

ABSTRACT
Phosphatidylinositol 3-phosphate (PtdIns3P) is a key player of membrane trafficking regulation, mostly synthesized by the PIK3C3 lipid kinase. The presence of PtdIns3P on endosomes has been demonstrated; however, the role and dynamics of the pool of PtdIns3P dedicated to macroautophagy/autophagy remains elusive. Here we addressed this question by studying the mobilization of PtdIns3P in time and space during autophagosome biogenesis. We compared different dyes known to specifically detect PtdIns3P by fluorescence microscopy analysis, based on PtdIns3P-binding FYVE and PX domains, and show that these transfected dyes induce defects in endosomal dynamics as well as artificial and sustained autophagosome formation. In contrast, indirect use of recombinant FYVE enabled us to track and discriminate endosomal and autophagosomal pools of PtdIns3P. We used this method to analyze localization and dynamics of PtdIns3P subdomains on the endoplasmic reticulum, at sites of pre-autophagosome associated protein recruitment such as the PtdIns3P-binding ZFYVE1/DFCP1 and WIPI2 autophagy regulators. This approach thus revealed the presence of a specific pool of PtdIns3P at the site where autophagosome assembly is initiated.
KEYWORDS: autophagosome, endosome, FYVE domain, phosphatidylinositol-3-phosphate, PX domain
Introduction
Lipids of the phosphoinositide family play a major role in signal transduction and membrane dynamics.1-3 Phosphatidylinositol 3-phosphate (PtdIns3P) is critical to early endosome function,4,5 and a role of PtdIns3P in autophagy has been documented recently.6,7 In both the endocytic and autophagic pathways PtdIns3P recruits proteins bearing FYVE, PX (PhoX homology), or PROPPIN (β-propellers that bind polyphosphoinositides) domains.7-9 An example of PtdIns3P function at endosomes is illustrated by the FYVE-domain-containing EEA1 (early endosome antigen 1) protein, which is essential for early endosome homotypic fusion.10 PtdIns3P is also required for intralumenal vesicle biogenesis in early endosomes;11,12 for this function, PtdIns3P recruits ESCRT complex proteins such as HGS/Hrs and TSG101.13 PtdIns3P also recruits the retromer complex to endosomal membranes to enable retrograde transport from endosomes to the trans-Golgi network.14
In the autophagic pathway PtdIns3P is necessary in early stages of autophagosome formation as well. The FYVE-domain-containing protein ZFYVE1/DFCP1 (zinc finger FYVE-type containing 1), which is located at the endoplasmic reticulum (ER), binds to PtdIns3P giving rise to a peculiar ER structure (or subdomain) called the omegasome15 that serves as a matrix for autophagosome biogenesis. The phagophore, a pre-autophagosomal double-membrane structure, emanates from the omegasome and, once elongated, seals and forms the mature autophagosome.16,17 The elongation step of the phagophore is dependent on PtdIns3P via its interaction with WIPI2, a PROPPIN-domain-containing protein. WIPI2 contributes to the positioning of the autophagy-related (ATG) ATG12–ATG5-ATG16L1 conjugation system, which acts upstream of the MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) conjugation system.18,19 In addition, PtdIns3P is involved in controlling late steps of autophagy such as the fusion with the lysosomal compartment and the autophagosome-lysosome reformation process.20-22
PtdIns3P synthesis is mostly ensured by the class III phosphatidylinositol 3-kinase (PtdIns3K). PIK3C3/VPS34 and its adaptor PIK3R4/VPS15/p150 form the core of a complex that is active in the endocytic pathway or at various stages of the autophagic pathway23,24 depending on its associated proteins. In the early stage of autophagy, the ATG proteins BECN1/Beclin1 and ATG14/ATG14L are mobilized with PIK3C3 and PIK3R4 to form the PIK3C3 complex I. Other proteins such as AMBRA1, VMP1, and NRBF2 are able to control the activity of the complex.25-28 In the late stage of autophagy or in the endocytic pathway, UVRAG replaces ATG14 to form the PIK3C3 complex II. The activity of complex II is inhibited by the protein RUBCN/Rubicon.29 However, BECN1 is not absolutely required for the enzymatic function of PIK3C3 in the endo-lysosomal pathway.30,31 Recent crystallographic studies have illuminated how the selectivity of the PIK3C3 complexes depend on membrane curvature.32,33 These studies have contributed to our current understanding of the role of PIK3C3 complexes in different steps of intracellular trafficking.
In the autophagy field little information is available on the dynamics of PtdIns3P lipid itself. Most studies have focused on the proteins that produce PtdIns3P or those that interact with PtdIns3P (such as ZFYVE1 and WIPI proteins). Using a ZFYVE1-FYVE domain, Ktistakis and colleagues demonstrated that PtdIns3P is a marker of the omegasome.15 Interestingly, using PX-mediated detection of PtdIns3P coupled to an electron microscopy freeze fracture technique, it has been shown that the topology of PtdIns3P is different between yeast and mammalian autophagosomes:34 PtdIns3P is mainly associated with the cytoplasmic outer leaflet of the autophagosome membrane in mammalian cells, whereas it is more abundant in the lumenal leaflet of the autophagosome in yeast.
Previous membrane-trafficking studies have focused primarily on the enzymes and regulators that produce PtdIns3P or proteins known to bind PtdIns3P rather than on PtdIns3P itself. An efficient way to detect PtdIns3P by microscopy is to transfect cells with fluorescently labeled PtdIns3P-binding domains. The most commonly used PtdIns3P-binding domain is the FYVE domain, which is a conserved peptide sequence of approximately 65 amino acids that forms a hydrophobic loop. This domain is frequently used in tandem (2xFYVE) or double tandem (4xFYVE) constructs. Another successfully used PtdIns3P-binding domain is the PX domain (which stands for PhoX homology).35,36 This domain is approximately 120 amino acids long and is found in several kinases, PtdIns3P kinases, and trafficking regulators such as soluble NSF attachment protein receptor proteins (SNAREs) and sorting nexins (SNXs).
A risk with the PtdIns3P-binding domain approach is that the introduction of fluorescent PtdIns3P domains into living cells will interfere with the recognition of endogenous proteins containing these domains and/or with the dynamic distribution of PtdIns3P or its turnover.5 As PtdIns3P is produced on both endosomal and pre-autophagosomal membranes, one must also be particularly careful when monitoring PtdIns3P by microscopy to distinguish one pool from the other. Despite these caveats, GFP-tagged FYVE domain and PX domain as well as domains tagged with glutathione S-transferase (GST, which is in turn recognized by a labeled anti-GST antibody) or with biotin have been successfully used in fluorescence and electron microscopy studies of endosomal compartments37-39 and mature autophagosomes40 to reveal the presence and partitioning of PtdIns3P on specialized trafficking organelles. The aims of the present study were, first, to compare the use of GFP-2XFYVE, GFP-P40PX (containing the PX domain of NCF4) and indirect GST-FYVE staining to monitor PtdIns3P by fluorescence microscopy in the autophagic and endocytic pathways and, second, to investigate the behavior of the PtdIns3P pool during the early stages of autophagosome formation.
