

Abstract
The formation of the proper number of nephrons requires a tightly regulated balance between renal progenitor cell self-renewal and differentiation. The molecular pathways that regulate the transition from renal progenitor to renal vesicle are not well understood. Here, we show that Sall1interacts with the nucleosome remodeling and deacetylase complex (NuRD) to inhibit premature differentiation of nephron progenitor cells. Disruption of Sall1-NuRD in vivo in knock-in mice (ΔSRM) resulted in accelerated differentiation of nephron progenitors and bilateral renal hypoplasia. Transcriptional profiling of mutant kidneys revealed a striking pattern in which genes of the glomerular and proximal tubule lineages were either unchanged or upregulated, and those in the loop of Henle and distal tubule lineages were downregulated. These global changes in gene expression were accompanied by a significant decrease in THP-, NKCC2- and AQP1-positive loop of Henle nephron segments in mutant ΔSRM kidneys. These findings highlight an important function of Sall1-NuRD interaction in the regulation of Six2-positive multipotent renal progenitor cells and formation of the loop of Henle.
KEY WORDS: Lgr5, NuRD, Sall1, Loop of Henle, Nephron progenitor
Summary: A Sall1 knock-in mouse mutant shows accelerated differentiation of nephron progenitors and impaired lineage determination of loop of Henle progenitors, implicating the Sall1-NuRD interaction as being vital for kidney development.
INTRODUCTION
The human kidney has on the order of one million nephrons (Nyengaard and Bendtsen, 1992), made up of an intricate system of specialized tubules that all descend from a pool of nascent progenitor cells. Regulation of the nephron progenitor cell is of the utmost importance to produce the full complement of nephrons. The nephron progenitor cells must self-renew to maintain the progenitor pool, and at the same time differentiate to form the renal vesicle (RV), the first epithelial tubule formation of the nephron. Improper maintenance of the progenitor pool or premature differentiation results in a depletion of nephron progenitors and renal hypoplasia. Six2 and Sall1 have been identified as transcription factors that inhibit differentiation to prevent premature differentiation (Basta et al., 2014; Self et al., 2006). However, our knowledge of pathways that regulate and balance the transition between nephron progenitor cell self-renewal and differentiation is still limited.
Although much progress has been made in determining gene networks that regulate early patterning and initial renal epithelial differentiation, a large gap remains in steps that regulate nephron segmentation (reviewed by Desgrange and Cereghini, 2015). The first evidence of a proximal/distal axis appears shortly after mesenchymal cells differentiate into epithelial cells in the renal vesicle, where WT1 marks the proximal lineage and Lhx1 marks the distal lineage. A distal, intermediate and proximal boundary is apparent in the S-shaped body, where Notch and WT1 mark proximal fates, while Hnf1b and the Iroquois family (Irx1, Irx2, Irx3) mark intermediate fates, and Lgr5 and Pou3f3 (Brn1) mark the intermediate/distal lineage. Deletion of Pou3f3 results in underdeveloped loops of Henle (Rieger et al., 2016), and deletion of Hnf1b in the cap mesenchyme results in the complete loss of proximal tubule, loop of Henle and distal tubule (Massa et al., 2013). Recently, Lgr5 was identified as a marker of progenitor cells for the thick ascending loop of Henle and distal convoluted tubule (Barker et al., 2012). However, we do not know how lineage-restricted progenitor cells of this nephron segment are specified.
Mutations in human SALL1 cause Townes Brocks Syndrome (TBS, OMIM #107408), an autosomal dominant disorder associated with multi-organ defects, including renal hypoplasia, cystic kidneys and renal agenesis (Kohlhase, 2000; Kohlhase et al., 1998). Recent studies have also identified SALL1 mutations in non-syndromic renal hypoplasia, further underscoring the importance of this gene for common birth defects of the kidney (Weber et al., 2006; Hwang et al., 2014). Sall1 encodes a multi-zinc-finger transcription factor that is required for normal kidney development in the mouse. It is highly expressed in multi-potent renal progenitor cells (Osafune et al., 2006) and cap mesenchyme (CM)-derived differentiating structures [pre-tubular aggregates (PTA), renal vesicles (RV), comma and S-shaped bodies] (Takasato et al., 2004). After initial outgrowth of the ureter, Sall1 functions in the nephron progenitor cells to inhibit premature differentiation of the progenitor cells into renal vesicles (Basta et al., 2014).
Sall family members alter gene expression by associating with the nucleosome remodeling and deacetylase (NuRD) complex via the first 12 amino acids of Sall1, termed the Sall repression motif (SRM) (Lauberth et al., 2007). The NuRD complex, consisting of at least eight protein subunits, is one of four major types of ATP-dependent chromatin remodeling complexes (reviewed by Lai and Wade, 2011; Basta and Rauchman, 2015). It is distinguished by the presence of two enzymatic functions: protein deacetylase activity (HDAC1/2) and ATP-dependent chromatin remodeling activity attributed to Mi2-α (CHD3) and Mi2-β (CHD4). Although HDACs are present in many other complexes, Mi2-β and metastases-associated protein (Mta1/2/3) family members are NuRD-defining subunits. NuRD regulates key developmental processes, such as stem cell maintenance and differentiation, cell proliferation and epithelial-to-mesenchymal transition (EMT) (Fujita et al., 2003; Luo et al., 2000; Yoshida et al., 2008; Basta and Rauchman, 2015). Despite its initial characterization as a co-repressor, recent data show that NuRD can either activate or repress target genes, depending on the context (Zhang et al., 2012; Yoshida et al., 2008; Gregory et al., 2010). In lymphocytes, Mi2-β uses distinct mechanisms to mediate opposing effects on growth and differentiation, depending on its association with the sequence-specific DNA-binding protein Ikaros (Zhang et al., 2012).
Because of its crucial developmental functions and its association with Sall1, we postulated that NuRD would also be required in renal progenitor cells. Indeed, our studies show that the NuRD-specific subunit Mi2-β is required in kidney development for proper progenitor cell maintenance (Denner and Rauchman, 2013). Furthermore, Mi2-β and Sall1 exhibit a strong genetic interaction in the kidney (Denner and Rauchman, 2013). We postulated that the interaction between Sall1 and NuRD is required for proper kidney development. To test this hypothesis, we engineered a mouse mutant with a three-amino acid mutation in the N-terminal Sall repression motif of Sall1 that disrupts the NuRD interaction domain (ΔSRM).
RESULTS
Disruption of Sall1-NuRD interaction in vivo causes renal hypoplasia
Our previous studies identified crucial residues in the SRM that mediate Sall1-NuRD association (Lauberth et al., 2007). Based on these findings, we designed a gene targeting strategy to specifically disrupt NuRD interaction with Sall1 by mutating three amino acids in the SRM (Fig. 1A). We performed GST pulldowns to verify that these mutations abrogated the interaction of Sall1 with NuRD components. GST fusion proteins for wild type and the Sall1 protein with the SRM mutation (hereafter referred to as ΔSRM) were expressed in COS-1 cells, which express all NuRD components endogenously, but not Sall1 or other Sall1 family proteins (Sall2-4). Precipitated complexes were analyzed by western blot. Wild-type GST-Sall1 pulled down NuRD complex components Hdac2, Mta2, Mbd3 and RbAp48. In contrast, the GST-ΔSRM Sall1 fusion protein failed to pull down any of the NuRD components (Fig. 1Ba). To exclude the possibility that the mutations affected dimerization or DNA binding of the ΔSRM mutant protein, we performed protein interaction assays and electromobility shift assays (EMSAs). The Sall1 dimerization domain is located in a glutamine rich region in exon II, 220 amino acids downstream of the SRM (Fig. 1A). This domain mediates homo- and hetero-dimerization between Sall proteins (Kiefer et al., 2003; Sweetman et al., 2002). Both Sall1-HA and ΔSRM-HA proteins pulled down Sall1-Flag protein when co-expressed in COS-1 cells (Fig. 1Bb). GST-Sall1 and GST-ΔSRM proteins pulled down Sall4 when co-expressed in COS-1 cells (Fig. 1Bc), indicating that a N-terminal domain distinct from the SRM is not affected by the point mutations in the SRM, and the ΔSRM protein can homo- and hetero-dimerize with Sall1 and Sall4, respectively.
Fig. 1.
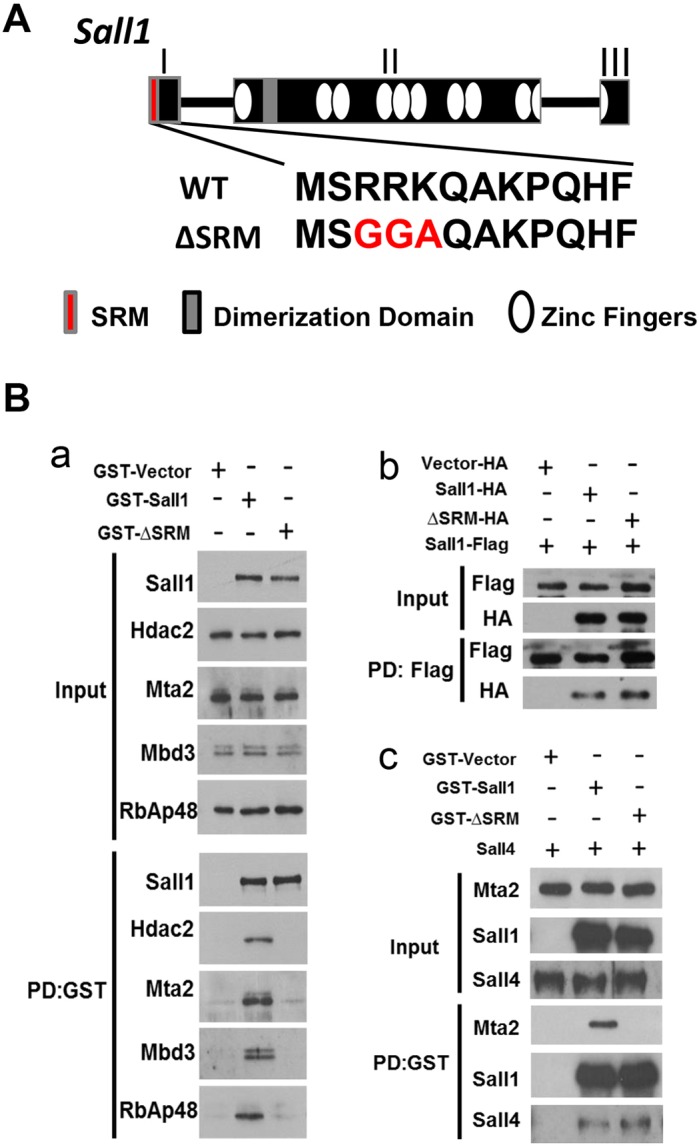
A three amino acid mutation in the SRM of Sall1 disrupts NuRD binding. (A) Schematic of the wild-type Sall1 locus with three exons (I-III). Zinc fingers are represented by white ovals; gray shaded area in exon II represents the glutamine-rich Sall family member interaction domain. The first 12 amino acids of Sall1 that interact with NuRD (Sall repression motif, SRM; shown in red) are encoded in exon I and are listed below. A three amino acid mutation (R3G, R4G, K5A) encodes ΔSRM. (B) (a) GST constructs of full-length wild-type Sall1 or ΔSRM were overexpressed in COS-1 cells, which express components of the NuRD complex endogenously, but do not express Sall1 or family members (Sall2-4). Cell lysates were precipitated with glutathione sepharose and analyzed by western blot (n=3). GST-Sall1 interacts with NuRD components Hdac2, Mta2, Mbd3 and RbAp48. However, a three amino acid mutation (ΔSRM-GST) abolishes the interaction with NuRD components Hdac2, Mta2, Mbd3 and RbAp48. (b) Flu (HA)-tagged Sall1 and ΔSRM were expressed in COS-1 cells with Flag-tagged Sall1. Cell lysates were precipitated with anti-Flag agarose and analyzed by western blot. Sall1-Flag interacts with both wild-type Sall1-HA and ΔSRM-HA (n=2). (c) GST constructs of full-length wild-type Sall1 or ΔSRM were overexpressed with Sall4 in COS-1 cells. Cell lysates were precipitated with glutathione sepharose and analyzed by western blot (n=2). ΔSRM-GST does not interact with the NuRD component Mta2; however, it still interacts with overexpressed Sall4.
