

Abstract
There are several different techniques for measuring telomere length, each with their own advantages and disadvantages. The traditional approach, Telomere Restriction Fragment (TRF) analysis, utilizes a DNA hybridization technique whereby genomic DNA samples are digested with restriction enzymes, leaving behind telomere DNA repeats and some sub-telomeric DNA. These are separated by agarose gel electrophoresis, transferred to a filter membrane and hybridized to oligonucleotide probes tagged with either chemiluminescence or radioactivity to visualize telomere restriction fragments. This approach, while requiring a larger quantity of DNA than other techniques such as PCR, can measure the telomere length distribution of a population of cells and allows measurement expressed in absolute kilobases. This manuscript demonstrates a modified DNA hybridization procedure for determining telomere length. Genomic DNA is first digested with restriction enzymes (that do not cut telomeres) and separated by agarose gel electrophoresis. The gel is then dried and the DNA is denatured and hybridized in situ to a radiolabeled oligonucleotide probe. This in situ hybridization avoids loss of telomere DNA and improves signal intensity. Following hybridization, the gels are imaged utilizing phosphor screens and the telomere length is quantified using a graphing program. This procedure was developed by the laboratories of Drs. Woodring Wright and Jerry Shay at the University of Texas Southwestern1,2. Here, we present a detailed description of this procedure, with some modifications.
Keywords: Cancer Research, Issue 125, Southern blot, terminal restriction fragments, leukocyte telomere length, telomere length, in-gel hybridization, DNA hybridization
Introduction
Telomeres, situated at the ends of chromosomes, are nucleotide repeats of the sequence TTAGGG (on human chromosomes) that play a critical role in protecting cellular genetic information3,4,5. Telomeres are several thousand base pairs in length and serve to protect the integrity of the rest of the chromosome during DNA replication. Replication of chromosomes is not perfect resulting in the ends of the chromosome not being completely copied. This inefficiency in replication is termed the "end-replication problem" and results in loss of some of the telomere repeats during each round of replication6,7. Telomere length can be restored after cell division by telomerase, an enzyme that replaces the lost telomere sequences8,9. Telomerase, however, is not activated in most human somatic cells and as a result, over time, telomeres will shorten, eventually resulting in cellular senescence10. Due to telomere shortening, telomeres are regarded as a biomarker of aging and risk of age-related diseases11,12. Additionally, telomere length can be affected by genetic, environmental, and lifestyle factors and other stimuli such as oxidative stress, indicating that telomere length can play a role as a biomarker in toxicological, epidemiological and behavioral studies13,14,15.
This article demonstrates a technique for measuring telomere length using a modified terminal restriction fragment (TRF) analysis adapted from Mender and Shay2 and Kimura et al.16, and similar to Herbert, Shay and Wright1 and Haussmann and Mauck17. Traditionally, TRF analysis involves a Southern blot procedure. Southern blots are considered the "gold-standard" for studying telomere length and are actively used for examining leukocyte telomere length in epidemiology studies18. This TRF analysis uses digested genomic DNA leaving behind the telomere repeats. The DNA fragments are then separated by gel electrophoresis, denatured and transferred to a nitrocellulose membrane. The telomere sequences are hybridized to a telomere oligonucleotide probe. Rather than transferring the separated telomeres to a membrane, other TRF techniques use in-gel hybridization techniques with and without denaturation of the DNA. The inclusion of denaturation is important when considering the detection of interstitial telomeres, which have been reported in some species19. Procedures that lack denaturing steps only detect the single stranded DNA overhangs at terminal telomeres and will not bind to interstitial telomere sequences18.
Besides TRF analysis, there are several other techniques for measuring telomere length that each have their own advantages and disadvantages. The Flow-FISH technique can measure mean telomere length of individual cells of a distinct cell type using flow cytometry16,20. While it can accurately tag telomeres with highly specific probes and reduce human error through automation, Flow-FISH is expensive, less efficient and requires fresh samples and a highly-skilled technician, limiting its ability for epidemiology studies16. In addition, this method does not account for potential changes in the number of chromosomes in the cell population. Another technique, qPCR, is widely accepted as a means of measuring telomere length for epidemiology studies16 due to its high-throughput, low-cost and requirement for small amounts of DNA compared to other methods. However, qPCR can only measure average telomeric DNA content relative to a single copy gene and is, thus, an indirect estimate of average telomere length. It also yields a high coefficient of variance in comparison studies, compared to southern blots, leading to questionable accuracy21.
