

ABSTRACT
Chromatin modification is traditionally assessed in biochemical assays that provide average measurements of static events given that the analysis requires components from many cells. Microscopy can visualize single cells, but the cell body and organelles can hamper staining and visualization of the nucleus. Normally, chromatin is visualized by immunostaining a fixed sample or by expressing exogenous fluorescently tagged proteins in a live cell. Alternative microscopy tools to observe changes of endogenous chromatin in real-time are needed. Here, we isolated transcriptionally competent nuclei from cells and used antibody staining without fixation to visualize changes in endogenous chromatin. This method allows the real-time addition of drugs and fluorescent probes to one or more nuclei while under microscopy observation. A high-resolution map of 11 endogenous nuclear markers of the histone code, transcription machinery and architecture was obtained in transcriptionally active nuclei by performing confocal and structured illumination microscopy. We detected changes in chromatin modification and localization at the single-nucleus level after inhibition of histone deacetylation. Applications in the study of RNA transcription, viral protein function and nuclear architecture are presented.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Nucleus, Chromatin, Histone, Acetylation, Microscopy
Summary: ChIP is the standard technique to study chromatin. We propose a high-resolution microscopy tool to visualize changes in histone modifications, RNA transcription and nuclear architecture at single-nucleus level in real time.
INTRODUCTION
The nucleus is the defining organelle of the eukaryotic cell and is responsible for containing, copying and transcribing genetic information in the form of nucleic acid. In 1928 Heitz first noted a distinction between the active and accessible euchromatin and the more inert heterochromatin (Heitz, 1928). Over time, those observations have deepened and generated a vast scientific literature about chromatin compartmentalization, and accessibility for DNA replication, repair and transcription (Harr et al., 2016). The current model of the histone code proposes that chromatin regulation is modulated by chemical modifications of the histones (Jenuwein and Allis, 2001). In general, acetylation and methylation of the histone tails indicate the status of active and silent chromatin. Although these epigenetic modifications are important for chromatin regulation, we know, in addition, that histone spatial localization, interaction with nuclear architecture and position of the modified residues also all determine DNA accessibility (Bannister and Kouzarides, 2011).
Given that epigenetics plays a critical role in cell development, oncogenesis and viral pathogenesis (Brien et al., 2016; Milavetz and Balakrishnan, 2015; Zhu et al., 2013), it is crucial that techniques be developed to monitor these chromatin alterations. We have developed a microscopy technique suitable for examining chromatin modifications in a single nucleus over time. Traditionally, chromatin modifications are investigated by chromatin immunoprecipitation (ChIP)-based assays. Despite the advantages, these biochemical genomic assays are unable to query changes in real-time in a single cell. These data provide averages of cells and are unable to acquire important spatial and temporal information on modifications contributing to chromatin regulation. Chemical fixation of the specimen is the method of choice for immunostaining protocols used in most microscopy techniques, but it impedes observations of dynamic events. Some new optical microscopy techniques can visualize the chromatin structure in a single cell (Lakadamyali and Cosma, 2015). Even though the resolution of diffraction-limited optical microscopy is sufficient for subcellular imaging, real-time visualization of the endogenous chromatin remains difficult and any increase in resolution or light detection would be valuable since molecules of interest may have few signals to detect and the chromatin structure is below the diffraction limit. In recent years, efforts have been made to push the envelope and study chromatin in living cells. Monoclonal fluorescently conjugated Fab fragments targeting histones have been used to track changes in real-time resulting in a promising approach (Kimura et al., 2015). Higher resolution images of chromatin can now be obtained with super resolution microscopy by transiently or stably expressing tagged histones in living cells (Klein et al., 2011; Ricci et al., 2015; Smeets et al., 2014; Wombacher et al., 2010). These studies have focused on imaging intact cells. While this has benefits, the cytoplasm and nuclear membranes act as barriers and limit the real-time addition of reagents or antibodies to the nucleus. Alternative microscopy approaches to quantitatively study changes of the endogenous chromatin in an unfixed nucleus are therefore welcomed, especially as high- and super-resolution techniques take hold.
We have developed a tool to visualize chromatin changes at the single-nucleus level. This method is based on a protocol to isolate transcriptionally competent nuclei from cells. Immunofluorescent staining of unfixed nuclei allowed us to generate images of endogenous proteins relevant to the histone code, transcription machinery and nuclear architecture. By using spinning disk confocal and instant structured illumination microscopy (iSIM), we mapped 11 chromatin and nuclear architectural markers at high resolution. We demonstrate that this technique can provide quantitative information on chromatin remodeling dynamics in single unfixed nuclei treated with histone deacetylase inhibitors. Finally, we show applications for studies of RNA transcription, chromatin structure at the site of integrated viral DNA and changes in the nuclear three-dimensional architecture.
RESULTS
System to image isolated and unfixed nuclei with high-resolution microscopy
We postulated that eliminating the plasma membrane, cytosol and other organelles would help with chromatin imaging in two ways: it would provide us with higher resolution images of the nucleus and also would permit the experimenter to interact with one or more nuclei in real-time by performing perfusion experiments. This would allow the rapid addition of drugs, fluorescent dyes and antibodies. This technique allowed us to visualize endogenous nuclear proteins and nucleic acids in transcriptionally competent unfixed nuclei (Fig. 1). Nuclei can be easily isolated from cells by using a buffer consisting of non-ionic detergents and salts (Claude, 1975; De Duve and Beaufay, 1981). This procedure yields nuclei that have been stripped of the lipids in the nuclear envelope, but maintain their structure inside the nuclear lamina (Greenberg and Bender, 2007; Marzluff et al., 1974). For this study, we used nuclei from TZM-bl cells, a HeLa-derived cell line engineered to contain separate integrated copies of the luciferase and β-galactosidase genes under control of the HIV-1 promoter and used broadly in HIV research. We incubated TZM-bl nuclei with primary antibodies against endogenous nuclear proteins, followed by secondary antibodies conjugated with fluorescent dyes (Fig. 1A). Stained nuclei were then visualized by fluorescence microscopy and images analyzed computationally. To exclude the possibility that the isolation of nuclei altered the integrity of the nucleus, we stained fixed TZM-bl cells and unfixed isolated nuclei under similar immunofluorescence conditions. We compared the staining of seven nuclear targets in fixed TZM-bl cells with unfixed isolated nuclei and did not observe gross differences in localization and topology (Fig. 1B,C; Fig. S1A–N). Scientists isolate nuclei with different procedures to study nuclear structure and function. Thus, the nuclei purification protocol is crucial to allowing correct interpretation of the observations. We sought to test whether a custom nuclei isolation protocol would maintain the nuclear organization observed with the commercial kit used for this work. For this purpose, we isolated nuclei from TZM-bl cells with a custom protocol (Farrell, 2010) and with the commercial kit in parallel. Nuclei isolated with either procedure were stained for acetylated H3, lamin B1, RNA and RNA polymerase II phosphorylated at S2 (Pol II pS2) and imaged by confocal microscopy. The two isolation procedures yielded nuclei with similar structure and architecture (Fig. S1O,P) and the lamina visualized with the support of a microcompressor (described in Fig. 7) appeared intact (Fig. S1Q,R).
Fig. 1.

Method to visualize and map endogenous nuclear proteins and nucleic acids in isolated nuclei. (A) Method workflow. (B) Differential interference contrast (DIC) image of a fixed TZM-bl cell (left) and Pol II pS2 immunostaining (right). (C) DIC image of an isolated nucleus (left) and immunostaining of Pol II pS2 (right). Nuclei isolated from TZM-bl cells were co-stained for the given nuclear targets and imaged by confocal microscopy (D–G) and iSIM (I–O). Deconvolved confocal equatorial z-series of representative nuclei are shown. Scale bars: 5 μm. (H) Total signal intensity from nuclei stained for CBFβ and from control nuclei stained with secondary antibody only (no-primary, n=3 per condition; bars indicate mean). ***P≤0.001 (Student's t-test, two-tailed, homoscedastic). (P) Representative map of nuclear proteins and RNA location in the nucleus as determined by these studies.
Fig. 7.

Utilization of a mechanical microcompressor to visualize proteins of the nuclear lamina. (A) Schematic of mechanical microcompression. (B) From left to right, deconvolved images of the bottom, equatorial and top confocal sections of a representative nucleus stained for β-actin (green) and Pol II pS2 (red). Confocal z-series of representative nuclei are shown. Scale bar: 5 μm.