Sall1 binds DNA through its C2H2 zinc fingers (Fig. 1A; Kanda et al., 2014; Yamashita et al., 2007; Lauberth et al., 2007), which are located C-terminal to the NuRD-binding domain. To determine whether the SRM missense mutations affect DNA binding, we performed EMSAs using probes corresponding to previously identified Sall1 genomic-binding sites (Kanda et al., 2014). We did not find any differences between Sall and ΔSRM DNA binding (Fig. S1A-E). We conclude that DNA binding is not affected by point mutations in the SRM.
To test whether Sall1-NuRD interaction is required in vivo for kidney development, we derived mutant mice (ΔSRM) in which exon I of Sall1 was altered to encode for R3G, R4G and K5A mutations in the ΔSRM-Sall1 protein (Fig. 2A; Lauberth et al., 2007). Embryos were born at near Mendelian frequency (27.9% wild type, 51.8% Δ/+ and 20.2% Δ/Δ, n=326 embryos). Both heterozygous and homozygous mutant ΔSRM embryos looked morphologically normal (Fig. 2B). However, homozygous ΔSRM mutant mice died within 4 weeks of birth. Their kidneys were hypoplastic, and in some mutants at 21 days of age the kidneys were pale with cysts visible (Fig. 2C). ΔSRM homozygous mutant embryos exhibited bilateral renal hypoplasia that was first apparent at E15 (Fig. 3A). Body weights at E13 and E16 were indistinguishable between wild-type and mutant embryos. At E16, kidney size adjusted for body weight is markedly reduced in the mutants (56.0±5.3 versus 26.1±5.3 mm2/g, P<0.05) (Fig. 3B).
Fig. 2.

A three amino acid mutation in the SRM of Sall1 causes renal hypoplasia. (A) Schematic of the Sall1 locus, the targeting vector and the ΔSRM mutant allele. The targeting vector introduced a three-amino acid mutation into exon I of Sall1, a LoxP site (black arrowhead) and a neomycin cassette (Neo) flanked by Frt sites (white arrowheads) after this region. Deletion of the neomycin cassette by Flp recombinase yields the targeted ΔSRM locus. (B) E11.5 wild-type and homozygous ΔSRM mutant embryos. (C) Weanling (21 day old) wild-type and homozygous ΔSRM mutant mice and their kidneys. The mutant kidney is hypoplastic with cysts visible.
Fig. 3.

ΔSRM mutant kidneys have a smaller nephrogenic zone. (A) Bright-field images of E15 wild-type and homozygous mutant ΔSRM kidneys showing renal hypoplasia evident at E15. Immunofluorescence for Sall1 of E16 wild-type and homozygous mutant ΔSRM kidneys. (B) Body weight (in g) does not differ in wild-type and mutant kidneys at E13 and E16. The kidney size (height×width in mm)/body weight (in g) ratio was calculated (mm2/g) for E16 wild-type (n=20) and homozygous mutant ΔSRM (n=10) kidneys. Mutant E16 kidneys normalized to body weight were significantly smaller than wild-type kidneys (56.0±5.3 versus 26.1±5.3, P<0.05, two-tailed t-test). (C) Histological analysis of E17 wild-type and homozygous ΔSRM mutant kidney; mutant kidneys have a smaller nephrogenic zone (bracket) than wild-type kidneys. Scale bars: 200 µm in C.
To exclude the possibility that our targeting strategy affected expression of ΔSRM, we examined the protein and mRNA levels in developing kidney. Immunofluorescence staining of E16 kidneys revealed that the expression level and localization of Sall1 protein was similar in wild-type and ΔSRM homozygous mutant kidneys (Fig. 3A). Furthermore, expression of wild-type Sall1 and mutant ΔSRM mRNA transcripts were not significantly different at E17 (qRT-PCR 0.90) relative to wild type. Histological analysis of E17 kidneys revealed a smaller nephrogenic zone in the homozygous ΔSRM mutant (Fig. 3C). We conclude that disruption of the interaction between Sall1 and the NuRD complex leads to severe bilateral renal hypoplasia.
Renal hypoplasia in ΔSRM mutants is not due to effects on ureter branching or proliferation of nephron progenitors
To investigate the developmental mechanism of renal hypoplasia in the ΔSRM mutants, we examined whether branching of the ureter or the ability of the nephron progenitor population to expand as the kidney grows was affected. The number of cytokeratin-positive ureteric bud tips was not significantly different in ΔSRM mutants compared with stage-matched wild-type controls at E13, E15, E18 or P2 (Fig. 4A,B). Thus, impaired branching morphogenesis did not account for renal hypoplasia. We tested whether reduced proliferation or increased apoptosis could account for renal hypoplasia. The number of phospho-histone H3/Six2 double-positive cells were not different in wild type and ΔSRM mutants at E13, E15 and E18, indicating no reduction in proliferation in the nephron progenitors (Fig. 4C,D). We analyzed apoptosis by counting the number of total TUNEL-positive cells per high power field. This analysis did not reveal an increase in total apoptosis in ΔSRM mutants at E13, E15 or E18 in the kidney. We also evaluated apoptosis specifically in the Six2-positive progenitor cells. Although we observed no difference in TUNEL-positive cells at E13 and E15 between wild-type and mutant kidney in the Six2-positive cells, there was an increase in TUNEL-positive cells at E18 in the mutant specifically in the Six2-positive nephron progenitor cells (Fig. 4E-G). Together, these results indicate that hypoplasia of ΔSRM mutant kidneys evident by E15 is not due to impaired proliferation or an increase in programmed cell death, suggesting an alternative mechanism to account for this observation. However, apoptosis in nephron progenitor cells at E18 is likely contributing to the severe degree of hypoplasia seen in postnatal kidneys.
Fig. 4.

Renal hypoplasia in ΔSRM mutants is not due to effects on ureter branching or proliferation of nephron progenitors. (A) Quantification of UB tips at different developmental stages in wild-type and mutant kidney. Cytokeratin+ UB tips are not reduced in the mutant at E13, E15, E18 or P2. (B) Representative images of E13 kidney used for counting UB tips, stained for cytokeratin and with DAPI. (C) Quantification of mitotic index calculated by counting pHH3+ Six2+ cells, divided by the total number of Six2+ cells per high-powered field (HPF). Mitotic index of Six2+ progenitor cells is not reduced at E13, E15 or E18 in mutant kidneys. (D) Representative images of E18 kidney used for quantification of mitotic index, stained for pHH3 and Six2, and with DAPI. (E) Quantification of the number of total TUNEL+ cells/HPF at E13, E15 and E18. Total TUNEL+ cells are not significantly different in wild-type and mutant kidney at these stages. (F) Quantification of TUNEL+ Six2+ cells/HPF in E13, E15 and E18 kidney. A significant number of Six2+ progenitor cells are undergoing apoptosis at E18 in the mutant compared with wild-type kidney (*P<0.05). (G) Representative images of E18 kidney used for quantification of TUNEL+ Six2+ cells. Scale bars: 50 µm. For A,C,E,F, n=6 sections from two embryos at each developmental stage and genotype was analyzed. Statistical analysis was carried out using a two-tailed t-test.
Nephron progenitor cell fate is altered in ΔSRM mutants
Our previous studies showed that Sall1 functions to restrain differentiation of nephron progenitor cells (Basta et al., 2014). We thus examined whether ΔSRM mutants exhibited accelerated differentiation by quantifying the number of Six2+ caps, renal vesicles (RVs) and ureteric bud tips (UB), and evaluating the ratio between caps or RVs to UB tips in wild-type and mutant kidneys. At E13 there was a 1.6-fold increase in the ratio of RVs/UB tips in ΔSRM mutant kidneys, 1.4-fold at E15 and a 2.6-fold increase at E18. This was accompanied by a 46% reduction in the ratio of caps/UB tips at E18 in the mutant (Fig. 5A,B), resulting in a decrease in the ratio of caps/UB tips and increase in the ratio of RVs/UB tips. By E18 we observed a noticeable change in the organization of Six2-positive cap mesenchyme in the mutant. In the mutant, the cap cells were poorly organized and aggregated as if induced to differentiate; many RVs were in ectopic locations toward the periphery of the kidney, rather than below (ventral to) the UB tips, as in the wild type. In contrast, in wild-type E18 kidneys the condensed mesenchyme was present at the periphery surrounding the UB tip in an organized cap (Fig. 5A,C). The NCAM+/Rcdh+ renal vesicle-like structures in the mutant developed lumens as expected for RVs, and the majority of these renal vesicles exhibited properly polarized WT1 and Lhx1 expression; however, some RVs toward the periphery of the kidney did not express Lhx1 (Fig. 5D). Unlike wild-type RVs, Six2 protein expression persisted at a relatively high level, comparable with that in cap mesenchyme. This pattern is reminiscent of that seen during cessation of nephrogenesis (Hartman et al., 2007; Chen et al., 2015; Rumballe et al., 2011). During nephron cessation, there is a burst of nephron induction that is accompanied by an extension of Wnt9b expression in UB tips, and an increase in expression of differentiation genes in the peripheral nephrogenic zone from P0 to P4. Concomitantly, progenitor gene expression declines. Multiple progenitor genes were prematurely downregulated in the mutant kidney at E17, as determined by RNA-seq (Cited1, −3.38; Pla2g7, −2.27; Meox2, −1.68; Crym, −1.42; Eya1, −1.27). In situ hydridization revealed that the Wnt9b expression domain was expanded from UB trunks to the tips in ΔSRM mutant kidneys (Fig. S2A). Among differentiation genes, the Wnt9b target Pax8 was significantly upregulated (RNA-seq 1.62) in the ΔSRM mutant, and this was confirmed by in situ hybridization at E13, E15 and E18 (Fig. 5E). Immunofluorescence for Pax8 demonstrated induced mesenchyme aggregated at the periphery in the mutant (Fig. 5F and Movies 1 and 2). However, we did not detect any significant difference in expression of Wnt4 between wild-type and mutant kidneys (Fig. S2B). Together, these results suggest that, in ΔSRM mutant kidneys, nephron progenitor cells are depleted due to accelerated differentiation in a process that resembles premature cessation of nephrogenesis.
