

ABSTRACT
Misfolded endoplasmic reticulum (ER) proteins are dislocated towards the cytosol and degraded by the ubiquitin–proteasome system in a process called ER-associated protein degradation (ERAD). During infection with human cytomegalovirus (HCMV), the viral US2 protein targets HLA class I molecules (HLA-I) for degradation via ERAD to avoid elimination by the immune system. US2-mediated degradation of HLA-I serves as a paradigm of ERAD and has facilitated the identification of TRC8 (also known as RNF139) as an E3 ubiquitin ligase. No specific E2 enzymes had previously been described for cooperation with TRC8. In this study, we used a lentiviral CRISPR/Cas9 library targeting all known human E2 enzymes to assess their involvement in US2-mediated HLA-I downregulation. We identified multiple E2 enzymes involved in this process, of which UBE2G2 was crucial for the degradation of various immunoreceptors. UBE2J2, on the other hand, counteracted US2-induced ERAD by downregulating TRC8 expression. These findings indicate the complexity of cellular quality control mechanisms, which are elegantly exploited by HCMV to elude the immune system.
KEY WORDS: ERAD, ER-associated protein degradation, E2, Ubiquitin, US2, Cytomegalovirus
Summary: Multiple ubiquitin E2 enzymes involved in cytomegalovirus US2-mediated ERAD of immunoreceptors are identified. UBE2G2 is required for ERAD, whereas UBE2J2 counteracts it by downregulating the crucial TRC8 ubiquitin ligase.
INTRODUCTION
Human cytomegalovirus (HCMV) is a member of the β-herpesviridae and carries the largest double-stranded (ds)DNA genome of the human herpesviruses family (McGeoch et al., 2006). HCMV is a common virus with a seroprevalence of 40–100%, depending on the socioeconomic status of the host population. Primary infection of healthy individuals usually is asymptomatic, but in pregnant women and immunocompromised patients, HCMV infection can cause serious disease (Griffiths et al., 2015). HCMV encodes multiple immunomodulatory proteins that facilitate interference with the immune system of the host, thereby facilitating HCMV to establish a lifelong infection (Dunn et al., 2003; Hansen et al., 2010; Mocarski, 2002).
The HLA-I antigen presentation pathway is a major target for the immune-evasive strategies of HCMV, resulting in its effective elusion from CD8-positive cytotoxic T cells (Noriega et al., 2012b; Schuren et al., 2016). At least five unique short (US) regions in the HCMV genome are known to encode proteins that specifically interfere with the expression of HLA-I molecules (van de Weijer et al., 2015). US3 retains newly synthesized HLA-I proteins in the ER and blocks tapasin-dependent peptide loading (Jones et al., 1996; Noriega et al., 2012a; Park et al., 2004). US6 interacts with the transporter associated with antigen processing (TAP) complex and induces conformational changes of TAP that prevent ATP binding, thereby inhibiting TAP-mediated peptide translocation into the ER (Ahn et al., 1997; Hengel et al., 1997; Hewitt et al., 2001; Lehner et al., 1997). US10 specifically targets HLA-G molecules for degradation (Park et al., 2010). US2 and US11 are type 1 transmembrane ER glycoproteins that cause retrograde transport, or dislocation, of newly synthesized HLA-I heavy chains from the ER into the cytosol for proteasomal degradation (Oresic et al., 2009; Wiertz et al., 1996a,b).
As a result of the above process, US2 and US11 hijack the cellular protein quality control pathway in the ER that recognizes misfolded proteins and targets them for degradation via the ubiquitin–proteasome system. This reaction is referred to as ER-associated protein degradation (ERAD). At the center of this process are multiprotein complexes that combine the various functions essential to this reaction, namely substrate recognition, dislocation, ubiquitylation and degradation (Christianson et al., 2011; Olzmann et al., 2013; Preston and Brodsky, 2017; Ye et al., 2001, 2004). Although US2 and US11 both target HLA-I for degradation, they utilize distinct protein complexes. US11 uses derlin-1 and the E3 ubiquitin ligase TMEM129 in cooperation with the E2 ubiquitin-conjugating enzymes UBE2J2 and UBE2K to dislocate HLA-I (Flierman et al., 2006; Lilley and Ploegh, 2004; van de Weijer et al., 2014; van den Boomen et al., 2014). US2, however, does not depend on these proteins but usurps the E3 ubiquitin ligase TRC8 (also known as RNF139) to mediate HLA-I downregulation (Stagg et al., 2009). On the cytosolic side, both US2 and US11 rely on the ATPase p97/VCP to shuttle HLA-I to the proteasome for degradation (Soetandyo and Ye, 2010; Ye et al., 2005). Besides HLA-I, US2 induces downregulation of multiple immunoreceptors to modulate cellular migration and immune signaling, whereas US11-mediated degradation is restricted to HLA-I (Hsu et al., 2015).
In US2-mediated degradation of HLA-I, the function of TRC8 as E3 ligase is well documented (Hsu et al., 2015; Stagg et al., 2009). However, no specific E2 ubiquitin-conjugating enzymes have been implicated in this process. Here, we constructed a lentiviral CRISPR/Cas9-based library targeting all known human E2 enzymes and used this resource to screen for E2 enzymes that regulate US2-mediated HLA-I downregulation. We identify UBE2G2 as an essential E2 enzyme for this process. Upon UBE2G2 depletion, HLA-I molecules are detected in an ER-resident US2- and TRC8-containing complex, possibly because ubiquitylation may be required for extraction of the class I molecules from the ER and their subsequent degradation. Interestingly, our screen also identifies UBE2J2 as a counteracting E2 enzyme, depletion of which further downregulates HLA-I in US2-expressing cells. In line with our findings for HLA-I, the immunoreceptors integrin-α1, -α2 and -α4, the interleukin (IL)12 receptor β1-subunit (IL12R-B1), and thrombomodulin are also degraded by US2 in a UBE2G2-dependent manner, whereas UBE2J2 counteracts this effect. In conclusion, we show that the E2 ubiquitin-conjugating enzymes UBE2G2 and UBE2J2 are broadly involved in regulating the downregulation of immunoreceptors targeted by HCMV US2.
RESULTS
A CRISPR/Cas9 library screen identifies the E2 ubiquitin-conjugating enzyme UBE2G2 to be essential for US2-mediated HLA-I downregulation
To identify E2 ubiquitin-conjugating enzymes required for US2-mediated degradation of HLA-I molecules, we established a lentiviral CRISPR/Cas9-based library targeting all known human E2 ubiquitin-conjugating enzymes. Approximately three guide RNAs (gRNAs) were used per E2 gene. U937 monocytic cells stably expressing an HLA-A2 molecule with an N-terminal eGFP tag were generated to allow monitoring of HLA-I expression levels by flow cytometry. Upon stable introduction of HCMV US2, the chimeric HLA-I molecules as well as endogenous HLA-I proteins were degraded efficiently (Fig. 1A). These cells were then transduced with the lentiviral E2-targeting CRISPR/Cas9 library, after which eGFP–HLA-A2 expression was evaluated by flow cytometry. Disruption of an E2 gene essential for US2 activity would rescue eGFP–HLA-A2 from degradation and thus increase eGFP–HLA-A2 levels (Fig. 1B).
Fig. 1.