Results
Detection of PtdIns3P membranes by fluorescence microscopy
Many studies have successfully used PtdIns3P binding domains labeled with GFP (or RFP) to detect intracellular PtdIns3P by fluorescence in fixed conditions or by video microscopy. Here we first assessed the ability of the GST-FYVE purified peptide to recognize endogenous PtdIns3P in fixed cells. GST-FYVE was used as the “primary antibody” and was labeled with a fluorescent anti-GST antibody (see methods). We compared GST-FYVE data with that obtained using transfected GFP-2XFYVE and GFP-P40PX. As expected, we showed that GFP-2xFYVE, GFP-P40PX, and GST-FYVE all labeled endogenous vesicles and membrane structures that were partially positive for EEA1, a well-characterized early endosome marker, or LC3B, a marker of autophagosomes (Fig. 1). The use of a mutant unable to bind PtdIns3P (GST-mut2x-FYVE, corresponding to the C215S mutation) confirmed the specificity of PtdIns3P fluorescent labeling by the GST-2xFYVE peptide (Fig. 1).
Figure 1.
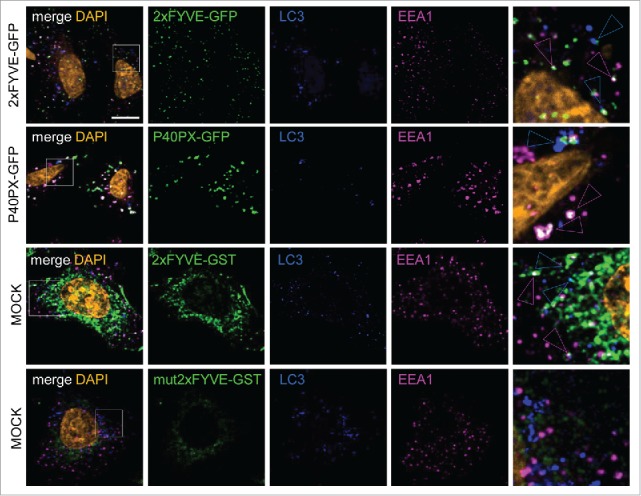
FYVE and PX PtdIns3P-binding domains detect both endosomal and autophagosomal pools of PtdIns3P. HeLa cells were transiently transfected with GFP-2xFYVE or GFP-P40PX expression vectors or were mock transfected. Mock-transfected cells were incubated after fixation with primary antibodies or GST-2xFYVE or GST-mut2xFYVE and then with appropriate secondary antibodies. Representative confocal microscopy images of tagged PtdIns3P-binding domain (green), LC3 (blue), EEA1 (magenta), and DAPI (orange) fluorescence. Blue and magenta arrowheads indicate codistribution/colocalization of tagged PtdIns3P-binding domains with LC3 or EEA1, respectively. Scale bar: 10 μm.
Presence of tagged PtdIns3P-binding domains affects PtdIns3P membrane stability in living cells
Comparison of the PtdIns3P staining obtained using GST-FYVE and GFP-2XFYVE or GFP-P40PX revealed differences in distribution, number, and size of PtdIns3P-positive vesicles. These differences may be due to the requirement for transfection of GFP-2XFYVE and GFP-P40PX. Indeed, the PtdIns3P binding domains are in contact with PtdIns3P membranes while the cells are alive when the GFP-tagged domains are used. In contrast, the GST-FYVE was used in a post-fixation step, effectively serving as a primary antibody, and thus should not interfere with PtdIns3P membrane dynamics and/or turnover.
We next compared the staining of PtdIns3P with that of endosomal vesicles in cells transfected or not with GFP-2xFYVE or GFP-P40PX, and we noticed a striking difference in the size of early endosomes (Fig. 2A and B). There was a massive increase in the volume of EEA1 structures in cells positive for GFP-2xFYVE or GFP-P40PX compared with nontransfected cells (Fig. 2C). This enlargement of endosomes could reflect an endosomal membrane instability due to permanent binding of FYVE or PX domains to PtdIns3P on endosomal platforms. Interestingly, this increase in early endosome volume was previously observed in cells lacking PIK3C3; as this kinase is the main contributor of endosomal PtdIns3P, these cells were in fact mostly devoid of PtdIns3P.41
Figure 2.
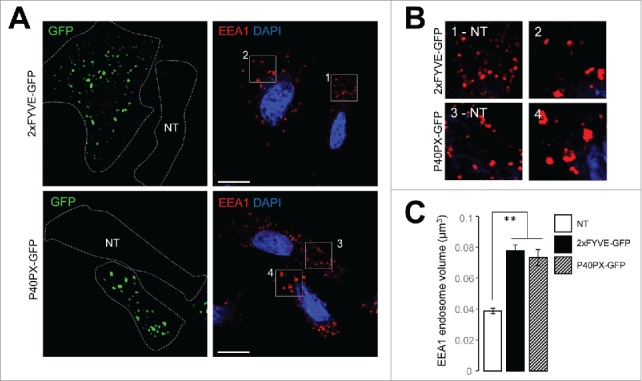
The presence of PtdIns3P-binding domains alters endosomal membrane dynamics. HeLa cells were transiently transfected with plasmids encoding GFP-2xFYVE or GFP-P40PX. (A) Representative confocal microscopy analysis of GFP-2xFYVE and GFP-P40PX (green), EEA1 (red), and DAPI (blue). Dashed lines indicate cell boundaries. NT indicates a cell that was not transfected. (B) Higher magnification of boxes from transfected and not transfected (NT) cells in panel (A) showing EEA1 fluorescence (red). (C) Quantification of endosome volume, calculated from EEA1 fluorescence (n = 25 cells). Means ± SEM **P < 0.01, unpaired 2-tailed t test. Scale bar: 10 μm.
We thus investigated whether the same kind of artificial perturbation was observed for the autophagic pathway, which depends directly on PIK3C3. In cells transfected with either GFP-2XFYVE or GFP-P40PX, we observed that the number of LC3B puncta, used as a marker of autophagosomal organelles, was increased in cells grown under fed or short-time starvation situations (Fig. 3A and B). This suggests that the level of autophagy is higher in cells expressing GFP-2XFYVE or GFP-P40PX constructs than in untransfected cells. This hypothesis was confirmed by biochemical analysis of LC3 lipidation and of the levels of SQSTM1/p62 (sequestosome 1), an autophagy substrate protein, in transfected cells treated or not with bafilomycin A1 (BAF.A1; an inhibitor of lysosomal degradation); the LC3-II level (the lipidated form of LC3, bound to autophagosomal membrane) was higher in GFP-2XFYVE- or GFP-P40PX-transfected cells compared with mock-treated cells, whereas the level of SQSTM1, classically used to monitor autophagy flux, was lower (Fig. 3D and E). These conclusions were confirmed by accumulation of LC3-II in the presence of BAF.A1 (Fig. 3D and E). Altogether these results suggest that the presence of GFP-2XFYVE or GFP-P40PX induce an artificial and sustained wave of autophagy. This autophagy was not due to modifications of components and/or regulators of PIK3C3, because levels of PIK3C3 (responsible for PtdIns3P synthesis), RAB5 (a regulator of PIK3C3 activity on endosomes), and ATG14 (a regulator of the role of PIK3C3 in autophagosome biogenesis) were not affected by the presence of GFP-2XFYVE or GFP-P40PX (Fig. 3D).
Figure 3.