Fig. 5.

Disruption of the Sall1-NuRD interaction causes accelerated differentiation of renal progenitor cells. (A) Sections of wild-type and mutant kidney at E13 and E18 stained for Six2 and cytokeratin, and with DAPI. The number of Six2+ caps surrounding UB tips looks similar in the wild type and mutant at E13. However, by E18 the number of Six2+ caps is reduced and Six2+ cells are in structures that resemble renal vesicles. (B) Quantification of the number of Six2+ caps/UB tip in E13, E15 and E18 kidney. The number of Six2+ caps/tip is reduced by E18 in the ΔSRM homozygous mutant (*P<0.05). Quantification of the number of renal vesicles (RVs)/UB tip in wild-type and mutant kidney at E13 and E15 (see C for E18). There are significantly more RVs/tip in the mutant at E13, E15 and E18 (*P<0.05, two-tailed t-test). Quantification performed on 10 non-sequential sections for each stage and genotype; E13, n=3; E15, n=2; E18 wild type, n=2, E18 mutant, n=4. (C) E18 wild-type and mutant kidney sections stained for Six2 and NCAM, and with DAPI. In the wild-type kidney, Six2+ cells are in caps surrounding the ureter. In the mutant, there are RV structures that are Six2+ NCAM+ towards the periphery of the kidney. (D) E18 kidney sections stained for Six2 and Rcdh, and with DAPI (left). Rcdh is found in the luminal side of RVs as well as in further differentiated structures. Examples of RVs are marked by asterisks in the wild-type and mutant kidney, although the Rcdh+ vesicles in the mutant kidney are also Six2+. E18 kidney sections stained for WT1 and Lhx1, and with DAPI (right). Properly polarized RVs are marked by asterisks in the wild-type and mutant kidney. The majority of RVs are properly polarized; however, we observed some toward the periphery of the kidney that do not properly polarize (arrow). (E) In situ hybridization for Pax8 at E13, E15 and E18. Pax8 mRNA expression is increased in developing differentiating structures at three developmental stages. Arrows at E18 indicate RVs in the wild type and mutant, with the RV in the mutant expressing strong Pax8 towards the periphery of the kidney. n=2 for each embryonic stage. (F) Immunofluorescence staining of E18 kidneys for Pax8 and NCAM, and with DAPI. At E18 in the wild type, Pax8 is detected in NCAM/Pax8 double-positive pre-tubular aggregates, developing RVs and comma/S-shaped bodies, with low expression in the UB; it is undetectable in the cap mesenchyme (arrowheads). In the mutant, Pax8 is detectable in the region of the cap mesenchyme ventral to the UB. These Pax8/NCAM double-positive structures are forming aggregates or RV-like structures toward the periphery of the kidney, indicative of induced mesenchyme and epithelial differentiation (four examples show the variation in phenotypes observed, with arrows indicating Pax8-positive aggregates in the first example). n=4 sections from three different embryos for each genotype. Scale bars: 50 µm in A,C,D,F; 25 µm in E.
Sall1-NuRD interaction is required for loop of Henle formation
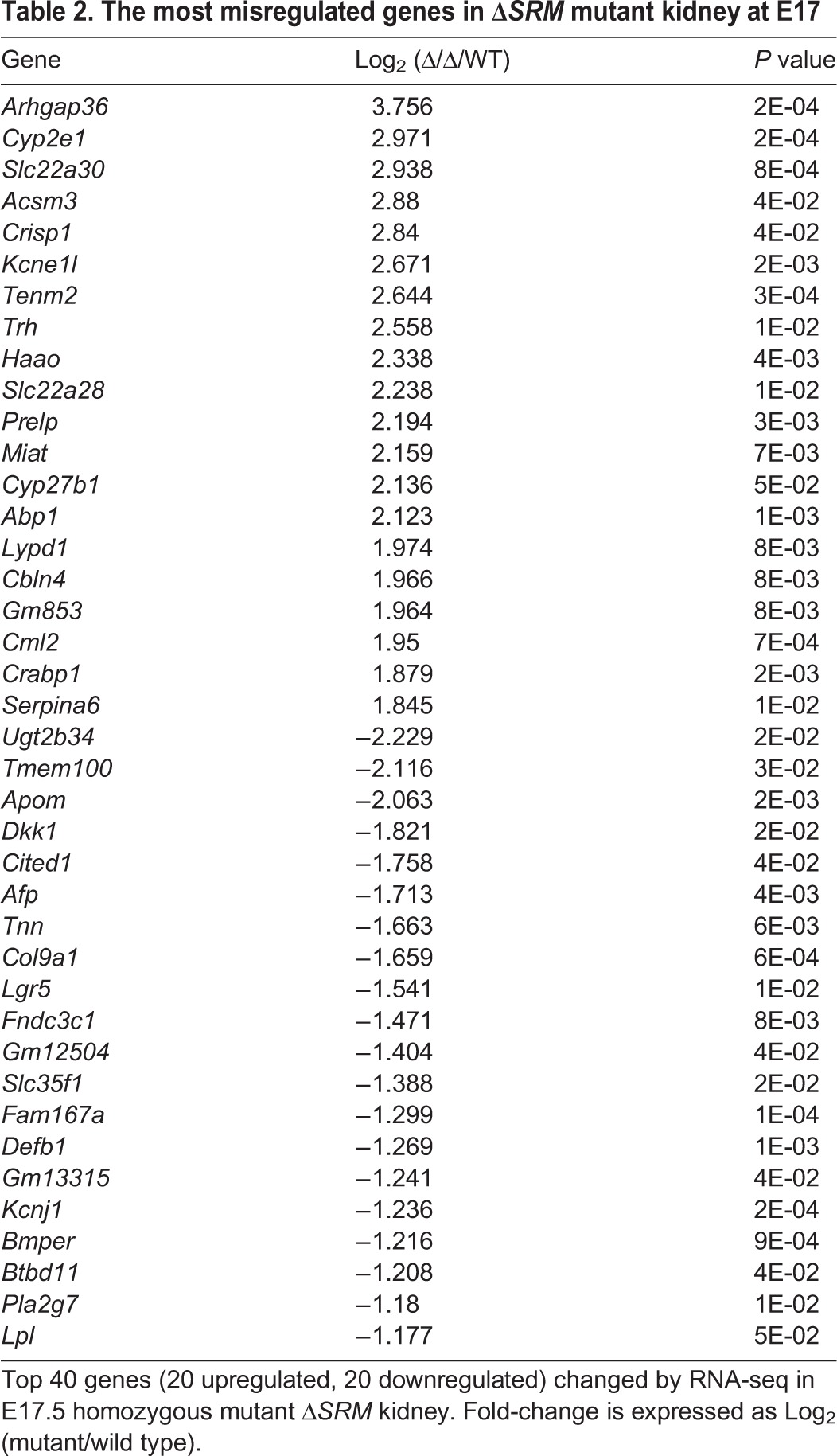
To gain insight into the molecular pathways that are coordinately regulated by Sall1 and NuRD, we performed transcriptional profiling of E17 wild-type and ΔSRM mutant kidneys by RNA-seq. The stage-matched comparison revealed 93 upregulated genes and 32 downregulated genes (≥2-fold). At the 1.5-fold level, there were 379 upregulated genes and 128 downregulated genes that reached statistical significance (P≤0.05). This finding supports our previous studies indicating that a major role of Sall1-NuRD is repression of gene expression (Lauberth and Rauchman, 2006; Basta et al., 2014). Gene ontology analysis using DAVID (https://david.ncifcrf.gov/) of genes changed at least 1.5-fold in the ΔSRM mutant was similar to what we found with the Sall1 mutant (Basta et al., 2014), with significant enrichment for the terms cellular adhesion and cell-cell interaction (Table 1). Analysis of the RNA-seq data revealed that genes expressed in proximal tubules were largely upregulated or unchanged, while those in the loop of Henle, thick ascending limb (TAL) and distal tubule segments were mostly downregulated (Desgrange and Cereghini, 2015) (Fig. 6A). Among the top ten downregulated genes was Lgr5, which was reduced 2.9-fold (Table 2, Fig. 6C). Lgr5 marks lineage-restricted progenitors in comma and S-bodies from E14 through birth that are dedicated to forming the thick ascending limb of the loop of Henle and the distal convoluted tubule (Barker et al., 2012). Interestingly, several other genes that are co-expressed in FACS purified Lgr5+ progenitors, including Jag1, Dkk1, Kcnj1, Slc12a1, Irx2 and Pou3f3 (Barker et al., 2012), were also significantly downregulated in the ΔSRM mutant kidney (Fig. 6A,D). Several of these genes are known to be important for formation of the loop of Henle and distal tubule. We further investigated the expression of genes expressed in the S-body marking the intermediate and distal fate of the nephron and found that as early as E13 Jag1, Lhx1, Lgr5, Pou3f3 and Irx2 all had reduced expression compared with wild-type E13 kidney. By E17, expression of these genes in mutant kidney were reduced even more significantly (Fig. 6D). Thus, analysis of global gene expression by RNA-seq supported a bias towards formation of proximal versus distal segments of the nephron in ΔSRM mutants.
Table 1.