A CRISPR/Cas9 library screen for E2 ubiquitin-conjugating enzymes identifies UBE2G2 and UBE2D3 as essential players in US2-mediated HLA-I downregulation. (A) Downregulation of eGFP–HLA-A2 and endogenous HLA-A3 mediated by US2 in U937 cells expressing eGFP–HLA-A2. (B) Schematic overview of the CRISPR/Cas9 library screen to identify E2 ubiquitin-conjugating enzymes essential for US2-mediated HLA-I downregulation. U937 cells are transduced with eGFP–HLA-A2 and subsequently transduced with an HCMV US2 expression vector. As a result, cells display low total eGFP–HLA-A2 expression levels, which can be monitored by means of the eGFP tag. Subsequently, cells are lentivirally transduced with CRISPR/Cas9 constructs targeting individual E2 ubiquitin-conjugating enzymes and selected to purity using puromycin. Cells are analyzed by flow cytometry at 10 dpi to assess total eGFP–HLA-A2 levels. (C) Quantification of the percentage eGFP–HLA-A2-positive US2-expressing cells upon transduction with CRISPR/Cas9 constructs targeting individual E2 ubiquitin-conjugating enzymes. gRNAs targeting UBE2D3, UBE2G2 and UBE2J2 are indicated. (D) CRISPR/Cas9-mediated knockout of E2 ubiquitin-conjugating enzymes UBE2G2 (gRNA #1) and UBE2D3 (gRNA #3) rescues expression of chimeric eGFP–HLA-A2 and endogenous HLA-A3 from US2-expressing cells. gRNAs targeting UBE2G1 (gRNA #1) were used as a negative control. eGFP–HLA-A2 and endogenous HLA-A3 surface levels were assessed at 10 dpi by flow cytometry. (E) Reconstitution of UBE2G2 or UBE2D3 expression in UBE2G2- and UBE2D3-knockout cells, respectively, rescues US2-mediated downregulation of HLA- I. The wild-type E2 (UBE2G2 wt or UBE2D3 wt) or a catalytically inactive E2 (UBE2G2 ci or UBE2D3 ci) was introduced in the corresponding polyclonal E2 knockout cells from C, after which flow cytometry analysis was performed to assess total eGFP–HLA-A2 levels and endogenous HLA-A3 surface levels at 7 dpi. (F) The expression of wild-type and catalytically inactive E2 ubiquitin-conjugating enzymes used in D was assessed via immunoblotting (IB). Transferrin receptor (TfR) was used as a loading control.
CRISPR gRNAs targeting UBE2D3 and UBE2G2 induced rescue of eGFP–HLA-A2 expression in US2-expressing cells (Fig. 1C). Expression of gRNAs targeting UBE2G2 induced the strongest rescue of eGFP–HLA-A2. The anti-UBE2G2 and anti-UBE2D3 gRNAs also increased the levels of endogenous HLA-A3 in these US2-expressing cells (Fig. 1D). By contrast, targeting the UBE2G2 homolog UBE2G1 with CRISPR gRNAs did not affect eGFP–HLA-A2 expression. To validate the involvement of UBE2G2 and UBE2D3, their expression was restored by introduction of gRNA-resistant E2 cDNA constructs into the gRNA-expressing cells (Fig. 1E). For both UBE2G2 and UBE2D3, reconstitution of protein expression restored US2-mediated HLA-A2–eGFP downregulation. For UBE2G2, a similar pattern was also observed for endogenous HLA-A3. On the other hand, introduction of catalytically inactive E2 mutants, in which the active cysteine residue was replaced with a serine, did not result in restoration of US2-mediated HLA-I downregulation. Expression of the HA-tagged wild-type (wt) and catalytically inactive (ci) E2 enzymes was confirmed by immunoblotting (Fig. 1F).
These results indicate that UBE2G2 and UBE2D3 affect US2-mediated degradation of HLA-I. Compared to UBE2G2, the effects of UBE2D3 depletion and reconstitution were limited, especially for the endogenous HLA-A3 molecule. Because UBE2G2 has a stronger effect on HLA-I rescue, mechanistic experiments in this study will focus on this E2 enzyme.
The E2 ubiquitin-conjugating enzyme UBE2J2 counteracts US2-mediated HLA-I downregulation
In contrast to UBE2G2 and UBE2D3, we noticed that targeting UBE2J2 with CRISPR gRNAs resulted in a further downregulation of eGFP–HLA-A2 (Fig. 1C) and endogenous HLA-A3 expression (Fig. 2A). The window to assess reduced eGFP–HLA-A2 levels was low, as US2 downregulates the chimeric eGFP–HLA-A2 molecule to near background levels (Fig. 1A). Downregulation of the endogenous HLA-A3 by US2 was less potent (Fig. 1A), allowing us to study the effect of UBE2J2 depletion in more detail. Indeed, gRNAs targeting UBE2J2 further decreased HLA-A3 levels (Fig. 2B). Depletion of the UBE2J2 homolog UBE2J1 did not affect HLA-A3 expression (Fig. 2B). Reconstitution of wt UBE2J2 expression abolished the enhanced downregulation of HLA-A3 molecules (Fig. 2C, left panel), showing that the gRNA effect was specific for UBE2J2. Expression of the exogenous UBE2J2 even resulted in slightly less downregulation of HLA-A3 by US2 (Fig. 2C, left panel), which could be due to overexpression of the cDNA. In US2-expressing cells with intact endogenous UBE2J2, introduction of this UBE2J2 cDNA increased HLA-A3 expression in a similar manner (Fig. 2D, left panel). These findings indicate that UBE2J2 counteracts US2-mediated downregulation of HLA-I.
Fig. 2.

UBE2J2 counteracts US2-mediated HLA-I downregulation. (A) Quantification of the percentage of cells showing enhanced downregulation of HLA-A3 upon transduction with CRISPR/Cas9 constructs targeting individual E2 ubiquitin-conjugating enzymes. gRNAs targeting UBE2J2 are indicated. (B) CRISPR/Cas9-mediated knockout of UBE2J2 decreases endogenous HLA-A3 surface expression in US2-expressing cells. The homolog UBE2J1 is shown as a negative control. Endogenous HLA-A3 surface levels were assessed at 10 dpi by flow cytometry. (C) Reconstitution of UBE2J2 expression in UBE2J2-knockout cells rescues HLA-I from enhanced US2-mediated downregulation. Wild-type UBE2J2 (wt), catalytically inactive UBE2J2 (ci), or soluble UBE2J2 (ΔTMD) was introduced in knockout cells from B (gRNA #1), after which flow cytometry analysis was performed to assess endogenous HLA-A3 surface levels at 7 dpi. (D) Ectopic expression of UBE2J2 increases HLA-A3 expression. Wild-type UBE2J2, catalytically inactive UBE2J2 or soluble UBE2J2 was ectopically expressed in cells similar to those from B, but lacking a gRNA targeting UBE2J2. Endogenous HLA-A3 surface levels were assessed using flow cytometry. (E) Expression of wild-type, catalytically inactive and ΔTMD UBE2J2, as used in C and D, were assessed by immunoblotting. Transferrin receptor (TfR) was used as a loading control. (F) Lysates of U937 cells expressing either wild-type UBE2J2 (wt), catalytically inactive UBE2J2 (ci), or soluble UBE2J2 (Δ) were prepared and immunoblotted for the indicated proteins.
