

Abstract
Ewing’s sarcoma is a highly aggressive malignant tumour most commonly affecting long bones in children and adolescents. It is part of the Ewing’s sarcoma family of tumours (ESFTs) that also include peripheral primitive neuroectodermal tumour and Askin’s tumours. ESFTs share common cytogenetic aberrations, antigenic profiles and proto-oncogene expression with an overall similar clinical course. In 99% of ESFTs, genetic translocation with molecular fusion involves the EWSR1 gene on 22q12. Approximately 30% of ESFTs are extraosseous, most commonly occurring in the soft tissues of extremities, pelvis, retroperitoneum and chest wall. Primary presentation in solid organs is very rare but has been described in multiple sites including the pancreas. Accurate diagnosis of a Ewing’s sarcoma in a solid organ is critical in facilitating correct treatment. We report the case of a 17-year-old girl with cytogenetically confirmed primary pancreatic Ewing’s sarcoma and provide a brief review of the published literature.
Keywords: Primary pancreatic Ewing’s sarcoma, Extraosseous Ewing’s sarcoma, Primitive neuroectodermal tumor, peripheral primitive neuroectodermal tumor (pPNET)
Background
Ewing’s sarcoma is an aggressive tumour of bone and soft tissue origin. It belongs to the Ewing’s sarcoma family of tumours (ESFTs) and has an incidence of approximately 6.5 per million of the population.1 Ewing’s sarcoma of bone was first described by James Ewing in 1921, and is the second most common primary childhood bone cancer. Tefft was the first to describe the extraosseous form of Ewing’s sarcoma in 1969, and this accounts for approximately 30% of ESFTs.2 3 Extraosseous ESFTs occur at a slightly older age and arise most commonly from the soft tissue of the extremities, pelvis, retroperitoneum and chest wall. Primary presentation in solid organs is rare but has been reported from various sites including lung, kidney, uterus, bladder, salivary glands, heart, vagina, prostate, stomach, oesophagus and pancreas.3
Histologically, Ewing’s sarcoma is a small round blue cell tumour composed of sheets of small monomorphic round cells with small nuclei and scanty cytoplasm. The morphology is not unique to this tumour, and the pathological differential diagnosis in a solid organ is wide and includes lymphoma, pancreatic endocrine tumour, pancreatoblastoma, hepatoblastoma, Wilms tumour, rhabdomyosarcoma and neuroblastoma among others.4 ESFTs, however, are characterised by a specific translocation involving EWSR1 gene in 99% of cases, and a successful tissue diagnosis requires application of immunohistochemistry and cytogenetic analysis on tumour tissue. These tumours remain asymptomatic for longer periods and show an 80% to 90% relapse rate with local therapy indicating that the disease was disseminated at the time of presentation.5 Improvements in multimodality treatment have resulted in significant improvement in outcomes of these tumours. We describe a case of a 17-year-old girl with primary Ewing’s sarcoma of the pancreatic head and discuss the available literature regarding this rare tumour.
Case presentation
A 17-year-old girl presented to our department with gradually progressive jaundice associated with generalised itching for 6 weeks. The patient also gave a history of loss of appetite and passing pale coloured stools. Patient had one episode of haematemesis containing approximately 100 to 150 mL of altered blood.
Investigations
Prior to referral, she had undergone an endoscopic retrograde cholangiopancreatography with common bile duct (CBD) stenting and a triphasic CT abdomen that identified a large enhancing mass of approximately 5.6×7.4 cm in the head and uncinate process of the pancreas with a small hypodense lesion measuring 9 mm in size in segment 2 of the liver, seen predominantly in portal venous phase. Serum tumour markers carcinoembryonic antigen and carbohydrate antigen (CA19-9) were within normal range. Following assessment in the outpatient department, an endoscopic ultrasound (EUS) was performed and revealed a heteroechoic well-circumscribed pancreatic head mass of 5.5×7.4 cm size with a dilated CBD (12 mm) and main pancreatic duct (8 mm) beyond the mass with a stent in situ in the CBD. The portal vein was separate from the mass but the superior mesenteric vein (SMV) was indented. EUS-guided fine needle aspiration taken from the mass showed small monomorphic tumour cells with scant cytoplasm staining positively for synaptophysin. Tumour cells were negative for beta-catenin and chromogranin staining. Based on these findings, a pancreatic neuroendocrine neoplasm was suspected. The serum chromogranin A level was 0.71 (normal range less than 36.4 ng/mL).