The TRF procedure has its own deficiencies that limit its use for some studies. TRF techniques require a large amount of DNA compared to other methods (between 2 and 3 µg per sample). This limits the technique's applications to examining telomere length of clinical samples for which sufficient genomic DNA can be collected, and cannot be used on degraded DNA samples. Additionally, the TRF analysis is costly and labor intensive, requiring 4-5 days to yield results. However, the TRF yields a low coefficient of variance granting its reproducibility. While this protocol is different than the traditional Southern blot (utilizing in situ gel hybridization), the two techniques are fundamentally the same.
The technique presented here is a combination of a Southern blot and in-gel hybridization; associating the DNA denaturing step from Southern blots with the in-gel hybridization. This combination has the added benefit of improved probe signal strength compared to the Southern blot and has consistently yielded quantifiable results within our lab. Additionally, the use of radioactive probes rather than chemiluminescent probes yields higher signal intensity and allows for visualization and quantification with a phosphorimager, making the analyses of the TRF user-friendly.
Protocol
1. Genomic DNA Extraction
Perform a genomic DNA extraction from the cells (here, cell line BJ-hTERT) to be analyzed using a commercial DNA extraction kit, according to the manufacturer's instructions.
Measure the DNA concentration with a spectrophotometer. If the DNA concentration is less than 100 µg/mL, precipitate the DNA with ethanol and resuspend in a volume of TE buffer (10 mM Tris-Cl; 0.1 mM EDTA (Ethylenediaminetetraacetic Acid); pH 8.0), to ensure a DNA concentration of 100-600 µg/mL).
2. DNA Integrity Assessment
NOTE: This portion of the protocol outlines a DNA integrity assessment using a gel electrophoresis system with a 11 cm x 14 cm gel. Other systems and gel sizes can also be used, but the voltage and DNA migration time may vary.
Prepare a 1% agarose gel by combining 1 g of electrophoresis grade agarose in 100 mL of 1x TAE buffer (40 mM Tris-base; 20 mM acetic acid; 2 mM EDTA; pH 8.5) and microwave until the agarose is dissolved and the solution is clear. Cool the solution at room temperature until it reaches 50 °C (approximately 15 - 20 min).
While agarose solution is cooling, prepare gel casting tray by adding casting end gates to the casting tray. To prevent agarose leakage, cool the end gates to 4 °C prior to attaching to the casting tray.
Once the agarose solution has cooled to 50 °C, add 10 µL of ethidium bromide (10 mg/mL in dH2O) and pour the agarose solution into the gel casting tray. Add the comb to the casting tray.
Allow the agarose solution to harden for 45 min at room temperature.
Prepare DNA samples for electrophoresis by diluting 25 ng of genomic DNA using 1x TE buffer to a final volume of 9 µL and add 1 µL of 10x Loading Dye (0.25% (w/v) bromophenol blue, 0.25% (w/v) xylene cyanol FF, 30% (v/v) glycerol). Mix well.
Prepare the gel electrophoresis system for DNA migration by filling the electrophoresis system with 1x TAE buffer so that the buffer level is several millimeters above the gel. Load the wells of the gel with the entirety of the DNA samples. Add 5 µL of a DNA ladder to a single well.
Run the DNA migration at 100 V for approximately 2 h or until the bromophenol blue dye is approximately half way through the gel.
Image the gel using an ultraviolet light transilluminator or equivalent. NOTE: DNA samples with good integrity have high molecular weight bands with no smearing (that is due to sheared or broken DNA). If the DNA bands are smeared, they have low integrity and are not suitable for the Southern blot analysis, and the DNA must be re-isolated with fresh cells.
3. Genomic DNA Digestion
- Perform the digestion of the genomic DNA in a final volume of 20-40 µL. NOTE: Volumes greater than 40 µL will overflow the wells of the agarose gel.
- Combine 3 µg of genomic DNA and the appropriate amount of 10x DNA digest buffer (supplied with the restriction enzymes) so that the final concentration is 1x, in a microtube. Add 1 µL of RsaI restriction enzyme (10,000 U/mL) and 1 µL of HinfI restriction enzyme (10,000 U/mL) to each tube. Adjust the final volume using sterile, deionized H2O. Mix well. Centrifuge briefly (10 - 15 s at 16,000 x g) to ensure that all components are in the bottom of the tube.
