

ABSTRACT
Rho GTPase family members are known regulators of directed migration and therefore play key roles in processes including development, the immune response and cancer metastasis. However, their individual contributions to these processes are complex. Here, we modify the activity of the two Rho GTPase family members Rac and Cdc42 by optogenetically recruiting specific guanine nucleotide exchange factor (GEF) DH or PH domains to defined regions of the cell membrane. We find that the localized activation of both GTPases produces lamellipodia in cells plated on a fibronectin substrate. By using a novel optotaxis assay, we show that biased activation can drive directional migration. Interestingly, in the absence of exogenous fibronectin, Rac activation is insufficient to produce stable lamellipodia or directional migration whereas Cdc42 activation is sufficient for these processes. We find that a remarkably small amount of fibronectin (<10 puncta per protrusion) is necessary to support stable GTPase-driven lamellipodia formation. Cdc42 bypasses the need for exogenous fibronectin by stimulating cellular fibronectin deposition under the newly formed lamellipodia.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Rac, Cdc42, Optogenetics, ILID, Fibronectin, Directed migration
Highlighted Article: Through an optogenetic approach, differences in the contributions of Rac and Cdc42 to directed migration are analyzed. Cdc42 can stabilize protrusions and directed migration via fibronectin secretion while Rac cannot.
INTRODUCTION
The ability of a cell to sense extracellular cues and respond with directed migration is central to many pathological and physiological conditions, such as development, wound healing, the immune response and cancer metastasis (Ridley et al., 2003). For directional migration to occur, a differential cue is first sensed through transmembrane receptors, then transduced and amplified into a differential intracellular signal, and eventually manifests into a physical force aligned with the extracellular gradient through cytoskeletal reorganization. One form of directional migration that has received recent attention is haptotaxis, where the cell is responding to substrate cues such as differing concentrations of extracellular matrix (ECM) components. The basic module of ECM sensing consists of trans-membrane receptors, mainly integrins, and the multimeric protein complexes that assemble at the cytoplasmic side where integrins cluster (Hu and Luo, 2013; Case and Waterman, 2015). We recently found that fibroblast haptotaxis requires biased lamellipodial dynamics that is achieved through protrusions formed by Arp2/3-branched actin (King et al., 2016). Furthermore, we linked the graded ECM-sensing module to the cytoskeletal dynamics and directed migration through a pathway involving the known adhesion signal transducer and Rac-specific guanine nucleotide exchange factor (GEF) Tiam1 and activation of the Rac GTPase.
Rho family GTPases are one of several downstream intracellular signaling nodes that spatially and temporally transduce extracellular cues into cytoskeletal remodeling (Heasman and Ridley, 2008). Two critical members of the family are Rac and Cdc42. While we showed that Rac is essential for haptotaxis in fibroblasts, other studies have shown that localized activity of Cdc42 is critical during directed migration (Yang et al., 2015). We therefore set out to further delineate the individual contributions of Rac and Cdc42 to directional migration.
Rac and Cdc42 signals both regulate actin dynamics through individual as well as overlapping downstream effectors. These pathways are also complicated by GTPase cross-regulation (Nishimura et al., 2005; Guilluy et al., 2011). For example, early work from Alan Hall's group showed that Cdc42 activity produced lamellipodia. Only upon inhibition of Rac did Cdc42 produce filopodia, pointing to a close interplay of the two proteins (Nobes and Hall, 1995). Further complicating the pathways, in migrating cells, Rac and Cdc42 activities are spatially overlapping with the highest activity at the leading edge where they regulate actin polymerization to drive the plasma membrane in the direction of motility (Ridley et al., 2003; Machacek et al., 2009; Yang et al., 2015). Furthermore, both GTPases are also involved in other motility-relevant processes such as endocytosis, exocytosis, ECM remodeling and microtubule dynamics (Fernandez-sauze et al., 2009; Gubar et al., 2013; Bretou et al., 2014; Wojnacki et al., 2014). The multi-faceted functions of Rho family activity as well as their interconnected signaling makes it difficult to study the cause-and-effect relationships of their individual roles in directed migration. Normally, directed migration is studied by supplying cells with a graded extracellular cue while genetically and/or pharmacologically manipulating intracellular components on a whole-cell basis. While these methods have produced informative results, much is left to be learned by directly manipulating the spatial and temporal activity of individual components within cells.
Cellular optogenetics provides the means to do just that. Optogenetics takes advantage of engineered light-sensitive proteins to manipulate the localization and/or activation state of a protein of interest. GEFs activate GTPases by aiding the exchange of GDP for GTP (Rossman et al., 2005). Each GEF has a functional domain (DH or PH, hereafter DH/PH) with a specific affinity for one or more GTPase. For example, several studies have shown that the DH/PH domains of Tiam1 and intersectin 1 (ITSN1) are highly specific for Rac and Cdc42, respectively (Worthylake et al., 2000; Hussain et al., 2001; Snyder et al., 2002; Jaiswal et al., 2013). Here, we employ an optogenetic system, the improved light-inducible dimer (iLID), to drive the DH/PH domains of Tiam1 and ITSN1 to the cell membrane to specifically activate Rac or Cdc42 in the presence of light to study protrusion and directional migration (Guntas et al., 2015).
RESULTS
Optogenetically biased GTPase activity is sufficient to spatially regulate lamellipodia protrusion and directed migration
Spatial and temporal Rho family GTPase activity functions to regulate cytoskeletal remodeling during cell polarization by producing lamellipodial protrusions at the front of the cell. As a proof of principal, we previously engineered a iLID system and used it to separately localize two GEF DH/PH domains to the plasma membrane where they function to activate specific Rho GTPases (Fig. 1A) (Guntas et al., 2015; Hallett et al., 2016; Zimmerman et al., 2016a). Here, we extend our experiments beyond this proof of principal to better understand the role of GTPase activity in directed migration.
Fig. 1.

Differential optogenetic stimulation of Rac and Cdc42 induces lamellipodia and directed migration on a FN substrate. (A) Schematic diagram of optogenetic switch to regulate GTPases by localizing specific GEF DH/PH domains to the cell membrane. T represents GTP-bound GTPase, while D represents GDP-bound GTPase. (B) Representative fluorescence micrographs of live IA32 fibroblasts being optogenetically activated. Top panels show optogenetic recruitment of RFP control, while the middle and bottom show recruitment of the Tiam1 and ITSN1 DH/PH domains over time. Right panels are kymographs along the yellow line. Blue arrows denote activated areas. Scale bars: 50 µm (fluorescence images); 5 µm and 250 s (kymographs). (C) Schematic diagram of the optotaxis chamber. (D) Image of the light gradient produced by the optotaxis chamber. (E) FMI graph for fibroblasts expressing Venus-iLID-CAAX and Tiam1-RFP to determine haptotaxis in response to light intensities at the positions demonstrated in D. Results are mean±95% c.i. (F) Rose plots, FMI graphs and velocity plots representing migration vectors for control fibroblasts, and fibroblasts expressing Tiam1- and ITSN1-Micro (with iLID-CAAX). The blue triangle represents the light gradient. Error bars are the 95% c.i. *P<0.05. In B,E and F, cells were plated on 10 µg/ml FN. Refer to Table S1 for experimental details.
iLID was expressed as a fusion protein with a yellow fluorescent protein (Venus) and a CAAX motif which localizes the protein to the plasma membrane. The iLID-binding partner, SspB_Micro (referred to here as Micro), was expressed as a fusion protein with tagRFPt (hereafter RFP) and the DH/PH domain of either Tiam1 or ITSN1 to target Rac and Cdc42 GTPases, respectively. We used a laser-scanning confocal microscope to stimulate a small region of interest (ROI) at the edge of the cell. Upon illumination with 488 nm light, the DH/PH domains were relocalized from the cytoplasm to the cell membrane within the ROI. In the cells expressing the Tiam1 DH/PH construct (abbreviated Tiam1-Micro throughout), a lamellipodial protrusion forms locally with respect to the ROI as we expected for Rac activation (Fig. 1B; Movie 1). However, stimulation of cells expressing the ITSN1 DH/PH construct (abbreviated ITSN1-Micro throughout), also formed lamellipodial protrusions within the stimulated ROI. While, canonically, Cdc42 activity induces filopodia it has also been shown to stimulate the formation of lamellipodia, likely through crosstalk with Rac (Nobes and Hall, 1995; Nishimura et al., 2005; Guilluy et al., 2011). Importantly, localization of control RFP-Micro (without any DH/PH component) left the morphology of the cell unchanged.