A positron emission tomography (PET) scan showed a large lobulated mildly DOTA NOC (68Ga-labelled (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)-1-Nal3-octreotide) avid mass in the pancreatic head and uncinate process closely abutting and compressing the CBD as well as the second and third portions of the duodenum (figure 1).
Figure 1.
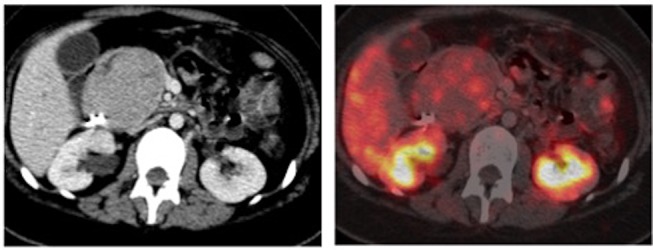
Whole-body 68Ga-labelled (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)-1-Nal3-octreotide PET CT scan showing a large mildly DOTA NOC avid mass lesion in the head and uncinate process of the pancreas abutting and compressing the CBD as well as the second and third portions of the duodenum.
Differential diagnosis
Initially, based on clinical and radiological evaluation, the differential diagnoses considered included solid pseudopapillary epithelial neoplasm and pancreatic neuroendocrine tumour.
Treatment
After a full discussion and consent, the patient underwent a pancreatoduodenectomy. Intraoperatively, there was a large firm mass in the head of the pancreas abutting the SMV. Two subcentimetre nodules in the left lobe of the liver were identified and confirmed as inflammatory cysts on intraoperative frozen section analysis. The CBD and pancreatic duct were dilated at 2.5 and 0.8 cm, respectively. Frozen section analysis of the pancreatic resection margin was negative for malignancy. The specimen was sent for histological examination, and on gross examination, the pancreatic head contained a 5.5×7.5 cm firm mass infiltrating the entire wall of the duodenum and extending as a polypoidal growth into the duodenal lumen, with ulceration of the overlying duodenal mucosa. The cut surface of the tumour was grey–white with few areas of congestion and softening suggestive of necrosis. On microscopic evaluation, H&E staining identified a small round blue cell tumour composed of sheets of small cells with ovoid, crenated nuclei with occasional nucleoli and nuclear grooves (figure 2A,B). The tumour cells were arranged in loosely cohesive sheets surrounding dilated delicate vascular channels with few areas of discohesion resulting in a vague pseudopapillary pattern. Along with areas of apoptosis, the tumour showed extensive haemorrhage and focal necrosis. Retrieved lymph nodes were free of tumour. The tumour exhibited a high mitotic activity with 10 mitoses counted in one random high power field and the proliferative index (Ki67) was 25%.
Figure 2.

Histopathological features of the tumour are shown. (A) Low power image showing tumour with adjacent pancreas (H&E staining). (B) High power magnification of tumour showing atypical small cells with scant cytoplasm. (C) Immunohistochemistry showing strongly CD99-positive cells. (D) Two-colour fluorescence in situ hybridization (FISH) assay probe showing EWSR1 break apart with one red and one green signal. (E) EWSR1-FLI1 dual fusion FISH probe showing FFRG signal pattern. (F) Reverse transcription (RT)-PCR image: lane A, RT-PCR with primers to EWSR1-FLI1 transcript; lane B, negative control (no target in reaction); lane C, negative control (negative complementary DNA); lane D, positive control (type 2).
On immunohistochemical analysis, the tumour cells were strongly positive for panCytokeratin AE1/AE3 and CD99 with diffuse nuclear staining for cyclin D1 (figure 2C). Beta-catenin showed membranous staining only with no nuclear expression.
Tumour cells were negative for synaptophysin, chromogranin, neuron-specific enolase, CD56, trypsin, DOG1, monoclonal WT1, polyclonal WT1 and desmin. Interface fluorescence in situ hybridization (FISH) analysis showed an EWSR1 rearranged signal pattern in 89 out of 100 nuclei examined indicating a clone with 22q12 rearrangement (figure 2D,E). Reverse transcription (RT)-PCR using RNA extracted from the tumour cells was positive for the EWSR1-FLI1 fusion transcript indicating a translocation t(11;22)(q24;q12) typical of Ewing’s sarcoma (figure 2F).