Incubate the digestions at 37 °C for ≥16 h. After the digestion, store the digested DNA at 4 °C or -20 °C for no longer than 2 days if needed17.
4. Genomic DNA Gel Electrophoresis
NOTE: This portion of the protocol outlines a procedure for using a 20 cm x 25 cm gel electrophoresis system. Different gel electrophoresis systems can be used, but several variables may need to be modified including voltage, time of migration and amount of electrophoresis buffer added to the system in order to yield optimal results.
Prepare 0.6% agarose solution by combining 2.25 g of gel electrophoresis grade agarose with 375 mL of electrophoresis buffer, either 1x TAE or 0.5x TBE (110 mM Tris base; 90 mM boric acid; 2.5 mM EDTA; pH 8.3). NOTE: For telomeres less than 8,000 kB, 0.5x TBE is recommended whereas 1x TAE is recommended for telomeres between 8,000 kB and 20,000 kB1.
Microwave the solution until the agarose has dissolved and the solution is clear. Allow the solution to cool for 15 - 20 min at room temperature or until the agarose reaches 50 °C.
While the agarose solution is cooling, prepare the gel electrophoresis system for gel casting as described in step 2.2. Pour the agarose gel solution into a gel casting tray. Place a comb into the gel. Allow the gel to harden for 45 min uncovered at room temperature.
Prepare the digested DNA samples for electrophoresis. For a 40 uL reaction, add 0.4 µL of 10x loading dye into each sample of digested DNA. Briefly centrifuge (10-15 s, 16,000 x g) to ensure that all of the solution is at the bottom of the tube.
Prepare the gel electrophoresis system for DNA migration. Remove the end gates and comb from the gel. Fill the gel electrophoresis system with 1x TAE or 0.5x TBE to the fill line, ensuring that the gel is completely submerged in the buffer.
Load the wells of the gel with the DNA samples such that there is a blank well between samples. Avoid causing bubbles or puncturing the gel. Add 10 µL of a DNA ladder into wells on the opposite ends of the gel. NOTE: The type of DNA ladder will depend on the length of the telomeres, which will vary from person to person and between cell sources.
Run the gel electrophoresis at 150 V for 30 min to quickly move the DNA into the gel; then adjust the voltage to 57 V and run for 18-24 h.
5. Gel Fluorescent Imaging and Hybridization
After 18-24 h, turn off the gel electrophoresis power source. Remove the gel casting tray with the gel from the electrophoresis buffer. Slice off the top right corner of the gel to serve as a reference for the orientation of the gel.
Prepare a piece of filter paper by cutting the paper to be a slightly larger size than the gel. Place the filter paper on top of the gel in the casting tray and allow the paper to soak up excess solution on the gel. Carefully remove the air bubbles between the filter paper and the gel using a pipette as a roller to squeeze the bubbles out through the sides.
Remove the gel from the casting tray by flipping the gel and filter paper over so that the paper is underneath the gel. Transfer the gel with the filter paper to a gel dryer and cover the gel with plastic wrap. Remove all air bubbles between the plastic wrap and gel using a pipette as a roller.
Dry the gel for 2 h at 50 °C.
Once dried, remove the plastic wrap and transfer the gel and filter paper to a glass dish containing 250 mL of deionized water (sufficient to cover the gel). Carefully remove the filter paper and incubate the gel at room temperature for 15 min. The gel will be thin, firm and easy to manipulate without tearing.
Prepare 5x SSC solution (750 mM NaCl; 75 mM sodium citrate) in deionized water.
Lay the gel on top of a nylon mesh (12 in x 12 in). Roll the nylon mesh and gel together making sure that the gel is not in contact with itself. Place the mesh and gel into a hybridization tube (e.g., 12 in tube with 1.5 in diameter). NOTE: When properly rolled up, the gel will be in contact with the nylon mesh on both sides.
Combine 100 mL of 5x SSC and 12 µL of green fluorescent nucleic acid stain, and place into the hybridization tube. Protect the solution from light and incubate for 30 min at 42 °C in a hybridization oven with rotation.