Mapping the location of endogenous proteins and nucleic acids in isolated nuclei
ChIP-based assays are able to resolve the interactions of proteins with the genome at a resolution measured in hundreds of base pairs and have been used to infer the spatial organization of chromatin in the nucleus (Guelen et al., 2008; Harr et al., 2016; Kosak et al., 2002; Peric-Hupkes et al., 2010; Pickersgill et al., 2006; Ugarte et al., 2015). To our knowledge, a map showing the microscopic organization of endogenous chromatin in an unfixed specimen is not available. We employed an arsenal of antibodies and dyes to stain nuclei for multiple targets and visualize the sub-nuclear compartmentalization of the lamina, chromatin and nucleolus. In order to obtain high-resolution images we took advantage of spinning disk confocal microscopy and iSIM. Lamin B1 signals were detected at the boundaries of the nuclear body and identified the nuclear lamina (Fig. 1D,F,O, and respective 3D reconstructions in Movies 1, 3, 9). Comparison of the localization of various proteins and modification in relation to lamin B1 and the nucleolus allowed us to assemble a map of nuclear geography. Histones H3 and its modifications [acetylated on K9 (H3K9ac), tri-methylated on K27 (H3K27me3) or tri-methylated on K9 (H3K9me3)] were detected in inner areas as well as at the periphery of the nucleus, where the interphase chromosome territories have been described (Cremer et al., 2006), but were not detectable in the nucleoli (Fig. 1D–F,L–N, and respective 3D reconstructions in Movies 1–3,6–8). H3K27me3, H3K9me3 and suppressor of variegation 3-9 homolog 1 (SUV39H1), markers for methylated chromatin, were found at the periphery of the nucleoli and near the nuclear lamina where heterochromatin is condensed (Aagaard et al., 1999; Boyle et al., 2001; Croft et al., 1999; Németh and Längst, 2011) (Fig. 1L,M, and respective 3D reconstructions in Movies 6,7). Histone deacetylase 3 (HDAC3) foci appeared to be diffused throughout the nuclei and localized at the nuclear periphery, the site of silenced heterochromatin (Yang et al., 2002) (Fig. 1E, and 3D reconstruction in Movie 2). Core-binding factor subunit β (CBFβ) and runt-related transcription factor 1 (RUNX1) formed heterodimeric complexes and colocalized in the interior of the nucleus (Fig. 1G and Movie 4). The RNA-selective dye preferentially stained inner regions of the nucleus occupied by the H3 and Pol II pS2 stains, with signal mostly found at the periphery of this region (Fig. 1I,N, and respective 3D reconstructions in Movies 5,8). We observed that acetylated H3 (H3K9ac), a marker for highly expressed chromatin, similarly stained the same inner subnuclear compartment and delimited the regions of active transcription known as euchromatin (Fig. 1D; Movie 1). Staining of the phosphorylated RNA polymerase II (Pol II pS2), a marker for active transcription, showed foci in inner areas of the nucleus, but not in the nucleoli, which is typical for nuclei in interphase (Dirks and Snaar, 1999; Mortillaro et al., 1996; Neugebauer and Roth, 1997) (Fig. 1C,F,I,O; Movies 3, 5, 9). As expected, the nucleoli, the site of ribosomal RNA synthesis, accumulated elevated levels of selective RNA dyes (Fig. 1I,N, and respective 3D reconstructions in Movies 5,8). We constructed a schematic of nuclear organization based on our microscopy observations (Fig. 1P). These results show that this application can visualize endogenous nuclear architectural elements, chromatin-associated proteins and transcription factors at a high resolution in unfixed isolated nuclei.
Imaging histone deacetylase enzymatic activity in isolated nuclei
We hypothesized that unfixed transcriptionally active nuclei could be used to study chromatin reorganization at single-nucleus level. The degree of histone acetylation is determined by the counteracting actions of histone acetyltransferases and histone deacetylases (HDACs). HDAC inhibitors (HDACi) impair HDAC enzymatic activity and unbalance the acetylation state towards hyperacetylation. Because HDACs control many aspects of cancer biology, addition of HDACi results in a higher global gene expression, activation of proteasome degradation pathways, impairment of protein–protein interactions, cell cycle arrest in G1 phase, induction of apoptosis and differentiation, and reduction of both cell migration and angiogenesis (Benedetti et al., 2015). As a proof of concept, we monitored the accumulation of the acetylated H3 proteins after treatment with suberanilohydroxamic acid (SAHA), a potent HDACi. SAHA acts by chelating zinc ions inside the pocket of the HDAC catalytic site (Marks et al., 2001). Because of its demonstrated efficacy as an HDACi, SAHA has been approved for therapies against cutaneous T-cell lymphoma and was reported to successfully reverse HIV-1 latency in patients on antiretroviral therapy (Archin et al., 2012).
TZM-bl cells were cultivated for 48 h in medium containing 1 µM SAHA and maintained in 1 µM SAHA during the nuclei purification, immunostaining and imaging procedures (Fig. 2A, 1 µM SAHA). In parallel, a subset of nuclei isolated from 1 µM SAHA-treated cells were processed in the absence of drug (Fig. 2A, 1 µM SAHA recovery). As a control, TZM-bl cells were cultivated in the presence of dimethyl sulfoxide (DMSO) and nuclei were isolated and processed without HDACi treatment (Fig. 2A, DMSO). We monitored the marker of chromatin acetylation H3K9ac in nuclei derived from the three conditions. Total H3K9ac fluorescence emitted by nuclei of the ‘1 µM SAHA’ and ‘1 µM SAHA recovery’ groups was higher than that from the ‘DMSO’ control group (Fig. 2B). Nuclei isolated from 1 µM SAHA-treated cells, but processed in the absence of HDACi showed less fluorescent signal than nuclei maintained in 1 µM SAHA, suggesting a loss of acetylation (Fig. 2B, 1 µM SAHA recovery). We took advantage of computational analysis to identify the number, position and intensity of H3K9ac signals in the images. H3K9ac foci were localized in confocal z-series of entire nuclei, and we were able to quantify the number and fluorescence intensity of H3K9ac signals. The largest number of H3K9ac signals were detected in the equatorial confocal z-series (Fig. 2C, highest peaks). For the nuclei represented in Fig. 2B, the sum of the H3K9ac signals detected in each nucleus was the following: DMSO=1254 signals, 1 µM SAHA=4506 signals and 1 µM SAHA recovery=2116 signals. This trend was confirmed when examining several nuclei (Fig. S2). We then measured the total intensity emitted by H3K9ac signals in nuclei and the average sum was the following: DMSO=47.7±15.8, 1 µM SAHA=390.9±81.4 and 1 µM SAHA recovery=198.3±31.2 (mean±s.d.; Fig. 2D). To verify the microscopy observations, fluorescence emitted from H3K9ac was measured by flow cytometry in 3000 nuclei per condition and plotted as histograms (Fig. 2E). The fluorescence peak of nuclei in recovery was between the DMSO and 1 µM SAHA conditions, validating the microscopy data. Finally, the accumulation of the acetylation marker H3K9ac in response to SAHA treatment was also confirmed biochemically by blotting nuclear protein extracts and staining for H3K9ac (Fig. 2F). β-actin was used as a loading control and H3K9ac protein amount was normalized to the ‘1 µM SAHA’ condition, set as 100% (DMSO=7%; 1 µM SAHA recovery=28%). These results demonstrated that isolated nuclei are active and sensitive to histone deacetylase enzymatic inhibition when separated from cells, and that they can also deacetylate histones when HDACi is withdrawn.
Fig. 2.

Examination of histone deacetylase inhibition in isolated nuclei. (A) Experimental workflow. TZM-bl cells were cultured for 48 h in medium containing 1 μM SAHA and processed for nuclei isolation, staining and microscopy (DMSO, untreated control). (B) Deconvolved confocal equatorial z-series of representative nuclei stained for acetylated H3 (H3K9ac). Scale bars: 5 μm. (C) Levels of H3K9ac signals in the confocal z-series for images shown in B. (D) Quantification of total H3K9ac signal fluorescence (DMSO, n=4; 1 μM SAHA, n=3; 1 μM SAHA recovery, n=4; bars indicate mean). (E) Quantification of H3K9ac fluorescence in isolated nuclei by flow cytometry. (F) Quantification of H3K9ac protein amount as determined by western blotting. **P≤0.01, ***P≤0.001 (Student's t-test, two-tailed, homoscedastic).
Visualizing histone acetylation at the single-nucleus level
In order to better understand the dynamics of histone acetylation, we monitored endogenous H3K9ac in isolated nuclei in time-lapse experiments after SAHA withdrawal. For this purpose, we cultivated TZM-bl cells in medium containing 1 µM SAHA, then nuclei isolation, staining and imaging was conducted in absence of SAHA. Fluorescence emitted by H3K9ac signals was monitored for 16 h (Fig. 3A). We tracked individual nuclei and observed a decrease of H3K9ac fluorescence intensity over time (Fig. 3B). We also noticed a reduction of the H3K9ac-labeled area in individual nuclei over time (Fig. 3A). To quantify this observation, nuclei isolated from SAHA-treated cells were stained for H3K9ac and lamin B1. The nuclear area was defined by the lamin B1 stain and the euchromatin area by the H3K9ac stain (Fig. S3A). We calculated the proportion of the H3K9ac-stained area in individual nuclei (H3K9ac area/lamin B1 area) at different time points. Values at the 0 h time point were set to 1. Interestingly, the H3K9ac-stained area of nuclei in recovery from the SAHA treatment decreased to 0.77±0.04, and was at 0.80±0.03 after 16 h (mean±s.d.; Fig. S3B). This is consistent with the progressive condensation of euchromatin observed during cell differentiation and gene inactivation (Ugarte et al., 2015). In order to validate the microscopy data we monitored the H3K9ac fluorescence by time-lapse flow cytometry. The H3K9ac mean fluorescence of ∼500 nuclei was measured at each time point of the time-lapse. At the beginning of the experiment (0 h time point) the mean fluorescence was ∼16,000 units and decreased to ∼13,000 after 9 h and was ∼9000 units 25 h later (Fig. 3C). To exclude the possibility that the decrease of H3K9ac fluorescence was caused by the spontaneous disassembly of the antibody–histone complex or degradation of the fluorochrome, we monitored fluorescence from IgG-coated beads stained in the conditions used for nuclei. The fluorescence emitted by H3K9ac-stained beads was measured by flow cytometry in a time-lapse experiment, and an average of ∼2000 beads were used at each time point. The initial mean fluorescence of 21,000 units modestly changed in the first 4 h and was ∼20,000 units 24 h later (Fig. 3D).
Fig. 3.

Visualization of histone acetylation at single-nucleus level. (A) Acetylated H3 (H3K9ac) visualized at the single-nucleus level during a time-lapse experiment in the absence of SAHA and (B) quantification of the total H3K9ac signals fluorescence in single nuclei (n=3, as represented by the three separate traces). (C) Mean±s.d. H3K9ac fluorescence emitted from isolated nuclei and measured by flow cytometry over time (500 nuclei per time point). (D) Mean±s.d. fluorescence from H3K9ac-stained IgG-coated beads as measured by flow cytometry over time (2000 beads per time point). (E) Time-lapse visualization of H3K9ac in representative nuclei treated ex vivo with 1 μM SAHA and (F) relative fluorescence quantification (hour 0, DMSO n=3, SAHA n=2; hour 0.5, DMSO n=3, SAHA n=3; bars indicate mean). (G) H3K9ac fluorescence after 1 μM SAHA treatment as measured by flow cytometry in isolated nuclei (3000 nuclei per time point and per condition). Deconvolved confocal equatorial z-series of representative TZM-bl nuclei are shown in A and E. Scale bars: 5 μm.