Outcome and follow-up
The postoperative course was uneventful and she was discharged on postoperative day 15 in a clinically stable condition. Postoperative fluorodeoxyglucose (FDG)-PET scan showed focal FDG uptake in the operative bed and few subcentimetre non-FDG avid abdominal lymph nodes (figure 3). On further follow-up, she completed 12 cycles of vincristine/cyclophosphamide/doxorubicin-based chemotherapy. Postchemotherapy PET scan showed mild focal FDG uptake in the operative bed, which was less FDG avid compared with the previous PET scan. There was no evidence of enlarged abdominal lymph nodes on postchemotherapy PET scan. As per the consensus at a multidisciplinary meeting, the patient received adjuvant radiotherapy, as there is no definitive way to distinguish the postoperative changes from the residual tumour.
Figure 3.
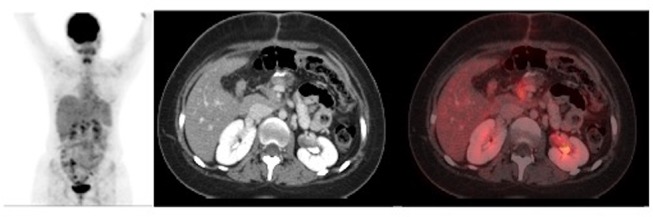
Postoperative fluorodeoxyglucose (FDG) positron emission tomography scan showing focal FDG uptake in the operative bed in the region of the head of pancreas and an ill-defined uptake along the resected margin of the pancreas without evidence of any separate primary lesion.
Discussion
Ewing’s sarcoma arises from primordial bone marrow-derived mesenchymal stem cells.6 ESFTs have common underlying characteristic genetic aberrations which in 99% of cases represents reciprocal chromosomal translocations involving the EWSR1 gene with a member of the Ewing’s tumour (ET)-family transcription factors, such as FLI1, ERG, ETV1, ETV4 and FEV.7 Translocation t(11;22)(q24;q12) results in the fusion product EWS-FLI1 which is found in more than 85% of the cases. A second translocation, found in approximately 10% of cases, is t(21; 22) (q22; q12). The fusion gene resulting from such translocations EWS-FLI1 produces a protein that drives the growth of these tumours through deregulation of transcription and apoptosis.7 Ewing’s family of tumours comprises Ewing’s sarcoma of bone, extraosseous Ewing’s sarcoma, peripheral primitive neuroectodermal tumour (pPNET) and Askin’s tumour. To our knowledge, only 27 cases of primary pancreatic Ewing’s sarcoma/pPNET have been described so far in the literature including the present one.8 9
Extraosseous Ewing’s sarcoma is more common in the second decade of life.8–10 In an evaluation of the Surveillance, Epidemiology and End Results Database, Applebaum et al evaluated over 2000 patients and compared osseous and extraosseous ESFTs. Patients with extraosseous Ewing’s sarcoma were slightly older (19.5 vs 16.3 years), had a slightly higher proportion of female and non-white patients compared with patients with osseous Ewing’s sarcoma and had axial primary sites.10 Nishizawa et al recently reviewed the literature of 25 primary pancreatic ESFTs of which 16 were cytogenetically confirmed. They found a mean age at time of diagnosis of 18.2 years and a median age of 20 years, with no gender predominance.8 Abdominal pain (68%) was the most common presenting finding followed by jaundice and anaemia.8 9 Endocrine disorders such as hyperglycaemia and precocious puberty were seen in one and two cases, respectively.9 These tumours have an expansive growth pattern in contrast to the highly infiltrative growth pattern of ductal adenocarcinoma and hence are generally present later in the disease course.8 Most of the tumours occurred in the head of the pancreas (68%).8 9 At the time of presentation, most of the tumours are large and bulky with an average tumour size of 88 mm reported by Nishizawa and colleagues.8 CT and MRI of the abdomen are important tools to characterise these tumours. They generally have similar attenuation to that of muscle (87% of cases).11 However, the density of the tumours on CT scan varies depending on the amount of the necrosis and generally appears isodense or hypodense on unenhanced CT scan. Calcification may be seen in up to 25%–30% of cases.11 On MRI, pancreatic ESFTs are isointense on T1-weighted images, and either isointense or hyperintense on T2-weighted images. They usually have ill-defined borders and irregular shapes with heterogeneous enhancement.11 FDG-PET has been found to be more useful than bone scintigraphy for detection of bone metastases. However, for detection of small lesions, especially pulmonary metastases, PET was found to be inferior to the helical CT due to low standardised uptake value in small lesions.12
