

Abstract
Iron-regulatory protein 1 (IRP1) belongs to a family of RNA-binding proteins that modulate metazoan iron metabolism. Multiple mechanisms are employed to control the action of IRP1 in dictating changes in the uptake and metabolic fate of iron. Inactivation of IRP1 RNA binding by iron primarily involves insertion of a [4Fe-4S] cluster by the cytosolic iron–sulfur cluster assembly (CIA) system, converting it into cytosolic aconitase (c-acon), but can also involve iron-mediated degradation of IRP1 by the E3 ligase FBXL5 that also targets IRP2. How CIA and FBXL5 collaborate to maintain cellular iron homeostasis through IRP1 and other pathways is poorly understood. Because impaired Fe-S cluster biogenesis associates with human disease, we determined the importance of FBXL5 for regulating IRP1 when CIA is impaired. Suppression of FBXL5 expression coupled with induction of an IRP1 mutant (IRP13C>3S) that cannot insert the Fe-S cluster, or along with knockdown of the CIA factors NUBP2 or FAM96A, reduced cell viability. Iron supplementation reversed this growth defect and was associated with FBXL5-dependent polyubiquitination of IRP1. Phosphorylation of IRP1 at Ser-138 increased when CIA was inhibited and was required for iron rescue. Impaired CIA activity, as noted by reduced c-acon activity, was associated with enhanced FBXL5 expression and a concomitant reduction in IRP1 and IRP2 protein level and RNA-binding activity. Conversely, expression of either IRP induced FBXL5 protein level, demonstrating a negative feedback loop limiting excessive accumulation of iron-response element RNA-binding activity, whose disruption reduces cell growth. We conclude that a regulatory circuit involving FBXL5 and CIA acts through both IRPs to control iron metabolism and promote optimal cell growth.
Keywords: iron metabolism, iron response element (IRE), iron–sulfur protein, protein degradation, RNA-binding protein, cytosolic iron–sulfur cluster assembly, FBXL5, IRP1, IRP2, iron-regulatory protein 1
Introduction
Iron-regulatory proteins (IRPs)2 are central regulators of cellular iron metabolism in vertebrates (1, 2). IRP1 and IRP2 are iron-regulated RNA-binding proteins that control the fate of mRNAs encoding critical modulators of iron metabolism (1, 2). IRP1 RNA binding is primarily controlled through insertion or loss of the Fe-S cluster, the so-called Fe-S switch mechanism. Insertion of a [4Fe-4S] cluster into the IRP1 apoprotein converts it to the cytosolic isoform of aconitase (c-acon) and alters iron metabolism by eliminating IRP1 RNA-binding activity. In some cell types, c-acon is present in vast excess compared with the RNA-binding form (3, 4). The fact that only a small portion of c-acon is recruited to IRP1 even in severe iron deficiency (3) suggests that unregulated conversion of this large pool of c-acon into the RNA-binding form would have negative consequences unless a compensatory mechanism limits IRP1 activation. Interestingly, IRP1 can be degraded when the Fe-S switch is inhibited, but the mechanistic links underlying this have not been fully explored (5–11). These studies suggest that the level to which IRP1 RNA-binding activity is increased in response to inhibition of Fe-S cluster biogenesis depends on the extent to which the apoprotein is degraded. The fact that the E3 ligase, FBXL5, which controls degradation of IRP2, also targets IRP1 suggests a role for this proteolytic mechanism in managing IRP1 accumulation in response to CIA inhibition (12, 13). Furthermore, the unanticipated finding that CIA inhibition alters IRP2 protein level suggests the existence of a regulatory circuit between Fe-S cluster biogenesis activity, FBXL5 action, and the steady-state level of each IRP (10). Taken together, CIA and FBXL5 appear to collaborate with IRP in sensing cellular iron status and needs to ensure that iron levels are optimal for cell viability.
Two dedicated systems exist for Fe-S cluster biogenesis (14). The mitochondrial iron–sulfur cluster (ISC) assembly system not only assembles Fe-S clusters for mitochondrial proteins, but its activity is required for the cytosolic iron–sulfur cluster assembly (CIA) system (15). The CIA system is essential for maturation of Fe-S proteins in the cytosol and nucleus (14, 16). Disruption of either the ISC or CIA system substantially impairs cell function with potentially lethal consequences (17, 18). In humans, highly debilitating diseases, including some anemias, ataxia, and encephalopathies, are linked to impaired Fe-S cluster biogenesis (17, 18). Whereas many recent studies have identified the major components of Fe-S biogenesis systems and the impact of inborn metabolic errors in their genes on cell function, much remains to be defined concerning the interrelationships between this key cellular process and the control of the metabolism of its central substrate, iron.
Recent studies have shed much light on the pathway of cytosolic Fe-S cluster biogenesis (14, 16). The heterotetramer composed of two copies of NUBP1 (also known as NBP35) and NUBP2 (also known as CFD1) serves as a scaffold for formation of transient Fe-S clusters that are transferred to IOP1, a key factor linking early and late steps of cytosolic Fe-S biogenesis. Subsequently, the CIA pathway divides into two branches with a complex containing FAM96B (also known as CIA2B) targeting general Fe-S cytosolic or nuclear apoproteins, whereas for the second branch, a complex including FAM96A (also known as CIA2A) targets IRP1 for cluster insertion. Knockdown of FAM96A causes a loss of IRP1 RNA-binding activity and protein level and an unexpected similar response of IRP2, whereas knockdown of FAM96B unexpectedly increased IRP2 without altering IRP1 (10). The mechanism(s) responsible for these unanticipated changes in IRP protein level and RNA-binding activity are not understood but further suggest a functional link between CIA and FBXL5.
In this study, the response of cells to CIA system inhibition was investigated, including asking what role FBXL5 has in limiting potentially deleterious consequences of overaccumulation of IRP1. Knockdown of FBXL5 itself failed to affect cell viability, but cell growth was strongly inhibited when this was combined with expression of an IRP1 mutant with constitutive RNA binding or if CIA activity was impaired by suppression of either NUBP2 or FAM96A. In response to CIA inhibition, IRP1 and IRP2 protein level and total IRP RNA-binding activity was reduced. Ser-138 phosphorylation of IRP1 increased when CIA was inhibited, and this phosphorylation event was required for complete iron rescue of cell growth. Interestingly, CIA inhibition or induction of either IRP1 or IRP2 was associated with increased FBXL5 protein level, demonstrating the existence of a feedback loop limiting the overaccumulation of either IRP. We conclude that an interactive regulatory network between the CIA system, both IRPs, and FBXL5 is critical for the appropriate management of the uptake and metabolic fate of iron in cell proliferation.
Results
A synthetic growth defect in human embryonic kidney (HEK) 293 cells is induced by uncoupling IRP1 from the Fe-S switch along with FBXL5 knockdown
Two approaches were used to determine the regulatory interrelationships between the Fe-S cluster biogenesis and FBXL5 mechanisms in the control of cellular iron metabolism by IRP1. First, two CIA factors, one acting early (NUBP2) and one acting late (FAM96A) in the CIA pathway, were knocked down with or without combined knockdown of FBXL5. NUBP2 is one of two scaffold proteins functioning as the initial site of formation of cytosolic Fe-S clusters before they are ultimately transferred to apo-Fe-S proteins functioning in the cytosol or nucleus (14, 16). FAM96A is downstream of NUBP2, inactivates apo-IRP1 by promoting cluster insertion, and has a stabilizing effect on IRP2 (10). Second, we determined the impact of FBXL5 knockdown in cells expressing either wild-type IRP1 or an Fe-S cluster mutant of IRP1 (IRP13C>3S) that is constitutively present in the RNA-binding form.
Using previously validated siRNA protocols (10), we determined the response of HEK 293 cells to the individual or combined knockdown of a CIA factor and FBXL5. Individual knockdown of NUBP2, FAM96A, and FBXL5 failed to affect cell number (Fig. 1A, lanes 2–4). In contrast, the combined knockdown of NUBP2 along with FBXL5 significantly impaired cell growth (Fig. 1A, lane 5). The same response was observed when FBXL5 and the CIA specificity factor for IRP1, FAM96A, were simultaneously knocked down (Fig. 1A, lane 6). A potential explanation for the reduction in cell growth in response to combined knockdown of CIA factors and FBXL5 could be iron toxicity caused by an excessive accumulation of IRE RNA-binding activity, presumably resulting in increased iron uptake (TfR) and reduced storage (ferritin). However, in contrast to what was expected, iron treatment blocked the decrease in cell viability in response to impaired CIA and FBXL5 function and was associated with an increase in FBXL5 protein level (Fig. 1B; see Fig. 7C for FBXL5 data). We conclude that FBXL5 protects cells from the negative consequences of reduced CIA activity.
Figure 1.