Reconstitution with a catalytically inactive UBE2J2 (ci) or a UBE2J2 mutant lacking its transmembrane domain (ΔTMD) did not counteract the enhanced HLA-I downregulation (Fig. 2C, middle and right panels). These results indicated that catalytic activity is required for this phenotype. Expression of the catalytically inactive mutant in the absence of gRNAs further stimulated US2-mediated HLA-A3 downregulation (Fig. 2D, middle panel), mimicking the phenotype of CRISPR/Cas9-mediated UBE2J2 disruption. This indicates that the catalytically inactive UBE2J2 acts as a dominant-negative mutant. Expression of the soluble mutant (ΔTMD) did not affect HLA-A3 expression levels (Fig. 2D, right panel). The expression levels of wild-type, catalytically inactive and soluble UBE2J2 were confirmed by immunoblotting (Fig. 2E). The effect of UBE2J2 counteracting US2-mediated HLA-I degradation was not caused by directly affecting US2 expression, as introduction of wild-type, catalytically inactive or soluble UBE2J2 did not alter US2 levels (Fig. 2F). Taken together, our data indicate that UBE2J2 counteracts US2-mediated downregulation of HLA-I through a mechanism that relies on its catalytic activity.
Depletion of UBE2G2 causes accumulation of HLA-I in the US2–TRC8 complex
Next, we investigated the effect of UBE2G2 and UBE2J2 depletion on the composition of the US2 ERAD complex. To this end, clonal UBE2G2- and UBE2J2-null cell lines were established (Fig. S1). Removal of UBE2G2 in these cells caused a growth defect, but was not lethal. HLA-I levels were increased in UBE2G2-null cells as compared to control cells (Fig. 3A, lane 1 and 2 versus lane 5), which was in agreement with the flow cytometry results from Fig. 1C. In addition, US2 protein levels were increased, likely due to the stabilization of the US2 ERAD complex in the absence of UBE2G2.
Fig. 3.

Loss of UBE2G2 locks the US2–TRC8 complex in a dislocation-incompetent state. (A) Two independent knockout clones of either UBE2G2 or UBE2J2 were established in U937 cells co-expressing eGFP–HLA-A2 and HA–US2. These clones were subjected to immunoblot analysis to assess expression levels of the indicated proteins. (B) ST2-HA-tagged US2 was immunoprecipitated (IP) by using StrepTactin beads from 1.0% digitonin lysates of cells in which TRC8 (lane 1–3), UBE2G2 (lane 4–6) or UBE2J2 (lane 7–9) were knocked out. US2-expressing U937 eGFP–HLA-A2 cells without knockout (lane 9) and cells without US2 (lane 10) were used as controls. Immunoprecipitated complexes were eluted using d-Desthiobiotin, after which immunoblot analysis was performed to detect the indicated proteins.
In line with our previous results, immunoblot analysis for UBE2J2-null cells revealed that HLA-I levels were further decreased as compared to control cells (Fig. 3A, lanes 3 and 4 versus lane 5). US2 protein levels were comparable to control conditions (lane 3) or slightly elevated (lane 4), suggesting that the strong increase in HLA-I degradation upon UBE2J2 knockout was not caused by increased US2 levels.
To investigate the effects of UBE2G2 and UBE2J2 depletion on the composition of the US2 ERAD complex, US2 was immunoprecipitated from cell lysates prepared by using digitonin, which preserves most membrane protein complexes. The E3 ubiquitin ligase TRC8 is part of this ERAD complex and is essential for HLA-I downregulation by US2 (Stagg et al., 2009). Knocking out TRC8 rescues US2-associated HLA-I (Fig. 3B, lanes 1–3 versus 10). In UBE2G2-null cells, HLA-I co-precipitated with US2 as well (lanes 4–6). In addition, increased amounts of TRC8 were co-precipitated from UBE2G2-null cells as compared to control cells (lane 10). Overall US2 expression varied slightly per condition and seemed higher in UBE2G2-null cells. Our data suggest that the US2–TRC8 ERAD complex is stabilized in the absence of UBE2G2, leading to the accumulation of HLA-I prior to the dislocation of US2–TRC8 from the ER membrane.
UBE2J2 counteracts HLA-I degradation by US2 via downregulation of TRC8
Upon UBE2J2 knockout (Fig. 3B, lanes 7–9), HLA-I was not detectable in the US2–TRC8 complex, consistent with the observation that HLA-I degradation is enhanced in these cells. Intriguingly, the amount of TRC8 present in the complex was increased upon UBE2J2 knockout (Fig. 3B, lanes 7–9) compared to control cells (lane 10), while US2 levels were elevated only slightly. This suggests that depletion of UBE2J2 may increase TRC8 levels. Because TRC8 is the rate-limiting factor for US2-mediated HLA-I degradation (Van den Boomen and Lehner, 2015), this TRC8 increase would explain the enhanced HLA-I downregulation. To investigate this, we assessed TRC8 levels in UBE2J2-knockout cells by direct quantitative immunoprecipitation of TRC8 (Fig. 4A), as the ligase could not be detected in lysate directly. Indeed, in US2-expressing cells, TRC8 expression levels were increased upon UBE2J2 knockout compared to what was observed in the control conditions (Fig. 4A). A minor increase in TRC8 expression could also be observed in cells without US2 (Fig. S4A, lane 3 versus 4).
Fig. 4.

Impaired UBE2J2 function enhances HLA-I downregulation by US2 via upregulation of TRC8. (A) US2-expressing TRC8-knockout cells were transduced with TRC8–Flag–ST2 or a control vector. To these cells, UBE2J2-targeting CRISPR gRNAs or empty vector were added. TRC8 levels were assessed by direct immunoprecipitation (IP). Upon UBE2J2 knockout, the expression of TRC8 increases. (B) US2-expressing TRC8-knockout cells were transduced with TRC8–Flag–ST2 or a control vector. To these cells, cDNA of wild-type (wt) or catalytically inactive (ci) HA-tagged UBE2J2 or a control vector were added. TRC8 levels were assessed by direct immunoprecipitation. Upon expression of catalytically inactive UBE2J2, the expression of TRC8 increases compared to that seen in control cells without HA–UBE2J2 or in cells with wild-type HA–UBE2J2. (C) Catalytically inactive UBE2J2 increases TRC8 levels in the US2 ERAD complex. A control vector, a vector expressing wild-type UBE2J2 (wt) or catalytically inactive UBE2J2 (ci) was transduced in U937 cells expressing eGFP–HLA-A2 and ST2–HA–US2. After G418 selection, cells were lysed in 1.0% digitonin lysis buffer, after which ST2–HA–US2 was immunoprecipitated by using StrepTactin beads. Immunoprecipitated complexes were eluted using d-Desthiobiotin, after which immunoblot analysis was performed for the proteins indicated. A short and a long exposure of the anti-HA immunoblot are shown.
We observed a similar increase in TRC8 expression in US2 cells expressing catalytically inactive UBE2J2 (Fig. 4B). These cells show stronger HLA-I downregulation compared to cells expressing US2 alone (Fig. 2C). Again, TRC8 expression was also elevated in cells without US2 (Fig. S4B, lane 4 versus 2), indicating that the effect of UBE2J2 on TRC8 is independent of US2. Expression levels of US2 (Fig. S4A,B) and UBE2G2 (Fig. 4C; Fig. S4A,B) were not clearly affected by expression of catalytically inactive or wild-type UBE2J2 constructs.
Catalytically inactive UBE2J2 not only raised total TRC8 expression, but also resulted in increased TRC8 levels present in the ERAD complex immunoprecipitated via US2 (Fig. 4C, lane 12 versus 10). By contrast, exogenous expression of wild-type UBE2J2 (lane 11 versus 10) caused decreased TRC8 association. Thus, UBE2J2 depletion or the expression of catalytically inactive UBE2J2 increases TRC8 expression levels, which in turn enhances US2-mediated HLA-I downregulation.
