

ABSTRACT
Sharpin, a multifunctional adaptor protein, regulates several signalling pathways. For example, Sharpin enhances signal-induced NF-κB signalling as part of the linear ubiquitin assembly complex (LUBAC) and inhibits integrins, the T cell receptor, caspase 1 and PTEN. However, despite recent insights into Sharpin and LUBAC function, a systematic approach to identify the signalling pathways regulated by Sharpin has not been reported. Here, we present the first ‘Sharpin interactome’, which identifies a large number of novel potential Sharpin interactors in addition to several known ones. These data suggest that Sharpin and LUBAC might regulate a larger number of biological processes than previously identified, such as endosomal trafficking, RNA processing, metabolism and cytoskeleton regulation. Importantly, using the Sharpin interactome, we have identified a novel role for Sharpin in lamellipodium formation. We demonstrate that Sharpin interacts with Arp2/3, a protein complex that catalyses actin filament branching. We have identified the Arp2/3-binding site in Sharpin and demonstrate using a specific Arp2/3-binding deficient mutant that the Sharpin–Arp2/3 interaction promotes lamellipodium formation in a LUBAC-independent fashion.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Sharpin, LUBAC, Arp2/3, Linear ubiquitylation, Interactome
Summary: We present the first Sharpin interactome and identify a new function for Sharpin in cytoskeleton function. Sharpin binds the Arp2/3 complex, which promotes lamellipodium formation.
INTRODUCTION
SHANK-associated RH domain interactor (Sharpin) is a multifunctional adaptor protein that is amplified (BioPortal.org; Jung et al., 2010) and overexpressed (Bii et al., 2015; De Melo and Tang, 2015; He et al., 2010) in many human cancers and promotes cancer cell proliferation, tumour formation and metastasis (Bii et al., 2015; He et al., 2010; Li et al., 2015; Zhang et al., 2014). The most-studied function of Sharpin is as part of the linear ubiquitin assembly complex (LUBAC) (Gerlach et al., 2011; Ikeda et al., 2011; Tokunaga et al., 2011), which also includes RBCK1 and the catalytic subunit RNF31. LUBAC was identified as a regulator of canonical nuclear factor (NF)-κB signalling (Gerlach et al., 2011; Tokunaga et al., 2009), but it has become evident that LUBAC regulates several other signalling pathways through a growing range of substrates (Chattopadhyay et al., 2016; Dubois et al., 2014; Rodgers et al., 2014; Zak et al., 2011). Consistent with these findings, LUBAC is linked to several diseases, such as autoinflammation, immunodeficiency, amylopectinosis, lymphangiectasia (Boisson et al., 2015; Lewis et al., 2015) and cancer (Yang et al., 2014, 2016).
In addition to its role in linear ubiquitylation, Sharpin also binds and inhibits integrins (Pouwels et al., 2013; Rantala et al., 2011), the T cell receptor (Park et al., 2016) and caspase 1 (Nastase et al., 2016) in a LUBAC-independent manner. Furthermore, Sharpin functionally interacts with PTEN (He et al., 2010), SHANK proteins (Lim et al., 2001) and EYA transcription factors (Landgraf et al., 2010).
The actin-related protein 2/3 (Arp2/3) complex is an actin nucleator that consists of seven subunits (Arp2, Arp3 and ArpC1–ArpC5) and specifically catalyses formation of branched actin filament structures, which play a role in several key cellular functions (Rotty et al., 2013). At the leading edge of migrating cells, Arp2/3 creates a dense dendritic meshwork of actin filaments that provides the protrusive force for lamellipodium formation and, thus, cell migration (Rogers et al., 2003; Wu et al., 2012). The Arp2/3 complex depends on nucleation-promoting factors (NPFs) such as WASP (also known as WASL) and WAVE1 (also known as WASF1) proteins for full activation, while other proteins inhibit formation of branched actin networks or cause their disassembly (Krause and Gautreau, 2014; Rotty et al., 2013). Importantly, NPFs themselves are subject to strict regulation and can for example be activated through interaction with the small GTPase RAC1 (Rotty et al., 2013).
Despite these discoveries in the linear ubiquitin field and on the range of Sharpin interactors, a systematic approach to map proteins and signalling pathways regulated by Sharpin and/or LUBAC has not been reported. Here, we present the first ‘Sharpin interactome’, which identifies many potential Sharpin interactors and suggests that Sharpin, and possibly LUBAC, could regulate a wide range of key biological processes. In addition, we describe a novel direct interaction between Sharpin and the Arp2/3 complex, which is mediated by the Sharpin ubiquitin-like (UBL) domain. By using a specific Arp2/3 binding-deficient Sharpin mutant, we describe a novel role for Sharpin in lamellipodium formation that depends on Sharpin interaction with the Arp2/3 complex but is independent of LUBAC function.
RESULTS
Mass spectrometry analyses identify several actin-associated proteins as novel Sharpin interactors
To identify new Sharpin-binding partners and systematically map Sharpin functions, GFP pulldowns from cells expressing GFP–Sharpin or GFP alone were analysed by mass spectrometry (MS). As Sharpin preferentially binds inactive integrins (Rantala et al., 2011), we aimed to identify proteins that associate with Sharpin in an integrin-dependent manner by using GFP–Sharpin-expressing cells that were either kept in suspension (integrins mostly inactive; GFP–Sharpin suspension dataset) or plated on fibronectin (integrins mostly active; GFP–Sharpin adherent dataset). Two biological repeats were performed and, in total, 3083 proteins were detected above a set detection threshold (Fig. S1A; Table S1).
To assist in the interrogation and visualisation, proteins were hierarchically clustered using normalised spectral counts as a measure of protein abundance (Fig. S1B). No major differences were observed between the GFP–Sharpin suspension and GFP–Sharpin adherent datasets (Fig. S1A,B) and, in addition, very few integrin-related proteins were identified (Table S1), which could be due to the fact that integrins are notoriously difficult to co-immunoprecipitate.
To list and score putative Sharpin interactors, three thresholds (low, medium and high confidence; see Materials and Methods) were used, reflecting the quality and the specificity of the binding to GFP–Sharpin (Table S1).
Importantly, we identified both other members of the LUBAC complex (RBCK1 and RNF31), as well as many proteins known to associate with LUBAC (Fig. S1C), confirming the validity of the screen. The most-established role of Sharpin and LUBAC is regulation of NF-κB function. While the NF-κB transcription factors were not identified, many proteins known to associate with NF-κB were, further validating the screen (Fig. S1D).
To define the gross composition of proteins recruited to Sharpin (medium threshold) in an unbiased manner, functional annotation clustering based on Gene Ontology (GO; Biological Process_5) was performed using DAVID (Huang da et al., 2009) and displayed as network-based enrichment maps (Merico et al., 2010) (Fig. 1A; Table S2). In total, 261 biological processes were over-represented in the GFP–Sharpin datasets. We identified some biological processes known to be regulated by Sharpin and LUBAC, such as regulation of cell death (Gerlach et al., 2011; Kumari et al., 2014; Nastase et al., 2016; Rickard et al., 2014), ubiquitin-based processes and cell signalling, the latter of which includes NF-κB signalling (Fig. 1A; Table S2). In addition, we identified several biological processes that have not been associated with Sharpin or LUBAC, such as protein transport, metabolic processes, regulation of the cell cycle and DNA/RNA-based processes, which could suggest that Sharpin and, possibly, LUBAC have a much broader function in cells than described until now. For hypothesis generation and future experimental design, proteins recruited to Sharpin and identified in the above biological process categories are short-listed and available in Table S2. Furthermore, all mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Vizcaíno et al., 2010, 2013) partner repository with the dataset identifier PXD004734.
Fig. 1.

Gene Ontology and protein network analyses reveal a putative role for Sharpin in regulation of the cytoskeleton. (A) GO-based functional annotation clustering analyses of the proteins recruited to Sharpin. Proteins enriched in the Sharpin pulldowns (medium threshold) were mapped onto the GO category Biological Process (GOTERM_BP_5) using DAVID. 261 GO terms that were overrepresented in the GFP–Sharpin datasets (P<0.05), were displayed as network-based enrichment maps (see Table S2 for details). Each node (circle) represents a GO term and each line connects GO terms that contain at least one common protein. Node area is proportional to the number of proteins that belong to a particular GO term. Nodes of this network were automatically organised using an algorithm that clusters nodes as a function of their connectivity and were then manually annotated. (B) Hierarchical clustering of the putative Sharpin binders (low threshold) annotated with the Gene Ontology term GO 0051493 (regulation of cytoskeleton organisation). (C) Proteins identified in the GFP–Sharpin datasets were mapped onto a literature-curated protein–protein interaction network and a sub-network containing the proteins within one interaction of the Arp2/3 complex was created. Putative Sharpin interactors in B and C were colour coded to illustrate the threshold of identification.
The GO analyses also suggest that Sharpin may regulate the cytoskeleton. To identify all cytoskeleton regulators among the proteins recruited to Sharpin, GO analyses using the low threshold list were performed (Table S3), and proteins belonging to the GO term ‘regulation of cytoskeleton organization’ were extracted and hierarchically clustered (Fig. 1B). From this list, we selected the Arp2/3 complex for further analysis, as two members of the Arp2/3 complex (ArpC2 and ArpC5), as well as many Arp2/3-associated proteins were identified in the screen (Fig. 1C). Some of these Arp2/3-associated proteins are well-established Arp2/3 regulators, such as GRB2 (Carlier et al., 2000) and Cdc42 (Rohatgi et al., 1999).