Synthetic growth defect in response to accumulation of the IRP1 apoprotein coupled with impaired FBXL5 expression. Cell viability was determined by trypan blue exclusion (A–F). Western blots are as described below. A and B, wild-type HEK 293 cells were transfected using either NT siRNA or siRNA targeting NuBP2 or Fam96a on days 0, 3, and 6 using an established protocol (11). For some experiments FBXL5 siRNA was then transfected into cells on day 6. Cells were then harvested, counted, and lysed on day 9. Western blots were performed for NuBP2 and tubulin. C–F, HEK 293 cells were transfected with NT or FBXL5-targeting siRNA for 48 h and then were treated without or with tet to induce IRP1WT or IRP13C>3S. G, IRP2 protein level was determined in HEK cells in the absence and presence of IRP13C>3S, without and with FBXL5 knockdown, by immunoblotting and normalized to the level of expression of tubulin. Western blots were performed on cell lysates using the 9E10 anti-Myc monoclonal antibody to detect Myc-tagged IRP1WT or IRP13C>3S or using antibodies directed against FBXL5, IRP2, or tubulin. Representative blots from n = 3 experiments are shown. Antibodies against Fam96a are not available. In B and E, some conditions included 80 μm FAC. For all panels, the 100% value represents the signal obtained from 1.0 × 106 cells. Results are expressed as mean ± S.E. (error bars) for n = 3–6 separate experiments for each panel. Representative immunoblots are shown in panels A, C, D, F, and G. Immunoblot results from multiple experiments were quantified by densitometry and are shown in the accompanying graphs.
Figure 7.

Stimulation of FBXL5 protein expression by induction of IRP1 or IRP2 or knockdown of FBXL5. A and B, HEK cells were treated with tet for 24 h to induce the expression of Myc-tagged IRP1WT or IRP13C>3S. A and B, the 24-h treatment without or with tet was followed by a 4-h treatment with various combinations of 8 μm FAC, 2.5 μm MG115, and 100 μm Df. A, cell lysates were probed for IRP2, FBXL5, and Myc-tagged-IRP1 (n = 4 experiments). B, HEK cells were treated with tet to induce the expression of IRP2FLAG for 24 h followed by a 4-h treatment with various combinations of 8 μm FAC, 2.5 μm MG115, and 100 μm Df. Cell lysates were then examined by immunoblotting using the indicated antibodies (n = 4). C, wild-type HEK 293 cells were transfected and harvested as in Fig. 1. Western blots using antibodies against FBXL5 and α-tubulin were conducted. For C, the samples used were from the same experiment as Fig. 5A, and the same α-tubulin blot is shown as a loading control for C and Fig. 5A. For all blots shown in this figure, representative blots from n = 3 experiments are shown, and tubulin was used as a loading control. Results are expressed as mean ± S.E. (error bars) for n = 3 experiments.
Given the link between CIA activity and the regulation of IRP1 RNA-binding activity, we sought to determine the extent to which the protective effect of FBXL5 was linked to IRP1 dysregulation. Cells capable of expressing Myc-tagged IRP13C>3S or, as a control, Myc-tagged IRP1WT were treated without or with tetracycline (tet) and transfected with either non-targeting (NT) or FBXL5-targeting siRNAs. In the absence of tet, when IRP13C>3S is not expressed, FBXL5 siRNA did not affect cell viability (Fig. 1C, lane 2). Similarly, induction of IRP13C>3S in the presence of an NT siRNA failed to impact cell growth but was associated with a strong increase in FBXL5 protein level (Fig. 1C, lane 3). However, when FBXL5 expression was knocked down in the presence of IRP13C>3S, cell viability was significantly reduced (Fig. 1C, lane 4). In contrast, simultaneous treatment with FBXL5 siRNA along with tet induction of IRP1WT was without effect on cell viability (Fig. 1D). Interestingly, when the level of FBXL5 expression was normalized to that of Myc-tagged IRP1WT or IRP13C>3S, the E3 ligase accumulated to a 3-fold higher level in the latter case (Fig. 1E). As was observed with combined knockdown of CIA and FBXL5, iron supplementation prevented the reduction in viability of HEK cells expressing IRP13C>3S in the presence of FBXL5 siRNA (Fig. 1F, lane 6) perhaps because iron stabilizes FBXL5 protein (12, 13) and increases its expression relative to cells treated with FBXL5 siRNA alone (Fig. 1F, compare lanes 5 and 6). Iron chelation with desferal failed to rescue the cells (results not shown). We conclude that when the Fe-S switch mechanism is not functional, as occurs when CIA is inhibited, the action of FBXL5 is critical in limiting the negative impact of excess IRP1 RNA-binding activity on cell function.
The impact of FBXL5 knockdown on the level of both IRP13C>3S and endogenous IRP2 was determined (Fig. 1, F and G). When compared with HEK cells treated with the NT siRNA, FBXL5 siRNA treatment significantly increased the level of IRP13C>3S protein (Fig. 1F, compare lanes 4 and 5). The endogenous level of IRP2 protein was also increased when FBXL5 was knocked down (Fig. 1G, compare lanes 2 and 1). The impact of IRP13C>3S expression on IRP2 protein level was also examined. Whereas induction of IRP13C>3S failed to statistically increase IRP2 protein level, the level of IRP2 protein trended upward (p = 0.083), such that there was no longer a significant impact of concurrent knockdown of FBXL5 on IRP2 protein level (compare lane 4 with lane 3 and lane 2 with lane 1 in Fig. 1G). This is consistent with the previous studies showing that the RNA-binding forms of IRP1 and IRP2 are both subject to FBXL5 regulation (12, 13). Taken together, these results suggest that in the face of reduced CIA activity, dysregulation of both IRPs may occur unless sufficient FBXL5 is present to prevent their overaccumulation.
Impact of CIA knockdown on IRP1 and IRP2 protein level
Because apoprotein mutants of IRP1 that cannot assemble an Fe-S cluster exhibit iron-dependent regulation of protein stability (6, 19), and knockdown of some CIA factors, such as FAM96A (10), reduces IRP1 protein level, the role of FBXL5 in controlling IRP1 protein level in response to knockdown of CIA factors was investigated. Knockdown of NUBP2 reduced IRP1 protein level by 20%, but this difference did not reach statistical significance at this time point (p = 0.086) (Fig. 2A, lanes 1 and 2). However, extended treatment from the standard 9-day to 12-day treatment with NUBP2 siRNA decreased IRP1 protein level without affecting the level of another Fe-S protein (GPAT) or tubulin (Fig. 2B, lanes 1 and 2). IRP2 responded similarly to IRP1. Extending treatment to 15 days resulted in loss of IRP1, IRP2, and GPAT but not tubulin (Fig. 2B, lanes 3 and 4). Knockdown of FAM96A reduced IRP1 protein level more effectively than was the case with NUBP2, with a nearly 50% reduction compared with cells treated with the NT siRNA, after the standard 9-day transfection protocol (Fig. 2A). This effect of FAM96A knockdown on IRP1 protein level was blocked by simultaneous transfection with FBXL5 siRNAs (Fig. 2A, compare lanes 6 and 3 with lane 1) (Fig. 7C contains FBXL5 protein expression data for this experiment).
Figure 2.

Impact of CIA knockdown on IRP1, IRP2, and GPAT protein level and cytosolic aconitase activity. A–C, Western blots for endogenous IRP1, IRP2, NuBP2, GPAT, or tubulin were performed on cell lysates as described under “Experimental procedures.” A, wild-type HEK 293 cells were transfected with NT, NuBP2, or Fam96a targeting siRNA and then with or without FBXL5 siRNA as described in the legend to Fig. 1. B, the siRNA transfections were extended such that the wild-type HEK 293 cells were retransfected on day 9 with harvest on day 12 (lanes 1 and 2) or retransfected on days 9 and 12 with harvest on day 15 (lanes 3 and 4). Representative blots from n = 3 experiments are shown. C, wild-type HEK 293 cells were treated as described for A. D, cells were transfected with NuBP2 or Fam96a siRNA, and cytosolic and mitochondrial fractions were obtained after digitonin permeabilization as described under “Experimental procedures” (11). The mean aconitase activities for the cytosolic and mitochondrial fractions were 5.38 and 1.31 milliunits/mg, respectively. Results were determined using a Student's two-tailed t test and are expressed as mean ± S.E. (error bars) for n = 3–6 separate experiments.
Because previous work has shown that IRP2 protein level (10) is reduced when FAM96A is knocked down, we asked whether a similar response occurred with NUBP2 knockdown and what role FBXL5 had in each scenario. IRP2 protein level declined by 40% upon knockdown of NUBP2 and by 80% when FAM96A was knocked down (Fig. 2C, lanes 2 and 3 versus lane 1). The reduction in IRP2 protein expression in response to knockdown of NUBP2 or FAM96A was completely blocked by co-transfection with FBXL5 siRNA (Fig. 2C, lanes 5 and 6) (see Fig. 6C for FBXL5 expression level). Taken together, it is apparent that the response of IRP1 and IRP2 to knockdown of NUBP2 or FAM96A occurred earlier than was the case for the cytosolic Fe-S protein GPAT (Fig. 2B). These results indicate that targeted degradation of both IRPs by FBXL5 represents an adaptive response necessary for maintenance of cell viability when cytosolic Fe-S biogenesis is impaired.
Figure 6.