Based on these findings, we hypothesized that a biased localization of the DH/PH domain would functionally bias cell migration towards higher light intensities. To test this, we designed a device to create a stable blue-light gradient. The device consists of a 467 nm LED light source projecting through a 3D-printed housing containing a graded neutral density filter, producing a graded light intensity. The housing is placed on top of a 35 mm glass-bottomed dish that is mounted on a microscope and imaged over 12–20 h (Fig. 1C,D; Fig. S1A,C). Although not optimal, the collected bright-field image produced by the light gradient was sufficient for whole-cell tracking purposes (Fig. S1C). We measured the gradient of light by analyzing line scans parallel to the direction of the gradient and found that, on average, the device produced a 2% gradient over 100 µm, the average diameter of a fibroblast (Fig. S1B). In pilot experiments, using Tiam1-Micro- and iLID-CAAX-expressing cells plated on fibronectin (FN)-coated glass, we found that cells expressing each protein at high levels would flatten out in all directions and become largely immobile. Based on this, we sorted a stable population of cells expressing low levels of the constructs by using fluorescence-activated cell sorting (FACS). To measure directional motility, we manually tracked all cells and calculated the forward migration index (FMI; net distance in relation to the gradient divided by the total path length; Fig. S1D) for each cell. For the purpose of this study, we considered a positive FMI with 95% confidence intervals not encompassing zero as directional migration. Consistent with our visual impression, we found that cells expressing high levels of the optogenetic components did not migrate while cells expressing low levels did (Fig. S1E). Although not directly tested, we assumed that the signaling produced by the switch in the cells expressing high levels was saturated across the entire cell, even at the side of the cell exposed to lower intensity light. Using the population expressing low levels, we found that the direction of cell migration was biased toward the higher intensity of light as determined by assessing the FMI. However, the bias was small. To find the optimal light intensity along the gradient, we imaged five equally spaced sections along 1 cm of the gradient, tracked cells in each, and found a biphasic response in FMI values (Fig. 1D,E). The area producing the optimal FMI was used in all subsequent experiments. Cells expressing either Tiam1- or ITSN1-Micro together with iLID-CAAX plated on FN-coated glass migrated directionally toward the higher intensity light, while the cells expressing the RFP-Micro control migrated randomly (Fig. 1F). Interestingly, cell velocities varied depending on the DH/PH. These data show that spatially biased GTPase activity is sufficient to induce directional cell migration. We named this phenomenon ‘optotaxis’.
Tiam1- and ITSN1-Micro specifically activate Rac and Cdc42
Since ITSN1-Micro localization induced lamellipodia, we wanted to ensure the specificity for Cdc42 with two separate approaches. First, we tested the effect of ITSN1-Micro activity in Cdc42-null fibroblasts. Fibroblastoid cells derived from embryonic stem cells containing one conditionally inactive Cdc42 allele (Cdc42f/−) were mock treated or treated with adenovirus encoding Cre recombinase (Adeno-Cre) (Czuchra et al., 2005). Through western blotting, we found that Adeno-Cre-treated cells contained no detectable Cdc42 at 5 days after infection (Fig. 2A). We therefore considered these cells as Cdc42−/−. By performing retroviral and lentiviral infection, both cell types were further transduced to stably express iLID-CAAX and ITSN1-Micro. Upon illumination of an ROI, we found that in both cell types ITSN1-Micro was localized to the ROI (Fig. 2B). Furthermore, in Cdc42f/− cells we found that localized ITSN1-Micro induced a lamellipodial protrusion, as we expected from previous experiments. However, although ITSN1-Micro localized to the ROI, Cdc42−/− cells failed to produce any substantial morphological change at the ROI. To quantify this difference, kymographs parallel to the edge of the cells within the ROI were traced for many cells and averaged for each cell type (Fig. 2C). On average, Cdc42f/− cells produced a ∼10 µm protrusion within the time of activation while Cdc42−/− cells produce a <1 µm protrusion.
Fig. 2.

Cdc42-null fibroblasts fail to protrude in response to optogenetic ITSN1 localization. (A) Representative western blot analysis of Cdc42 protein levels in MEFs containing a single floxed Cdc42 allele (Cdc42f/−) and either untreated or transduced with Adeno-Cre virus (to give Cdc42−/− cells). (B) Representative fluorescence micrographs of live fibroblasts from A being optogenetically activated to recruit ITSN1-micro to a ROI. Right panels are kymographs along the yellow line. Blue arrows denote activated areas. Scale bars: 50 µm (fluorescence images); 5 µm and 250 s (kymographs). (C) Mean distance for the ITSN1 DH/PH-induced protrusions for the denoted genotype as determined from the kymographs. *P<0.05. Refer to Table S2 for experimental details.
To further test the specificity of Tiam1- and ITSN1-Micro, we imaged a yellow fluorescent protein (Venus or YPET) fused to GTPase-binding domains (GBDs) during localization of each DH/PH. The GTPase-binding domains of the Rac/Cdc42 effector PAK (pGBD) and the Cdc42 effector WASP (wGBD) have both previously been used to monitor GTPase activity during optogenetic GTPase stimulation (Levskaya et al., 2009; O'Neill et al., 2016). Each GBD binds with a higher affinity to the activated GTPase and therefore acts as a translocation biosensor. We found a corresponding local increase in pGBD but not wGBD signal upon Tiam1-Micro localization (Fig. S2A,B). Upon ITSN1 localization, both pGBD and wGBD signals increased (Fig. S2C,D). Comparing the kinetics of DH/PH localization to GBD localization, GTPase activation and effector binding occurs within seconds of DH/PH localization. To ensure that the biosensor signal increases that we observed were not due to an increase in volume in the activated area, we also imaged soluble Venus upon stimulation and found no corresponding increases in signal (Fig. S2A–D). Taken together, these results suggest that Tiam1- and ITSN1-Micro specifically act on Rac and Cdc42, respectively, and that the lamellipodia induced by ITSN1-Micro are most likely due to crosstalk from Cdc42 to Rac.
Arp2/3 is necessary for optogenetically induced lamellipodia and optotaxis
Previous work from our group and others has shown that fibroblasts require the Arp2/3 complex for lamellipodia formation (Suraneni et al., 2012; Wu et al., 2012). Since localization of both DH/PH domains led to the formation of lamellipodia and biased directional migration, we hypothesized that perturbing Arp2/3 activity would inhibit the optogenetic induction of lamellipodia and phototactic fidelity. By using fibroblasts harboring a conditional allele of the Arpc2 gene, we produced cells genetically null for the p34 subunit (encoded by the Arpc2 gene) of the Arp2/3 complex (Asokan et al., 2014; Rotty et al., 2015). We illuminated small regions of interest in parental (Arpc2+) and null cells expressing iLID-CAAX and either Tiam1- or ITSN1-Micro. As expected, Tiam1-Micro localized to the membrane within the ROI and produced a lamellipodium in the parental line, but this treatment did not produce detectable morphological changes in the null cells (Fig. 3A; Movie 2). However, while ITSN1-Micro localization produced lamellipodia formation in the parental line, we observed that local filopodia formation occurred in the p34-null cells with exposure to light (Fig. 3D; Movie 2). For each condition, at least four cells were illuminated. Fig. 3A and Fig. 3D are representative of the response for each condition. These findings are consistent with ITSN1 localization to the membrane inducing filopodia formation through Cdc42 activity. This further suggests that ITSN1 may induce lamellipodia formation in the parental cells through GTPase crosstalk, with Arp2/3-containing lamellipodia overwhelming the smaller filopodial structures.
Fig. 3.

Arp2/3 activity is necessary for Rac- and Cdc42-induced lamellipodia and optotaxis. (A) Representative fluorescence micrographs depicting optogenetic recruitment of Tiam1 DH/PH in ArpC2 competent (f/f) and null (−/−) MEFs. Blue arrows denote the area of activation. The enlarged images depict areas of activation. (B) Rose plots, FMI graphs and velocity plots representing migration vectors for Tiam1-Micro- and iLID-CAAX-expressing fibroblasts in the presence of DMSO or 150 µM CK666. Blue triangles represent the light gradient. (C) Representative immunofluorescence images of fixed fibroblasts from B. (D) Fluorescence micrographs depicting optogenetic recruitment of ITSN1 DH/PH in ArpC2 competent and null MEFs. Blue arrows denote areas of activation. The enlarged images depict areas of activation. Yellow arrowheads denote filopodia. (E) Rose plots, FMI graphs and velocity plots representing migration vectors for control, and ITSN1-Micro- and iLID-CAAX-expressing fibroblasts in the presence of DMSO or 150 µM CK666. Blue triangles represent the light gradient. (F) Representative immunofluorescence images of fixed fibroblasts from B. Error bars are the 95% c.i. *P<0.005; ns, not significant. All cells were plated on 10 µg/ml FN. Scale bars: 50 µm. Refer to Table S1 for experimental details.
We next tested the necessity of Arp2/3 in Tiam1- and ITSN1-based optotaxis by treating fibroblasts with CK666 (an Arp2/3 inhibitor) while performing the previously described optotaxis assay. We found that cells inhibited by CK666 migrated randomly in both cases while control DMSO-treated cells migrated in the direction of the more intense light (Fig. 3B,E). To ensure the efficacy of CK666, each dish of cells was fixed, permeabilized and stained for the p34 subunit of Arp2/3 and phalloidin. Cells treated with CK666 almost entirely lacked p34-positive lamellipodial structures (Fig. 3C,F). These data suggest that Arp2/3 activity is necessary for differentially localized Rac or Cdc42 activity to bias migration direction. In the case of ITSN1-Micro localization, it also implies that a biased direction of filopodia formation is not sufficient to bias directionality of whole-cell migration.
