

ABSTRACT
Nonsense-mutation-containing messenger ribonucleoprotein particles (mRNPs) transit through cytoplasmic foci called P-bodies before undergoing nonsense-mediated mRNA decay (NMD), a cytoplasmic mRNA surveillance mechanism. This study shows that the cytoskeleton modulates transport of nonsense-mutation-containing mRNPs to and from P-bodies. Impairing the integrity of cytoskeleton causes inhibition of NMD. The cytoskeleton thus plays a crucial role in NMD. Interestingly, disruption of actin filaments results in both inhibition of NMD and activation of premature termination codon (PTC) readthrough, while disruption of microtubules causes only NMD inhibition. Activation of PTC readthrough occurs concomitantly with the appearance of cytoplasmic foci containing UPF proteins and mRNAs with nonsense mutations but lacking the P-body marker DCP1a. These findings demonstrate that in human cells, PTC readthrough occurs in novel ‘readthrough bodies’ and requires the presence of UPF proteins.
KEY WORDS: Readthrough body, Nonsense-mediated mRNA decay, UPF protein, Cytoskeleton, P-body
Summary: The cytoskeleton transports premature termination codon-containing mRNAs to be degraded by nonsense-mediated decay or to be read through in specific cytoplasmic foci called readthrough bodies.
INTRODUCTION
The cell cytoplasm notably contains a set of proteins forming the cytoskeleton. The cytoskeleton has three main components: (1) actin filaments consisting of actin subunits and actin-binding proteins, (2) microtubules assembled from tubulin units and microtubule-associated proteins, and (3) intermediate filaments (Fletcher and Mullins, 2010). Each element of the cytoskeleton plays a specific role. RNA transport has been mainly associated with actin filaments and microtubules, largely because the intermediate filaments are less dynamic and unable to self-organize (Aylett et al., 2011).
No association between the cytoskeleton and mRNAs harboring a premature termination codon (PTC) has yet been shown, but PTC-containing messenger ribonucleoprotein particles (PTC-mRNPs) are generated in the nucleus and exported to the cytoplasm. There, they are degraded by a surveillance mechanism called nonsense-mediated mRNA decay (NMD) (Fatscher et al., 2015; Hug et al., 2016; Karousis et al., 2016; Kervestin and Jacobson, 2012; Lejeune, 2017; Mühlemann and Lykke-Andersen, 2010; Popp and Maquat, 2014; Rebbapragada and Lykke-Andersen, 2009). In mammalian cells, several sets of factors are involved in NMD: UPF proteins [UPF1, UPF2, UPF3 (also called UPF3a) and UPF3X (also called UPF3b)], SMG proteins (SMG1, SMG5, SMG6, SMG7, SMG8 and SMG9), and components of the exon junction complex (EJC) (Chang et al., 2007; Yamashita et al., 2009). NMD not only targets mRNAs that have acquired a PTC by mutation but also regulates the expression pathways of certain ‘natural NMD substrate’ genes when a PTC arises after specific splicing events or under specific cellular conditions such as amino acid starvation or deprivation (He et al., 2003; Lelivelt and Culbertson, 1999; Mendell et al., 2004; Rehwinkel et al., 2005; Viegas et al., 2007). NMD is a cytoplasmic mechanism occurring soon after PTC-mRNP export from the nucleus (Singh et al., 2007; Trcek et al., 2013). PTC-mRNPs transit through P-bodies before undergoing NMD (Durand et al., 2007). This transport might be facilitated by the cytoskeleton, although this has not yet been demonstrated. P-bodies are not organelles per se, as they are not limited by a membrane and look more like aggregates of degradative enzymes and RNAs (Cougot et al., 2004; Ingelfinger et al., 2002; Sheth and Parker, 2003; van Dijk et al., 2002). The function of P-bodies in mammalian cells remains unclear. It has been proposed that P-bodies might store enzymes capable of degrading some RNAs or be sites of RNA decay, as shown in yeast (Aizer et al., 2014; Sheth and Parker, 2003).
Although NMD can reduce the level of a PTC-mRNA by over 95% (as compared to the level of the corresponding wild-type mRNA), the efficiency of NMD depends on the target (Kuzmiak and Maquat, 2006). A proportion of each PTC-mRNA, depending on its sensitivity to NMD, thus remains available either for degradation (without translation) via the general mRNA decay pathway (You et al., 2007) or for translation into truncated proteins (Anczuków et al., 2008; Dorard et al., 2011). In the presence of various agents (e.g. aminoglycosides), PTC-mRNAs can even be translated to full-length proteins via a PTC-readthrough mechanism. Compounds facilitating PTC readthrough essentially ‘force’ the translational machinery to introduce an amino acid at the PTC position. PTC readthrough can be concomitant with NMD inhibition (Correa-Cerro et al., 2005; Gonzalez-Hilarion et al., 2012) or not (Welch et al., 2007). PTC readthrough remains largely uncharacterized, notably as regards the molecular events favoring complete translation of the open reading frame (ORF), thanks to incorporation of a given tRNA, over recruitment of release factors to the ribosome A site. The work described here offers new insights into the mechanism of NMD. The results demonstrate that NMD requires the cytoskeleton and provide evidence that nonsense codon readthrough occurs in a specific cellular environment distinct from that of normal translation. They also show that UPF protein NMD factors are required for readthrough in human cells, in contrast to what has been found in other species, such as yeast (Harger and Dinman, 2004; Salas-Marco and Bedwell, 2005; Wang et al., 2001).
RESULTS
Role of the cytoskeleton in NMD
To demonstrate a possible link between the cytoskeleton and NMD, cytoskeleton-destabilizing reagents were used to disrupt the cytoskeleton prior to assessing the efficiency of NMD: cytochalasin D was used to prevent polymerization of actin filaments, and colchicine to inhibit that of microtubules. Cytoskeleton-stabilizing reagents (promoting polymerization or preventing de-polymerization) were also tested: jasplakinolide (JPK) (for actin filaments) and Taxotere (for microtubules). Cells derived from cystic fibrosis (CF) patients and harboring a nonsense mutation in the CF transmembrane conductance regulator (CFTR) gene were incubated for 48 h in the presence of each drug. The CF cell lines used were 6CFSMEo- and IB3, characterized by the F508del mutation (a deletion of three nucleotides encoding the phenylalanine residue at position 508) on one CFTR allele and, respectively, a mutation at codon 2 (Q2X) or 1282 (W1282X, where X represents a stop codon) on the other allele, which are mutations that have been shown to act as PTCs (Cozens et al., 1992; da Paula et al., 2005; Gonzalez-Hilarion et al., 2012). Previous studies have demonstrated, at both the RNA and protein levels, very low to no CFTR expression in these cell lines (da Paula et al., 2005; Farinha et al., 2004; Gonzalez-Hilarion et al., 2012; Tucker et al., 2012). Cytoskeletal disruption was assessed by immunostaining of the cytoskeletal structure (Fig. 1 and data not shown for IB3 cells). In both cell lines, the agents used were found to modify actin filaments or microtubule structure (according to the target of the agent) as compared to DMSO-treated control cells. The immunostaining patterns obtained were in keeping with the mode of action of each tested drug. Under physiological conditions, actin was detected primarily in the cytoplasm. Polymerization-blocking cytochalasin D caused it to aggregate in the cytoplasm, whereas treatment with JPK stabilized actin at the cell membrane (Fig. 1A and data not shown for IB3 cells). In DMSO-treated cells, tubulin immunostaining revealed the presence of tubulin in cytoplasmic fibers (Fig. 1B and data not shown for IB3 cells). Upon colchicine treatment, the tubulin fibers were lost, whereas Taxotere treatment stabilized tubulin fiber structure by inhibiting microtubule depolymerization.
Fig. 1.

Cellular distribution of the cytoskeleton under cytoskeleton inhibitor treatment. 6CFSMEo- cells were incubated with DMSO, cytochalasin D (CytoD), JPK, colchicine (COL) or Taxotere (TAX). After 48 h, the cells were fixed and permeabilized, and incubated with phalloidin to stain actin (A) or with anti-tubulin antibody followed by an Alexa Fluor 594-conjugated secondary antibody (red) for tubulin staining (B). Finally, their nuclei were visualized in blue with Hoechst 33342 stain. These results are representative of two independent experiments.
After a 48 h exposure to cytochalasin D, JPK, colchicine or Taxotere, the amount of endogenous CFTR mRNA was more than twice as high as in cells incubated with DMSO alone (Fig. 2A). Interestingly, the tested cytoskeleton disruptors inhibited NMD more effectively than amlexanox, a previously reported NMD inhibitor (Gonzalez-Hilarion et al., 2012). To rule out an indirect transcriptional effect, the level of CFTR pre-mRNA was measured in both cell lines. No significant variations were detected in the CFTR pre-mRNA level relative to the level of GAPDH mRNA (Fig. S1A). The effects of all cytoskeleton disruptors on the level of wild-type CFTR mRNA were also assessed in 16HBE14o- cells, which harbor no PTC in the CFTR gene (Cozens et al., 1994) (Fig. S1B). None of the treatments was found to influence the level of wild-type CFTR. This supports the idea that an intact cytoskeleton is required for NMD (Fig. 2A).