We hypothesized that chromatin modifications can be examined at single-nucleus level in isolated nuclei. In a second set of experiments, we induced chromatin acetylation by perfusing isolated nuclei with SAHA. For this experiment, TZM-bl cells were cultivated in presence of DMSO and used for nuclei isolation. Nuclei were then stained for the acetylation marker H3K9ac and treated with 1 µM SAHA or DMSO (Fig. 3E). Images of nuclei were acquired at the beginning of the treatment. As expected, H3K9ac fluorescence in the SAHA-treated and untreated nuclei was comparable (Fig. 3E, 0 h time point). Images acquired 30 min later (Fig. 3E, 0.5 h time point) showed an increase in H3K9ac fluorescence in the SAHA-treated nuclei, but not in the untreated controls whose fluorescence intensity decreased. Computational analysis of the images assessed that the average fluorescence emitted from nuclei maintained in 1 µM SAHA increased over time (Fig. 3F, 1 µM SAHA), while the fluorescence signal emitted from untreated nuclei decreased (Fig. 3F, untreated). Flow cytometry was used to verify the microscopy observations (Fig. 3G). For this purpose, nuclei isolated from TZM-bl cells cultured in DMSO were stained for acetylated H3 and treated with 1 µM SAHA (Fig. 3G, 0 h time point). We measured the H3K9ac mean fluorescence by flow cytometry in time-lapse and normalized the fluorescence of SAHA-treated nuclei to that of the untreated control at each time point (3000 nuclei per time point/per condition). We observed a fluorescence increase 2 h after beginning the SAHA treatment that positively correlated with time (Spearman's rank correlation coefficient r=0.95, P≤0.001).
Visualizing chromatin reorganization in isolated nuclei
We next sought to use isolated nuclei to examine chromatin reorganization in response to HDAC inhibition. For this purpose we cultivated TZM-bl cells in the presence of 1 µM SAHA or DMSO. Nuclei were isolated from either condition and stained for H3K9ac, Pol II pS2 and lamin B1. Images of isolated nuclei were acquired by multicolor spinning disk confocal microscopy and lamin B1 was used to determine the nuclear boundaries (Fig. 4A). SAHA treatment significantly increased the levels of H3K9ac (Fig. 4A,D) and Pol II pS2 (Fig. 4A,E). By taking advantage of software-assisted identification of the fluorescent signals, we obtained the coordinates of H3K9ac and Pol II pS2 signals. Then we identified the colocalization of H3K9ac and Pol II pS2 signals and plotted their coordinates (yellow dots, Fig. 4B). The H3K9ac and Pol II pS2 coordinates that did not colocalize were color coded in green and red, respectively (Fig. 4B). Acetylated H3 and Pol II pS2 and the colocalizing signals appeared to be diffusely localized in nuclei isolated from DMSO-treated cells (Fig. 4A,B, upper panels). Conversely, the topology of the H3K9ac-stained euchromatin was different in nuclei purified from SAHA treated cells. Acetylated H3 signals concentrated in an area delimited by discrete clusters of at least three contiguous H3K9ac signals (Fig. 4B, lower panel, green dots). Pol II pS2 signals were detected mostly within the H3K9ac-delimited stain and colocalized with H3K9ac signals forming linear clusters parallel to the euchromatin periphery (Fig. 4B, lower panel, yellow dots). The colocalization percentage of H3K9ac and Pol II pS2 signals in the nuclei presented in Fig. 4A increased following SAHA treatment (Fig. 4C). The proportion of acetylated H3 not associated with Pol II pS2 signal also increased. The proportion of Pol II pS2 not associated with H3K9ac decreased. On average, the number of colocalizing H3K9ac and Pol II pS2 signals significantly increased by ∼2-fold in the SAHA-treated condition (Fig. 4F).
Fig. 4.

Visualization of chromatin reorganization in isolated nuclei. (A) Deconvolved images of nuclei isolated from TZM-bl cells treated with 1 μM SAHA and stained for acetylated H3 (H3K9ac, green), phosphorylated RNA polymerase II (Pol II pS2, red) and lamin B1 (blue). DMSO, untreated control. (B) Localization maps of images in A showing the position of H3K9ac (green dots), Pol II pS2 (red dots) and colocalizing H3K9ac and Pol II pS2 signals (yellow dots). (C) Quantification of H3K9ac and Pol II pS2 colocalization percentage for the nuclei shown in A. (D) Total number of H3K9ac and (E) Pol II pS2 and (F) colocalizing H3K9ac and Pol II pS2 signals detected in equatorial z-series (DMSO, n=4; SAHA, n=3; bars indicate mean). *P≤0.05, **P≤0.01 (Student's t-test, two-tailed, homoscedastic). (G) Time-lapse images of H3K9ac stain reorganization after 10 μM SAHA treatment; minutes are indicated at bottom left corner of each time point (images are not deconvolved). (H) Intensity profile of H3K9ac fluorescence detected along the diameter of the nucleus shown in G. (I) H3K9ac signal intensity measured in regions of interest at the periphery (solid lines) and center (dashed lines) of the H3K9ac stain; colors indicate five individual nuclei. Scale bars: 5 μm.
In order to determine whether our system would allow us to observe chromatin reorganization at the single-nucleus level, we monitored the H3K9ac topology following SAHA treatment. For this purpose, we conducted a time-lapse experiment acquiring images of H3K9ac-stained nuclei with 10 min intervals. At the beginning of the observation (minute 0), H3K9ac signals were diffuse. SAHA was then perfused onto nuclei after 1 h (minute 60) and H3K9ac fluorescence was monitored for additional 2 h (Fig. 4G). We observed that H3K9ac fluorescence gradually increased after SAHA treatment followed by a decrease of signal intensity in the center of the staining area and accumulation at the periphery. In the example illustrated in Fig. 4G and Movie 10, the H3K9ac stain gradually reorganized to the periphery. After 40 min (minute 180), the majority of signals were detected at the periphery of the staining area. Fig. 4H shows the intensity profiles measured along the diameter of the H3K9ac staining area at minute 140 and 180. At minute 180, the signal intensity in the center decreased and slightly increased at the periphery (Fig. 4H). We plotted the intensity measured in the center (dashed lines) and at the periphery (solid lines) at each time point and found that from minute 160 until the end of the observation, the fluorescence measured in the center was significantly lower than that at the periphery, denoting a change of H3K9ac localization after SAHA treatment (Fig. 4I).
Imaging RNA transcription in isolated nuclei
We sought to visualize transcription by staining isolated nuclei with an RNA-selective dye suitable for time-lapse microscopy. We hypothesized that transcription could be detected as an increase of RNA stain fluorescence intensity and that the treatment with α-amanitin, an RNA polymerase II and III inhibitor, would reduce RNA transcription. To verify this hypothesis, nuclei were pre-stained with SYTO® RNA Select™ green fluorescent cell stain (RNA SYTO, Thermo Fisher Scientific), then suspended in a solution containing RNA SYTO and visualized by time-lapse microscopy. Images of stained nuclei were acquired at the beginning of the experiment (Fig. 5A, 0 h time point). Nuclei were then supplemented with 5 µg ml−1 α-amanitin (Sigma-Aldrich) and visualized 1.25 h later (Fig. 5A, 1.25 h time point). As expected, at the beginning of the observation the RNA intensity was similar in both conditions (Fig. 5A, 0 h time point). Nuclei maintained in the absence of α-amanitin showed an increase of RNA SYTO stain in the nucleoli and at the periphery of the stain, and newly formed RNA structures were detected (Fig. 5A, untreated, white arrow). Conversely, isolated nuclei treated with α-amanitin showed only a modest increase in RNA stain over time (Fig. 5A, α-amanitin). We quantified the RNA stain intensity at the beginning and at the end of the time-lapse and set the values measured at the 0 h time point to 1 (Fig. 5B). In the absence of α-amanitin, the RNA fluorescence increased ∼50% in 1.25 h and only ∼20% in the α-amanitin treatment, showing a significant reduction in RNA transcription. To validate the results obtained by microscopy, we performed a flow cytometry time-lapse experiment with similar staining and time-lapse conditions. The RNA mean fluorescence was measured at the beginning of the time-lapse (Fig. 5C, 0 h time point) followed by treatment with α-amanitin. Untreated nuclei were used as control, and RNA fluorescence was measured from 3000 nuclei per condition at each time point. The mean fluorescence of the untreated sample was normalized to the α-amanitin condition at each time point (Fig. 5C). At 15 min after the beginning of the experiment, the untreated nuclei mean fluorescence was ∼15% higher than the α-amanitin-treated control (0.25 h time point) and ∼13% higher 1 h later (1.25 h time point), demonstrating that we can monitor α-amanitin-mediated inhibition of transcription in single nuclei.
Fig. 5.

Imaging RNA transcription in isolated nuclei. (A) RNA transcription measured by accumulation of RNA SYTO stain intensity detected in single nuclei. The white arrow indicates areas of newly formed signal. Deconvolved confocal equatorial z-series of representative TZM-bl nuclei are shown. Scale bar: 5 μm. (B) Quantification of mean±s.d. RNA SYTO stain intensity from microscopy images (untreated, n=7; α-amanitin, n=5). **P≤0.01 (Student's t-test, two-tailed, homoscedastic). (C) RNA SYTO stain uptake in transcriptionally active TZM-bl nuclei quantified by flow cytometry over time (n=3000 nuclei per condition at each time point).