Exogenous FN is necessary for Tiam1- but not ITSN1-induced optotaxis
We previously found that the Arp2/3 complex and lamellipodia were necessary for haptotaxis and that a haptotactic gradient of FN reinforced Tiam1-Micro induced lamellipodial protrusions in the direction of higher FN concentrations (King et al., 2016). We were therefore interested in whether a FN substrate was necessary for Tiam1- and ITSN1-Micro-induced optotaxis. To test this, we coated glass-bottomed dishes with either 10 µg/ml FN back-filled with 0.01% poly-L-lysine (poly-LL) or 0.01% poly-LL alone, and performed optotaxis assays for both Tiam1- or ITSN1-Micro in iLID-CAAX-expressing cells. Poly-LL allows cells to adhere to the glass substrate through electrostatic interactions with cell glycocalyx but without specific receptor–ECM interactions. In the case of Tiam1-Micro-induced optotaxis, cells on FN migrated directionally towards the higher intensity light, while cells on poly-LL migrated randomly (Fig. 4A). Interestingly, ITSN1-Micro-expressing cells moved directionally independently of the substrate on which they were plated (Fig. 4B). In both cases, cells on poly-LL migrated slightly faster (Fig. 4A,B). These results raised two connected yet separate questions. First, why is differential Tiam1-Micro localization (Rac activity) insufficient to directionally bias migration in the absence of a FN substrate, and second, why is differential ITSN1-Micro localization (Cdc42 activity) sufficient to directionally bias cell migration independently of an ECM substrate.
Fig. 4.
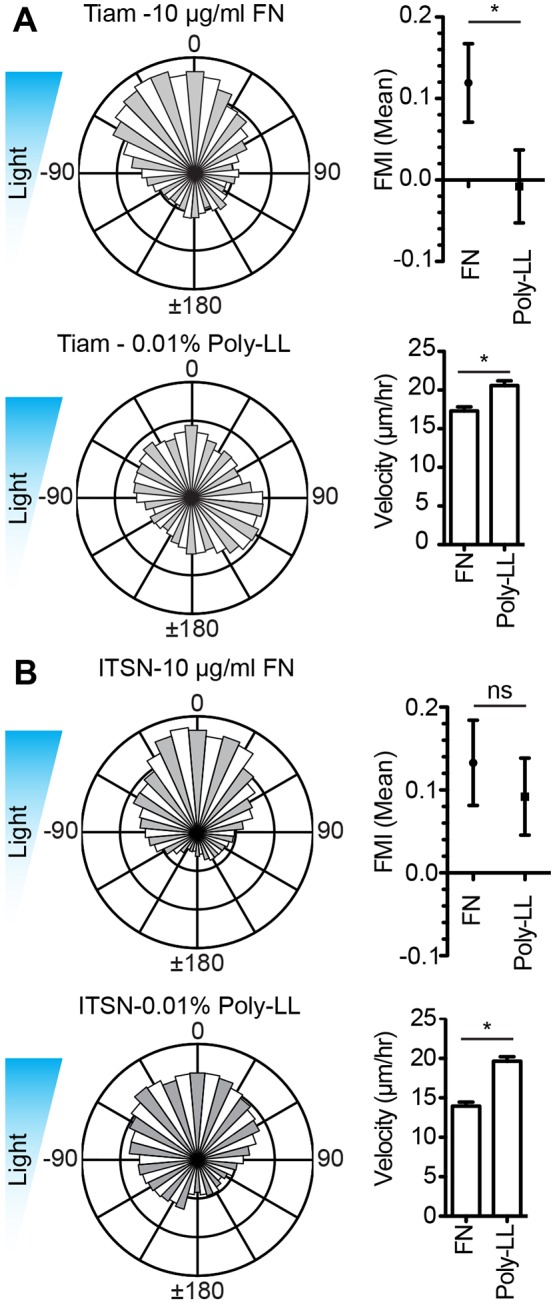
FN substrate is necessary for Tiam1- but not ITSN1-induced optotaxis. (A) Rose plots, FMI graphs and velocity plots representing migration vectors for Tiam1-Micro- and iLID-CAAX-expressing fibroblasts plated on FN or poly-L-lysine (poly-LL). (B) Rose plots, FMI graphs and velocity plots representing migration vectors for Tiam1-Micro- and iLID-CAAX-expressing fibroblasts plated on fibronectin or poly-L-lysine. Blue triangles represent the light gradient. Error bars are the 95% c.i. *P<0.005. Refer to Table S1 for experimental details.
A small amount of FN is sufficient to reinforce Tiam1-Micro-induced protrusions
To address our first question, we used Tiam1-Micro and iLID-CAAX-expressing fibroblasts to perform single-cell activation assays on FN- or poly-LL-coated dishes. We first ensured that pre-coating with poly-LL blocked surface adsorption of FN from the serum component of our medium (10% fetal bovine serum) (Fig. S3A). In these conditions, we found that cells plated on 10 µg/ml FN formed stable protrusions upon light activation within the ROI (Fig. 5A; Movie 3). By using kymography, we found that, on average, the induced protrusions extended ∼12 µm during the period of activation (Fig. 5A,B; Movie 3). However, cells activated on poly-LL-coated substrates failed to form an effective protrusion and instead formed a series of ruffle-like lamellipodial protrusions that were quickly retracted and the overall distance protruded during activation was negligible (less than 5 µm; Fig. 5A,B; Movie 3). To determine how much FN was required to support stable protrusions, we coated the dishes with a 10-fold dilution series of FN, back-filled with poly-LL and performed the same assay. We hypothesized that a linear relationship would exist between the coating concentration and protrusion distance. However, we found that on any coating concentration greater than or equal to 10−3 µg/ml FN, cells protruded to a similar average distance as cells on 10 µg/ml FN. At any concentration less than 10−3 µg/ml, cells failed to form an effective protrusion (Fig. 5B). Importantly, extracellular staining of FN on cells plated on poly-LL revealed that cells produced and secreted their own FN, but it was not found in regions beyond the cell perimeter (Fig. S3C). This suggests that de novo protrusions produced via the optogenetic activation of Rac are dependent on exogenously supplied FN present under the area of the new protrusion.
Fig. 5.

Tiam1-induced protrusions require very low amounts of substrate FN for stability. (A) Representative fluorescence micrographs depicting optogenetic recruitment of Tiam1 DH/PH to a ROI in a fibroblast plated on 10 µg/ml FN or poly-L-lysine (poly-LL). Right panels are kymographs along the yellow line. Blue arrows denote activated areas. Scale bars: 50 µm (fluorescence images); 5 µm and 250 s (kymographs). (B) Mean distance for the Tiam1 DH/PH-induced protrusions for the denoted FN concentration as determined from the kymographs. (C) Relationship between FN coating concentrations and FN substrate concentrations. Right axis denotes the concentrations as the number of FN puncta that the average protrusion would contact before retracting. *P<0.05. Refer to Table S2 for experimental details.
The surprising ability of a cell to form substantial protrusions at such low coating concentrations of FN prompted us to test the relationship between FN coating concentration and the concentration of FN adhered to the glass. We imaged a series of 10-fold dilutions of FN coatings by using Cy5-labeled FN. At higher concentrations, the Cy5 FN signal was evenly distributed across the glass. However, at lower concentrations, labeled FN puncta were distributed far enough apart to accurately count them (Fig. S3B). Therefore, by imaging glass coated with 10-fold dilutions of unlabeled FN supplemented with 10−4 µg/ml Cy5 FN, we counted individual FN puncta on the glass across a range of coating concentrations. Importantly, we found that in the range of concentrations tested, the concentration of bound FN on the glass scaled linearly with the solution concentration used for coating. Furthermore, we determined that at a 10−3 µg/ml FN coating concentration (the lowest concentration at which a cell will form a stable Tiam1-Micro-induced protrusion) an induced protrusion will encounter less than ten FN puncta on average (Fig. 5C; Fig. S3B). At 10−4 µg/ml (the first concentration at which cells fail to form and effective protrusion) induced protrusions will encounter less than one FN puncta. While we were unable to determine how many molecules of FN are in a single puncta, we hypothesize that they consist of dimers or small higher-order oligomers or polymers (Schwarzbauer and DeSimone, 2011).