Fig. 2.

Cytoskeleton disruptors influence NMD or readthrough. (A) Cytoskeleton disruptors inhibit NMD. 6CFSMEo- cells (above) and IB3 cells (below) were incubated with DMSO (negative control), cytochalasin D (CytoD), JPK, colchicine (COL), Taxotere (TAX) or amlexanox (positive control) for 48 h. The level of CFTR mRNA was measured by qRT-PCR and normalized to the level of GAPDH mRNA. The five leftmost lanes represent two-fold serial dilutions of RNA from Calu-3 cells overexpressing CFTR mRNA. A histogram representation of the results is presented to the right of each gel (mean±s.d.; n=3). (B) Actin inhibitors activate PTC readthrough of nonsense-mutation-containing CFTR mRNA. 6CFSMEo- cells (above) or IB3 cells (below) were incubated with DMSO, CytoD, JPK, COL, TAX or amlexanox for 48 h before analysis of protein content by western blotting. The Ku80 protein was detected as a loading control. The three leftmost lanes represent two-fold serial dilutions of protein extract from Calu3 cells. (C) Actin inhibitors promote PTC readthrough on PTC-containing mRNA introduced by transfection. 6CFSMEo- cells were transfected with an expression vector encoding YFP-tagged GPx1 46 Ter or Flag-tagged SRSF7 as a reference plasmid. The transfected cells were incubated with DMSO, CytoD, JPK, COL, TAX or amlexanox for 48 h before analysis of protein content by western blotting. Flag-tagged SRSF7 protein was used as the loading control. The three leftmost lanes represent two-fold serial dilutions of protein extract from 6CFSMEo- cells transfected with an expression vector encoding GPx1 Norm (wild type). * indicates non-specific protein species. These results are representative of three independent experiments.
To make sure the above measurements of NMD were performed on viable cells, cell viability was assessed via propidium iodide staining, and the apoptosis rate was assessed by measuring Annexin V staining on cells exposed for 48 h to each of the cytoskeleton disruptors used. None of the four treatments was found to affect the viability of IB3 or 6CFSMEo- cells (Fig. S1C) or to increase their apoptosis (Fig. S1D). Thus, the NMD inhibition observed in Fig. 2A is consistent with the view that it is caused by cytoskeletal disruption and did not reflect a cytotoxic or apoptotic response to exposure to a tested drug (Fig. S1C,D).
To see whether the tested cytoskeleton disruptors inhibit only the NMD of nonsense-mutation-containing substrates or also that of ‘natural NMD substrates’ (Mendell et al., 2004; Sureau et al., 2001; Viegas et al., 2007), their effect was tested on three natural NMD substrates (Fig. S1E). None of the four cytoskeleton disruptors had any significant effect on levels of the chosen natural NMD substrate mRNAs. Inhibition of NMD without any effect on the regulation of natural NMD substrate mRNA levels has likewise been reported for the NMD inhibitor amlexanox (Gonzalez-Hilarion et al., 2012).
To rule out the possibility that the NMD inhibition observed with cytoskeleton inhibitors might result indirectly from inhibition of translation, 6CFSMEo- cells were incubated as described above (Fig. 2A), except that a modified amino acid was added to the medium in order to assay the levels of newly synthesized protein. In contrast to the response observed with cycloheximide (a translation inhibitor), translation proved to be unaffected by treatment with any tested drug (Fig. S1F). These results indicate that the NMD inhibition observed after treatment with cytochalasin D, JPK, colchicine or Taxotere was not an indirect effect due to altered translation.
Cytochalasin D and JPK promote PTC readthrough
Protein synthesis from PTC-mRNAs was also studied in the presence of cytoskeletal disruption, as NMD inhibitors such as G418 and amlexanox have been found to promote PTC readthrough (Correa-Cerro et al., 2005; Gonzalez-Hilarion et al., 2012). IB3 and 6CFSMEo- cells were incubated with one of the four cytoskeleton-disrupting agents or with amlexanox (as a positive control) before performing CFTR protein analysis (Fig. 2B). Full-length CFTR protein was detected in cells treated with amlexanox (as reported in Gonzalez-Hilarion et al., 2012), cytochalasin D or JPK. No full-length CFTR protein was detected after colchicine or Taxotere treatment. These findings indicate that disruption of actin filaments promotes readthrough on PTC-containing CFTR mRNAs, while disruption of microtubules does not. To get some idea of how general the differential effects of actin and tubulin disruption on readthrough might be, 6CFSMEo- cells were transfected with an expression construct encoding an YFP-tagged version of the glutathione peroxidase (GPx) 1 protein from mRNA that was either wild-type or carrying a PTC at codon 46 (46Ter-GPx1 mRNA) (Fig. 2C) and analyzed as above. Consistent with the observations depicted in Fig. 2B, PTC readthrough was observed on GPx1 Ter mRNA in the presence of amlexanox, cytochalasin D or JPK, but not upon treatment with colchicine or Taxotere. As well as the full-length GPX1 protein, the truncated GPX1 protein was also detected in the presence of cytochalasin D, JPK and amlexanox. Our findings suggest that these three drugs at least facilitate translation of the GPx1 Ter mRNA. Overall, they indicate that PTC readthrough is promoted when actin filament formation is disrupted, but not upon microtubule disruption (Fig. 2B,C). Since canonical translation is not affected by treatment with cytoskeleton-disrupting agents (Fig. S1F), these results suggest that the translation occurring during readthrough is distinct from the translation occurring during canonical translation. These observations imply that PTC readthrough may require a specific intracellular localization.
When actin filaments are disrupted, UPF1 and UPF3X localize to cytoplasmic foci that are distinct from P-bodies
Previous studies have shown that inhibition of NMD can cause changes in the cellular distribution of NMD factors and substrates (Durand et al., 2007). To address this question and assess whether disrupting the cytoskeleton causes intracellular redistribution of NMD factors and substrates, 6CFSMEo- cells were transfected with an expression vector encoding an YFP–UPF1 or YFP–UPF3X protein (human proteins) (Fig. S2) and treated 1 day later with each of the four cytoskeleton-disrupting agents. The DMSO control showed a homogeneous cytoplasmic distribution of UPF1, consistent with previous findings (Durand et al., 2007; Lykke-Andersen et al., 2000; Mendell et al., 2002; Serin et al., 2001; Unterholzner and Izaurralde, 2004). After treatment with cytochalasin D, JPK, colchicine, or Taxotere, YFP–UPF1 was found to accumulate in cytoplasmic granules that appeared not to show a concentration of actin or tubulin (Fig. 1; Fig. S2A,B). UPF3X localization (mainly nuclear under physiological conditions) was altered only by JPK treatment: the protein accumulated in a subset of cytoplasmic granules that likewise did not show a concentration of actin or tubulin (Fig. S2C,D).
To test the possible involvement of the cytoskeleton in transporting PTC-mRNPs, physical interactions between NMD factors and cytoskeletal proteins were assessed in 6CFSMEo- cells under physiological conditions and under cytoskeleton disruption (Fig. 3A). In the presence of DMSO alone, UPF1, UPF2 and UPF3X were found to co-immunoprecipitate with actin or tubulin, indicating an interaction between the cytoskeleton and NMD proteins. To test whether RNAs were required for this interaction, the immunoprecipitation was repeated in the presence of RNase (Fig. 3B). In the absence of RNA, the interaction between the cytoskeletal and UPF proteins was lost. When actin or tubulin was immunoprecipitated after treatment with cytoskeleton disruptors, the UPF proteins failed to co-immunoprecipitate with the cytoskeleton (Fig. 3). Loss of the interaction between cytoskeleton and NMD factors upon cytoskeleton disruption is consistent with the observed failure of the UPF proteins to colocalize with actin or tubulin (Fig. S2).
Fig. 3.

Cytoskeleton inhibitors interfere with the interaction between NMD factors and the cytoskeleton. (A) 6CFSMEo- cells were incubated with DMSO, cytochalasin D (CytoD), JPK, colchicine (COL) or Taxotere (TAX) for 48 h before protein extraction and immunoprecipitation (IP) with anti-actin, anti-tubulin, or, as a negative control, anti-Flag antibodies. Western blotting was performed to detect the presence of UPF1, UPF2, UPF3X, tubulin and actin. (B) The interaction between NMD factors and the cytoskeleton requires RNA. Protein-containing lysate of 6CFSMEo- cells was incubated with 10 mg/ml BSA or RNase A before immunoprecipitation with anti-actin, anti-tubulin, or anti-flag antibody (negative control). The level of UPF1, UPF2, UPF3X, tubulin, or actin protein was analyzed by western blotting. The three leftmost lanes of each western blot represent two-fold serial dilutions of protein extract from 6CFSMEo- cells. These results are representative of two independent experiments.