Degradation of immunoreceptors by US2 is dependent on UBE2G2 and counteracted by UBE2J2
HCMV US2-mediated protein degradation is not limited to HLA-I; US2 also modulates the expression of various other cell surface receptors, including integrin-α2 (ITGA2), integrin-α4 (ITGA4) and thrombomodulin (THBD) (Hsu et al., 2015). Similar to HLA-I, the degradation of these cell surface receptors is catalyzed by the E3 ubiquitin ligase TRC8 (Hsu et al., 2015). We tested whether the degradation of these cell surface receptors induced by US2 is also dependent on UBE2G2, and whether their downregulation can be counteracted by UBE2J2. Indeed, TRC8 and UBE2G2 depletion restored the surface expression of these receptors in US2-expressing U937 and THP-1 cells (Fig. 5A,B), whereas UBE2J2 depletion further decreased the expression compared to that in control US2-expressing cells (Fig. 5A,B). When looking at HLA-I downregulation, we noticed a difference between the two cell lines used. In contrast to U937 cells, which express the HLA-A3 allele, the HLA-A2-expressing THP-1 cells displayed only partial rescue of this HLA-A protein upon introduction of anti-UBE2G2 gRNAs. Other US2 substrates, including integrin α1 (ITGA1), ITGA2, ITGA4, the IL12 receptor β1-subunit (IL12R-B1), and thrombomodulin were upregulated more potently in the THP-1 cells (Fig. 5B,C). The elevated expression of HLA class I, ITGA2, ITGA4 and THBD upon UBE2G2 knockout was confirmed by immunoblotting (Fig. 5D). Thus, UBE2G2 and UBE2J2 not only regulate US2-induced downregulation of HLA-I, but also that of other cell surface receptors, including several integrins and thrombomodulin.
Fig. 5.

UBE2G2 and UBE2J2 regulate US2-induced immunoreceptor downregulation. (A,B) U937 (A) or THP-1 (B) cells expressing US2 were lentivirally transduced with a CRISPR/Cas9 vector targeting TRC8 (gRNA #1), UBE2G2 (gRNA #1) or UBE2J2 (gRNA #1). At 2 days after infection, gRNA-expressing cells were selected using puromycin. Cell surface expression of integrin-α4 (ITGA4), thrombomodulin (THBD) and HLA-I (HLA-A2 for THP-1 cells, HLA-A3 for U937 cells) was assessed by flow cytometry at 10 days (U937) or 15 days (THP-1) post infection. (C) THP-1 cells expressing US2 were lentivirally transduced with two CRISPR gRNAs (#4 and #5) targeting UBE2G2. At 2 days after infection, gRNA-expressing cells were selected using puromycin, and expression of integrin α1 (ITGA1), integrin α2 (ITGA2) or IL12 receptor β1-subunit (IL12R-B1) was assessed by flow cytometry at 7 dpi. (D) Lysates from cells used in B were prepared and subjected to immunoblotting analysis for total protein expression levels of ITGA2, ITGA4, thrombomodulin, and HLA-I (HCA2). US11-expressing cells were included as a control. Actin was used as a loading control. The asterisk marks an unspecific band.
DISCUSSION
HCMV US2 facilitates proteasomal degradation of HLA-I and various other immunoreceptors through a pathway that is poorly characterized (Hsu et al., 2015; Stagg et al., 2009; Wiertz et al., 1996b). Here, we identify multiple E2 ubiquitin-conjugating enzymes involved in US2-mediated immunoreceptor downregulation. Knocking out UBE2G2 in US2-expressing cells by using the CRISPR/Cas9 system rescues HLA-I expression. Loss of UBE2G2 stabilizes HLA-I in a complex that also contains US2 and TRC8, suggesting that UBE2G2 is required for the dislocation to occur. We were unable to show association of UBE2G2 with the US2–TRC8 complex, possibly due to the weak and transient nature of the interaction between E2 ubiquitin-conjugating enzymes and the E3 ubiquitin ligases in vivo, as has been reported for other E2–E3 interactions (Duncan et al., 2010; Kleiger et al., 2009; Yin et al., 2009).
In our CRISPR/Cas9 screen, we noticed that the knockout of a second E2 enzyme, UBE2D3, also rescued HLA-I expression. UDE2D3 appears to be an essential gene for U937 cell survival, as we were not able to establish clonal knockout cell lines. UBE2D3 depletion only moderately counteracted US2-mediated HLA-I downregulation in U937 cells, although this low rescue may have been impacted by the lethality of knocking out this E2 enzyme.
Our studies consistently demonstrated a partial rescue of HLA-A2 expression upon depletion of UBE2G2; HLA-A3, as well as other US2 substrates were rescued more strongly. This difference was most apparent when HLA-A-specific antibodies were used in cell lines that carry a single HLA-A locus product. The weaker rescue phenotype observed for HLA-A2 suggests that another E2 enzyme, possibly UBE2D3, may contribute to the downregulation of this HLA protein.
Previous functional studies have associated UBE2G2 with several ER-resident E3 ubiquitin ligases. For example, UBE2G2 interacts with HRD1 (also known as SYVN1) (Kikkert et al., 2004) and TEB4 (also known as MARCH6) (Hassink et al., 2005) to promote K48-linked polyubiquitylation in vitro. The E3 ligase gp78 (also known as AMFR) also cooperates with UBE2G2 (Liu et al., 2014). A dedicated UBE2G2-binding region (G2BR) has been identified for gp78 that increases its affinity for UBE2G2 by ∼50-fold. The strong interaction between gp78 and UBE2G2 allows for efficient ubiquitylation and degradation of the CD3δ subunit (Das et al., 2009; Chen et al., 2006). Both gp78 and HRD1 act with UBE2G2 to catalyze cholera toxin dislocation (Bernardi et al., 2010). UBE2G2 has also been proposed to be involved in the turnover of HMGCR (Miao et al., 2010). HMGCR degradation is facilitated by the E3 ligases gp78 (Jo et al., 2011, 2013) and TRC8 (Chen et al., 2006; Jo et al., 2011) as well as the UBE2G2-recruiting protein AUP1 (Jo et al., 2013). Although this suggests that UBE2G2 cooperates with the E3s TRC8 and gp78 to facilitate degradation of HMGCR, a direct role for UBE2G2 has not been established in these studies. Our data show that UBE2G2 is a crucial player in US2-induced dislocation of HLA-I as well as degradation of other immunoreceptors, all of which also require the E3 ubiquitin ligase TRC8 (Stagg et al., 2009). We therefore propose that TRC8 and UBE2G2 indeed cooperate in ubiquitylating ERAD substrates.
Surprisingly, UBE2J2 is able to counteract US2-induced HLA-I degradation. When knocking out UBE2J2, we observed an unexpected further downregulation of eGFP–HLA-A2 as well as endogenous HLA-A3. This phenotype is reversed upon reconstitution of UBE2J2 expression by means of cDNA expression. In contrast to UBE2G2, little is known about the tail-anchored E2 ubiquitin-conjugating enzyme UBE2J2 and its role in protein degradation. Its murine homolog plays a role in ERAD of unassembled T-cell receptor subunits (Tiwari and Weissman, 2001).
