

Abstract
High-grade epithelial ovarian carcinomas containing mutated BRCA1 or BRCA2 (BRCA1/2) homologous recombination (HR) genes are sensitive to platinum-based chemotherapy and PARP inhibitors (PARPi), while restoration of HR function due to secondary mutations in BRCA1/2 has been recognized as an important resistance mechanism. We sequenced core HR pathway genes in 12 pairs of pretreatment and postprogression tumor biopsy samples collected from patients in ARIEL2 Part 1, a phase II study of the PARPi rucaparib as treatment for platinum-sensitive, relapsed ovarian carcinoma. In 6 of 12 pretreatment biopsies, a truncation mutation in BRCA1, RAD51C, or RAD51D was identified. In five of six paired postprogression biopsies, one or more secondary mutations restored the open reading frame. Four distinct secondary mutations and spatial heterogeneity were observed for RAD51C. In vitro complementation assays and a patient-derived xenograft, as well as predictive molecular modeling, confirmed that resistance to rucaparib was associated with secondary mutations.
Significance
Analyses of primary and secondary mutations in RAD51C and RAD51D provide evidence for these primary mutations in conferring PARPi sensitivity and secondary mutations as a mechanism of acquired PARPi resistance. PARPi resistance due to secondary mutations underpins the need for early delivery of PARPi therapy and for combination strategies.
Introduction
The hallmark of synthetic lethality is the requirement for two complementary hits that, although tolerated individually, result in cancer cell death when they occur together. A prime example is the observation that cells tolerate PARP inhibition or homologous recombination (HR) impairment individually, but cancer cells with impaired HR are killed by PARP inhibitors (PARPi), reflecting drug-induced inhibition of PARP1 catalytic activity, trapping of PARP1 at sites of DNA damage, and/or alterations in the balance between error-free and error-prone repair pathways (1–5).
An exquisite proof of synthetic lethality comes from high-grade epithelial ovarian carcinomas with mutated BRCA1 or BRCA2 (BRCA1/2) that are sensitive to platinum-based chemotherapy and PARPi (6–9). Furthermore, somatic reversion mutations in either BRCA1/2 following exposure of ovarian carcinoma to platinum-based chemotherapy or PARPi are identified as a mechanism of resistance. First reported in 2008 in a human pancreatic cell line and human ovarian carcinoma (10, 11), secondary mutations that restore the wild-type BRCA2 open reading frame were detected in clinical ovarian carcinoma, with a higher rate in women with platinum-resistant ovarian carcinoma who had prior chemotherapy (12, 13). More recently, examination of multiple tumor deposits at autopsy revealed additional evidence of BRCA2 reversion mutations and intrapatient heterogeneity with 12 distinct reversion events observed in a single patient with end-stage BRCA2-mutant ovarian carcinoma who had received multiple chemotherapy regimens (14). To date, most of the secondary BRCA1/2 mutations are documented after platinum-chemotherapy exposure, with only limited reports after PARPi (15).
Identification of patients with wild-type BRCA1/2 but HR-defective ovarian carcinoma is important, as these patients may potentially respond to PARPi therapy. Germline or somatic mutations in core HR genes beyond BRCA1/2, although individually rare, collectively occur in 7% to 8% of ovarian carcinomas (16, 17) and have been shown in vitro and in patients to underpin responses to PARPi (9, 18). These mutations are being increasingly identified in the clinic due to use of germline multiplex genetic testing and tumor sequencing (16, 19). In addition to BRCA1/2, the RAD51 paralogues RAD51C and RAD51D are well-established core HR pathway genes in which germline mutations increase ovarian cancer susceptibility (20–24). We recently reported that the PARPi rucaparib is active in ovarian carcinoma with RAD51C or RAD51D mutations, with three partial responses (PR) and two cases of prolonged stable disease (SD) of 8.3- and 11.0-month duration among five evaluable patients treated with rucaparib (9). It is unknown if somatic reversion mutations are a mechanism of acquired resistance in cancers driven by mutations in HR genes beyond BRCA1/2.