Integrin binding and myosin contractility are necessary for stable Tiam1-Micro-induced protrusions and directed migration
Integrins are the primary FN receptors on cells; specifically, β1-, β3- and β5-containing integrins. To ensure that the Tiam1-Micro-induced protrusion stability, provided by FN, was due to interactions with integrins, we took advantage of the integrin inhibitor cilengitide (an inhibitor specific for αvβ3, αvβ5 and α5β1 integrins) (Mas-Moruno et al., 2010). In contrast to in untreated cells, Tiam1-Micro and iLID-CAAX-expressing fibroblasts plated on 10−3 µg/ml FN-coated glass and treated with 10 µM cilengitide consistently failed to form an effective protrusion within the activated ROI (Fig. 6A). Since we previously found that stable lamellipodia were necessary for Tiam1-Micro-induced optotaxis (Fig. 3), we tested whether cilengitide affected optotaxis in these cells. We found that Tiam1-Micro- and iLID-CAAX-expressing cells on 10 µg/ml FN treated with 10 µM cilengitide were unable to migrate directionally while DMSO-treated control cells had a biased migration towards the more intense light (Fig. 6B). Importantly, treated cells had a similar velocity to that of control cells.
Fig. 6.

Tiam1-induced lamellipodia require active integrin and myosin. (A) Mean distance for the Tiam1 DH/PH-induced protrusions for the denoted FN concentration back-filled with poly-L-lysine as determined from kymographs. Cilengitide (Cil) was used to treat cells at 10 µM. (B) Rose plots, FMI graphs and velocity plots representing migration vectors for Tiam1-Micro- and iLID-CAAX-expressing fibroblasts plated on FN and mock treated or treated with 10 µM cilengitide. (C) Mean distance for the Tiam1 DH/PH-induced protrusions on the denoted concentration of FN back-filled with poly-L-lysine as determined from kymographs. Y-27632 and blebbistatin was used to treat cells at 15 µM. (D) Rose plots, FMI graphs and velocity plots representing migration vectors for Tiam1-Micro- and iLID-CAAX-expressing fibroblasts plated on FN and mock treated or treated with 15 µM Y-27632. Error bars are the 95% c.i. *P<0.05; **P<0.005. Refer to Tables S1 and S2 for experimental details.
The full cascade of integrin signaling is not only dependent on ligand binding but also on tension across the molecule produced by actin-myosin contractility (Burridge and Guilluy, 2016). We therefore tested the role of actin-myosin contractility in stabilizing Tiam1-Micro-induced protrusions by directly inhibiting myosin with blebbistatin and indirect inhibition with the ROCK inhibitor Y-27632. As expected, upon treatment with either inhibitor, cells had an enlarged footprint, formed large lamellipodia and had elongated ‘tails’ at the rear of the cell (Medeiros et al., 2006; Koestler et al., 2008). However, we found that for both inhibitors, the cells predominantly failed to form effective protrusions within the activated ROIs when plated on 10−3 µg/ml FN (Fig. 6C). Since all previous conditions which inhibited Tiam1-Micro-induced stable protrusion also inhibited optotaxis, we hypothesized that the inhibition of myosin contractility would also inhibit optotaxis. In these experiments, we were limited to the use of Y-27632 since blebbistatin is rendered inactive by blue light (Sakamoto et al., 2005). We found that on 10 µg/ml FN with Y-27632, cells migrated randomly while cells in control conditions migrated directionally (Fig. 6D). As previously observed (King et al., 2016), Y-27632 increased cell velocity. Taken together, these data suggest that specific integrin adhesions and myosin contractility are necessary for Tiam1-induced lamellipodia stability and phototactic fidelity.
ITSN1-Micro induces protrusions in the absence of FN yet requires integrin binding and myosin contractility
While Tiam1-Micro-induced optotaxis and protrusion required a FN substrate, ITSN1-Micro-induced optotaxis did not. To extend our observations, we tested the ability of ITSN1-Micro to produce stable lamellipodia on a 10 μg/ml FN or a 0.01% poly-LL substrate as above. By using a 12×12 µm ROI, we found that ITSN1-Micro induced stable lamellipodia on a FN substrate with an average protrusion distance of ∼20 µm. However, on poly-LL, lamellipodia would form up to the edge of the ROI and then fail to protrude further (Fig. 7A,B). We hypothesized that the induced lamellipodia required input from either the light or feedback from surface-bound FN to form a stable lamellipodia. We therefore extended the ROI to 12×24 µm. Under these conditions the cells produced stable lamellipodia that protruded to the same extent as those for cells plated on FN (Fig. 7A,B).
Fig. 7.

Integrin and myosin activity are necessary for ITSN1-induced lamellipodia and optotaxis while a FN substrate is not. (A) Representative fluorescence micrographs depicting optogenetic recruitment of ITSN1 DH/PH to a ROI in a fibroblast plated on 10 µg/ml FN or poly-L-lysine (0 μg/ml FN). The size of the ROI was modified as indicated so that the long axis of the ROI is perpendicular to the cell edge. Right panels are kymographs along the yellow line. White boxes denote activated ROIs. Scale bars: 50 µm (fluorescence images); 5 µm and 250 s (kymographs). (B) Mean distance for the ITSN1 DH/PH-induced protrusions on the denoted concentration of FN back-filled with poly-L-lysine as determined from kymographs. The size of the ROI was modified as indicated so that the long axis of the ROI is perpendicular to the cell edge. (C) Mean distance for the ITSN1 DH/PH-induced protrusions on the denoted concentration of FN back-filled with poly-L-lysine as determined from kymographs. Cilengitide (Cil) was used to treat cells at 10 µM. (D) Mean distance for the ITSN1 DH/PH-induced protrusions on the denoted concentration of FN back-filled with poly-L-lysine as determined from kymographs. Y-27632 and blebbistatin was used to treat cells at 15 µM. (E) Rose plots, FMI graphs and velocity plots representing migration vectors for ITSN1-Micro- and iLID-CAAX-expressing fibroblasts plated on poly-L-lysine and mock treated or treated with 10 µM cilengitide. (F) Rose plots, FMI graphs and velocity plots representing migration vectors for ITSN1-Micro- and iLID-CAAX-expressing fibroblasts plated on poly-L-lysine and mock treated or treated with 15 µM Y-27632. Error bars are the 95% c.i. *P<0.05; **P<0.005; ns, not significant. Refer to Tables S1 and S2 for experimental details.
As previously shown (Figs 4,5), we found that Tiam1-Micro-induced protrusion and optotaxis required an integrin–FN interaction as well as myosin contractility. Since ITSN1-Micro-induced protrusions and optotaxis did not require an exogenous FN substrate, we hypothesized that inhibiting integrins and myosin would not affect these processes. We first tested this by inducing local protrusions in the presence of cilengitide, blebbistatin and Y-27632. Contrary to our expectations, ITSN1-Micro- and iLID-CAAX-expressing cells plated on poly-LL failed to produce effective protrusion in the presence of each inhibitor (Fig. 7C,D). When plated on 10 µg/ml FN, in the presence of cilengitide, the cells produced varied results; most cells failed to protrude but some formed stable protrusions. This led to an average kymograph trace that fell between protrusion and ruffling (Fig. 7C). Interestingly, like cilengitide, Y-27632 only inhibited a subset of cells on 10 µg/ml FN, while blebbistatin inhibited effective protrusion formation in the majority of cells (Fig. 7D). We further examined our hypothesis by testing the ability of ITSN1-Micro- and iLID-CAAX-expressing cells to phototax in the presence of cilengitide or Y-27632. We found that both compounds inhibited directional migration of the cells on poly-LL (Fig. 7E,F). These results suggest that although ITSN1-Micro activity can induce a stable protrusion and bias directional motility in the absence of FN-coated glass, integrin and myosin activities were still necessary.
ITSN1-Micro induces FN deposition and stabilizes lamellipodia
Clustered integrins form nascent adhesions that mature into focal adhesions under the tension of myosin contractility (Rottner et al., 1999; Riveline et al., 2001). To visualize these structures, we imaged Venus–paxillin, an adhesion component, while inducing ITSN1-Micro-based protrusions on poly-LL. To our surprise, we found that paxillin puncta formed under the newly induced protrusions, and that these puncta resembled nascent and focal adhesions (Fig. 8A; Movie 4). Since these structures should only be triggered by integrin–ECM engagement and no exogenous surface-bound FN was present, we hypothesized that ITSN1 stimulation was causing cells to locally deposit FN, thus causing integrin engagement, leading to stabilization of the protrusions. We reasoned that there are two possible FN sources: FN provided to the cells from the serum in the medium or endogenously produced cellular FN. By western blot analysis, we estimated that our medium contained ∼10 µg/ml of FN (Fig. S4A). We therefore depleted the FN from our serum by gelatin–Sepharose extraction. We verified the depletion by western blot analysis (Fig. S4B). We then repeated the protrusion assays in cells expressing ITSN1-Micro and iLID-CAAX grown in medium containing the FN-depleted serum. We found that in this absence of serum FN, cells produced similar stable protrusions on substrates coated with FN or poly-LL (Fig. S4C).
Fig. 8.