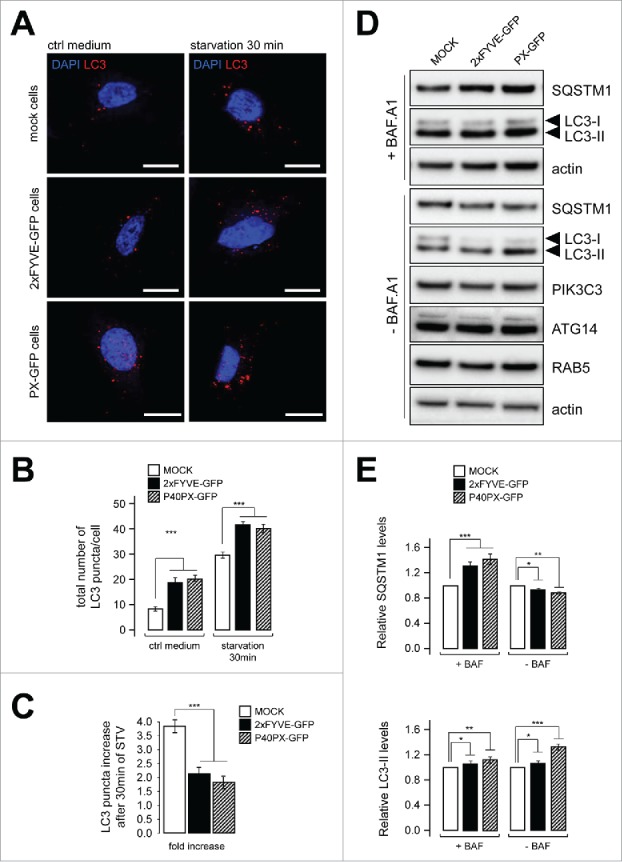
Transfection of tagged PtdIns3P-binding domains promotes artificial and sustained autophagy. HeLa cells were transiently transfected with GFP-2xFYVE, GFP-P40PX, or mock vectors. (A) Confocal microscopy images of LC3 (red) and DAPI (blue) fluorescence. Scale bar: 10 μm. (B) Quantification of LC3 puncta per cell in normal medium (ctrl medium) and in starvation (EBSS, 30 min) (n = 20 cells). (C) Fold increase of LC3 puncta response in starved cells compared with control. (D) Western blot analysis of the autophagy flux markers SQSTM1 and LC3, of PIK3C3 and ATG14, and of the endosome marker RAB5 in cells transfected with plasmids encoding GFP-2xFYVE, GFP-P40PX, or mock vectors, with or without bafilomycin A1 (BAF.A1, an inhibitor of autophagosome-lysosome fusion) treatment. Actin was used as a loading control. (E) Quantification of western blots shown in (D). Relative amounts of SQSTM1 and LC3-II are shown compared with mock-transfected cells. Means ± SEM **P < 0.01, ***P < 0.001 unpaired 2-tailed t test.
Finally, although our observations indicate that basal autophagy was boosted by the presence of PtdIns3P-binding domains, we found that the response of the autophagic machinery was less efficient after brief starvation in the presence of GFP-2XFYVE or GFP-P40PX than its absence: the number of LC3 puncta was higher in mock-transfected cells than in cells overexpressing GFP-2XFYVE or GFP-P40PX (Fig. 3C), suggesting that even if basal autophagy was increased by the presence of PtdIns3P-binding domain proteins (Fig. 3A, B, and D), the starvation-induced stress response was weaker (or slower). The effects on endosome stability and the impaired autophagy response strongly suggest that the use of FYVE or P40PX reporters in living cells induces artificial stabilization of intracellular PtdIns3P platforms, thus reducing membrane dynamics at endosomes and autophagosomes.
Indirect probe GST-FYVE detects endosomal and autophagosomal pools of PtdIns3P
We assessed the use of GST-FYVE by analyzing the global behavior of PtdIns3P-positive structures distinguished as endosomal by EEA1 staining or autophagosomal by LC3 staining in cells treated with drugs known to inhibit PIK3C3 activity, 3-methyladenine (3MA) and wortmannin, or transfected with siRNAs designed to deplete cells of PIK3C3, BECN1, or ATG14 (Fig. 4A). As expected, 3MA and wortmannin treatments, as well as the inhibition of PIK3C3 expression, led to a drastic decrease in total PtdIns3P signal, whereas depletion of BECN1 or ATG14 had a small—but significant—effect (Fig. 4B and C). By analyzing the PtdIns3P-EEA1 and PtdIns3P-LC3 codistribution, we showed that inhibition of BECN1 or ATG14 expression only affected the autophagy-related PtdIns3P pool, whereas the endosomal pool of PtdIns3P was not affected by the absence of either of the autophagy-related regulators (Fig. 4D and E). These results show that while BECN1 and ATG14 are required for the PtdIns3P synthesis by PIK3C3 that promotes autophagosome biogenesis, neither BECN1 nor ATG14 is required for early endosomal PtdIns3P pool synthesis by PIK3C3.
Figure 4.
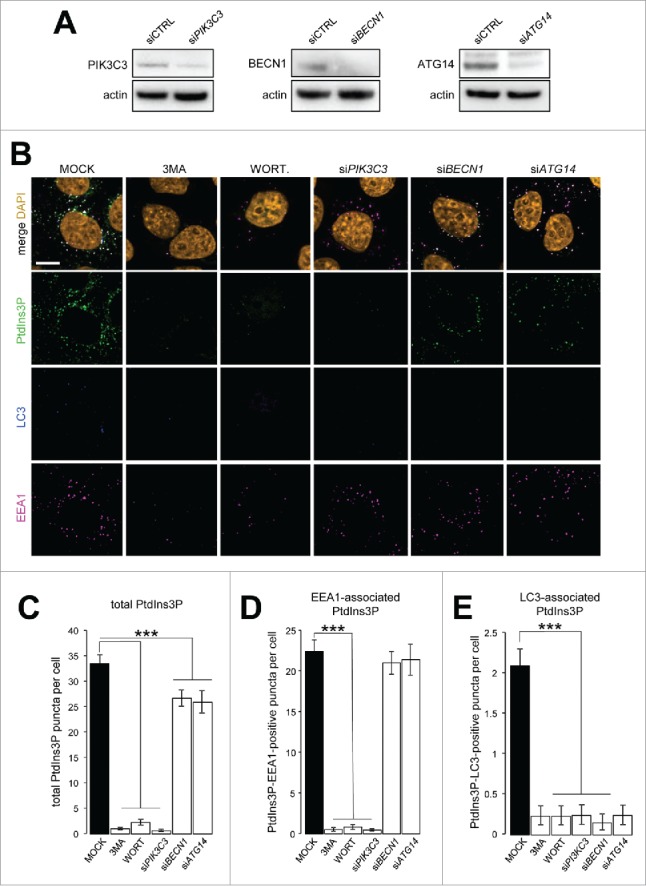
Discrimination of endosomal and autophagosomal pools of PtdIns3P. HeLa cells were transiently transfected with siRNAs designed to inhibit expression of PIK3C3, BECN1, or ATG14 or with nontargeting siRNA (siCTRL) as a negative control or were treated with 10 mM 3MA or 100 nM wortmannin (WORT). (A) Western blot analysis of PIK3C3, BECN1, and ATG14 after siRNA transfection. Actin was used as a loading control. (B) Confocal microscopy analysis of PtdIns3P (detected by GST-2xFYVE fluorescence, artificially shown in green), LC3 (blue), EEA1 (magenta), and DAPI (orange). (C-E) Quantification of (C) total PtdIns3P, (D) EEA1-associated PtdIns3P (PtdIns3P and EEA1 positive), and (E) LC3-associated PtdIns3P (PtdIns3P and LC3 positive) puncta per cell (n = 30 cells). Means ± SEM ***P < 0.001, unpaired 2-tailed t test.