Here, we investigated whether secondary mutations in genes other than BRCA1/2 can arise as a mechanism of resistance post exposure to the PARPi rucaparib.
Results
To study acquired PARPi resistance, we profiled pretreatment tumor samples and postprogression biopsies from 12 patients with platinum-sensitive, relapsed high-grade epithelial ovarian carcinoma treated with rucaparib in ARIEL2 Part 1. Samples were assessed using targeted next-generation sequencing (NGS) with Foundation Medicine's T5 assay, which sequences 287 cancer-related genes, including core HR pathway genes (Supplementary Fig. S1; Supplementary Table S1; ref. 9). In 6 of the 12 cases, a deleterious mutation causing early protein termination in an HR pathway gene (four in BRCA1 and one each in RAD51C and RAD51D; five germline and one somatic mutation) was detected in either an archival tumor sample (n = 6) and/or screening biopsy sample (n = 4) prior to initiation of rucaparib treatment (Table 1). All six patients with HR pathway genes mutated in their ovarian carcinoma derived clinical benefit from rucaparib (four with a confirmed RECIST PR and two with SD; progression-free survival (PFS) ranged from 9.6 to 22.0 months). In a seventh case, a somatic CDK12 mutation (c.264delC) was identified with no additional mutations detected in the postprogression biopsy. CDK12 has been reported to affect transcription of multiple HR genes, although it is yet to be established as a core HR pathway gene (25, 26).
Table 1. Clinical and molecular features of twelve ovarian carcinoma cases with postprogression biopsies.
| Subject ID |
No. of prior treatment regimens |
RECIST, CA-125 response | PFS (mo) | Archival tumor sample | Pretreatment tumor sample | Postprogression tumor biopsy sample | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||||||||||
| Esti- mated tumor puritya |
HR gene mutations detected | Esti- mated tumor puritya |
HR gene mutations detected | Esti- mated tumor puritya |
HR gene mutations detected | |||||||||||||
|
|
|
|
||||||||||||||||
| HGVS nomenclature | Coverage | Mutation frequency |
LOH | HGVS nomenclature | Cover- age |
Mutation frequency |
LOH | HGVS nomenclature | Cover- age |
Mutation frequency |
LOH | |||||||
| 1 | 2 | Partial response, CA-125 response | 25.2 | 34% | BRCA1:c.1175 1214del, p.(L392Qfs*5) GL | 897 | 67% | Yes | 60% | BRCA1:c.1175 1214del, p.(L392Qfs*5) GL | 855 | 84% | Yes | 56% | BRCA1:c.1175 1214del, p.(L392Qfs*5) GL | 991 | 68% | No |
| BRCA1:c.1129 1251del, p.(S377 N417del) 2° MUT | 1388 | 31% | No | |||||||||||||||
|
| ||||||||||||||||||
| 2 | 1 | Stable disease, CA-125 NE | 22 | 14% | BRCA1:c.5346G>A, p.(W1782*) GL | 524 | 57% | Yes | 73% | BRCA1:c.5346G>A,p.(W1782*) GL | 542 | 77% | Yes | 56% | BRCA1:c.5346G>A, p.(W1782*) GL | 534 | 70% | Yes |
|
| ||||||||||||||||||
| 3 | 1 | Partial response, CA-125 response | 16.4 | 91% | BRCA1:c.1892 1893del, p.(L631Qfs*4) SOM | 246 | 80% | Yes | 50% | BRCA1:c.1892 1893del, p.(L631Qfs*4) SOM | 796 | 33% | Yes | 20% | BRCA1:c.1892 1893del, p.(L631Qfs*4) SOM | 1108 | 10% | No |
| BRCA1:c.1279 2139del, p.(E427 S713del) 2° MUT | 1215 | 7% | No | |||||||||||||||