Localizing HIV-1 Tat and HDAC3 proteins in isolated nuclei
After reverse transcription, the HIV-1 genome is integrated irreversibly into the host DNA. The HIV-1 Tat protein is expressed early during infection and binds to the nascent promoter-proximal viral RNA to transactivate viral transcription (Karn, 2011). Because of this property, Tat is an excellent marker to localize integrated HIV-1 DNA. How HIV-1 Tat, in concert with host factors, regulates the HIV-1 LTR promoter is still a matter of ongoing research. To examine Tat function microscopically, we created an expression vector coding for HIV-1 Tat fused to enhanced GFP (Tat–GFP). We also built a similar construct, Tat-K41A–GFP containing a single amino acid change in the HIV-1 Tat sequence (K41A). The K41A mutation maintains the wild-type Tat–RNA binding, but impairs Tat recruitment of pTEFb and transactivation of the HIV-1 LTR (Rice and Carlotti, 1990; Wei et al., 1998; Yang et al., 1996). Tat–GFP and Tat-K41A–GFP plasmids were transfected into TZM-bl cells that are genetically engineered to contain integrated copies of the luciferase gene under the control of the HIV-1 promoter. Tat activity was assayed by measuring relative light units following transfection (RLU, Fig. 6A). In a similar assay, Tat proteins fused to a Flag epitope, Flag–Tat and Flag–Tat-K41A, yielded similar results verifying that the EGFP tag did not affect Tat activity (Fig. S4). We hypothesized that Tat recruitment to the HIV-1 promoter should negatively correlate with staining of chromatin suppressive markers, whereas Tat-K41A should colocalize to the viral promoter without perturbing suppressed chromatin. To test our hypothesis, we looked at the association of Tat with the HDAC3, a marker for inactive chromatin that interacts with the HIV-1 LTR (Malcolm et al., 2007). For this purpose, either Tat–GFP or Tat-K41A–GFP constructs were transfected in TZM-bl cells. Nuclei isolated from transfected cells were stained for HDAC3 and imaged by confocal microscopy (Fig. 6B). As would be expected, Tat-K41A–GFP signals were found to be associated with HDAC3 signals, whereas Tat–GFP signals were not (Fig. 6B, white arrows, insets).
Fig. 6.
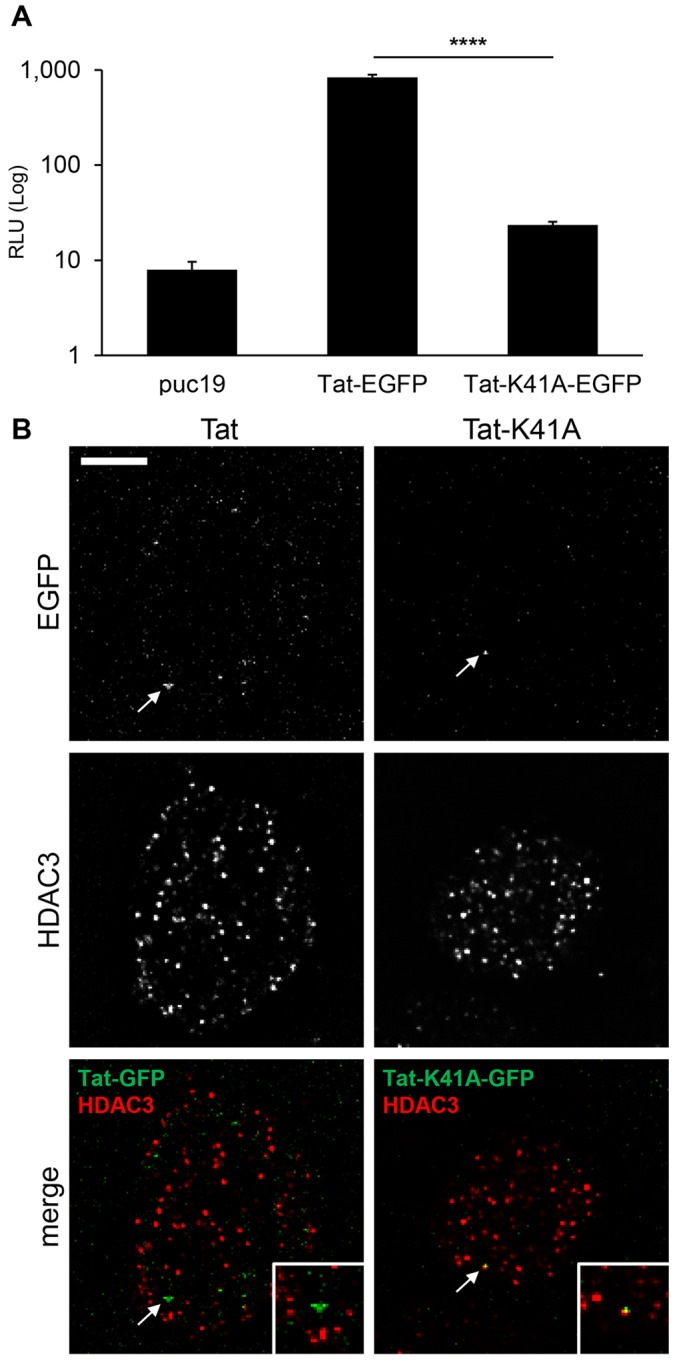
Localization of HIV-1 Tat and HDAC3 proteins in isolated nuclei. (A) Tat activity measured in a TZM-bl-based reporter assay by assessing the relative light units (RLU) produced. Results are mean±s.d. (n=3) ****P≤0.0001 (Student's t-test, two-tailed, homoscedastic). (B) Nuclei isolated from TZM-bl cells transfected with HIV-1 Tat–GFP or the mutant Tat-K41A–GFP were stained for HDAC3. Tat–GFP signals are indicated by white arrows; colocalizing Tat and HDAC3 are signals in yellow. Boxes in the merge panels magnify the areas indicated by the arrows. Deconvolved confocal equatorial z-series of representative nuclei are shown. Scale bar: 5 μm.
Visualization of the architecture of the nuclear lamina
In order to improve the imaging of proteins of the nuclear lamina in isolated nuclei, we took advantage of a mechanical microcompressor that has been previously used to achieve highly detailed visualization of cellular structures by gentle flattening and immobilization of the specimen (Yan et al., 2014; Zinskie et al., 2015). Mechanical microcompression of the nuclei increased the area of contact between the nuclear surface and the glass slide (Fig. 7A). Nuclei were stained for β-actin and Pol II pS2, immobilized in a microcompressor and imaged by confocal microscopy (Fig. 7B). The bottom and top confocal z-series of the nuclei showed intricate β-actin filamentous structures on the nuclear surface at better resolution than experiments performed without microcompression, demonstrating the usefulness of this approach (Fig. S5). Pol II pS2 signals were detected at the nuclear equator similar to Figs 1C,F,I,O and 4A.
DISCUSSION
We have developed a microscopy tool to visualize endogenous ‘NUClear Live Events by Optical Microscopy’ that we hereafter name NUCLEO-M. Cells are harvested and gently lysed by a non-ionic detergent in a one-step procedure allowing the separation of the nucleus from smaller organelles and cellular debris by centrifugation, and purifying nuclei that lack the nuclear membrane and pore complexes. This worked ideally for us, as efficient delivery of molecules and antibodies (∼150 kDa) to the nucleus is prohibited by the nuclear membranes and the size exclusion limit of the nuclear pores (∼60 kDa). The nuclei isolation procedure did not notably affect the nuclear integrity and chromatin organization. The topology of seven nuclear targets detected in isolated nuclei stained without fixation was comparable to that observed in cells stained after fixation (Fig. 1B,C; Fig. S1A–N). We also compared our findings in isolated nuclei with other studies reporting the localization of nuclear proteins in whole cells and verified, when available, that the nuclear structure and topology were unperturbed by the nuclear isolation. Finally, a custom nuclei isolation protocol was tested to compare the results with those obtained upon use of the commercial kit used in this work (Farrell, 2010). Staining of four nuclear targets showed that integrity and structure of the nuclei was maintained (Fig. S1O-S1R), further validating our system and providing an alternative method to future users that can be customized to a specific application.
The method presented in this work can be complementary to ChIP, which has been the standard technique to analyze chromatin changes. Our application allowed us to visualize a panel of 11 nuclear endogenous targets for the first time in unfixed isolated nuclei (Fig. 1; Movies 1–9). Our results confirm previous observations in fixed and live cells and demonstrate that, after isolation from the cell, nuclei can be used to investigate the dynamics of nuclear events in a single nucleus. This included visualization of histone acetylation (Figs 2 and 3), chromatin reorganization (Fig. 4), RNA transcription (Fig. 5), interaction of viral proteins with chromatin (Fig. 6) and nuclear architecture (Fig. 7). We selected chromatin markers that have been previously reported to play a key role in cell development, response to pathogens and cancer. We were able to detect markers for the histone code such as acetylated and methylated H3 histones, the chromatin ‘writer’ SUV39H1 and the ‘eraser’ HDAC3 proteins (Fig. 1E,M). We could also detect endogenous CBFβ proteins, usually present in a low amount in a cell (Miyagi et al., 2014), showing the high sensitivity of this technique (Fig. 1G,H). Staining of the core binding factors in nuclei isolated from peripheral blood mononuclear cells showed similar localization patterns to those observed in TZM-bl nuclei and demonstrated that this method can be applied to other cell types including primary cells from human patients (Fig. S6). By using a combination of co-staining experiments, we generated a compartmentalization map that supports the current model of nuclear architecture (Harr et al., 2016) (Fig. 1P). Knowledge about the spatial organization of the nucleus has largely been gleaned from genomic studies. Biochemical assays coupled with next-generation sequencing have been used to analyze chromatin organization in detail (Belton et al., 2012; Dekker et al., 2013; Dixon et al., 2012; Gómez-Díaz and Corces, 2014; Lieberman-Aiden et al., 2009; Risca et al., 2017; Simonis et al., 2007; Stevens et al., 2017). Others have generated molecular maps of the regions of the chromatin interacting with the nuclear lamina (Guelen et al., 2008; Peric-Hupkes et al., 2010; Pickersgill et al., 2006; Ugarte et al., 2015). In these studies, it was shown that genes in regions of the chromatin in lamina-associated domains (LADs) are expressed at very low levels, which is typical of heterochromatin. We observed that endogenous markers for heterochromatin (H3K37me3, H3K9me3, SUV39H1, HDAC3) were enriched near the nuclear lamina in unfixed nuclei, validating the information previously inferred from biochemical experiments. The current model for chromatin distribution in differentiated cell nuclei proposes that areas of active transcription are confined to a sub-compartment of the nucleus traditionally described as euchromatin (Harr et al., 2016). This compartment is considered dynamic, as genes can move in and out of the euchromatin depending on the active or inactive status of their promoters. Because of this, gene expression is finely controlled by acetylation and methylation of the histone tails. In agreement with this model, we detected acetylated and methylated modifications of the histone H3 in the area occupied by the euchromatin (Fig. 1). Interestingly, we observed that acetylated H3 (H3K9ac) and RNA were enriched at the periphery of the euchromatin, suggesting the existence of zones of highly active transcription. One can speculate that the accumulation of transcription machinery in one spot, in close proximity to the nuclear envelope, might have been evolutionarily favorable for efficient processing and export of mRNA.