Detection of PtdIns3P at the ER membrane during autophagosome biogenesis
The ER membrane appears to be a major site for autophagosome biogenesis and/or early autophagy associated signaling.42,43 We detected the 2 main PtdIns3P synthesis regulators, namely BECN1 and ATG14, at the ER membrane soon after autophagy initiation by nutritional deprivation (Fig. 5A). Results described above indicated that the use of GST-FYVE in fixed cells enabled detection of endogenous PtdIns3P without affecting its turnover or stability and that it could be used to track autophagy-associated PtdIns3P. Using GST-FYVE, we demonstrated that PtdIns3P colocalized with ATG14 and LC3 (Fig. 5B).
Figure 5.
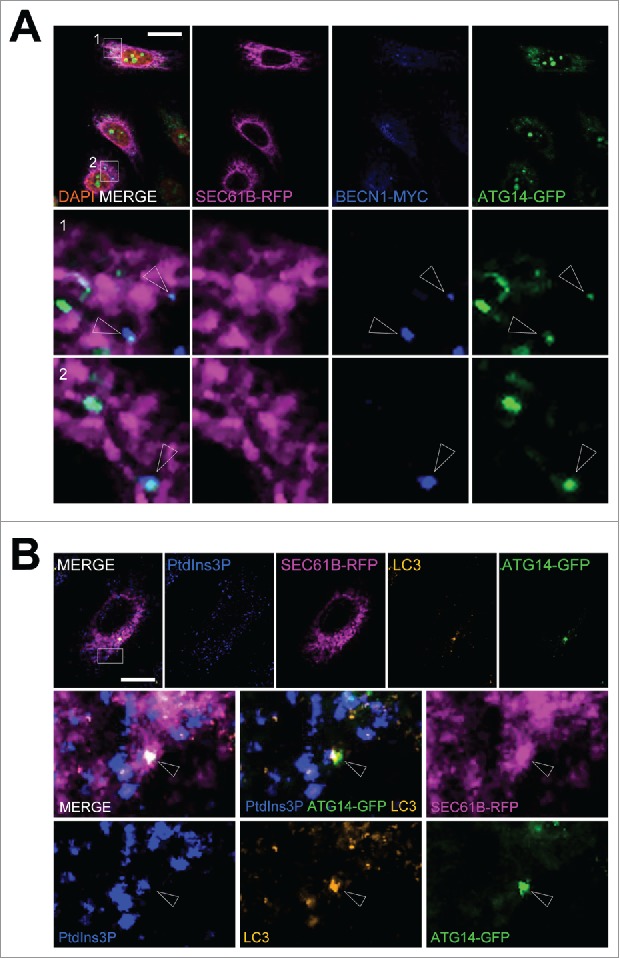
PtdIns3P is observed at ER sites positive for PIK3C3 regulators during early autophagy response to stress. HeLa cells were transiently transfected with a plasmid encoding RFP-SEC61B, MYC-BECN1, and GFP-ATG14 or with RFP-SEC61B and GFP-ATG14, and starved in EBSS medium for 15 min to induce autophagy. (A) Confocal microscopy analysis of RFP-SEC61B (magenta), MYC-BECN1 (blue), and GFP-ATG14 (green). Arrowheads indicate colocalization between BECN1 and ATG14 at ER platforms. (B) Confocal microscopy analysis of PtdIns3P (detected by GST-2xFYVE fluorescence, artificially shown in blue), RFP-SEC61B (magenta), LC3 (orange), and GFP-ATG14 (green). Arrowheads indicate colocalization of PtdIns3P, ATG14, and LC3 at ER platforms. Scale bars: 10 μm.
We were also able to detect PtdIns3P on or in the immediate vicinity of the ER membrane (Fig. 6A). Some of these ER-PtdIns3P structures, but not all, were positive for the autophagosomal marker LC3 (Fig. 6A) after 5 min of starvation. We then used GST-FYVE to monitor the dynamics of the autophagy-associated pool of PtdIns3P during the very early steps of autophagy induction. We analyzed and quantified PtdIns3P-associated fluorescence after autophagy induction by starvation: We showed that the PtdIns3P signal strongly, and immediately, increased once cells were subjected to nutritional stress (Fig. 6B and 6C); the PtdIns3P signal returned to a basal level after 60 min of starvation. Interestingly, PtdIns3P rapidly colocalized with the ER membrane marker after induction of stress, and with slower kinetics colocalized with LC3 (Fig. 6D). These results illustrate the dynamic mobilization of PtdIns3P to the ER membranes soon after autophagy induction and the presence of PtdIns3P on forming (or formed) autophagosomes labeled by LC3.
Figure 6.
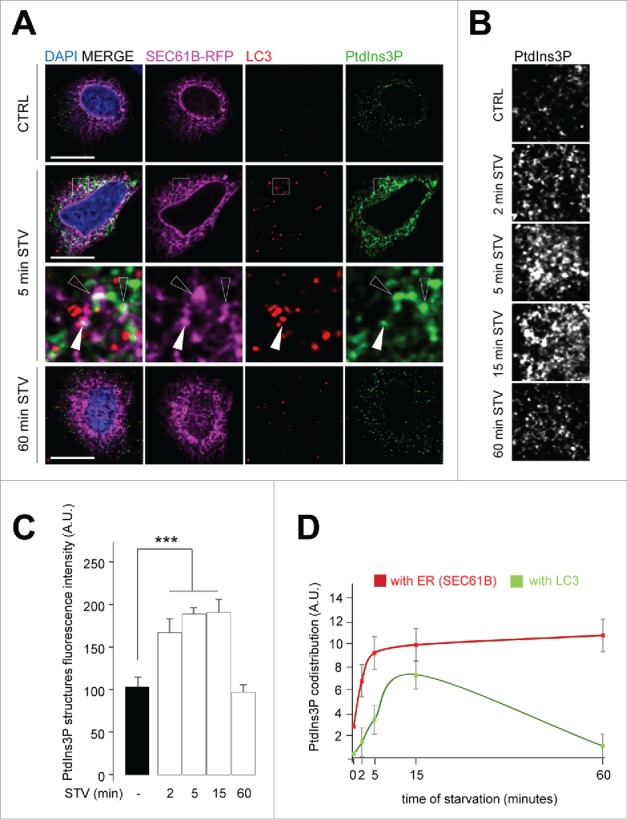
Kinetics of autophagy-related PtdIns3P pool localization at the ER membrane and autophagosome biogenesis. HeLa cells were transiently transfected with a plasmid encoding RFP-SEC61B and grown in normal medium (ctrl) or starved in EBSS medium for the indicated periods (2, 5, 15, 60 min STV). (A) Confocal microscopy analysis of RFP-SEC61B (magenta), LC3 (red), PtdIns3P (detected by GST-2x-FYVE fluorescence, artificially shown in green), and DAPI (blue). Empty arrowheads indicate SEC61B and PtdIns3P codistribution, white arrowheads mark SEC61B, PtdIns3P, and LC3 codistribution. (B) Confocal microscopy analysis of PtdIns3P (detected by GST-2xFYVE fluorescence, artificially shown in white), as a function of time in starvation medium (2–60 min STV). (C) Quantification of PtdIns3P-positive structure fluorescence in starved conditions (2–60 min STV) normalized to number of structures in normal medium (−) (n = 30 cells). A.U., arbitrary units. (D) Quantification of PtdIns3P- and SEC61B-positive puncta (indicative of PtdIns3P ER codistribution) and of PtdIns3P- and LC3-positive puncta (indicative of PtdIns3P localization to autophagic structures) as a function of starvation time. Means ± SEM ***P < 0.001, unpaired 2-tailed t test. Scale bars: 10 μm.