|
| ||||||||||||||||||
| 4 | 2 | Partial response, CA-125 NE | 12.8 | 62% | BRCA1:c.2043dup, p.(N682*) GL | 768 | 73% | Yes | Tissue not suitable for analysis | 58% | BRCA1:c.2043dup, p.(N682*) GL | 870 | 60% | No | ||||
| BRCA1:c.[1835 1964del; 2043dup], p.(R612 S681delins27) 2° MUT | 823 | 15% | No | |||||||||||||||
| BRCA1:c.4096+1G>A 2° MUT | 742 | 9% | No | |||||||||||||||
|
| ||||||||||||||||||
| 5 | 1 | Partial response, CA-125 response | 9.6 | 80% | RAD51C:c.577C>T, p.(R193*) GL | 656 | 92% | Yes | 36% | RAD51C:c.577C>T, p.(R193*) GL | 711 | 73% | Yes | 66% | RAD51C:c.577C>T, p.(R193*) GL | 1050 | 63%b | No |
| RAD51C:c.577 579delinsTGG, p.(R193W) 2° MUT | 1050 | 10%b | No | |||||||||||||||
| RAD51C:c.577C>A, p.(=) 2° MUT | 1050 | 7%b | No | |||||||||||||||
| RAD51C:c.574 577delinsGGCG, p.(H192 R193delinsGG) 2° MUT | 1021 | 3%b | No | |||||||||||||||
| RAD51C:c.577 578delinsTT, p.(R193L) 2° MUT | 1050 | 2%b | No | |||||||||||||||
|
| ||||||||||||||||||
| 6 | 2 | Stable disease, CA-125 response | 11 | 91% | RAD51D:c.770 776del, p.(G258Sfs*50) GL | 247 | 89% | Yes | Tissue not suitable for analysis | 91% | RAD51D:c.770 776delinsA, p.(S257 R259delinsK) 2° MUTc | 418 | 90% | Yes | ||||
|
| ||||||||||||||||||
| 7 | 1 | Partial response, CA-125 response | 16.7 | 85% | CDK12:c.264delC, p.(F89Sfs*3) SOM | 901 | 83% | Yes | 50% | CDK12:c.264delC, p.(F89Sfs*3) SOM | 712 | 50% | Yes | 74% | CDK12:c.264delC, p.(F89Sfs*3) SOM | 1027 | 75% | Yes |
|
| ||||||||||||||||||
| 8 | 1 | Progressive disease, CA-125 NE | 3.7 | 30% | Nil found | Tissue not suitable for analysis | 39% | Nil found | ||||||||||
|
| ||||||||||||||||||
| 9 | 3 | Stable disease, CA-125 NE | 5 | 50% | Nil found | 60% | Nil found | 43% | Nil found | |||||||||
|
| ||||||||||||||||||
| 10 | 2 | Stable disease, CA-125 no response | 3.5 | 73% | Nil found | Tissue not suitable for analysis | 52% | Nil found | ||||||||||
|
| ||||||||||||||||||
| 11 | 2 | Stable disease, CA-125 NE | 18.2 | 90% | Nil found | 78% | Nil found | 66% | Nil found | |||||||||
|
| ||||||||||||||||||
| 12 | 1 | Stable disease, CA-125 no response | 14.5 | 20% | Nil found | 10% | Nil found | 20% | Nil found | |||||||||
NOTE: The number of prior chemotherapeutic regimens, best response to treatment from ARIEL2 Part 1, PFS from ARIEL2 Part 1, and details of the mutations detected in known HR DNA repair genes are shown. Seven of 12 cases were found to contain a mutation in either an HR gene (BRCA1 or RAD51C/D) or a gene that affects transcription of multiple HR genes (CDK12), five of which contained secondary mutations in the postprogression biopsy. For five cases, no HR or related gene mutation was observed in archival, pretreatment, or postprogression tumor material. Number of prior regimens and best response by RECIST/GCIG CA-125 criteria are shown. RECIST, CA-125, and PFS data based on the data cutoff date of April 29, 2016. LOH status was determined from variant allele frequency. Transcripts used for variant annotation: BRCA1, NM_007294.3; RAD51C, NM_058216.2; RAD51D, NM_002878.3; and CDK12, NM_016507.3.
