P ≤ .05.
BAL protein contents were assessed as an indicator of epithelial permeability (Figure 4B). Lavage protein contents were elevated in all hyperoxia-exposed groups compared to controls but more dramatically in spc–mthGR mice. However, the increased protein contents in BAL obtained from spc–mthGR mice were attenuated by riboflavin supplementation. Independent effects of transgene and exposure and an interaction between transgene and exposure were detected.
GSH levels were increased in lung tissues from WT mice fed riboflavin-supplemented diets and maintained in RA. In spc–mthGR mice exposed to RA; however, no effect of riboflavin supplementation on GSH levels was detected. Hyperoxia decreased GSH levels in spc–mthGR mice fed either diet but no effect of riboflavin supplementation on the GSH levels in the WT animals was detected (Table 1). An effect of transgene and an interaction between transgene and diet were detected.
TABLE 1.
GSH Contents in Lung Tissue Homogenates
| Diet | RA
|
O2
|
||
|---|---|---|---|---|
| WT | spc–mthGR | WT | spc–mthGR | |
| Control | 0.87 ± 0.06 | 0.95 ± 0.04 | 0.94 ± 0.12 | 0.66 ± 0.10 |
| Riboflavin | 1.28 ± 0.12# | 0.75 ± 0.15*,# | 1.43 ± 0.30*,# | 0.76 ± 0.10* |
Note. Transgenic spc–mthGR mice and WT littermates were fed either a control or high-riboflavin diet beginning at 3 weeks of age, and at 6 weeks of age were exposed to RA or >95% O2 for 84 hours. GSH concentrations were measured in 10% lung homogenates as described in Methods. Data were non-normal, and were transformed to square root and analyzed by 3-way ANOVA. An individual effect of transgene and an interaction between transgene and diet were detected. Two-way ANOVA with modified t test post hoc was performed with exposure as a fixed factor. An individual effect of transgene was detected.
Difference compared to WT, same diet.
difference between diets, same genotype; n = 5–22; P ≤ .05.
GSSG levels were greater in lung tissues from mice exposed to hyperoxia than in room air-exposed controls. As previously reported14, spc–mthGR mice had lower GSSG levels in lung tissue homogenates than did WT mice in either RA or O2 (Table 2). Riboflavin supplementation did not result in further decreases in GSSG levels. Independent effects of transgene and O2 were detected.
TABLE 2.
GSSG Contents in Lung Tissue Homogenates
| Diet | RA
|
O2
|
||
|---|---|---|---|---|
| WT | spc–mthGR | WT | spc–mthGR | |
| Control | 31.11 ± 6.01 | 4.69 ± 1.54* | 53.51 ± 8.94 | 16.71 ± 4.68* |
| Riboflavin | 29.49 ± 0.61 | 7.24 ± 1.91* | 59.11 ± 7.16 | 23.27 ± 9.71*,# |
Note. Transgenic spc–mthGR mice and WT littermates were fed either a control or high-riboflavin diet beginning at 3 weeks of age and at 6 weeks of age were exposed to RA or >95% O2 for 84 hours. GSSG concentrations in 10% lung homogenates were measured as described in Methods. Data were non-normal, and were transformed to square root and were analyzed by 3-way ANOVA. Individual effects of exposure and transgene were detected. Two-way ANOVA with modified t tests post hoc was performed with exposure as a fixed factor. An individual effect of transgene was detected.
Difference compared to WT, same diet.
difference between diets, same genotype; n = 5–22; P ≤ .05.
DISCUSSION
Although overt riboflavin deficiencies are rare in humans, the effects of increased expression of enzymes like GR, which requires FAD as a cofactor, could be limited by the availability of this essential component. In the present studies, dietary riboflavin supplementation did not increase GR activities in the lung tissues from mitochondrially targeted GR-overexpressing transgenic mice. These data suggest that either the mice were not functionally riboflavin deficient or that dietary supplementation with riboflavin did not result in the generation of a readily available coenzyme (Figure 1). As expected, riboflavin supplementation did not alter the levels of human GR protein expression in the lung, as indicated in Figure 3.
The suppression of GR activities in the riboflavin-supplemented, RA-exposed spc–mthGR mice is intriguing. It is possible the availability of sufficient quantities of FAD enabled normal regulatory mechanisms to repress transgene-mediated increases in GR activities. Another possibility is that the mitochondria-specific nature of the transgene expression precludes the availability of excess riboflavin to enter the mitochondrial compartment in healthy tissues.