We next assessed whether the cytoplasmic foci showing an accumulation of UPF1 and UPF3X under cytoskeleton disruption were P-bodies. To address this, the cellular localization of UPF1 and UPF3X was compared with that of the P-body marker DCP1a (Cougot et al., 2004) (Fig 4; Fig. S3). Following colchicine or Taxotere treatment, colocalization of UPF1 foci and DCP1a foci exceeded 80%, indicating that UPF1 concentrates in P-bodies under these conditions. After cytochalasin D or JPK treatment, UPF1 foci and DCP1a foci showed only ∼10% colocalization. This indicates that under these conditions, UPF1 concentrates in cytoplasmic foci that are distinct from P-bodies. Similar results were obtained when the stained protein was endogenous UPF1, rather than a tagged version produced in transfected cells (Fig. S4). These results suggest that when actin filaments are compromised, a fraction of the UPF1 is sequestered in cytoplasmic foci that are not P-bodies, and that actin filaments may be required for UPF1 exit from these new cytoplasmic foci. The localization pattern of UPF3X upon JPK treatment was similar to that of UPF1; UPF3X was found to concentrate at foci showing only ∼10% colocalization with P-bodies (Fig. S3).
Fig. 4.

In 6CFSMEo- cells, UPF1 protein is concentrated partially or totally in P-bodies after cytoskeleton inhibitor treatment. Cells were transfected with constructs expressing YFP–UPF1 and GPx1 Ter and then incubated with DMSO, cytochalasin D (CytoD), JPK, colchicine (COL) or Taxotere (TAX) for 48 h. After fixation and permeabilization, the cells were incubated first with anti-DCP1a primary antibody and then with the Alexa Fluor 594-conjugated secondary antibody (red). Finally, the nuclei were visualized in blue by Hoechst 33342 staining. Green arrowheads indicate UPF1 cytoplasmic foci; red arrowheads indicate the P-body marker DCP1a; orange arrowheads indicate colocalization of UPF1 and DCP1a. The histogram represents the percentage of colocalization between UPF1 foci and DCP1a foci. Cells (mean±s.d.; n=10) from three different experiments were counted for each condition.
The specific cellular distribution of UPF proteins observed under cytoskeleton disruption raises the possibility that cytoskeleton disruption might also affect the cellular distribution of PTC-mRNPs. To test this possibility, the cellular localization of GPx1 Ter PTC-mRNAs (Moriarty et al., 1998) was analyzed after treatment with cytochalasin D, JPK, colchicine or Taxotere (Fig 5; Fig. S5). The localization patterns observed were the same as for the UPF1 and UPF3X proteins (Fig 4; Fig. S3). In contrast, the wild-type version of GPx1 mRNA (GPx1 Norm) did not localize to cytoplasmic foci after disruption of cytoskeleton (Fig. S6). The redistribution observed with GPx1 Ter PTC-mRNA thus appears specific to PTC-mRNAs.
Fig. 5.

In 6CFSMEo- cells, the NMD factor UPF1 colocalizes with NMD substrates under cytoskeleton disruptor treatment. 6CFSMEo- cells were transfected with constructs expressing GPx1 Ter and YFP–UPF1 and then incubated with DMSO, cytochalasin D (CytoD), JPK, colchicine (COL) or Taxotere (TAX) for 48 h. A fluorescence in situ hybridization (FISH) assay was performed to detect the cellular localization of GPx1 Ter mRNA. Nuclei are stained in blue with Hoechst 33342 stain. Green arrowheads indicate UPF1 cytoplasmic foci; red arrowheads indicate mRNA cytoplasmic GPx1 mRNA foci; orange arrowheads indicate colocalization of the UPF1 factor and NMD substrates. The histogram represents the percentage of colocalization between UPF1 foci and GPx1 Ter. Cells (mean±s.d.; n=10) from three different experiments were counted for each condition.
Consistent with the above observations, GPx1 Ter mRNA colocalization with DCP1a was high after colchicine or Taxotere treatment, and low after cytochalasin D or JPK treatment (Fig. S7). These results indicate that NMD substrates and NMD factors are sequestered in P-bodies when microtubules are disrupted but concentrate primarily in other, novel, cytoplasmic foci when actin filaments are disrupted.
When readthrough is activated, PTC-mRNPs predominantly localize to the novel cytoplasmic foci
The identity of the novel foci containing UPF1, UPF3X and PTC-mRNAs after actin filament disruption was investigated by comparing the cellular localization of the UPF proteins with those of various organelle markers. No colocalization was observed between UPF1 or UPF3X and the stress granule marker protein eIF3β (Brown et al., 2011) or the autophagosome marker protein LC3B (also known as MAP1LC3B) (Tanida et al., 2008) (Figs S8, S9). This indicates that upon actin filament disruption, NMD factors and substrates concentrate in cytoplasmic foci that are distinct from P-bodies, stress granules and autophagosomes, and these foci are therefore a new category of cytoplasmic foci.
The fact that the new cytoplasmic foci were detected after cytochalasin D or JPK treatment suggests that they could be related to the readthrough process, since readthrough was also observed after cytochalasin D or JPK treatment (Figs 2B,C, 4–6; Figs S3 and S5–7). To confirm this hypothesis, the cellular localization of UPF1 was studied after treatment with G418, a reference readthrough-inducing molecule (Gatti, 2012; Manuvakhova et al., 2000; Shalev et al., 2013) (Fig. 6), or amlexanox (Fig. S10). When readthrough was induced with G418 or amlexanox, a limited fraction of the UPF1 was found sequestered in P-bodies, but most of it was found sequestered in another type of cytoplasmic foci containing no DCP1a. Under G418 treatment, furthermore, PTC-mRNAs fully colocalized with UPF1-containing cytoplasmic foci. In contrast, wild-type mRNA failed to concentrate in cytoplasmic foci under G418 treatment (Fig. S11). Overall, these results suggest that upon readthrough activation, PTC-mRNPs are moderately concentrated in P-bodies but are predominantly found in a new type of cytoplasmic foci.
Fig. 6.

G418 causes UPF1 to localize to cytoplasmic foci containing NMD substrates but not the P-body marker DCP1a. 6CFSMEo- cells were transfected with constructs expressing YFP-UPF1 or GPx1 Ter before treatment with G418 at 400 µg/ml for 48 h. Left panel, cells were incubated sequentially with anti-DCP1a antibody and Alexa Fluor 594-conjugated secondary antibody (red). Right panel, cells were incubated with a GPx1 fluorescence in situ hybridization (FISH) probe. The percentage of colocalization between UPF1 and DCP1a (left) or between UPF1 and GPx1 Ter mRNA (right) is presented in histograms at the bottom of the figure. Cells (mean±s.d.; n=10) from three different experiments were counted for each condition. Nuclei were stained with Hoechst 33342 solution (blue). Green arrowheads indicate UPF1 cytoplasmic foci; red arrowheads indicate P-bodies or foci containing GPx1 mRNA; orange arrowheads indicate colocalization foci.
Further evidence supporting the existence of readthrough-related foci that are distinct from P-bodies was obtained by measuring the distance between the new cytoplasmic foci and P-bodies with ImageJ software (Fig. S12A). Under colchicine or Taxotere treatment, the DCP1a and the UPF1 foci were mainly found to overlap. Upon G418 or cytochalasin D treatment, the DCP1a foci and UPF1 foci appeared to be separated from each other, the distance between them usually being less than 2 µm.
To study the dynamics of the new cytoplasmic foci relative to P-bodies, a fluorescence recovery after photobleaching (FRAP) assay was performed under different conditions. 6CFSMEo- cells were transfected with two expression vectors: one encoding RFP–DCP1a, as a marker of P-bodies, and one encoding YFP–UPF1, so as to observe the dynamics of the new cytoplasmic foci (Fig. S12B). Upon cytochalasin D treatment, no fluorescence recovery was observed after photobleaching. This indicates that both DCP1a and UPF1 require the cytoskeleton to move, respectively, to P-bodies or to the new cytoplasmic foci. Under G418 treatment, some fluorescence was recovered by both cytoplasmic foci, indicating that both proteins could still move. In P-bodies, the maximum recovery (∼20%) was reached in 20 s, as previously reported (Andrei et al., 2005). A quite similar fluorescence recovery rate was observed for the new cytoplasmic foci (Fig. S12B, green curves). The two structures thus show similar dynamics.
To rule out the possibility that the new cytoplasmic foci might be an artifact related to use of a drug, an siRNA against actin was designed. The use of this siRNA in 6CFSMEo- cells promoted inhibition of NMD, as shown by an increase in CFTR mRNA (Fig. S13A). It was also found to activate readthrough, as shown by detection of full-length CFTR protein (Fig. S13A). Consistent with these results, cytoplasmic foci containing UPF1 and GPx1 Ter mRNA but not DCP1a were detected upon actin knockdown (Fig. S13B).