Next we used GST-FYVE to analyze the local distribution of PtdIns3P at the ER membrane after induction of autophagy during the time period in which autophagosome biogenesis from ER/omegasome domains occurs. ZFYVE1 is an ER marker of pre-autophagosome assembly.15 Using GST-FYVE we detected PtdIns3P at sites that were also stained using ZFYVE1-GFP (Fig. 7A). Interestingly, we were able to detect structures containing ZFYVE1 that colocalized with or were in the immediate vicinity of PtdIns3P on the ER with (Fig. 7A, insert 2) and without LC3 (Fig. 7A, insert 1). The sites without LC3 are most likely corresponding to a step of omegasome-to-phagophore transition in which ZFYVE1, but not LC3, has been recruited to a PtdIns3P-containing domain of the ER. Similar results were obtained using the WIPI2 protein as a marker of autophagosome biogenesis initiation: WIPI2, which binds PtdIns3P through a PROPPIN domain, was found to be specifically recruited to PtdIns3P-positive zones of the ER. We detected structures positive for PtdIns3P, WIPI2, and LC3 (Fig. 7B, insert 2), as well as structures in which LC3 was absent (Fig. 7B, insert 1), a situation that was predicted by the function of WIPI2 itself, which is to direct the ATG12–ATG5-ATG16L1 triad to the omegasome domain to promote final LC3 lipidation and recruitment to the future autophagosomal membrane.17,19
Figure 7.
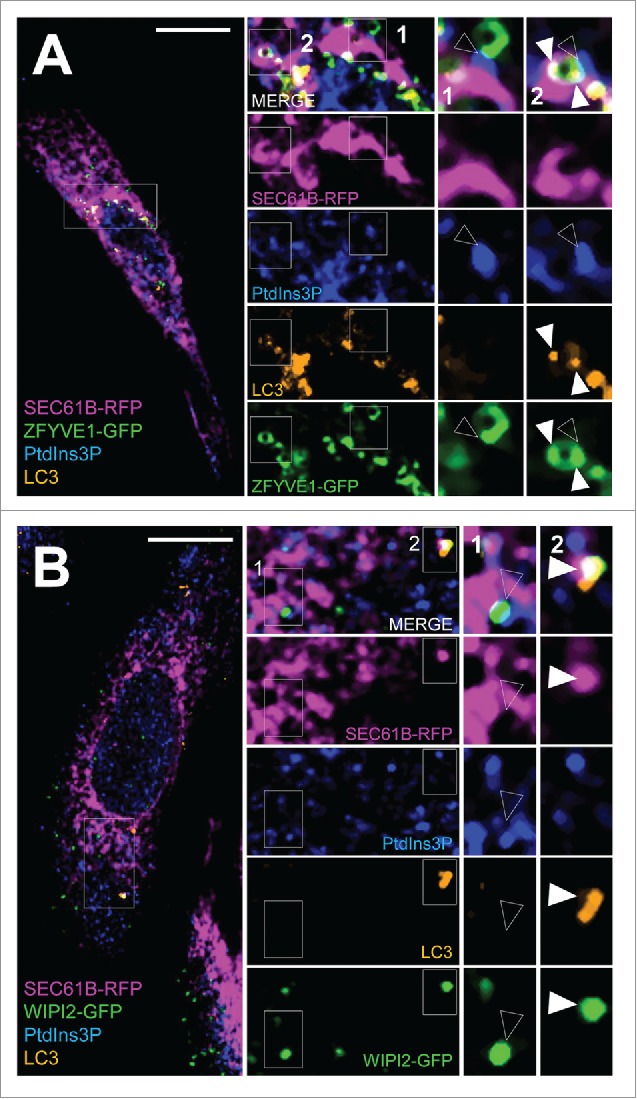
In situ tracking of pre-autophagosome structures at the ER membrane. HeLa cells were transiently transfected with RFP-SEC61B and with GFP-ZFYVE1 or GFP-WIPI2 and were starved in EBSS medium for 15 min. (A) Confocal microscopy analysis of RFP-SEC61B (magenta), LC3 (orange), PtdIns3P (detected by GST-2xFYVE fluorescence, artificially shown in blue) and GFP-ZFYVE1 or (B) GFP-WIPI2 (green). Empty arrowheads indicate SEC61B, PtdIns3P, and ZFYVE1/DFCP1 (or WIPI2) codistribution (early-autophagic PtdIns3P pool). White arrowheads mark SEC61B, PtdIns3P, ZFYVE1 (or WIPI2) and LC3 codistribution (total autophagic PtdIns3P pool). Scale bars: 10 μm.
Discussion
The local synthesis of PtdIns3P at a given membrane is critical for autophagy initiation as well as in endosomal membrane dynamics. To understand how enzymes that synthesize different pools of PtdIns3P and proteins that specifically bind to PtdIns3P cooperate to modulate biological functions associated with PtdIns3P requires detection of the PtdIns3P itself on membranes. This has proven more difficult than detection of the proteins responsible for PtdIns3P synthesis and turnover: Indeed, BECN1, ATG14, and PIK3C3 have been reported to participate in the early steps of autophagy (as illustrated in Fig. 5). Previous analyses of PtdIns3P on pre-autophagosomal membranes have been through the use of proteins with PtdIns3P-binding domains, such as WIPI2 or ZFYVE1. Here we address both the technical issue of PtdIns3P fluorescence-based monitoring in cells and evaluate the presence of PtdIns3P in the early steps of autophagosome biogenesis at the ER membrane.
We first showed that transfection of vectors for expression of PtdIns3P-binding domains linked to GFP (GFP-2XFYVE and GFP-P40PX) into living cells altered PtdIns3P membrane dynamics most probably by stabilizing these domains and by inhibiting normal turnover of PtdIns3P. This was notably illustrated by the increase in early endosome volume (Fig. 2) and an enhancement of autophagy level (Fig. 3) in cells transfected with GFP-2XFYVE or GFP-P40PX. In contrast, the indirect use of GST-FYVE to detect PtdIns3P in fixed cells enabled us to discriminate, by the use of specific co-markers, the endosomal and autophagosomal pools of PtdIns3P (Fig. 4). By using this technique we showed the presence of PtdIns3P domains on ER membrane soon after autophagy induction. PtdIns3P colocalized with ATG14 (Fig. 5), a key player of autophagy induction and a master regulator of autophagic PtdIns3P synthesis, as well as with BECN1, a component of the PtdIns3P synthesis machinery. Moreover we showed that autophagy induction by starvation was immediately followed by the appearance of ER-associated PtdIns3P, which subsequently colocalized with pre-autophagosomal markers such as ZFYVE1 and WIPI2, and then with LC3 (Fig. 6 and 7).