Sharpin and the Arp2/3 complex interact directly in cells and in vitro
We confirmed the Sharpin–Arp2/3 interaction by using several techniques. A proximity ligation assay (PLA) (Söderberg et al., 2006) in HeLa cervical cancer cells showed PLA signals for endogenous Sharpin and Arp2 that were similar to PLA signals for the known interaction pairs α2-integrin–Sharpin and Arp2–actin (Fig. 2A), indicating interaction between Sharpin and the Arp2/3 complex. The specificity of the PLA was confirmed using antibodies against GFP (Fig. 2A) and irrelevant cytoplasmic proteins (Fig. S2A). In addition, the Arp2–Sharpin PLA signal was significantly reduced in Sharpin-silenced cells (Fig. S2B–E). The interaction between endogenous Sharpin and the Arp2/3 complex was also verified in co-immunoprecipitation experiments from HEK-293 cell extracts with two different Sharpin antibodies (Fig. 2B; Arp2 was detected as a representative for the whole Arp2/3 complex). In addition, in HEK-293 cells, endogenous Arp2/3 was pulled down with overexpressed GFP–Sharpin, but not GFP alone (Fig. 2C). Furthermore, we performed fluorescence resonance energy transfer (FRET) measurements between co-expressed mCherry–Sharpin and Arp3–GFP (Fig. 2D) or GFP–Sharpin and Arp3–TagRFP (Fig. 2E) by using fluorescence lifetime imaging microscopy (FLIM). The reduced GFP fluorescence lifetime upon co-expression of Sharpin and Arp3 showed that Sharpin and the Arp2/3 complex interact directly. To further demonstrate the direct interaction, we performed an in vitro pulldown assay, showing that recombinant GST–Sharpin (Fig. S2F; Rantala et al., 2011), but not GST alone, was able to pull down a purified bovine Arp2/3 complex (Fig. 2F). Therefore, we conclude that Sharpin and the Arp2/3 complex interact directly.
Fig. 2.

Sharpin and the Arp2/3 complex interact in cells. (A) PLA with indicated antibody pairs in HeLa cells (rb-GFP and mo-GFP represent mouse and rabbit antibodies against GFP). DAPI indicates nuclei. The graph shows average number of PLA signals (spots) per cell (n=10 images from a representative experiment of n=3 experiments). (B) Co-immunoprecipitation (CoIP) of endogenous Arp2 and Sharpin using two different Sharpin antibodies [mouse (mo) and rabbit (rb)] from HEK-293 cells (representative of three experiments). (C) Pulldown experiments to determine the interaction between GFP or GFP–Sharpin and endogenous Arp2 in HEK-293 cells (representative from three experiments). (D,E) HeLa cells overexpressing the indicated proteins were subjected to FRET analysis by FLIM. Fluorescence lifetimes, mapping spatial FRET in cells, are depicted using a pseudo-colour scale [red–yellow, normal lifetime; yellow–blue, FRET (reduced lifetime)]. Graphs show quantification of FRET efficiency [n=27–31 cells (D) and 12 cells (E)]. (F) Pulldown experiment to determine the interaction between recombinant GST or GST–Sharpin and purified bovine Arp2/3 complex. Numerical data are Arp2 levels normalised to GST (n=3 experiments). (G) Pyrene-actin polymerisation assay in the presence of Arp2/3, GST–VCA and GST–Sharpin. Samples of pyrene-actin (4 µM) were polymerised in the presence of the indicated combinations of GST–Sharpin (11 µM), Arp2/3 (40 nM) and GST–VCA (30 nM) (n=3 experiments). All numerical data (except in G) are mean±s.e.m. ***P<0.001; n.s., not significant. Scale bars: 10 μm.
To address whether Sharpin regulates Arp2/3 activity, we performed an established actin polymerisation assay (Fig. 2G), revealing that recombinant GST–Sharpin does not affect actin polymerisation induced by Arp2/3, either in the absence or presence of VCA (the WASP verprolin-cofilin-acidic homology domain), which activates Arp2/3 activity (Prehoda et al., 2000). In conclusion, Sharpin and the Arp2/3 complex interact in cells and in vitro, but Sharpin does not seem to affect Arp2/3 activity directly in vitro.
Interaction between Sharpin and Arp2/3 depends on an intact cytoskeleton and Arp2/3 activity
To determine whether the actin cytoskeleton is required for the Sharpin–Arp2/3 interaction in cells, we performed PLA and FRET-FLIM experiments, similar to those in Fig. 2A,E, in the presence or absence of the actin polymerisation inhibitor cytochalasin D (CytD). Treatment with CytD strongly reduced the PLA signal between endogenous Sharpin and Arp2, as well as FRET between GFP–Sharpin and Arp3–TagRFP (Fig. S3A,C), showing that the interaction between Sharpin and the Arp2/3 complex depends on an intact cytoskeleton. In addition, 30 min treatment with the Arp2/3 inhibitor CK666 (Nolen et al., 2009) also disrupted interaction between Sharpin and Arp2/3 (Fig. S3A,C), but not between Sharpin and α2-integrin (Fig. S3B), suggesting that Arp2/3 activity is required for the Sharpin–Arp2/3 interaction. Sharpin acts as a negative regulator of integrin activity (Rantala et al., 2011). The disturbed interaction between Arp2/3 and Sharpin after CK666 treatment is not due to differences in integrin activity, however, as these treatments did not affect integrin activity, as shown using total internal reflection fluorescence (TIRF) imaging (Fig. S3D) and fluorescence-activated cell sorting (FACS) analyses (Fig. S3E).
Identification of a specific Arp2/3-binding deficient Sharpin mutant
The Sharpin protein has three conserved functional domains; an N-terminal pleckstrin homology (PH) superfold (Lim et al., 2001; Stieglitz et al., 2012), a central UBL domain that binds to most known Sharpin-interacting proteins (De Franceschi et al., 2015) and a C-terminal NPL4 zinc finger domain (NZF) that mediates interaction with the T cell receptor (Park et al., 2016) and contributes to LUBAC function (Ikeda et al., 2011). To address which domain mediates the interaction with Arp2/3 we used GFP–Sharpin fragments spanning the entire protein (De Franceschi et al., 2015). FRET-FLIM experiments using Arp3–TagRFP and wild-type (WT) Sharpin or the fragment of Sharpin conjugated to GFP in HeLa cells (Fig. 3A) and pull-down of endogenous Arp2 with these Sharpin proteins from HEK-293 cells (Fig. 3B) showed that the Arp2/3 complex binds to the UBL domain of Sharpin, further confirming the UBL domain as a protein interaction hub.
Fig. 3.

Mapping the Arp2/3 interaction site of Sharpin. (A) HeLa cells overexpressing the indicated proteins were subjected to FRET analysis by FLIM. The graph shows quantification of FRET efficiency (n=62–99 cells, from three experiments). (B) Pulldown experiments to determine the interaction between overexpressed GFP-Sharpin WT or its fragments and endogenous Arp2 from HEK-293 cells. Blotting for RNF31 confirmed the interaction of RNF31 with the Sharpin UBL domain. Numerical data are Arp2 levels in the pulldown, normalised to Arp2 levels in the extract and GFP levels in the pulldown (three experiments). (C) HeLa cells, overexpressing the indicated proteins were subjected to FRET analysis by FLIM. Fluorescence lifetimes, mapping spatial FRET in cells, are depicted using a pseudo-colour scale [red–yellow, normal lifetime; yellow–blue, FRET (reduced lifetime)]. Scale bar: 10 μm. The graph shows quantification of FRET efficiency (n=36–74 cells from three experiments; donor alone was set to 0 for each individual GFP–Sharpin construct). For statistical analyses, FRET efficiencies were compared to GFP–Sharpin WT. (D) Pulldown experiments to determine the interaction between overexpressed GFP–Sharpin WT and V240A/L242A and endogenous Arp2 from HEK-293 cells. Numerical data are Arp2 levels in the pulldown, normalised to Arp2 levels in the extract and GFP levels in the pulldown (three experiments). All numerical data are mean±s.e.m. ***P<0.001; n.s., not significant.
Using a panel of point mutations of the Sharpin UBL domain, we recently showed that the integrin and RNF31-binding domains in the UBL domain partially overlap (De Franceschi et al., 2015). FRET-FLIM experiments between these GFP–Sharpin point mutants and Arp3–TagRFP identified three mutants (V240A/L242A, V267A and L276A) that abolished Arp2/3 binding (Fig. 3C). The other mutants (E260A/L261A, L261A/F263A and I272A) interacted with the Arp2/3 complex, although binding was reduced compared to WT (Fig. 3C). Importantly, the V240A/L242A mutant seems to be specifically unable to bind Arp2/3 as it does not interfere with Sharpin-mediated integrin inhibition and NF-κB activation (De Franceschi et al., 2015). The V267A and L276A mutations, on the other hand, also abolish the ability of Sharpin to regulate integrins and NF-κB (De Franceschi et al., 2015). The decreased ability of GFP–Sharpin V240A/L242A to interact with endogenous Arp2 in pulldown assays (Fig. 3D), as compared to GFP-Sharpin WT, confirmed the important role of these residues in binding the Arp2/3 complex. In conclusion, these experiments map the Arp2/3 interaction site in the Sharpin UBL domain and, importantly, identify a Sharpin mutant (V240A/L242A) that is specifically unable to bind the Arp2/3 complex.