Gene ontology analysis of genes changed in ΔSRM mutant kidney
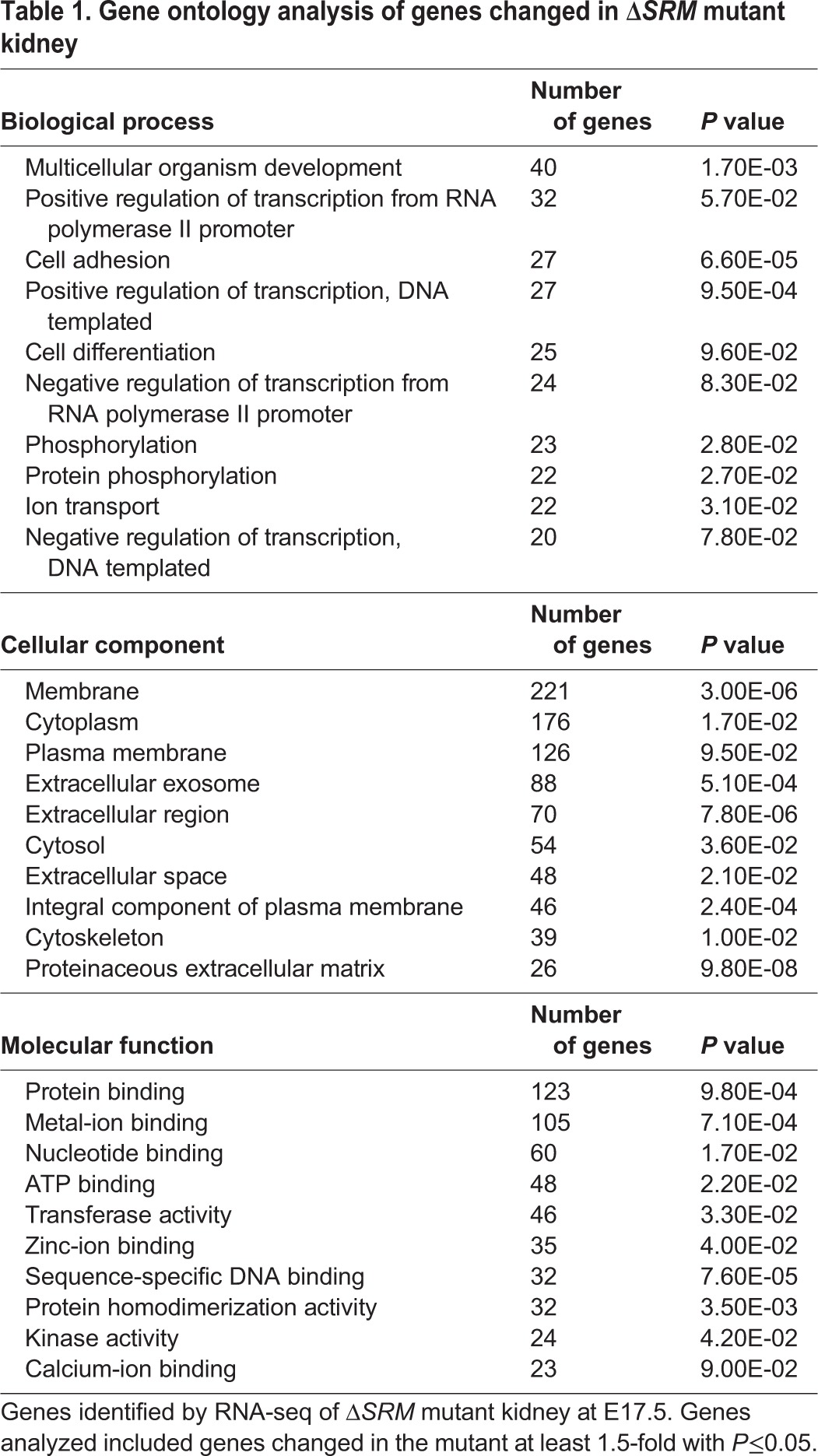
Fig. 6.

Expression of loop of Henle and distal tubule markers are decreased in the ΔSRM mutant at E17. (A) RNA-seq data represented by log2 fold change (mutant/wild type) for genes expressed in terminally differentiated nephron segments. The majority of genes expressed in glomeruli and proximal tubule have no change or are upregulated. However, those genes expressed in Henle's loop (HL), the thick ascending limb of Henle's loop (TAL) and the distal convoluted tubule are all downregulated. (B) The segments of the nephron. Colors correspond to the gene expression for each segment in A. (C) Section in situ hybridization for Lgr5 at E18 reveals reduced mRNA expression in the mutant in the intermediate region of S-shaped bodies (arrows). Scale bar: 25 µm. (D) qRT-PCR for genes expressed in the intermediate and distal regions of the S-body in wild-type and mutant ΔSRM mutant kidney at E13, E15 and E17. At E13, when S-bodies are beginning to form, genes such as Dkk1, Lgr5, Irx2, Tfap2b, Jag1 and Pou3f3 all have reduced expression in the mutant kidney. Data are expressed as fold-change in expression relative to wild-type controls at each time point. RT-PCR was performed in triplicate; E13, n=10 kidneys/cDNA pool; E15, n=5 kidneys/cDNA pool; E17, n=2 kidneys from independent embryos/cDNA pool.
Table 2.
The most misregulated genes in ΔSRM mutant kidney at E17
The role of Sall1 in terminally differentiated nephron structures is not known, and we investigated whether Sall1 was expressed in mature nephron segments. We analyzed Sall1 expression in P0 wild-type kidney and observed strong Sall1 expression in LTL-positive proximal tubule, THP-positive thick ascending limb, NKCC2-positive thick ascending limb and PNA-positive distal tubule. Sall1 was expressed in AQP1-positive thick and thin descending limb, but to a lesser extent, and we observed no expression in cytokeratin-positive ureter or collecting duct (Fig. 7). As genes expressed in the S-body marking the intermediate and distal segments of the nephron were reduced in the ΔSRM mutant, we hypothesized that these segments may not develop normally in the mutant. We stained 80 μm sections from E18 kidneys and obtained optical sections over several cell diameters in both sagittal and transverse planes using confocal microscopy. These studies showed that, while medullary collecting ducts are present in the mutant, there is a near total loss of NKCC2/AQP1-positive loops of Henle in the inner medulla (Fig. 8, Movies 3-6). To confirm this finding, we analyzed P2 wild-type and mutant kidneys for proteins expressed in terminally differentiated nephron structures: glomeruli (WT1), proximal tubules (LTL), thick and thin descending limb (AQP1), thick ascending limb (NKCC2 and THP), distal tubule (PNA) and collecting duct (cytokeratin) (Fig. 9A). Quantification of these structures revealed a statistically significant reduction of all of these structures in the mutant compared with wild-type kidneys. However, we observed a markedly disproportionate reduction of THP- and NKCC2-positive structures in the thick ascending limb of the loop of Henle in the inner medulla (Fig. 9B,C). These data suggest that Sall1-NuRD interaction is required for the proper formation of the loop of Henle. The significant reduction of Lgr5 expression at E17 could be due to reduced formation of these loop of Henle precursors or their loss due to apoptosis. To distinguish these possibilities, we quantified TUNEL-positive cells in comma and S-bodies. We did not detect any differences in the number of TUNEL-positive cells per comma/S-bodies between wild-type and mutant kidneys at E15 (1.25 versus 1.51, P=0.35). However, in situ hybridization revealed that Lgr5 mRNA expression was significantly reduced in S-bodies at E18 (Fig. 6C). Together, these results suggest that there is reduced formation of Lgr5-positive loop of Henle precursors in ΔSRM mutant kidneys.
Fig. 7.

Sall1 expression in terminally differentiated nephron segments. Sections from P0 wild-type kidney stained for terminally differentiated nephron markers: LTL, proximal tubule; THP, thick ascending limb; PNA, distal tubule; AQP1, thick and thin descending limb; NKCC2, thick ascending limb; cytokeratin, ureter and collecting duct; and Sall1. Scale bars: 100 μm.
Fig. 8.

ΔSRM mutant kidneys have significant loss of loops of Henle. (A-D) Sagittal sections (80 µm) of E18 kidney from wild type and mutant were stained for AQP1, NKCC2 and LTL, and with DAPI (A,B) or for AQP1, NKCC2 and cytokeratin, and with DAPI (C,D), and imaged using confocal microscopy. Images are 3D projections from ∼50 µm z stacks. Loops of Henle are stained green (both AQP1 and NKCC2 primary antibodies are rabbit polyclonal antibodies and are detected with the same Alexa 488 antibody); proximal tubules are stained in red (from LTL) (A,B); collecting ducts are stained red (from cytokeratin) (C,D). (A,B) Proximal tubules (red) are present in the cortex and outer medulla, and Loops of Henle (green) descend into the inner medulla. Proximal tubule/descending loop junctions are observed (yellow) in the wild-type kidney. In the mutant kidney, proximal tubules (red) and proximal/descending limb junctions (yellow) are detected, but very few loops of Henle (green) descend into the inner medulla. (C,D) The same pattern of loops of Henle (green) descending into the inner medulla is observed in the wild type, whereas very few loops are seen in the mutant; however, cytokeratin-positive collecting ducts and papilla are present in the mutant inner medulla, indicating proper patterning. The loops present in the mutant in the deep cortex/outer medullary region appear largely cystic and misshapen (C,D).
Fig. 9.

Thick ascending limb segments of the loop of Henle are disproportionately fewer in number in ΔSRM homozygous mutant kidney at P2. (A) Sections from P2 wild-type and homozygous mutant ΔSRM kidney stained for terminally differentiated nephron markers: LTL, proximal tubule; THP, thick ascending limb (TAL); PNA, distal tubule; AQP1, thick and thin descending limb; NKCC2, thick ascending limb; cytokeratin, ureter and collecting duct. (B) Quantification of the number of terminally differentiated nephron structures/high-powered field (HPF) in the inner medulla (IM) and outer medulla/deep cortex region (OM). The gray dashed line in A indicates the separation between IM and OM/deep cortex for quantification. The numbers represented are the average±s.e.m. All nephron structures were statistically significantly fewer in number in P2 mutant (*P<0.05, two-tailed t-test). The % area stained by DAPI (DAPI % area) of the HPF was calculated and did not differ between wild-type and mutant sections analyzed for quantification of nephron structures. (C) Fold-change in the number of nephron structures (mutant/wild type)/HPF. All nephron structures are reduced in number by at least 25%; however, those structures in the loop of Henle marked by AQP1 [inner medullary (IM) thin descending limb], THP (thick ascending limb) and NKCC2 (thick ascending limb) were all reduced in number by at least 80-90%. Scale bars: 100 μm. Quantification performed on 10 non-sequential sections for each genotype (n=2).
Overall, this study demonstrates that Sall1 and NuRD act cooperatively to regulate the fate of two progenitor cell populations in the developing kidney: Sall1-NuRD acts to restrict differentiation of multipotent Six2+ cells and is important in mediating lineage delineation of the Lgr5+ nephron precursor into thick ascending limb of the loop of Henle.
DISCUSSION
In order to form a functional kidney with a full complement of nephrons, a balance between nephron progenitor self-renewal and differentiation must be tightly regulated. Reduction in progenitor cell self-renewal or an increase in differentiation can deplete the progenitor cell population prematurely, resulting in renal hypoplasia, a common cause of childhood kidney failure. Although our knowledge of genes and pathways that control renal organogenesis has increased substantially, our understanding of molecular mechanisms that regulate this crucial nephron progenitor cell fate decision is limited.