Interestingly, UBE2J2 has previously been described to be involved in MHC class I downregulation in the context of herpesviral infection. During infection with γ-herpesvirus 68 (γ-HV68), UBE2J2 is recruited by the viral murine K3 (mK3) E3 ubiquitin ligase and facilitates the degradation of murine MHC class I molecules (Wang et al., 2009). The HCMV protein US11 also cooperates with UBE2J2 to downregulate HLA-I. Similar to US2, US11 causes accelerated ER-associated degradation of HLA-I, but the proteins do so through different degradation pathways. US11 acts in concert with the cellular E3 ubiquitin ligase TMEM129 and recruits UBE2J2 to ubiquitylate and dislocate HLA-I from the ER membrane (van de Weijer et al., 2014; van den Boomen et al., 2014). How UBE2J2 balances HLA-I downregulation during HCMV infection, in which both the UBE2J2-dependent US11 as well as the UBE2J2-counteracted US2 proteins are present, is currently unknown.
The counteracting effect of UBE2J2 on US2-mediated HLA-I degradation may be related to downregulation of the ubiquitin E3 ligase TRC8. We observe increased levels of TRC8 in both clonal UBE2J2-knockout cells and cells expressing catalytically inactive UBE2J2. TRC8 is a crucial player in US2-mediated ERAD, not only for HLA-I degradation (Stagg et al., 2009), but also for degradation of other cell surface receptors, including α- and β-integrins, the NK cell-activating receptor CD112 (also known as nectin-2), thrombomodulin and the IL-12 receptor β1 subunit (Hsu et al., 2015). In the absence of US2, UBE2J2 depletion or expression of a catalytically inactive UBE2J2 also results in increased TRC8 expression levels, indicating that UBE2J2 might play a general role in the turnover of TRC8. We thus propose that the increased downregulation of HLA-I upon UBE2J2 depletion is primarily caused by increased TRC8 levels in these cells (Fig. 6). Expression of a catalytically inactive UBE2J2 mutant also enhances HLA-I downregulation, mimicking the UBE2J2-knockout phenotype. These results suggest that the catalytic activity of UBE2J2 is required for the downregulation of TRC8 and hence for the increased HLA-I downregulation.
Fig. 6.
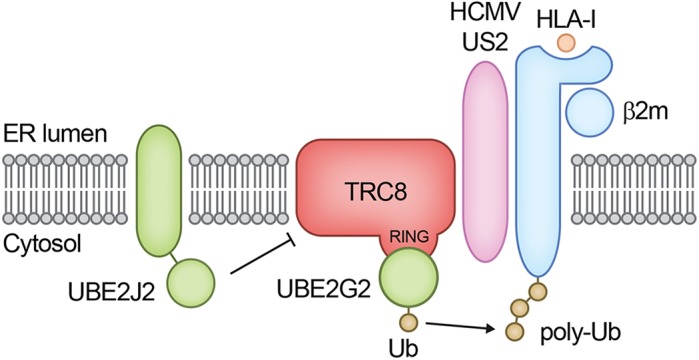
Schematic overview of the ubiquitylation step in US2-mediated HLA-I downregulation. US2 engages β2m-associated HLA-I and directs it to the E3 ubiquitin ligase TRC8. In cooperation with UBE2G2, TRC8 facilitates polyubiquitylation of HLA-I. Next, p97 catalyzes the extraction from the ER membrane into the cytosol for proteasomal degradation. UBE2J2 depletion increases TRC8 expression levels in the presence of US2, and in this way, enhances US2-mediated HLA-I downregulation.
Remarkably, expression of US2 alone was sufficient to induce a dramatic decrease in TRC8 levels. This observation suggests that US2 also induces degradation of TRC8. This reaction is potentially aided by UBE2J2. Why US2 limits TRC8 expression while it relies on this E3 ligase to dispose of HLA-I and other immunoreceptors remains to be determined.
Counteracting mechanisms, such as TRC8 downregulation by UBE2J2 and US2, highlight an interesting regulatory pathway in which ERAD factors themselves influence turnover of the ERAD- and ER stress machinery. A similar phenomenon has been described for HERP (also known as HEY2), a key organizer of ERAD complexes in the ER (Leitman et al., 2014). HERP is upregulated under ER stress, when the unfolded protein response (UPR) is activated. Once stress is relieved, HERP levels are reduced again via ERAD, in this case via UBE2G2- and gp78-mediated ubiquitylation (Yan et al., 2014). IRE1α, a sensor for the unfolded protein response, is also upregulated under conditions of ER stress. This ER-resident transmembrane protein is degraded by an ERAD complex centered around the E3 ubiquitin ligase HRD1, in complex with SEL1L (Sun et al., 2015). For TRC8, no such downregulation has been described to date, although this E3 ligase is known to catalyze auto-ubiquitylation and cause its own downregulation upon sterol abundance (Lee et al., 2010). The existence of negative-feedback loops in the life cycle of ERAD- and ER stress-related factors underlines the complexity of these protein turnover pathways. The present study sheds light on the regulation of HCMV-induced immunoreceptor degradation, as well as a feedback loop within this ERAD pathway, a process that involves a multitude of acting and counteracting E2 ubiquitin-conjugating enzymes.
MATERIALS AND METHODS
Cell culture and lentiviral infection
U937 human monocytic cells and 293T human embryonic kidney cells were obtained from ATCC (American Type Culture Collection) and grown in RPMI medium (Lonza) supplemented with glutamine (Gibco), penicillin/streptomycin (Gibco) and 10% fetal bovine serum (Biowest). For individual transductions using lentiviruses, virus was produced in 24-well plates using standard lentiviral production protocols and third-generation packaging vectors. The supernatant containing virus was harvested 3 days post transfection and stored at −80°C. For lentiviral transductions, 50 µl supernatant containing virus supplemented with 8 µg ml−1 polybrene (Santa Cruz Biotechnology) was used to infect ∼20,000 U937 cells by spin infection at 1000 g for 2 h at 33°C. Fully-supplemented medium was added after centrifugation to reduce polybrene concentration.
Generation of the CRISPR/Cas9-based library and selection of clonal E2-knockout cell lines
CRISPR gRNAs and Cas9 were expressed from a lentiviral vector, as described previously (van de Weijer et al., 2014). In short, this vector carried a CRISPR gRNA under control of a U6 promoter as well as a Cas9 gene that was N-terminally fused to a puromycin-resistance cassette by means of a T2A sequence. The region immediately downstream of the U6 promoter contains a cassette with a BsmBI restriction site on each side to allow cloning of gRNA target sites followed by the gRNA scaffold and a terminator consisting of five T residues. gRNAs targeting all known human E2s were designed using an online CRISPR design tool (http://crispr.mit.edu/). gRNA sequences are listed in Table S1. The genomic target sites for the gRNAs used in the validation studies and/or in the generation of clonal knockout cell lines are listed in Table S2.
U937 cells stably co-expressing eGFP–Myc–HLA-A2 and HA–US2 or 3xST2–HA–US2 [ST2 is Strep(II)] were transduced with this CRISPR/Cas9 system. At 2 days post infection (dpi), transduced cells were selected by using 2 μg ml−1 puromycin and allowed to recover. Additional CRISPR gRNAs targeting UBE2G2 were expressed from a pKLV lentiviral backbone containing a U6 sgRNA as well as a pGK Puro-2A-BFP cassette. This vector was expressed in THP1 cells already containing US2-IRES–mCherry with hygromycin resistance as well as Cas9 under blasticidin resistance.