Localisation of Sharpin and the Arp2/3 complex in cells
Both Sharpin (Rantala et al., 2011) and the Arp2/3 complex (Rogers et al., 2003; Wu et al., 2012) were reported to localise to lamellipodia. We also observed both proteins in lamellipodia of HeLa cells (Fig. S4A,B) and NCI-H460 lung cancer cells (Fig. S4C). However, the fluorescence intensities suggested that, relative to their cytoplasmic levels, endogenous Arp2 is more enriched in lamellipodia than Sharpin. To quantify this, we measured endogenous Sharpin and Arp2 levels in lamellipodia and the cytoplasm of NCI-H460 cells with line scans, confirming that Arp2 accumulates more in lamellipodia (Fig. S4C). This might suggest that Sharpin is cytoplasmic and that Sharpin localisation to lamellipodia is due to cytosolic thickening. To address this, we determined the colocalisation of Sharpin and p65 (also known as RELA; an NF-κB subunit that is mostly cytoplasmic in unstimulated cells) and analysed their enrichment in lamellipodia (Fig. S4D). These data show that Sharpin and p65 accumulate to similar levels in lamellipodia and, therefore, Sharpin in lamellipodia is likely cytoplasmic.
If Sharpin localisation in lamellipodia is a result of cytoplasmic thickening, Sharpin would not be expected to localise to lamellipodia in cells that form flat lamellipodia. Therefore, we analysed accumulation of Sharpin and Arp2 in lamellipodia of U2OS osteosarcoma cells, which showed that, while Arp2 was clearly enriched in such flat lamellipodia, Sharpin accumulation was very limited (Fig. S4E), consistent with Sharpin being a cytoplasmic protein.
Fluorescence recovery after photobleaching (FRAP) experiments demonstrated that Sharpin in lamellipodia is very dynamic (Fig. S4F, Movie 1). However, unlike the Arp2/3 complex, which is incorporated into the actin network at the lamellipodium tip (Lai et al., 2008), GFP–Sharpin fluorescence recovered evenly throughout the length of the lamellipodium, consistent with simple diffusion of Sharpin in the cytoplasm. Finally, unlike Arp2, Sharpin was not enriched at actin comets induced by phosphatidylinositol phosphate 5-kinase (PIP5KI) (Fig. S4G; Rozelle et al., 2000), consistent with Sharpin being cytoplasmic. All in all, these localisation studies show that Sharpin is a cytoplasmic protein and, unlike the Arp2/3 complex, not specifically enriched in lamellipodia.
Sharpin promotes lamellipodium formation
Both Sharpin and Arp2/3 play a role in lamellipodium formation (Rantala et al., 2011; Rogers et al., 2003; Rotty et al., 2013; Wu et al., 2012). Consistent with these studies, we observed that Sharpin and Arp3-silenced NCI-H460 lung cancer cells (Fig. S2G,H) formed significantly fewer lamellipodia compared to control silenced cells based on cortactin (Fig. 4A) and filamentous actin (F-actin) staining (Fig. S5A). A similar phenotype was observed in NCI-H460 cells treated for 30 min with CK666 (Fig. 4A). The more pronounced phenotype of the CK666-treated cells compared to Arp3 silencing is most likely due to the strong inhibition of Arp2/3 activity by CK666 (Nolen et al., 2009), while residual Sharpin and Arp3 are present in siRNA-transfected cells (Fig. S2G,H). Importantly, silencing of RNF31 (Fig. S2G,H) did not affect lamellipodium formation (Fig. 4A), suggesting that Sharpin promotes lamellipodium formation independently of its role in LUBAC function. Live-cell imaging of control silenced NCI-H460 cells expressing fluorescently tagged Lifeact (Riedl et al., 2008) to visualise F-actin, showed that the lamellipodia are highly dynamic (Movie 2). Consistent with the still images (Fig. 4A), Sharpin-silenced NCI-H460 cells formed fewer lamellipodia (Movie 3). Instead, the surface of Sharpin-silenced cells was covered with dynamic filopodia (Movie 3), similar to cells with reduced Arp2/3 levels (Movie 4; Beli et al., 2008; Wu et al., 2012).
Fig. 4.

Sharpin promotes lamellipodium formation. (A) Control-, Sharpin-, Arp3- and RNF31-silenced NCI-H460 cells, as well as control silenced NCI-H460 cells treated with 100 μM CK666 for 30 min were stained for cortactin. The graph depicts the percentage of cells with cortactin-positive lamellipodia (n=4–9 experiments, >150 cells per condition per experiment). (B) WT and two Sharpin knockout NCI-H460 cell lines immunostained for cortactin and F-actin. The graph depicts the percentage of cells with cortactin- and F-actin-positive lamellipodia (n=4 experiments, >125 cells per condition per experiment). (C) Control- and Sharpin-silenced U2OS cells were grown under normal conditions, serum starved overnight, or released into serum-containing medium for 120 min after overnight serum starvation, and subsequently stained for cortactin. The graph depicts the percentage of cells with cortactin-positive lamellipodia (n=4 experiments, 20–79 cells per condition per experiment). (D) Control- and Sharpin-silenced NCI-H460 cells attached to linear micropatterns were treated for 30 min with 100 μM CK666 and then released by adding medium containing serum. Cells were fixed without CK666 treatment, after CK666 treatment and at different time points after release, followed by staining for cortactin (the position of the linear micropatterns is indicated with green dotted lines). The graph shows the percentage of cells with cortactin-positive lamellipodia (n=3 experiments, 12–40 cells per condition per experiment). All numerical data are mean±s.e.m. DAPI was used to stain nuclei. *P<0.05; **P<0.01; ***P<0.001; n.s., not significant. Scale bars: 10 μm.
To confirm the role of Sharpin in lamellipodium formation and to rule out off-target effects, we created two monoclonal Sharpin-knockout NCI-H460 cell lines by using CRISPR (Fig. S2I). Consistent with Sharpin facilitating lamellipodium formation, we observed a substantial reduction in lamellipodium formation upon loss of Sharpin (Fig. 4B). Importantly, quantification of lamellipodium formation in control and Sharpin-silenced U2OS cells (Fig. S2J) confirmed the role of Sharpin in lamellipodium formation in another cell line and showed that Sharpin also plays a role in the formation of prominent flat lamellipodia (Fig. 4C).
To investigate lamellipodium dynamics in more detail, we plated control and Sharpin-silenced NCI-H460 cells on line-shaped micropatterns coated with 50 μg/ml fibronectin and 5 μg/ml Alexa Fluor 488-conjugated fibrinogen and investigated lamellipodium formation after release from CK666 based on cortactin (Fig. 4D) and F-actin staining (Fig. S5B). Consistent with the cells plated on regular coverslips (Fig. 4A), Sharpin silencing strongly reduced the number of cells with lamellipodia on linear micropatterns under unperturbed conditions. Inhibition of Arp2/3 with CK666 abolished formation of lamellipodia in control silenced cells and further reduced the low number of lamellipodia in Sharpin-silenced cells. As reported previously (Brayford et al., 2016; Haynes et al., 2015), lamellipodia reformed within minutes after CK666 release in control silenced cells. However, such ruffles did not appear in Sharpin-silenced cells after CK666 washout (Fig. 4D; Fig. S5B), suggesting that Sharpin facilitates the formation of lamellipodia rather than stabilising existing ones.
Sharpin promotes lamellipodium formation through interaction with Arp2/3
To prove that the loss of lamellipodia in Sharpin-silenced and -knockout cells is specifically due to loss of Sharpin, we performed rescue experiments in NCI-H460 cells (Fig. 5). Expression of GFP–Sharpin WT (insensitive to Sharpin siRNA1; Rantala et al., 2011) restored lamellipodium formation (Fig. 5), showing that reduced lamellipodium formation in Sharpin-silenced and -knockout cells is not due to off-target effects. Importantly, lamellipodium formation was not rescued by the Arp2/3-binding deficient (Fig. 3C,D) GFP–Sharpin (V240A/L242A) (Fig. 5), showing that Sharpin binding to the Arp2/3 complex is essential for Sharpin to promote lamellipodium formation. On the other hand, GFP–Sharpin mutants L261A/F263A and I272A, which are unable to bind RNF31 and support LUBAC function (De Franceschi et al., 2015), rescued lamellipodium formation (Fig. 5A), further supporting the notion that Sharpin supports lamellipodium formation independently of LUBAC.
Fig. 5.

Sharpin promotes lamellipodium formation through interaction with the Arp2/3 complex. (A) Sharpin-silenced NCI-H460 cells or (B) Sharpin KO2 NCI-H460 cells overexpressing the indicated proteins were stained for cortactin. Graphs depict the number of GFP-positive cells with cortactin-positive lamellipodia, relative to GFP–Sharpin WT, for (A) Sharpin-silenced (n=4 experiments, >30 cells per condition per experiment) and (B) Sharpin-knockout cells (n=3 or 4 experiments, 19–56 cells per condition per experiment). All numerical data are mean±s.e.m. *P<0.05, **P<0.01. Scale bars: 10 μm.
All in all, these experiments show that Sharpin promotes the lamellipodium formation through interaction with the Arp2/3 complex, independently of LUBAC function.
Sharpin promotes stimulus-induced lamellipodium formation
To address whether Sharpin regulates integrin-mediated lamellipodium formation, we plated WT and Sharpin-knockout NCI-H460 cells for 3 h on 5 µg/ml fibronectin and quantified lamellipodium formation, showing that absence of Sharpin strongly inhibited formation of integrin-dependent lamellipodia in these spreading cells (Fig. S5C). We also observed reduced numbers of integrin-induced lamellipodia upon Sharpin silencing (Fig. S5D), but the effect was smaller, consistent with the modest silencing efficiency in NCI-H460 cells (Fig. S2G,H).