Remove the nylon mesh/gel from the hybridization tube, roll open and place flat into a glass dish containing 250 mL of deionized water, and gently remove nylon mesh. Image the gel using a phosphorimager. Use the fluorescent settings: 520 BP, 40 Blue and 488 nm. Set the photomultiplier tube (PMT) to 375, the focal plane to +3 mm and the sensitivity to normal. Save the image to a file.
6. Hybridization
- Prepare the radioactive telomere probe.
- Combine 2 µL (2 ng) of telomere probe (5- (CCC TAA)4 3'), 2.5 µL of 10x polynucleotide kinase reaction buffer, 3 µL of γ32P-dATP (6,000 Ci/mmol), 4 µL of T4 polynucleotide kinase and DNase free, sterile deionized water to a final volume of 20 µL in a microcentrifuge tube. Caution: Follow facilities' radioactive safety and regulations when using radioactivity.
Briefly centrifuge (10-30 s at 16,000 x g) and incubate in water bath at 37 °C for 1 h. Centrifuge for 10-30 s at 16,000 x g and incubate at 65 °C for 20 min to stop the reaction.
Using a G-25 chromatography microspin column, mix the contents of the column by vortexing, then centrifuge for 1 min at 700 x g at room temperature. Discard the filtrate.
Load the radioactive probe solution into the G-25 chromatography column and centrifuge for 2 min at 700 x g. Save the filtrate. NOTE: Retain the filtrate as this contains the radioactive telomere probe. The radioactive probe can be stored at 4 °C if needed.
Prepare 250 mL of denaturing solution composed of 1.5 M NaCl and 0.5 M NaOH in sterile deionized water.
Place the gel into a glass dish containing 250 mL of denaturing solution and incubate for 15 min at room temperature. Remove the denaturing solution and replace with 250 mL of sterile deionized water. Incubate for 10 min on an orbital shaker (40 - 50 rpm) at room temperature.
Prepare 250 mL of neutralization solution composed of 1.5 NaCl and 0.5 M Tris-hydrochloric acid, pH 8.0 in sterile deionized water.
Remove the deionized water and replace with 250 mL neutralization solution and incubate for 15 min at room temperature.
During the incubation in the neutralization solution, prepare 50 mL hybridization solution composed of 5x SSC, 5x Denhardt's solution (0.1% non-ionic synthetic polymer; 0.1% polyvinyl pyrrolidine; 0.1% bovine serum albumin), 10 mM disodium phosphate (Na2HPO4) and 1 mM sodium pyrophosphate tetrabasic decahydrate (Na4P2O7 . 10 H2O) in sterile deionized water.
Roll the gel into nylon mesh and place into the hybridization tube (step 5.7). Add 20 mL of the hybridization solution and incubate in a hybridization oven at 42 °C for 10 min with rotation.
Remove the hybridization solution and replace with 20 mL of fresh hybridization solution. Add the entirety of the telomere probe and incubate in a hybridization oven at 42 °C overnight with rotation.
7. Gel Washing, Phosphor Screen Exposure, and Radio Imaging
Prepare 500 mL of 2x SSC solution (300 mM NaCl; 30 mM sodium citrate) in sterile deionized water.
Prepare 250 mL of 0.1x SSC (15 mM NaCl; 1.5 mM sodium citrate) and 0.1% Sodium Dodecyl Sulfate (SDS) in deionized water.
Remove the gel and mesh from the hybridization tube and place into a glass dish containing 250 mL of 2x SSC. Unroll and remove the nylon mesh from the dish. Place the glass dish onto an orbital shaker and incubate for 15 min at room temperature.
Remove the 2x SSC and replace with 250 mL of 0.1x SSC and 0.1% SDS solution, and incubate for 15 min on an orbital shaker at room temperature.
During the incubation, expose the Phosphor screen to the image eraser for 15 min.
Remove the 0.1x SSC and 0.1% SDS solution and replace with 250 mL of 2x SSC. Incubate for 15 min on an orbital shaker.
Remove the gel from 2x SSC solution and either wrap the gel in plastic wrap or place into a heat sealable pouch. Remove all bubbles and air pockets between the gel and the plastic wrap or plastic pouch. Place the gel into an exposure cassette with the Phopshor screen. Expose the Phosphor screen to the gel overnight.