Our study validated a simple technique to visualize endogenous PtdIns3P without the need for transfection, thus avoiding effects of transfection or the PtdIns3P binding reagents on lipid stability and turnover. Although it is now generally agreed that PtdIns3P synthesis is one of the initial and necessary steps in autophagy initiation, our analysis provides the first evidence for the appearance and kinetics of such a PtdIns3P autophagy-specific pool at the site of autophagosome assembly. We observed that ER-associated PtdIns3P appearance was a rapid process directly associated with the specific recruitment of regulatory proteins, such as ZFYVE1 and WIPI2, at the site of phagophore assembly. The observed ER areas found to be positive for PtdIns3P in early steps of the autophagic response could thus be the omegasome domain described previously,44 and our data highlight the importance of PtdIns3P itself in the steps of autophagosome biogenesis.
Materials and Methods
Cell culture, transfection, and treatments
HeLa cells were obtained from ATCC (CCL2) and maintained in Minimum Essential Medium, supplemented with GlutaMAX (Gibco, 41090–028) 10% fetal bovine serum (Gibco, 16000044), and nonessential amino acids (Gibco, 11140035) at 37°C and 5% CO2. For starvation assays, cells were washed 3 times and maintained in Earle's balanced salt solution (EBSS, Gibco, 24010043) for the indicated times. To analyze the autophagic flux, cells were treated with 20 nM of BAF.A1 (Sigma, B1793), an inhibitor of autophagosome-lysosome fusion. Cells were transiently transfected with plasmid vectors or siRNA using the Fugene HD (Promega, E2312) or the Lipofectamine RNAiMAX kit (Invitrogen, 13378150) according to the manufacturer's instructions. Cells were incubated with siRNA for 72 h and efficient silencing was confirmed by western blot. Plasmids were transiently expressed for 24 h before analysis.
Reagents and antibodies
The following compounds were used in autophagy assays: 3-methyladenine (10 mM; Sigma, M9281), wortmannin (50 nM; Sigma, W1628). Primary antibodies used for immunofluorescence included: anti-LC3 rabbit polyclonal antibody (1:200 dilution; MBL, PM036), mouse monoclonal antibody (1:200 dilution; MBL, M152–3), anti-EEA1 mouse monoclonal antibody (1:200 dilution; BD Transduction Laboratories, 610456), and anti-MYC mouse monoclonal antibody (1:200 dilution; Sigma, M5546). Fluorescently labeled secondary antibodies, all diluted 1:200 and all from Invitrogen, included Alexa Fluor 546 donkey anti-mouse (A10036), Alexa Fluor 647 donkey anti-mouse (A31571), Alexa Fluor 546 donkey anti-rabbit (A10040), and Alexa Fluor 647 donkey anti-rabbit (A31573). For anti-GST detection, we used Alexa Fluor 649-labeled goat anti-GST (Rockland-Inc., 600–143–200). Primary antibodies used for western blotting included anti-SQSTM1 guinea pig polyclonal antibody (1:2000 dilution; Progen, GP62-C), anti-LC3 rabbit polyclonal antibody (1:10000 dilution; Sigma, L7543), anti-PIK3C3 rabbit polyclonal antibody (1:1000 dilution; Cell Signaling Technology, 4263), anti-ATG14 rabbit polyclonal antibody (1:1000 dilution; Sigma, A6358), anti-RAB5 mouse monoclonal antibody (1:1000 dilution; Synaptic Systems, 108011), anti-BECN1 mouse monoclonal antibody (1:1000 dilution; BD Transduction Laboratories, 612113), and anti-actin mouse monoclonal antibody (1:60000 dilution; Merck Millipore, MAB1501). Secondary antibodies used for western blot included horseradish peroxidase (HRP)-conjugated anti-mouse (1:20000 dilution; Bio-Rad, 1706516), anti-rabbit (1:10000 dilution; GE Healthcare, NA9340V), and anti-guinea pig (1:10000 dilution; Sigma, A5545).
Plasmids and RNAi
Plasmids encoding GST-2xFYVE, GST-mut-2xFYVE (C215S), and GFP-2xFYVE were kind gifts from H. Stenmark (University of Oslo, Oslo, Norway) and J. Gruenberg (University of Geneva, Geneva, Switzerland). GFP-P40PX (including amino acids 1–148 of NCF4) was obtained from Addgene (19010, deposited by Michael Yaffe). RFP-SEC61B/Sec61β was a kind gift from T. Rapoport (Harvard University, Cambridge, MA, USA), and GFP-ZFYVE1 was a gift from N. Ktistakis (Cambridge University, Cambridge, UK). MYC-BECN1 was a kind gift from M.I. Vaccaro (University of Buenos Aires, Buenos Aires, Argentina), and GFP-ATG14 was a kind a gift from N. Mizushima (University of Tokyo, Tokyo, Japan).
Gene silencing was achieved using commercially validated siRNAs designed to target human PIK3C3 (Qiagen, SI00040950 and SI00040971), human BECN1 (Qiagen, S100055573 and S100055580), and human ATG14 (Qiagen, S104266878 and S100455574).
Western blotting analysis
HeLa cells cultured on 60-mm plates were washed twice with ice-cold phosphate-buffered saline (PBS; Gibco, 14190–094) and lysed on ice in lysis buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40 (Thermo Scientific, 88124)) containing a protease and phosphatase inhibitors cocktail (Thermo Scientific, 78442). Protein concentration was determined using a BCA protein assay kit (Thermo Scientific, 23225) with bovine serum albumin as a standard. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF; GE Healthcare Life Sciences, 10600023) membranes and probed with the indicated primary and HRP-conjugated secondary antibodies. Immunoreactive protein bands were visualized using a chemiluminescent HRP substrate kit (Immobilon Western; Merck Millipore, WBKLS0500) and were imaged with the ChemiDoc MP Imaging System (Bio-Rad). Densitometric quantification was performed using the Image Lab software (Bio-Rad).
Immunofluorescence microscopy analysis
Cells grown on coverslips were fixed with 4% paraformaldehyde for 20 min at room temperature, incubated in blocking solution (10% fetal bovine serum in PBS) for 30 min, and then incubated with the specified primary antibodies in permeabilization/blocking buffer (PBS with 10% fetal bovine serum and 0.05% saponin (Sigma Aldrich, 8047) for 1 h at room temperature. Subsequently, cells were incubated with the appropriate Alexa Fluor secondary antibodies for 1 h at room temperature. Coverslips were mounted using Mowiol 4–88 reagent (Merck Millipore, 475904) with 4,6-diamino-2-phenylindole (DAPI, Merck Millipore, 508741) to visualize nuclei. Images were acquired by confocal laser-scanning microscopy (Zeiss LSM-700) and analyzed with Zeiss Zen lite and ImageJ (NIH) softwares. The number of endosomes and their volumes (expressed as µm3) were normalized to the number of cells in each field. The colocalization between PtdIns3P and EEA1 and between PtdIns3P and LC3 puncta was calculated as the number of pixels overlapping in the 2 channels per cell. The PtdIns3P fluorescence intensity was calculated using ImageJ.
