

Abstract
The receptor tyrosine kinase family consisting of Tyro3, Axl, and Mer (TAM) is one of the most recently identified receptor tyrosine kinase families. TAM receptors are up-regulated postnatally and maintained at high levels in adults. They all play an important role in immunity, but Axl has also been implicated in cancer and therefore is a target in the discovery and development of novel therapeutics. However, of the three members of the TAM family, the Axl kinase domain is the only one that has so far eluded structure determination. To this end, using differential scanning fluorimetry and hydrogen-deuterium exchange mass spectrometry, we show here that a lower stability and greater dynamic nature of the Axl kinase domain may account for its poor crystallizability. We present the first structural characterization of the Axl kinase domain in complex with a small-molecule macrocyclic inhibitor. The Axl crystal structure revealed two distinct conformational states of the enzyme, providing a first glimpse of what an active TAM receptor kinase may look like and suggesting a potential role for the juxtamembrane region in enzyme activity. We noted that the ATP/inhibitor-binding sites of the TAM members closely resemble each other, posing a challenge for the design of a selective inhibitor. We propose that the differences in the conformational dynamics among the TAM family members could potentially be exploited to achieve inhibitor selectivity for targeted receptors.
Keywords: enzyme inhibitor, protein dynamic, protein kinase, protein stability, protein structure
Introduction
Tyro3, Axl, and Mer constitute the TAM2 RTK family (for a review, see Lemke (1); Ref. 2). They play their most important role in the immune system in addition to their roles in the nervous, reproductive, and vascular systems (3). All three TAM receptors are activated by the growth arrest-specific protein 6 (GAS6) (4–6). Protein S is another ligand for Tyro3 and Mer that has little or no affinity for Axl (3, 5). The activation mechanism involves binding of the extracellular domains of TAM receptors expressed on the surface of phagocytes to the C-terminal domain of GAS6/Protein S, inducing kinase dimerization and activation. The N-terminal end of GAS6/Protein S recognizes the phosphatidylserine externalized by apoptotic cell membranes. Thus, GAS6/Protein S-mediated activation of TAM family members plays an important role in phagocytosis (7–9). The removal of apoptotic cells suppresses inflammation, possibly contributing to the roles Mer and Axl play in the negative regulation of inflammatory responses and maintaining the balance of innate immunity (3, 8). TAM knock-out mice, although viable initially, eventually develop broad-spectrum autoimmune disease characterized by lymphoproliferation (10). These mice are born with normal retinas, but most of their photoreceptors die soon after birth (11). In humans, there are about a dozen variants of Mer linked to inherited retinitis pigmentosa and retinal dystrophy (12, 13). Tyro3 or Axl knock-out mice show defects in platelet aggregation (14). In general, dysfunction of TAM family member receptors is thought to contribute to human autoimmune conditions such as rheumatoid arthritis and inflammatory bowel disease.
Overexpression or up-regulation of TAM family members has been observed in many cancers, although it is not clear whether the TAM RTK enzymatic activity is the driver of oncogenicity (14). TAM activity seems to play roles in angiogenesis, evasion of apoptosis, and metastases (14, 15). Axl has been shown to play a role in the metastasis of breast carcinoma (16), and increased Axl activity is correlated with poor prognosis in pancreatic cancer and glioma (17, 18). Axl activation has also been identified as a mechanism of acquired resistance to EGFR inhibitors in mutant EGFR lung cancer models (19). Elevated expression of Axl or GAS6 was observed in inhibitor-resistant mutant EGFR-driven lung cancer patients, which led to the recognition of Axl as a promising therapeutic target. It has been actively pursued for the design and discovery of small-molecule inhibitors (20, 21) with the first Axl-selective inhibitor, BGB324 from BergenBio, entering clinical trials in 2013 (22).
The N-terminal ectodomain of the TAM family members is composed of two immunoglobulin-related domains responsible for ligand recognition followed by two fibronectin type III repeats (Fig. 1a) (23). It is connected via a single-pass transmembrane polypeptide chain to the cytoplasmic kinase domain. The extracellular domains of Axl and Tyro3 in complex with GAS6 have been structurally characterized (23, 24) and so have the kinase domains of Tyro3 (25) and Mer (26, 27). An atomic resolution structure of the Axl kinase domain has so far been elusive despite the focus to develop inhibitors of Axl kinase activity to treat various cancers. Available structures of Mer and Tyro3, by virtue of shared sequences in the ATP-binding site, provide seemingly reasonable surrogates for the Axl kinase domain. However, the influence of overall differences in their sequences on the conformational equilibrium is unpredictable. Here we report a crystal structure of the Axl kinase domain in complex with a macrocyclic inhibitor, compound 1. The protein structure shows two conformational states, including the one that meets all known structural constraints for an active kinase domain. This provides the first glimpse of the active conformation of a TAM family member because previously reported structures of Tyro3 and Mer kinase domain are in their inactive states. The structure also suggests a role for the intracellular juxtamembrane region in enzyme activity that is further supported by mutagenesis and biochemical characterization of the Axl intracellular domain. We probed the dynamics of the Axl and Mer kinase domains by thermal stability and hydrogen-deuterium exchange mass spectrometry (HDX-MS) measurements. The results suggest that the Axl kinase domain is significantly more dynamic than Mer. Given the similarities between the ATP-binding sites of Axl and Mer kinases, differences in the dynamics of the two proteins may offer an avenue to explore for selective Axl inhibitors.
Figure 1.

Domain architecture and kinase domain sequences of TAM family of RTKs. a, domain architecture of Axl. Domains are highlighted in color: Ig in cyan, fibronectin in orange, transmembrane chain in red, and kinase domain in green. The region between the transmembrane and the kinase domain (residues 473–535) is the intracellular juxtamembrane region. The numbers indicate the domain boundaries. b, sequence alignment of the TAM family kinase domains to the crystallization construct for Axl with the observed secondary structure elements in the Axl crystal structure. TT denotes the β-turns. Triangles denote the reported or inferred Tyr phosphorylation sites in the Axl sequence.
Results
The cytoplasmic region of Axl (residues 473–894) consists of a membrane-proximal juxtamembrane region followed by the kinase domain and a C-terminal tail (Fig. 1). Numerous protein constructs encompassing the kinase domain and varying lengths of the juxtamembrane region were tested for crystallizability but without success. This is in contrast to the Mer and Tyro3 kinase domains, which have been structurally characterized (25–27). To understand the reasons behind the differences in the crystallizability of Axl and Mer kinase domains, we measured the thermal stabilities of both proteins. Despite sharing 69% sequence identity (Fig. 1), the isolated kinase domain of Mer showed a higher melting temperature than the Axl kinase domain as measured by differential scanning fluorimetry (Fig. 2). An approximate 10 °C difference in their measured melting temperatures suggests lower stability and likely a greater dynamic character of the Axl kinase that interfere with crystallization. To explore whether a small-molecule inhibitor could help increase the protein stability, we made a complex of Axl kinase domain with compound 1, a macrocyclic inhibitor of Mer and Axl (28). Compound 1 is a very potent inhibitor of both kinases with a measured inhibitory constant, Ki, of 130 ± 10 pm for Axl. The Ki value for Mer was determined to be <50 pm (the detection limit for the assay). The addition of the inhibitor stabilized both the Axl and Mer kinase domains (Fig. 2). Consistent with this observation, the complex of Axl kinase domain with compound 1 co-crystallized (Table 1 and supplemental Fig. S1), whereas the apoprotein remained recalcitrant to crystallization efforts.
Figure 2.