Image the exposed Phosphor screen using a phosphorimager.
8. Telomere Length Quantification
Open the phosphorimager file containing the fluorescent image of the green fluorescent nucleic acid-stained gel (Figure 1).
Select 'Tools' and then 'Pixel Distance'. Using the mouse, draw a line from the bottom of the gel well to the middle of the first marker band and record the distance. Repeat this procedure for each marker band (Figure 2). These distances will be used in the graphing program as described below.
Open the phosphorimage of the radiolabeled gel (Figure 3). Select 'Object Manager' and then 'Grid'. Set the Grid to 1 column and 150 rows. Adjust the grid so that it stretches from the top of the gel to the bottom of the gel. Adjust the width of the grid to equal the width of the lane, clicking on the edge of the grid and dragging the width of the grid to equal the width of the lane.
Copy this grid (so that the width and height of the boxes remain the same) and move the second grid to an empty lane (for background purposes). Continue to copy and move additional grids for additional sample lanes. NOTE: When finished, there should be grids for each sample lane and one grid of an empty lane for background (Figure 4).
Select 'Analysis" and then 'Volume Report'. Select all of the grids for the report. Once the volume report is displayed, double click on the report to activate the spreadsheet program. Save the spreadsheet file. The data for this file will be under the 'Volume' lane.
Open an appropriate graphing program and create a new data and table. Select 'Y: Enter and plot a single Y value for each point' then select 'Create'.
Label the first column (Column X) as 'Measured Distance' and label the second column (Column Y) as 'Molecular Weight'. Enter the marker molecular weights of each marker band in column Y. Enter the migration distances obtained in step 8.2 corresponding to each molecular weight marker.
Go to 'Analysis' and select 'Analyze' and then 'nonlinear regression (curve fit)' and select 'OK'. At the next screen, select 'Exponential and then 'One phase decay'. Select 'Range' tab and enter '150' in 'number of points that define the curve' and check 'Create a table of XY coordinates to export or copy the curve to another program'. NOTE: The result, 'nonlinear fit of Data 1', contains the molecular weight for each distance in the Y row.
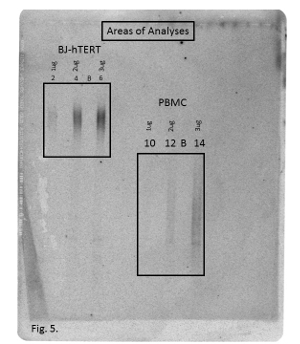
For analysis of the sample lanes, select an area of analysis that is above and below the actual telomere bands. Do not use the entire length of the lane (Figure 5). Determine the grid locations for the area of analysis of each lane.
Select 'File', 'New' 'Create new Table and Graph'. Select 'Y: Enter and plot a single Y value for each point' then select 'Create'. Copy and paste the background lane volume values (intensity values) from the spreadsheet file into the first column (X) and the volume values (intensity values) from the sample lane into the first column Y. If there are multiple sample lanes, copy each set of volume values from the spreadsheet file into additional Y columns in the graphing program file.
Delete the values from the background column (X) and each sample lane (Y) that are outside the areas of analysis determined in step 8.8. NOTE: The data table should only contain values in those grid locations corresponding to the areas of analysis.
- Select Analysis, then 'Analyze' and 'transform'. Click 'OK'. Select 'Transform Y values using Y=Y-X'. This will transform the data in Data 2 (transformed intensity value (Y) = sample intensity (Y) - background intensity (X)).
- Select Analysis then 'Analyze', 'Column Analysis' and 'Column statistics'. Select OK. The results tab 'Col. Stats of Transform of Data 2' shows under the 'Sum' row, ∑(Inti).
- Select 'File', 'New', 'Create New Table and Graph'. Select 'Y: Enter and plot a single Y value for each point' then select 'Create'. Copy and paste the Y column data from the results page 'Nonlin fit of Data 1, Curve' into the new X column. Copy and paste the Y column data from the results page 'Transform of Data 2' into the new Y column. Select Analysis then 'Analyze' and 'Transform'. Click 'OK'. Select 'Transform Y values using Y=Y/X'.
- Select Analysis then 'Analyze', 'Column Analysis' and 'Column statistics'. Select OK. The results tab 'Col. Stats of Transform of Data 3' shows under the 'Sum' row, ∑(Inti/MWi). MW=molecular weight.