Intrinsic properties of nephron progenitor cells that affect gene regulatory networks are crucial in determining whether a cell remains in the stem cell niche to self-renew or exits the niche to differentiate (Chen et al., 2015; Brown et al., 2011, 2015; Park et al., 2012). Tissue-restricted transcription factors must cooperate with large chromatin-modifying complexes to direct rapid changes in gene expression to regulate stem cell fate in developing organs. In the kidney, Six2 and Sall1 are transcription factors expressed in nephron progenitor cells that inhibit or restrain differentiation (Basta et al., 2014; Self et al., 2006). However, the identity of chromatin remodeling complexes that cooperate with these tissue-restricted transcription factors in the kidney is poorly understood. Using a knock-in mouse strategy, our studies reveal that Sall1 cooperates with the nucleosome remodeling and deacetylase (NuRD) complex to regulate the fate of multipotent Six2-positive nephron progenitor cells and thereby significantly impact nephron endowment at birth.
The NuRD chromatin remodeling complex is ubiquitously expressed and has crucial functions in embryonic stem cells and progenitor cells. A key aspect of how NuRD acts in different tissues and contexts is via its interaction with tissue-restricted transcription factors. In addition to Sall family proteins, several other tissue-restricted transcription factors have the conserved first 12 amino acids of the Sall repression motif and disrupting their interaction with NuRD leads to developmental defects in mice and humans (de Ligt et al., 2012; Kiefer et al., 2008; Liu et al., 2014; Verstappen et al., 2008; Wieczorek et al., 2013; Willemsen et al., 2013; Waldron et al., 2016; Mori and Bruneau, 2004; Garnatz et al., 2014; Roche et al., 2008; Wang et al., 2011).
We have previously shown that Sall1 controls the balance between self-renewal and differentiation of nephron progenitor cells (Basta et al., 2014). When Sall1 is knocked out in the mouse, Six2-positive nephron progenitor cells are depleted due to rapid differentiation into renal vesicles. This results in growth arrest and severely hypoplastic kidneys. Conditional deletion of Sall1 in Six2-positive cells produced a similar phenotype, indicating that Sall1 is required cell-autonomously to restrain differentiation of nephron progenitor cells (Kanda et al., 2014; J.M.B. and M.R., unpublished). Our previous work showed that the NuRD-specific component Mi2-β (Chd4) is required to maintain renal progenitor cells in a state of self-renewal (Denner and Rauchman, 2013).
As Sall1 and NuRD physically associate and both are required for maintenance of renal progenitors, we hypothesized that the functional interaction between Sall1 and NuRD would be important for kidney development and the regulation of renal progenitor cells. To test this hypothesis, we made a Sall1 mouse mutant that specifically disrupted the interaction between Sall1 and NuRD. Embryos exhibited renal hypoplasia by E15, which was not accompanied by a reduction in UB branching, a decrease in proliferation of nephron progenitors or an increase of apoptosis in nephron progenitors until late in development. However, a notable finding was an increase in renal vesicles evident as early as E13, leading us to conclude that the interaction between Sall1 and NuRD was important for restraining the progenitor cells from differentiating prematurely into renal vesicles. This phenotype is similar, but less severe than that in Sall1 null homozygous mutants, indicating that Sall1 must also use NuRD-independent mechanisms to regulate the propensity of nephron progenitors to undergo differentiation. Our studies also suggest a related role for Sall1 in determining the timing of the burst of differentiation associated with nephron cessation.
At the level of gene regulation, two models could explain the occurrence of unrestrained differentiation of nephron progenitor cells. One model posits that the Sall1-NuRD interaction is required to activate or maintain expression of genes such as Six2, Fgf9 and Fgf20 (Self et al., 2006; Barak et al., 2012), which promote self-renewal and retention in the stem cell niche. An alternative model is that Sall1 is required to repress differentiation genes to prevent formation of renal vesicles. Sall1 and Six2 physically interact and co-occupy nephron progenitor gene loci to positively regulate their expression (Kanda et al., 2014). However, direct repression of differentiation genes by Sall1 appears to be independent of Six2 (Kanda et al., 2014). In ΔSRM, Six2 and Fgf9/20 expression is not altered, supporting the second model whereby Sall1 and NuRD cooperate to repress the nephron differentiation gene expression program. Consistent with this model, we observed upregulation of Pax8, a Wnt9b target gene induced in renal vesicles. In addition, binding in a genomic region downstream of the Pax8 gene may suggest that it is a direct Sall1 target (Fig. S1E). In contrast, we did not find increased expression of Wnt4, a known inducer of mesenchymal-epithelial transition (MET) and RV formation in the kidney (Stark et al., 1994). Similarly, Wnt4 expression is not ectopically expressed in Sall1-null mutants that also exhibit robust premature differentiation (Basta et al., 2014; Kanda et al., 2014). How can ectopic RV formation occur in the absence of increased Wnt4? Wnt4 is thought to induce MET and RV formation by activating β-catenin-independent, non-canonical signaling pathways (Tanigawa et al., 2011). Our RNA-seq data revealed that multiple genes involved in non-canonical Wnt signaling are upregulated in the ΔSRM mutant kidney (Ror2, Fzd2, Dvl1 and Prickle1), as well as enrichment for Rho GTPase activator activity (P<0.03). We hypothesize that loss of Sall1-NURD interaction leads to increased responsiveness to the Wnt4 ligand, resulting in accelerated differentiation. This model can be tested in future studies using ChIP-seq to determine where Sall1 and NuRD components colocalize at these genes, and regulate their expression and function.
Six2 expression is normally downregulated as cap mesenchyme cells transition to pre-tubular aggregates and renal vesicles. However, we observed Six2 protein expression in renal vesicles in the mutant ΔSRM kidney equal to expression levels observed in the cap mesenchyme. This is reminiscent of the nephron cessation process, in which signals initiate a burst of differentiation, cap mesenchyme markers are downregulated and renal progenitor cells are depleted. During nephron cessation, Six2 expression is downregulated in the cap and is instead expressed in the renal vesicles (Rumballe et al., 2011). Similar to what others have observed with nephron cessation, this transition in Six2 expression was accompanied by a loss of genes expressed in the cap mesenchyme in the ΔSRM mutant (RNA-seq: Cited1, −3.38; Meox2, −1.68; Crym1,−1.42). Although these changes are typically evident at P2 in wild-type mouse kidneys, in ΔSRM mutant kidneys they occurred 3-4 days earlier in utero, severely blunting the developmental window when many nephrons are formed (Short et al., 2014). These findings suggest that disruption of the Sall1 interaction with NuRD results in renal hypoplasia due in part to premature cessation of nephrogenesis. Chen et al. discovered that young and old Cited1-positive progenitors displayed different rates of self-renewal and exit from the nephron stem cell niche. Older cells are more likely to leave the niche and differentiate. Age-dependent differences in progenitor cell behavior could be the result of an internal clock that determines when a cell will stop self-renewing. However, because this ‘clock’ can be reset by transplanting ‘old’ cells into a younger niche, the environment is also crucial to the decision to stay or exit the niche (Chen et al., 2015). Cell-cell contact between young and old cells tends to create an environment that favors retention in the niche. A possible explanation for the ΔSRM phenotype is that Six2-positive nephron progenitors in this mutant are behaving like ‘old’ cells. As development proceeds, ‘old’ cells accumulate more quickly in ΔSRM mutant than in the wild type, altering the niche environment to favor exit over retention. This culminates in early cessation of nephrogenesis. Our transcriptional profiling identified that the most significant pathway altered in ΔSRM mutant kidneys involved cell-adhesion and cell-cell interactions. Differences in global gene expression between old and young Cited1 progenitors also showed enrichment for genes involved in cell adhesion, as well as chromatin modifiers (Chen et al., 2015). This suggests that disruption of cell-cell or cell-matrix interactions in the cap mesenchyme contributes to premature differentiation in the ΔSRM mutant.
In embryonic stem cells, NuRD acts to fine tune the level of expression of the pluripotency network genes, allowing a permissive environment for differentiation, while still enabling cells to self-renew (Reynolds et al., 2012). We speculate that, in the developing kidney, NuRD may have a similar function to enable Six2 progenitors to self-renew, yet still be capable to respond to a Wnt9b inductive signal. Canonical Wnt signaling is required both to promote self-renewal and to induce differentiation of nephron progenitors (Karner et al., 2011). The NuRD-specific component Mi2β has been shown to function as a modulator of β-catenin-dependent gene transcription (Kim et al., 2012; Major et al., 2008). Similarly, prior studies have shown that Sall1 associates with β-catenin and regulates canonical Wnt signaling in the kidney (Karner et al., 2011; Sato et al., 2004). We hypothesize that NuRD, through its association with Sall1 may function to interpret the response to canonical Wnt signaling to balance self-renewal and differentiation of nephron progenitors.
The nephron is a complex epithelial structure with distinct regional identities. Nephron segments comprise distinct cell types that perform unique physiological functions in the mature kidney. How does regional specification of the nephron occur in the developing kidney? Multipotent Six2/Cited1+ progenitors in cap mesenchyme contribute to cells along the entire axis of the nephron, from the proximal to the distal tubule (Boyle et al., 2008; Kobayashi et al., 2014). This indicates that lineage-restricted precursors that define specific epithelial phenotypes are not found in this progenitor cell population. Rather progeny of these multipotent cells must give rise to committed precursors in immature nephrons that in turn generate specialized segments of the mature nephron. The molecular identity of these committed segment-specific cell populations and the mechanism regulating their differentiation is not well understood.
Our studies reveal that formation of the loop of Henle depends on the association of Sall1 and NuRD in the developing kidney. In the ΔSRM mutant, there is a reduction in all nephron segments because accelerated differentiation leads to loss of nephron progenitor cells. However, the almost complete absence of tubules that express Tamm-Horsfall protein (THP, UMOD) and NKCC2, unique markers of the loop of Henle, is disproportionate to the loss of all other nephron segments. The reduction in loop of Henle formation was >80% compared with a ∼30-40% reduction in other regions of the nephron. This indicates that specification of this nephron segment is selectively impaired and its loss is not simply a consequence of the depletion of multipotent Six2-positive nephron progenitor cells. Lgr5, an epithelial stem cell marker, identifies progenitor cells in the comma and S-shaped bodies that gives rise to the thick ascending limb and distal convoluted tubule. Isolation of Lgr5-positive cells identified a subset of genes that are co-expressed in the comma and S-shaped body (Barker et al., 2012). Our transcriptional profiling revealed Lrg5 as one of the most highly downregulated genes in the ΔSRM mutant kidney at E17.5. In addition, Lgr4, Jag1, Dkk1, Pou3f3 and Slc12a1 were all significantly downregulated in the mutant kidney. Early epithelial structures in the developing kidney, beginning at the renal vesicle stage, exhibit polarization of gene expression, prefiguring segmentation of the nephron. Initially, proximal and distal regions can be discerned in the renal vesicle, but in more mature epithelial structures (comma and S-bodies), an intermediate region, which will give rise to the loop of Henle becomes evident. In ΔSRM mutants, we found that genes expressed in the intermediate region that are required for loop of Henle formation, Lgr5, Pou3f3 and Irx1/2/3, were significantly reduced at E13-E15. Sall1 binds genomic regions in the vicinity of Lgr5, Pou3f3 and Tfap2b (Fig. S1B-D), suggesting it may directly regulate the genes that specify loop of Henle precursors. Together, these data suggest that Sall1 and NuRD are required to specify lineage-restricted progenitors of the loop of Henle.