Histopathological analysis is the cornerstone of diagnosis of small round blue cell tumour. The consideration of overall clinical presentation, imaging analysis, morphological appearances, immunohistochemistry and cytogenetic analysis is needed to correctly characterise these tumours, which is important as the treatment and prognosis differ considerably among these tumours.13
All tumours of the ESFT group express the MIC gene product universally which markers such as CD99 can detect. The detection of a chimeric gene also helps to confirm the diagnosis of extraosseous Ewing’s sarcoma. Interphase FISH and RT-PCR can be used to detect the translocation t(11;22)(q24;q12) which is seen in more than 85% of extraosseous Ewing’s sarcoma (EES)/pPNETs. While FISH can be performed easily on formalin-fixed tissues, RT-PCR is better performed on fresh or frozen tissues rather than formalin-fixed ones.14 Ewing’s sarcoma of bone and extraosseous Ewing’s sarcoma can both metastasise to the pancreas.15 In the present case, the postoperative FDG-PET scan did not show any evidence of a separate primary lesion in the bone or elsewhere (figure 3). So corroborating the clues in the clinical, radiological, histological, immunohistochemical and cytogenetic findings, a diagnosis of primary Ewing’s sarcoma of the pancreas was made.
Extraosseous Ewing’s sarcoma has an aggressive clinical course with early distant metastases, indicating the need for systemic chemotherapy in these patients.9 The current treatment strategy primarily consists of multiagent induction chemotherapy followed by local therapy and adjuvant therapy.9 The preferred local therapy is surgical excision whenever negative resection margin can be achieved. For patients with unresectable tumours, or those with positive resection margin and/or gross residual tumour, radiotherapy is used for local control of the disease. Recently chemotherapy protocols based on the line of treatment protocols for bone origin Ewing’s sarcoma family tumours have been suggested for ESFTs instead of malignant mesenchymal tumours treatment-based protocols.16 Chemotherapy protocols used are vincristine, cyclophosphamide and doxorubicin or dactinomycin (VDC) based. Addition of ifosfamide and etoposide (IE) to the standard chemotherapy regimen has shown improved overall survival and event-free survival in the patients with non-metastatic Ewing’s sarcoma family tumours of bone but not in those with metastatic tumours.17 The standard regimen prescribes 17 cycles of VDC-based chemotherapy alternating with IE every 3 weeks, administered over 48 weeks.16 17 An intensified regimen with 11 cycles of chemotherapy (VDC+IE) over 30 weeks administering the same chemotherapeutic dose did not improve the overall and disease-free survival in patients with non-metastatic ESFT.16 The radiotherapy dose advocated for extraosseous tumour either as primary local therapy or rescue local therapy is 45 Gy to the initial tumour volume plus a 2-cm margin area followed by a boost of 5.4 Gy to a total dose of 50.4 Gy.16 Combined modality of treatment has significantly improved the outcome compared with surgery and/or radiation.16 Overall reported 5-year survival rates range from 61% to 77% in patients with extraskeletal Ewing’s sarcoma although tumour dissemination at the time of diagnosis is associated with bad prognosis.10 18
Learning points.
Primary pancreatic Ewing’s sarcoma is a very rare tumour but should be considered in the differential diagnosis of an unusual pancreatic tumour especially in young adults.
Establishing the correct diagnosis requires a multidisciplinary approach involving close cooperation between clinical, radiological and pathological teams with accurate application of immunohistochemistry and cytogenetic analysis.
A multimodality approach to treatment has improved the outcome of this tumour significantly in patients with localised disease, but the overall prognosis of this tumour still remains poor.
Footnotes
Contributors: AG conceptualised the manuscript, acquired data and wrote the manuscript.
SR acquired data and edited the manuscript.
BH participated in clinical pathological work, acquired data and edited the manuscript.
SHS conceptualised the manuscript, acquired data and edited the manuscript.
All authors have seen and approved the final version of the manuscript.