FBXL5-dependent regulation of IRP1 in response to impaired CIA. A, HEK 293 cells that can express Myc-tagged IRP1WT or IRP13C>3S were transfected with an HA-ubiquitin (HA-UB) expression construct for 20 h. Cells were then treated with tet to induce expression of Myc-IRP1WT or Myc-IRP13C>3S for 36 h followed by a 4-h treatment with various combinations of 8 μm FAC, proteasome inhibitor MG115 (2.5 μm), or 100 μm Df. Cell lysates were subjected to immunoprecipitation (IP) with the 9E10 anti-Myc monoclonal antibody followed by immunoblotting (IB) for HA-ubiquitin or Myc-IRP1. B, HEK 293 cells capable of expressing IRP1WT were transfected with either NT siRNA or NuBP2 siRNA followed by FBXL5 siRNA using the 9-day protocol as in Fig. 1. On day 9, cells were treated with tet for 12 h prior to harvesting. During the last 4 h of tet induction, cells were treated with various combinations of 8 μm FAC and 2.5 μm MG115. Cells lysates were then immunoprecipitated and examined by Western blotting using the indicated antibodies (n = 3). C, HEK 293 cells were transfected with either NT siRNA or NuBP2 siRNA on days 0, 3, and 6. On day 6, all cells were transfected with the HA-ubiquitin expression construct, and cells in lane 3 were also transfected with FBXL5 siRNA. Cell extracts were then immunoprecipitated using an antibody that recognizes endogenous IRP1. The immunoprecipitates were then examined by Western blotting for HA-ubiquitin or IRP1 (n = 3). D, wild-type HEK 293 cells were plated for 24 h and transfected with the HA-ubiquitin expression plasmid for 20 h. Cells could grow for another 24 h and were treated with 100 μm Df for 12 h followed by treatment with 2.5 μm MG115 and 8 μm FAC for 1, 2, and 12 h (n = 3). Cell extracts were immunoprecipitated and probed for endogenous IRP1 as described in C.
Knockdown of NUBP2 or FAM96A decreases the cytosolic aconitase activity of IRP1
To further understand how impaired CIA assembly affects IRP1 function, the impact of NUBP2 and FAM96A knockdown on c-acon activity was assessed. Knockdown of NUBP2 or FAM96A decreased c-acon activity by 52 and 60%, respectively (Fig. 2D). The activity of mitochondrial aconitase was not significantly affected. Interestingly, the observation that NUBP2 knockdown decreased c-acon activity indicates that it is a bona fide CIA component in mammalian cells. This result demonstrates that like FAM96A (10), NUBP2 has an important role in human CIA activity, including regulating the function of IRP1.
The impact of CIA factor knockdown on IRE RNA-binding activity
The impact of CIA factor knockdown on IRP protein level suggests that RNA-binding activity is also altered. IRP RNA-binding activity was determined by electrophoretic mobility shift assay (EMSA). Because IRP1 and IRP2 co-migrate in human cells, the sum of IRP1 plus IRP2 RNA-binding activity was determined first. The RNA-binding activity obtained in as-isolated cell lysates (Spontaneous) and the total cellular RNA binding (Total) was determined after activation of the latent pool of IRP1 with 2-mercaptoethanol (Fig. 3A). An antibody specific to IRP1 was used to quantify IRP1-specific binding by gel supershift (Fig. 3B).
Figure 3.

IRP RNA-binding activity as a function of CIA and FBXL5 knockdown. Wild-type HEK 293 cells were transfected with NT or NuBP2- or Fam96A-targeting siRNAs in the absence or presence of FBXL5 targeting siRNA. IRP RNA-binding activity was determined by EMSA including antibody supershift for IRP1. A, summed IRP (IRP1 + IRP2) (top graph) and 2-ME-inducible “total” RNA-binding activity (bottom graph) was determined in cells transfected with various siRNAs. For the top graph, the RNA-binding values were as follows: NT siRNA, 88 fmol/mg; NuBP2 siRNA, 64 fmol/mg; Fam96a siRNA, 56 fmol/mg; FBXL5, 109 fmol/mg; NuBP2 and FBXL5 siRNA, 82 fmol/mg; Fam96a and FBXL5 siRNA, 85 fmol/mg. For the total 2-ME-inducible RNA binding, the RNA-binding values were as follows: NT siRNA, 155 fmol/mg; NuBP2 siRNA, 116 fmol/mg; Fam96a siRNA, 83 fmol/mg; FBXL5 188 fmol/mg; NuBP2 and FBXL5 siRNA, 145 fmol/mg; Fam96a and FBXL5 siRNA, 127 fmol/mg. B, the same extracts used in A were subjected to an EMSA-supershift assay using an antibody specific to IRP1 (43). Spontaneous IRP and 2-ME-inducible RNA-binding activity was determined after incubation with anti-IRP1 antibody. The amount of RNA bound in the non-supershifted (SS) band was also quantified and is taken to represent IRP2. An antibody for IRP2 supershifts was not available. For all panels, representative results from n = 3 experiments are shown for EMSA (A and B). Results are expressed as mean ± S.E. (error bars).
Knockdown of NUBP2 or FAM96A reduced the sum of IRP1 plus IRP2 IRE-binding activity by about 27 and 36%, respectively (Fig. 3A). Simultaneous knockdown of FBXL5 largely blocked these effects (Fig. 3A, compare lanes 5 and 6 with lanes 2 and 3, respectively). 2-ME-inducible RNA-binding activity (IRP1 plus IRP2) showed a generally similar response (Fig. 3A). IRP1 RNA-binding activity, measured by supershift with an antibody specific to IRP1, showed a substantial reduction in RNA-binding activity in response to NUBP2 siRNA or FAM96A siRNA (Fig. 3B). FBXL5 siRNA did not reverse the reduction in IRP1 RNA-binding activity induced by knockdown of NUBP2 or FAM96A. 2-ME-induced IRP1 RNA-binding activity showed a similar response. The lack of impact of FBXL5 knockdown in reversing the decline in RNA-binding activity of IRP1 when CIA was inhibited contrasted with the partial rescue of IRP1 protein level noted in Fig. 2A, suggesting accumulation of a form of IRP1 that neither binds RNA or is an aconitase. Previous work in HEK cells or when IRP1 was expressed in yeast observed that IRP1 can accumulate in a form that neither binds RNA nor exhibits aconitase activity (20, 21). In contrast, the non-supershifted band, which represents IRP2 RNA-binding activity, did exhibit an FBXL5-dependent response. IRP2 RNA binding was reduced upon knockdown of NUBP2 or FAM96A, but when FBXL5 was simultaneously knocked down, RNA binding increased significantly (Fig. 3B, bottom histogram, compare lanes 5 and 6 with lanes 2 and 3, respectively).
It should be noted that, upon knockdown of CIA components in HEK cells, the fraction of IRP1 present in the RNA-binding form, measured as spontaneous over 2-ME-inducible RNA binding, was not increased in our study. This contrasts with earlier studies in HeLa cells where knockdown of Fe-S biogenesis activity increased the fraction of IRP1 in the RNA-binding form (5, 9–11) but is similar to the impact of longer-term reduction in Fe-S biogenesis in liver, which included induction of FBXL5 expression (8). Importantly, our previous studies (22) in HeLa cells found a level of IRP1 RNA-binding activity that was about 10-fold higher than what we observed in the current study with HEK cells, suggesting a different set point for regulation of IRPs in HeLa cells. To address this, we determined the relative abundance of FBXL5, IRP1, and IRP2 at the protein level in these cell lines. HEK cells were found to express substantially higher levels of FBXL5 as compared with HeLa cells in the absence or presence of added iron (Fig. 4). Conversely, HEK cells expressed a lower level of both IRP1 and IRP2 protein compared with HeLa cells. Taken together, these observations suggest that cell type–specific differences in FBXL5 expression influence the steady-state level of RNA binding by both IRPs as well as their response to changes in Fe-S biogenesis activity.
Figure 4.
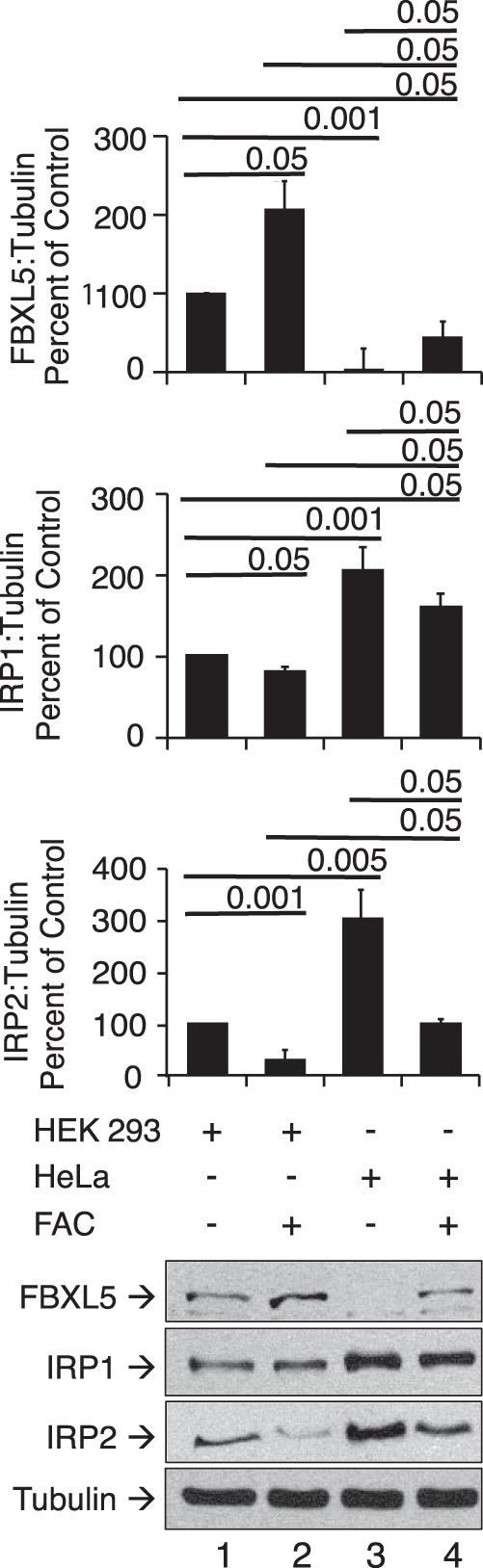
Expression level of IRPs and FBXL5 in HEK 293 and HeLa Cells. The protein level of FBXL5, IRP1, IRP2, and tubulin was determined in HEK cells or HeLa cells grown in the absence or presence of 8 μm FAC for 4 h. For all panels, representative results for n = 3–4 experiments are shown in the Western blots. Results are expressed as mean ± S.E. (error bars).