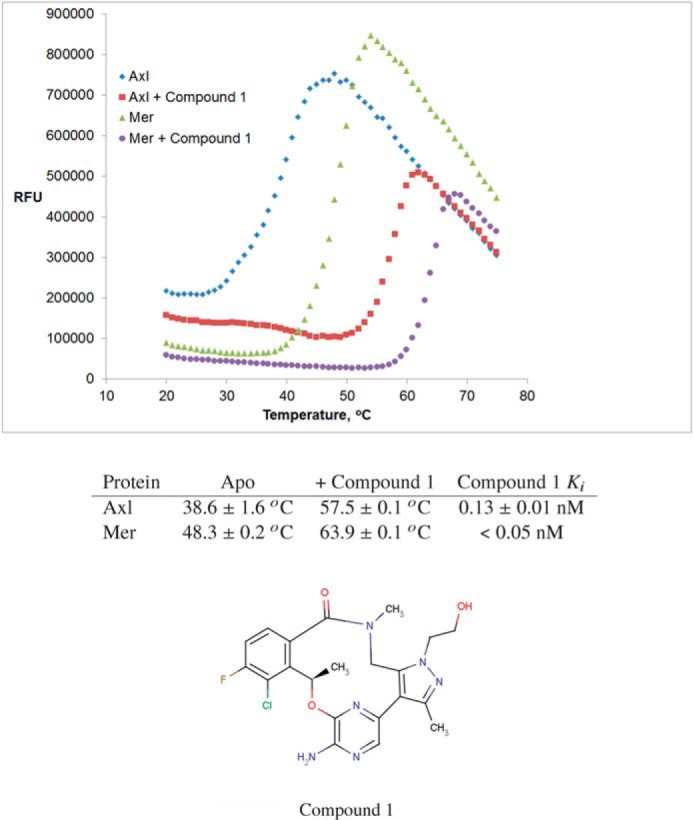
Thermal stability profiles of Axl and Mer kinase domains and Ki values for compound 1-mediated inhibition of the two proteins. Relative fluorescence units (RFU) are plotted against temperature. Average protein melting temperatures (deduced as the midpoint of the thermal transition) and the standard deviation from the triplicate measurements are reported in the table. The compound 1 inhibitory constant, Ki, is an average and the standard deviation of two independent measurements.
Table 1.
X-ray data collection and refinement statistics
Values in parentheses indicate highest resolution shell. r.m.s., root mean square.
| Axl | Mer | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 81.7, 100.7, 81.9 | 51.7, 92.3, 69.3 |
| α, β, γ (°) | 90, 94, 90 | 90, 100.8, 90 |
| Resolution (Å) | 63–2.84 (2.99–2.84) | 92–2.13 (2.25–2.13) |
| Rmerge | 0.095 (0.565) | 0.063 (0.425) |
| I/σI | 12.0 (2.4) | 11.1 (2.5) |
| Completeness (%) | 99.3 (99.8) | 98.7 (98.5) |
| Redundancy | 3.4 (3.3) | 3.3 (3.4) |
| Refinement | ||
| Resolution (Å) | 60–2.84 | 90–2.13 |
| No. of reflections | 31,145 | 35,087 |
| Rwork/Rfree | 0.211/0.24 | 0.212/0.257 |
| No. of atoms | ||
| Protein | 8,874 | 4,254 |
| Water | 0 | 174 |
| Ligand | 128 | 64 |
| B-factors | ||
| Protein | 57 | 42 |
| Ligand | 51 | 29 |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.008 | 0.006 |
| Bond angles (°) | 1.0 | 0.8 |
| Ramachandran analysis | ||
| Favored (%) | 91.0 | 93.5 |
| Allowed (%) | 8.7 | 6.3 |
| Disallowed (%) | 0.3 | 0.2 |
| Protein Data Bank code | 5U6B | 5U6C |
Overall structure
The Axl protein construct that crystallized in complex with the inhibitor comprised residues 514–818. The crystals consisted of four molecules in the asymmetric unit. As expected, the structure conforms to the overall kinase domain architecture with a smaller N-terminal lobe (N-lobe) consisting largely of a β-sheet and a single α-helix (the catalytically important C-helix) and a larger C-terminal lobe (C-lobe) that is predominantly helical (Fig. 3a). The interlobe cleft represents the ATP-binding site, which is occupied by the small-molecule inhibitor compound 1 in the structure presented here.
Figure 3.

Axl crystal structure and the role of the juxtamembrane region in enzyme activity. a, the backbone trace of the Axl kinase domain. Compound 1 is shown in ball-and-stick representation. The structure on the left with orange backbone is the inactive state, whereas the structure on the right with the blue backbone is an active conformation. Functionally important structural elements are highlighted in color: C-helix in cyan, activation loop in green, and juxtamembrane region in magenta. Chain breaks are shown using black lines. Some of the catalytically important residues, Asp690 (from the DFG motif), Lys567, and Glu585, are highlighted. Asp690 orientation shows DFG-in conformation. Lys567 and Glu585 form an ion-pair interaction as in the active state (right) but not in the inactive state (left). b, different orientations of the juxtamembrane regions in the inactive (left) and active (right) conformations of the Axl kinase. The color code is the same as in a. Leu526 from the juxtamembrane region packs against Leu583 and Val587 of the C-helix in the active state.
The structure shows the kinase domain in two distinct conformations: molecules A and C are clearly in the inactive state, whereas molecules B and D meet most of the known constraints for the active conformation of the kinase domains (29). Henceforth, we will focus on molecules A and B as representatives of the two conformational states. Differences in these two conformations are mainly localized to the N-lobe and C-terminal end of the juxtamembrane region (residues 514–529) that is part of the crystallization construct (Fig. 3 and supplemental Fig. S2). Unsurprisingly, the C-lobes (except the activation loop) are nearly identical in the two conformations.
In the inactive state (molecule A), the C-helix is rotated away from the rest of the kinase domain. This rotation allows the β-sheet of the N-lobe (especially strands 3, 4, and 5) to come closer to the C-lobe (supplemental Fig. S2). The electron density for most of the activation loop downstream of the DFG motif is missing and hence is not modeled. The Asp690 of the DFG motif is oriented in the ATP-binding cleft, whereas the Phe691 side chain is oriented in the hydrophobic core facing the C-helix in what is often referred to as the DFG-in conformation (Fig. 3a).
In the active state (molecule B), the activation loop is mostly ordered and shows an extended conformation that is consistent with what has been observed in other active kinase structures. This conformation of the activation loop allows for the recognition of the substrate peptide. The extended conformation may be partially stabilized by its interaction with the C-lobe of the neighboring molecule in the crystal lattice. The C-helix is rotated closer to the N-lobe with Glu585 forming a salt bridge with the conserved catalytically important Lys567. This interaction is one of the hallmarks of active kinases (30). The C-helix must bend to make this specific interaction. Crystal packing interactions do not influence the C-helix conformation; the closest neighboring molecule is about 10 Å from the C-helix. The C-helix is in close contact with a helix from the juxtamembrane region. This may suggest a role for the juxtamembrane region in transition to the active state of the enzyme (Fig. 3b).
Although our crystallization construct has only 16 residues (514–529) of the juxtamembrane region, they take on very different conformations in the two states of the protein. This region encompasses a short helix of about 10 residues in the active state (molecule B), and it is oriented such that it packs against the C-helix and helps it adopt the catalytically competent conformation. The orientation of the juxtamembrane helix is very similar to that observed in other RTKs like active c-MET (31) and EPHB2 (32). Various residues from the two helices are in van der Waals interactions contributing to the hydrophobic core of the N-lobe. For example, Leu526 from the juxtamembrane region is in close contact with Leu583 and Val587 from the C-helix (Fig. 3b). In the inactive state (molecule A), the juxtamembrane helix is partially unwound at its N-terminal end and is oriented 45° away from its active conformation, allowing the C-helix to rotate out of its active state. In the active and inactive conformations, juxtamembrane orientation is not influenced by the crystal packing interactions. In view of these structural observations, we probed the potential role of the juxtamembrane region in the enzyme activity.