We also investigated the potential application of using isolated nuclei to study the nuclear architecture. The nuclear lamina is a protein matrix that mechanically supports the nuclear scaffold against mechanical stress (Clubb and Locke, 1998; Holaska et al., 2004; Holaska and Wilson, 2007; Shimi et al., 2015). β-actin has been shown to be a component of the nuclear envelope in several cell types (Hoffman, 2009) and we were able to detect β-actin at high resolution in equatorial sections of the nucleus with a ring-like staining (Fig. 7). We used a mechanical compressor, which allows the gentle compression of the specimen between two glass coverslips and has previously been used to immobilize single prokaryotic and eukaryotic cells and visualize dynamic cellular physiological events (Zinskie et al., 2015). With this device, we were able to immobilize and flatten isolated nuclei. This strategy allowed us to increase the contact of the nuclear lamina with the glass slide enabling the visualization of filamentous actin structures at high resolution (Fig. 7B). This method can be further exploited to study changes in the nuclear architecture during cell development or during the onset of cell malignancies. The presence of a large area of sample in close proximity to the coverslip should also make it possible to perform total internal reflection fluorescence (TIRF) microscopy on isolated nuclei to further increase the signal-to-noise ratio for single-molecule studies of transcription or for the visualization of cargo through the nuclear lamina.
An intriguing application of the tool presented in this work is the possibility to study chromatin remodeling in time-lapse experiments (Figs 2 and 3). We demonstrated that isolated nuclei are sensitive to the HDACi SAHA. In fact, nuclei accumulated a higher amount of H3K9ac proteins when maintained in solutions supplemented with SAHA (Fig. 2B–F). In the experiment schematized in Fig. 2A, nuclei were maintained in SAHA-containing solutions for ∼8 h from the first step of nuclei isolation. We showed that, within that time frame, nuclei deprived of SAHA underwent the process of deacetylation (SAHA recovery) and the H3K9ac total signals fluorescence decreased ∼2-fold (Fig. 2D), suggesting that HDACs are active in isolated nuclei. Notably, H3K9ac total signal fluorescence in nuclei in recovery was 4-fold higher than in the DMSO control (Fig. 2D). In fact, nuclei in the ‘DMSO’ and ‘SAHA recovery’ conditions were isolated and processed in the absence of SAHA. Thus, this increase was solely due to the effect of SAHA supplied to cells in the culture medium. We observed similar differences in the total number of H3K9ac signals detected in nuclei (Fig. S2) denoting a positive correlation between the total fluorescence and number of signals detected. Similar differences were quantified in H3K9ac protein blot (Fig. 2F). In the western blot analysis, the H3K9ac protein levels in nuclei in recovery was about 4-fold lower than in the SAHA-treated nuclei. Overall, the trends observed by microscopy, flow cytometry and western blot are in agreement, and confirmed what others have observed in fixed cells (Bártová et al., 2005), demonstrating than our tool can be an effective alternative to the microscopy techniques currently in use.
The acetylation dynamics were further explored in time-lapse experiments (Fig. 3). Following withdrawal of SAHA, we observed a 2.7-fold reduction of total H3K9ac signals fluorescence in 16 h by microscopy (Fig. 3B) and 1.8-fold reduction in 25 h by flow cytometry (Fig. 3C). Despite showing similar trends in H3K9ac reduction, the different experimental approaches generated different fold changes that may be attributed to different experimental conditions. However, the overall trends remained the same. We verified that the reduction of H3K9ac stain is not due to the spontaneous antibody–target dissociation or loss of fluorescence. In fact, most of the primary anti-H3K9ac antibody remained associated to IgG-coated beads for 24 h (Fig. 3D). Therefore, these data suggest that HDACs are active and can remove acetyl groups from histones in isolated nuclei. HDACs appear capable of displacing the acetyl group from H3, despite the presence of the antibody. We were also able to observe acetylation of histones (Fig. 3E–G). In those experiments, we inhibited histone deacetylation by supplying SAHA to isolated nuclei. We observed a 1.6-fold increase of H3K9ac fluorescence in 30 min in our microscopy experiments. In the flow cytometry experiment, the SAHA effect started between 1 and 2 h after beginning the treatment, and the increase of H3K9ac fluorescence correlated with time (Fig. 3G), validating the microscopy observations and further suggesting that histone acetyl transferases are active in isolated nuclei for several hours.
State of the art immunofluorescence techniques allow the visualization of many targets in a single cell (Lin et al., 2016), but require fixation. The method described in this work can be used to observe chromatin reorganization by directly visualizing multiple endogenous targets without fixing the sample. This is of great interest to the study of the epigenetics of cell development and disease progression. For instance, cancerous cells have strikingly different chromatin organization compared to that in quiescent cells (Ferraro, 2016). Viruses can also induce chromatin remodeling and either activate transcription of genes important for their replication or silence the host restriction machinery (Milavetz and Balakrishnan, 2015). We treated cells with SAHA and looked at the spatial organization of chromatin in isolated unfixed nuclei (Fig. 4). Interestingly, SAHA changed the localization of active euchromatin, labeled in our experiments by H3K9ac. H3K9ac signals formed a ring-like conformation in TZM-bl nuclei (Fig. 4A,B, bottom panels), confirming what has been observed elsewhere by us (Fig. 2B) and by others (Bártová et al., 2008). The alignment of H3K9ac and Pol II pS2 signals at the periphery of euchromatin indicated the zones of active mRNA transcription and elongation (Komarnitsky et al., 2000; West and Corden, 1995). Clusters of acetylated chromatin and active RNA polymerase may be indicative of the ‘transcription factories’ that Cook and colleagues hypothesized over a decade ago (Cook, 1999; Faro-Trindade and Cook, 2006). Interestingly, we were able to capture chromatin reorganization by treating isolated nuclei with SAHA and to observe H3K9ac signals localizing at the periphery of euchromatin where active transcription occurs (Fig. 4G,H,I; Movie 10). Moreover, our microscopy observations support the current model of chromatin organization (Fedorova and Zink, 2008; Schneider and Grosschedl, 2007) that postulates that active genes move to sites of active transcription and not vice versa (Osborne et al., 2004). In accordance with this model, we observed that Pol II pS2 signals distributed to the inner areas of the nucleus and were unaffected by the HDACi treatment. H3K9ac signal topology instead strikingly changed when histone deacetylases were inhibited (Fig. 4). In related experiments, we also observed that SAHA reduced the number of colocalizing H3K9ac and lamin B1 signals (Fig. S7). These data are in agreement with work from the van Steensel group showing that trichostatin A, a potent HDACi, induces loss of chromatin lamina binding at the nuclear periphery and a relocation of chromatin from the periphery to inner areas of the nucleus (Pickersgill et al., 2006).
Chromatin remodeling controls transcription and we sought to visualize this in our system. We verified that isolated nuclei are transcriptionally competent by using the selective SYTO dye to stain RNA. We then used α-amanitin to inhibit RNA transcription and found that untreated nuclei significantly accumulated RNA over time and new RNA structures could be visualized (Fig. 5A, untreated). The inhibition of transcription exerted by α-amanitin was detected at the single-nucleus level, and significantly lowered the synthesis of RNA, showing the effectiveness of our method to follow changes in RNA content in a single nucleus (Fig. 5A,B, α-amanitin). Overall, the observed accumulation of RNA was relatively modest, owing to the large mass of RNA present in the nucleolus at the beginning of the experiment, but was validated on a larger scale by flow cytometry (Fig. 5C). The absence of a nuclear membrane is an advantage for our experiments, which require antibody delivery without fixation and rapid perfusion of dyes and drugs inside the nucleus. However, it is likely that soluble nuclear factors such as nucleotides, acetyl-CoA, proteins and other macromolecules present inside the nucleus are lost during the nuclei isolation procedure. Similarly, we speculate that the antibodies, dyes and drugs used for our experiments can diffuse out of the nuclei during time-lapse experiments. We tried to address this issue in two ways. First, we used vehicle controls in the experiments employing a histone deacetylase inhibitor or a RNA polymerase inhibitor as a control for the nuclear functions in absence of inhibitors. Secondly, in the experiments involving measurement of RNA transcription, we supplemented the storage buffer with SYTO dye to maintain a source of dye to stain the RNA during the time lapse. It is recommended to other users to supplement reagents in time-lapse experiments and verify nuclear functions with appropriate controls.
We extended the capability of this new method to relevant questions in virology. In the last decade, several studies have employed live microscopy to discern the mechanisms of action of HIV-1 proteins and their dynamics in a living cell (Baumgärtel et al., 2011; Chen et al., 2014, 2016; Jouvenet et al., 2008, 2009; Sardo et al., 2015). HIV-1 expression during spreading infection and latency is regulated by epigenetic changes (Hakre et al., 2011). HIV-1 Tat protein is expressed early during the infection, localizes to the integrated viral DNA by interacting with the nascent RNA transcript and recruits several host factors to transactivate viral transcription and increase gene expression (Easley et al., 2010). Biochemical evidence showed that Tat recruits host factors, but the mechanism, timing and means of the recruitment have not been visualized in live cells. As a proof of concept, we utilized isolated nuclei to visualize the association of GFP-tagged Tat proteins with HDAC3 (Fig. 6). HDAC3 is known to be recruited to the HIV-1 LTR promoter in latently infected cells and to suppress viral gene expression (Marban et al., 2007). Owing to its nature as a transcriptional activator, we hypothesized that wild-type Tat could be visualized at sites of active transcription. We found that HDAC3, a marker of transcriptionally suppressed chromatin, colocalized with an HIV-Tat mutant defective for transactivation (Tat-K41A) whereas the wild-type form of Tat did not (Fig. 6B). These observations will be further extended to other chromatin markers and HIV-1 promoter regulators, such as RUNX1 and CBFβ (Klase et al., 2014), to study the mechanisms that finely control the HIV-1 LTR during infection and design strategies to interfere with these mechanisms. Additionally, this experiment shows the utility of this new technique to study changes in chromatin modification and structure at a given locus. HIV-1 Tat is useful for localizing the integrated HIV-1 provirus, but other techniques, such as fluorescently labeling loci by use of CRISPR/Cas9, could be used to examine other specific areas of interest (Chen et al., 2013; Tanenbaum et al., 2014).