Impact of knockdown of CIA factors on TfR1 and ferritin protein level
To relate the impact of knockdown of CIA factors and FBXL5 on IRP action, the expression of TfR1 and ferritin heavy chain protein was determined. Like what was shown when FAM96A was knocked down (10), knockdown of NUBP2 resulted in a paradoxical IRE-independent increase in TfR and decline in H-ferritin protein level (Fig. 5, A and B, lanes 2 and 3), although IRP1 and IRP2 protein level (see Fig. 2) and total as well as IRP1 RNA-binding activity (Fig. 3) were decreased upon treatment with CIA targeting siRNAs. To extend this finding, the impact of simultaneous knockdown of FBXL5 was examined. Interestingly, TfR1 protein level was further increased in response to simultaneous knockdown of FBXL5 (Fig. 5A, lanes 5 and 6) when total IRE-binding activity (Fig. 3A) and IRP2 protein level increased (Fig. 2C). These findings suggest that cells act to limit IRP action in response to CIA inhibition.
Figure 5.
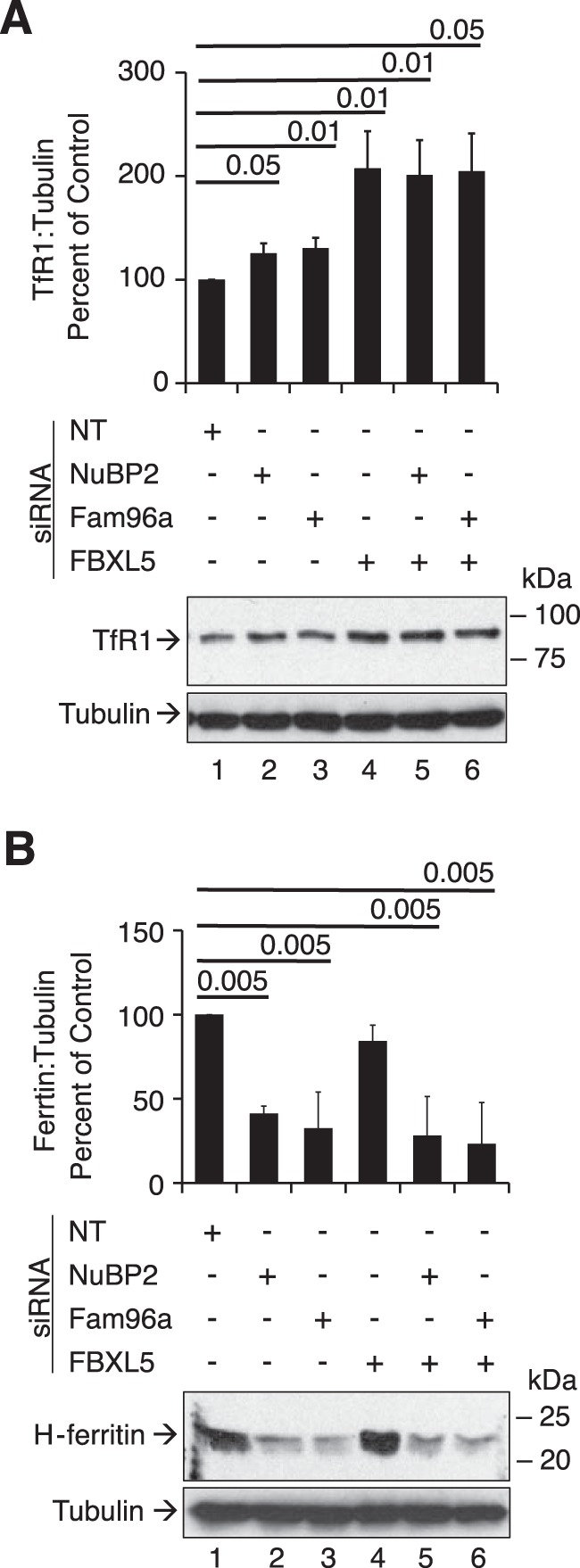
Impact of CIA knockdown on TfR1 and ferritin protein expression. HEK 293 cells were transfected with various siRNAs as described in the legend to Fig. 1. A, TfR1 protein expression was determined in cell lysates by immunoblotting with an antibody against TfR1. B, ferritin heavy chain protein expression was determined in the same lysates by immunoblotting with antibody against H-ferritin. α-Tubulin was used as a control. Representative blots from n = 3 experiments are shown. A two-tailed Student's t test was used to determine statistical significance of three independent experiments.
FBXL5 targets IRP1 for proteasomal degradation
Because previous studies demonstrated that IRP13C>3S is stabilized by depletion of FBXL5 (12), we reasoned that this E3 ligase was required for control of IRP1 protein stability when CIA is impaired or in response to iron overload. Cells capable of expressing Myc-tagged IRP1WT or IRP13C>3S were transiently transfected with a HA-tagged ubiquitin construct (23). The ubiquitination state of IRP1 was assessed by immunoprecipitation of Myc-tagged IRP1 followed by immunoblotting for the HA epitope. IRP1WT exhibited a significant level of mono- or perhaps oligoubiquitinated species, the abundance of which was not detectably affected by iron status or the proteasome inhibitor MG115 (Fig. 6A, lanes 3–6). In contrast, a substantial amount of polyubiquitinated IRP13C>3S was detected, the abundance of which was iron-regulated (Fig. 6A, lanes 9–12). The polyubiquitinated species of IRP13C>3S increased upon treatment of cells with MG115 and iron, whereas desferal eliminated it. Polyubiquitination of IRP13C>3S was substantially inhibited by FBXL5 knockdown (results not shown); others have noted that FBXL5 overexpression increases the polyubiquitination of IRP13C>3S (13). We conclude that the apoprotein form of IRP1 is the preferred substrate for iron-dependent FBXL5-mediated polyubiquitination.
IRP1 is polyubiquitinated when CIA is impaired
The failure to detect polyubiquitination of transfected IRP1WT in HEK cells may relate to the fact that in some cell types, <5% of IRP1 is present in the apoprotein (RNA-binding) form (3, 4, 21). To address this, HEK cells were treated with NUBP2 siRNA to decrease formation of c-acon from IRP1 and co-transfected with the HA-ubiquitin expression plasmid. NUBP2 knockdown led to increased accumulation of polyubiquitinated IRP1WT (Fig. 6B, lane 2). Inhibition of the proteasome with MG115 substantially increased the level of polyubiquitinated IRP1WT (Fig. 6B, lane 3). Simultaneous knockdown of FBXL5 nearly eliminated NUBP2-dependent polyubiquitination of IRP1WT (Fig. 6B, lane 4). We conclude that FBXL5-dependent polyubiquitination of IRP1WT is strongly increased when CIA is impaired due to enhanced accumulation of the RNA-binding (apoprotein) form of IRP1.
Polyubiquitination of endogenous HEK cell IRP1 increases in response to CIA inhibition
To determine the relationship between CIA activity and polyubiquitination of endogenous IRP1, the impact of NUBP2 knockdown in the presence of added iron was assessed in HEK cells. Knockdown of NUBP2 stimulated IRP1 polyubiquitination over that which was observed in cells treated with the non-targeting siRNA (Fig. 6C, lane 2). Simultaneous knockdown of FBXL5 reduced the level of polyubiquitination (Fig. 6C, lane 3).
To further examine the polyubiquitination of endogenous IRP1, HEK cells were transfected with the HA-ubiquitin expression plasmid and treated with desferal for 12 h followed by the addition of ferric ammonium citrate (FAC) and MG115 for 1, 2, or 12 h to determine whether the expanded pool of IRP1 apoprotein induced by desferal was subject to iron-dependent degradation. Endogenous IRP1 was immunoprecipitated and immunoblotted with an anti-HA antibody. A strong time-dependent increase in polyubiquitination of endogenous IRP1 was observed (Fig. 6D). Taken together, these findings demonstrate that FBXL5-dependent polyubiquitination of endogenous IRP1 is enhanced when CIA is impaired or in response to acute iron treatment of iron-deficient cells.
IRP1 can interfere with FBXL5-dependent control of IRP2
Given the fact that both IRPs are substrates for FBXL5 and co-immunoprecipitation studies suggest a direct interaction between ligase and substrate (12, 13), we asked whether increased expression of IRP1WT and IRP13C>3S influenced the accumulation of endogenous IRP2 protein and whether this was affected by iron status or proteasomal activity (Fig. 7A). Interestingly, IRP13C>3S but not IRP1WT substantially inhibited the down-regulation of IRP2 protein by iron (Fig. 7A, compare lanes 3 and 8). These findings suggest that the dysregulation of cellular iron metabolism in response to CIA inhibition is in part a consequence of increased accumulation of the IRP1 apoprotein coupled with reduced ability of FBXL5 to down-regulate IRP2.