The new cytoplasmic foci are the site of readthrough in the cell
Although we were unable to detect RPL13, eIF5A or PABP protein in the new cytoplasmic foci (data not shown), likely because readthrough would occur at a much lower level than canonical translation, we hypothesized that these foci might be the place where readthrough occurs. To validate this hypothesis, we examined whether readthrough proteins might be synthesized in the new cytoplasmic bodies. For this, the GPx1 Ter gene was fused C-terminally to the sequence encoding the Neptune tag under the control of a TET-inducible promoter. The resulting construct was expressed in 6CFSMEo- cells after transient transfection, together with an expression vector encoding YFP–UPF1 and another encoding RFP–DCP1a. The transfected cells were incubated with cytochalasin D for 48 h before addition of doxycycline to the medium to promote synthesis of the C-terminally Neptune-tagged version of GPx1 Ter. It was reasoned that the appearance of Neptune fluorescence should show where PTC readthrough occurs. Pictures were taken with a confocal microscope every 2 min over a 1 h period starting about 10 min after doxycycline addition (Movie 1; Fig. 7). The results show that the Neptune signal was stronger in samples exposed to doxycycline than it was prior to doxycycline exposure. The presence of a Neptune signal in the absence of doxycycline reflects some leakage of the promotor, as previously reported (Zeng et al., 1998). Interestingly, the Neptune signal was confined to certain cytoplasmic foci, and the location of these foci was compared to that of DCP1a and UPF1. All newly detected Neptune signals were observed in cytoplasmic foci containing UPF1 but not DCP1a. This confirms that readthrough occurs in the new cytoplasmic foci. In addition, the translation observed in the new cytoplasmic foci appeared to concern only PTC-containing mRNAs, since no translation of a wild-type GPx1–Neptune-encoding mRNA was found to occur there (Movie 2). When the experiment was performed in the presence of the translation inhibitor cycloheximide, no Neptune signals were detected in the cells (Movie 3). We thus coined the term ‘readthrough bodies’ to designate the cytoplasmic foci where readthrough occurs.
Fig. 7.
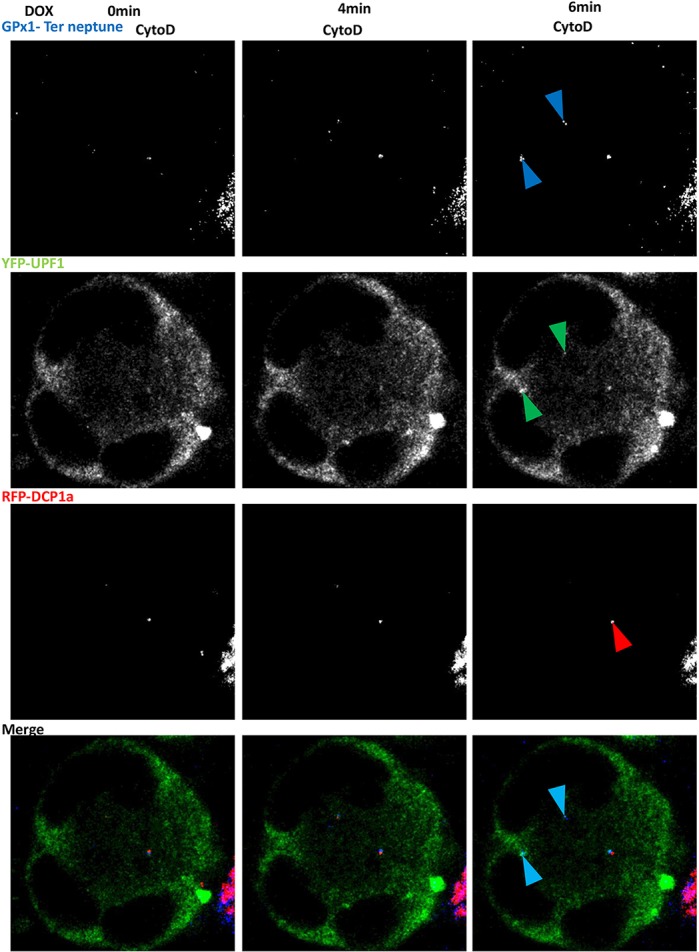
Readthrough GPx1 Ter–Neptune proteins colocalize with UPF1 under cytochalasin D treatment. 6CFSMEo- cells transfected with plasmids encoding YFP–UPF1 (green), RFP–DCP1a (red), and GPx1 Ter–Neptune (blue) were treated with cytochalasin D (CytoD). After 48 h, doxycycline was added to the medium and pictures were taken every 2 min for 1 h under confocal laser scanning microscopy. Pictures taken at 0, 4 and 6 min after adding doxycycline are shown here. Blue arrowheads indicate the GPx1 protein; green arrowheads indicate the UPF1 protein; red arrowheads indicate P-bodies; cyan arrowheads indicate colocalization foci.
UPF proteins are required for PTC readthrough
The presence of UPF1 in readthrough bodies suggests that UPF1 could play a role in readthrough. To test this possibility, 6CFSMEo- cells were transfected with a control siRNA or with an siRNA targeting endogenous UPF1 (Mendell et al., 2002). The cells were then incubated with DMSO, cytochalasin D or G418 before extracting proteins and detection of full-length CFTR protein by western blotting. As shown in Fig. 8A, and consistent with the results of Fig. 2B,C, the CFTR protein was detected in control-siRNA-treated cells upon incubation with cytochalasin D or G418, but not after incubation with DMSO (lanes 2 and 3 compared to lane 1). In UPF1-knockdown cells, the CFTR protein was not detected, even in the presence of cytochalasin D or G418 (lanes 7–9). Hence, UPF1 is required for nonsense mutation readthrough. To confirm the specificity of downregulation by the UPF1-targeting siRNA, cells were transfected with a construct expressing an siRNA-resistant isoform of the UPF1 protein. The siRNA-resistant isoform of UPF1 was found not to interfere with the production of readthrough CFTR under cytochalasin D or G418 treatment, as demonstrated in the presence of the control siRNA (lanes 4–6). The effect of the UPF1-targeting siRNA was inhibited in the presence of the siRNA-resistant isoform of UPF1, since full-length CFTR was detected in cells treated with cytochalasin D or G418 (lanes 10–12). This validates the conclusion that UPF1 is necessary for PTC readthrough.
Fig. 8.

Downregulation of UPF proteins inhibits PTC readthrough. 6CFSMEo- cells were transfected with (A) control or UPF1 siRNA, with or without a plasmid expressing an siRNA-resistant UPF1 isoform (UPF1R), (B) control or UPF2 siRNA, or (C) siRNA control or UPF3X siRNA. At 24 h after transfection, the cells were incubated with DMSO, cytochalasin D (CD) or 400 µg/ml G418 for 48 h before estimation of the protein content by western blotting. Levels of CFTR, UPF1, UPF2 and tubulin (as loading control) were estimated. The three leftmost lanes represent two-fold serial dilutions of protein extract from Calu-3 cells. These results are representative of three independent experiments.
To determine whether UPF1 is the only NMD factor required for readthrough, UPF2 or UPF3X was knocked down with siRNA and readthrough was induced with cytochalasin D or G418 (Fig. 8B,C). A significant decrease in the level of UPF2 or UPF3X protein was found to fully inhibit readthrough induced by cytochalasin D or G418. Overall, the results of Fig. 8 demonstrate that readthrough requires the UPF proteins in human cells.
Since readthrough is inhibited when UPFs are downregulated, another way to confirm that readthrough occurs in readthrough bodies is to check whether readthrough bodies persist in the presence of a UPF-targeting siRNA in cells incubated with readthrough molecules (Fig. S13). In the presence of a control siRNA, both G418 and cytochalasin D were found to promote stabilization of the NMD substrate GPx1 Ter mRNA and its concentration in P-bodies and readthrough bodies (Fig. S13B–F). When the UPF proteins were knocked down with siRNAs (Fig. S13B,D,E), labeled GPx1 Ter mRNA became detectable in DMSO-treated cells because of the resulting NMD inhibition (see Fig. S13F for the efficiency of NMD inhibition). As previously reported (Durand et al., 2007), the GPx1 Ter signal appeared to be homogeneously distributed throughout the cytoplasm in DMSO-treated cells. In the presence of G418 or cytochalasin D, GPx1 Ter mRNA did not concentrate in P-bodies or readthrough bodies. This is consistent with the view that the UPF proteins are required to form readthrough bodies (Fig S13D,E).