To our knowledge, ours is the first report that provides high-resolution visualization of endogenous nuclear proteins and nucleic acids in an unfixed sample. We generated an extensive localization map of structural proteins, proteins of the histone code and transcription factors in isolated nuclei that supports the current model of nuclear compartmentalization. Our studies demonstrate the utility of this novel system to investigate the dynamics of chromatin remodeling, RNA transcription, viral protein function and nuclear architecture at the single-nucleus level. We believe that this new tool will find applications in different fields of biology.
MATERIALS AND METHODS
Nuclei isolation and staining
TZM-bl cells were harvested by trypsinization and transcriptionally competent nuclei were isolated by chemical lysis and stored following the procedure for cell suspension lines recommended by the manufacturer (nuclei isolation kit: Nuclei EZ Prep, Sigma-Aldrich, Nuc-101, St. Louis, MO). Nuclei were stained for 1 h on ice in 5% fetal bovine serum in 1× -phosphate-buffered saline (5% FBS/PBS) solution containing primary antibodies diluted at the concentrations reported in Table S1. Nuclei were washed twice in 5% FBS/PBS and stained for 1 h on ice with appropriate secondary antibodies conjugated with fluorescent dyes (Table S1). Nuclei were finally washed twice in 5% FBS/PBS and resuspended in the storage buffer provided by the kit (Nuclei EZ Prep, Sigma-Aldrich, Nuc-101) and used for imaging experiments. For experiments on RNA, RNA stain was performed on ice for 20 min in 5% FBS/PBS solution containing 50–500 nM SYTO® RNA Select™ green fluorescent cell stain (Thermo Fisher Scientific, Waltham, MA), followed by two washes in 5% FBS/PBS and resuspension in storage buffer (from the kit as above).
As an alternative method (Fig. S1O–R), nuclei were isolated from TZM-bl cells by chemical lysis with an hypotonic lysis buffer (10 mM Tris-HCl pH 7.4, 10 mM NaCl, 5 mM MgCl2 and 0.5% NP-40) and stored in glycerol storage buffer (50 mM Tris-HCl pH 8.0, 30% glycerol, 2 mM MgCl2, 0.1 mM EDTA) as previously described (Farrell, 2010).
Cells fixation and staining
TZM-bl cells were seeded on a chambered coverglass (Nunc™ Lab-Tek™ Chambered Coverglass, Thermo Fisher Scientific, Waltham, MA) and fixed after 24 h with 4% paraformaldehyde for 10 min at room temperature, washed with 1× PBS and permeabilized with PBS and 0.25% Triton X-100 for 10 min. Following washes with PBS and 0.1% Triton X-100 (PBST), cells were treated with PBST plus 3% BSA (PBST/BSA) blocking solution for 45 min at room temperature. Cells were then incubated with primary antibodies diluted in PBST/BSA for 1 h at room temperature, washed with PBST and incubated with secondary antibodies diluted in PBST/BSA for 30 min followed by washes with PBST and PBS. Total RNA was stained by 50–500 nM SYTO® RNA Select™ Green fluorescent cell stain (Thermo Fisher Scientific, Waltham, MA).
Microscopy, imaging acquisition, processing and analysis
Isolated nuclei and TZM-bl cells seeded on chambered coverglass (Nunc™ Lab-Tek™ Chambered Coverglass, Thermo Fisher Scientific, Waltham, MA) were imaged with a Zeiss Axiovert (Jena, Germany) microscope with a Zeiss alpha Plan-Fluar 100×, 1.45 NA oil objective and a spinning disk confocal scan head (Yokogawa CSU-X1, Tokyo, Japan). A 488 nm laser source with 488/25 nm, 617/73 nm and 692/40 nm emission filters were used for the illumination of Alexa Fluor® 488 dye (Thermo Fisher Scientific), SYTO® RNA Select™ Green Fluorescent Cell Stain (Thermo Fisher Scientific) and PerCP-Cy5.5 (BD Biosciences, San Jose, CA), respectively. A 561 nm laser source with 568/25 nm emission filters was used for illumination of Alexa Fluor® 555 dye (Thermo Fisher Scientific, Waltham, MA, USA). Digital images were captured sequentially by a high-speed EM-CCD camera with 100 ms exposure time (Photometrics Evolve™ 512, Tucson, AZ). Confocal z-stacks of the entire nucleus or of the nuclear equator were acquired with a z-step size of 0.4–1 µm. Time-lapse experiments were automated by software (Slidebook 6.0.9, 3i, Denver, CO).
A home-built instant structured illumination microscope, iSIM (York et al., 2013), was used to acquire super-resolution images of TZM-bl nuclei. In this microscope, super-resolution images are acquired in a single exposure via multiplication of a sharp excitation pattern with the fluorescently labeled sample and analog image processing of the resulting fluorescence, prior to camera-based detection. Resolution was further improved by deconvolution with the Richardson–Lucy algorithm. Two-color images were collected by exciting the sample at 488 nm (for Alexa Fluor® 488 dye) and 561 nm (for Alexa Fluor® 555 dye); notch filters were used to reject pump light. Samples were collected at 98 ms frame rates, at 0.2 µm per plane. A 60×, 1.45 NA objective (Olympus, Tokyo, Japan) was used to collect data; the final pixel size after ∼117× magnification was 55.5 nm.
Images of TZM-bl cells and individual nuclei were analyzed with ImageJ 1.49v (NIH, Bethesda, MD). Look-up tables and Localize software thresholding were established on no-primary antibody controls or untransfected controls in order to suppress the background fluorescence. The coordinates, intensity and position of diffraction-limited spots were identified by the Localize software (courtesy of Daniel Larson, NIH, Bethesda, MD, USA). Colocalizing signals within a distance of three pixels were identified with the use of a custom Excel (Microsoft) macro (Sardo et al., 2015; available from corresponding author upon request). Raw confocal images were used for analysis. Deconvolution of confocal images was performed with a constrained iterative algorithm (Slidebook 6.0.9, 3i, Denver, CO). Three-dimensional reconstructions of confocal and iSIM z-series stacks were made with ImageJ 1.49v. Plot profiles, and regions of interests were also delineated with ImageJ 1.49v.
Flow cytometry
Stained nuclei were resuspended in 5% FBS/PBS and the fluorescence emitted by either Alexa Fluor® 488 dye or SYTO® RNA Select™ Green fluorescent cell stain was measured with a custom flow cytometer equipped with a 488 nm laser (FACSort DxP12, Cytek, Fremont, CA). The digital signal was analyzed by FlowJo (Ashland, OR, USA).
Cell culture, plasmid construction, transfection and luciferase assay
TZM-bl cells (NIH-AIDS Reagent Program, catalog number 8129, NIH, Bethesda, MD) were maintained in Dulbecco's modified Eagle's medium (DMEM; HyClone, GE Healthcare Life Sciences, Chicago, IL) supplemented with 10% FBS, 100 units ml−1 penicillin, 100 µg ml−1 streptomycin and 0.3 mg ml−1 L-glutamine (PSG, Gibco, Thermo Fisher Scientific). Cells were grown at 37°C with 5% CO2. For the histone deacetylase inhibition experiment, 3×106 TZM-bl cells were seeded on 55 cm2 tissue culture dishes and grown for 48 h in DMEM-FBS-PSG containing 1 µM suberanilohydroxamic acid diluted in DMSO (SAHA, Sigma-Aldrich).
The CMVPRO-Tat-EGFP and CMVPRO-TatK41A-EGFP constructs were generated by using the Gibson Assembly cloning kit (New England BioLabs, Ipswich, MA). Wild-type and mutant sequences of the Tat gene were PCR amplified from the Flag–Tat and Flag–Tat-K41A plasmids, respectively, using primers TJO3 (5′-TACCGGACTCAGATCATGGAGCCAGTAGATCCTAG-3′) and TJO4 (5′-TGGTGGCGACCGGTGGATCGAATGGATCTGTCTCTG-3′). PCR fragments were introduced into the XhoI/BamHI site of EB3-EGFP [a gift from Prof Kenneth Myers (Addgene #56474, deposited by Michael Davidson)] generating a Tat–EGFP fusion sequence (indicated in the text as Tat–GFP). Both Tat–GFP and Tat-K41A–GFP constructs were verified by sequencing.
Tat–GFP-expressing nuclei were prepared with the following procedure: 3×106 TZM-bl cells were seeded on a 55 cm2 tissue culture dishes; 24 h later plasmids Tat–GFP or Tat-K41A–GFP were delivered to cells with TransFectin Lipid Reagent (Bio-Rad, Hercules, CA). Nuclei were isolated at 24 h post transfection (nuclei isolation kit as above).
For the luciferase assay, 25,000 TZM-bl cells were seeded in a 96-well plate and transfected 24 h later with Transfectin Reagent (Bio-Rad). Luciferase was harvested 24 h after transfection with the Bright-Glo™ Luciferase Assay System (Promega, Fitchburg, WI), and luciferase activity was measured by a microplate reader (Synergy, BioTek, Winooski, VT).
Western blot analysis
Isolated nuclei were lysed with RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, pH 8.0) and total nuclear proteins blotted as previously described (Klase et al., 2014). Anti-histone H3 (acetyl K9) antibody (Abcam, ab61231, Cambridge, UK) was used at 1:1000 dilution and anti-β-actin antibody was used at 1:3000 (Sigma-Aldrich, A1978). Secondary horseradish peroxidase (HRP)-conjugated anti-mouse-IgG antibody (Bio-Rad, 170-6516) was used at 1:1000 dilution. The intensity of the bands detected in the blots was measured by ImageJ 1.49v.