Expression of IRP1 or IRP2 increases FBXL5 protein level in an iron-dependent manner
The finding (Fig. 1C) that induction of IRP13C>3S led to accumulation of FBXL5 protein suggested a feedback mechanism to control IRP RNA-binding activity. To further investigate the relationship between IRP expression and the accumulation of FBXL5 protein, the question of whether IRP13C>3S and IRP1WT were equipotent in inducing FBXL5 was examined. tet induction of IRP1WT led to a slight accumulation of FBXL5 protein (Fig. 7A, lane 2). The addition of iron increased the level of FBXL5 more significantly, whereas the addition of MG115 had no effect when iron was present (Fig. 7A, lanes 3 and 4). In contrast, FBXL5 expression was strongly induced by IRP13C>3S in the absence of added iron (Fig. 7A, lane 7). About 8-fold more FBXL5 protein was detected in cells expressing IRP13C>3S relative to IRP1WT in the absence of added iron (Fig. 7A, compare lanes 7 and 2). As was the case with IRP1WT, MG115 did not further increase FBXL5 protein level in cells simultaneously treated with iron (Fig. 7A, compare lane 9 with lane 8). Desferal blocked the induction of FBXL5 by either IRP1 allele.
To determine whether the induction of FBXL5 was specific to IRP1, a HEK cell line that expresses FLAG-tagged IRP2 in a tet-inducible manner was used (24). FBXL5 expression increased about 2-fold in response tet-induced expression of IRP2 (Fig. 7B). FBXL5 and IRP2 expression were reciprocally regulated by iron as expected. The level of FBXL5 appeared to be increased by the addition of FAC, whereas that of IRP2 was reduced (Fig. 7B). The addition of MG115 to the FAC treatment had little additional impact on the FBXL5 (Fig. 7B). Desferal eliminated FBXL5 protein expression while further increasing IRP2 protein (Fig. 7B, lane 5). Overall, the pattern of response of FBXL5 to induction of IRP2 with or without treatment with iron, desferal, or MG115 was quite like the same experiments with IRP13C>3S. In sum, these findings suggest that the iron-dependent induction of FBXL5 expression in response to accumulation of apo-IRP1 or IRP2 serves as a mechanism to limit overaccumulation of IRP RNA-binding activity.
Enhanced FBXL5 expression in response to CIA inhibition
The reduction in IRP2 protein level when CIA factors NUBP2 and FAM96A (10) are knocked down (Fig. 2C) suggests that cytosolic iron is increased, which is in line with increased TfR1 expression (Fig. 5A) and binding of iron-loaded transferrin to cells with impaired CIA activity (10, 11). Consistent with this, a strong impact of CIA knockdown on FBXL5 protein level was observed (Fig. 7C), reflecting the impact of iron (Fig. 7A). NUBP2 knockdown resulted in a nearly 2.3-fold increase in FBXL5 protein accumulation relative to cells treated with NT siRNA (Fig. 7C, lane 2). FAM96A siRNA treatment resulted in a 3.3-fold increase (Fig. 7C, lane 3). The relative impact of knockdown of these two CIA factors mirrors their impact on IRP2 and IRP1 protein abundance, IRP RNA binding, and c-acon activity. FAM96A knockdown had the stronger effect, followed by NUBP2. Furthermore, a reciprocal impact of the knockdown of CIA factors on FBXL5 relative to IRP protein levels was observed (compare Fig. 2 (A and C) with Fig. 7C). FBXL5 protein level is increased, whereas that of both IRP1 and IRP2 is decreased when CIA factor expression is impaired. These findings support the concept that FBXL5 is an integral component of the adaptive processes influencing the IRP-dependent control of cellular iron metabolism when Fe-S biogenesis is dysregulated.
Phosphorylation of IRP1 at Ser-138 is required to maintain cellular growth rate in response to CIA inhibition
Previous studies have suggested that Ser-138 phosphorylation of IRP1 targets the RNA-binding form and favors its accumulation due to destabilization of the Fe-S cluster in c-acon (6, 21, 25). Hence, we reasoned that induction of Ser-138 phosphorylation might represent an adaptive response to CIA inhibition, perhaps by promoting TfR accumulation and iron uptake. Knockdown of NUBP2 resulted in a >3-fold increase in Ser-138-phosphorylated IRP1 (Fig. 8A, lane 2 versus lane 1). The abundance of Ser-138-phosphorylated IRP1 was not affected by knockdown of FBXL5 as expected based on previous studies with Ser-138 phosphomutants (6). Interestingly, CIA inhibition resulted in a level of Ser-138 phosphorylation of IRP1 similar to that observed in response to the addition of the strong PKC agonist phorbol 12-myristate 13-acetate (Fig. 8A, lane 4).
Figure 8.

Ser-138 phosphorylation of IRP1 in response to CIA inhibition. A, wild-type HEK 293 cells were transfected and harvested as in Fig. 1. Endogenous IRP1 was immunoprecipitated from cell lysates using a pan-IRP1 antibody and immunoblotted for Ser-138–phosphorylated IRP1 using Ser-138–phosphospecific IRP1 antibodies (6) (n = 3). B, wild-type HEK 293 cells were transfected with either NT or FBXL5 siRNA for 48 h. During the last 24 h of siRNA treatment, cells were treated with tet to induce the expression of Myc-tagged IRP13C>3S or IRP13C>3S/S138A. Cell viability was determined by trypan blue exclusion. Cell lysates were subjected to Western blotting using antibodies against FBXL5, tubulin, and the Myc epitope on IRP13C>3S or IRP13C>3S/S138A (n = 3). Error bars, S.E.
To determine whether Ser-138 phosphorylation of IRP1 played a role in the coordination of iron metabolism and cell proliferation, the impact of the non-phosphorylatable mutant IRP13C>3S/S138A on the response of HEK cells to FBLX5 knockdown was determined. As noted previously, coordinate induction of IRP13C>3S and knockdown of FBXL5 impaired cell growth, and this response was reversed by iron (Fig. 8B). In contrast, in the presence of IRP13C>3S/S138A and FBXL5 knockdown, cell viability was not rescued by iron (Fig. 8B, compare lanes 6 and 3). These findings support the concept that induction of Ser-138 phosphorylation of IRP1 is an adaptive response to maintain cell viability in the face of inhibition of Fe-S cluster biogenesis.
Discussion
Our findings describe key roles for a regulatory network involving FBXL5 and both IRP1 and IRP2 that responds to changes in CIA activity and is central to the regulation of iron metabolism during cell proliferation. This conclusion is based on the following observations (Fig. 9). First, a CIA-FBXL5 regulatory circuit was shown to be a critical factor controlling cellular iron metabolism. Induction of FBXL5 expression is a required component of the cellular adaptive response to CIA inhibition. Second, a negative feedback loop between IRP1 and FBXL5 was shown to be essential for maintenance of cellular iron metabolism. Furthermore, FBXL5 expression was enhanced in response to induction of either IRP1 or IRP2. Third, our findings support the concept that differences in CIA activity and FBXL5 expression are used to selectively control the action of IRP1 to meet unique iron needs of specific cell types under diverse physiological scenarios. Fourth, the CIA system and FBXL5 respond to perturbations of the same or interconnected cellular pool(s) of iron that appears to be necessary to properly coordinate the uptake and metabolic fate of iron. Fifth, the requirement for phosphorylation of IRP1 at Ser-138 to maintain optimal cell proliferation in response to CIA inhibition suggests the existence of novel signaling pathways that act to limit the dysregulation of iron metabolism. Taken together, our studies support the concept that the CIA-FBXL5-IRP axis is central for integrating a range of cellular regulatory pathways and coordinates changes in iron status with central aspects of cell physiology, including growth rate.
Figure 9.
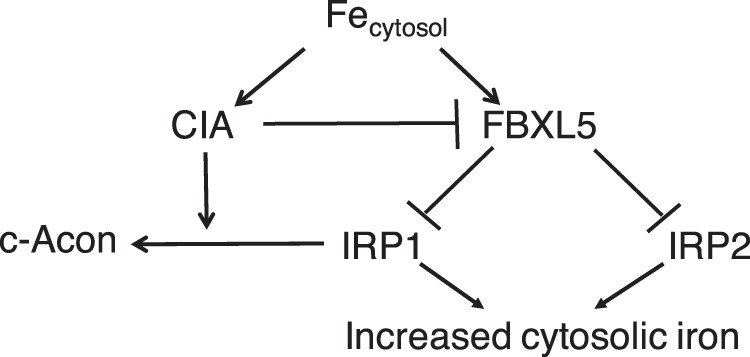
Model of regulatory processes controlling IRP1. IRP1 RNA-binding activity can be inactivated through insertion of an Fe-S cluster by the CIA pathway or by targeted polyubiquitination and degradation initiated by the ubiquitin E3 ligase FBXL5. Thus, IRP1 senses changes in cellular iron levels either through supply of iron for CIA followed by incorporation of an Fe-S cluster or by the iron-dependent activation of FBXL5, leading to degradation of the RNA-binding form of IRP1. FBXL5 protein stability is controlled by a constitutive and iron-induced mechanism (12, 13, 30). Both CIA and FBXL5 require iron, and as cells progress from an iron-deficient to an iron-replete state, inhibition of IRP1 prevents iron overload. Iron-dependent activation of these mechanisms leads to a suppression of IRP1, which reduces iron uptake. Presumably, the CIA system “acquires” iron more avidly than does FBXL5, but this remains to be determined. CIA acts to inhibit FBXL5, as evidenced by RNAi knockdown of CIA components, leading to increased expression of the ligase. The model predicts that as the CIA system increases in activity, it suppresses FBXL5, perhaps by limiting iron for formation of holo-FBXL5 or by generation of a signal indicative of increased demand for Fe-S biogenesis. In this condition, suppression of FBXL5 would enhance cellular iron uptake to meet the needs for increased Fe-S biogenesis. Once the CIA system is saturated with iron, activation of FBXL5 occurs, which then suppresses IRP1.