To determine whether Sharpin affects receptor signalling-induced lamellipodium formation, we serum starved control or Sharpin-silenced U2OS cells overnight and subsequently induced lamellipodium formation through serum stimulation for 120 min [lamellipodium formation was not very prominent after 15, 30 and 60 min (data not shown)]. In control cells, serum starvation abolished lamellipodia, and subsequent release into serum-containing medium induced a burst of lamellipodia (Fig. 4C). In Sharpin-silenced U2OS cells, which make fewer lamellipodia than control cells under unperturbed conditions, we also observed reduced induction of lamellipodium formation after serum release (Fig. 4C), suggesting that Sharpin also regulates receptor signalling-induced lamellipodia.
Finally, to address whether Sharpin also regulates RAC1-driven lamellipodium formation, we expressed constitutively active GFP-RAC1(Q61L) or GFP alone in WT and the two Sharpin-knockout NCI-H460 cell lines (KO1 and KO2). In WT cells, we observed the typical shape associated with overexpression of constitutively active RAC1 (Fig. S6A) – large flat round cells with very profound lamellipodia all around the edge. GFP–RAC1(Q61L) induced similar shape changes in both Sharpin-knockout cell lines, as judged by cell area (Fig. S6A), indicating that Sharpin is dispensable for RAC1-driven lamellipodium formation. We confirmed these results using control or Sharpin-silenced HeLa cells, in which GFP-RAC1(Q61L) induced similar shape changes (Fig. S6B; roundness was also measured for these cells).
The Sharpin-Arp2/3 interaction promotes cell migration
Arp2/3-dependent lamellipodium formation promotes cell migration, as for example assayed using wound healing assays (Liu et al., 2013; Suraneni et al., 2012). In support of those studies, we also observed decreased wound healing upon silencing of Arp3 in HeLa cells (Fig. S6C). Sharpin silencing, on the other hand, did not affect wound healing under these conditions (Fig. S6C). We have previously observed in several cell lines that Sharpin depletion affects cell migration due to elevated integrin activity (Rantala et al., 2011). Therefore, Sharpin might regulate cell migration through different partners, that is, inhibition of integrins and stimulation of Arp2/3-dependent lamellipodium formation, which in this wound healing assay seem to cancel each other out.
To address the role of the Sharpin–Arp2/3 interaction in cell migration independently from its role in integrin inactivation, we overexpressed GFP alone, GFP–Sharpin WT or GFP–Sharpin V240A/L242A in immortalized mouse embryonic fibroblasts (MEFs) isolated from Sharpin-deficient [cpdm (chronic proliferative dermatitis)] mice (Rantala et al., 2011). Under these conditions, overexpression of GFP–Sharpin WT increased cell migration compared to that seen upon overexpression of GFP alone (Fig. 6). Importantly, cpdm MEFs overexpressing GFP–Sharpin V240A/L242A, which is unable to bind the Arp2/3 complex (Fig. 3C,D) and support lamellipodium formation (Fig. 5) but is fully capable of inhibiting integrins (De Franceschi et al., 2015), did not migrate significantly faster than those overexpressing GFP alone (Fig. 6). On the other hand, GFP–Sharpin V240A/L242A-overexpressing cpdm MEFs did migrate slower than their counterparts overexpressing GFP–Sharpin WT, suggesting that, under these conditions, Sharpin promotes cell migration through interaction with the Arp2/3 complex, rather than through integrin inhibition.
Fig. 6.

Sharpin promotes cell migration through interaction with the Arp2/3 complex. (A) Quantification of migration speed and (B) representative cell tracks (4.5 h) of cpdm MEFs overexpressing GFP alone, WT GFP–Sharpin or GFP–Sharpin V240A/L242A on 5 μg/ml fibronectin [n=24, 34 and 34 cells, respectively, from three, four and four experiments (6–10 cells from each experiment)]. All numerical data are mean±s.e.m. *P<0.05; **P<0.01; n.s., not significant.
Taken together, we identify the Arp2/3 complex as a novel Sharpin interactor and show that this interaction plays a role in Arp2/3-dependent lamellipodium formation and could regulate cell migration.
DISCUSSION
Despite rapid developments in the linear ubiquitin field and the clear disease relevance of Sharpin and LUBAC, a comprehensive overview of Sharpin and LUBAC functions is missing. Here, we report the first Sharpin interactome, which potentially links Sharpin and LUBAC to many new pathways, suggesting that Sharpin and LUBAC have a much broader function than reported. In addition, we establish a novel LUBAC-independent function for Sharpin in lamellipodium formation through interaction with the Arp2/3 complex.
Although the vast majority of Sharpin studies focus on LUBAC, several LUBAC-independent functions have been described for Sharpin (Nastase et al., 2016; Park et al., 2016; Rantala et al., 2011). The LUBAC-independent role for Sharpin in lamellipodium formation through interaction with the Arp2/3 complex described here strengthens the increasingly appreciated notion that Sharpin has important functions outside of its role in LUBAC.
Our observation that Sharpin does not accumulate specifically in lamellipodia (Fig. 4) suggests that Sharpin interacts with cytoplasmic Arp2/3 (i.e. Arp2/3 not associated with the cytoskeleton). This seems inconsistent with the dependency of the Sharpin–Arp2/3 interaction on an intact cytoskeleton (Fig. S3A,C). However, disruption of the cytoskeleton with CytD potentially leads to deactivation of Arp2/3, which could also disrupt the Arp2/3–Sharpin interaction (Fig. S3A,C). Alternatively, CytD-mediated disruption of the actin cytoskeleton might affect Sharpin function, for example by affecting post-translational modification of Sharpin or disturbing the interaction between Sharpin and another protein. Consistent with the cytoplasmic Sharpin–Arp2/3 interaction, NPFs were absent from the Sharpin interactome (Table S1) and Sharpin did not affect Arp2/3 activation directly in vitro (Fig. 2G). However, we cannot rule out that post-translational modifications of Sharpin or other proteins that have yet to be identified could mediate the Sharpin–Arp2/3 interplay in cells. For example, lamellipodium formation could be regulated by a multiprotein interaction, such as a Sharpin–integrin–Arp2/3 complex. Alternatively, the Sharpin–Arp2/3 interaction could enhance signalling to the Arp2/3 complex or modulate the Arp2/3–cortactin interaction, consistent with the role for Sharpin in formation of signal-induced lamellipodia (Fig. 4C; Fig. S5B,C). Sharpin does not regulate cortactin stability, however, as cortactin levels are largely unaffected in the absence of Sharpin (Fig. S2E,I)
One potential mechanism through which Sharpin could promote lamellipodium formation is stabilisation of the active Arp2/3 conformation, which is consistent with our observations that Arp2/3 levels are modestly reduced upon Sharpin silencing or knockout (Fig. S2E,H,I) and that the Sharpin–Arp2/3 interaction depends on Arp2/3 activity (Fig. S3A,C). Reduced Arp2/3 levels are unlikely to completely explain the Sharpin phenotype, however, as reducing Arp3 levels by 60% using siRNA in NCI-H460 cells (Fig. S2H) reduced lamellipodium formation by ∼50% (Fig. 4A), while lamellipodium formation in Sharpin KO1 NCI-H460 cells, which show modestly reduced Arp2 levels (24±10%, mean±s.e.m.; Fig. S2I), is reduced by ∼75% (Fig. 4B).
Lamellipodia induced by constitutively active RAC [GFP–RAC(Q61L)] were not affected by the absence of Sharpin (Fig. S6A,B), which could suggest that Sharpin regulates lamellipodium formation upstream of RAC. However, GFP–RAC(Q61L)-induced lamellipodia in NCI-H460 cells were fully resistant to 6 h Arp2/3 inhibition with CK666 (data not shown), suggesting that GFP–RAC(Q61L)-induced lamellipodia are hyperstable. Therefore, while lamellipodia formation is strongly reduced in the absence of Sharpin (Figs 4 and 5; Fig. S5), such lamellipodia could become hyperstable in the presence of GFP–RAC(Q61L), resulting in large round flat cells with profound lamellipodia despite strongly decreased lamellipodium formation rates.
Irrespective of the molecular mechanism, we show that the Sharpin–Arp2/3 interaction is physiologically relevant as it promotes lamellipodium formation (Fig. 5). This might have implications in wound healing and metastasis, although this needs further investigation. The Arp2/3 complex critically regulates several other cellular processes in addition to cell migration (Rotty et al., 2013), but whether Sharpin plays a role in these remains to be established. Interestingly, the Sharpin interactome contains several proteins involved in endocytic trafficking (Table S2), suggesting a role for Sharpin in this Arp2/3-dependent process.
We assigned three different thresholds to objectively score the Sharpin interactors (Table S1). Our ‘low threshold’ is commonly used in mass spectrometry studies. Importantly, most of the biological functions identified in the Gene Ontology analyses (Fig. 1A) are also represented in the high threshold list. For example, the proteasome has a very prominent role in the high threshold list, suggesting that Sharpin either binds to or is degraded by the proteasome. Furthermore, in addition to RNF31, two other E3 ubiquitin ligases were identified (RNF114 and STUB1), which could suggest that Sharpin regulates other ubiquitin ligases than LUBAC. Alternatively, Sharpin could be a substrate for these ubiquitin ligases. Interestingly, several Sharpin ubiquitylation sites have been identified (Wagner et al., 2016; http://www.phosphosite.org), although their significance remains unknown.
All in all, we identified several cellular functions that have not been linked to Sharpin and LUBAC (Fig. 1A; Table S2). Therefore, it is tempting to speculate that Sharpin and, potentially, LUBAC have a much broader function than the mostly immunological roles that have been described to date. Our identification of a novel LUBAC-independent role for Sharpin in the regulation of the cytoskeleton shows that the Sharpin interactome can lead to important new insights. We hope that the research community will use this resource to better understand Sharpin and LUBAC function and how these proteins link to cancer and immune-related disease.