Acknowledgements
We thank Daniel Larson (NIH, Bethesda, MD, USA) for providing the Localize software for image analysis and Ryan Burdick, Wei-Shau Hu and Vinay Pathak (NIH, Frederick, MD, USA) for providing image analysis tools. We thank Kenneth Myers, James Holaska, Jason Heindl (University of the Sciences, Philadelphia, PA, USA) and Elizabeth Anderson (NIH, Bethesda, MD, USA) for discussion and critical reading of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: L.S., C.J., Z.A.K.; Methodology: L.S., Z.A.K.; Software: L.S.; Validation: L.S.; Formal analysis: L.S., A.L., S.K., L.B.; Investigation: L.S., S.K., L.R., W.E., T.J., H.V.; Resources: H.S., C.J., Z.A.K.; Writing - original draft: L.S.; Writing - review & editing: L.S., A.L., S.K., H.V., H.S., C.J., Z.A.K.; Visualization: L.S.; Supervision: Z.A.K.; Project administration: Z.A.K.; Funding acquisition: C.J., Z.A.K.
Funding
This work was supported by University of the Sciences internal funds, a National Institutes of Health (NIH)/NIDA Avenir award to Z.A.K., and a grant from the W. W. Smith Charitable Trust to Z.A.K. and C.J. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.205823.supplemental
References
- Aagaard L., Laible G., Selenko P., Schmid M., Dorn R., Schotta G., Kuhfittig S., Wolf A., Lebersorger A., Singh P. B. et al. (1999). Functional mammalian homologues of the Drosophila PEV-modifier Su(var)3-9 encode centromere-associated proteins which complex with the heterochromatin component M31. EMBO J. 18, 1923-1938. 10.1093/emboj/18.7.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin N. M., Liberty A. L., Kashuba A. D., Choudhary S. K., Kuruc J. D., Crooks A. M., Parker D. C., Anderson E. M., Kearney M. F., Strain M. C. et al. (2012). Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482-485. 10.1038/nature11286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister A. J. and Kouzarides T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381-395. 10.1038/cr.2011.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bártová E., Pacherník J., Harničarová A., Kovařík A., Kovaříková M., Hofmanová J., Skalniková M., Kozubek M. and Kozubek S. (2005). Nuclear levels and patterns of histone H3 modification and HP1 proteins after inhibition of histone deacetylases. J. Cell Sci. 118, 5035-5046. 10.1242/jcs.02621 [DOI] [PubMed] [Google Scholar]
- Bártová E., Krejči J., Harničarová A., Galiová G. and Kozubek S. (2008). Histone modifications and nuclear architecture: a review. J. Histochem. Cytochem. 56, 711-721. 10.1369/jhc.2008.951251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgärtel V., Ivanchenko S., Dupont A., Sergeev M., Wiseman P. W., Kräusslich H.-G., Bräuchle C., Müller B. and Lamb D. C. (2011). Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 13, 469-474. 10.1038/ncb2215 [DOI] [PubMed] [Google Scholar]
- Belton J.-M., McCord R. P., Gibcus J. H., Naumova N., Zhan Y. and Dekker J. (2012). Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268-276. 10.1016/j.ymeth.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti R., Conte M. and Altucci L. (2015). Targeting histone deacetylases in diseases: where are we? Antioxid Redox Signal. 23, 99-126. 10.1089/ars.2013.5776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle S., Gilchrist S., Bridger J. M., Mahy N. L., Ellis J. A. and Bickmore W. A. (2001). The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 10, 211-219. 10.1093/hmg/10.3.211 [DOI] [PubMed] [Google Scholar]
- Brien G. L., Valerio D. G. and Armstrong S. A. (2016). Exploiting the epigenome to control cancer-promoting gene-expression programs. Cancer Cell 29, 464-476. 10.1016/j.ccell.2016.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Gilbert L. A., Cimini B. A., Schnitzbauer J., Zhang W., Li G.-W., Park J., Blackburn E. H., Weissman J. S., Qi L. S. et al. (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479-1491. 10.1016/j.cell.2013.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Grunwald D., Sardo L., Galli A., Plisov S., Nikolaitchik O. A., Chen D., Lockett S., Larson D. R., Pathak V. K. et al. (2014). Cytoplasmic HIV-1 RNA is mainly transported by diffusion in the presence or absence of Gag protein. Proc. Natl. Acad. Sci. USA 111, E5205-E5213. 10.1073/pnas.1413169111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Rahman S. A., Nikolaitchik O. A., Grunwald D., Sardo L., Burdick R. C., Plisov S., Liang E., Tai S., Pathak V. K. et al. (2016). HIV-1 RNA genome dimerizes on the plasma membrane in the presence of Gag protein. Proc. Natl. Acad. Sci. USA 113, E201-E208. 10.1073/pnas.1518572113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claude A. (1975). The coming of age of the cell. Science 189, 433-435. 10.1126/science.1098146 [DOI] [PubMed] [Google Scholar]
- Clubb B. H. and Locke M. (1998). Peripheral nuclear matrix actin forms perinuclear shells. J. Cell. Biochem. 70, 240-251. [DOI] [PubMed] [Google Scholar]
- Cook P. R. (1999). The organization of replication and transcription. Science 284, 1790-1795. 10.1126/science.284.5421.1790 [DOI] [PubMed] [Google Scholar]
- Cremer T., Cremer M., Dietzel S., Müller S., Solovei I. and Fakan S. (2006). Chromosome territories--a functional nuclear landscape. Curr. Opin. Cell Biol. 18, 307-316. 10.1016/j.ceb.2006.04.007 [DOI] [PubMed] [Google Scholar]
- Croft J. A., Bridger J. M., Boyle S., Perry P., Teague P. and Bickmore W. A. (1999). Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119-1131. 10.1083/jcb.145.6.1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Duve C. and Beaufay H. (1981). A short history of tissue fractionation. J. Cell Biol. 91, 293s-299s. 10.1083/jcb.91.3.293s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J., Marti-Renom M. A. and Mirny L. A. (2013). Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet. 14, 390-403. 10.1038/nrg3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks R. W. and Snaar S. (1999). Dynamics of RNA polymerase II localization during the cell cycle. Histochem. Cell Biol. 111, 405-410. 10.1007/s004180050374 [DOI] [PubMed] [Google Scholar]
- Dixon J. R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J. S. and Ren B. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376-380. 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley R., Van Duyne R., Coley W., Guendel I., Dadgar S., Kehn-Hall K. and Kashanchi F. (2010). Chromatin dynamics associated with HIV-1 Tat-activated transcription. Biochim. Biophys. Acta 1799, 275-285. 10.1016/j.bbagrm.2009.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faro-Trindade I. and Cook P. R. (2006). Transcription factories: structures conserved during differentiation and evolution. Biochem. Soc. Trans. 34, 1133-1137. 10.1042/BST0341133 [DOI] [PubMed] [Google Scholar]
- Farrell R. E. (2010). Analysis of nuclear RNA. In RNA Methodologies Laboratory Guide for Isolation and Characterization, chapter 16, pp. 349 Amsterdam; Boston: Elsevier/Academic Press. [Google Scholar]
- Fedorova E. and Zink D. (2008). Nuclear architecture and gene regulation. Biochim. Biophys. Acta 1783, 2174-2184. 10.1016/j.bbamcr.2008.07.018 [DOI] [PubMed] [Google Scholar]
- Ferraro A. (2016). Altered primary chromatin structures and their implications in cancer development. Cell. Oncol. (Dordr) 39, 195-210. 10.1007/s13402-016-0276-6 [DOI] [PubMed] [Google Scholar]
- Gómez-Díaz E. and Corces V. G. (2014). Architectural proteins: regulators of 3D genome organization in cell fate. Trends Cell Biol. 24, 703-711. 10.1016/j.tcb.2014.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg M. E. and Bender T. P. (2007). Identification of newly transcribed RNA. Curr. Protoc. Mol. Biol. 78, 4.10.1–4.10.12. 10.1002/0471142727.mb0410s78 [DOI] [PubMed] [Google Scholar]
- Guelen L., Pagie L., Brasset E., Meuleman W., Faza M. B., Talhout W., Eussen B. H., de Klein A., Wessels L., de Laat W. et al. (2008). Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature 453, 948-951. 10.1038/nature06947 [DOI] [PubMed] [Google Scholar]
- Hakre S., Chavez L., Shirakawa K. and Verdin E. (2011). Epigenetic regulation of HIV latency. Curr. Opin. HIV AIDS 6, 19-24. 10.1097/COH.0b013e3283412384 [DOI] [PubMed] [Google Scholar]
- Harr J. C., Gonzalez-Sandoval A. and Gasser S. M. (2016). Histones and histone modifications in perinuclear chromatin anchoring: from yeast to man. EMBO Rep. 17, 139-155. 10.15252/embr.201541809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz E. (1928). Das Heterochromatin der Moose. Jahrb Wiss Botanik 69, 762-818. [Google Scholar]
- Hoffman W. A. (2009). Cell and molecular biology of nuclear actin. International review of cell and molecular biology . Elsevier Inc. 273, 219-263. 10.1016/S1937-6448(08)01806-6 [DOI] [PubMed] [Google Scholar]
- Holaska J. M. and Wilson K. L. (2007). An emerin “proteome”: purification of distinct emerin-containing complexes from HeLa cells suggests molecular basis for diverse roles including gene regulation, mRNA splicing, signaling, mechanosensing, and nuclear architecture. Biochemistry 46, 8897-8908. 10.1021/bi602636m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holaska J. M., Kowalski A. K. and Wilson K. L. (2004). Emerin caps the pointed end of actin filaments: evidence for an actin cortical network at the nuclear inner membrane. PLoS Biol. 2, e231 10.1371/journal.pbio.0020231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T. and Allis C. D. (2001). Translating the histone code. Science 293, 1074-1080. 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- Jouvenet N., Bieniasz P. D. and Simon S. M. (2008). Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 454, 236-240. 10.1038/nature06998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N., Simon S. M. and Bieniasz P. D. (2009). Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 106, 19114-19119. 10.1073/pnas.0907364106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karn J. (2011). The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 6, 4-11. 10.1097/COH.0b013e328340ffbb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H., Hayashi-Takanaka Y., Stasevich T. J. and Sato Y. (2015). Visualizing posttranslational and epigenetic modifications of endogenous proteins in vivo. Histochem. Cell Biol. 144, 101-109. 10.1007/s00418-015-1344-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klase Z., Yedavalli V. S. R. K., Houzet L., Perkins M., Maldarelli F., Brenchley J., Strebel K., Liu P. and Jeang K.-T. (2014). Activation of HIV-1 from latent infection via synergy of RUNX1 inhibitor Ro5-3335 and SAHA. PLoS Pathog. 10, e1003997 10.1371/journal.ppat.1003997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein T., Löschberger A., Proppert S., Wolter S., van de Linde S. and Sauer M. (2011). Live-cell dSTORM with SNAP-tag fusion proteins. Nat. Methods 8, 7-9. 10.1038/nmeth0111-7b [DOI] [PubMed] [Google Scholar]