FBXL5 responds to CIA activity
We observed a strong induction of FBXL5 when either of the CIA components, NUBP2 or FAM96A, was impaired. The induction of FBXL5 presumably was a factor contributing to the decline in IRP1 and IRP2 protein expression and probably contributes to the “stabilizing effect” of FAM96A expression on IRP2 expression (10). Further support for this relationship came from the observation that the increased FBXL5 expression in response to induction of either IRP1 or IRP2 was iron-dependent. These data are consistent with a model where FBXL5 and CIA respond to the same cytosolic iron pool (8, 26). Paradoxically, although IRE binding is low and the cytosolic labile iron pool appears to be elevated, as evidenced by accumulation of FBXL5, cells with impaired CIA appear to be in a state of iron acquisition and/or utilization as the iron storage protein ferritin is decreased and the iron import protein TfR1 is elevated, as noted here and elsewhere (10). Our findings support this paradoxical observation and further demonstrate the importance of FBXL5 in regulating the RNA-binding activity of IRP1 and IRP2 in response to impaired CIA, presumably because excessive induction of IRP would be deleterious. The significant increase in total spontaneous IRE-binding activity when FBXL5 and CIA factors are simultaneously suppressed, compared with cells with only CIA knockdown, supports this view. We favor the view that limiting the level of IRP1 and IRP2 RNA-binding activity by FBXL5 is crucial for the adaptive response to CIA inhibition, but it is important to note that FBXL5 has been reported to have other protein targets, and their dysregulation may contribute to the phenotypes we observed (27).
A negative feedback loop between IRPs and FBXL5 is essential for optimal cell growth
Insertion and removal of Fe-S has widely been accepted as the dominant mechanism for regulating IRP1 RNA-binding activity (28). However, like IRP2, which lacks the ability to insert a Fe-S, IRP1 can also be controlled by the iron-dependent E3 ubiquitin ligase FBXL5 (12, 13). We therefore sought to determine conditions under which FBXL5-mediated degradation of IRP1 is important for regulating cellular iron metabolism. Interestingly, a decrease in cell viability was observed when FBXL5 expression was suppressed in combination with either impaired CIA or cells expressing a mutant form of IRP1 that cannot insert an Fe-S cluster, IRP13C>3S. The reduction in cell number in response to FBXL5 suppression was presumably due to iron-dependent dysregulation of IRP1 and IRP2 as total IRP RNA-binding activity was increased. Our findings suggest that FBXL5 is needed to limit IRP1 action when Fe-S biogenesis is impaired presumably to prevent unacceptably high levels of IRE RNA-binding activity. Notable examples in support of this concept include the shiraz zebrafish mutant, where overactivation of IRP1 causes lethality; the human equivalent of the shiraz zebrafish mutant developing sideroblastic-like microcytic anemia associated with cellular iron overload and decreased IRP1 expression; and Fbxl 5−/− mice that die in utero unless rescued by simultaneous ablation of IRP2 (29–31). Conversely, recent work in liver deficient in the mitochondrial Fe-S biogenesis factor frataxin demonstrated that IRP1 is essential for adaptive processes when Fe-S biogenesis is impaired (8). In that study, IRP1 RNA binding was initially activated but subsequently declined due to significant loss of IRP1 protein that was associated with induction of FBXL5 (8). Taken together, these studies indicate that a delicate balance exists between IRP1, IRP2, and FBXL5 essential for cell viability when Fe-S biogenesis is impaired. Dysregulation of this balance as produced by FBXL5 knockdown when CIA is impaired reduces cell growth and may be lethal in some circumstances.
FBXL5 regulates both IRP1 and IRP2 when CIA is impaired
Previous studies have demonstrated that when Fe-S biogenesis components, such as FAM96A, are impaired, IRP1 protein levels decrease significantly (9, 32–37). Our work further demonstrates knockdown of the CIA decrease IRP1 levels in an FBXL5-dependent manner. Surprisingly, although IRP2 does not possess a Fe-S cluster, we noted a decrease in IRP2 protein with CIA knockdown, a result that has previously been observed with the knockdown of FAM96A (10). We also observed that impairment of the CIA components NUBP2 and FAM96A had a greater effect on IRP2 protein stability than IRP1, presumably due in part to the pool of c-aconitase being resistant to FBXL5-mediated degradation, as observed in frataxin-deficient liver (8). In support of FBXL5 regulation of IRP1 during impaired Fe-S cluster biogenesis, we also observed FBXL5-dependent polyubiquitination of IRP1 with impaired NUBP2 function. This supports work completed by others demonstrating that overexpression of FBXL5 induces the polyubiquitination of IRP1 (12, 13). Further, support for both IRP utilizing the same protein degradation machinery came from the observation that overexpression of apo-IRP1, presented as IRP13C>3S, interferes with IRP2 degradation. Our findings further support a key interaction of IRP1 and FBXL5 in maintaining cell viability/growth.
Role of Ser-138 phosphorylation of IRP1 in the cellular response to CIA inhibition
Previous studies demonstrated that Ser-138 of IRP1 is a protein kinase C phosphorylation site (38). On the basis of studies with phosphomimetic mutants of Ser-138 (e.g. IRP1S138E), phosphorylation of IRP1 allows its conversion to c-acon (6, 7, 25), but the Fe-S cluster is much more unstable and, unlike IRP1S138S, undergoes spontaneous non-oxidative demetallation, allowing its preferential accumulation in the RNA-binding form in cells (6, 21). Ser-138 phosphomutants of IRP1 are more sensitive to iron-mediated protein degradation (6, 7). In the context of CIA inhibition, the strong increase in Ser-138 phosphorylation of IRP1 is predicted to promote the increased TfR and reduced ferritin expression noted in the current study and is consistent with the fact that iron supplementation reversed the inhibitory effect of CIA and FBXL5 knockdown in cell growth. Finally, it remains to be determined whether phosphorylation of IRP1 reflects activation of broad scale signaling pathways that respond to CIA activity perhaps through an Fe-S cluster–regulated kinase or phosphatase as is the case with the bacterial kinase NreB (39).
Summary and future directions
Our studies demonstrate that both Fe-S cluster insertion and protein degradation are critical components of a regulatory network controlling the level of IRP1 RNA-binding activity. Whereas previous studies have shown that IRP1 is subject to these modes of regulation (40, 41), the work described here identified novel interactions between the Fe-S biogenesis and protein degradation pathways that control IRP1 action. Several issues need to be addressed in future studies. First, over the continuum of iron levels that cells experience, from deficient to overloaded, is there a hierarchical role for the iron-dependent processes of the Fe-S switch versus protein degradation that control IRP1? Second, do tissue-specific differences in oxygenation selectively influence the control of IRP1 by FBXL5 versus the CIA system? FBXL5 requires oxygen to function. In contrast, CIA components with labile Fe-S clusters may be less active at the oxygen levels that are optimal for FBXL5. Thus, in contrast to iron, oxygen would appear to have opposing effects on the Fe-S switch versus protein degradation pathways that may contribute to tissue-specific control of IRP1 action. Third, how is IRP2 regulation integrated with that of IRP1 in relation to CIA and FBXL5 action? Considering the two branches of the CIA pathway, how does inhibition of the FAM 96B branch lead to increased IRP2 protein level without affecting IRP1 (10)? Does inhibition of the FAM 96B pathway, which delivers Fe-S clusters to nearly all cytosolic and nuclear apo-Fe-S proteins, increase FBXL5 expression, as is the case when the IRP-1 targeting FAM 96A branch is inhibited? Is the response of FBXL5 to inhibition of both arms of the CIA pathway, as shown here with knockdown of the upstream factor NuBP2, the same when FAM 96B is impaired? Fourth, what is the mechanism through which FBXL5 senses CIA activity? Is it merely changes in iron availability influencing assembly of the hemerythrin center in FBXL5, or is there some molecular species in the CIA pathway whose iron-dependent accumulation influences FBXL5 action? What is the protein kinase that phosphorylates IRP1 when CIA is inhibited? Future studies will address these questions.
Experimental procedures
Mammalian cell culture and reagents
HEK 293 cells were grown in DMEM with 10% FBS from Gibco. HeLa cells capable of expressing HFE were grown as described but in the absence of neomycin or antibiotics (22). HEK cells capable of tet-induced (1 μg/ml tet) expression of Myc-tagged IRP13C>3S, IRP13C>3S/S138A, or Myc-tagged IRP1WT were maintained as described (6). Cell viability was determined by trypan blue. Protein concentration was determined by a BCA assay (Thermo Fisher). Wild-type HEK cells refer to the parental non-transfected cell line. HEK cells expressing FLAG-tagged IRP2 were a generous gift of Elizabeth Leibold (University of Utah) (24).