The increased GR activation in the lung tissues of riboflavin-supplemented animals (Figure 2) supports the hypothesis that in these mice the lung tissues are functionally riboflavin deficient. Erythrocyte GR activation is measured routinely as an indicator of suboptimal riboflavin status and a value greater than 1.3 is considered the breakpoint for riboflavin deficiency [18]. GR activation in erythrocytes has been assessed in adults with chronic lung disease and an increase in activation (>1.3) was noted, suggesting that reoccurring systemic inflammation may lead to suboptimal riboflavin status [17]. Although it is not possible to extrapolate GR activation between lung tissue homogenates and the reports of erythrocyte GR activation, the GR activation in the lungs of animals supplemented with riboflavin strongly suggests that the GR enzyme activity in lung homogenates was riboflavin limited. Erythrocyte GR activation was not measured in this study primarily because the riboflavin status was most likely to be deficient in the tissue in which the transgene was expressed and assessments in the lung tested our primary hypothesis.
No changes in lung injury, as assessed by right lung/body weight ratios or lavage protein levels, were observed by the addition of riboflavin to the diets of WT mice (Figure 4A). Our data did indicate a modest decrease in lavage protein concentrations in riboflavin-supplemented spc–mthGR mice. This observation was not correlated with lower right lung to body weight ratios; therefore, the biological relevance of these findings are uncertain (Figure 4B).
Lung GSH levels were increased by riboflavin supplementation (Table 1) in both RA and hyperoxia-exposed WT mice. Changes in GSH contents in biological systems are usually secondary to increased synthesis by induction of the rate limiting enzyme γ-glutamylcysteine ligase [19]. In the present study, we did not investigate the mechanism(s) for increased GSH in lung tissues but we speculate that the increased expression is not a direct effect of riboflavin supplementation on GSH synthesis. These increases were not accompanied by decreases in GSSG contents (Table 2). Similar to our previous report, we detected increases in lung GSSG levels in both strains of hyperoxia-exposed mice compared to RA and substantially lower levels of GSSG in the lungs of spc–mthGR mice were detected compared to WT. Riboflavin supplementation did not alter GSSG levels suggesting that GR activities were not enhanced in vivo.
Our data indicate that regulation of GR activity in vivo is more complex than simple cofactor availability. Functionally, the regulation of GR activities are related to tertiary structure, the availability of cofactors, the formation of both intracellular and intercellular disulfide bonds, the binding of cofactors, and the availability of NADPH as a substrate to provide reducing equivalents. In the spc–mthGR mice, a more reduced environment in lung tissues, as reflected by lower GSSG levels in the transgenic lines, could interfere with the stability of essential disulfide bonds necessary for tertiary structure and enzymatic activity. Thus in an excessively reduced environment the additional GR protein produced by transgene expression could be rendered functionally inactive. It is also possible that in an artificially reduced environment, especially in the mitochondria, the inappropriate reduction of disulfide bonds might result in changes in the structure and function of other essential proteins.
In conclusion, our findings indicate that enhanced GR activities in the mitochondria of lung type II cells do not protect adult mice from hyperoxic lung injury. Furthermore, that the addition of riboflavin to the diets of spc–mthGR mice neither enhances GR activities nor offers protection from hyperoxic lung injury. These data indicate that the lack of protection against hyperoxic lung injury previously observed in spc–mthGR mice was not due to riboflavin deficiency. Modulation of GR activities, especially in the mitochrondria, is not an appropriate or effective therapy to protect against the deleterious effects of hyperoxic lung injury.