MATERIALS AND METHODS
Antibodies
These antibodies were used: rabbit RNF31/HOIP [ab46322, Abcam; 1:1000 western blotting (WB)], rabbit GST (91G1, Cell Signaling Technology; 1:1000 WB), rabbit GFP (A11122, Molecular Probes; 1:1000 WB; 1:100 PLA), mouse GFP (ab1218, Abcam; 1:100 PLA), mouse Sharpin [ab69507, Abcam; 1:100 regular immunofluorescence (IF) and PLA; 1 µg immunoprecipitation], rabbit Sharpin (14626-1-AP, Proteintech; 1:1000 WB; 1 µg immunoprecipitation), mouse cortactin (p80/85) (05-180, Merck Millipore; 1:300 IF, 1:1000 WB), rabbit Arp2 (ab47654, Abcam; 1:1000 WB, 1:100 IF), rabbit Arp3 (58182, One World Lab; 1:500 WB), rabbit paxillin (SC-5574, Santa Cruz Biotechnology; 1:100 TIRF), mouse GAPDH (5G4MaB6C5, HyTest; 1:20.000 WB, 1:100 PLA), mouse β-actin (A2228, Sigma; 1:1000 WB, 1:100 PLA), rabbit α2-integrin (ab1936, Chemicon; 1:100 PLA), rabbit phospho-p44/42 MAPK (Erk1/2) (4370, Cell Signaling Technology; 1:100 PLA), rat 9EG7 (553715, BD Biosciences; 1:100 FACS), mouse P5D2 (Hybridoma bank; 1:20 FACS and TIRF), mouse 12G10 (ab30394, Abcam; 1:100 FACS and TIRF), rabbit p65 (8242, Cell Signaling Technology; 1:100 PLA), rabbit Arpc3 (57646, One World Lab; 1:1000 WB).
These secondary antibodies were Alexa Fluor 488- or Alexa Fluor 555-conjugated IgGs (Invitrogen; IF), horseradish peroxidase (HRP)-conjugated IgGs (GE Healthcare; WB), DyLight 680- or 800-conjugated anti-mouse and rabbit IgGs (Thermo Scientific; WB).
Plasmids and siRNAs
Construction of GST–Sharpin and siRNA1-insensitive GFP–Sharpin (Rantala et al., 2011), and Sharpin mutant plasmids (De Franceschi et al., 2015) has been previously described. Arp3–GFP was expressed from Addgene Plasmid #8462 deposited by Matthew Welch (Welch et al., 1997). Arp3–TagRFP was constructed using primers introducing EcoRI and BamHI sites, followed by cloning into pTagRFP-N (Evrogen). For construction of pmCherry, we replaced GFP in GFP-C1 (Clontech) with mCherry using primers introducing NheI and EcoRI restriction sites. For mCherry–Sharpin, the siRNA1-resistant Sharpin coding sequence was cloned into pmCherry using primers introducing EcoRI and BamHI restriction sites. The mEmerald-Lifeact expression plasmid (mEmerald-Lifeact-7) was Addgene plasmid #54148 (deposited by Michael Davidson). pcDNA3-EGFP-Rac1-Q61L was Addgene plasmid #12981 deposited by Gary Bokoch (Subauste et al., 2000). Laura Machesky (CRUK Beatson Institute, Glasgow, UK) kindly donated pRK5-Myc-PIP5KIb (Rozelle et al., 2000). pSpCas9(BB)-2A-GFP (PX458) was Addgene plasmid #48138 deposited by Feng Zhang (Ran et al., 2013).
These siRNAs were used: Sharpin [Hs_SHARPIN_1 HP siRNA (Qiagen)], Arp3 [siGENOME Human ACTR3 siRNA (Dharmacon)], RNF31 [siGENOME RNF31 siRNA (Dharmacon)] and control siRNA [AllStars negative control siRNA (Qiagen)].
Synthetic peptides and recombinant proteins
Recombinant GST and GST–Sharpin were produced in E. coli Rosetta BL21DE3 and purified according to the manufacturer's instructions (BD Biosciences). The Arp2/3 complex from bovine brain and the GST-tagged human WASP VCA domain were from Cytoskeleton (Cytoskeleton, Inc.). Rabbit skeletal muscle actin was purified as described previously (Pollard and Cooper, 1984).
Cells and transfections
HeLa cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS), 1% L-glutamine, 1% MEM non-essential amino acids, 1% sodium pyruvate, 2% HEPES and 1% penicillin-streptomycin. HEK-293 and U2OS cells were grown in DMEM with 1% penicillin-streptomycin, 10% FBS and 1% L-glutamine. NCI-H460 cells were grown in RPMI1640 with 10% FBS, 1% penicillin-streptomycin, 1% L-glutamine, 1% MEM non-essential amino acids, 1% sodium pyruvate and 1% glucose. The generation and maintenance of cpdm MEF cells has been described previously (Rantala et al., 2011). All cell lines were regularly tested for contaminations and, except cpdm MEFs, were from American Type Culture Collection (ATCC). Plasmid transfections were performed using Lipofectamine 2000 (HeLa and HEK-293 cells) and Lipofectamine 3000 (NCI-H460, U2OS) (Life Technologies). siRNA transfections were performed using Hiperfect (Qiagen).
Sharpin-knockout cell lines created with CRISPR
Sharpin-knockout NCI-H460 cell lines were created using CRISPR genome engineering by excising a defined 488 bp region in the Sharpin-encoding gene using two guide RNAs. These guide RNAs (Sigma-Aldrich; 5′-TGGCTGTGCACGCCGCGGTG-3′, and 5′-TCAGTTTCCTACACCATCCG-3′) were designed with MIT CRISPR Designer (http://crispr.mit.edu/) and cloned individually in pSpCas9(BB)-2A-GFP (PX458) as described previously (Ran et al., 2013). Both plasmids were co-transfected into NCI-H460 cells and, 4 days later, GFP-positive cells were sorted with a FACSaria IIu Cell Sorter (BD Biosciences). Subsequently, we screened for clones that lack the intervening DNA sequence by PCR (forward primer 5′-GTGTCCATTTGTGGGCAAAG-3′ and reverse primer 5′-GGCACTGACCATTCTGTCCT-3′) to ensure that the gene is disrupted. Subsequent blotting of two cell lines with the appropriate 488 bp deletion in the Sharpin gene confirmed successful Sharpin knockout. WT control cells went through the same sorting procedure but did not have the deletion and showed normal levels of Sharpin expression.
Mass spectrometry
GFP pulldowns were performed using GFP-Trap beads (ChromoTek) according to the manufacturer's instructions, with cells kept in suspension or plated on fibronectin (10 μg/ml; Sigma) for 1 h before lysis. Protein samples were separated by SDS-PAGE and, following staining with InstantBlue (Expedeon), gel lanes were sliced and subjected to in-gel digestion with trypsin as described previously (Shevchenko et al., 1996) with modifications (Byron et al., 2015).
Digested samples were analysed by liquid chromatography tandem MS (LC-MS/MS) using an UltiMate® 3000 Rapid Separation LC (Dionex Corporation) coupled to an Orbitrap Elite (Thermo Fisher Scientific) mass spectrometer. Peptide mixtures were separated using a gradient from 92% A (0.1% formic acid in water) and 8% B (0.1% formic acid in acetonitrile) to 33% B, in 44 min at 300 nl/min, using a 75 mm×250 μm internal diameter 1.7 μM BEH C18 analytical column (Waters). Peptides were automatically selected for fragmentation by data-dependent analysis.
Protein identification was performed with the Proteome Discoverer (1.4) connected to in-house Mascot (v. 2.4) software. Data were searched against the SwissProt database (release 2015_08). Carbamidomethylation of cysteine was set as a fixed modification and oxidation of methionine was allowed as a variable modification. Only tryptic peptides were considered, with up to one missed cleavage permitted. Monoisotopic precursor mass values were used, and only doubly and triply charged precursor ions were considered. Data were validated in Scaffold (version 3.6) using a threshold of identification of at least 50% probability at the peptide level, at least 99% probability at the protein level and assignment of at least two unique, validated peptides. These acceptance criteria resulted in an estimated protein false discovery rate of 0.1% for all datasets. Data were converted using PRIDE Converter (version 2.5.5) (Barsnes et al., 2009) and validated using PRIDE Inspector (version 2.5.2) (Perez-Riverol et al., 2016).
Two biological replicates were performed for each GFP pulldown. Relative protein abundance was calculated by using the unweighted spectral count of a given protein normalised to the total number of spectra observed in the entire sample and to the molecular mass of that protein (normalised spectral count). To list and score the putative Sharpin interactors, three thresholds were used, reflecting the quality and the specificity of the binding to GFP–Sharpin. Low confidence was assigned to 690 proteins detected with at least four spectra and that were enriched twofold in the GFP–Sharpin datasets (suspension or adherent) over control. To provide lists of putative Sharpin binders that are likely to contain fewer false positives, two other thresholds were used. A medium confidence was assigned to 297 proteins detected with at least five spectra and enriched fourfold in the GFP–Sharpin datasets (suspension or adherent) over control, while a high confidence was assigned to 48 proteins detected with at least ten spectra and enriched fourfold in both GFP–Sharpin datasets.