To calculate the average telomere length (TL) for each sample, use the formula TL=∑(Inti)/ ∑(Inti/MWi).



Representative Results
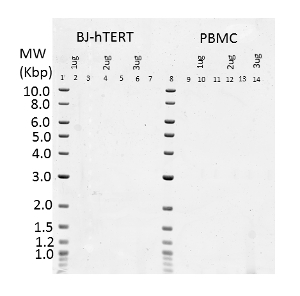
Three concentrations of DNA (1, 2 and 3 µg) isolated from the cell line BJ-hTERT (telomerase immortalized human foreskin fibroblasts)22 and human peripheral blood mononuclear cells (PBMCs) were analyzed for telomere length. Table 1 shows the lane assignments for each DNA sample. Figure 1 shows a nucleic acid fluorescent stain of the gel. The molecular weight standards are clearly visible. The distance from the top of the gel to each molecular weight standard was determined (Figure 2). Figure 3 shows the phosphorimage of the gel following hybridization with the telomere probe. The telomere bands are shown as smears in each lane. Grids of 150 boxes were calculated for each lane plus one background lane (Figure 4). The areas of analysis corresponding to the areas of the gel containing the telomere bands for each set of samples is shown in Figure 5. Separate background lanes were selected for each set of samples to ensure that the background intensities were similar for each sample set (lanes marked B in Figure 5). Table 2 shows the telomere restriction fragment lengths for each sample. The average telomere length in the hTERT immortalized cell line (BJ-hTERT) is higher than that seen in human PBMCs (compare lanes 2, 4 and 6 with lanes 10, 12, and 14 in Figure 3). These results also demonstrate that the amount of genomic DNA analyzed in each lane is critical to obtaining a detectable signal following hybridization. For samples with telomeres that are fairly uniform (i.e., the BJ-hTERT cell line) as little as 1 µg was detectable using this procedure (although the signal was very faint). However, for samples with telomeres that have a larger variance (i.e., human PBMCs), the minimum amount of DNA providing a reliable signal is 2 µg.
Figure 1: Green Fluorescent Nucleic Acid Stained Gel. Phosphorimage of the dried agarose gel stained with green fluorescent nucleic acid stain showing the location of the DNA ladder bands (lanes 1 and 8). Please click here to view a larger version of this figure.
Figure 2: Determination of the Marker Band Migration Distance. Blue arrows indicate the distance measured from the top of the gel to each DNA ladder band. Please click here to view a larger version of this figure.
Figure 3: Phosphorimage of DNA Samples Probed with the Telomere Probe. Lanes 2, 4 and 6 contain BJ-hTERT DNA (1, 2, and 3 µg, respectively). Lanes 10, 12 and 14 contain human PBMC DNA (1, 2 and 3 µg, respectively). Please click here to view a larger version of this figure.
Figure 4: Grid Location. Screenshot showing the location of 150 count grids for each analyzed lane. Please click here to view a larger version of this figure.
Figure 5: Areas of Analysis. Phosphorimage of telomere bands showing the selected areas of analysis for each set of samples. Lanes 2, 4 and 6 contain BJ-hTERT DNA; lanes 10, 12 and 14 contain human PBMC DNA. B = background lanes. Please click here to view a larger version of this figure.
| Lane | Sample |
| 1 | DNA Ladder |
| 2 | BJ-hTERT 1µg |
| 3 | Blank |
| 4 | BJ-hTERT 2 µg |
| 5 | Blank |
| 6 | BJ-hTERT 3µg |
| 7 | Blank |
| 8 | DNA Ladder |
| 9 | Blank |
| 10 | PBMCs 1µg |
| 11 | Blank |
| 12 | PBMCs 2µg |
| 13 | Blank |
| 14 | PBMCs 3µg |
| DNA Samples separated on 0.6% agarose gel |
Table 1: DNA Agarose Sample Assignments.
| Sample (Lane) | ∑(Inti) | ∑(Inti/MWi) | Average Telomere Length = ∑(Inti)/∑(Inti/MWi) |
| BJTert 1ug (1) | 268691 | 20596 | 13.04578559 |
| BJTert 2ug (2) | 1357000 | 103204 | 13.14871517 |
| BJTert 3ug (3) | 1962000 | 148411 | 13.22004434 |
| PBMC 1ug (4) | -416337 | -142585 | 2.91992145 |
| PBMC 2ug (5) | 286653 | 62845 | 4.561269791 |
| PBMC 3ug (6) | 1880000 | 524123 | 3.586944286 |
Table 2: Average Telomere Length Calculations.