Aconitase activity
HEK 293 cells were harvested and fractionated using digitonin treatment as described previously (5, 42). Cytosolic and mitochondrial aconitase activity was measured as described (21). In brief, cells were permeabilized with digitonin and fractionated into cytosolic and mitochondrial fractions by spinning lysates at 15,000 × g for 10 min. Aconitase activity was determined by measuring the conversion of isocitrate to cis-aconitate at 240 nm.
siRNA knockdown experiments
All siRNA transfection mixtures were prepared under low light using Opti-MEM (Invitrogen), Lipofectamine 2000 (Invitrogen), and 30 nm siRNA. siRNA transfection mixtures were prepared in 0.5 ml of Opti-MEM as described previously and plated with 0.5 × 106 HEK 293 cells in 3.5 ml antibiotic-free media (DMEM + 10% FBS) for the indicated times (12). FBXL5 siRNA transfection mixture was added to Myc-tagged IRP1–expressing cell lines 48 h prior to harvesting. To increase the extent to which CIA component expression was reduced, cells were transfected on day 0 and retransfected on days 3 and 6 with CIA siRNA as described previously (11). FBXL5 siRNA was added with CIA siRNA on day 6. Fam96a Silencer® Select predesigned individual siRNA (catalog no. s38637) was from Ambion. NuBP2 individual siRNA (D-004705-02-0005), non-targeting siRNA (D-001210-03-05), and FBXL5 siRNA (D-012424-04-0010) were from Dharmacon.
IRP1 ubiquitination state
Cells were plated in DMEM + 10% FBS 24 h prior to transfection. Cells were transfected with a HA-ubiquitin expression plasmid for 20 h (23) before induction of IRP1 with 1 μg/ml tetracycline for 36 h in DMEM + 10% FBS. Cells were then treated with various combinations of FAC, desferal (Df), and proteasome inhibitor MG115 (Calbiochem) for 4 h. To assess the effect of FBXL5 on Myc-tagged IRP1 ubiquitination species, cells were cotransfected with the HA-ubiquitin expression plasmid and FBXL5 siRNA in 25% Opti-MEM and 75% DMEM + 10% FBS for 48 h. Myc-tagged IRP1 was induced during the last 24 h of incubation using tet as described (6).
To evaluate IRP ubiquitination state, cells were lysed as described (6) with the addition of 10 mm N-ethylmaleimide, 3 mm iodoacetamide, and 0.5 mm dithiothreitol to inhibit deubiquitination (44, 45). IRP1 was immunoprecipitated from 0.5 mg of cell lysate by incubation with 5 μg of anti-Myc monoclonal antibody (9E10) for 12 h at 4 °C with orbital shaking. During the last hour of incubation, 30 μl of 5% BSA-blocked protein L beads was added (Thermo Fisher). Samples were centrifuged at 1500 × g at 4 °C for 2 min. The protein L beads were washed with 1 ml of 0.1% Triton X-100, 50 mm Tris, pH 7.4, 300 mm NaCl, 5 mm EDTA. The beads were washed three more times in this manner followed by resuspension in 0.1% SDS, 0.1% sodium deoxycholate in PBS, pH 7.4, buffer. The final wash was in PBS alone. Protein was treated with Laemmli reducing sample buffer and heated at 70 °C for 12 min. The samples were then run in an 8% SDS-polyacrylamide gel.
Antibodies and immunoblotting
Myc-tagged IRP1 was determined using anti-Myc antibody (9E10). IRP1 monoclonal antibodies (clone 295B) were made against recombinant rabbit IRP1 by Neoclone (Madison, WI). Other antibodies were obtained as follows: α-tubulin (T5168) and anti-FLAG M2 (F3165) (Sigma); human NUBP2 (H-79) antibody, ferritin heavy chain (H-53), and IRP2 antibody (7H6) (Santa Cruz Biotechnology); anti-HA (clone 3F10) (Roche Applied Science); and human TfR1 antibody (H68.4) (Zymed Laboratories Inc.). Mouse and human FBXL5 antibodies were generated in collaboration with the laboratory of Rick Bruick (46). Rabbit phosphospecific antibodies for Ser-138 of IRP1 have been described (6). Rabbit anti-GPAT antibody was generously provided by Oliver Stehling and Roland Lill (11). A plasmid encoding rat NUBP2 cDNA (MRN1768-98079773) (Thermo Scientific) was transfected into HEK 293 cells and used to confirm the molecular weight of human NUBP2 in our Western blot analyses (data not shown). Samples were electrophoresed through an 8 or 12% SDS-polyacrylamide gel and transferred to nitrocellulose at 60 mA for 12 h. After incubation with antibodies, proteins were detected using SuperSignal West Pico (Thermo Scientific). Blots were exposed to Blue Ultra X-ray film (GeneMate), and multiple exposures were used and quantified by densitometry.
To prevent excessive overcrowding of figures, the molecular weight markers are shown when a given target protein is analyzed for the first time (e.g. NuBP2 in Fig. 1A) as a representative example of where the markers migrate.
RNA-binding assays
IRP RNA-binding activity in HEK cell lysates was determined by EMSA using a 32P-labeled RNA of the first 65 nucleotides of the rat l-ferritin 5′-UTR with a specific radioactivity of 10,000–20,000 dpm/fmol (47). RNA binding was determined in the absence and presence of 2% 2-mercaptoethanol to assess the so-called spontaneously active and latent pools, respectively, of IRP1. To separate human IRP1 from IRP2, gel supershift assays were performed. Cell lysate (20 μg of protein) was diluted into RNA-binding buffer (38) containing anti-IRP1 IgG (10 μg) without or with 2% 2-ME. After 90 min at 4 °C, [32P]RNA was added, and the 4 °C incubation continued for 10 min in the absence of 2-ME or 20 min in the presence of 2% 2-ME.
Statistical analyses
Means of data sets were compared by the two-tailed Student's t test.
Author contributions
N. B. J., K. M. D., and C. P. N. performed the experiments. N. B. J., K. M. D., and R. S. E. interpreted the data. N. B. J. and R. S. E. designed the study. N. B. J. and R. S. E. wrote the paper with input from all authors.
Acknowledgments
We acknowledge Roland Lill, Oliver Stehling, William Walden, Richard Bruick, and Shigeki Miyamoto for helpful discussions and Elizabeth Leibold for use of HEK cells expressing FLAG-tagged IRP2.
This work was supported by National Institutes of Health Grant RO1 DK66600, United States Department of Agriculture Hatch Project WIS01667, and the Iron Metabolism Research Fund. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- IRP
- iron-regulatory protein
- FAC
- ferric ammonium citrate
- FBXL5
- F box L5
- GPAT
- glycerol-3-phosphate acyltransferase
- HEK
- human embryonic kidney
- IRE
- iron-responsive element
- NT
- non-targeting
- tet
- tetracycline
- c-acon
- cytosolic aconitase
- ISC
- iron–sulfur cluster
- CIA
- cytosolic iron–sulfur cluster assembly
- TfR
- transferrin receptor
- 2-ME
- 2-mercaptoethanol
- Df
- desferal.
References
- 1. Anderson C. P., Shen M., Eisenstein R. S., and Leibold E. A. (2012) Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta 1823, 1468–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hentze M. W., Muckenthaler M. U., Galy B., and Camaschella C. (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142, 24–38 [DOI] [PubMed] [Google Scholar]
- 3. Chen O. S., Schalinske K. L., and Eisenstein R. S. (1997) Dietary iron intake modulates the activity of iron regulatory proteins (IRPs) and the abundance of ferritin and mitochondrial aconitase in rat liver. J. Nutr. 127, 238–248 [DOI] [PubMed] [Google Scholar]
- 4. Meyron-Holtz E. G., Ghosh M. C., Iwai K., LaVaute T., Brazzolotto X., Berger U. V., Land W., Olivarierre-Wilson H., Grinberg A., Love P., and Rouault T. A. (2004) Genetic ablations of iron regulatory protein 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 23, 386–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biederbick A., Stehling O., Rösser R., Niggemeyer B., Nakai Y., Elsässer H. P., and Lill R. (2006) Role of human mitochondrial Nfs1 in cytosolic iron-sulfur protein biogenesis and iron regulation. Mol. Cell Biol. 26, 5675–5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clarke S. L., Vasanthakumar A., Anderson S. A., Pondarré C., Koh C. M., Deck K. M., Pitula J. S., Epstein C. J., Fleming M. D., and Eisenstein R. S. (2006) Iron-responsive degradation of iron-regulatory protein 1 does not require the Fe-S cluster. EMBO J. 25, 544–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fillebeen C., Chahine D., Caltagirone A., Segal P., and Pantopoulos K. (2003) A phosphomimetic mutation at Ser-138 renders iron regulatory protein 1 sensitive to iron-dependent degradation. Mol. Cell Biol. 23, 6973–6981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martelli A., Schmucker S., Reutenauer L., Mathieu J. R., Peyssonnaux C., Karim Z., Puy H., Galy B., Hentze M. W., and Puccio H. (2015) Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab. 21, 311–322 [DOI] [PubMed] [Google Scholar]
- 9. Song D., and Lee F. S. (2008) A role for IOP1 in mammalian cytosolic iron-sulfur protein biogenesis. J. Biol. Chem. 283, 9231–9238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stehling O., Mascarenhas J., Vashisht A. A., Sheftel A. D., Niggemeyer B., Rösser R., Pierik A. J., Wohlschlegel J. A., and Lill R. (2013) Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 18, 187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stehling O., Netz D. J., Niggemeyer B., Rösser R., Eisenstein R. S., Puccio H., Pierik A. J., and Lill R. (2008) Human Nbp35 is essential for both cytosolic iron-sulfur protein assembly and iron homeostasis. Mol. Cell Biol. 28, 5517–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salahudeen A. A., Thompson J. W., Ruiz J. C., Ma H. W., Kinch L. N., Li Q., Grishin N. V., and Bruick R. K. (2009) An E3 ligase possessing an iron responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vashisht A. A., Zumbrennen K. B., Huang X., Powers D. N., Durazo A., Sun D., Bhaskaran N., Persson A., Uhlen M., Sangfelt O., Spruck C., Leibold E. A., and Wohlschlegel J. A. (2009) Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paul V. D., and Lill R. (2015) Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 1853, 1528–1539 [DOI] [PubMed] [Google Scholar]