Abbreviations: NE, CA-125 not evaluable; GL, germline mutation; SOM, somatic mutation; 2° MUT, secondary mutation; LOH, loss of heterozygosity.
Tumor purity was estimated based on the genome-wide copy-number analysis by Foundation Medicine.
The reported RAD51C mutation frequencies were estimated from biopsy core 1 using Foundation Medicine's T5 assay. To investigate tumor heterogeneity in more detail, this sample was sequenced using a single amplicon assay (see Fig. 2).
RAD51D secondary mutation was detected only in the nonresponding splenic lesion, whereas the primary mutation was detected only in the responding liver lesion (see Supplementary Table S1).
In five of six cases with HR pathway gene mutations, postprogression biopsy samples contained at least one secondary mutation that was not detected in the pretreatment carcinomas. These secondary mutations restored the open reading frame of the HR genes and thus potentially restored HR function and conferred resistance to rucaparib (Table 1). Secondary mutations were identified only in postprogression cases with HR gene mutations (P = 0.015, Fisher exact test). In case 2, the only case in which no secondary HR gene mutation was detected in the postprogression biopsy sample, the possibility of a reversion mutation to wild-type sequence was unlikely, because the wild-type allele frequency observed was not higher than expected based on the estimated tumor purity. The secondary mutations detected in the BRCA1-mutated cases were large in-frame deletions (ranging from 123 to 861 bp) that restored the open reading frame either by deleting the primary frameshift mutation (cases 1 and 3) or by shifting the reading frame back into the correct state (case 4). The phasing of primary and secondary mutations in case 4 could not be established with the Foundation Medicine T5 assay, because the secondary mutation (c.1835_1964del) was 80 bp away from the primary mutation (c.2043dup); therefore, we performed colony PCR that confirmed cis configuration of these mutations (Supplementary Table S2; Supplementary Fig. S2). We have also detected another secondary mutation in the postprogression biopsy in case 4 that is a known splice-site mutation (c.4096+1G>A) downstream of the primary frameshift mutation. mRNA analysis has previously shown that this variant results in a shortened BRCA1 isoform that lacks a large portion of exon 10, the BRCA1-Δ11q isoform, which would also splice out the primary mutation (27). This shortened isoform has been implicated as a potential mechanism of PARPi and platinum resistance (28).
In case 5 with a germline RAD51C mutation (c.577C>T, p.R193*), a postprogression biopsy of an enlarging groin lymph node contained four distinct secondary mutations that all restored the open reading frame of RAD51C (Table 1; Fig. 1A–D). The functional capacity of the primary mutation and four identified secondary mutations was investigated in vitro using the OVCAR8 ovarian carcinoma cell line. First, CRISPR/Cas9-mediated knockout (KO) of RAD51C resulted in increased cisplatin and rucaparib sensitivity that could be reverted to the level of parental cell resistance by introduction of wild-type RAD51C cDNA (Fig. 1E; Supplementary Fig. S3). Conversely, introduction of RAD51C cDNA with the primary mutation failed to convey resistance, whereas RAD51C cDNA containing the secondary mutations did confer resistance to rucaparib, as well as to multiple other PARPi (olaparib, niraparib, talazoparib, and veliparib) and platinum compounds (cisplatin and carboplatin; Fig. 1E; Supplementary Fig. S3 and Supplementary Table S3).
Figure 1.