All authors agree to be accountable for the accuracy and integrity of the manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Schutte WP, Knight PJ. Precocious puberty because of a pancreatic neuroectodermal tumor. J Pediatr Surg 2006;41:1916–8. 10.1016/j.jpedsurg.2006.05.046 [DOI] [PubMed] [Google Scholar]
- 2. Tefft M, Vawter GF, Mitus A. Paravertebral "round cell" tumors in children. Radiology 1969;92:1501–9. 10.1148/92.7.1501 [DOI] [PubMed] [Google Scholar]
- 3. Colovic RB, Grubor NM, Micev MT, et al. Perigastric extraskeletal Ewing’s sarcoma: a case report. World J Gastroenterol 2009;15:245–7. 10.3748/wjg.15.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Danner DB, Hruban RH, Pitt HA, et al. Primitive neuroectodermal tumor arising in the pancreas. Mod Pathol 1994;7:200–4. [PubMed] [Google Scholar]
- 5. Carvajal R, Meyers P. Ewing’s sarcoma and primitive neuroectodermal family of tumors. Hematol Oncol Clin North Am 2005;19:501–25. 10.1016/j.hoc.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 6. Suvà ML, Riggi N, Stehle JC, et al. Identification of cancer stem cells in Ewing’s sarcoma. Cancer Res 2009;69:1776–81. 10.1158/0008-5472.CAN-08-2242 [DOI] [PubMed] [Google Scholar]
- 7. Jain S, Xu R, Prieto VG, et al. Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol 2010;3:416–28. [PMC free article] [PubMed] [Google Scholar]
- 8. Nishizawa N, Kumamoto Y, Igarashi K, et al. A peripheral primitive neuroectodermal tumor originating from the pancreas: a case report and review of the literature. Surg Case Rep 2015;1:1–8. 10.1186/s40792-015-0084-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bose P, Murugan P, Gillies E, et al. Extraosseous Ewing’s sarcoma of the pancreas. Int J Clin Oncol 2012;17:399–406. 10.1007/s10147-011-0311-6 [DOI] [PubMed] [Google Scholar]
- 10. Applebaum MA, Worch J, Matthay KK, et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 2011;117:3027–32. 10.1002/cncr.25840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robbin MR, Murphey MD, Jelinek JS, et al. Imaging of soft tissue Ewing sarcoma and primitive neuroectodermal tumor. Radiology 1998;209:311. [Google Scholar]
- 12. Györke T, Zajic T, Lange A, et al. Impact of FDG PET for staging of Ewing sarcomas and primitive neuroectodermal tumours. Nucl Med Commun 2006;27:17–24. 10.1097/01.mnm.0000186608.12895.69 [DOI] [PubMed] [Google Scholar]
- 13. Schmidt D, Herrmann C, Jürgens H, et al. Malignant peripheral neuroectodermal tumor and its necessary distinction from Ewing’s sarcoma. a report from the Kiel pediatric tumor registry. Cancer 1991;68:2251–9. [DOI] [PubMed] [Google Scholar]
- 14. Yamaguchi U, Hasegawa T, Morimoto Y, et al. A practical approach to the clinical diagnosis of Ewing’s sarcoma/primitive neuroectodermal tumour and other small round cell tumours sharing EWS rearrangement using new fluorescence in situ hybridisation probes for EWSR1 on formalin fixed, paraffin wax embedded tissue. J Clin Pathol 2005;58:1051–6. 10.1136/jcp.2004.025502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bertucci F, Araujo J, Giovannini M. Pancreatic metastasis from osteosarcoma and Ewing sarcoma: literature review. Scand J Gastroenterol 2013;48:4–8. 10.3109/00365521.2012.711852 [DOI] [PubMed] [Google Scholar]
- 16. Granowetter L, Womer R, Devidas M, et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: a Children’s Oncology Group Study. J Clin Oncol 2009;27:2536–41. 10.1200/JCO.2008.19.1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694–701. 10.1056/NEJMoa020890 [DOI] [PubMed] [Google Scholar]
- 18. Eralp Y, Bavbek S, Başaran M, et al. Prognostic factors and survival in late adolescent and adult patients with small round cell tumors. Am J Clin Oncol 2002;25:418–24. 10.1097/00000421-200208000-00020 [DOI] [PubMed] [Google Scholar]