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Cantin AM, North SL, Hubbard RC, Crystal RG. Normal alveolar epithelial lining fluid contains high levels of glutathione. J Appl Physiol. 1987;63:152–157. doi: 10.1152/jappl.1987.63.1.152. [DOI] [PubMed] [Google Scholar]
- 2.Kosower NS, Kosower EM. The glutathione status of cells. Int Rev Cytol. 1978;54:109–160. doi: 10.1016/s0074-7696(08)60166-7. [DOI] [PubMed] [Google Scholar]
- 3.Biswas SK, Rahman I. Environmental toxicity, redox signaling and lung inflammation: the role of glutathione. Mol Asp Med. 2009;30:60–76. doi: 10.1016/j.mam.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutherland MW, Glass M, Nelson J, Lyen Y, Forman HJ. Oxygen toxicity: loss of lung macrophage function without metabolite depletion. J Free Radic Biol Med. 1985;1:209–214. doi: 10.1016/0748-5514(85)90120-5. [DOI] [PubMed] [Google Scholar]
- 5.Reed DJ. Glutathione: toxicological implications. Annu Rev Pharmacol Toxicol. 1990;30:603–631. doi: 10.1146/annurev.pa.30.040190.003131. [DOI] [PubMed] [Google Scholar]
- 6.Smith CV, Hansen TN, Martin NE, McMicken HW, Elliott SJ. Oxidant stress responses in premature infants during exposure to hyperoxia. Pediatr Res. 1993;34:360–365. doi: 10.1203/00006450-199309000-00024. [DOI] [PubMed] [Google Scholar]
- 7.O’Donovan DJ, Fernandes CJ. Mitochondrial glutathione and oxidative stress: implications for pulmonary oxygen toxicity in premature infants. Mol Genet Metab. 2000;71:352–358. doi: 10.1006/mgme.2000.3063. [DOI] [PubMed] [Google Scholar]
- 8.Beeh KM, Beier J, Koppenhoefer N, Buhl R. Increased glutathione disulfide and nitrosothiols in sputum supernatant of patients with stable COPD. Chest. 2004;126:1116–1122. doi: 10.1378/chest.126.4.1116. [DOI] [PubMed] [Google Scholar]
- 9.Kehrer JP, Paraidathathu T. Enhanced oxygen toxicity following treatment with 1,3-bis(2-chloroethyl)-1-nitrosourea. Fundam Appl Toxicol. 1984;4:760–767. doi: 10.1016/0272-0590(84)90097-6. [DOI] [PubMed] [Google Scholar]
- 10.Walther UI, Czermak A, Muckter H, Walther SC, Fichtl B. Decreased GSSG reductase activity enhances cellular zinc toxicity in three human lung cell lines. Arch Toxicol. 2003;77:131–137. doi: 10.1007/s00204-002-0421-z. [DOI] [PubMed] [Google Scholar]
- 11.Tipple TE, Welty SE, Rogers LK, Hansen TN, Choi YE, Kehrer JP, Smith CV. Thioredoxin-related mechanisms in hyperoxic lung injury in mice. Am J Respir Cell Mol Biol. 2007;37:405–413. doi: 10.1165/rcmb.2006-0376OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Donovan DJ, Katkin JP, Tamura T, et al. Gene transfer of mitochondrially targeted glutathione reductase protects H441 cells from t-butyl hydroperoxide-induced oxidant stresses. Am J Respir Cell Mol Biol. 1999;20:256–263. doi: 10.1165/ajrcmb.20.2.3367. [DOI] [PubMed] [Google Scholar]
- 13.O’Donovan DJ, Katkin JP, Tamura T, Smith CV, Welty SE. Attenuation of hyperoxia-induced growth inhibition in H441 cells by gene transfer of mitochondrially targeted glutathione reductase. Am J Respir Cell Mol Biol. 2000;22:732–738. doi: 10.1165/ajrcmb.22.6.3836. [DOI] [PubMed] [Google Scholar]
- 14.Heyob KM, Rogers LK, Welty SE. Glutathione reductase targeted to type II cells does not protect mice from hyperoxic lung injury. Am J Respir Cell Mol Biol. 2008;39:683–688. doi: 10.1165/rcmb.2008-0112OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jopperi-Davis KS, Park MS, Rogers LK, Backes CH, Jr, Lim IK, Smith CV. Compartmental inhibition of hepatic glutathione reductase activities by 1,3-bis(2-chloroethyl)-N-nitrosourea (BCNU) in Sprague-Dawley and Fischer-344 rats. Toxicol Lett. 2004;147:219–228. doi: 10.1016/j.toxlet.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 16.Rogers LK, Gupta S, Welty SE, Hansen TN, Smith CV. Nuclear and nucleolar glutathione reductase, peroxidase, and transferase activities in livers of male and female Fischer-344 rats. Toxicol Sci. 2002;69:279–285. doi: 10.1093/toxsci/69.1.279. [DOI] [PubMed] [Google Scholar]
- 17.Gariballa S, Forster S, Powers H. Riboflavin status in acutely ill patients and response to dietary supplements. JPEN J Parenter Enteral Nutr. 2009;33:656–661. doi: 10.1177/0148607109336602. [DOI] [PubMed] [Google Scholar]
- 18.Hoey L, McNulty H, Strain JJ. Studies of biomarker responses to intervention with riboflavin: a systematic review. Am J Clin Nutr. 2009;89:1960S–1980S. doi: 10.3945/ajcn.2009.27230B. [DOI] [PubMed] [Google Scholar]
- 19.Rahman I. Regulation of glutathione in inflammation and chronic lung diseases. Mutat Res. 2005;579:58–80. doi: 10.1016/j.mrfmmm.2005.02.025. [DOI] [PubMed] [Google Scholar]