- 15. Lill R. (2009) Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838 [DOI] [PubMed] [Google Scholar]
- 16. Sharma A. K., Pallesen L. J., Spang R. J., and Walden W. E. (2010) Cytosolic iron-sulfur cluster assembly (CIA) system: factors, mechanism, and relevance to cellular iron regulation. J. Biol. Chem. 285, 26745–26751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rouault T. A. (2012) Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis. Model. Mech. 5, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sheftel A., Stehling O., and Lill R. (2010) Iron-sulfur proteins in health and disease. Trends Endocrinol. Metab. 21, 302–314 [DOI] [PubMed] [Google Scholar]
- 19. Wang J., Fillebeen C., Chen G., Biederbick A., Lill R., and Pantopoulos K. (2007) Iron-dependent degradation of apo-IRP1 by the ubiquitin-proteasome pathway. Mol. Cell Biol. 27, 2423–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brown N. M., Kennedy M. C., Antholine W. E., Eisenstein R. S., and Walden W. E. (2002) Detection of a [3Fe-4S] cluster intermediate of cytosolic aconitase in yeast expressing iron regulatory protein 1: insights into the mechanism of Fe-S cluster cycling. J. Biol. Chem. 277, 7246–7254 [DOI] [PubMed] [Google Scholar]
- 21. Deck K. M., Vasanthakumar A., Anderson S. A., Goforth J. B., Kennedy M. C., Antholine W. E., and Eisenstein R. S. (2009) Evidence that phosphorylation of iron regulatory protein 1 at serine 138 destabilizes the [4Fe-4S] cluster in cytosolic aconitase by enhancing 4Fe-3Fe cycling. J. Biol. Chem. 284, 12701–12709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roy C. N., Blemings K. P., Deck K. M., Davies P. S., Anderson E. L., Eisenstein R. S., and Enns C. A. (2002) Increased IRP1 and IRP2 RNA binding activity accompanies a reduction of the labile iron pool in HFE-expressing cells. J. Cell Physiol. 190, 218–226 [DOI] [PubMed] [Google Scholar]
- 23. Treier M., Staszewski L. M., and Bohmann D. (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell 78, 787–798 [DOI] [PubMed] [Google Scholar]
- 24. Zumbrennen K. B., Wallander M. L., Romney S. J., and Leibold E. A. (2009) Cysteine oxidation regulates the RNA-binding activity of iron regulatory protein 2. Mol. Cell Biol. 29, 2219–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown N. M., Anderson S. A., Steffen D. W., Carpenter T. B., Kennedy M. C., Walden W. E., and Eisenstein R. S. (1998) Novel role of phosphorylation in Fe-S cluster stability revealed by phosphomimetic mutations at Ser-138 of iron regulatory protein 1. Proc. Natl. Acad. Sci. U.S.A. 95, 15235–15240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ma C., Kotaria R., Mayor J. A., Eriks L. R., Dean A. M., Walters D. E., and Kaplan R. S. (2004) The mitochondrial citrate transport protein: probing the secondary structure of transmembrane domain III, identification of residues that likely comprise a portion of the citrate transport pathway, and development of a model for the putative TMDIII-TMDIII` interface. J. Biol. Chem. 279, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 27. Viñas-Castells R., Frías Á., Robles-Lanuza E., Zhang K., Longmore G. D., García de Herreros A., and Díaz V. M. (2014) Nuclear ubiquitination by FBXL5 modulates Snail1 DNA binding and stability. Nucleic Acids Res. 42, 1079–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Haile D. J., Rouault T. A., Harford J. B., Kennedy M. C., Blondin G. A., Beinert H., and Klausner R. D. (1992) Cellular regulation of the iron-responsive element binding protein: disassembly of the cubane iron-sulfur cluster results in high-affinity RNA binding. Proc. Natl. Acad. Sci. U.S.A. 89, 11735–11739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Camaschella C., Campanella A., De Falco L., Boschetto L., Merlini R., Silvestri L., Levi S., and Iolascon A. (2007) The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 110, 1353–1358 [DOI] [PubMed] [Google Scholar]
- 30. Moroishi T., Nishiyama M., Takeda Y., Iwai K., and Nakayama K. I. (2011) The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 14, 339–351 [DOI] [PubMed] [Google Scholar]
- 31. Wingert R. A., Galloway J. L., Barut B., Foott H., Fraenkel P., Axe J. L., Weber G. J., Dooley K., Davidson A. J., Schmidt B., Paw B. H., Shaw G. C., Kingsley P., Palis J., Schubert H., et al. (2005) Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature 436, 1035–1039 [DOI] [PubMed] [Google Scholar]
- 32. Mochel F., Knight M. A., Tong W. H., Hernandez D., Ayyad K., Taivassalo T., Andersen P. M., Singleton A., Rouault T. A., Fischbeck K. H., and Haller R. G. (2008) Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am. J. Hum. Genet. 82, 652–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pondarré C., Antiochos B. B., Campagna D. R., Clarke S. L., Greer E. L., Deck K. M., McDonald A., Han A. P., Medlock A., Kutok J. L., Anderson S. A., Eisenstein R. S., and Fleming M. D. (2006) The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum. Mol. Genet. 15, 953–964 [DOI] [PubMed] [Google Scholar]
- 34. Shan Y., and Cortopassi G. (2012) HSC20 interacts with frataxin and is involved in iron-sulfur cluster biogenesis and iron homeostasis. Hum. Mol. Genet. 21, 1457–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sheftel A. D., Stehling O., Pierik A. J., Elsässer H. P., Mühlenhoff U., Webert H., Hobler A., Hannemann F., Bernhardt R., and Lill R. (2010) Humans possess two mitochondrial ferredoxins, Fdx1 and Fdx2, with distinct roles in steroidogenesis, heme, and Fe/S cluster biosynthesis. Proc. Natl. Acad. Sci. U.S.A. 107, 11775–11780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shi Y., Ghosh M. C., Tong W. H., and Rouault T. A. (2009) Human ISD11 is essential for both iron-sulfur cluster assembly and maintenance of normal cellular iron homeostasis. Hum. Mol. Genet. 18, 3014–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Song D., Tu Z., and Lee F. S. (2009) Human ISCA1 interacts with IOP1/NARFL and functions in both cytosolic and mitochondrial iron-sulfur protein biogenesis. J. Biol. Chem. 284, 35297–35307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eisenstein R. S., Tuazon P. T., Schalinske K. L., Anderson S. A., and Traugh J. A. (1993) Iron-responsive element binding protein: phosphorylation by protein kinase C. J. Biol. Chem. 268, 27363–27370 [PubMed] [Google Scholar]
- 39. Unden G., Nilkens S., and Singenstreu M. (2013) Bacterial sensor kinases using Fe-S cluster binding PAS or GAF domains for O2 sensing. Dalton Trans. 42, 3082–3087 [DOI] [PubMed] [Google Scholar]
- 40. Goessling L. S., Daniels-McQueen S., Bhattacharyya-Pakrasi M., Lin J.-J., and Thach R. E. (1992) Enhanced degradation of the ferritin repressor protein during induction of ferritin messenger RNA translation. Science 256, 670–673 [DOI] [PubMed] [Google Scholar]
- 41. Haile D. J., Rouault T. A., Tang C. K., Chin J., Harford J. B., and Klausner R. D. (1992) Reciprocal control of RNA-binding and aconitase activity in the regulation of the iron-responsive element binding protein: role of the iron-sulfur cluster. Proc. Natl. Acad. Sci. U.S.A. 89, 7536–7540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Drapier J. C., and Hibbs J. B. Jr. (1996) Aconitases: a class of metalloproteins highly sensitive to nitric oxide synthesis. Methods Enzymol. 269, 26–36 [DOI] [PubMed] [Google Scholar]
- 43. Schalinske K. L., Blemings K. P., Steffen D. W., Chen O. S., and Eisenstein R. S. (1997) Iron regulatory protein 1 is not required for the modulation of ferritin and transferrin receptor expression by iron in a murine pro-B lymphocyte cell line. Proc. Natl. Acad. Sci. U.S.A. 94, 10681–10686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mabb A. M., Wuerzberger-Davis S. M., and Miyamoto S. (2006) PIASy mediates NEMO sumoylation and NF-κB activation in response to genotoxic stress. Nat. Cell Biol. 8, 986–993 [DOI] [PubMed] [Google Scholar]
- 45. Song D., and Lee F. S. (2011) Mouse knock-out of IOP1 protein reveals its essential role in mammalian cytosolic iron-sulfur protein biogenesis. J. Biol. Chem. 286, 15797–15805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thompson J. W., Salahudeen A. A., Chollangi S., Ruiz J. C., Brautigam C. A., Makris T. M., Lipscomb J. D., Tomchick D. R., and Bruick R. K. (2012) Structural and molecular characterization of iron-sensing hemerythrin-like domain within F-box and leucine-rich repeat protein 5 (FBXL5). J. Biol. Chem. 287, 7357–7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goforth J. B., Anderson S. A., Nizzi C. P., and Eisenstein R. S. (2010) Multiple determinants within iron-responsive elements dictate iron regulatory protein binding and regulatory hierarchy. RNA 16, 154–169 [DOI] [PMC free article] [PubMed] [Google Scholar]