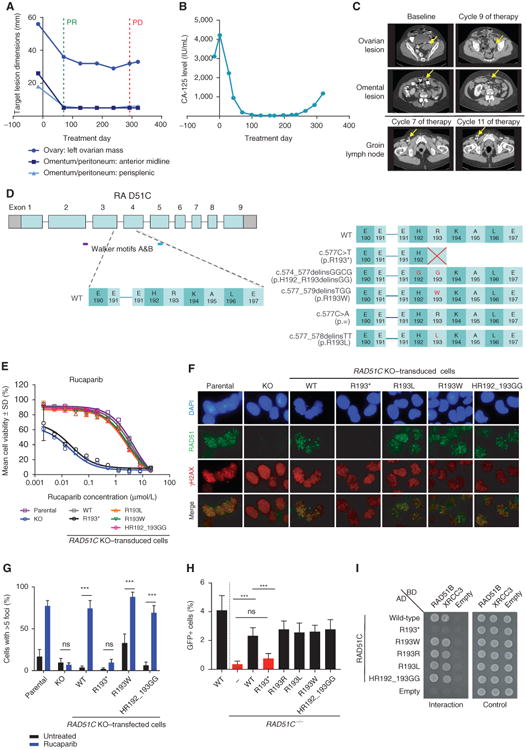
Identification and functional assessment of RAD51C secondary mutations identified in the postprogression biopsy in case 5. A, RECIST measurements of three metastatic sites, which were monitored in the patient identified to have a germline RAD51C mutation (c.577C>T). None of these lesions progressed during rucaparib treatment. After 11 months of rucaparib treatment, the patient developed a new enlarged groin lymph node, which was biopsied. B, Serum CA-125 levels monitored during the ARIEL2 Part 1 trial in the patient with the germline RAD51C mutation (c.577C>T). C, CT scans prior to (Baseline), during (Cycles 7 and 9), and following (Cycle 11) treatment of the patient with the germline RAD51C mutation (c.577C>T). D, Diagram of the predicted RAD51C protein sequence changes caused by the primary (c.577C>T) and the secondary mutations (c.577_579delinsTGG, c.577C>A, c.574_577delinsGGCG, and c.577_578delinsTT) detected in the progressing groin lymph node biopsy from the patient with the germline RAD51C mutation (c.577C>T). Examination of the parental OVCAR8, OVCAR8 RAD51C KO clone 2-130, and OVCAR8 RAD51C KO clone 2-130 transduced with wild-type (WT), primary mutant, or secondary mutant RAD51C transcripts using (E) cell viability assay after treatment with rucaparib for 6 days and (F) γH2AX and RAD51 foci formation 48 hours after rucaparib (10 μmol/L) exposure: γH2AX foci are observed at the sites of DNA damage, and RAD51 foci are observed at the sites of HR pathway repair. G, Quantification of RAD51 foci formation in geminin-positive cells (mean ± SEM). OVCAR8 RAD51C KO cells were transfected with plasmids expressing the WT, primary mutant, or secondary mutant RAD51C transcripts. The response of these cells to 10 μmol/L rucaparib was compared after 48 hours with the parental OVCAR8 cell line or OVCAR8 RAD51C KO clone. n = 8 fields of view (4 fields of view from 2 independent experiments) for each cell type and treatment. ***, P < 0.001. H, RAD51C secondary mutants restore HR as well as WT RAD51C in RAD51C-mutant cells. RAD51C−/− MCF10A cells containing the DR-GFP reporter were infected with an I-SceI expressing lentivirus and cultured for 48 hours. GFP+ cells were quantified by flow cytometry. n > 4 independent experiments. ***, P < 0.001. I, RAD51C secondary mutants restore RAD51C-R193* Y2H interactions with RAD51C binding partners RAD51B and XRCC3. RAD51C and the corresponding mutants were cloned into the Y2H plasmids expressing the GAL4 activating domain (AD), whereas RAD51B and XRCC3 were cloned into GAL4 binding domain (BD) expressing plasmids. Empty AD and BD vectors were used as negative controls. A Y2H interaction was observed as growth on medium lacking histidine, leucine, and tryptophan (interaction), whereas equal cell loading was observed on medium lacking leucine and tryptophan (control). ns, not significant; PD, progressive disease; PR, partial response.