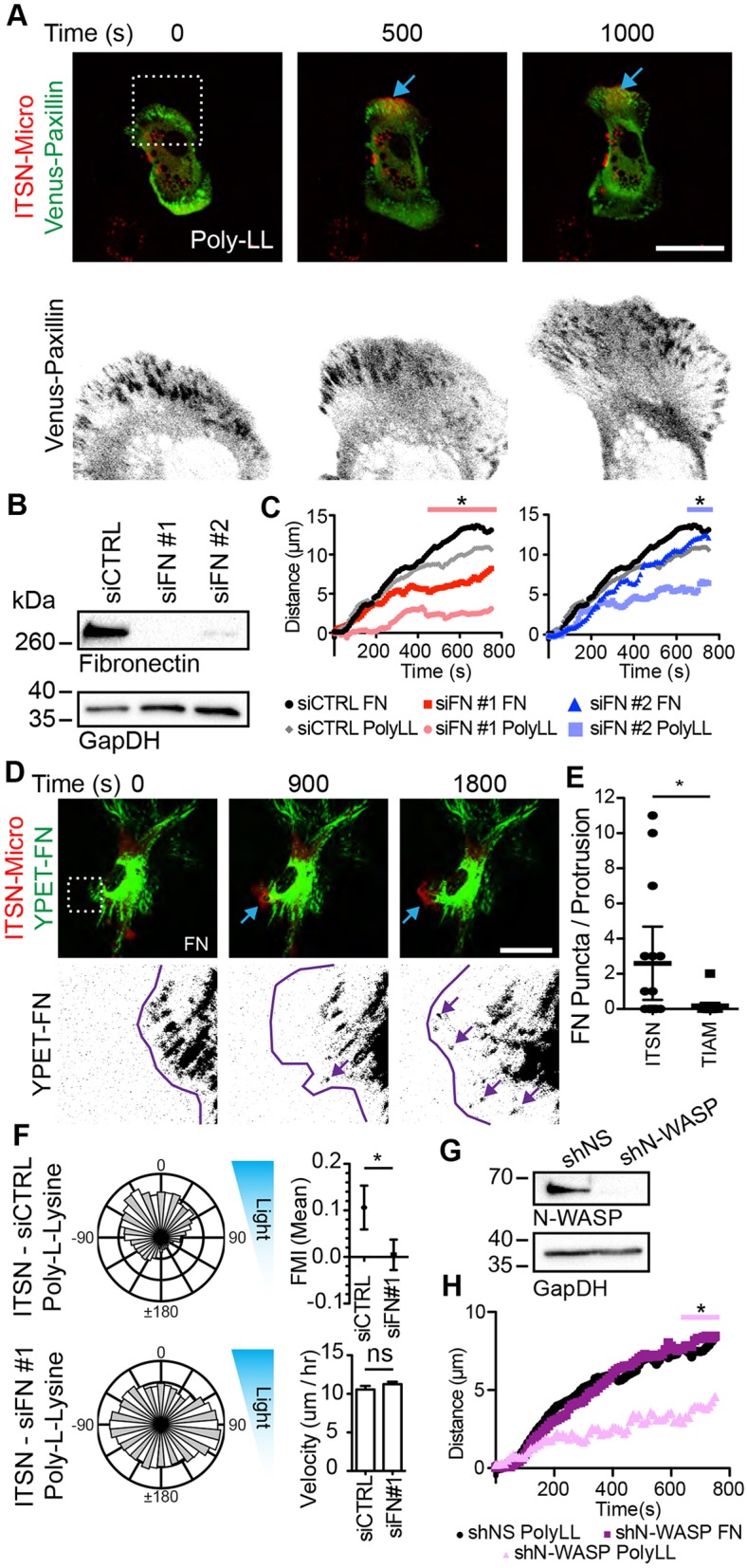
ITSN1 recruitment stimulates cellular FN deposition to stabilize lamellipodia. (A) Representative fluorescence micrographs depicting optogenetic recruitment of ITSN1 DH/PH in a fibroblast co-expressing Venus–paxillin and Halo–iLID-CAAX. The dotted white box in top panel represents the enlarged area displayed in bottom panels. Blue arrows denote the area of activation. (B) Representative western blot analysis of cells treated with control siRNA (siCTRL) or FN siRNA (siFN) #1 or #2. (C) Mean distance for the ITSN1 DH/PH-induced protrusions on FN or poly-L-lysine (PolyLL) in fibroblasts treated with the denoted siRNA as determined from kymographs. (D) Representative fluorescence micrographs depicting optogenetic recruitment of ITSN1 DH/PH in a fibroblast co-expressing YPET–FN and Halo–iLID-CAAX. The dotted white box in top panel represents the enlarged area displayed in the bottom panels. Blue arrows denote the area of activation. The purple line represents the cell edge. Purple arrows demark deposited YPET–FN. (E) Plot of the number of YPet–FN puncta counted under each induced protrusion. (F) Rose plots, FMI graphs and velocity plots representing migration vectors for ITSN1-Micro- and iLID-CAAX-expressing fibroblasts plated on poly-L-lysine and treated with the denoted siRNA. (G) Representative western blot of cells treated with the denoted shRNA (shNS is a non-specific shRNA). (H) Mean distance for the ITSN1 DH/PH-induced protrusions on FN or poly-L-lysine in fibroblasts treated with the denoted shRNA. Error bars are the 95% c.i. *P<0.05. Scale bars: 50 µm. Refer to Tables S1 and S2 for experimental details.
To test the possibility that cellular FN was critical for protrusion stability, we depleted the endogenously produced FN with two distinct siRNAs. Cells stably expressing ITSN1-Micro and iLID-CAAX were transfected with FN siRNAs as well as a nonspecific control siRNA. As shown by western blotting, FN was not detectable in cells transfected with siRNA #1, while a faint band was present for siRNA #2 (Fig. 8B). We examined the contribution of serum FN and cellular FN to the adherence and spreading of these cells by plating cells on poly-LL in the presence and absence of serum (Fig. S4D). In the presence of serum, compared to control cells, FN siRNA-treated cells spread to a similar extent but contained a reduced FN signal as determined by immunofluorescence imaging. In the absence of serum, the majority of FN siRNA-treated cells failed to spread effectively and contained no FN immunofluorescence signal, while control cells were well spread and contained a punctate FN signal. To test the effect of endogenous FN depletion on ITSN1-induced protrusion, we repeated the induced protrusion assay (Fig. 8C). Cells transfected with control siRNA produced protrusions on both FN and poly-LL substrates as expected. However, cells transfected with FN siRNA #1 efficiently protruded on FN substrate but failed to form effective protrusions on poly-LL. Cells transfected with FN siRNA #2 (with incomplete knockdown) produced stable protrusions on a FN substrate, and a mix of protrusion and ruffling on poly-LL.
The FN depletion data suggest that, in response to ITSN1-Micro translocation, the protrusions of a cell are stabilized by the deposition of cellular FN. To directly investigate this, we transfected cells with a construct encoding FN fused to YPet and induced ITSN1- or Tiam1-Micro-based protrusions on a FN substrate. In many (but not all cells), we visualized YPet–FN puncta forming under newly induced ITSN1-Micro protrusions (Fig. 8D,E; Movie 5). These puncta were absent from all but one Tiam1-Micro-induced protrusion. Since we found above that only a small number of FN molecules were necessary to stabilize a protrusion, we suspect that the FN deposition in the other cells was endogenous (unlabeled) or below our detection limit.
We further investigated the role of cellular FN in ITSN1-Micro-induced directed migration on poly-LL using the FN-depleted cells. ITSN1-Micro and iLID-CAAX-expressing cells transfected with control siRNA or FN siRNA #1 were plated on poly-LL-coated dishes and subjected to the optotaxis assay. We found that control siRNA-transfected cells migrated with a directional bias while FN siRNA cells migrated randomly (Fig. 8F). Taken together, these data suggest that activation of Cdc42 with the ITSN1 DH/PH domain induces cellular FN deposition, which stabilizes protrusions and leads to biased directional migration in the light-gradient optotaxis experiment.
The nucleation-promoting factor (NPF), N-WASP (also known as WASL) has been shown to enhance exocytic efficiency by inducing Arp2/3-dependent actin polymerization at the site of the exocytic event (Ory and Gasman, 2011). N-WASP is a direct effector of Cdc42. We therefore depleted N-WASP by using shRNA and tested the ability of ITSN1-Micro localization to induce protrusions on FN and poly-LL (Fig. 8G). Compared to cells expressing a non-specific shRNA control, the N-WASP shRNA-expressing cells failed to form an effective protrusion when plated on poly-LL (Fig. 8H). However, on FN, the N-WASP-depleted cells maintained their ability to efficiently protrude. These data support the hypothesis that localized Cdc42 activity induces deposition of FN through an N-WASP-dependent mechanism, supporting efficient lamellipodial protrusion and directed migration.