Discussion
Following electrophoresis, the genomic DNA smear is higher than 800 bp. This can occur if the restriction enzymes are faulty or if additional enzymes (as described above) are needed. A second possible explanation is that the protein is still attached to the DNA. When determining the DNA concentration by spectrophotometer, the 260/280 ratio should be around 1.8. If this ratio is <1.5, there is protein contamination which can interfere with restriction enzyme digestion. Either the DNA sample would need to be re-isolated, or the protein would need to be removed with a proteinase K digestion and phenol:chloroform extraction followed by ethanol precipitation. The use of a commercial DNA isolation kit routinely results in DNA with a 260/280 ratio of 1.8-1.85.
Following the exposure of the gel to a phosphor screen, there are no telomere fragments detected. This can result from a poor radio-labelled telomere probe, improper hybridization conditions or insufficient quantity of starting genomic DNA. When preparing the telomere probe, at the end of the centrifugation of the G-25 column, there should be readily detectable radioactivity in the flow-through (representing the radio-labelled telomere probe). If radioactivity is not detected, then there was a problem with the preparation of the probe. It is also important to ensure that the hybridization temperature does not exceed 42 °C (to prevent melting of the probe:template complex) and that the hybridization buffer completely immerses the gel in the hybridization chamber. The minimum amount of genomic DNA that provides detectable telomeres is 2.0-3.0 µg. Smaller amounts of starting material can result in very weak to no signal following hybridization.
This protocol requires 2-3 µg for each telomere length determination. If the samples are to be run in duplicate or triplicate, then 6-12 µg of DNA is required. This prevents this procedure from being used on samples with limited DNA quantity. The procedure is fairly laborious requiring 3-4 days to complete. The number of samples analyzed at one time is limited to the number of gel electrophoresis rigs and the size of the agarose gel comb. As a result, it is less than ideal for studies employing large numbers of samples (such as epidemiological studies).
DNA hybridization techniques of telomeres separated by gel electrophoresis remains the gold standard for determining telomere length. No other procedure directly measures telomere length resulting in actual telomere sizes.
There are several critical steps in this procedure. First, the DNA sample must have good integrity, i.e., not sheared. DNA that is sheared can result in false telomere length results. It is also important that the DNA digestions be complete. The digestion of genomic DNA should result in a DNA smear of 800 bp or less2. To ensure proper digestion, some procedures have described a total of 6 restriction enzymes (rather than just RsaI and Hinf1 as described in this procedure). The additional restriction enzymes include HhaI, MspI, HaeIII and AluI2. A second critical step is allowing the agarose gel to run long enough to get proper separation of the digested DNA. If there is better separation, then there is better precision in the analyses. The gel run time should be long enough so that DNA fragments representing 1 Kbp are located at the bottom of the gel (Figure 1). Depending on the gel size and the voltage of the electrophoresis, this can require 12 to 24 h. A third critical step is the hybridization. There must be sufficient hybridization buffer in the chamber tube to immerse the entire gel when the tube is horizontal. Insufficient levels of hybridization buffer can result in uneven hybridization of the telomere probe.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors acknowledge the support of the University of Pittsburgh Cancer Institute Biobehavioral Oncology shared facility that is supported in part by award P30CA047904.