Juxtamembrane region and the enzyme activity
We cloned, expressed, and purified the wild-type and L526A mutant forms of Axl intracellular domain (residues 473–894) with co-expression of YopH phosphatase to eliminate in situ phosphorylation. Nevertheless, both the proteins were partially phosphorylated, the wild-type Axl more so than the L526A mutant. The time course of the autophosphorylation of both proteins was followed using mass spectrometry. The unphosphorylated wild-type protein was significantly diminished in the first 5 min of the experiment and completely depleted in an hour, whereas the unphosphorylated mutant protein persisted past the 1st h of the experiment (supplemental Fig. S3).
Next we biochemically characterized preactivated (by autophosphorylation) wild-type and L526A Axl. L526A Axl was ∼2-fold less active than wild-type enzyme by apparent kcat with 5FAM-Lys-Lys-Lys-Lys-Glu-Glu-Ile-Tyr-Phe-Phe-Phe-NH2 (5FAM-FL-Peptide30) substrate by a mobility shift assay without significantly affecting ATP Km (supplemental Fig. S4) but exhibited more than 4-fold higher Km for a peptide substrate (Abltide) by a continuous ATP-regenerating assay (supplemental Fig. S5) in which ATP consumption in the kinase reaction results in NADH oxidation in the presence of the coupled enzyme system.
Phosphorylation sites
A systematic and exhaustive study of Axl phosphorylation is not available; however, the major autophosphorylation sites of Mer have been identified (33). If the findings from the Mer phosphorylation study can be extrapolated to Axl, the corresponding autophosphorylation sites in Axl would be Tyr698, Tyr702, and Tyr703 in the activation loop (Fig. 4). Tyrosine residues at positions 702 and 703 have been shown to be phosphorylated upon GAS6 binding and the consequent activation of the enzyme (34). In our experimental electron density maps, all three Tyr residues are unequivocally unphosphorylated in the active conformation (molecule B). Tyr702 is the structural equivalent of phosphorylated Tyr1235 of active c-MET (31) and occupies a similar electropositive environment, so it is conceivable that the phosphorylation of Tyr702 would stabilize the extended conformation of the activation loop through electrostatic interactions with Arg671 (from the highly conserved catalytically important HRD motif), Lys695, and Arg704 (Fig. 4b). Phosphorylated Tyr698 may play a similar role via its interactions with Lys666, Arg667, and Lys696 (Fig. 4b). Tyr703 is largely solvent-exposed, and it is unclear what role its phosphorylation would play in activation. It is plausible that it destabilizes the inactive conformation of the enzyme. It may also contribute to substrate recognition. Interestingly, mutation of the corresponding Tyr754 to Phe in Mer abolishes the enzyme activity (33).
Figure 4.

Axl phosphorylation sites. a, phosphorylation sites in the context of the overall structure. b, close-up view of the phosphorylation sites on the activation loop.
There are three additional phosphorylation sites reported for the intracellular portion of Axl RTK: Tyr779, Tyr821, and Tyr866 (35). Of these, Tyr779 is present in the protein construct used for structural studies (Fig. 4a). Although close to the surface, it is largely solvent-inaccessible. However, probing the Axl structure using HDX-MS (see below) suggests that this region of protein is amenable to deuterium exchange and hence may be dynamic. Tyr779 phosphorylation reportedly contributes to PI3K recognition by Axl (35).
We also crystallized Mer kinase domain spanning residues 571–864 in complex with compound 1 (Table 1 and supplemental Fig. S1). The Mer co-crystal structure has a single molecule per asymmetric unit. The overall structure resembles the inactive conformation of the Axl kinase domain described above with an r.m.s.d. of 0.8 Å. This is also the conformation observed previously in the Tyro3 kinase domain structure (25). Molecule A (inactive) of Axl superimposes onto Tyro3 with an r.m.s.d. of 0.9 Å.
Ligand recognition
Despite significant conformational differences in the N-lobes of the active and inactive states of Axl, the shape of the ATP-binding site is more or less invariant. As a result, the mode of ligand binding is essentially identical, not only between the two conformational states of Axl (Fig. 5) but also between the Axl and Mer kinase domains (supplemental Fig. S1). Key specific interactions are provided by the backbone peptides from the hinge region of the protein: Pro621, Phe622, and Met623. Additionally, Asp627 serves as an acceptor in its hydrogen bonding interaction with the hydroxyl from the ligand and the backbone peptide donor to the oxygen of the ligand (Fig. 5). The catalytically important Lys567 and Asp690 of the DFG motif show some differences in the two states. However, they do not contribute to any specific protein–ligand interactions.
Figure 5.
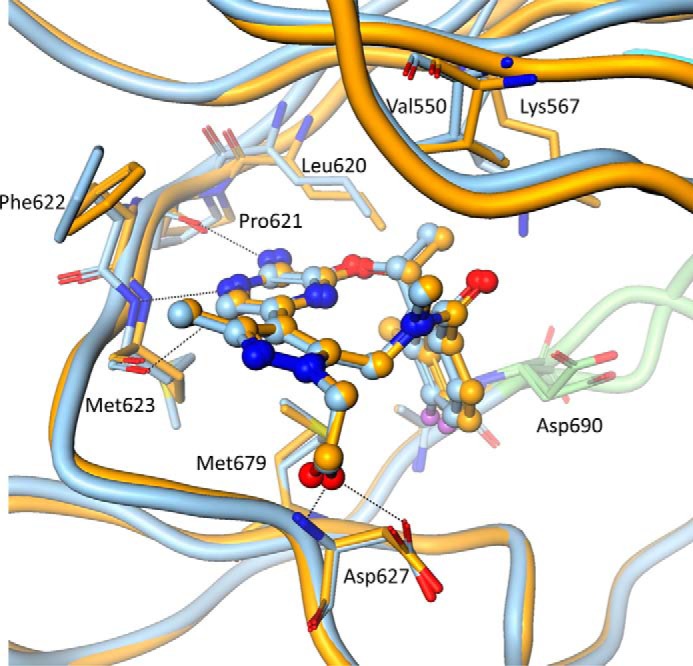
The compound binding mode. Molecules A (orange) and B (blue) are superposed to show that the mode of compound recognition is nearly identical in the inactive and the active states. Dotted lines show specific interactions between the compound and the protein. Some of the side chains in the compound-binding site are highlighted.
Compound 1 is similar to the ALK inhibitor lorlatinib, which is currently under clinical investigation as a targeted therapy for ALK-driven lung cancer patients. Likewise, the ALK–lorlatinib complex closely resembles the active Axl–compound 1 complex reported here (supplemental Fig. S6). Lorlatinib is a potent inhibitor of ALK with a cell IC50 of 1.3 nm (36). However, the L1198F mutation renders ALK resistant to lorlatinib-mediated inhibition. Replacement of Leu with a bulkier residue, Phe, at the hinge location of the kinase domain results in steric clash with the nitrile group of the inhibitor, resulting in loss of potency (37). Similarly, both Axl and Mer have Phe at the equivalent hinge location (622 and 673, respectively), explaining the selectivity of lorlatinib for ALK over Axl and Mer. To further test this hypothesis, we expressed and purified F622L Axl and measured lorlatinib binding to the wild-type and F622L Axl using isothermal titration calorimetry. Consistent with the model, F622L Axl recognizes lorlatinib with over 45-fold stronger binding affinity than the wild-type Axl (Fig. 6).