Cells were single-cell sorted by fluorescence-activated cell sorting (FACSAriaII, BD Biosciences). The knockout status of the clonal cell lines was confirmed by flow cytometry and immunoblotting (UBE2G2) or genomic target site sequencing (UBE2J2). For genomic target site sequencing of UBE2J2, genomic DNA was isolated using the Quick-DNA Miniprep kit (Zymo Research), and the specific region containing the gRNA-target site was amplified by PCR using primers 5′-CCGACGTCTCTATACTGCCC-3′ and 5′-GGCCCTTTCTGTTTTGTTTCC-3′. Subsequently a nested PCR was performed using primers 5′-CCGACGTCTCTATACTGCCC-3′ and 5′-GACACAGCCTGCAAAACGGG-3′ to generate more-specific PCR products. PCR products were prepared according to the manufacturer's guidelines, and subjected to deep-sequencing using the MiSeq Reagent Nano Kit v2 for 500 cycles. Deep sequencing data were analyzed using the Varscan algorithm and are presented in Fig. S2.
Antibodies
Primary antibodies used in our studies were: mouse anti-HLA-I HC HC10 monoclonal antibody (mAb), 1:400; mouse anti-HLA-I HC HCA2 mAb, 1:25; phycoerythrin (PE)-conjugated mouse anti-HLA-A2 mAb (clone BB7.2, no. 558570, BD Pharmingen), 1:20; human anti-HLA-A3 OK2F3 mAb (LUMC, Leiden, the Netherlands), 1:40; mouse anti-TfR H68.4 mAb (no. 13-68xx, Invitrogen), 1:5000; mouse anti-FLAG-M2 mAb (no. F1804, Sigma-Aldrich), 1:10,000; rat anti-HA 3F10 mAb (no. 11867423001, Roche) 1:1000; rabbit anti-UBE2G2 mAb (EPR9248, no. ab174296, Abcam), 1:1000; rabbit anti-TRC8 polyclonal antibody (pAb) (H89, no. sc-68373, Santa Cruz Biotechnology), 1:1000; mouse anti-CD141 (THBD) mAb (clone 1A4, no. 559780, BD Pharmingen), 1:320; mouse anti-CD49b (ITGA2) mAb (clone 12F1, no. 555668, BD Pharmingen), 1:1000; mouse anti-CD49d (ITGA4) mAb (clone 9F10, no. 555502, BD Pharmingen), 1:1000; rabbit anti-ITGA4 mAb (EPR1355Y, no. ab81280, Abcam), 1:1000; mouse anti-IL12 receptor β1 subunit (CD212) mAb (clone 2.4E6, no. 556064, BD Pharmingen), 1:1000; and rabbit anti-ITGA2 mAb (EPR17338, no. ab181548, Abcam), 1:1000.
Secondary antibodies used were: F(ab′)2 goat anti-human-IgG+IgM(H+L) conjugated to PE (no. 109-116-127, Jackson ImmunoResearch) 1:500; F(ab′)2 goat anti-mouse-IgG conjugated to PE (no. R0480, Dako) 1:500; goat anti-mouse-IgG conjugated to AlexaFluor647 (no. A21236, Invitrogen/Thermo) 1:500; goat anti-mouse IgG(H+L) conjugated to horseradish peroxidase (HRP; no. 170-6516, Bio-Rad), 1:10,000; goat anti-rabbit-IgG(H+L) conjugated to HRP (no. 4030-05, Southern Biotech), 1:10,000; mouse anti-rabbit-IgG(L) conjugated to HRP (no. 211-032-171, Jackson Immunoresearch), 1:10,000; goat anti-mouse-IgG(L) conjugated to HRP (no. 115-035-174, Jackson Immunoresearch), 1:10,000; and goat anti-rat-IgG(L) conjugated to HRP (no. 112-035-175, Jackson Immunoresearch), 1:10,000.
Plasmids and cDNAs
Several different lentiviral vectors were used in the present studies. The N-terminally eGFP- and Myc-tagged human HLA-A2 vector present in the lentiviral pHRSincPPT-SGW vector was kindly provided by Dr Paul Lehner and Dr Louise Boyle (University of Cambridge, Cambridge, UK). HCMV US2 was N-terminally tagged with either an HA tag only, or three ST2 tags followed by an HA tag. The original leader was replaced by the human (h)CD8 leader sequence in the tagged US2 constructs. US2 and tagged variants were expressed from a dual promoter lentiviral vector, which also included expression of BlastR-T2A-mAmetrine via the hPGK promotor. For E2 rescue and overexpression experiments, gRNA-resistant HA-tagged E2 ubiquitin-conjugating enzymes were generated and cloned into a dual promoter lentiviral vector, which also included expression of ZeoR-T2A-mAmetrine via the hPGK promotor, as described previously (van de Weijer et al., 2014). UBE2D3 and UBE2G2 were tagged N-terminally; UBE2J2 was tagged C-terminally. Catalytically inactive E2 mutants were generated using PCR-based site-directed mutagenesis with primers carrying the mutation desired: UBE2D3 C85S, UBE2G2 C89S, UBE2J2 C94S. TRC8 was tagged C-terminally with a FLAG and ST2 tag. The resulting TRC8–FLAG–ST2 was still functional (Fig. S3). All constructs were verified by standard Sanger sequencing (Macrogen, The Netherlands).
Flow cytometry
Cells were washed in FACS buffer (PBS containing 0.5% BSA and 0.02% sodium azide) and analyzed on a FACSCanto II (BD Bioscience). Flow cytometry data were analyzed using FlowJo software.
Immunoblotting
Cells were lysed in 1% Triton X-100 lysis buffer (1.0% Triton X-100, 20 mM MES, 100 mM NaCl, 30 mM Tris-HCl pH 7.5) containing 1 mM Pefabloc SC (Roche) and 10 μM leupeptin (Roche). Nuclei and cell debris were pelleted at 12,000 g for 20 min at 4°C. Post-nuclear lysates were denatured in Laemmli sample buffer and incubated at room temperature for 30 min. Proteins were separated by SDS-PAGE and transferred to PVDF membranes (Immobilon-P, Millipore). Membranes were probed with the indicated antibodies. Reactive bands were detected by ECL (Thermo Scientific Pierce), and exposed to Amersham Hyperfilm ECL films (GE Healthcare).
Co-immunoprecipitations
Cells were lysed in digitonin lysis buffer [1% digitonin (Calbiochem), 50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 150 mM NaCl] containing 1 mM Pefabloc SC (Roche) and 10 μM leupeptin (Roche). Lysates were incubated for 90 min at 4°C. Nuclei and cell debris were pelleted 12,000 g for 20 min at 4°C. Post-nuclear supernatants were incubated overnight with StrepTactin beads (GE Healthcare). After four washes in 0.1% digitonin lysis buffer, proteins were eluted in elution buffer (2.5 mM desthiobiotin, 150 mM NaCl, 100 mM Tris-HCl pH 8, 1 mM EDTA) for 30 min on ice. The eluate was separated from the beads using 0.45 μm Spin-X filter column (Corning Costar), and subsequently denatured in Laemmli sample buffer containing DTT. Immunoblotting was performed as described above.