DISCUSSION
In this work, we utilized an optogenetic approach to investigate specific roles of Rac and Cdc42 signaling in directed migration. We found that local control of Rac and Cdc42 activity induced protrusions that were sufficient to drive directed whole-cell migration. The stability of these protrusions was highly dependent on a small number of integrin–FN interactions. Furthermore, our findings suggest that activation of Cdc42 elicits deposition of FN that is sufficient to support stable protrusion and directed migration while Rac activation does not.
Cellular optogenetics provided us with a unique tool in this study, empowering our experimental design to answer novel questions about the role of well-studied GTPases. Here, by optogenetically targeting multiple endogenous GTPases, we linked their specific activity to differences in protrusion and directed migration. Furthermore, spatial and temporal control of their activity allowed us to investigate the contributions of adhesion to individual protrusions, thus revealing critical, but unappreciated, aspects of GTPase function during migration. Our optotaxis experiment using a gradient of light allowed us to place these results in the context of whole-cell migration. The ability to precisely control protein activity in space and time with optogenetics will undoubtedly provide many novel biological insights in future studies.
Our findings demonstrate that both Rac and Cdc42 activities, through Arp2/3-based protrusions, are sufficient to drive directed whole-cell migration over long timescales (>12 h). Consistent with previous studies, local activation of Rac produced lamellipodia protrusions that were dependent on the Arp2/3 complex. Somewhat more surprising was the fact that Cdc42 activation also led to the formation of lamellipodia, likely due to crosstalk between Cdc42 and Rac (Nishimura et al., 2005; Guilluy et al., 2011). The ability of ITSN1-Micro to generate filopodia in the absence of Arp2/3 complex and its inability to generate significant morphological change in the absence of Cdc42 bolsters this argument. Regardless of the precise mechanisms by which each GTPase produces lamellipodia, our novel optotaxis assay shows that lamellipodia are sufficient to drive directional migration. We directly tested this link between GTPase-driven lamellipodia and directed migration through the inhibition of Arp2/3 activity. In these conditions, neither asymmetric Rac nor Cdc42 activity produced lamellipodial protrusion or directed migration, demonstrating the need for lamellipodia. Interestingly, ITSN1-Micro-induced filopodia production was not sufficient to drive directed migration. Future studies will be needed to dissect the possible role(s) of filopodia in directed migration.
One of our most striking findings was the stark difference in Rac- and Cdc42-driven protrusion and directed migration in the absence of an ECM substrate. On poly-LL, localized Rac activity failed to produce stable lamellipodia, and asymmetric Rac activation in the optotaxis assay did not produce directed migration. On the other hand, localized Cdc42 activity produced lamellipodia and led to directed migration on poly-LL. These results prompted us to address why differential Tiam1-Micro localization (Rac activity) is insufficient to directionally bias migration in the absence of a FN substrate while differential ITSN1-Micro localization (Cdc42 activity) is sufficient without an ECM substrate.
The need for FN to stabilize Rac-induced protrusions raises several interesting issues about the role of adhesion during protrusion. We reasoned that two possible phenomena might be involved in this effect: physical adhesion and integrin signaling. Owing to the molecular interconnection between adhesion and signaling, it is difficult to experimentally decouple the two functions of integrins. Although more studies are needed to fully unlink signaling from adhesion, we postulate that induced protrusions are reinforced by integrin signaling feedback rather than physical stabilization. We found that Rac-induced protrusions were supported by a small number of FN molecules of less than ten FN puncta per protrusion. We find it unlikely that this small number of FN–integrin interactions would be sufficient to provide physical stabilization to the newly formed structure. Additional support for our hypothesis comes from the inhibition of myosin contractility. In the presence of either blebbistatin or Y-27632, endogenous lamellipodia become exaggerated, possibly due to myosin no longer providing tension to retract them (Medeiros et al., 2006; Koestler et al., 2008). We therefore initially hypothesized that Rac-induced protrusions would be larger in the presence of contractility inhibitors. To our surprise, we found the opposite; myosin inhibition caused ruffling upon Rac activation. These data suggest that contractility is necessary to provide tension on integrins to trigger the full signaling feedback and support new protrusions rather than simply provide a retraction force. Further experiments will be required to interrogate the nature of the integrin signaling required to stabilize protrusions.
Surprisingly, we found that biased Cdc42 activity was sufficient to drive lamellipodia and directed migration in the absence of an exogenous FN substrate. The appearance of focal adhesion-like structures under ITSN1-induced protrusions on poly-LL, and the sensitivity of protrusion and directed migration to cilengitide strongly suggested that integrin signaling was occurring on poly-LL. Our data indicate that Cdc42 activity not only induces lamellipodia protrusions, but signals to the cell to locally deposit cellular FN that triggers this integrin signaling and leads to new protrusion. To our knowledge, this is the first report linking Cdc42 activity to FN deposition. However, there is support for this notion in the literature. Cdc42 has well-established roles in endocytosis and exocytosis in other contexts (Harris and Tepass, 2010; Ory and Gasman, 2011). Furthermore, full-length endogenous ITSN1 is a known regulator of endocytosis and exocytosis and is thought to function through the Cdc42 effector, N-WASP (Malacombe et al., 2006; Gubar et al., 2013). N-WASP is known to enhance the efficiency of exocytosis through Arp2/3-nucleated actin polymerization at the exocytic site (Ory and Gasman, 2011). Indeed, Lengfeld et al. showed a link between Cdc42 and collagen (another ECM protein) release in vascular smooth muscle cells (Lengfeld et al., 2012). We therefore hypothesize that localization of the ITSN1 DH/PH domain has two congruent effects that are driven by Cdc42 activity and lead to protrusion and directed migration in the absence of FN: (1) it leads to the localized formation of lamellipodia through Cdc42 crosstalk with Rac; and (2) it promotes local Cdc42 activity that activates additional effectors beyond those involved in protrusion and, in turn, initiates local FN deposition to support newly formed lamellipodia. In support of this hypothesis, we found that Cdc42 is necessary for any ITSN1-Micro-induced morphological change to occur. Furthermore, the Cdc42 effector N-WASP is necessary for efficient ITSN1-Micro-induced protrusion on poly-LL. However, N-WASP was not necessary for protrusion on a FN substrate, reinforcing the notion of the involvement of N-WASP in exocytosis but not lamellipodia formation (Snapper et al., 2001). In addition, endogenous FN is necessary for efficient ITSN1-Micro-induced protrusion and directed migration in the absence of exogenous FN. By depositing FN as the cell protrudes, the cell effectively lays down its own tracks to reinforce and support both protrusion and directed migration. This may represent a novel pathway by which cells remodel the ECM to produce an autonomous feedback, reinforcing directed migration. Future studies will be directed at further delineating this pathway and how it might be used in other directed migration events.
MATERIALS AND METHODS
Materials, reagents and cell culture
Cdc42f/− cells were a gift from Cord Brakebusch (Biotech Research and Innovation Centre, University of Copenhagen, Denmark). Lentiviral iLID plasmids are available from Addgene. pBabe-Puro iLID-CAXX was produced by Gibson assembly. iLID-CAAX was amplified from pLL7.0 Venus-iLID-CAAX by the following primers: Fwd, 5′-TAGGCGCCGGCCGGATCCGCCACCATGGGGGAGTTTCTGGCAACCACAC-3′ and Rev, 5′-CACTGTGCTGGCGAATTCCTACATAATTACACACTTTGTCTTTGACTTC-3′. pBabe-Puro was digested with BamHI and EcoRI and assembled with the PCR product by Gibson assembly. pHLSec2-FN-YPet was Addgene plasmid #65421 [deposited by Harold Erickson (Ohashi and Erickson, 2011)]; mVenus-Paxillin-22 was Addgene plasmid #56620 (deposited by Michael Davidson). Venus-wRBD was a gift from N. Gautam (Division of Biology and Biomedical Sciences, Washington University, USA). YPet-pGBD was a gift from Klaus Hahn (Department of Pharmacology, University of North Carolina at Chapel Hill, USA). Anti-FN antibody was purchased from Abcam (ab2413) and was used at 1:500 for both western blotting and immunofluorescence. Anti-GAPDH antibody was purchased from Ambion (Clone 6C5) and was used at 1:10,000 dilution for western blotting. Anti-N-WASP antibody was purchased from Cell Signaling Technology (#4848S) and was diluted 1:1000 for western blotting. Anti-Cdc42 antibody was purchased from BD Biosciences (#610929) and was diluted 1:500 for western blotting. siRNAs were purchased from Qiagen (FN siRNA #1-SI01004066; FN siRNA #2-SI01004080). siRNA transfection was performed with Lipofectamine RNAiMAX (Thermo Fisher) according to the manufacturer's protocol. Lentiviral shRNA for N-WASP was used as in King et al. (2016). Drug treatments were performed with 150 µM CK666 (Millipore), 10 µM cilengitide (Selleck Chemical), 15 µM Y-27632 (Sigma) and 15 μM blebbistatin (Sigma). Mouse IA32 fibroblasts and conditional Arpc2 mutant mouse embryonic fibroblasts (MEFs) (generated in our laboratory; Rotty et al., 2015) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) FBS (HyClone), and 292 µg/ml L-glutamine. Cells were cultured at a constant 37°C and 5% (v/v) CO2. Conditional Arpc2 mutant MEFs were rendered null as previously reported (Asokan et al., 2014; Rotty et al., 2015). Cells were transiently transfected in six-well cell culture dishes using 1 μg of total DNA at equal ratios for multi-component transfections with NanoJuice (EMD Millipore) transfection reagent, as recommended by the manufacturer.