Figure 6.

Binding of the wild-type and F622L Axl with lorlatinib measured by ITC. The original titration data (top curve) and integrated data (lower curve) in which Lorlatinib was titrated into the wild-type (a) or F622L protein (b) are shown. c, thermodynamic properties of the interaction. Average and standard deviation from three independent measurements are reported. DP, differential power.
HDX-MS
The inability to crystallize apo-Axl along with its lower thermal stability indicated a more structurally dynamic Axl protein. We conducted HDX-MS analysis to profile structural dynamics. To identify regions of Axl flexibility and probe the associated differences between Axl and Mer dynamics, we applied a short-duration (10-s) deuterium incubation profile (Fig. 7 and supplemental Fig. S7). A few regions, including the activation loop (residues 690–712), within Axl generated high deuterium uptake in excess of 60%. Most notably, the Axl N terminus up to C-helix (residue 575) indicated highly flexible portions with extensive deuterium uptake (60 to >90%) by these peptides. These data are consistent with the N-lobe and the activation loop taking on different orientations in the two conformational states of Axl observed in the crystal structure.
Figure 7.

HDX-MS profiles of Axl and Mer. a, deuterium exchange of the N-lobes of Axl and Mer is color-coded by the extent/percentage of peptide deuterium uptake from blue (<10%) to red (>90%). (The profiles for the entire kinase domains are reported in supplemental Fig. S5.) b, perturbation of deuterium uptake upon compound 1 in complex with Axl and Mer relative to the apoprotein. Changes in deuterium exchange are illustrated by blue bars (−10–30%) and purple bars (−30%), indicating measurable suppression in deuterium uptake upon ligand binding.
A comparative HDX examination of the corresponding Mer profile highlighted some of the differences between the two. It should be noted that 69% sequence identity between Axl and Mer kinase domains prevented direct HDX comparisons of equivalent peptides. Regardless, a noteworthy difference in Axl and Mer HDX profiles is evident in the N-lobe where uptake differences were <50% for a number of peptides. Specifically, Met602–Lys607 from two and Val616–Lys619 from three Mer peptides show lower deuterium incorporation than the equivalent region in Axl (Met551–Asn556 and Val564–Lys567). These regions form the core of the N-lobe β-sheet that contains the catalytically important lysine. Given that the N-lobe structures of the kinases are very similar, increases in deuterium exchange at 10 s reflect the greater dynamic nature of this region in Axl relative to Mer. Subsequent HDX-MS experiments on both proteins in the presence of compound 1 clearly show that ligand binding resulted in substantial suppression of deuterium uptake especially in the parts of their N-lobes that are proximal to the ligand-binding sites (Fig. 7b).
Discussion
Structures of TAM RTKs in inactive states closely resemble one another, which is not surprising given that they are closely related to each other. They also bear a distant homology to the macrophage-stimulating receptor RON and hepatocyte growth factor receptor (c-MET) kinases (38), and this is again reflected in the similarity of their structures. The r.m.s.d. between the inactive states (molecule A) of Axl and RON (39) is 0.9 Å. The active conformation of Axl (molecule B) closely resembles (among all kinases in the Protein Data Bank) the dually phosphorylated c-MET kinase structure (31, 40) with an r.m.s.d. of 1.2 Å. Additionally, the juxtamembrane regions of active c-MET and active Axl show a very similar orientation relative to the kinase domain.
In a number of RTK families, the juxtamembrane region plays an important role in either negative regulation of the kinase activity (as in Eph, PDGF receptor, and muscle-specific kinase (MUSK)) (41) or activation by facilitating dimerization as is the case in EGFR (42, 43). To our knowledge, no role in the enzyme activity has been ascribed to the juxtamembrane region of the TAM RTKs. The Axl structure presented here provides the first clue of such a role; hence, we pursued it by biochemically characterizing the intracellular domain (residues 473–894) of wild-type and L526A Axl. The mutation L526A in the juxtamembrane region was designed to impair the interaction between the juxtamembrane region and the C-helix of the protein (Fig. 3b) and probe its impact on the enzyme activity. The autophosphorylation of the mutant enzyme was significantly slower than that of the wild type, which supported the notion that the juxtamembrane region has a role in activation. Once activated, the mutant enzyme showed a 4-fold increased Km value for the substrate peptide relative to the wild-type Axl. Both of these observations are consistent with the possibility that the substrate recognition may be impaired by the L526A mutation. It is not obvious how the juxtamembrane region plays a role in substrate recognition. It is plausible that through its interaction with the C-helix in the active state the juxtamembrane region stabilizes the extended conformation of the activation loop that serves as the substrate recognition site.
Although Mer and Tyro3 kinase domains have been structurally characterized before, previous structures show the inactive conformation of the TAM family kinases. Molecules B and D of the Axl structure reported here provide a model for an active TAM kinase domain. This is the first visualization of the active state of a member of the TAM RTK family, and it provides a model to help design type I kinase inhibitors of TAM family members. Stability and HDX results also suggest that protein dynamics may play an important role in designing selective inhibitors of Axl.
Lower thermal stability of the Axl kinase implicitly suggests that it may be especially dynamic, sampling multiple conformational states. Corresponding HDX data revealed regions of enhanced Axl deuterium exchange reflective of the extended dynamics of the N-lobe of Axl relative to that of Mer. The magnitude of deuterium differentiation of highly dynamic regions is typically most evident at short exchange times. Deuterium exchange profiling of Axl dynamics may be more measurably differentiated at even shorter exchange times than we could achieve (<10 s). Extended Axl dynamics provide the opportunity for a broader spectrum of conformational variability, which is reflected in the two distinct conformations of the kinase present in the asymmetric unit of Axl. Given that these differences in the dynamics are observed around the ATP/inhibitor-binding pocket, it could potentially provide an opportunity to design a specific inhibitor of Axl activity that is selective over Mer.
Axl overexpression and activation are implicated in a variety of human cancers (44). Axl up-regulation confers resistance to targeted therapies in multiple cancers (19, 45–48) and is also implicated in cell motility and invasion (49). Hence, Axl kinase activity remains a target to develop cancer therapeutics (50–53). Given the dynamic nature of the Axl kinase domain, it is conceivable that it samples numerous conformations in the solution state, some of which may either be inaccessible to other TAM members or be higher energy conformers of Mer and Tyro3. Although it is unclear what such a conformation may look like, we speculate that a ligand against such a unique conformation of Axl could display the desired specificity for Axl inhibition and selectivity over Mer and Tyro3. However, challenges are significant in experimentally characterizing transiently populated conformational states by current methods, and this characterization is a necessary prerequisite for structure-based drug design. Molecular dynamics could be a useful tool to computationally identify potentially stable intermediate states. Our results encourage more extensive screening efforts to discover leads targeting unique conformational states of Axl, yielding inhibitor molecules with a desired selectivity profile over other TAM RTKs. Stable complexes of such inhibitors with Axl can then enable structure-based drug design.
Experimental procedures
Axl and Mer kinase domain expression and purification
The cDNAs encoding the various constructs of Axl (intracellular domain residues 473–894, wild-type and F622L kinase domain residues 514–818, and kinase domain residues 505–811 used for the compound 1 Ki determination) and Mer (residues 571–864, I650M) were synthesized and cloned into pKRIC-N5 and pRSFDuet1, respectively. pKRIC-N5 is a modified version of insect cell expression cloning vector pFastbac 1, and pRSFDuet1 was purchased from Novagen. Both constructs contain an N-terminal His tag followed by the tobacco etch virus (TEV) protease cleavage site.