Acknowledgements
We thank Jasper Soppe, Ferdy van Diemen and Luan Nguyen from the Medical Microbiology department at UMC Utrecht for their assistance in deep sequencing.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.L.v.d.W., A.B.C.S., R.J.L., E.J.H.J.W.; Methodology: M.L.v.d.W., A.B.C.S.; Validation: M.L.v.d.W., A.B.C.S., D.J.H.v.d.B., P.J.L., R.J.L., E.J.H.J.W.; Investigation: M.L.v.d.W., A.B.C.S., D.J.H.v.d.B.; Resources: A.M., F.H.J.C.; Writing - original draft: M.L.v.d.W., A.B.C.S.; Writing - review & editing: M.L.v.d.W., A.B.C.S., D.J.H.v.d.B., P.J.L., R.J.L., E.J.W.; Supervision: R.J.L., E.J.H.J.W.; Project administration: R.J.L., E.J.H.J.W.; Funding acquisition: P.J.L., R.J.L., E.J.H.J.W.
Funding
A.B.C.S. is funded by the Graduate Programme of the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Netherlands Organization for Scientific Research; NWO) (project number 022.004.018), and by a Wellcome Trust PRF (Principal Research Fellowship) to P.J.L. with grant no. 101835/Z/13/Z. Deposited in PMC for release after 6 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.206839.supplemental
References
- Ahn K., Gruhler A., Galocha B., Jones T. R., Wiertz E. J. H. J., Ploegh H. L., Peterson P. A., Yang Y. and Früh K. (1997). The ER-luminal domain of the HCMV glycoprotein US6 inhibits peptide translocation by TAP. Immunity 6, 613-621. 10.1016/S1074-7613(00)80349-0 [DOI] [PubMed] [Google Scholar]
- Bernardi K. M., Williams J. M., Kikkert M., van Voorden S., Wiertz E. J., Ye Y. and Tsai B. (2010). The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol. Biol. Cell 21, 140-151. 10.1091/mbc.E09-07-0586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Mariano J., Tsai Y. C., Chan A. H., Cohen M. and Weissman A. M. (2006). The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl. Acad. Sci. USA 103, 341-346. 10.1073/pnas.0506618103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson J. C., Olzmann J. A., Shaler T. A., Sowa M. E., Bennett E. J., Richter C. M., Tyler R. E., Greenblatt E. J., Harper J. W. and Kopito R. R. (2011). Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 14, 93-105. 10.1038/ncb2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R., Mariano J., Tsai Y. C., Kalathur R. C., Kostova Z., Li J., Tarasov S. G., McFeeters R. L., Altieri A. S., Ji X. et al. (2009). Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol. Cell 34, 674-685. 10.1016/j.molcel.2009.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan L. M., Nathan J. A. and Lehner P. J. (2010). Stabilization of an E3 Ligase–E2–ubiquitin complex increases cell surface MHC class I expression. J. Immunol. 184, 6978-6985. 10.4049/jimmunol.0904154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn W., Chou C., Li H., Hai R., Patterson D., Stolc V., Zhu H. and Liu F. (2003). Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 100, 14223-14228. 10.1073/pnas.2334032100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flierman D., Coleman C. S., Pickart C. M., Rapoport T. A. and Chau V. (2006). E2-25K mediates US11-triggered retro-translocation of MHC class I heavy chains in a permeabilized cell system. Proc. Natl. Acad. Sci. USA 103, 11589-11594. 10.1073/pnas.0605215103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths P., Baraniak I. and Reeves M. (2015). The pathogenesis of human cytomegalovirus. J. Pathol. 235, 288-297. 10.1002/path.4437 [DOI] [PubMed] [Google Scholar]
- Hansen S. G., Powers C. J., Richards R., Ventura A. B., Ford J. C., Siess D., Axthelm M. K., Nelson J. A., Jarvis M. A., Picker L. J. et al. (2010). Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 328, 102-106. 10.1126/science.1185350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassink G., Kikkert M., van Voorden S., Lee S.-J., Spaapen R., van Laar T., Coleman C. S., Bartee E., Früh K., Chau V. et al. (2005). TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem. J. 388, 647-655. 10.1042/BJ20041241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel H., Koopmann J.-O., Flohr T., Muranyi W., Goulmy E., Hämmerling G. J., Koszinowski U. H. and Momburg F. (1997). A viral ER-resident glycoprotein inactivates the MHC-encoded peptide transporter. Immunity 6, 623-632. 10.1016/S1074-7613(00)80350-7 [DOI] [PubMed] [Google Scholar]
- Hewitt E. W., Gupta S. S. and Lehner P. J. (2001). The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J. 20, 387-396. 10.1093/emboj/20.3.387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu J.-L., van den Boomen D. J. H., Tomasec P., Weekes M. P., Antrobus R., Stanton R. J., Ruckova E., Sugrue D., Wilkie G. S., Davison A. J. et al. (2015). Plasma membrane profiling defines an expanded class of cell surface proteins selectively targeted for degradation by HCMV US2 in cooperation with UL141. PLoS Pathog. 11, e1004811 10.1371/journal.ppat.1004811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y., Lee P. C. W., Sguigna P. V. and DeBose-Boyd R. A. (2011). Sterol-induced degradation of HMG CoA reductase depends on interplay of two Insigs and two ubiquitin ligases, gp78 and Trc8. Proc. Natl. Acad. Sci. USA 108, 20503-20508. 10.1073/pnas.1112831108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo Y., Hartman I. Z. and DeBose-Boyd R. A. (2013). Ancient ubiquitous protein-1 mediates sterol-induced ubiquitination of 3-hydroxy-3-methylglutaryl CoA reductase in lipid droplet-associated endoplasmic reticulum membranes. Mol. Biol. Cell 24, 169-183. 10.1091/mbc.E12-07-0564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T. R., Wiertz E. J., Sun L., Fish K. N., Nelson J. A. and Ploegh H. L. (1996). Human cytomegalovirus US3 impairs transport and maturation of major histocompatibility complex class I heavy chains. Proc. Natl. Acad. Sci. USA 93, 11327-11333. 10.1073/pnas.93.21.11327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikkert M., Doolman R., Dai M., Avner R., Hassink G., van Voorden S., Thanedar S., Roitelman J., Chau V. and Wiertz E. (2004). Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 279, 3525-3534. 10.1074/jbc.M307453200 [DOI] [PubMed] [Google Scholar]
- Kleiger G., Saha A., Lewis S., Kuhlman B. and Deshaies R. J. (2009). Rapid E2-E3 assembly and disassembly enable processive ubiquitylation of cullin-RING ubiquitin ligase substrates. Cell 139, 957-968. 10.1016/j.cell.2009.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. P., Brauweiler A., Rudolph M., Hooper J. E., Drabkin H. A. and Gemmill R. M. (2010). The TRC8 ubiquitin ligase is sterol regulated and interacts with lipid and protein biosynthetic pathways. Mol. Cancer Res. 8, 93-106. 10.1158/1541-7786.MCR-08-0491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner P. J., Karttunen J. T., Wilkinson G. W. G. and Cresswell P. (1997). The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing-dependent peptide translocation. Proc. Natl. Acad. Sci. USA 94, 6904-6909. 10.1073/pnas.94.13.6904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitman J., Shenkman M., Gofman Y., Shtern N. O., Ben-Tal N., Hendershot L. M. and Lederkremer G. Z. (2014). Herp coordinates compartmentalization and recruitment of HRD1 and misfolded proteins for ERAD. Mol. Biol. Cell 25, 1050-1060. 10.1091/mbc.E13-06-0350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley B. N. and Ploegh H. L. (2004). A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429, 834-840. 10.1038/nature02592 [DOI] [PubMed] [Google Scholar]