Gene Ontology analyses were performed using DAVID (version 6.8) (Huang da et al., 2009) and the Gene Ontology map was created using the Cytoscape plugin ‘Enrichment Map’ (Merico et al., 2010). Proteins were hierarchically clustered on the basis of uncentred Pearson correlation using Cluster 3.0 (CClustering Library, version 1.50) (de Hoon et al., 2004) and visualised using JavaTreeView (version 1.1.6r2) (Saldanha, 2004). Protein–protein interaction network analyses were performed in Cytoscape (version 3.4.0) (Smoot et al., 2011). Proteins were mapped onto a merged human interactome consisting of protein–protein interactions reported in the Protein Interaction Network Analysis platform Homo sapiens network (Wu et al., 2009) integrated within Cytoscape using PINA4MS (version 2.0.1).
Accession numbers
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD004734.
Immunoblottings, immunoprecipitations and pulldowns
All immunoblottings, immunoprecipitations and GFP-bead (ChromoTek) pulldowns were undertaken as described previously (Pouwels et al., 2013).
For GST pulldown experiments glutathione–Sepharose 4B beads (GE Healthcare) were washed twice with binding buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 0.5 mM EDTA and 2 mM MgCl2) and subsequently incubated with 10 μg GST or GST–Sharpin in binding buffer (1 h rotation at 4°C). After washing, beads were incubated with binding buffer with 2 μM purified Arp2/3 complex and 0.1 mM ATP (1.5 h rotation at 4°C). Subsequently, beads were washed and suspended into loading buffer. Samples were analysed using immunoblotting.
Immunofluorescence
NCI-H460 or HeLa cells were fixed with 4% paraformaldehyde and stained. Immunofluorescence images in Fig. S4 (except Fig. S4C,D) were obtained with a 3i Marianas Spinning disk confocal microscope, equipped with Yokogawa CSU-W1 scanner (Intelligent Imaging Innovations), ORCA-Flash4.0 v2 sCMOS Camera (Hamamatsu Photonics), and a Plan-Apochromat 63×1.4 NA oil objective. Images in Fig. S4G were processed using the super-resolution radial fluctuations (SRRF) ImageJ plugin (Gustafsson et al., 2016). Images in Figs 4 and 5, Figs S4C,D and S5 were obtained using a Zeiss AxioVert 200 M inverted wide-field microscope equipped with a Plan-NEOFLUAR 63×1.25 NA oil objective (Zeiss) and Orca-ER camera (Hamamatsu Photonics). All image processing was performed using Fiji image analysis software (Schindelin et al., 2012).
Total internal reflection fluorescence microscopy
HeLa cells, freshly adherent to 3 cm glass-bottom dishes (MatTek Corporation) coated with 5 μg/ml fibronectin, were stained for active (12G10) or total (P5D2) integrins, along with paxillin, F-actin (Atto Phalloidin) and DAPI. Samples were imaged using a Carl Zeiss Laser-TIRF 3 Imaging System equipped with a 63×1.46 NA oil objective (alpha Plan-Apochromat, DIC) and Hamamatsu ImageEM C9100-13 emccd camera (Hamamatsu Photonics). All image analysis was performed with Fiji software.
Fluorescence recovery after photobleaching
GFP–Sharpin-expressing NCI-H460 cells on glass-bottom dishes (MatTek Corporation), in Ham's F12 nutrient mixture with 10% FBS and 1% HEPES, were subjected to FRAP imaging with 1 s intervals using the 3i Marianas Spinning disk confocal microscope, equipped as described above. After imaging for ∼10 s, cellular areas were bleached, followed by imaging for 90–120 s. FRAP data were analysed using the SlideBook 6 (Intelligent Imaging Innovations) FRAP analysis module. Movies and still images were prepared using Fiji software.
Live-cell imaging
NCI-H460 cells expressing mEmerald-Lifeact, plated for 4 h in Ham's F12 nutrient mixture with 10% FBS and 1% HEPES on glass-bottom dishes (MatTek Corporation) coated with 5 µg/ml fibronectin, were imaged at 37°C at 5 frames/min for 5–10 min with a Carl Zeiss Laser-TIRF 3 Imaging System equipped as described above. Movies were prepared using Fiji software.
For live-cell imaging, cpdm MEFs overexpressing GFP alone, or GFP-tagged WT or V240A/L242A Sharpin were plated sparsely in regular medium with 5% HEPES onto eight-well µ-Slides (Ibidi) coated with 5 µg/ml fibronectin. At 6 h after plating cells were imaged every 10 min under phase-contrast for at least 8 h, with GFP images taken every 10 frames. Imaging was undertaken using a 3i Marianas Spinning disk confocal microscope, equipped as described above, except that a 10× objective was used.
For the wound healing assays, equal amounts of HeLa cells were plated on an IncuCyte ImageLock™ 96-well plate (Essen BioScience). The next day a wound was made in the confluent monolayer using an Essen BioScience WoundMaker™ and wound closure was imaged every 2 hours using an IncuCyte Zoom™ System (Essen BioScience) with a 10× objective.
Pyrene-actin polymerisation assay
For the pyrene-actin mixture, 5% of pyrene-labelled actin was mixed with non-labelled G-actin in G-buffer (5 mM Tris-HCl pH 7.5 with 0.2 mM DTT, 0.2 mM CaCl2 and 0.2 mM ATP) to a final concentration of 20 µM. 11 µM of GST–Sharpin was mixed with 40 nM Arp2/3 and 30 nM VCA (or equal volume of VCA-buffer) in 50 mM Tris-HCl pH 7.5 with 150 mM NaCl, 3 mM DTT and 10% glycerol in the presence of 1× initiation mix (1 mM EGTA, 5 mM MgCl2, 0.25 mM ATP and NaCl to the total 100 mM in the final sample). Polymerisation of actin filaments was monitored at 22°C with excitation at 365 nm (excitation slit=10 nm) and emission at 407 nm (emission slit=20 nm) after addition of 4 µM pyrene-actin. Measurements were carried out on an Agilent Cary Eclipse Fluorescence Spectrophotometer with BioMelt Bundle System (Agilent Technologies). Origin 7.5 software (OriginLab Corp.) was used for data analyses.
FACS
HeLa cells, treated for 30 min with DMSO, 10 μM CytD (Sigma-Aldrich) or 100 μM CK666 (Sigma-Aldrich), were detached and fixed with 4% paraformaldehyde. Cells were stained for active β1-integrin (12G10) or total β1-integrin (P5D2). Samples were analysed using FACSCalibur with CellQuest software (BD Biosciences) and non-commercial Flowing Software ver. 2.5 (Perttu Terho; Turku Centre for Biotechnology, Finland; www.flowingsoftware.com). The Integrin Activation Index was calculated by dividing the background-corrected active cell-surface integrin levels by total cell-surface integrin levels.
Proximity ligation assays
PLAs were carried out according to a previously described protocol (Söderberg et al., 2006). Images were taken with a Carl Zeiss LSM780 laser scanning confocal microscope equipped with 63×1.2 W Corr Apochromat objective (Zeiss). PLA signals per cell were calculated by dividing the amount of PLA signal dots in one field of view, determined using Cell Profiler software (Carpenter et al., 2006), by the number of cells, as determined by counting nuclei.
FRET measurements by FLIM
HeLa cells were transfected with donor alone [GFP–Sharpin constructs (WT, fragments or point mutants) or Arp3–GFP] in control samples, or with donor together with the acceptor (Arp3–TagRFP or mCherry–Sharpin). As an additional control, cells were transfected with Arp3–GFP and mCherry alone. After 24 h, cells were fixed and mounted with Mowiol 4-88 (Sigma-Aldrich). GFP fluorescence lifetime was measured by using a fluorescence lifetime imaging attachment (Lambert Instruments) on a Zeiss AXIO Observer D1 inverted microscope (Zeiss). For sample excitation, a sinusoidally modulated 3W, 497 nm LED at 40 MHz under epi-illumination was used. Cells were imaged using the 63× NA 1.4 oil objective (excitation, BP470/40; beam splitter, FT495; emission, BP525/50). The phase and modulation were determined using the manufacturer's software from images acquired at 12 phase settings. Fluorescein at 0.01 mM, pH 9 was used as a lifetime reference standard. The apparent FRET efficiency was calculated using the measured lifetimes of each donor-acceptor pair (τDA) and the average lifetime of the donor-only (τD) samples, according to:
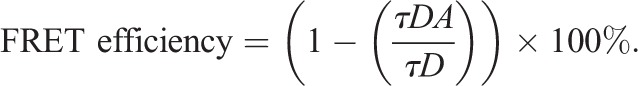 |
Micropatterns
Linear micropatterns with a width of 9 μm were produced on glass coverslips as described previously (Azioune et al., 2009) and coated with 50 μg/ml fibronectin and 5 μg/ml fibrinogen (Alexa Fluor 488 conjugate; Thermo Scientific). NCI-H460 cells were seeded in antibiotic-free medium for 7 h, followed by fixing and staining as described above. Cells were observed using a Zeiss AxioVert 200 M inverted wide-field microscope equipped with Plan-NEOFLUAR 100× 1.30 oil objective (Zeiss) and Hamamatsu Orca-ER camera (Hamamatsu Photonics). All image processing was done using Fiji software.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 5.03 for Windows (GraphPad Software). The Student's t-test was used for normally distributed data (Shapiro-Wilk normality test α=0.05). For all other data the Mann–Whitney test was used. P<0.05 was considered significant.