Expression of Axl was done in Sf21 cells with co-expression of YopH to eliminate the in situ phosphorylation of Axl during expression. Cells were harvested 72 h after infection, and the pellet was stored at −80 °C. Expression of Mer was carried out in Escherichia coli BL21(DE3), also expressing PTPN1, in terrific broth medium and induced by isopropyl 1-thio-β-d-galactopyranoside overnight at 15 °C.
To purify Axl, the cell pellet was resuspended in a lysis buffer containing 50 mm Tris-Cl, pH 7.4, 300 mm NaCl, 10% glycerol, 1 mm TCEP, and protease inhibitor tablets (Roche Applied Science). Cells were lysed by stirring the suspension at 4 °C for 45 min, and the clear lysate was obtained by centrifugation. His-tagged protein was purified by batch binding with Invitrogen ProBond resin for 2 h at 4 °C with slow rotation using a Roto-Shaker Genie. His-tagged protein was treated with TEV protease overnight at 4 °C, and the untagged protein was purified by reverse nickel affinity chromatography followed by size exclusion Superdex 75 and anion exchange chromatographies. Pure protein was concentrated to 9.5 mg/ml and stored in small aliquots at −80 °C in the storage buffer (20 mm Hepes, pH 7.5, 50 mm NaCl, 10% glycerol, and 1 mm TCEP).
To purify Mer, the cell pellet was resuspended in a lysis buffer containing 50 mm Tris, pH 8.0, 500 mm NaCl, 5% glycerol, 2 mm TCEP, and protease inhibitor tablets (Roche Applied Science). Cells were lysed by a microfluidizer, and the clear lysate was obtained by centrifugation. His-tagged protein was purified by batch binding with Invitrogen ProBond resin. The protein was then treated with TEV protease and λ-phosphatase overnight at 4 °C, and the untagged protein was purified by reverse nickel affinity chromatography followed by size exclusion Superdex 75 chromatography in a buffer containing 20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 5% glycerol, and 2 mm TCEP. Peak fractions were pooled and concentrated to 28 mg/ml. The protein was stored in small aliquots at −80 °C.
Protein purity was assessed by SDS-PAGE and intact mass spectroscopy. Protein identity and post-translational modifications were confirmed by intact mass spectroscopy.
Biochemical characterization
Time dependence of enzyme autophosphorylation was studied using mass spectrometry as described previously (54) at room temperature in the presence of 2 mm ATP. Enzyme kinetics of wild-type and L526A Axl (residues 473–894), preactivated by autophosphorylation with MgATP, were measured using an ATP-regenerating coupled spectrophotometric enzyme assay with kinase-catalyzed production of ADP from ATP that accompanies phosphoryl transfer to the Abltide peptide substrate (Glu-Ala-Ile-Tyr-Ala-Ala-Pro-Phe-Ala-Lys-Lys-Lys) derived from the C terminus of Abl kinase. Initial reaction velocities of kinase reactions were determined by following the decrease in absorbance at 340 nm (ϵ = 6.22 cm−1 mm−1) coupled with phosphorylation of Abltide peptide substrate using a Molecular Devices (Sunnyvale, CA) Spectromax Plus plate reader. Reactions contained 26 nm wild-type Axl (adjusted for active site content of 26% as determined by titration with potent inhibitors) or 21 nm L526A variant (active site content, 10%), 0.0625–8 mm Abltide, 1 mm ATP, 20 mm MgCl2, 0.33 mm NADH, 1 mm phosphoenolpyruvate, 20 units/ml pyruvate kinase, 30 units/ml lactate dehydrogenase, 1 mm sodium orthovanadate, and 1 mm DTT and were conducted at 37 °C. These Axl constructs were also characterized kinetically by the mobility shift assay as described below using 1.3 nm wild type and 2.6 nm L526A variant (kcat = 0.63 and 0.27 s−1, respectively, using 3 μm 5FAM-FL-Peptide30). Enzymes were first optimally preactivated by incubating 5 μm Axl, 2 mm ATP, 20 mm MgCl2, Phosphatase Inhibitor Mixture Set II from EMD Millipore (1:100), and 1 mm DTT in 100 mm Hepes buffer, pH 7.3, for 5 min (wild type) or 2 h (L526A mutant) at room temperature. Similar phosphorylation levels of both protein constructs were verified by intact protein mass quadrupole TOF mass spectrometry. All enzyme reactions were conducted in duplicate. Initial reaction velocities were fit to a Michaelis-Menten equation with GraphPad Prism (GraphPad Software, San Diego, CA) to derive kcat and Km values.
Ki determination
Compound 1-mediated inhibition of Axl activity was tested with the Caliper LabChip EZ Reader II assay (Caliper Life Science, Hopkinton, MA), a mobility shift assay that combines the basic principles of capillary electrophoresis in a microfluidic environment. Activated Axl was found to be 100% catalytically competent by active-site titration using a tight-binding inhibitor. The protein was highly active (kcat = 1.17 ± 0.01 s−1 and Km,ATP = 70.4 ± 1.8 μm using 3 μm 5FAM-FL-Peptide30). The kinase-catalyzed production of ADP from ATP that accompanies phosphoryl transfer to 5FAM-FL-Peptide30 was monitored.
Axl reaction solutions (50 μl) contained 0.5 nm Axl kinase domain, 2% DMSO (±inhibitor), 10 mm MgCl2, 0.01% Tween 20, 1 mm DTT, 3 μm 5FAM-FL-Peptide30, and 120 μm ATP in 100 mm HEPES buffer at pH 7.3. The reactions were initiated with 120 μm ATP after 15-min preincubation of Axl with the inhibitor at 25 °C. The reactions were stopped after 30 min at 25 °C by the addition of 50 μl of 200 mm EDTA.
Unactivated Mer (residues 571–864) was found to be 56% catalytically competent by active-site titration using a tight-binding inhibitor. It was catalytically active (kcat = 0.014 ± 0.000 s−1 and Km,ATP = 32.9 ± 2.5 μm using 3 μm 5FAM-FL-Peptide30). Mer reaction solutions (50 μl) contained 30 nm Mer, 2% DMSO (±inhibitor), 10 mm MgCl2, 0.01% Tween 20, 1 mm DTT, 3 μm 5FAM-FL-Peptide30, and 2000 μm ATP in 100 mm HEPES buffer at pH 7.3. The reactions were initiated with 3 μm 5FAM-FL-Peptide30 after 35-min preincubation of inhibitor, Mer, and ATP at room temperature. The reactions were stopped after 60 min at 25 °C by the addition of 50 μl of 80 mm EDTA.
The reaction mixture was applied to a Caliper LabChip EZ Reader II instrument, and the product/substrate peptide peaks were separated. The kinase reaction was quantitated by the product ratio calculated from peak heights of product (P) and substrate (S) peptides (P/(P + S)). Fractional velocity data were fit to the equation for tight-binding competitive inhibition (Morrison equation) to determine Ki values (55, 56). The R2 values of the fit were greater than 0.99. Two independent experiments were performed for each Ki determination.
Protein thermal stability measurements
Unphosphorylated protein (1 mg/ml) was incubated with compound 1 at a 1:3 molar ratio on ice for 60 min. For apoprotein, DMSO vehicle was used. After incubation, protein was diluted to 0.2 mg/ml in the storage buffer containing 20 mm Hepes, pH 7.5, 50 mm NaCl, 10% glycerol, and 1 mm TCEP. 20 μl of protein and 1 μl of 200× SYPRO Orange were added to a 384-well measuring plate and overlaid with mineral oil. Fluorescence measurements were done using FluoDia T70.