PtdIns3P detection by 2xFYVE-GST
Purified 2xFYVE-GST and C215S FYVE-GST mutant peptides were produced by a classical GST-fusion protein purification protocol, using pGEX-5X-1 vector (GE Healthcare, 27–4584–01) cloning. BL21 bacteria strain maxi-cultures were lysed and subjected to glutathione beads binding for GST purification. The glutathione beads were finally treated with benzamidine-coated beads (GE Healthcare Life Science, 17512301) to remove the factor Xa and dialyzed for the final purification in a 75 mM KAc, 30 mM HEPES, pH 7.4, 5 mM MgCl2 solution. For fluorescence analysis on cells fixed with 4% paraformaldehyde (Electron Miscroscopy Sciences, 15714), the purified peptides were used during the primary round of the immunofluorescence protocol (along with other primary antibodies used in this study, for 1 h at room temperature) at a final concentration of 20 µg/mL, in a 0.05% saponin buffer (see above) and revealed by an Alexa Fluor 649-conjugated anti-GST antibody (Rockland Inc., 600–143–200). Of note, the use of a methanol-mediated fixation did not allow any specific detection of PtdIns3P by the 2xFYVE-GST method.
Statistical analysis
Quantitative results represent 3 independent experiments and values given are means ± SEM. Statistical analysis was performed using a 2-tailed, equal variance Student t test. P-values of < 0.05 (*), < 0.01 (**), < 0.001 (***) were determined to be statistically significant. All statistical analyses were performed with GraphPad Prism software.
Abbreviations
- ATG
autophagy related
- BECN1
Beclin 1
- DAPI
4,6-diamino-2-phenylindole
- EEA1
early endosome antigen 1
- ER
endoplasmic reticulum
- HRP
horseradish peroxidase
- LC3
microtubule associated protein 1 light chain 3
- PBS
phosphate-buffered saline
- PIK3C3
class III phosphatidylinositol 3-kinase
- PtdIns3P
phosphatidylinositol 3-phosphate
- PROPPIN
β-propellers that bind polyphosphoinositides
- PX
PhoX homology
- SQSTM1
sequestosome 1
- SEM
standard error of the mean
- siRNA
small interfering RNA
- WIPI2
WD repeat domain, phosphoinositide interacting 2
- ZFYVE1
zinc finger FYVE-type containing 1
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We warmly thank M.I. Vaccaro, H. Stenmark, J. Gruenberg, T. Rapoport, N. Ktistakis and N. Mizushima for kindly sharing reagents with us.
References
- [1].Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature [Internet]. 2006. [cited 2015 Mar 25];443:651-7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17035995. doi: 10.1038/nature05185. PMID:17035995. [DOI] [PubMed] [Google Scholar]
- [2].Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev [Internet]. 2013. [cited 2016 Sep 19];93:1019-137. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23899561. doi: 10.1152/physrev.00028.2012. PMID:23899561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Marat AL, Haucke V. Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J [Internet]. 2016;35(6):561-79. [cited 2016 Feb 24]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/26888746. doi: 10.15252/embj.201593564. PMID:26888746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol [Internet]. 2004;5:317-23. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15071556. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- [5].Schink KO, Tan K-W, Stenmark H. Phosphoinositides in Control of Membrane Dynamics. Annu Rev Cell Dev Biol [Internet]. 2016;32:143-71. [cited 2016 Sep 13]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/27576122. doi: 10.1146/annurev-cellbio-111315-125349. PMID:27576122. [DOI] [PubMed] [Google Scholar]
- [6].Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol [Internet]. 2009;186:773-82. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19797076. doi: 10.1083/jcb.200907014. PMID:19797076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lystad AH, Simonsen A. Phosphoinositide-binding proteins in autophagy. FEBS Lett [Internet]. 2016[cited 2016 Sep 19];590:2454-68. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27391591. doi: 10.1002/1873-3468.12286. PMID:27391591. [DOI] [PubMed] [Google Scholar]
- [8].Simonsen A, Stenmark H. PX domains: attracted by phosphoinositides. Nat Cell Biol [Internet]. 2001[cited 2016 Sep 19];3:E179-82. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11483972. doi: 10.1038/35087112. PMID:11483972. [DOI] [PubMed] [Google Scholar]
- [9].Stenmark H, Aasland R, Driscoll PC. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett [Internet]. 2002. [cited 2016 Sep 19]; 513:77-84. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11911884. doi: 10.1016/S0014-5793(01)03308-7. PMID:11911884. [DOI] [PubMed] [Google Scholar]
- [10].Simonsen A, Lippé R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature [Internet]. 1998. [cited 2016 Sep 19];394:494-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9697774. doi: 10.1038/28879. PMID:9697774. [DOI] [PubMed] [Google Scholar]
- [11].Futter CE, Collinson LM, Backer JM, Hopkins CR. Human VPS34 is required for internal vesicle formation within multivesicular endosomes. J Cell Biol [Internet]. 2001. [cited 2016 Sep 19];155:1251-64. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11756475. doi: 10.1083/jcb.200108152. PMID:11756475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Petiot A, Faure J, Stenmark H, Gruenberg J. PI3P signaling regulates receptor sorting but not transport in the endosomal pathway. J Cell Biol [Internet]. 2003. [cited 2016 Sep 19];162:971-9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12975344. doi: 10.1083/jcb.200303018. PMID:12975344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Scott CC, Vacca F, Gruenberg J. Endosome maturation, transport and functions. Semin Cell Dev Biol [Internet]. 2014;31:2-10. Available from: https://doi.org/10.1016/j.semcdb.2014.03.034. doi: 10.1016/j.semcdb.2014.03.034. PMID:24709024. [DOI] [PubMed] [Google Scholar]
- [14].Skånland SS, Wälchli S, Utskarpen A, Wandinger-Ness A, Sandvig K. Phosphoinositide-regulated retrograde transport of ricin: crosstalk between hVps34 and sorting nexins. Traffic [Internet]. 2007. [cited 2016 Sep 19];8:297-309. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17319803. doi: 10.1111/j.1600-0854.2006.00527.x. PMID:17319803. [DOI] [PubMed] [Google Scholar]
- [15].Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol [Internet]. 2008;182:685-701. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18725538. doi: 10.1083/jcb.200803137. PMID:18725538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ktistakis NT, Tooze SA. Digesting the Expanding Mechanisms of Autophagy. Trends Cell Biol [Internet]. 2016. [cited 2016 Sep 19];26:624-35. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27050762. doi: 10.1016/j.tcb.2016.03.006. PMID:27050762. [DOI] [PubMed] [Google Scholar]
- [17].a Lamb C, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol [Internet]. 2013;14:759-74. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=24201109. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- [18].Proikas-Cezanne T, Takacs Z, Dönnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci [Internet]. 2015. [cited 2016 Sep 19];128:207-17. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25568150. doi: 10.1242/jcs.146258. PMID:25568150. [DOI] [PubMed] [Google Scholar]