Acknowledgements
We thank K. Ikkala, M. Skaldin, P. Laasola and A. Augenlicht for technical assistance and the Cell Imaging Core at Turku Centre for Biotechnology for assistance with microscopy and flow cytometry. We thank J.D. Humphries, A. Byron, S. Warwood and D. Knight for help with the acquisition and analysis of the mass-spectrometry data, and Matthew Welch (UC Berkeley), Laura Machesky (CRUK Beatson Institute), the Bokoch lab (The Scripps Research Institute), Feng Zhang (Broad Institute) and Michael Davidson (Florida State University) are acknowledged for plasmids.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.H.K., S.I.S., P.J.L., M.H., J.P.; Methodology: M.H.K., S.I.S., G.J., U.B., M.M., M.H., J.P.; Validation: M.H.K., S.I.S., G.J., J.P.; Formal analysis: M.H.K., S.I.S., G.J., T.D., E.K., J.P.; Investigation: M.H.K., S.I.S., G.J., U.B., T.D., E.K., J.P.; Resources: G.J., J.P.; Writing - original draft: M.H.K., S.I.S., G.J., J.P.; Writing - review & editing: M.H.K., S.I.S., G.J., P.J.L., M.H., J.P.; Visualization: G.J., J.P.; Supervision: P.J.L., M.H., J.P.; Project administration: J.P.; Funding acquisition: J.P.
Funding
This work was supported by the University of Turku (Turun Yliopisto) Graduate School, the Turku doctoral programme of molecular medicine (TuDMM) (M.H.K. and T.D.) and Drug research doctoral programme (DRDP) (S.I.S. and M.M.), the Sigrid Juséliuksen Säätiö (P.L.), the Wellcome Trust (grant 092015; M.J.H.), Cancer Research UK (grant C13329/A21671; M.J.H.) and the Academy of Finland (J.P.). Deposited in PMC for release after 6 months.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD004734 (https://www.ebi.ac.uk/pride/archive/projects/PXD004734).
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.200329.supplemental
References
- Azioune A., Storch M., Bornens M., Théry M. and Piel M. (2009). Simple and rapid process for single cell micro-patterning. Lab. Chip 9, 1640-1642. 10.1039/b821581m [DOI] [PubMed] [Google Scholar]
- Barsnes H., Vizcaíno J. A., Eidhammer I. and Martens L. (2009). PRIDE converter: making proteomics data-sharing easy. Nat. Biotechnol. 27, 598-599. 10.1038/nbt0709-598 [DOI] [PubMed] [Google Scholar]
- Beli P., Mascheroni D., Xu D. and Innocenti M. (2008). WAVE and Arp2/3 jointly inhibit filopodium formation by entering into a complex with mDia2. Nat. Cell Biol. 10, 849-857. 10.1038/ncb1745 [DOI] [PubMed] [Google Scholar]
- Bii V. M., Rae D. T. and Trobridge G. D. (2015). A novel gammaretroviral shuttle vector insertional mutagenesis screen identifies SHARPIN as a breast cancer metastasis gene and prognostic biomarker. Oncotarget 6, 39507-39520. 10.18632/oncotarget.6232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B., Laplantine E., Dobbs K., Cobat A., Tarantino N., Hazen M., Lidov H. G. W., Hopkins G., Du L., Belkadi A. et al. (2015). Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J. Exp. Med. 212, 939-951. 10.1084/jem.20141130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayford S., Bryce N. S., Schevzov G., Haynes E. M., Bear J. E., Hardeman E. C. and Gunning P. W. (2016). Tropomyosin promotes lamellipodial persistence by collaborating with Arp2/3 at the leading edge. Curr. Biol. 26, 1312-1318. 10.1016/j.cub.2016.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byron A., Askari J. A., Humphries J. D., Jacquemet G., Koper E. J., Warwood S., Choi C. K., Stroud M. J., Chen C. S., Knight D. et al. (2015). A proteomic approach reveals integrin activation state-dependent control of microtubule cortical targeting. Nat. Commun. 6, 6135 10.1038/ncomms7135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier M.-F., Nioche P., Broutin-L'Hermite I., Boujemaa R., Le Clainche C., Egile C., Garbay C., Ducruix A., Sansonetti P. and Pantaloni D. (2000). GRB2 links signaling to actin assembly by enhancing interaction of neural wiskott-aldrich syndrome protein (N-WASp) with actin-related protein (ARP2/3) complex. J. Biol. Chem. 275, 21946-21952. 10.1074/jbc.M000687200 [DOI] [PubMed] [Google Scholar]
- Carpenter A. E., Jones T. R., Lamprecht M. R., Clarke C., Kang I. H., Friman O., Guertin D. A., Chang J. H., Lindquist R. A., Moffat J. et al. (2006). CellProfiler: Image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7, R100 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S., Kuzmanovic T., Zhang Y., Wetzel J. L. and Sen G. C. (2016). Ubiquitination of the transcription factor IRF-3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity 44, 1151-1161. 10.1016/j.immuni.2016.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franceschi N., Peuhu E., Parsons M., Rissanen S., Vattulainen I., Salmi M., Ivaska J. and Pouwels J. (2015). Mutually exclusive roles of SHARPIN in integrin inactivation and NF-κB signaling. PLoS ONE 10, e0143423 10.1371/journal.pone.0143423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon M. J., Imoto S., Nolan J. and Miyano S. (2004). Open source clustering software. Bioinformatics 20, 1453-1454. 10.1093/bioinformatics/bth078 [DOI] [PubMed] [Google Scholar]
- De Melo J. and Tang D. (2015). Elevation of SIPL1 (SHARPIN) increases breast cancer risk. PLoS ONE 10, e0127546 10.1371/journal.pone.0127546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois S. M., Alexia C., Wu Y., Leclair H. M., Leveau C., Schol E., Fest T., Tarte K., Chen Z. J., Gavard J. et al. (2014). A catalytic-independent role for the LUBAC in NF-kappaB activation upon antigen receptor engagement and in lymphoma cells. Blood 123, 2199-2203. 10.1182/blood-2013-05-504019 [DOI] [PubMed] [Google Scholar]
- Gerlach B., Cordier S. M., Schmukle A. C., Emmerich C. H., Rieser E., Haas T. L., Webb A. I., Rickard J. A., Anderton H., Wong W. W. et al. (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591-596. 10.1038/nature09816 [DOI] [PubMed] [Google Scholar]
- Gustafsson N., Culley S., Ashdown G., Owen D. M., Pereira P. M. and Henriques R. (2016). Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 7, 12471 10.1038/ncomms12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes E. M., Asokan S. B., King S. J., Johnson H. E., Haugh J. M. and Bear J. E. (2015). GMFbeta controls branched actin content and lamellipodial retraction in fibroblasts. J. Cell Biol. 209, 803-812. 10.1083/jcb.201501094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L., Ingram A., Rybak A. P. and Tang D. (2010). Shank-interacting protein-like 1 promotes tumorigenesis via PTEN inhibition in human tumor cells. J. Clin. Invest. 120, 2094-2108. 10.1172/JCI40778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Huang W., Sherman B. T. and Lempicki R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44-57. 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- Ikeda F., Deribe Y. L., Skånland S. S., Stieglitz B., Grabbe C., Franz-Wachtel M., van Wijk S. J., Goswami P., Nagy V., Terzic J. et al. (2011). SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 471, 637-641. 10.1038/nature09814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J., Kim J. M., Park B., Cheon Y., Lee B., Choo S. H., Koh S. S. and Lee S. (2010). Newly identified tumor-associated role of human sharpin. Mol. Cell. Biochem. 340, 161-167. 10.1007/s11010-010-0413-x [DOI] [PubMed] [Google Scholar]
- Krause M. and Gautreau A. (2014). Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 15, 577-590. 10.1038/nrm3861 [DOI] [PubMed] [Google Scholar]
- Kumari S., Redouane Y., Lopez-Mosqueda J., Shiraishi R., Romanowska M., Lutzmayer S., Kuiper J., Martinez C., Dikic I., Pasparakis M. et al. (2014). Sharpin prevents skin inflammation by inhibiting TNFR1-induced keratinocyte apoptosis. Elife 3, 10.7554/eLife.03422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F. P. L., Szczodrak M., Block J., Faix J., Breitsprecher D., Mannherz H. G., Stradal T. E., Dunn G. A., Small J. V. and Rottner K. (2008). Arp2/3 complex interactions and actin network turnover in lamellipodia. EMBO J. 27, 982-992. 10.1038/emboj.2008.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf K., Bollig F., Trowe M.-O., Besenbeck B., Ebert C., Kruspe D., Kispert A., Hanel F. and Englert C. (2010). Sipl1 and Rbck1 are novel Eya1-binding proteins with a role in craniofacial development. Mol. Cell. Biol. 30, 5764-5775. 10.1128/MCB.01645-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis M. J., Vyse S., Shields A. M., Boeltz S., Gordon P. A., Spector T. D., Lehner P. J., Walczak H. and Vyse T. J. (2015). UBE2L3 polymorphism amplifies NF-kappaB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am. J. Hum. Genet. 96, 221-234. 10.1016/j.ajhg.2014.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Lai Y., Cao Y., Du T., Zeng L., Wang G., Chen X., Chen J., Yu Y., Zhang S. et al. (2015). SHARPIN overexpression induces tumorigenesis in human prostate cancer LNCaP, DU145 and PC-3 cells via NF-kappaB/ERK/Akt signaling pathway. Med. Oncol. 32, 444 10.1007/s12032-014-0444-3 [DOI] [PubMed] [Google Scholar]
- Lim S., Sala C., Yoon J., Park S., Kuroda S., Sheng M. and Kim E. (2001). Sharpin, a novel postsynaptic density protein that directly interacts with the shank family of proteins. Mol. Cell. Neurosci. 17, 385-397. 10.1006/mcne.2000.0940 [DOI] [PubMed] [Google Scholar]
- Liu Z., Yang X., Chen C., Liu B., Ren B., Wang L., Zhao K., Yu S. and Ming H. (2013). Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol. Rep. 30, 2127-2136. 10.3892/or.2013.2669 [DOI] [PubMed] [Google Scholar]