Isothermal titration calorimetry (ITC)
The experiments were performed using a MicroCal PEAQ-ITC automated system (Malvern Instruments) at 25 °C in 20 mm Hepes, pH 7.5, 50 mm NaCl, 10% glycerol, 1 mm TCEP, and 1% DMSO (ITC buffer). A 10 μm concentration of either Axl or F622L Axl kinase domains (residues 514–818) in the sample cell was titrated with 200 μm lorlatinib from the injection syringe. Lorlatinib was added in 12 stepwise injections of 3 μl with 150-s interval between injections. A reference power of 5 cal/s was used. In control experiments, the ITC buffer was titrated with 200 μm lorlatinib from the injection syringe as described above. Data were analyzed by a one set of sites binding model to obtain binding affinity (Kd), stoichiometry (n), and binding enthalpy (ΔH) using MicroCal PEAQ-ITC Analysis Software (Malvern Instruments). The analysis software also calculated Gibbs free energy (ΔG) and binding entropy (−TΔS).
Co-crystallization and structure determination
Axl (residues 514–818) was concentrated to 9 mg/ml for crystallization. Diffraction quality crystals were obtained at 21 °C in 5–7 days from hanging drops containing 1.2 μl of the protein–ligand complex (1:3 ratio of protein:ligand) and 1.2 μl of the reservoir solution (0.1 m Tris, pH 8.5, 0.2 m MgCl2, 30% (w/v) PEG-4000, and 1–2% (v/v) 1-butanol). Crystals belong to the space group P21 with cell dimensions a = 81.9 Å, b = 101.1 Å, c = 82.2 Å, β = 93.9°. Crystals were mounted into Hampton loops and frozen via stream freezing in the mother liquor with 15% glycerol. These crystals diffracted to 2.8 Å at the Advanced Photon Source (Industrial Macromolecular Crystallography Association beamline 17-ID). The Axl complex structure was determined by molecular replacement in Phaser (57) using the inhibitor-bound Mer structure as the starting model. The structure was iteratively refined using CNX and Coot (58). The final round of refinement was carried out using BUSTER (59).
Mer (residues 571–864, I650M) protein at a concentration of 25–30 mg/ml was introduced to 1 mm (final concentration) compound 1 and incubated at 4 °C for a minimum of 4 h. The complex was then filtered with a low-protein-binding 0.45-μm membrane to remove particulates and set up for crystallization using a Phoenix (Art Robbins) robot. Sitting drops at a 1:1 ratio (protein:well solution) with well solution containing 0.2 m magnesium chloride, 28–35% PEG-600, and 0.1 m Tris, pH 8.7–9.0, were incubated at 13 °C for ∼5–7 days before crystals resembling large, rectangular blocks grew. Mer crystallized in space group P21 with cell dimensions a = 51.7 Å, b = 92.3 Å, c = 69.3 Å, β = 100.8°. The well solution acted as a cryoprotectant so crystals were removed directly from drops and quickly dipped into liquid nitrogen. The structure was determined using the phosphoaminophosphonic acid-adenylate ester (ANP)-bound structure of Mer (Protein Data Bank code 2P0C) as the starting model. Rigid body refinement followed by iterative refinement cycles in CNX and Coot (58) provided the final model.
HDX-MS
Kinase domain constructs of both Axl (residues 514–818) and Mer (residues 571–864, I650M) were diluted to a working concentration of 0.5 mg/ml (∼15 μm) with dilution buffer (20 mm Tris, 50 mm NaCl, and 2 mm TCEP, pH 7.2). HDX exchange was performed on a Fusion Lumos Orbitrap (Thermo Fisher Scientific) equipped with a Pal HDX3 autosampler (Leap Technologies) and a combination of Vanquish capillary pumps (Dionex). The autosampler sample plate was held at 4 °C where exchange was initiated upon addition of 20 μl of D2O solution (20 mm Tris and 50 mm NaCl) to 4 μl of protein sample. Deuterium exchange was performed for 10 s, and then 20 μl of sample was transferred to the chilled quench plate with a prechilled syringe and added to 20 μl of 4 °C cold quench buffer (3.2 m guanidine hydrochloride and 0.8% formic acid) to a final pH 2.5. Samples were then injected into a chilled box (1 °C) that housed the sample loop, protease column, and trap/analytical columns. Samples were first run across a 2.1 × 30-mm immobilized protease type XIII/pepsin column (w/w, 1:1; NovaBioAssay) at 200 μl/min using a 0.1% formic acid mobile phase. The digested protein was subsequently trapped and desalted for 2 min on an Acquity UPLC BEH 300 C18 (2.1 × 5-mm) trap column (Waters). Peptides were eluted from the trap to a Kinetex 1.3-μm C18 (2.1 × 30 mm; Phenomenex) analytical column using a 5.5-min gradient of organic solvent (8–37% acetonitrile and 0.1% formic acid) running at 150 μl/min. When initially collecting non-deuterated MS2 spectra for identifying peptides, the electrospray source was set at 300 °C, and MS2 was collected by both high-energy collision dissociation and electron-transfer/high-energy collision dissociation. All samples were collected in triplicate with blanks between each sample to minimize potential sample carryover. Peptide lists were generated using Proteome Discoverer (version 2.1, Thermo Fisher Scientific). Low-confidence peptides were removed from the peptide list (mass tolerance <5 ppm), and peptide pools were constructed for subsequent deuterium analysis. During collection of deuterium exchange, the electrospray source was lowered to 200 °C and transfer rf was set at 30 V to minimize potential deuterium back-exchange. For deuterium incorporation analysis, MS spectra only were acquired, and samples were processed in HD Examiner 2.0 (Sierra Analytics) for deuterium incorporation. Low-confidence peptides and peptides less than six residues in length were removed. Final peptide pools for Axl comprised 275 peptides (93.5% sequence coverage), and Mer provided 326 peptides (98.3% sequence coverage). Results were from an average of triplicate runs and are presented as heat maps generated in HD Examiner.
Author contributions
K. S. G. conceived and coordinated the study, solved the crystal structures, interpreted data, and wrote the paper. N. G. conducted the stability and ITC studies and crystallized Axl. B. B. conducted HDX-MS studies. J. F. cloned and purified the proteins and conducted stability studies. R. A. F. crystallized Mer. S. T., M. X., and B. W. M. contributed to and interpreted kinetic analyses of Axl and Mer as well as ligand binding to these proteins. T. W. J. identified and provided the Axl inhibitor. A. S. coordinated the study and contributed to the manuscript. All authors reviewed the results, contributed to the manuscript, and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We gratefully acknowledge Nicole Grable for protein expression, Oleg Brodsky for assistance in interpretation and presentation of FluoDia data, and Advanced Photon Source Industrial Macromolecular Crystallography Association Collaborative Access Team staff for help in data collection.
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S7.
The atomic coordinates and structure factors (codes 5U6B and 5U6C) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TAM
- Tyro3, Axl, and Mer
- RTK
- receptor tyrosine kinase
- GAS6
- growth arrest-specific protein 6
- EGFR
- EGF receptor
- HDX
- hydrogen-deuterium exchange
- N-lobe
- N-terminal lobe
- C-lobe
- C-terminal lobe
- r.m.s.d.
- root mean square deviation
- ALK
- anaplastic lymphoma kinase
- TEV
- tobacco etch virus
- TCEP
- tris(2-carboxyethyl)phosphine
- ITC
- isothermal titration calorimetry.