It has been suggested that a gradient of canonical Wnt activity at the S-body stage is crucial for proper segmentation of the nephron (Lindstrom et al., 2015). Lgr5 is both a mediator and a target of Wnt activity. Both Sall1 and NuRD has been shown to modulate Wnt signaling (Karner et al., 2011; Sato et al., 2004; Major et al., 2008). We hypothesize that the two phenotypes observed in ΔSRM mutants, accelerated differentiation of nephron progenitor cells and impaired lineage determination of loop of Henle progenitors, could be attributable to related molecular mechanisms whereby Sall1 and NuRD function cooperatively to interpret Wnt signals at target genes.
MATERIALS AND METHODS
Protein interaction assays
For Sall1 and ΔSRM/NuRD interaction assays, GST-Sall1 fusion proteins were cloned into pEBG and overexpressed in COS-1 cells (ATCC CRL-1650). After 48 h, cells were lysed and precipitated with glutathione sepharose. Lysates were analyzed by western blot using primary and HRP-labeled secondary antibodies (Table S1). For Sall1 and ΔSRM homo-dimerization assays, Sall1 was cloned into Flag-tagged pCDNA3 and HA-tagged pCDNA3, and ΔSRM was cloned into HA-tagged pCDNA3. Constructs were overexpressed in COS-1 cells and cell lysates were precipitated with anti-Flag agarose (Sigma Aldrich) and analyzed by western blot. For Sall1 and ΔSRM hetero-dimerization assays, GST-Sall1 fusion proteins were overexpressed as described above, in addition to Sall4, which was cloned into pCDNA3 and overexpressed in COS-1 cells. Lysates were precipitated with glutathione sepharose and analyzed by western blot.
Generation of ΔSRM homozygous mutant mice
A targeting vector (pSV-FLP-Cre), containing four DNA base pair mutations in exon I of Sall1 encoding for a triple amino acid mutation [R3R4(G)K5(A)] was generated by recombineering using a BAC clone (pBeloBAC11) containing exon I and II of the Sall1 locus. The targeting vector was linearized and electroporated into Scc10 cells. Clones were screened using Southern blot analysis. Positively targeted clones were injected into 129-SvJ blastocysts (Mouse Genetics Core, Washington University, MO, USA). Chimeric mice were bred with ICR mice to obtain germline transmission. Progeny were bred with ROSA26-FLPeR mice (Jackson Laboratory 003946) to excise the neomycin cassette from the Sall1 locus. Wild-type and mutant alleles were detected by PCR genotyping using the following primers: 5′-CTGATGTTTGAGCCAGCATG-3′ and 5′-AAGTGGGAACGAGAGTTTGG-3′. Mutations in ΔSRM mice were verified by sequencing. All experiments were performed with approval of the Saint Louis University IACUC.
Histology and immunohistochemistry
Embryonic kidney size was determined by measuring the height and width of the kidney (height×width in mm), and related this to the overall weight of the embryo (in g). Embryonic kidneys were fixed with 4% PFA overnight and embedded in paraffin wax, sectioned at 4 μm and stained using Harris' Hematoxylin and Eosin Y (St Louis University Research Microscopy and Histology Core). For immunofluorescence, 7 µm frozen sections were washed with ice-cold 100% methanol, boiled in 10 mM citric acid (pH 6) for 20 min and incubated with primary antibodies (Table S1). Reactivity was detected using fluorescently labeled secondary antibodies (Table S1). Sections were counterstained with DAPI (Sigma Aldrich), mounted in Mowiol 4-88 (Poly Sciences) and digital images acquired using a Leica DM5000B epifluorescence microscope and Leica DFC365FX camera. Minimum and maximum values for each channel were set manually to represent structures stained by antibodies rather than for the purpose of relative intensity comparisons. The total brightness was adjusted globally in Photoshop to allow display of signal range in figures.
Quantification of UB branching
Embryonic kidneys were immunostained and the number of cytokeratin-positive UB tips was counted on six non-sequential sections (20× magnification) from two independent embryos for each stage and genotype. Results are reported as the average number of tips per section±s.e.m. Statistical analysis using standard t-tests was performed.
Mitotic index of Six2-positive cells
Mitotic index was determined by staining embryonic kidneys for pHH3 and Six2. Nuclei were stained using DAPI. The ratio of pHH3+Six2+/total Six2+ cells was calculated. At least 2000 nuclei for each stage and genotype were counted on six non-sequential sections (20× magnification) from two independent embryos.
Apoptosis
Apoptosis was determined by performing TUNEL analysis using the ApopTag Red In Situ Apoptosis Detection Kit (Millipore). The average total number of TUNEL+ cells/section (20×magnifications) or the average TUNEL+/Six2+ cells/section were calculated from six non-sequential sections from two different embryos for each stage. Results were reported as the total TUNEL/high power field (HPF) or TUNEL+/Six2+ cells/HPF±s.e.m. Statistical analysis using standard t-tests was performed. The average number of TUNEL-positive cells in NCAM-positive comma/S-bodies was calculated from at least six non-sequential sections from E15 kidneys.
Quantification of Caps/Tip and RVs/Tip
Embryonic day (E) 13, E15 and E18 kidneys were immunostained for Six2, cytokeratin or NCAM, and with DAPI. For each section, the number of cytokeratin+ UB tips, Six2+ caps and NCAM+ RVs were counted. For each stage and genotype, 10 non-sequential sections at 20×magnification were counted. Results were reported as the average ratio of the number of caps or RVs divided by the number of UB tips for each section±s.e.m. Statistical analysis using standard t-tests was performed.
RNA-sequencing
Total RNA was isolated from three E17.5 kidneys for each genotype using an RNeasy Mini Kit (Qiagen) with on the column DNAse I treatment. Polyadenylated mRNA was purified from 4-5 µg total RNA using Dynabeads mRNA Direct (Life Technologies). Construction of barcoded sequencing libraries was performed using the Ion Total RNA-seq v2 kits (Life Technologies) according to the manufacturer's instructions. Sequencing was performed on an Ion Torrent Proton with a mean read lengths of 85-110 nucleotides, and reads were aligned to the mouse mm10 genome using the TMAP aligner map4 algorithm. Soft-clipping at both 5′ and 3′ ends of the reads was permitted during alignment to accommodate spliced reads, with a minimum seed length of 20 nucleotides. Genome-wide strand-specific nucleotide coverages were calculated from the aligned bam files for each sample using the ‘genomecoveragebed’ program in BEDTools (Quinlan and Hall, 2010) and the nucleotide coverage for all non-redundant exons for each gene were summed using custom R scripts (http://www.R-project.org). Normalization factors were calculated by averaging the total exon coverage for all replicates and dividing this average by the total exon coverage for each individual sample. The total coverage for each gene in each replicate was then multiplied by these factors after adding an offset of 1 to each gene to preclude division by 0 in subsequent calculations. The averages and P values of the coverage values for all genes in the individual groups were calculated using Microsoft Excel. The expression values for each gene are the normalized strand-specific total nucleotide coverage for each gene.
Quantitative real-time PCR (qRT-PCR)
Total RNA was isolated from embryonic kidney tissue using an RNeasy Mini Kit with DNAse I treatment on the column (Qiagen). cDNA was prepared using the High Capacity RNA-to-cDNA kit (Life Technologies). Primer sequences are in Table S2. qRT-PCR was performed using a Quant Studio 3 (Applied Biosystems) Thermocycler and SYBR Green PCR Master Mix (Life Technologies) as described previously (Kiefer et al., 2012). Real-time reactions were performed in triplicate and relative expression was calculated using the delta CT method and normalized to Gapdh or Hprt1 control transcripts (Kiefer et al., 2012).
Quantification of terminal nephron segments
Postnatal day 2 (P2) kidneys were immunostained and terminal nephron segments were counted on ten non-sequential sections (10× magnification) from two independent embryos for each genotype. The data represent the average±s.e.m. Statistical analysis using standard t-tests was performed. The percentage of the area stained with DAPI was measured using Image J analysis.
Thick section immunofluorescence and confocal imaging
E18 kidneys were fixed in 4% PFA overnight and transferred to 20% sucrose. Frozen kidneys were mounted to obtain sagittal 80 µm sections, washed in ice-cold 100% methanol and antigen retrieval performed. Sections were blocked for 1 h at room temperature (10% NGS, 0.4% Triton X-100) and incubated in primary antibody in 1% BSA for 2.5 days at room temperature, washed for 8 h at room temperature and incubated with fluorescent secondary antibodies and DAPI in 1% BSA for 1.5 days at 4°C. After washing for 8 h at room temperature, sections were mounted in Mowiol for confocal imaging using a Leica SP8 TCS confocal microscope using the DAPI diode laser (405 nm) and white light laser. Lasers were set to 405 nm at 20% power, 488 nm at 7% power, 550 nm at 10% power and 594 nm at 4% power. Detectors were set to collect fluorescence over the following ranges: 416-481 nm, 501-543 nm, 558-669 nm and 626-722 nm. Hybrid detectors were set at ∼80% gain and PMT for DAPI was set at 700 V. The pinhole was set to 1 airy unit. Images represent the average of three line scans. Whole kidneys were imaged with a HCX PL APO CS 10×/0.40 dry objective using the Mosaic Stitch Feature of LasX using the statistical method and default parameters selected. Maximum intensity projections of the axial (z) slices was calculated and used for each image. Whole-kidney images represent at least 40 µm in the axial (z) dimension. 3D images (both still shots and movies) were created using the 3D module of LasX. Additional higher resolution scans were performed using a HC PL APO CS2 20×/0.75 oil, HC PL APO CS2 40×/1.30 oil or HC PL APO CS2 63×/1.40 oil lens. Minimum and maximum values for each channel were set manually to represent structures stained by antibodies rather than for the purpose of relative intensity comparisons. The total brightness and contrast was reset globally in ImageJ to allow display of signal range in figures. It was set equally for paired (mutant versus wild type) specimens.
Whole-mount and section in situ hybridization
Whole-mount in situ hybridization was performed using digoxigenin-labeled antisense riboprobes for Pax8 (nucleotides from ATG, 6-704), Wnt4 (67-1013), Wnt9b (486-1076) and Lgr5 (2498-3206) (Kiefer et al., 2008). Section in situ hybridization was performed as described previously (Little et al., 2007) on 25 μm frozen sections using digoxigenin-labeled riboprobes. After incubation with digoxigenin-alkaline phosphatase antibody (1:2500), signal was visualized using the alkaline phosphatase substrate BM purple (Roche).