DISCUSSION
In cells, the cytoskeleton ensures transport of various cellular structures and molecules such as proteins and nucleic acids, including mRNAs (Bassell et al., 1999; Fletcher and Mullins, 2010; Lopez de Heredia and Jansen, 2004). We thus hypothesized that PTC-mRNAs might be transported by the cytoskeleton to their degradation sites in the cytoplasm. In support of this hypothesis, we demonstrate here that disrupting actin or tubulin filaments leads to inhibition of NMD (Figs 1,2A) and that the observed inhibition is not an indirect effect due to altered transcription or increased cell death (Fig. S1). In addition, we show that cytoskeleton disruptors cause stronger NMD inhibition than amlexanox, without affecting the translation mechanism (Fig. S1F) or the regulation of natural NMD substrates (Fig. S1E). Our results thus show that cytoskeleton disruptors constitute, alongside translation inhibitors and apoptosis inducers (Jia et al., 2015; Popp and Maquat, 2015), a new family of effective NMD inhibitors.
Since translation appears unaffected by cytoskeleton inhibitors, and since we and others have previously shown that some NMD inhibitors can also activate readthrough (Correa-Cerro et al., 2005; Gonzalez-Hilarion et al., 2012), we investigated whether full-length proteins can be synthesized from PTC-mRNAs after cytoskeleton disruption (Fig. 2B,C). Interestingly, our results demonstrate readthrough in the presence of actin filament disruptors but not in the presence of microtubule disruptors, suggesting that readthrough requires microtubules and that the absence of actin filaments favors PTC readthrough.
Although we do not favor it, we cannot exclude the hypothesis that the activation of PTC readthrough and/or the inhibition of NMD by cytoskeleton disruptors might be an indirect effect. For example, NMD inhibition might be caused by failure of an essential NMD factor to be transported to P-bodies. Alternatively, the alteration of actin filaments by cytochalasin D or JPK might lead to the absence of a factor responsible for PTC recognition on PTC-mRNAs, thus causing increased PTC readthrough. In-depth analysis of the protein composition of PTC-mRNPs in cells treated with cytochalasin D or JPK should provide information supporting or ruling out these possibilities and should contribute to better understanding of the molecular role of the cytoskeleton in NMD and PTC readthrough.
We further show that P-bodies are the structures where UPF1 concentrates when the microtubules are disrupted (Fig. 4), consistent with sequestration of NMD factors in P-bodies in some cases of NMD inhibition (Durand et al., 2007). When actin filaments are disrupted, on the other hand, UPF1 and UPF3X concentrate to some extent in P-bodies, but are mostly found in another type of cytoplasmic foci that is distinct from stress granules and autophagosomes (Fig. 4; Figs S3, S4, S9). PTC-mRNAs also concentrate in P-bodies upon microtubule disruption and but are also mostly found in the new type of UPF1-containing cytoplasmic foci upon actin filament disruption (Fig. 5; Figs S5, S7). The fact that this cellular distribution pattern is not observed with wild-type mRNAs (Fig. S6) indicates that PTC-mRNPs are specifically addressed to P-bodies and to the new cytoplasmic foci.
Since the same conditions appear to favor both PTC readthrough and the appearance of the new cytoplasmic foci, we examined whether the latter might be the place where readthrough occurs. Consistent with this idea, the new cytoplasmic foci were detected when amlexanox or the reference readthrough molecule G418 was used to elicit PTC readthrough (Fig. 6; Fig. S10). In addition, newly synthesized readthrough proteins were detected in the new cytoplasmic foci and not in P-bodies. This demonstrates that readthrough occurs in the newly discovered bodies, which we have therefore named readthrough bodies (Fig. 7; Movie 1). Our results indicate that readthrough and the translation of wild-type mRNAs occur in distinct environments (Movie 2). They suggest that readthrough requires specific factors and is subject to dedicated regulation, requiring further investigation. In support of this idea, UPF1 and UPF3X are found in readthrough bodies, and downregulation of UPF1, UPF2 or UPF3X results in inhibition of readthrough (Fig. 8; Fig. S13). These results reveal a new function for UPFs: a role in PTC readthrough. This role is an additional argument in favor of separation of readthrough from translation, since downregulation of UPF proteins has no impact on translation (Isken et al., 2008) but does affect PTC readthrough. Our observation that downregulation of UPF proteins abolishes readthrough highlights an additional difference in the roles played by UPF proteins in yeast and mammals. The situation in yeast remains somewhat controversial: some studies have demonstrated increased readthrough, attributed to defective translation termination, in yeast cells lacking of one of the UPF proteins (Salas-Marco and Bedwell, 2005; Wang et al., 2001), while another has concluded that UPF proteins have no involvement in readthrough in yeast (Harger and Dinman, 2004). The present study on human cells leads to a very different conclusion: that in mammals, these proteins play a role in PTC readthrough, since downregulation of UPF1, UPF2 or UPF3X abolishes this process (Fig. 8). The mechanism of readthrough remains largely unexplored, and no specific proteins involved in this process had been previously identified in mammals. Our finding that the absence of UPF proteins impairs readthrough and not translation sheds some light on the molecular mechanism of readthrough.
Recently, readthrough of physiological stop codons has been demonstrated in the case of four genes in human cells (Loughran et al., 2014). Further studies will be necessary to determine whether UPF proteins are also required for readthrough of physiological stop codons. It will be also interesting to identify the cellular site of physiological stop codon readthrough, in order to determine whether readthrough bodies are specialized solely in PTC readthrough or whether stop codon readthrough also occurs there.
The relationship between P-bodies and readthrough bodies needs to be further studied. One question is: do readthrough bodies derive from P-bodies through addition/removal of certain factors, or are they pre-existing entities that exchange certain mRNPs with P-bodies? The latter hypothesis seems more plausible, since P-bodies are detected in the presence and absence of the cytoskeleton and thus appear to be independently stable. In addition, both structures are found in close vicinity to each other (Fig. S12). A clear demonstration of transfer will require identifying a specific and stable marker of readthrough bodies, just as the stability of P-bodies under physiological conditions can be demonstrated by monitoring the cellular localization of a permanent P-body component such as DCP1a.
Finally, our results show that cytoskeleton disruptors can inhibit NMD and/or activate PTC readthrough. Some molecules, such as Taxotere and colchicine, are used to treat pathologies (cancer and gout, respectively, in the case of these agents). Given the effects of these molecules on NMD and readthrough, it would be interesting to investigate their use in, for example, treatment of cancers related to the presence of a nonsense mutation in a tumor suppressor gene. Such applications would constitute a new type of targeted therapy for cancers.
MATERIALS AND METHODS
Chemicals
Cytochalasin D and jasplakinolide (JPK) were obtained from Enzo Life Sciences. Colchicine was purchased from Sigma-Aldrich (Lyon, France) and Taxotere was obtained from Selleck Chemicals. Amlexanox was purchased from Sequoia Research. Each molecule was dissolved in DMSO except G418 (GIBCO), which was dissolved in water. Cytochalasin D, JPK and Taxotere were each used at 1 µM, colchicine at 10 µM, amlexanox at 25 µM, and G418 at 400 µg/ml.
Cell culture
IB3 cells (ATCC-CRL-2777) harboring a nonsense mutation in exon 20 of the CFTR gene were grown in LHC-8 (without gentamicin) supplemented with 10% fetal bovine serum (FBS) and Zell shield (Minerva Biolabs, Berlin, Germany) to prevent mycoplasma contamination. 6CFSMEo- cells (Cozens et al., 1992) harboring a nonsense mutation in exon 1 of the CFTR gene were grown in α-MEM supplemented with 10% FBS, 1 mM L-glutamine and Zell shield. Calu-3 cells (ATCC-HTB-55) overexpressing CFTR protein were grown on RPMI with 10% FBS and Zell shield. Cells were incubated with the specified molecule or with DMSO as a negative control for 48 h at 37°C under 5% CO2.
qRT-PCR analysis
Quantitative RT-PCR (qRT-PCR) was performed as described previously (Gonzalez-Hilarion et al., 2012). The primer sequences used in this study were for: GAPDH (sense, 5′-CATTGACCTCAACTACATGG-3′; antisense 5′-GCCATGCCAGTGAGCTTCC-3′), CFTR (sense, 5′-GGCCAGAGGGTGGGCCTCTT-3′; antisense, 5′AGGAAACTGTTCTATCACAG-3′), CFTR pre-mRNA (sense, 5′-TTGATGCCTAGAGGGCAGAT-3′; antisense, 5′-TGTCAAAGGGATTGGGAGGG-3′), SC35 (also known as SRSF2) (sense, 5′-CCTCTTAAGAAAATGCTGCGGTCTC-3′; antisense, 5′-ATCAGCCAAATCAGTTAAAATCTGC-3′), NAT9 (sense, 5′-ATTGTGCTGGATGCCGAGA-3′; antisense, 5′-ACCTAGCGTGGTCACTCCGTA-3′), and TBL2 (sense, 5′- GCAGTCATTTACCACATGC-3′; antisense, 5′-TATTGTTTCTGCTTCTTGGAT-3′).