References
- 1. Lemke G. (2013) Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 5, a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Prasad D., Rothlin C. V., Burrola P., Burstyn-Cohen T., Lu Q., Garcia de Frutos P., and Lemke G. (2006) TAM receptor function in the retinal pigment epithelium. Mol. Cell. Neurosci. 33, 96–108 [DOI] [PubMed] [Google Scholar]
- 3. Lemke G., and Rothlin C. V. (2008) Immunobiology of the TAM receptors. Nat. Rev. Immunol. 8, 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Varnum B. C., Young C., Elliott G., Garcia A., Bartley T. D., Fridell Y. W., Hunt R. W., Trail G., Clogston C., Toso R. J., Yanagihara D., Bennett L., Sylber M., Merewether L. A., Tseng A., et al. (1995) Axl receptor tyrosine kinase stimulated by the vitamin K-dependent protein encoded by growth-arrest-specific gene 6. Nature 373, 623–626 [DOI] [PubMed] [Google Scholar]
- 5. Stitt T. N., Conn G., Gore M., Lai C., Bruno J., Radziejewski C., Mattsson K., Fisher J., Gies D. R., Jones P. F., Lai C., Long G. L., Lemke G., Mattsson K., Masiakowski P., et al. (1995) The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 80, 661–670 [DOI] [PubMed] [Google Scholar]
- 6. Nagata K., Ohashi K., Nakano T., Arita H., Zong C., Hanafusa H., and Mizuno K. (1996) Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 271, 30022–30027 [DOI] [PubMed] [Google Scholar]
- 7. Wu Y., Tibrewal N., and Birge R. B. (2006) Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol. 16, 189–197 [DOI] [PubMed] [Google Scholar]
- 8. Scott R. S., McMahon E. J., Pop S. M., Reap E. A., Caricchio R., Cohen P. L., Earp H. S., and Matsushima G. K. (2001) Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211 [DOI] [PubMed] [Google Scholar]
- 9. Lew E. D., Oh J., Burrola P. G., Lax I., Zagórska A., Través P. G., Schlessinger J., and Lemke G. (2014) Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 3, e03385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu Q., and Lemke G. (2001) Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 293, 306–311 [DOI] [PubMed] [Google Scholar]
- 11. Lu Q., Gore M., Zhang Q., Camenisch T., Boast S., Casagranda F., Lai C., Skinner M. K., Klein R., Matsushima G. K., Earp H. S., Goff S. P., and Lemke G. (1999) Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 398, 723–728 [DOI] [PubMed] [Google Scholar]
- 12. Gal A., Li Y., Thompson D. A., Weir J., Orth U., Jacobson S. G., Apfelstedt-Sylla E., and Vollrath D. (2000) Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat. Genet. 26, 270–271 [DOI] [PubMed] [Google Scholar]
- 13. Ostergaard E., Duno M., Batbayli M., Vilhelmsen K., and Rosenberg T. (2011) A novel MERTK deletion is a common founder mutation in the Faroe Islands and is responsible for a high proportion of retinitis pigmentosa cases. Mol. Vis. 17, 1485–1492 [PMC free article] [PubMed] [Google Scholar]
- 14. Graham D. K., DeRyckere D., Davies K. D., and Earp H. S. (2014) The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 14, 769–785 [DOI] [PubMed] [Google Scholar]
- 15. Linger R. M., Keating A. K., Earp H. S., and Graham D. K. (2008) TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 100, 35–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gjerdrum C., Tiron C., Høiby T., Stefansson I., Haugen H., Sandal T., Collett K., Li S., McCormack E., Gjertsen B. T., Micklem D. R., Akslen L. A., Glackin C., and Lorens J. B. (2010) Axl is an essential epithelial-to-mesenchymal transition-induced regulator of breast cancer metastasis and patient survival. Proc. Natl. Acad. Sci. U.S.A. 107, 1124–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koorstra J. B., Karikari C. A., Feldmann G., Bisht S., Rojas P. L., Offerhaus G. J., Alvarez H., and Maitra A. (2009) The Axl receptor tyrosine kinase confers an adverse prognostic influence in pancreatic cancer and represents a new therapeutic target. Cancer Biol. Ther. 8, 618–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hutterer M., Knyazev P., Abate A., Reschke M., Maier H., Stefanova N., Knyazeva T., Barbieri V., Reindl M., Muigg A., Kostron H., Stockhammer G., and Ullrich A. (2008) Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 14, 130–138 [DOI] [PubMed] [Google Scholar]
- 19. Zhang Z., Lee J. C., Lin L., Olivas V., Au V., LaFramboise T., Abdel-Rahman M., Wang X., Levine A. D., Rho J. K., Choi Y. J., Choi C. M., Kim S. W., Jang S. J., Park Y. S., et al. (2012) Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 44, 852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Verma A., Warner S. L., Vankayalapati H., Bearss D. J., and Sharma S. (2011) Targeting Axl and Mer kinases in cancer. Mol. Cancer Ther. 10, 1763–1773 [DOI] [PubMed] [Google Scholar]
- 21. Baladi T., Abet V., and Piguel S. (2015) State-of-the-art of small molecule inhibitors of the TAM family: the point of view of the chemist. Eur. J. Med. Chem. 105, 220–237 [DOI] [PubMed] [Google Scholar]
- 22. Sheridan C. (2013) First Axl inhibitor enters clinical trials. Nat. Biotechnol. 31, 775–776 [DOI] [PubMed] [Google Scholar]
- 23. Heiring C., Dahlbäck B., and Muller Y. A. (2004) Ligand recognition and homophilic interactions in Tyro3: structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 279, 6952–6958 [DOI] [PubMed] [Google Scholar]
- 24. Sasaki T., Knyazev P. G., Clout N. J., Cheburkin Y., Göhring W., Ullrich A., Timpl R., and Hohenester E. (2006) Structural basis for Gas6-Axl signalling. EMBO J. 25, 80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Powell N. A., Kohrt J. T., Filipski K. J., Kaufman M., Sheehan D., Edmunds J. E., Delaney A., Wang Y., Bourbonais F., Lee D. Y., Schwende F., Sun F., McConnell P., Catana C., Chen H., et al. (2012) Novel and selective spiroindoline-based inhibitors of Sky kinase. Bioorg. Med. Chem. Lett. 22, 190–193 [DOI] [PubMed] [Google Scholar]
- 26. Huang X., Finerty P. Jr., Walker J. R., Butler-Cole C., Vedadi M., Schapira M., Parker S. A., Turk B. E., Thompson D. A., and Dhe-Paganon S. (2009) Structural insights into the inhibited states of the Mer receptor tyrosine kinase. J. Struct. Biol. 165, 88–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu J., Yang C., Simpson C., Deryckere D., Van Deusen A., Miley M. J., Kireev D., Norris-Drouin J., Sather S., Hunter D., Korboukh V. K., Patel H. S., Janzen W. P., Machius M., Johnson G. L., et al. (2012) Discovery of novel small molecule Mer kinase inhibitors for the treatment of pediatric acute lymphoblastic leukemia. ACS Med. Chem. Lett. 3, 129–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bailey S., Burke B., Collins M., Cui J., Deal J., Hoffmann R., Huang Q., Johnson T. W., Kania R., Kath J., Le P., McTigue M., Palmer C. L., Richardson P., and Sach N. W. (September 12, 2013) Macrocyclic Derivatives for the Treatment of Proliferative Diseases. WO Patent 2013132376 A1
- 29. Huse M., and Kuriyan J. (2002) The conformational plasticity of protein kinases. Cell 109, 275–282 [DOI] [PubMed] [Google Scholar]