Electromobility shift assay
Gel shift assays were performed according to the LightShift Chemiluminescent EMSA kit protocol (Thermo Scientific). COS-1 cells were transfected with 1 µg GST-vector, GST-Sall1 or GST-ΔSRM, and after 48 h nuclear extracts were prepared using NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific). DNA probes (Table S3) were synthesized (Invitrogen), end labeled with biotin and annealed by heating to 95°C for 5 min then cooling to room temperature. Nuclear extract (5 µg) was added to 1× binding buffer, 2.5% glycerol, 5 mM MgCl2, 50 ng/µl Poly (dI•dC), 0.5% NP-40 and 20fmol biotin-labeled probe; the reaction was incubated for 20 min at room temperature. Competition reactions were performed with the same binding conditions with 4pmol unlabeled probe, and supershifts were performed by adding 1 µg Sall1 polyclonal antibody (Abcam) for 20 min after the initial binding reaction. Binding reactions were run on a 5% native polyacrylamide gel in 0.5× TBE, transferred to positively charged nylon and crosslinked. Labeled DNA was detected following the Nucleic Acid Detection Module (Thermo Scientific).
Sall1 ChIP data analysis
The fastq files were downloaded from the DNA Data Bank of Japan (Kanda et al., 2014) (DDBJ) trace archives (http://trace.ddbj.nig.ac.jp/DRASearch/submission?acc=DRA000957) and aligned to the Mus musculus GRCm38 genome (University of California Santa Cruz mm10, without mitochondrial sequences) using Torrent Mapping Alignment Program (TMAP) (https://github.com/iontorrent/TMAP/blob/master/doc/tmap-book.pdf). The TMAP parameters used were: -g 0 –o 2 stage1 map4 –min-seed-length 20. After the sequences were aligned, the aligned bam files were sorted and indexed, and alignment statistics were generated using samtools (samtools sort; samtools index; samtools idxstats) (Li et al., 2009). A custom Perl script was used to determine the percentage of reads aligned to the genome and fold genome coverage. sgr files containing the chromosome, position and score were generated using the bedtool, genomecoveragebed, with the –d parameter, to report 1-based coordinates (Quinlan and Hall, 2010). Owing to the size of the murine genome, the sgr files were generated one chromosome at a time, rather than for the entire genome. The input for Sall1, RR006515, was used to normalize the Sall1 data, RR006513 and the IgG control, RR006514, and to calculate enrichment via a custom R script (http://www.R-project.org) (Dorsett and Misulovin, 2017) to adjust for differences in chromatin isolation, amplification and sequencing. The enriched sgr files for Sall1 and the IgG control were loaded into the Integrated Genome Browser and the IgG control was subtracted from Sall1 (Nicol et al., 2009). Binding peaks were determined using the threshold function, set at fourfold and a 100 bp minimum run.
Acknowledgements
The authors thank Ziva Misulovin for RNA-seq library preparation; Dr Dale Dorsett and Kathie Mihindukulasuriya of the Saint Louis University Genomics Core for bioinformatics analysis; the Saint Louis University Research Microscopy and Histology Core for confocal imaging expertise; and Lisa Stout for technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.M.B., D.R.D., M.R.; Methodology: J.M.B., L.R., D.R.D., G.R.K.; Software: G.R.K.; Validation: J.M.B., L.R.; Formal analysis: J.M.B., L.R., D.R.D., G.R.K.; Investigation: J.M.B., M.R.; Resources: M.R.; Data curation: J.M.B., G.R.K.; Writing - original draft: J.M.B., M.R.; Writing - review & editing: J.M.B., L.R., M.R.; Supervision: M.R.; Project administration: M.R.; Funding acquisition: M.R.
Funding
This work was supported by the March of Dimes Foundation (6-FY13-127) and the and National Institute of Diabetes and Digestive and Kidney Diseases (DK098563). Deposited in PMC for release after 12 months.
Data availability
RNA-seq data are available in Gene Expression Omnibus under accession number GSE102583.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.148692.supplemental
References
- Barak H., Huh S.-H., Chen S., Jeanpierre C., Martinovic J., Parisot M., Bole-Feysot C., Nitschké P., Salomon R., Antignac C. et al. (2012). FGF9 and FGF20 maintain the stemness of nephron progenitors in mice and man. Dev. Cell 22, 1191-1207. 10.1016/j.devcel.2012.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N., Rookmaaker M. B., Kujala P., Ng A., Leushacke M., Snippert H., Van De Wetering M., Tan S., Van Es J. H., Huch M. et al. (2012). Lgr5(+ve) stem/progenitor cells contribute to nephron formation during kidney development. Cell Rep. 2, 540-552. 10.1016/j.celrep.2012.08.018 [DOI] [PubMed] [Google Scholar]
- Basta J. and Rauchman M. (2015). The nucleosome remodeling and deacetylase complex in development and disease. Transl. Res. 165, 36-47. 10.1016/j.trsl.2014.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basta J. M., Robbins L., Kiefer S. M., Dorsett D. and Rauchman M. (2014). Sall1 balances self-renewal and differentiation of renal progenitor cells. Development 141, 1047-1058. 10.1242/dev.095851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle S., Misfeldt A., Chandler K. J., Deal K. K., Southard-Smith E. M., Mortlock D. P., Baldwin H. S. and De Caestecker M. (2008). Fate mapping using Cited1-CreERT2 mice demonstrates that the cap mesenchyme contains self-renewing progenitor cells and gives rise exclusively to nephronic epithelia. Dev. Biol. 313, 234-245. 10.1016/j.ydbio.2007.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C., Blank U., Adams D. C., Karolak M. J., Fetting J. L., Hill B. L. and Oxburgh L. (2011). Isolation and culture of cells from the nephrogenic zone of the embryonic mouse kidney. J. Vis. Exp. 20, 2555. 10.3791/2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C., Muthukrishnan S. D. and Oxburgh L. (2015). A synthetic niche for nephron progenitor cells. Dev. Cell 34, 229-241. 10.1016/j.devcel.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Brunskill E. W., Potter S. S., Dexheimer P. J., Salomonis N., Aronow B. J., Hong C. I., Zhang T. and Kopan R. (2015). Intrinsic age-dependent changes and cell-cell contacts regulate nephron progenitor lifespan. Dev. Cell 35, 49-62. 10.1016/j.devcel.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ligt J., Willemsen M. H., Van Bon B. W., Kleefstra T., Yntema H. G., Kroes T., Vulto-Van Silfhout A. T., Koolen D. A., De Vries P., Gilissen C. et al. (2012). Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921-1929. 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- Denner D. R. and Rauchman M. (2013). Mi-2/NuRD is required in renal progenitor cells during embryonic kidney development. Dev. Biol. 375, 105-116. 10.1016/j.ydbio.2012.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrange A. and Cereghini S. (2015). Nephron patterning: lessons from Xenopus, Zebrafish, and mouse studies. Cells 4, 483-499. 10.3390/cells4030483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett D. and Misulovin Z. (2017). Measuring sister chromatid cohesion protein genome occupancy in Drosophila melanogaster by ChIP-seq. Methods Mol. Biol. 1515, 125-139. 10.1007/978-1-4939-6545-8_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita N., Jaye D. L., Kajita M., Geigerman C., Moreno C. S. and Wade P. A. (2003). MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell 113, 207-219. 10.1016/S0092-8674(03)00234-4 [DOI] [PubMed] [Google Scholar]
- Garnatz A. S., Gao Z., Broman M., Martens S., Earley J. U. and Svensson E. C. (2014). FOG-2 mediated recruitment of the NuRD complex regulates cardiomyocyte proliferation during heart development. Dev. Biol. 395, 50-61. 10.1016/j.ydbio.2014.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory G. D., Miccio A., Bersenev A., Wang Y., Hong W., Zhang Z., Poncz M., Tong W. and Blobel G. A. (2010). FOG1 requires NuRD to promote hematopoiesis and maintain lineage fidelity within the megakaryocytic-erythroid compartment. Blood 115, 2156-2166. 10.1182/blood-2009-10-251280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman H. A., Lai H. L. and Patterson L. T. (2007). Cessation of renal morphogenesis in mice. Dev. Biol. 310, 379-387. 10.1016/j.ydbio.2007.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D.-Y., Dworschak G. C., Kohl S., Saisawat P., Vivante A., Hilger A. C., Reutter H. M., Soliman N. A., Bogdanovic R., Kehinde E. O. et al. (2014). Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 85, 1429-1433. 10.1038/ki.2013.508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda S., Tanigawa S., Ohmori T., Taguchi A., Kudo K., Suzuki Y., Sato Y., Hino S., Sander M., Perantoni A. O. et al. (2014). Sall1 maintains nephron progenitors and nascent nephrons by acting as both an activator and a repressor. J. Am. Soc. Nephrol. 25, 2584-2595. 10.1681/ASN.2013080896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner C. M., Das A., Ma Z., Self M., Chen C., Lum L., Oliver G. and Carroll T. J. (2011). Canonical Wnt9b signaling balances progenitor cell expansion and differentiation during kidney development. Development 138, 1247-1257. 10.1242/dev.057646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer S. M., Ohlemiller K. K., Yang J., Mcdill B. W., Kohlhase J. and Rauchman M. (2003). Expression of a truncated Sall1 transcriptional repressor is responsible for Townes-Brocks syndrome birth defects. Hum. Mol. Genet. 12, 2221-2227. 10.1093/hmg/ddg233 [DOI] [PubMed] [Google Scholar]
- Kiefer S. M., Robbins L., Barina A., Zhang Z. and Rauchman M. (2008). SALL1 truncated protein expression in Townes-Brocks syndrome leads to ectopic expression of downstream genes. Hum. Mutat. 29, 1133-1140. 10.1002/humu.20759 [DOI] [PubMed] [Google Scholar]
- Kiefer S. M., Robbins L. and Rauchman M. (2012). Conditional expression of Wnt9b in Six2-positive cells disrupts stomach and kidney function. PLoS ONE 7, e43098 10.1371/journal.pone.0043098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. J., Khalid O., Vo S., Sun H.-H., Wong D. T. W. and Kim Y. (2012). A novel regulatory factor recruits the nucleosome remodeling complex to wingless integrated (Wnt) signaling gene promoters in mouse embryonic stem cells. J. Biol. Chem. 287, 41103-41117. 10.1074/jbc.M112.416545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A., Mugford J. W., Krautzberger A. M., Naiman N., Liao J. and Mcmahon A. P. (2014). Identification of a multipotent self-renewing stromal progenitor population during mammalian kidney organogenesis. Stem. Cell Rep. 3, 650-662. 10.1016/j.stemcr.2014.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase J. (2000). SALL1 mutations in Townes-Brocks syndrome and related disorders. Hum. Mutat. 16, 460-466. [DOI] [PubMed] [Google Scholar]