Western blot analysis
Proteins were extracted in the following lysis buffer: 50 mM Tris-HCl pH 7.0, 20 mM EDTA and 5% SDS. The CFTR protein was analyzed by separating on a 3–8% precast NuPAGE gel (Life technologies) and transfer to nitrocellulose membrane by i-blot transfer (Life technologies). Migration of all the other proteins was carried out in a 10% SDS-PAGE gel. After migration, the proteins were transferred to a nitrocellulose membrane and incubated with primary antibodies overnight at 4°C before incubation with a secondary antibody [111-035-003 (rabbit) or 115-035-003 (mouse), Jackson Immuno Research, West Grove, PA] at 4°C for 2 h. Finally, the proteins were observed with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce-Biotechnology, Rockford, IL, USA). The primary antibodies used were: rabbit anti-UPF1 antibody at 1:5000 (ab86057, Abcam, Paris, France), rabbit anti-UPF2 antibody at 1:5000 [antibody raised against the following synthetic peptide: H2N-MPAERKKPASMEEKDC-CONH2 (Eurogentec, Angers, France)], rabbit anti-UPF3X antibody at 1:5000 (ARP40998_T100, Aviva Systems Biology, San Diego, CA), rabbit or mouse anti-actin antibody at 1:1000 (ab8227/ab3280, Abcam), rabbit anti-tubulin antibody at 1:1000 (PM054, Epitomics, Burlingame, CA), anti-human CFTR C-terminus monoclonal antibody (clone 24-1) at 1:1000 (R&D Systems, Abingdon, UK), rabbit anti-GFP antibody at 1:1000 (G1544, Sigma-Aldrich, Lyon, France), rabbit anti-flag at 1:1000 (F1804, Sigma-Aldrich) or rabbit anti-Ku80 (also known as XRCC5) antibody at 1:1000 (Epitomics).
Immunocytochemistry
6CFSMEo- cells were incubated on 12-mm coated slides and transfected with 250 ng plasmids expressing YFP–UPF1 or YFP–UPF3X and pCMV-GPx1 Ter. Coating was performed with a solution made of LHC-8 medium containing 0.1 mg/ml BSA, 0.03 mg/ml collagen I, rat tail (Gibco) and 0.01 mg/ml human fibronectin (Sigma-Aldrich). At 24 h after transfection, the cells were treated with 1 µM cytochalasin D, 1 µM JPK, 10 µM colchicine, 1 µM Taxotere or DMSO for 48 h. They were then fixed with 10% formalin for 10 min at room temperature, washed three times with Dulbecco's PBS, and permeabilized with 70% ethanol for 1 h at 4°C. Fixed cells were incubated with primary antibodies [anti-actin (Abcam, ab3280, dilution 1:1000), anti-tubulin (Epitomics, PM054, dilution 1:1000), anti-DCP1a (Sigma-Aldrich, D5444, dilution 1:1000), anti-eIF3β (Santa Cruz Biotechnology, sc-374156, dilution 1:50), anti-LC3B (Novus Biologicals, NB600-1384, dilution 1:200) or phalloidin (Molecular Probes – Life Technologies)] for 1 h at 4°C, washed three times with 0.05% Tween-20 in PBS, and incubated with goat anti-rabbit-IgG antibody conjugated to Alexa Fluor 594 or 488 (Life Technologies) for 1 h at 4°C. Finally, the cells were incubated with Hoechst 33342 stain for 2 min at room temperature before observation under a ZEISS AxioImager Z1 ApoTome microscope (Obj PLAN APOCHROMAT ×40, NA 1.40 oil immersion; Filter Sets FS49-DAPI, FS38-FITC and FS43-TexasRed).
Immunoprecipitations
6CFSMEo- cells were lysed in lysis buffer [1% Triton X-100, 150 mM NaCl, 10 mM Tris-HCl pH 7.4, 1 mM EDTA pH 8.0, 1 mM EGTA PH8.0, 0.2 mM sodium orthovanadate, 0.5% NP-40, Halt™ protease, phosphatase inhibitor cocktail (Thermo Scientific)]. To eliminate nonspecific interactions, protein G (for mouse antibody) and A (for rabbit antibody) agarose beads were added to the cell extracts and incubated for 30 min at 4°C. The cell extracts were then incubated with mouse anti-actin antibody (Abcam, ab3280, dilution 1:1000) or rabbit tubulin antibody (Epitomics, PM054, dilution 1:1000). After 2 h at 4°C protein G and A agarose beads were added to the cell extracts and incubated for 2 h at 4°C. The beads were then washed four times with lysis buffer before eluting proteins from beads with 2× sample loading buffer.
Knockdown with siRNA
ICAFectin™ 442 reagent (In Cell Art, Nantes, France) was used to transfect 6CFSMEo- cells with 100 nM siRNA [siRNA control (Eurogentec), siRNA UPF1 (5′-AAGATGCAGTTCCGCTCCATTTT-3′), siRNA UPF2 (5′-GAAGTTGGTACGGGCACTC-3′), siRNA UPF3X (5′-GGAGAAGCGAGTAACCCTG-3′) (Sigma Aldrich) or siRNA actin (5′-CCCACAACGTGCCCATTTA-3′) (Thermo Scientific)], and pCMV-GPX1 Norm or pYFP-GPx1 Ter. At 24 h post transfection, the cells were incubated with DMSO or with 1 µM cytochalasin D or G418 for an additional 48 h.
Image analysis
Images were analyzed with the ImageJ software (NIH). Spots from green and red channels were detected using a local maxima detection and thresholding plugin, and counted using a particle analysis function. Logical combination of obtained masks enabled further counting of spots showing both red and green fluorescence.
Translation efficiency
6CFSMEo- cells were incubated with DMSO, 1 µM cytochalasin D, 1 µM JPK, 10 µM colchicine or 1 µM Taxotere for 48 h or with 200 µg/ml cycloheximide (as a positive control) for 4 h before harvest. Cells were then treated as previously described (Gonzalez-Hilarion et al., 2012).
Cell viability and apoptosis rate measurements
Cell viability and the apoptosis rate were measured as previously described (Jia et al., 2015).
Fluorescence in situ hybridization assays
6CFSMEo- cells were transfected with construct pCMV-GPx1 Ter and constructs expressing YFP–UPF1 or YFP–UPF3X. After 24 h, the cells were exposed to DMSO, 1 µM cytochalasin D, 1 µM JPK, 10 μM colchicine or 1 µM Taxotere for 48 h and then fixed in 10% formalin solution for 10 min at room temperature and permeabilized in 70% ethanol at 4°C for 1 h. The cells were then washed twice with 2× SSC before exposure to prehybridization buffer (125 µg/ml tRNA, 500 µg/ml herring DNA, 2xSSC, 1% BSA, 10% dextran sulfate, 50% formamide, 10 mM vanadyl ribonucleoside complexes) for 1 h at 37°C. Next, the cells were incubated in hybridization buffer (prehybridization buffer with 0.1 ng/µl Texas Red or Cy3-labeled probes; sequence, 5′-ACAGCAGGGCTTCTATATCGGGTTCGATGTCGATGGTGCGAAAGCGCCTG-3′) overnight at 4°C, washed twice with 2× SSC containing 10% formamide and once with 2× SSC+0.1% Triton X-100, and twice with 1× SSC and finally incubated with Hoechst 33342 stain for 2 min at room temperature.
Construction of the plasmid expressing GPx1–Neptune
The GPx1Ter gene was amplified by PCR from plasmid pCMV-GPx1 Ter, with the following primers: sense, 5′-AGCCTCCGCGGCCCCGAATTATGTCTGCTGCTCGGCTCTCCGC-3′; antisense, 5′-TCTTCGCCCTTAGACACCATGGGGTTGCTAGGCTGCTTGGACA,3′; Neptune-tag cDNA was amplified by PCR from the vector pmNeptune-N1 (generous gift from Prof. Roger Tsien, Department of Pharmacology, Howard Hughes Medical Institute Laboratories at the University of California, San Diego, USA), using the following primers: sense, 5′-TGTCCAAGCAGCCTAGCAACCCCATGGTGTCTAAGGGCGAAGAGCT-3′; antisense, 5′ATCTTATCATGTCTGGATCTTACTTGTACAGCTCGTCCATG-3′. Both amplicons were used as templates to amplify, by PCR, the GPx1 Ter-Neptune construct. The Infusion cloning kit (Clontech, Mountain View, CA,) was then used to introduce this last amplicon into the pSTAR vector (Zeng et al., 1998) linearized with the BamHI and EcoRI restriction enzymes.