- 30. Nolen B., Taylor S., and Ghosh G. (2004) Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 31. Rickert K. W., Patel S. B., Allison T. J., Byrne N. J., Darke P. L., Ford R. E., Guerin D. J., Hall D. L., Kornienko M., Lu J., Munshi S. K., Reid J. C., Shipman J. M., Stanton E. F., Wilson K. J., et al. (2011) Structural basis for selective small molecule kinase inhibition of activated c-Met. J. Biol. Chem. 286, 11218–11225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wiesner S., Wybenga-Groot L. E., Warner N., Lin H., Pawson T., Forman-Kay J. D., and Sicheri F. (2006) A change in conformational dynamics underlies the activation of Eph receptor tyrosine kinases. EMBO J. 25, 4686–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ling L., Templeton D., and Kung H. J. (1996) Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 271, 18355–18362 [DOI] [PubMed] [Google Scholar]
- 34. Pao-Chun L., Chan P. M., Chan W., and Manser E. (2009) Cytoplasmic ACK1 interaction with multiple receptor tyrosine kinases is mediated by Grb2: an analysis of ACK1 effects on Axl signaling. J. Biol. Chem. 284, 34954–34963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Braunger J., Schleithoff L., Schulz A. S., Kessler H., Lammers R., Ullrich A., Bartram C. R., and Janssen J. W. (1997) Intracellular signaling of the Ufo/Axl receptor tyrosine kinase is mediated mainly by a multi-substrate docking-site. Oncogene 14, 2619–2631 [DOI] [PubMed] [Google Scholar]
- 36. Johnson T. W., Richardson P. F., Bailey S., Brooun A., Burke B. J., Collins M. R., Cui J. J., Deal J. G., Deng Y. L., Dinh D., Engstrom L. D., He M., Hoffman J., Hoffman R. L., Huang Q., et al. (2014) Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(metheno)pyrazolo[4,3-h][2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 57, 4720–4744 [DOI] [PubMed] [Google Scholar]
- 37. Shaw A. T., Friboulet L., Leshchiner I., Gainor J. F., Bergqvist S., Brooun A., Burke B. J., Deng Y. L., Liu W., Dardaei L., Frias R. L., Schultz K. R., Logan J., James L. P., Smeal T., et al. (2016) Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N. Engl. J. Med. 374, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Manning G., Whyte D. B., Martinez R., Hunter T., and Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 39. Wang J., Steinbacher S., Augustin M., Schreiner P., Epstein D., Mulvihill M. J., and Crew A. P. (2010) The crystal structure of a constitutively active mutant RON kinase suggests an intramolecular autophosphorylation hypothesis. Biochemistry 49, 7972–7974 [DOI] [PubMed] [Google Scholar]
- 40. Northrup A. B., Katcher M. H., Altman M. D., Chenard M., Daniels M. H., Deshmukh S. V., Falcone D., Guerin D. J., Hatch H., Li C., Lu W., Lutterbach B., Allison T. J., Patel S. B., Reilly J. F., et al. (2013) Discovery of 1-[3-(1-methyl-1H-pyrazol-4-yl)-5-oxo-5H-benzo[4,5]cyclohepta[1,2-b]pyridin-7-yl]-N-(pyridin-2-ylmethyl)methanesulfonamide (MK-8033): A Specific c-Met/Ron dual kinase inhibitor with preferential affinity for the activated state of c-Met. J. Med. Chem. 56, 2294–2310 [DOI] [PubMed] [Google Scholar]
- 41. Hubbard S. R. (2004) Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 5, 464–471 [DOI] [PubMed] [Google Scholar]
- 42. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., and Kuriyan J. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Red Brewer M., Choi S. H., Alvarado D., Moravcevic K., Pozzi A., Lemmon M. A., and Carpenter G. (2009) The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell 34, 641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wu X., Liu X., Koul S., Lee C. Y., Zhang Z., and Halmos B. (2014) AXL kinase as a novel target for cancer therapy. Oncotarget 5, 9546–9563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yao T. W., Zhang J., Prados M., Weiss W. A., James C. D., and Nicolaides T. (2017) Acquired resistance to BRAF inhibition in BRAFV600E mutant gliomas. Oncotarget 8, 583–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mahadevan D., Cooke L., Riley C., Swart R., Simons B., Della Croce K., Wisner L., Iorio M., Shakalya K., Garewal H., Nagle R., and Bearss D. (2007) A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 26, 3909–3919 [DOI] [PubMed] [Google Scholar]
- 47. Liu L., Greger J., Shi H., Liu Y., Greshock J., Annan R., Halsey W., Sathe G. M., Martin A. M., and Gilmer T. M. (2009) Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 69, 6871–6878 [DOI] [PubMed] [Google Scholar]
- 48. Hong C. C., Lay J. D., Huang J. S., Cheng A. L., Tang J. L., Lin M. T., Lai G. M., and Chuang S. E. (2008) Receptor tyrosine kinase AXL is induced by chemotherapy drugs and overexpression of AXL confers drug resistance in acute myeloid leukemia. Cancer Lett. 268, 314–324 [DOI] [PubMed] [Google Scholar]
- 49. Kimani S. G., Kumar S., Davra V., Chang Y. J., Kasikara C., Geng K., Tsou W. I., Wang S., Hoque M., Boháč A., Lewis-Antes A., De Lorenzo M. S., Kotenko S. V., and Birge R. B. (2016) Normalization of TAM post-receptor signaling reveals a cell invasive signature for Axl tyrosine kinase. Cell Commun. Signal. 14, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tan L., Zhang Z., Gao D., Luo J., Tu Z. C., Li Z., Peng L., Ren X., and Ding K. (2016) 4-Oxo-1,4-dihydroquinoline-3-carboxamide derivatives as new Axl kinase inhibitors. J. Med. Chem. 59, 6807–6825 [DOI] [PubMed] [Google Scholar]
- 51. Mollard A., Warner S. L., Call L. T., Wade M. L., Bearss J. J., Verma A., Sharma S., Vankayalapati H., and Bearss D. J. (2011) Design, synthesis and biological evaluation of a series of novel Axl kinase inhibitors. ACS Med. Chem. Lett. 2, 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rho J. K., Choi Y. J., Kim S. Y., Kim T. W., Choi E. K., Yoon S. J., Park B. M., Park E., Bae J. H., Choi C. M., and Lee J. C. (2014) MET and AXL inhibitor NPS-1034 exerts efficacy against lung cancer cells resistant to EGFR kinase inhibitors because of MET or AXL activation. Cancer Res. 74, 253–262 [DOI] [PubMed] [Google Scholar]
- 53. Feneyrolles C., Spenlinhauer A., Guiet L., Fauvel B., Daydé-Cazals B., Warnault P., Chevé G., and Yasri A. (2014) Axl kinase as a key target for oncology: focus on small molecule inhibitors. Mol. Cancer Ther. 13, 2141–2148 [DOI] [PubMed] [Google Scholar]
- 54. Timofeevski S. L., McTigue M. A., Ryan K., Cui J., Zou H. Y., Zhu J. X., Chau F., Alton G., Karlicek S., Christensen J. G., and Murray B. W. (2009) Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors. Biochemistry 48, 5339–5349 [DOI] [PubMed] [Google Scholar]
- 55. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 56. Murphy D. J. (2004) Determination of accurate KI values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design. Anal. Biochem. 327, 61–67 [DOI] [PubMed] [Google Scholar]
- 57. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O., Vonrhein C., and Womack T. (2016) BUSTER, Global Phasing Ltd., Cambridge, UK [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.