- [19].Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze S a. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell [Internet]. 2014; 55:238-52. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=24954904. doi: 10.1016/j.molcel.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cebollero E, van der Vaart A, Zhao M, Rieter E, Klionsky DJ, Helms JB, Reggiori F. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr Biol [Internet]. 2012. [cited 2016 Sep 19];22:1545-53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22771041. doi: 10.1016/j.cub.2012.06.029. PMID:22771041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Munson MJ, Allen GF, Toth R, Campbell DG, Lucocq JM, Ganley IG. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J [Internet]. 2015. [cited 2016 Sep 19]; 34:2272-90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26139536. doi: 10.15252/embj.201590992. PMID:26139536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pankiv S, Alemu EA, Brech A, Bruun J-A, Lamark T, Overvatn A, Bjørkøy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol [Internet]. 2010. [cited 2016 Sep 19]; 188:253-69. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20100911. doi: 10.1083/jcb.200907015. PMID:20100911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Backer JM. The intricate regulation and complex functions of the Class III phosphoinositide 3-kinase Vps34. Biochem J [Internet]. 2016. [cited 2016 Sep 14]; 473:2251-71. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27470591. doi: 10.1042/BCJ20160170. PMID:27470591. [DOI] [PubMed] [Google Scholar]
- [24].Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol [Internet]. 2010;20:355-62. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20356743. doi: 10.1016/j.tcb.2010.03.002. PMID:20356743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Calvo-Garrido J, King JS, Muñoz-Braceras S, Escalante R. Vmp1 regulates PtdIns3P signaling during autophagosome formation in Dictyostelium discoideum. Traffic [Internet]. 2014. [cited 2016 Sep 21];15:1235-46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25131297. doi: 10.1111/tra.12210. PMID:25131297. [DOI] [PubMed] [Google Scholar]
- [26].Cianfanelli V, De Zio D, Di Bartolomeo S, Nazio F, Strappazzon F, Cecconi F. Ambra1 at a glance. J Cell Sci [Internet]. 2015. [cited 2016 Sep 14]; 128:2003-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26034061. doi: 10.1242/jcs.168153. [DOI] [PubMed] [Google Scholar]
- [27].Molejon MI, Ropolo A, Lo Re A, Boggio V, Vaccaro MI. The VMP1-Beclin 1 interaction regulates autophagy induction. Sci Rep [Internet]. 2013. [cited 2016 Sep 21];3:1055. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23316280. doi: 10.1038/srep01055. PMID:23316280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ohashi Y, Soler N, García Ortegón M, Zhang L, Kirsten ML, Perisic O, Masson GR, Burke JE, Jakobi AJ, Apostolakis AA, et al.. Characterization of Atg38 and NRBF2, a fifth subunit of the autophagic Vps34/PIK3C3 complex. Autophagy [Internet]. 2016;12(11):2129-44. [cited 2016 Sep 21]:0. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27630019. doi: 10.1080/15548627.2016.1226736. PMID:27630019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al.. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol [Internet] 2009;11:385-96. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19270696. doi: 10.1038/ncb1846. PMID:19270696. [DOI] [PubMed] [Google Scholar]
- [30].He S, Ni D, Ma B, Lee J-H, Zhang T, Ghozalli I, Pirooz SD, Zhao Z, Bharatham N, Li B, et al.. PtdIns(3)P-bound UVRAG coordinates Golgi-ER retrograde and Atg9 transport by differential interactions with the ER tether and the beclin 1 complex. Nat Cell Biol [Internet]. 2013. [cited 2016 Sep 14];15:1206-19. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24056303. doi: 10.1038/ncb2848. PMID:24056303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci [Internet]. 2006;119:259-70. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16390869. doi: 10.1242/jcs.02735. PMID:16390869. [DOI] [PubMed] [Google Scholar]
- [32].Baskaran S, Carlson L-A, Stjepanovic G, Young LN, Kim DJ, Grob P, Stanley RE, Nogales E, Hurley JH. Architecture and dynamics of the autophagic phosphatidylinositol 3-kinase complex. Elife [Internet]. 2014. [cited 2016 Sep 19]; 3. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25490155. doi: 10.7554/eLife.05115. PMID:25490155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rostislavleva K, Soler N, Ohashi Y, Zhang L, Pardon E, Burke JE, Masson GR, Johnson C, Steyaert J, Ktistakis NT, et al.. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science [Internet]. 2015;350(6257):aac7365 [cited 2016 Sep 19]; 350:aac7365. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26450213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cheng J, Fujita A, Yamamoto H, Tatematsu T, Kakuta S, Obara K, Ohsumi Y, Fujimoto T. Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries. Nat Commun [Internet]. 2014. [cited 2016 Sep 19];5:3207 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24492518. [DOI] [PubMed] [Google Scholar]
- [35].Munson M, Ganley I. Determination of Cellular Phosphatidylinositol-3-phosphate (PI3P) Levels Using a Fluorescently Labelled Selective PI3P Binding Domain (PX). BIO-PROTOCOL [Internet]. 2016. [cited 2017 Feb 15];6:e1903 Available from: http://www.ncbi.nlm.nih.gov/pubmed/28127574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cheng J, Fujita A, Yamamoto H, Tatematsu T, Kakuta S, Obara K, Ohsumi Y, Fujimoto T. Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries. Nat Commun [Internet]. 2014. [cited 2017 Feb 15];5:3207 Available from: http://www.nature.com/doifinder/10.1038/ncomms4207. [DOI] [PubMed] [Google Scholar]
- [37].Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J [Internet]. 2000. [cited 2017 Mar 9];19:4577-88. Available from: http://emboj.embopress.org/cgi/doi/10.1093/emboj/19.17.4577. doi: 10.1093/emboj/19.17.4577. PMID:10970851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ketel K, Krauss M, Nicot A-S, Puchkov D, Wieffer M, Müller R, Subramanian D, Schultz C, Laporte J, Haucke V. A phosphoinositide conversion mechanism for exit from endosomes. Nature [Internet]. 2016. [cited 2017 Mar 9];529:408-12. Available from: http://www.nature.com/doifinder/10.1038/nature16516. doi: 10.1038/nature16516. [DOI] [PubMed] [Google Scholar]
- [39].Munson MJ, Allen GF, Toth R, Campbell DG, Lucocq JM, Ganley IG. mTOR activates the VPS34-UVRAG complex to regulate autolysosomal tubulation and cell survival. EMBO J [Internet]. 2015. [cited 2017 Mar 9];34:2272-90. Available from: http://emboj.embopress.org/cgi/doi/10.15252/embj.201590992. doi: 10.15252/embj.201590992. PMID:26139536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cheng J, Fujita A, Yamamoto H, Tatematsu T, Kakuta S, Obara K, Ohsumi Y, Fujimoto T. Yeast and mammalian autophagosomes exhibit distinct phosphatidylinositol 3-phosphate asymmetries. Nat Commun [Internet]. 2014. [cited 2016 Sep 14];5:3207 Available from: http://www.ncbi.nlm.nih.gov/pubmed/24492518. [DOI] [PubMed] [Google Scholar]
- [41].Morel E, Chamoun Z, Lasiecka ZM, Chan RB, Williamson RL, Vetanovetz C, Dall'Armi C, Simoes S, Point Du Jour KS, McCabe BD, et al.. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun [Internet]. 2013. [cited 2015 Aug 21];4:2250 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3905799&tool=pmcentrez&rendertype=abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Burman C, Ktistakis NT. Autophagosome formation in mammalian cells. Semin Immunopathol [Internet]. 2010. [cited 2016 Sep 13];32:397-413. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20740284. doi: 10.1007/s00281-010-0222-z. [DOI] [PubMed] [Google Scholar]
- [43].Carlsson SR, Simonsen a. Membrane dynamics in autophagosome biogenesis. J Cell Sci [Internet]. 2015;128:193-205. Available from: http://jcs.biologists.org/cgi/doi/10.1242/jcs.141036.. [DOI] [PubMed] [Google Scholar]
- [44].Roberts R, Ktistakis NT. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem [Internet]. 2013;55:17-27. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd = Retrieve&db = PubMed&dopt = Citation&list_uids = 24070468. doi: 10.1042/bse0550017. PMID:24070468. [DOI] [PubMed] [Google Scholar]