Live imaging
Live imaging was performed under a laser scanning confocal microscope (Zeiss Axio Observer Z1; LSM 880). Cells were maintained at 37°C in the incubation chamber. Images were taken in lambda mode (Scan Modul Quasar 34 channels) and Spectral Unmixing (Zen, Zeiss software) was used to separate signals of different wavelengths (corresponding, respectively, to YFP, dsRed and Neptune). Observations were made with a 63× P1N APO DIC II, NA 1.4 oil immersion objective, at the frequency of one image every 2 min.
FRAP assay
FRAP experiments were performed under a Zeiss LSM880 confocal microscope. A 488 nm argon laser was used to bleach YFP–UPF1 spots, and a 561 nm diode was used to bleach the RFP–DCP1a spots (200 iterations at 100% power). The sequence was programmed into the Zen (Zeiss) software. For each experiment, we first recorded two images, bleached the sample, and then recorded images every 2 s for at least 100 s. We imaged several cells, and in each cell, we bleached several bodies.
Sequences of images were analyzed with the ImageJ software (NIH). For each time point, we measured the mean intensity of regions of interest around GFP or RFP foci. Curves were normalized because the intensity was not exactly the same and bleaching was not total in every foci (intensity before bleaching was set to 1, with offset at 0). The graphical representation of the curves was made with MATLAB®.
Measuring the distance between P-bodies and readthrough bodies
Distance analysis was performed with ImageJ. A script was written to measure, for each green foci, the distance of the nearest red foci (available upon request). To do this, we first needed to threshold the images to obtain binary masks of green and red foci. Then, we drew regions of interest with increasing diameters around the different green body. By successive measurements, we determined at which diameter of the region of interest a red body appeared. We analyzed several foci for each condition, and a boxplot representation of the results was made with MATLAB®.
Acknowledgements
The authors wish to thank Prof. Lynne Maquat, Prof. Wanjin Hong, Dr Roger Y. Tsien, Prof. Pascale Fanen, Dr Laurent Heliot, Dr Edouard Bertrand and Dr James Stévenin for reagents and Prof. Yves Lemoine and Prof. Alex Duval for helpful discussions. The authors thank the BioImaging Center Lille for access to instruments. We dedicate this article to the memory of Prof. Dieter C. Gruenert.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.J., S.G.-H., E.W., D.C.G., F. Lejeune; Methodology: J.J., S.G.-H., E.W., F. Lejeune; Software: E.W.; Formal analysis: E.W., D.C.G., F. Lejeune; Investigation: J.J., S.G.-H., E.W., F. Lejeune; Writing - original draft: D.C.G., F. Lejeune; Writing - review & editing: J.J., S.G.-H., E.W., F. Lafont, D.T., F. Lejeune; Supervision: F. Lafont, F. Lejeune; Project administration: F. Lejeune; Funding acquisition: F. Lafont, D.C.G., D.T., F. Lejeune
Funding
J.J. received funding from the Association Française contre les Myopathies and from Association Vaincre la Mucoviscidose. F. Lejeune is an Institut National de la Santé et de la Recherche Médicale (INSERM) researcher, and received funding for this project from Association Vaincre la Mucoviscidose, Association Française contre les Myopathies, Cancéropôle Nord Ouest and Fondation ARC pour la Recherche sur le Cancer. D.C.G. was supported by funds from Pennsylvania Cystic Fibrosis Inc., Cystic Fibrosis Research Inc. and the National Institutes of Health (grant DK104681). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.198176.supplemental
References
- Aizer A., Kalo A., Kafri P., Shraga A., Ben-Yishay R., Jacob A., Kinor N. and Shav-Tal Y. (2014). Quantifying mRNA targeting to P-bodies in living human cells reveals their dual role in mRNA decay and storage. J. Cell Sci. 127, 4443-4456. 10.1242/jcs.152975 [DOI] [PubMed] [Google Scholar]
- Anczuków O., Ware M. D., Buisson M., Zetoune A. B., Stoppa-Lyonnet D., Sinilnikova O. M. and Mazoyer S. (2008). Does the nonsense-mediated mRNA decay mechanism prevent the synthesis of truncated BRCA1, CHK2, and p53 proteins? Hum. Mutat. 29, 65-73. 10.1002/humu.20590 [DOI] [PubMed] [Google Scholar]
- Andrei M. A., Ingelfinger D., Heintzmann R., Achsel T., Rivera-Pomar R. and Lührmann R. (2005). A role for eIF4E and eIF4E-transporter in targeting mRNPs to mammalian processing bodies. RNA 11, 717-727. 10.1261/rna.2340405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylett C. H. S., Löwe J. and Amos L. A. (2011). New insights into the mechanisms of cytomotive actin and tubulin filaments. Int. Rev. Cell Mol. Biol. 292, 1-71. 10.1016/B978-0-12-386033-0.00001-3 [DOI] [PubMed] [Google Scholar]
- Bassell G. J., Oleynikov Y. and Singer R. H. (1999). The travels of mRNAs through all cells large and small. FASEB J. 13, 447-454. [DOI] [PubMed] [Google Scholar]
- Brown J. A. L., Roberts T. L., Richards R., Woods R., Birrell G., Lim Y. C., Ohno S., Yamashita A., Abraham R. T., Gueven N. et al. (2011). A novel role for hSMG-1 in stress granule formation. Mol. Cell. Biol. 31, 4417-4429. 10.1128/MCB.05987-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.-F., Imam J. S. and Wilkinson M. F. (2007). The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 76, 51-74. 10.1146/annurev.biochem.76.050106.093909 [DOI] [PubMed] [Google Scholar]
- Correa-Cerro L. S., Wassif C. A., Waye J. S., Krakowiak P. A., Cozma D., Dobson N. R., Levin S. W., Anadiotis G., Steiner R. D., Krajewska-Walasek M. et al. (2005). DHCR7 nonsense mutations and characterisation of mRNA nonsense mediated decay in Smith-Lemli-Opitz syndrome. J. Med. Genet. 42, 350-357. 10.1136/jmg.2004.022749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cougot N., Babajko S. and Seraphin B. (2004). Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165, 31-40. 10.1083/jcb.200309008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozens A. L., Yezzi M. J., Chin L., Simon E. M., Finkbeiner W. E., Wagner J. A. and Gruenert D. C. (1992). Characterization of immortal cystic fibrosis tracheobronchial gland epithelial cells. Proc. Natl. Acad. Sci. USA 89, 5171-5175. 10.1073/pnas.89.11.5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozens A. L., Yezzi M. J., Kunzelmann K., Ohrui T., Chin L., Eng K., Finkbeiner W. E., Widdicombe J. H. and Gruenert D. C. (1994). CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 10, 38-47. 10.1165/ajrcmb.10.1.7507342 [DOI] [PubMed] [Google Scholar]
- da Paula A. C., Ramalho A. S., Farinha C. M., Cheung J., Maurisse R., Gruenert D. C., Ousingsawat J., Kunzelmann K. and Amaral M. D. (2005). Characterization of novel airway submucosal gland cell models for cystic fibrosis studies. Cell. Physiol. Biochem. 15, 251-262. 10.1159/000087235 [DOI] [PubMed] [Google Scholar]
- Dorard C., de Thonel A., Collura A., Marisa L., Svrcek M., Lagrange A., Jego G., Wanherdrick K., Joly A. L., Buhard O. et al. (2011). Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat. Med. 17, 1283-1289. 10.1038/nm.2457 [DOI] [PubMed] [Google Scholar]
- Durand S., Cougot N., Mahuteau-Betzer F., Nguyen C.-H., Grierson D. S., Bertrand E., Tazi J. and Lejeune F. (2007). Inhibition of nonsense-mediated mRNA decay (NMD) by a new chemical molecule reveals the dynamic of NMD factors in P-bodies. J. Cell Biol. 178, 1145-1160. 10.1083/jcb.200611086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farinha C. M., Mendes F., Roxo-Rosa M., Penque D. and Amaral M. D. (2004). A comparison of 14 antibodies for the biochemical detection of the cystic fibrosis transmembrane conductance regulator protein. Mol. Cell. Probes 18, 235-242. 10.1016/j.mcp.2004.03.005 [DOI] [PubMed] [Google Scholar]
- Fatscher T., Boehm V. and Gehring N. H. (2015). Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell. Mol. Life Sci. 72, 4523-4544. 10.1007/s00018-015-2017-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher D. A. and Mullins R. D. (2010). Cell mechanics and the cytoskeleton. Nature 463, 485-492. 10.1038/nature08908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti R. A. (2012). SMRT compounds correct nonsense mutations in primary immunodeficiency and other genetic models. Ann. N. Y. Acad. Sci. 1250, 33-40. 10.1111/j.1749-6632.2012.06467.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Hilarion S., Beghyn T., Jia J., Debreuck N., Berte G., Mamchaoui K., Mouly V., Gruenert D. C., Deprez B. and Lejeune F. (2012). Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 7, 58 10.1186/1750-1172-7-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harger J. W. and Dinman J. D. (2004). Evidence against a direct role for the Upf proteins in frameshifting or nonsense codon readthrough. RNA 10, 1721-1729. 10.1261/rna.7120504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F., Li X., Spatrick P., Casillo R., Dong S. and Jacobson A. (2003). Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol. Cell 12, 1439-1452. 10.1016/S1097-2765(03)00446-5 [DOI] [PubMed] [Google Scholar]