References
- Herbert BS, Shay JW, Wright WE. Analysis of telomeres and telomerase. Curr Protoc Cell Biol. 2003. p. 16. Chapter 18 Unit 18. [DOI] [PubMed]
- Mender I, Shay JW. Telomere Restriction Fragment (TRF) Analysis. Bio Protoc. 2015;5(22) doi: 10.21769/bioprotoc.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH. Telomeres and Telomerase: The Means to the End (Nobel lecture) Angew Chem Int Ed Engl. 2010;49(41):7405–7421. doi: 10.1002/anie.201002387. [DOI] [PubMed] [Google Scholar]
- Greider CW. Telomerase Discovery: The Excitement of Putting Together Pieces of the Puzzle (Nobel lecture) Angew Chem Int Ed Engl. 2010;49(41):7422–7439. doi: 10.1002/anie.201002408. [DOI] [PubMed] [Google Scholar]
- Szostak JW. DNA Ends: Just the Beginning (Nobel lecture) Angew Chem Int Ed Engl. 2010;49(41):7386–7404. doi: 10.1002/anie.201000635. [DOI] [PubMed] [Google Scholar]
- Watson JM, Riha K. Telomeres, Aging, and Plants: From Weeds to Methuselah - A Mini-Review. Gerontology. 2011;57(2):129–136. doi: 10.1159/000310174. [DOI] [PubMed] [Google Scholar]
- Lu W, Zhang Y, Liu D, Songyang Z, Wan M. Telomeres-Structure, Function, and Regulation. Exp Cell Res. 2013;319(2):133–141. doi: 10.1016/j.yexcr.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNeil DE, Bensoussan HJ, Autexier C. Telomerase Regulation from Beginning to the End. Genes (Basel) 2016;7(9):64. doi: 10.3390/genes7090064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouquerel E, Opresko P. Convergence of The Nobel Fields of Telomere Biology and DNA Repair. Photochem Photobiol. 2016. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Bernadotte A, Mikhelson VM, Spivak IM. Markers of Cellular Senescence. Telomere Shortening as a Marker of Cellular Senescence. Aging (Albany NY) 2016;8(1):3–11. doi: 10.18632/aging.100871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opresko PL, Shay JW. Telomere-Associated Aging Disorders. Ageing Res Rev. 2016. pp. 1568–1637. Epub ahead of print. [DOI] [PMC free article] [PubMed]
- Chiodi I, Mondello C. Telomere and Telomerase Stability in Human Diseases and Cancer. Front Biosci (Landmark Ed) 2016;1(21):203–224. doi: 10.2741/4385. [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Epel ES, Lin J. Human Telomere Biology: A Contributory and Interactive Factor in Aging, Disease Risks, and Protection. Science. 2015;350(6265):1193–1198. doi: 10.1126/science.aab3389. [DOI] [PubMed] [Google Scholar]
- Lindqvist D, et al. Psychiatric Disorders and Leukocyte Telomere Length: Underlying Mechanisms Linking Mental Illness with Cellular Aging. Neurosci Biobehav Rev. 2015;55:333–364. doi: 10.1016/j.neubiorev.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodelon C, Savage SA, Gadalla SM. Telomeres in Molecular Epidemiology Studies. Prog Mol Biol Transl Sci. 2014;125(113) doi: 10.1016/B978-0-12-397898-1.00005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, et al. Measurement of telomere length by the Southern blot analysis of terminal restriction fragment lengths. Nat Protoc. 2010;5(9):1596–1607. doi: 10.1038/nprot.2010.124. [DOI] [PubMed] [Google Scholar]
- Haussmann MF, Mauck RA. TECHNICAL ADVANCES: New strategies for telomere-based age estimation. Mol Ecol Resour. 2008;8(2):264–274. doi: 10.1111/j.1471-8286.2007.01973.x. [DOI] [PubMed] [Google Scholar]
- Nussey DH, et al. Measuring telomere length and telomere dynamics in evolutionary biology and ecology. Methods Ecol Evol. 2014;5(4):299–310. doi: 10.1111/2041-210X.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delany ME, Krupkin AB, Miller MM. Organization of telomere sequences in birds: evidence for arrays of extreme length and for in vivo shortening. Cytogenet Cell Genet. 2000;90(1-2):139–145. doi: 10.1159/000015649. [DOI] [PubMed] [Google Scholar]
- Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protoc. 2006;1(5):2365–2376. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- Sanders JL, Newman AB. Telomere length in epidemiology: a biomarker of aging, age-related disease, both, or neither. Epidemiol Rev. 2013;35:112–131. doi: 10.1093/epirev/mxs008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh D, Fouquerel E, Murphy CT, Wang H, Opresko PL. Telomeres are partly shielded from ultraviolet-induced damage and proficient for nucleotide excision repair of photoproducts. Nat Commun. 2015;6:8214. doi: 10.1038/ncomms9214. [DOI] [PMC free article] [PubMed] [Google Scholar]