- Komarnitsky P., Cho E. J. and Buratowski S. (2000). Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452-2460. 10.1101/gad.824700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosak S. T., Skok J. A., Medina K. L., Riblet R., Le Beau M. M., Fisher A. G. and Singh H. (2002). Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science 296, 158-162. 10.1126/science.1068768 [DOI] [PubMed] [Google Scholar]
- Lakadamyali M. and Cosma M. P. (2015). Advanced microscopy methods for visualizing chromatin structure. FEBS Lett. 589, 3023-3030. 10.1016/j.febslet.2015.04.012 [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E., van Berkum N. L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B. R., Sabo P. J., Dorschner M. O. et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289-293. 10.1126/science.1181369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.-R., Fallahi-Sichani M., Chen J.-Y. and Sorger P. K. (2016). Cyclic immunofluorescence (CycIF), a highly multiplexed method for single-cell imaging. Curr. Protoc. Chem. Biol. 8, 251-264. 10.1002/cpch.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm T., Chen J., Chang C. and Sadowski I. (2007). Induction of chromosomally integrated HIV-1 LTR requires RBF-2 (USF/TFII-I) and Ras/MAPK signaling. Virus Genes 35, 215-223. 10.1007/s11262-007-0109-9 [DOI] [PubMed] [Google Scholar]
- Marban C., Suzanne S., Dequiedt F., de Walque S., Redel L., Van Lint C., Aunis D. and Rohr O. (2007). Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 26, 412-423. 10.1038/sj.emboj.7601516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks P., Rifkind R. A., Richon V. M., Breslow R., Miller T. and Kelly W. K. (2001). Histone deacetylases and cancer: causes and therapies. Nat. Rev. Cancer 1, 194-202. 10.1038/35106079 [DOI] [PubMed] [Google Scholar]
- Marzluff W. F. Jr, Murphy E. C. Jr and Huang R. C. C. (1974). Transcription of the genes for 5S ribosomal RNA and transfer RNA in isolated mouse myeloma cell nuclei. Biochemistry 13, 3689-3696. 10.1021/bi00715a011 [DOI] [PubMed] [Google Scholar]
- Milavetz B. I. and Balakrishnan L. (2015). Viral epigenetics. Methods Mol. Biol. 1238, 569-596. 10.1007/978-1-4939-1804-1_30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi E., Kao S., Yedavalli V. and Strebel K. (2014). CBFbeta enhances de novo protein biosynthesis of its binding partners HIV-1 Vif and RUNX1 and potentiates the Vif-induced degradation of APOBEC3G. J. Virol. 88, 4839-4852. 10.1128/JVI.03359-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortillaro M. J., Blencowe B. J., Wei X., Nakayasu H., Du L., Warren S. L., Sharp P. A. and Berezney R. (1996). A hyperphosphorylated form of the large subunit of RNA polymerase II is associated with splicing complexes and the nuclear matrix. Proc. Natl. Acad. Sci. USA 93, 8253-8257. 10.1073/pnas.93.16.8253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Németh A. and Längst G. (2011). Genome organization in and around the nucleolus. Trends Genet. 27, 149-156. 10.1016/j.tig.2011.01.002 [DOI] [PubMed] [Google Scholar]
- Neugebauer K. M. and Roth M. B. (1997). Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes Dev. 11, 1148-1159. 10.1101/gad.11.9.1148 [DOI] [PubMed] [Google Scholar]
- Osborne C. S., Chakalova L., Brown K. E., Carter D., Horton A., Debrand E., Goyenechea B., Mitchell J. A., Lopes S., Reik W. et al. (2004). Active genes dynamically colocalize to shared sites of ongoing transcription. Nat. Genet. 36, 1065-1071. 10.1038/ng1423 [DOI] [PubMed] [Google Scholar]
- Peric-Hupkes D., Meuleman W., Pagie L., Bruggeman S. W. M., Solovei I., Brugman W., Gräf S., Flicek P., Kerkhoven R. M., van Lohuizen M. et al. (2010). Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol. Cell 38, 603-613. 10.1016/j.molcel.2010.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickersgill H., Kalverda B., de Wit E., Talhout W., Fornerod M. and van Steensel B. (2006). Characterization of the Drosophila melanogaster genome at the nuclear lamina. Nat. Genet. 38, 1005-1014. 10.1038/ng1852 [DOI] [PubMed] [Google Scholar]
- Ricci M. A., Manzo C., García-Parajo M. F., Lakadamyali M. and Cosma M. P. (2015). Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160, 1145-1158. 10.1016/j.cell.2015.01.054 [DOI] [PubMed] [Google Scholar]
- Rice A. P. and Carlotti F. (1990). Mutational analysis of the conserved cysteine-rich region of the human immunodeficiency virus type 1 Tat protein. J. Virol. 64, 1864-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risca V. I., Denny S. K., Straight A. F. and Greenleaf W. J. (2017). Variable chromatin structure revealed by in situ spatially correlated DNA cleavage mapping. Nature 541, 237-241. 10.1038/nature20781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardo L., Hatch S. C., Chen J., Nikolaitchik O., Burdick R. C., Chen D., Westlake C. J., Lockett S., Pathak V. K. and Hu W.-S. (2015). Dynamics of HIV-1 RNA near the plasma membrane during virus assembly. J. Virol. 89, 10832-10840. 10.1128/JVI.01146-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R. and Grosschedl R. (2007). Dynamics and interplay of nuclear architecture, genome organization, and gene expression. Genes Dev. 21, 3027-3043. 10.1101/gad.1604607 [DOI] [PubMed] [Google Scholar]
- Shimi T., Kittisopikul M., Tran J., Goldman A. E., Adam S. A., Zheng Y., Jaqaman K. and Goldman R. D. (2015). Structural organization of nuclear lamins A, C, B1, and B2 revealed by superresolution microscopy. Mol. Biol. Cell 26, 4075-4086. 10.1091/mbc.E15-07-0461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis M., Kooren J. and de Laat W. (2007). An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods 4, 895-901. 10.1038/nmeth1114 [DOI] [PubMed] [Google Scholar]
- Smeets D., Markaki Y., Schmid V. J., Kraus F., Tattermusch A., Cerase A., Sterr M., Fiedler S., Demmerle J., Popken J. et al. (2014). Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenetics Chromatin 7, 8 10.1186/1756-8935-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T. J., Lando D., Basu S., Atkinson L. P., Cao Y., Lee S. F., Leeb M., Wohlfahrt K. J., Boucher W., O'Shaughnessy-Kirwan A. et al. (2017). 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59-64. 10.1038/nature21429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum M. E., Gilbert L. A., Qi L. S., Weissman J. S. and Vale R. D. (2014). A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635-646. 10.1016/j.cell.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte F., Sousae R., Cinquin B., Martin E. W., Krietsch J., Sanchez G., Inman M., Tsang H., Warr M., Passegue E. et al. (2015). Progressive chromatin condensation and H3K9 methylation regulate the differentiation of embryonic and hematopoietic stem cells. Stem Cell Rep. 5, 728-740. 10.1016/j.stemcr.2015.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P., Garber M. E., Fang S.-M., Fischer W. H. and Jones K. A. (1998). A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451-462. 10.1016/S0092-8674(00)80939-3 [DOI] [PubMed] [Google Scholar]
- West M. L. and Corden J. L. (1995). Construction and analysis of yeast RNA polymerase II CTD deletion and substitution mutations. Genetics 140, 1223-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wombacher R., Heidbreder M., van de Linde S., Sheetz M. P., Heilemann M., Cornish V. W. and Sauer M. (2010). Live-cell super-resolution imaging with trimethoprim conjugates. Nat. Methods 7, 717-719. 10.1038/nmeth.1489 [DOI] [PubMed] [Google Scholar]
- Yan Y., Jiang L., Aufderheide K. J., Wright G. A., Terekhov A., Costa L., Qin K., McCleery W. T., Fellenstein J. J., Ustione A. et al. (2014). A microfluidic-enabled mechanical microcompressor for the immobilization of live single- and multi-cellular specimens. Microsc. Microanal. 20, 141-151. 10.1017/S1431927613014037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Herrmann C. H. and Rice A. P. (1996). The human immunodeficiency virus Tat proteins specifically associate with TAK in vivo and require the carboxyl-terminal domain of RNA polymerase II for function. J. Virol. 70, 4576-4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.-M., Tsai S.-C., Wen Y.-D., Fejér G. and Seto E. (2002). Functional domains of histone deacetylase-3. J. Biol. Chem. 277, 9447-9454. 10.1074/jbc.M105993200 [DOI] [PubMed] [Google Scholar]
- York A. G., Chandris P., Nogare D. D., Head J., Wawrzusin P., Fischer R. S., Chitnis A. and Shroff H. (2013). Instant super-resolution imaging in live cells and embryos via analog image processing. Nat. Methods 10, 1122-1126. 10.1038/nmeth.2687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Adli M., Zou J. Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P. L. et al. (2013). Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642-654. 10.1016/j.cell.2012.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinskie J. A., Shribak M., Bruist M. F., Aufderheide K. J. and Janetopoulos C. (2015). A mechanical microcompressor for high resolution imaging of motile specimens. Exp. Cell Res. 337, 249-256. 10.1016/j.yexcr.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]