- Hug N., Longman D. and Cáceres J. F. (2016). Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44, 1483-1495. 10.1093/nar/gkw010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelfinger D., Arndt-Jovin D. J., Luhrmann R. and Achsel T. (2002). The human LSm1-7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA 8, 1489-1501. [PMC free article] [PubMed] [Google Scholar]
- Isken O., Kim Y. K., Hosoda N., Mayeur G. L., Hershey J. W. B. and Maquat L. E. (2008). Upf1 phosphorylation triggers translational repression during nonsense-mediated mRNA decay. Cell 133, 314-327. 10.1016/j.cell.2008.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia J., Furlan A., Gonzalez-Hilarion S., Leroy C., Gruenert D. C., Tulasne D. and Lejeune F. (2015). Caspases shutdown nonsense-mediated mRNA decay during apoptosis. Cell Death Differ. 22, 1754-1763. 10.1038/cdd.2015.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karousis E. D., Nasif S. and Mühlemann O. (2016). Nonsense-mediated mRNA decay: novel mechanistic insights and biological impact. Wiley Interdiscip. Rev. RNA 7, 661-682. 10.1002/wrna.1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kervestin S. and Jacobson A. (2012). NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 13, 700-712. 10.1038/nrm3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiak H. A. and Maquat L. E. (2006). Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol. Med. 12, 306-316. 10.1016/j.molmed.2006.05.005 [DOI] [PubMed] [Google Scholar]
- Lejeune F. (2017). Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 10.5483/BMBRep.2017.50.4.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelivelt M. J. and Culbertson M. R. (1999). Yeast Upf proteins required for RNA surveillance affect global expression of the yeast transcriptome. Mol. Cell. Biol. 19, 6710-6719. 10.1128/MCB.19.10.6710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Heredia M. and Jansen R.-P. (2004). mRNA localization and the cytoskeleton. Curr. Opin. Cell Biol. 16, 80-85. 10.1016/j.ceb.2003.11.002 [DOI] [PubMed] [Google Scholar]
- Loughran G., Chou M.-Y., Ivanov I. P., Jungreis I., Kellis M., Kiran A. M., Baranov P. V. and Atkins J. F. (2014). Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 42, 8928-8938. 10.1093/nar/gku608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J., Shu M.-D. and Steitz J. A. (2000). Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103, 1121-1131. 10.1016/S0092-8674(00)00214-2 [DOI] [PubMed] [Google Scholar]
- Manuvakhova M., Keeling K. and Bedwell D. M. (2000). Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 6, 1044-1055. 10.1017/S1355838200000716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell J. T., ap Rhys C. M. and Dietz H. C. (2002). Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science 298, 419-422. 10.1126/science.1074428 [DOI] [PubMed] [Google Scholar]
- Mendell J. T., Sharifi N. A., Meyers J. L., Martinez-Murillo F. and Dietz H. C. (2004). Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 36, 1073-1078. 10.1038/ng1429 [DOI] [PubMed] [Google Scholar]
- Moriarty P. M., Reddy C. C. and Maquat L. E. (1998). Selenium deficiency reduces the abundance of mRNA for Se-dependent glutathione peroxidase 1 by a UGA-dependent mechanism likely to be nonsense codon-mediated decay of cytoplasmic mRNA. Mol. Cell. Biol. 18, 2932-2939. 10.1128/MCB.18.5.2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlemann O. and Lykke-Andersen J. (2010). How and where are nonsense mRNAs degraded in mammalian cells? RNA Biol. 7, 28-32. 10.4161/rna.7.1.10578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp M. W.-L. and Maquat L. E. (2014). The dharma of nonsense-mediated mRNA decay in mammalian cells. Mol. Cells 37, 1-8. 10.14348/molcells.2014.2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp M. W. and Maquat L. E. (2015). Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat. Commun. 6, 6632 10.1038/ncomms7632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbapragada I. and Lykke-Andersen J. (2009). Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr. Opin. Cell Biol. 21, 394-402. 10.1016/j.ceb.2009.02.007 [DOI] [PubMed] [Google Scholar]
- Rehwinkel J., Letunic I., Raes J., Bork P. and Izaurralde E. (2005). Nonsense-mediated mRNA decay factors act in concert to regulate common mRNA targets. RNA 11, 1530-1544. 10.1261/rna.2160905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas-Marco J. and Bedwell D. M. (2005). Discrimination between defects in elongation fidelity and termination efficiency provides mechanistic insights into translational readthrough. J. Mol. Biol. 348, 801-815. 10.1016/j.jmb.2005.03.025 [DOI] [PubMed] [Google Scholar]
- Serin G., Gersappe A., Black J. D., Aronoff R. and Maquat L. E. (2001). Identification and characterization of human orthologues to Saccharomyces cerevisiae Upf2 protein and Upf3 protein (Caenorhabditis elegans SMG-4). Mol. Cell. Biol. 21, 209-223. 10.1128/MCB.21.1.209-223.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalev M., Kandasamy J., Skalka N., Belakhov V., Rosin-Arbesfeld R. and Baasov T. (2013). Development of generic immunoassay for the detection of a series of aminoglycosides with 6′-OH group for the treatment of genetic diseases in biological samples. J. Pharm. Biomed. Anal. 75, 33-40. 10.1016/j.jpba.2012.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth U. and Parker R. (2003). Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300, 805-808. 10.1126/science.1082320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G., Jakob S., Kleedehn M. G. and Lykke-Andersen J. (2007). Communication with the exon-junction complex and activation of nonsense-mediated decay by human Upf proteins occur in the cytoplasm. Mol. Cell 27, 780-792. 10.1016/j.molcel.2007.06.030 [DOI] [PubMed] [Google Scholar]
- Sureau A., Gattoni R., Dooghe Y., Stévenin J. and Soret J. (2001). SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 20, 1785-1796. 10.1093/emboj/20.7.1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I., Ueno T. and Kominami E. (2008). LC3 and Autophagy. Methods Mol. Biol. 445, 77-88. 10.1007/978-1-59745-157-4_4 [DOI] [PubMed] [Google Scholar]
- Trcek T., Sato H., Singer R. H. and Maquat L. E. (2013). Temporal and spatial characterization of nonsense-mediated mRNA decay. Genes Dev. 27, 541-551. 10.1101/gad.209635.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker T. A., Fortenberry J. A., Zsembery A., Schwiebert L. M. and Schwiebert E. M. (2012). The DeltaF508-CFTR mutation inhibits wild-type CFTR processing and function when co-expressed in human airway epithelia and in mouse nasal mucosa. BMC Physiol. 12, 12 10.1186/1472-6793-12-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L. and Izaurralde E. (2004). SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell 16, 587-596. 10.1016/j.molcel.2004.10.013 [DOI] [PubMed] [Google Scholar]
- van Dijk E., Cougot N., Meyer S., Babajko S., Wahle E. and Seraphin B. (2002). Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 21, 6915-6924. 10.1093/emboj/cdf678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas M. H., Gehring N. H., Breit S., Hentze M. W. and Kulozik A. E. (2007). The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the Nonsense Mediated Decay pathway. Nucleic Acids Res. 35, 4542-4551. 10.1093/nar/gkm461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Czaplinski K., Rao Y. and Peltz S. W. (2001). The role of Upf proteins in modulating the translation read-through of nonsense-containing transcripts. EMBO J. 20, 880-890. 10.1093/emboj/20.4.880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch E. M., Barton E. R., Zhuo J., Tomizawa Y., Friesen W. J., Trifillis P., Paushkin S., Patel M., Trotta C. R., Hwang S. et al. (2007). PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87-91. 10.1038/nature05756 [DOI] [PubMed] [Google Scholar]
- Yamashita A., Izumi N., Kashima I., Ohnishi T., Saari B., Katsuhata Y., Muramatsu R., Morita T., Iwamatsu A., Hachiya T. et al. (2009). SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev. 23, 1091-1105. 10.1101/gad.1767209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You K. T., Li L. S., Kim N.-G., Kang H. J., Koh K. H., Chwae Y.-J., Kim K. M., Kim Y. K., Park S. M., Jang S. K. et al. (2007). Selective translational repression of truncated proteins from frameshift mutation-derived mRNAs in tumors. PLoS Biol. 5, e109 10.1371/journal.pbio.0050109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q., Tan Y. H. and Hong W. (1998). A single plasmid vector (pSTAR) mediating efficient tetracycline-induced gene expression. Anal. Biochem. 259, 187-194. 10.1006/abio.1998.2645 [DOI] [PubMed] [Google Scholar]