Creating stable cell lines expressing iLID-CAAX with Tiam1–RFP or ITSN1–RFP
Lentivirus and retrovirus production and infection were performed as previously described (Cai et al., 2008). IA32 cells were co-infected with the virus encoding Venus–iLID-CAAX and Tiam1–RFP or ITSN1–RFP. Cell populations were sorted based on expression by FACS using a Bio-Rad S3 cell sorter or puromycin selection at 2 µg/ml.
Adeno-Cre viral transduction of Cdc42f/− cells
Regular titer (1×1010–7×1010 pfu/ml) Ad5CMVCre was purchased from University of Iowa Viral Vector Core Facility. Cdc42f/− cells were plated in 6 cm tissue culture dishes and treated with 1:1000 Ad5CMVCre virus for 48 h. Cells were grown for an additional 72 h before being assayed by western blot and/or used for experiments.
Western blotting
Western blotting was performed as previously described (Haynes et al., 2015).
Coating glass with substrate
Purified human plasma FN (Corning 354008) was brought to the desired concentration in PBS. 3.5 cm Mattek dishes were coated with 900 µl of FN solution and incubated at 37°C for 1 h. Dishes were then washed with PBS once and treated with 900 µl of 0.01% poly-L-lysine solution (Sigma) for 1 h at 37°C. Dishes were then rinsed in PBS once and 95% ethanol once and allowed to dry completely. All dishes were used within 24 h of coating.
Optotaxis setup, image acquisition and analysis
To produce an intensity gradient of blue light, we produced a device consisting of three main pieces: a housing, a 467 nm LED light source (Thor Labs) and a gradient neutral density filter (Thor Labs, N.D. =0.04 to 4.0). The housing was custom designed using TinkerCAD and 3D printed on a Printrbot Metal Plus printer (.stl files for device are available upon request). It positions the LED 3 cm above the neutral density filter. The neutral density filter is positioned within the housing so that the projected area starts 10 mm from the clear end. The LED is powered by a 9 V battery in line with a 1 kΩ resistor. This setup projects a 1×1 cm area of graded light intensity. The housing is placed over a 3.5 cm Mattek culture dish and fits into an OlympusVivaView FL microscope with a 20× objective and a motorized magnification changer set to ×1. Cells were imaged every 15 min for 20 h. Cells of interest were initially imaged by fluorescence microscopy to ensure protein expression and then imaged every 15 min for up to 24 h by bright-field imaging. The bright-field image was collected from the graded LED light source and was used to measure the gradient slope. The bright-field images were then analyzed to track the cell trajectories over time with the Manual Tracking ImageJ plugin. Trajectories are processed using the Chemotaxis tool (Ibidi) ImageJ plugin to quantify the FMI and velocity, and make rose plots. Rose plots were redrawn using the Matlab SecPlot plugin. Drug treatments were performed 1 h before beginning imaging.
Immunofluorescence
Samples were fixed in a cold 4% (w/v) paraformaldehyde solution for 5 min. Some samples were permeabilized with 0.1% Triton X-100 for 5 min. Samples were blocked in 10% (w/v) BSA in PBS for 30 min at room temperature. Primary and secondary antibodies were diluted in 1% (w/v) BSA in PBS and incubated with samples at room temperature for 1 h. See Table S3 for full antibody details. Samples were mounted with Fluoromount G (Electron Microscopy Sciences)
Confocal microscopy and single cell optical activation
Fixed and live cell imaging was performed on an Olympus FV1000 scanning confocal microscope equipped with an environmental chamber. Cell images were collected using a 0.17 NA 40× objective or for single molecule FN imaging a 0.17 N.A. 100× objective. Single cell activation was performed as described in Zimmerman et al., 2016b. In short, the time controller software was used to create a timeline of imaging and activation. For all protrusion dynamics experiments, images were acquired every 10 s with constant ROI illumination in between. While imaging paxillin and FN, images were acquired every 60 s with activation occurring at ten evenly spaced intervals between images. Drug treatments were performed 20 min before beginning imaging.
Kymography analysis
Kymographs were produced from single-cell activation image series. A kymograph was produced along a line running perpendicular to the cell edge in the center of the activated ROI by using the Fiji (ImageJ) kymography plugin. Kymographs were then traced in Fiji software, and the position of the cell edge at each time point was interpolated by using Microsoft Excel. Protrusion data was then normalized to the initial five images obtained before activation, and averaged and plotted with Prism statistical software.
FBS FN depletion
25 ml of gelatin sepharose 4B medium slurry (GE Healthcare) was pelleted and washed with PBS. 50 ml of FBS was incubated with medium overnight at 4°C. Medium were then separated from FBS by gravity flow column chromatography. The column was washed with two column volumes of PBS and the protein was eluted off the column with 8 M urea in PBS. The depletion of FN from FBS was verified by western blotting.
Statistics
Statistics were performed by using the Graphpad Prism statistical software package. FMI and velocity values were compared by t-test or ANOVA depending on the number of comparisons. Kymograph statistics were performed by comparing values of the kymographs at 100 s time intervals starting from 750 s and working backward. ANOVAs were performed for each set of time points and a Dunnet's post-test was performed to compare each sample to the control.
Acknowledgements
We thank members of the Bear and Kuhlman laboratories for useful feedback on this project. We thank N. Gautam and Klaus Hahn for GBD plasmids.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.E.B., S.P.Z., S.B.A., B.K.; Methodology: S.P.Z., S.B.A.; Formal analysis: S.P.Z.; Writing - original draft: S.P.Z.; Writing - review & editing: J.E.B., S.B.A., B.K.; Supervision: J.E.B., B.K.; Project administration: J.E.B., B.K.; Funding acquisition: J.E.B., B.K.
Funding
This work was supported by the National Institutes of Health (DA036877, GM093208 to B.K.; GM083035, GM110155 to J.E.B.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.205948.supplemental
References
- Asokan S. B., Johnson H. E., Rahman A., King S. J., Rotty J. D., Lebedeva I. P., Haugh J. M. and Bear J. E. (2014). Mesenchymal chemotaxis requires selective inactivation of myosin II at the leading edge via a noncanonical PLCγ/PKCα pathway. Dev. Cell 31 747-760. 10.1016/j.devcel.2014.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretou M., Jouannot O., Fanget I., Pierobon P., Larochette N., Gestraud P., Guillon M., Emiliani V., Gasman S., Desnos C. et al. (2014). Cdc42 controls the dilation of the exocytotic fusion pore by regulating membrane tension. Mol. Biol. Cell 25, 3195-3209. 10.1091/mbc.E14-07-1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K. and Guilluy C. (2016). Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 343, 14-20. 10.1016/j.yexcr.2015.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai L., Makhov A. M., Schafer D. A. and Bear J. E. (2008). Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell 134, 828-842. 10.1016/j.cell.2008.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case L. B. and Waterman C. M. (2015). Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 17, 955-963. 10.1038/ncb3191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuchra A., Wu X., Meyer H., Hengel J., Van, Schroeder T., Geffers R., Rottner K. and Brakebusch C. (2005). Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid. 16, 4473-4484. 10.1091/mbc.E05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-sauze S., Grall D., Cseh B. and Van Obberghen-schilling E. (2009). Regulation of fibronectin matrix assembly and capillary morphogenesis in endothelial cells by Rho family GTPases. Exp. Cell Res. 315, 2092-2104. 10.1016/j.yexcr.2009.03.017 [DOI] [PubMed] [Google Scholar]
- Gubar O., Morderer D., Tsyba L., Croisé P., Houy S., Ory S., Gasman S. and Rynditch A. (2013). Intersectin: the crossroad between vesicle exocytosis and endocytosis. Front. Endocrinol. 4 109 10.3389/fendo.2013.00109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C., Garcia-Mata R. and Burridge K. (2011). Rho protein crosstalk: another social network? Trends in Cell Biol. 21, 718-726. 10.1016/j.tcb.2011.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntas G., Hallett R. A., Zimmerman S. P., Williams T., Yumerefendi H., Bear J. E. and Kuhlman B. (2015). Engineering an improved light-induced dimer (iLID) for controlling the localization and activity of signaling proteins. Proc. Natl. Acad. Sci. 112, 112-117. 10.1073/pnas.1417910112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett R. A., Zimmerman S. P., Yumerefendi H., Bear J. E. and Kuhlman B. (2016). Correlating in vitro and in vivo activities of light-inducible dimers: a cellular optogenetics guide. ACS Synthetic Biol. 5, 53-64. 10.1021/acssynbio.5b00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K. P. and Tepass U. (2010). Cdc42 and vesicle trafficking in polarized cells. Traffic 11, 1272-1279. 10.1111/j.1600-0854.2010.01102.x [DOI] [PubMed] [Google Scholar]