- Kohlhase J., Wischermann A., Reichenbach H., Froster U. and Engel W. (1998). Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat. Genet. 18, 81-83. 10.1038/ng0198-81 [DOI] [PubMed] [Google Scholar]
- Lai A. Y. and Wade P. A. (2011). Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer 11, 588-596. 10.1038/nrc3091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauberth S. M. and Rauchman M. (2006). A conserved 12-amino acid motif in Sall1 recruits the nucleosome remodeling and deacetylase corepressor complex. J. Biol. Chem. 281, 23922-23931. 10.1074/jbc.M513461200 [DOI] [PubMed] [Google Scholar]
- Lauberth S. M., Bilyeu A. C., Firulli B. A., Kroll K. L. and Rauchman M. (2007). A phosphomimetic mutation in the Sall1 repression motif disrupts recruitment of the nucleosome remodeling and deacetylase complex and repression of Gbx2. J. Biol. Chem. 282, 34858-34868. 10.1074/jbc.M703702200 [DOI] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome P. R. O. J. et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078-2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom N. O., Lawrence M. L., Burn S. F., Johansson J. A., Bakker E. R., Ridgway R. A., Chang C. H., Karolak M. J., Oxburgh L., Headon D. J. et al. (2015). Integrated beta-catenin, BMP, PTEN, and Notch signalling patterns the nephron. Elife 3, e04000 10.7554/eLife.04000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little M. H., Brennan J., Georgas K., Davies J. A., Davidson D. R., Baldock R. A., Beverdam A., Bertram J. F., Capel B., Chiu H. S. et al. (2007). A high-resolution anatomical ontology of the developing murine genitourinary tract. Gene Expr. Patterns 7, 680-699. 10.1016/j.modgep.2007.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z. L. F., Ruan K., Zhang J., Mej Y., Wu J. and Shi Y. (2014). Structural and functional insights into the human borjeson-forssman-lehmann syndrome associated protein PHF6. J. Biol. Chem. 289, 10069-10083. 10.1074/jbc.M113.535351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Su F., Chen D., Shiloh A. and Gu W. (2000). Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377-381. 10.1038/35042612 [DOI] [PubMed] [Google Scholar]
- Major M. B., Roberts B. S., Berndt J. D., Marine S., Anastas J., Chung N., Ferrer M., Yi X., Stoick-Cooper C. L., Von Haller P. D. et al. (2008). New regulators of Wnt/beta-catenin signaling revealed by integrative molecular screening. Sci. Signal. 1, ra12 10.1126/scisignal.2000037 [DOI] [PubMed] [Google Scholar]
- Massa F., Garbay S., Bouvier R., Sugitani Y., Noda T., Gubler M.-C., Heidet L., Pontoglio M. and Fischer E. (2013). Hepatocyte nuclear factor 1beta controls nephron tubular development. Development 140, 886-896. 10.1242/dev.086546 [DOI] [PubMed] [Google Scholar]
- Mori A. D. and Bruneau B. G. (2004). TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealed. Curr. Opin. Cardiol. 19, 211-215. 10.1097/00001573-200405000-00004 [DOI] [PubMed] [Google Scholar]
- Nicol J. W., Helt G. A., Blanchard S. G. Jr, Raja A. and Loraine A. E. (2009). The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25, 2730-2731. 10.1093/bioinformatics/btp472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyengaard J. R. and Bendtsen T. F. (1992). Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat. Rec. 232, 194-201. 10.1002/ar.1092320205 [DOI] [PubMed] [Google Scholar]
- Osafune K., Takasato M., Kispert A., Asashima M. and Nishinakamura R. (2006). Identification of multipotent progenitors in the embryonic mouse kidney by a novel colony-forming assay. Development 133, 151-161. 10.1242/dev.02174 [DOI] [PubMed] [Google Scholar]
- Park J.-S., Ma W., O'brien L. L., Chung E., Guo J.-J., Cheng J.-G., Valerius M. T., Mcmahon J. A., Wong W. H. and Mcmahon A. P. (2012). Six2 and Wnt regulate self-renewal and commitment of nephron progenitors through shared gene regulatory networks. Dev. Cell 23, 637-651. 10.1016/j.devcel.2012.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R. and Hall I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841-842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds N., Latos P., Hynes-Allen A., Loos R., Leaford D., O'shaughnessy A., Mosaku O., Signolet J., Brennecke P., Kalkan T. et al. (2012). NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell 10, 583-594. 10.1016/j.stem.2012.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger A., Kemter E., Kumar S., Popper B., Aigner B., Wolf E., Wanke R. and Blutke A. (2016). Missense mutation of POU domain class 3 transcription factor 3 in Pou3f3L423P mice causes reduced nephron number and impaired development of the thick ascending limb of the loop of Henle. PLoS ONE 11, e0158977 10.1371/journal.pone.0158977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche A. E., Bassett B. J., Samant S. A., Hong W., Blobel G. A. and Svensson E. C. (2008). The zinc finger and C-terminal domains of MTA proteins are required for FOG-2-mediated transcriptional repression via the NuRD complex. J. Mol. Cell Cardiol. 44, 352-360. 10.1016/j.yjmcc.2007.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumballe B. A., Georgas K. M., Combes A. N., Ju A. L., Gilbert T. and Little M. H. (2011). Nephron formation adopts a novel spatial topology at cessation of nephrogenesis. Dev. Biol. 360, 110-122. 10.1016/j.ydbio.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A., Kishida S., Tanaka T., Kikuchi A., Kodama T., Asashima M. and Nishinakamura R. (2004). Sall1, a causative gene for Townes-Brocks syndrome, enhances the canonical Wnt signaling by localizing to heterochromatin. Biochem. Biophys. Res. Commun. 319, 103-113. 10.1016/j.bbrc.2004.04.156 [DOI] [PubMed] [Google Scholar]
- Self M., Lagutin O. V., Bowling B., Hendrix J., Cai Y., Dressler G. R. and Oliver G. (2006). Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. EMBO J. 25, 5214-5228. 10.1038/sj.emboj.7601381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Short K. M., Combes A. N., Lefevre J., Ju A. L., Georgas K. M., Lamberton T., Cairncross O., Rumballe B. A., Mcmahon A. P., Hamilton N. A. et al. (2014). Global quantification of tissue dynamics in the developing mouse kidney. Dev. Cell 29, 188-202. 10.1016/j.devcel.2014.02.017 [DOI] [PubMed] [Google Scholar]
- Stark K., Vainio S., Vassileva G. and Mcmahon A. P. (1994). Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nature 372, 679-683. 10.1038/372679a0 [DOI] [PubMed] [Google Scholar]
- Sweetman D., Smith T., Farrell E. R., Chantry A. and Munsterberg A. (2002). The conserved glutamine rich region of chick csal1 and csal3 mediates protein interactions with other spalt family members - implications for Townes-Brocks syndrome. J. Biol. Chem. 278, 6560-6566. 10.1074/jbc.M209066200 [DOI] [PubMed] [Google Scholar]
- Takasato M., Osafune K., Matsumoto Y., Kataoka Y., Yoshida N., Meguro H., Aburatani H., Asashima M. and Nishinakamura R. (2004). Identification of kidney mesenchymal genes by a combination of microarray analysis and Sall1-GFP knockin mice. Mech. Dev. 121, 547-557. 10.1016/j.mod.2004.04.007 [DOI] [PubMed] [Google Scholar]
- Tanigawa S., Wang H., Yang Y., Sharma N., Tarasova N., Ajima R., Yamaguchi T. P., Rodriguez L. G. and Perantoni A. O. (2011). Wnt4 induces nephronic tubules in metanephric mesenchyme by a non-canonical mechanism. Dev. Biol. 352, 58-69. 10.1016/j.ydbio.2011.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstappen G., Van Grunsven L. A., Michiels C., Van De Putte T., Souopgui J., Van Damme J., Bellefroid E., Vandekerckhove J. and Huylebroeck D. (2008). Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum. Mol. Genet 17, 1175-1183. 10.1093/hmg/ddn007 [DOI] [PubMed] [Google Scholar]
- Waldron L., Steimle J. D., Greco T. M., Gomez N. C., Dorr K. M., Kweon J., Temple B., Yang X. H., Wilczewski C. M., Davis I. J. et al. (2016). The cardiac TBX5 interactome reveals a chromatin remodeling network essential for cardiac septation. Dev. Cell 36, 262-275. 10.1016/j.devcel.2016.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Meng R., Hayes V., Fuentes R., Yu X., Abrams C. S., Heijnen H. F. G., Blobel G. A., Marks M. S. and Poncz M. (2011). Pleiotropic platelet defects in mice with disrupted FOG1-NuRD interaction. Blood 118, 6183-6191. 10.1182/blood-2011-06-363580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S., Moriniere V., Knuppel T., Charbit M., Dusek J., Ghiggeri G. M., Jankauskiene A., Mir S., Montini G., Peco-Antic A. et al. (2006). Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J. Am. Soc. Nephrol. 17, 2864-2870. 10.1681/ASN.2006030277 [DOI] [PubMed] [Google Scholar]
- Wieczorek D., Bögershausen N., Beleggia F., Steiner-Haldenstätt S., Pohl E., Li Y., Milz E., Martin M., Thiele H., Altmüller J. et al. (2013). A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet 22, 5121-5135. 10.1093/hmg/ddt366 [DOI] [PubMed] [Google Scholar]
- Willemsen M. H., Nijhof B., Fenckova M., Nillesen W. M., Bongers E. M. H. F., Castells-Nobau A., Asztalos L., Viragh E., Van Bon B. W. M., Tezel E. et al. (2013). GATAD2B loss-of-function mutations cause a recognisable syndrome with intellectual disability and are associated with learning deficits and synaptic undergrowth in Drosophila. J. Med. Genet 50, 507-514. 10.1136/jmedgenet-2012-101490 [DOI] [PubMed] [Google Scholar]
- Yamashita K., Sato A., Asashima M., Wang P.-C. and Nishinakamura R. (2007). Mouse homolog of SALL1, a causative gene for Townes-Brocks syndrome, binds to A/T-rich sequences in pericentric heterochromatin via its C-terminal zinc finger domains. Genes Cells 12, 171-182. 10.1111/j.1365-2443.2007.01042.x [DOI] [PubMed] [Google Scholar]
- Yoshida T., Hazan I., Zhang J., Ng S. Y., Naito T., Snippert H. J., Heller E. J., Qi X., Lawton L. N., Williams C. J. et al. (2008). The role of the chromatin remodeler Mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev. 22, 1174-1189. 10.1101/gad.1642808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Jackson A. F., Naito T., Dose M., Seavitt J., Liu F., Heller E. J., Kashiwagi M., Yoshida T., Gounari F. et al. (2012). Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat. Immunol. 13, 86-94. 10.1038/ni.2150 [DOI] [PMC free article] [PubMed] [Google Scholar]