- Liu W., Shang Y., Zeng Y., Liu C., Li Y., Zhai L., Wang P., Lou J., Xu P., Ye Y. et al. (2014). Dimeric Ube2g2 simultaneously engages donor and acceptor ubiquitins to form Lys48-linked ubiquitin chains. EMBO J. 33, 46-61. 10.1002/embj.201385315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch D. J., Rixon F. J. and Davison A. J. (2006). Topics in herpesvirus genomics and evolution. Virus Res. 117, 90-104. 10.1016/j.virusres.2006.01.002 [DOI] [PubMed] [Google Scholar]
- Miao H., Jiang W., Ge L., Li B. and Song B. (2010). Tetra-glutamic acid residues adjacent to Lys248 in HMG-CoA reductase are critical for the ubiquitination mediated by gp78 and UBE2G2. Acta Biochim. Biophys. Sin. (Shanghai). 42, 303-310. 10.1093/abbs/gmq022 [DOI] [PubMed] [Google Scholar]
- Mocarski E. S. (2002). Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol. 10, 332-339. 10.1016/S0966-842X(02)02393-4 [DOI] [PubMed] [Google Scholar]
- Noriega V. M., Hesse J., Gardner T. J., Besold K., Plachter B. and Tortorella D. (2012a). Human cytomegalovirus US3 modulates destruction of MHC class I molecules. Mol. Immunol. 51, 245-253. 10.1016/j.molimm.2012.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noriega V., Redmann V., Gardner T. and Tortorella D. (2012b). Diverse immune evasion strategies by human cytomegalovirus. Immunol. Res. 54, 140-151. 10.1007/s12026-012-8304-8 [DOI] [PubMed] [Google Scholar]
- Olzmann J. A., Kopito R. R. and Christianson J. C. (2013). The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 5, a013185 10.1101/cshperspect.a013185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oresic K., Ng C. L. and Tortorella D. (2009). TRAM1 participates in human cytomegalovirus US2- and US11-mediated dislocation of an endoplasmic reticulum membrane glycoprotein. J. Biol. Chem. 284, 5905-5914. 10.1074/jbc.M807568200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B., Kim Y., Shin J., Lee S., Cho K., Früh K., Lee S. and Ahn K. (2004). Human cytomegalovirus inhibits tapasin-dependent peptide loading and optimization of the MHC class I peptide cargo for immune evasion. Immunity 20, 71-85. 10.1016/S1074-7613(03)00355-8 [DOI] [PubMed] [Google Scholar]
- Park B., Spooner E., Houser B. L., Strominger J. L. and Ploegh H. L. (2010). The HCMV membrane glycoprotein US10 selectively targets HLA-G for degradation. J. Exp. Med. 207, 2033-2041. 10.1084/jem.20091793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston G. M. and Brodsky J. L. (2017). The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem. J. 474, 445-469. 10.1042/BCJ20160582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuren A. B. C., Costa A. I. and Wiertz E. J. H. J. (2016). Recent advances in viral evasion of the MHC Class I processing pathway. Curr. Opin. Immunol. 40, 43-50. 10.1016/j.coi.2016.02.007 [DOI] [PubMed] [Google Scholar]
- Soetandyo N. and Ye Y. (2010). The p97 ATPase dislocates MHC class I heavy chain in US2-expressing cells via a Ufd1-Npl4-independent mechanism. J. Biol. Chem. 285, 32352-32359. 10.1074/jbc.M110.131649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg H. R., Thomas M., van den Boomen D., Wiertz E. J. H. J., Drabkin H. A., Gemmill R. M. and Lehner P. J. (2009). The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 186, 685-692. 10.1083/jcb.200906110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S., Shi G., Sha H., Ji Y., Han X., Shu X., Ma H., Inoue T., Gao B., Kim H. et al. (2015). IRE1α is an endogenous substrate of endoplasmic-reticulum-associated degradation. Nat. Cell Biol. 17, 1546-1555. 10.1038/ncb3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S. and Weissman A. M. (2001). Endoplasmic reticulum (ER)-associated degradation of T cell receptor subunits. Involvement of ER-associated ubiquitin-conjugating enzymes (E2s). J. Biol. Chem. 276, 16193-16200. 10.1074/jbc.M007640200 [DOI] [PubMed] [Google Scholar]
- van de Weijer M. L., Bassik M. C., Luteijn R. D., Voorburg C. M., Lohuis M. A. M., Kremmer E., Hoeben R. C., LeProust E. M., Chen S., Hoelen H. et al. (2014). A high-coverage shRNA screen identifies TMEM129 as an E3 ligase involved in ER-associated protein degradation. Nat. Commun. 5, 3832 10.1038/ncomms4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Weijer M. L., Luteijn R. D. and Wiertz E. J. H. J. (2015). Viral immune evasion: lessons in MHC class I antigen presentation. Semin. Immunol. 27, 125-137. 10.1016/j.smim.2015.03.010 [DOI] [PubMed] [Google Scholar]
- Van den Boomen D. J. H. and Lehner P. J. (2015). Identifying the ERAD ubiquitin E3 ligases for viral and cellular targeting of MHC class I. Mol. Immunol. 68, 106-111. 10.1016/j.molimm.2015.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boomen D. J. H., Timms R. T., Grice G. L., Stagg H. R., Skødt K., Dougan G., Nathan J. A. and Lehner P. J. (2014). TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. USA 111, 11425-11430. 10.1073/pnas.1409099111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Herr R. A., Rabelink M., Hoeben R. C., Wiertz E. J. H. J. and Hansen T. H. (2009). Ube2j2 ubiquitinates hydroxylated amino acids on ER-associated degradation substrates. J. Cell Biol. 187, 655-668. 10.1083/jcb.200908036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiertz E. J. H. J., Jones T. R., Sun L., Bogyo M., Geuze H. J. and Ploegh H. L. (1996a). The human cytomegalovirus US11 gene product dislocates MHC class I heavy chains from the endoplasmic reticulum to the cytosol. Cell 84, 769-779. 10.1016/S0092-8674(00)81054-5 [DOI] [PubMed] [Google Scholar]
- Wiertz E. J. H. J., Tortorella D., Bogyo M., Yu J., Mothes W., Jones T. R., Rapoport T. A. and Ploegh H. L. (1996b). Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384, 432-438. 10.1038/384432a0 [DOI] [PubMed] [Google Scholar]
- Yan L., Liu W., Zhang H., Liu C., Shang Y., Ye Y., Zhang X. and Li W. (2014). Ube2g2-gp78-mediated HERP polyubiquitylation is involved in ER stress recovery. J. Cell Sci. 127, 1417-1427. 10.1242/jcs.135293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Meyer H. H. and Rapoport T. A. (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652-656. 10.1038/414652a [DOI] [PubMed] [Google Scholar]
- Ye Y., Shibata Y., Yun C., Ron D. and Rapoport T. A. (2004). A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429, 841-847. 10.1038/nature02656 [DOI] [PubMed] [Google Scholar]
- Ye Y., Shibata Y., Kikkert M., van Voorden S., Wiertz E. and Rapoport T. A. (2005). Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 102, 14132-14138. 10.1073/pnas.0505006102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Q., Lin S.-C., Lamothe B., Lu M., Lo Y.-C., Hura G., Zheng L., Rich R. L., Campos A. D., Myszka D. G. et al. (2009). E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658-666. 10.1038/nsmb.1605 [DOI] [PMC free article] [PubMed] [Google Scholar]