- Merico D., Isserlin R., Stueker O., Emili A. and Bader G. D. (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5, e13984 10.1371/journal.pone.0013984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastase M.-V., Zeng-Brouwers J., Frey H., Hsieh L. T.-H., Poluzzi C., Beckmann J., Schroeder N., Pfeilschifter J., Lopez-Mosqueda J., Mersmann J. et al. (2016). An essential role for SHARPIN in the regulation of caspase 1 activity in sepsis. Am. J. Pathol. 186, 1206-1220. 10.1016/j.ajpath.2015.12.026 [DOI] [PubMed] [Google Scholar]
- Nolen B. J., Tomasevic N., Russell A., Pierce D. W., Jia Z., McCormick C. D., Hartman J., Sakowicz R. and Pollard T. D. (2009). Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature 460, 1031-1034. 10.1038/nature08231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y., Jin H.-S., Lopez J., Lee J., Liao L., Elly C. and Liu Y.-C. (2016). SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat. Immunol. 17, 286-296. 10.1038/ni.3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y., Xu Q.-W., Wang R., Uszkoreit J., Griss J., Sanchez A., Reisinger F., Csordas A., Ternent T., Del-Toro N. et al. (2016). PRIDE inspector toolsuite: Moving toward a universal visualization tool for proteomics data standard formats and quality assessment of ProteomeXchange datasets. Mol. Cell. Proteomics 15, 305-317. 10.1074/mcp.O115.050229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T. D. and Cooper J. A. (1984). Quantitative analysis of the effect of acanthamoeba profilin on actin filament nucleation and elongation. Biochemistry 23, 6631-6641. 10.1021/bi00321a054 [DOI] [PubMed] [Google Scholar]
- Pouwels J., De Franceschi N., Rantakari P., Auvinen K., Karikoski M., Mattila E., Potter C., Sundberg J. P., Hogg N., Gahmberg C. G. et al. (2013). SHARPIN regulates uropod detachment in migrating lymphocytes. Cell. Rep. 5, 619-628. 10.1016/j.celrep.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehoda K. E., Scott J. A., Mullins R. D. and Lim W. A. (2000). Integration of multiple signals through cooperative regulation of the N-WASP-Arp2/3 complex. Science 290, 801-806. 10.1126/science.290.5492.801 [DOI] [PubMed] [Google Scholar]
- Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A. and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281-2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantala J. K., Pouwels J., Pellinen T., Veltel S., Laasola P., Mattila E., Potter C. S., Duffy T., Sundberg J. P., Kallioniemi O. et al. (2011). SHARPIN is an endogenous inhibitor of beta1-integrin activation. Nat. Cell Biol. 13, 1315-1324. 10.1038/ncb2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard J. A., Anderton H., Etemadi N., Nachbur U., Darding M., Peltzer N., Lalaoui N., Lawlor K. E., Vanyai H., Hall C. et al. (2014). TNFR1-dependent cell death drives inflammation in sharpin-deficient mice. Elife 3, 10.7554/eLife.03464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z. et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605-607. 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers M. A., Bowman J. W., Fujita H., Orazio N., Shi M., Liang Q., Amatya R., Kelly T. J., Iwai K., Ting J. et al. (2014). The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J. Exp. Med. 211, 1333-1347. 10.1084/jem.20132486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers S. L., Wiedemann U., Stuurman N. and Vale R. D. (2003). Molecular requirements for actin-based lamella formation in drosophila S2 cells. J. Cell Biol. 162, 1079-1088. 10.1083/jcb.200303023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T. and Kirschner M. W. (1999). The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97, 221-231. 10.1016/S0092-8674(00)80732-1 [DOI] [PubMed] [Google Scholar]
- Rotty J. D., Wu C. and Bear J. E. (2013). New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol. 14, 7-12. 10.1038/nrm3492 [DOI] [PubMed] [Google Scholar]
- Rozelle A. L., Machesky L. M., Yamamoto M., Driessens M. H., Insall R. H., Roth M. G., Luby-Phelps K., Marriott G., Hall A. and Yin H. L. (2000). Phosphatidylinositol 4,5-bisphosphate induces actin-based movement of raft-enriched vesicles through WASP-Arp2/3. Curr. Biol. 10, 311-320. 10.1016/S0960-9822(00)00384-5 [DOI] [PubMed] [Google Scholar]
- Saldanha A. J. (2004). Java treeview–extensible visualization of microarray data. Bioinformatics 20, 3246-3248. 10.1093/bioinformatics/bth349 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A., Wilm M., Vorm O., Jensen O. N., Podtelejnikov A. V., Neubauer G., Shevchenko A., Mortensen P. and Mann M. (1996). A strategy for identifying gel-separated proteins in sequence databases by MS alone. Biochem. Soc. Trans. 24, 893-896. 10.1042/bst0240893 [DOI] [PubMed] [Google Scholar]
- Smoot M. E., Ono K., Ruscheinski J., Wang P.-L. and Ideker T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431-432. 10.1093/bioinformatics/btq675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.-G. et al. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995-1000. 10.1038/nmeth947 [DOI] [PubMed] [Google Scholar]
- Stieglitz B., Haire L. F., Dikic I. and Rittinger K. (2012). Structural analysis of SHARPIN, a subunit of a large multi-protein E3 ubiquitin ligase, reveals a novel dimerization function for the pleckstrin homology superfold. J. Biol. Chem. 287, 20823-20829. 10.1074/jbc.M112.359547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste M. C., Von Herrath M., Benard V., Chamberlain C. E., Chuang T. H., Chu K., Bokoch G. M. and Hahn K. M. (2000). Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and fas. J. Biol. Chem. 275, 9725-9733. 10.1074/jbc.275.13.9725 [DOI] [PubMed] [Google Scholar]
- Suraneni P., Rubinstein B., Unruh J. R., Durnin M., Hanein D. and Li R. (2012). The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 197, 239-251. 10.1083/jcb.201112113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F., Sakata S.-i., Saeki Y., Satomi Y., Kirisako T., Kamei K., Nakagawa T., Kato M., Murata S., Yamaoka S. et al. (2009). Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol. 11, 123-132. 10.1038/ncb1821 [DOI] [PubMed] [Google Scholar]
- Tokunaga F., Nakagawa T., Nakahara M., Saeki Y., Taniguchi M., Sakata S.-i., Tanaka K., Nakano H. and Iwai K. (2011). SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature 471, 633-636. 10.1038/nature09815 [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A., Côté R., Reisinger F., Barsnes H., Foster J. M., Rameseder J., Hermjakob H. and Martens L. (2010). The proteomics identifications database: 2010 update. Nucleic Acids Res. 38, D736-D742. 10.1093/nar/gkp964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaíno J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J. et al. (2013). The PRoteomics IDEntifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 41, D1063-D1069. 10.1093/nar/gks1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S. A., Satpathy S., Beli P. and Choudhary C. (2016). SPATA2 links CYLD to the TNF-alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 35, 1868-1884. 10.15252/embj.201694300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M. D., DePace A. H., Verma S., Iwamatsu A. and Mitchison T. J. (1997). The human Arp2/3 complex is composed of evolutionarily conserved subunits and is localized to cellular regions of dynamic actin filament assembly. J. Cell Biol. 138, 375-384. 10.1083/jcb.138.2.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Vallenius T., Ovaska K., Westermarck J., Mäkelä T. P. and Hautaniemi S. (2009). Integrated network analysis platform for protein-protein interactions. Nat. Methods 6, 75-77. 10.1038/nmeth.1282 [DOI] [PubMed] [Google Scholar]
- Wu C., Asokan S. B., Berginski M. E., Haynes E. M., Sharpless N. E., Griffith J. D., Gomez S. M. and Bear J. E. (2012). Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 148, 973-987. 10.1016/j.cell.2011.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Schmitz R., Mitala J., Whiting A., Xiao W., Ceribelli M., Wright G. W., Zhao H., Yang Y., Xu W. et al. (2014). Essential role of the linear ubiquitin chain assembly complex in lymphoma revealed by rare germline polymorphisms. Cancer. Discov. 4, 480-493. 10.1158/2159-8290.CD-13-0915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Kelly P., Shaffer A. L. III, Schmitz R., Yoo H. M., Liu X., da Huang W., Webster D., Young R. M., Nakagawa M. et al. (2016). Targeting non-proteolytic protein ubiquitination for the treatment of diffuse large B cell lymphoma. Cancer. Cell. 29, 494-507. 10.1016/j.ccell.2016.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak D. E., Schmitz F., Gold E. S., Diercks A. H., Peschon J. J., Valvo J. S., Niemisto A., Podolsky I., Fallen S. G., Suen R. et al. (2011). Systems analysis identifies an essential role for SHANK-associated RH domain-interacting protein (SHARPIN) in macrophage toll-like receptor 2 (TLR2) responses. Proc. Natl. Acad. Sci. USA 108, 11536-11541. 10.1073/pnas.1107577108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Huang H., Zhou H., Du T., Zeng L., Cao Y., Chen J., Lai Y., Li J., Wang G. et al. (2014). Activation of nuclear factor kappaB pathway and downstream targets survivin and livin by SHARPIN contributes to the progression and metastasis of prostate cancer. Cancer 120, 3208-3218. 10.1002/cncr.28796 [DOI] [PubMed] [Google Scholar]