- Haynes E. M., Asokan S. B., King S. J., Johnson H. E., Haugh J. M. and Bear J. E. (2015). GMFβ controls branched actin content and lamellipodial retraction in fibroblasts. J. Cell Biol. 209, 803-812. 10.1083/jcb.201501094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman S. J. and Ridley A. J. (2008). Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 9, 690-701. 10.1038/nrm2476 [DOI] [PubMed] [Google Scholar]
- Hu P. and Luo B.-H. (2013). Integrin bi-directional signaling across the plasma membrane. J. Cell. Physiol. 228, 306-312. 10.1002/jcp.24154 [DOI] [PubMed] [Google Scholar]
- Hussain N. K., Jenna S., Glogauer M., Quinn C. C., Wasiak S., Guipponi M., Antonarakis S. E., Kay B. K., Stossel T. P., Lamarche-Vane N. et al. (2001). Endocytic protein intersectin-l regulates actin assembly via Cdc42 and N-WASP. Nat. Cell Biol. 3, 927-932. 10.1038/ncb1001-927 [DOI] [PubMed] [Google Scholar]
- Jaiswal M., Dvorsky R. and Ahmadian M. R. (2013). Deciphering the molecular and functional basis of Dbl family proteins: a novel systematic approach toward classification of selective activation of the Rho family proteins. J. Biol. Chem. 288, 4486-4500. 10.1074/jbc.M112.429746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King S. J., Asokan S. B., Haynes E. M., Zimmerman S. P., Rotty J. D., Alb J. G., Tagliatela A., Blake D. R., Lebedeva I. P., Marston D. et al. (2016). Lamellipodia are crucial for haptotactic sensing and response. J. Cell Sci. 129, 2329-2342. 10.1242/jcs.184507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koestler S. A., Auinger S., Vinzenz M., Rottner K. and Small J. V. (2008). Differentially oriented populations of actin filaments generated in lamellipodia collaborate in pushing and pausing at the cell front. Nat. Cell Biol. 10, 306-313. 10.1038/ncb1692 [DOI] [PubMed] [Google Scholar]
- Lengfeld J., Wang Q., Zohlman A., Salvarezza S., Morgan S., Ren J., Kato K., Rodriguez-Boulan E. and Liu B. (2012). Protein kinase C δ regulates the release of collagen type I from vascular smooth muscle cells via regulation of Cdc42. Mol. Biol. Cell 23, 1955-1963. 10.1091/mbc.E11-06-0531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levskaya A., Weiner O. D., Lim W. A. and Voigt C. A. (2009). Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997-1001. 10.1038/nature08446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machacek M., Hodgson L., Welch C., Elliott H., Pertz O., Nalbant P., Abell A., Johnson G. L., Hahn K. M. and Danuser G. (2009). Coordination of Rho GTPase activities during cell protrusion. Nature 461, 99-103. 10.1038/nature08242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malacombe M., Ceridono M., Calco V., Chasserot-Golaz S., McPherson P. S., Bader M.-F. and Gasman S. (2006). Intersectin-1L nucleotide exchange factor regulates secretory granule exocytosis by activating Cdc42. EMBO J. 25, 3494-3503. 10.1038/sj.emboj.7601247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas-Moruno C., Rechenmacher F. and Kessler H. (2010). Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anti-Cancer Agents Med. Chem. 10, 753-768. 10.2174/187152010794728639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros N. A., Burnette D. T. and Forscher P. (2006). Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat. Cell Biol. 8, 216-226. 10.1038/ncb1367 [DOI] [PubMed] [Google Scholar]
- Nishimura T., Yamaguchi T., Kato K., Yoshizawa M., Nabeshima Y., Ohno S., Hoshino M. and Kaibuchi K. (2005). PAR-6-PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat. Cell Biol. 7, 270-277. 10.1038/ncb1227 [DOI] [PubMed] [Google Scholar]
- Nobes C. D. and Hall A. (1995). Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53-62. 10.1016/0092-8674(95)90370-4 [DOI] [PubMed] [Google Scholar]
- Ohashi T. and Erickson H. P. (2011). Fibronectin aggregation and assembly: the unfolding of the second fibronectin type III domain. J. Biol. Chem. 286, 39188-39199. 10.1074/jbc.M111.262337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill P. R., Kalyanaraman V. and Gautam N. (2016). Subcellular optogenetic activation of Cdc42 controls local and distal signaling to drive immune cell migration. Mol. Biol. Cell 27, 1442-1450. 10.1091/mbc.E15-12-0832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory S. and Gasman S. (2011). Rho GTPases and exocytosis: what are the molecular links? Semin. Cell Dev. Biol. 22, 27-32. 10.1016/j.semcdb.2010.12.002 [DOI] [PubMed] [Google Scholar]
- Ridley A. J., Schwartz M. A., Burridge K., Firtel R. A., Ginsberg M. H., Borisy G., Parsons J. T. and Horwitz A. R. (2003). Cell migration: integrating signals from front to back. Science 302, 1704-1709. 10.1126/science.1092053 [DOI] [PubMed] [Google Scholar]
- Riveline D., Zamir E., Balaban N. Q., Schwarz U. S., Ishizaki T., Narumiya S., Kam Z., Geiger B. and Bershadsky A. D. (2001). Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J. Cell Biol. 153, 1175-1185. 10.1083/jcb.153.6.1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossman K. L., Der C. J. and Sondek J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167-180. 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- Rottner K., Hall A. and Small J. V. (1999). Interplay between Rac and Rho in the control of substrate contact dynamics. Curr. Biol. 9, 640-648. 10.1016/S0960-9822(99)80286-3 [DOI] [PubMed] [Google Scholar]
- Rotty J. D., Wu C., Haynes E. M., Suarez C., Winkelman J. D., Johnson H. E., Haugh J. M., Kovar D. R. and Bear J. E. (2015). Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell 32, 54-67. 10.1016/j.devcel.2014.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T., Limouze J., Combs C. A., Straight A. F. and Sellers J. R. (2005). Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry 44, 584-588. 10.1021/bi0483357 [DOI] [PubMed] [Google Scholar]
- Schwarzbauer J. E. and DeSimone D. W. (2011). Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb. Perspect. Biol. 3, a005041 10.1101/cshperspect.a005041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper S. B., Takeshima F., Antón I., Liu C.-H., Thomas S. M., Nguyen D., Dudley D., Fraser H., Purich D., Lopez-Ilasaca M. et al. (2001). N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nat. Cell Biol. 3, 897-904. 10.1038/ncb1001-897 [DOI] [PubMed] [Google Scholar]
- Snyder J. T., Worthylake D. K., Rossman K. L., Betts L., Pruitt W. M., Siderovski D. P., Der C. J. and Sondek J. (2002). Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat. Struct. Biol. 9, 468-475. 10.1038/nsb796 [DOI] [PubMed] [Google Scholar]
- Suraneni P., Rubinstein B., Unruh J. R., Durnin M., Hanein D. and Li R. (2012). The Arp2/3 complex is required for lamellipodia extension and directional fibroblast cell migration. J. Cell Biol. 197, 239-251. 10.1083/jcb.201112113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojnacki J., Quassollo G., Marzolo M. and Cáceres A. (2014). Rho GTPases at the crossroad of signaling networks in mammals: Impact of Rho-GTPases on microtubule organization and dynamics. Small GTPases 5, e28430 10.4161/sgtp.28430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthylake D. K., Rossman K. L. and Sondek J. (2000). Crystal structure of Rac1 in complex with the guanine nucleotide exchange region of Tiam1. Nature 408, 682-688. 10.1038/35047014 [DOI] [PubMed] [Google Scholar]
- Wu C., Asokan S. B., Berginski M. E., Haynes E. M., Sharpless N. E., Griffith J. D., Gomez S. M. and Bear J. E. (2012). Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell 148, 973-987. 10.1016/j.cell.2011.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. W., Collins S. R. and Meyer T. (2015). Locally excitable Cdc42 signals steer cells during chemotaxis. Nat. Cell Biol. 18, 191-201. 10.1038/ncb3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman S. P., Hallett R. A., Bourke A. M., Bear J. E., Kennedy M. J. and Kuhlman B. (2016a). Tuning the binding affinities and reversion kinetics of a light inducible dimer allows control of transmembrane protein localization. Biochemistry 55, 5264-5271. 10.1021/acs.biochem.6b00529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman S. P., Kuhlman B. and Yumerefendi H. (2016b). Engineering and application of LOV2-based Photoswitches. Method. Enzymol. 580, 169-190. 1st edn, Peptide, Protein and Enzyme Design. 1st edn. Elsevier Inc 10.1016/bs.mie.2016.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]