RAD51 foci formation assays confirmed proficient HR repair in cells complemented with wild-type RAD51C or any of the secondary RAD51C mutations tested, but not with the primary RAD51C mutation (Fig. 1F and G; Supplementary Fig. S4). Clear evidence for HR restoration was obtained using a standard HR reporter assay, in which a single genomic double-strand break is generated by the I-SceI endonuclease to induce HR (Fig. 1H; Supplementary Fig. S5). RAD51C−/− MCF10A cells had substantially reduced HR repair compared with wild-type cells or RAD51C−/− cells expressing wild-type RAD51C. Although the RAD51C primary mutation R193* failed to complement the HR defect of RAD51C−/− cells, expression of all four RAD51C secondary mutants was able to restore HR comparable with wild-type RAD51C. We further demonstrated that RAD51C secondary mutants restore RAD51C R193* yeast-two-hybrid (Y2H) interactions with binding partners RAD51B and XRCC3 (Fig. 1I; Supplementary Fig. S6).
In order to assess the relative functional capacity of the RAD51C variants in vivo, a patient-derived xenograft (PDX) was generated from the postprogression lymph node biopsy from case 5. Deep amplicon sequencing of RAD51C exon 4 was performed on two adjacent core biopsies of the lymph node, one of which had given rise to the PDX, and on tumors from three recipient-first passage (T1) mice (Fig. 2A). Although the first core contained all four RAD51C secondary mutations, only two of these were detected in the second core (Fig. 2B). Furthermore, microscopic spatial heterogeneity was observed between adjacent 40-μm sections within the second core (Supplementary Fig. S7). The portion of the second core biopsy used to generate the PDX predominantly contained the c.574_577delinsGGCG mutation, as did PDX tumors from all three T1 mice, which had been implanted with tissue from the same core (Fig. 2A and B).
Figure 2.
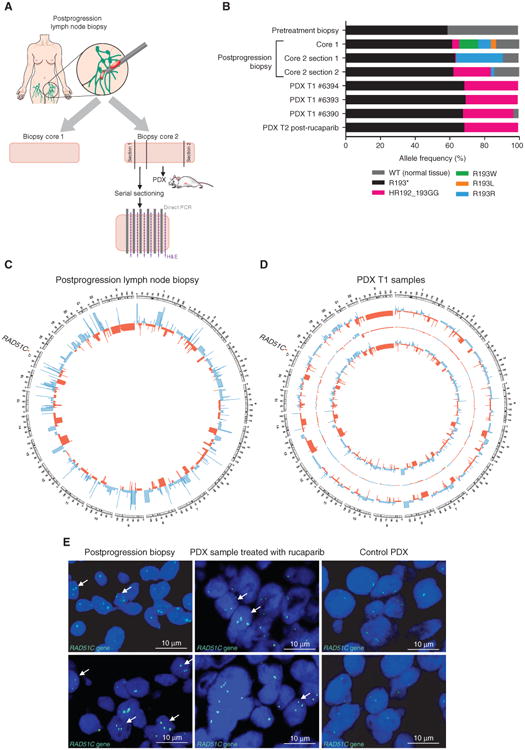
Tumor heterogeneity analysis of the postprogression biopsy with secondary RAD51C mutations. A, Model of the postprogression groin lymph node biopsy cores collected for analysis. Two postprogression core biopsies of the enlarging lymph node were obtained. Core 1 was used for genomic DNA analysis. Two ends of core 2, annotated as sections 1 and 2, were used for separate DNA extractions and subsequent analysis. The middle section of core 2 was used to generate the PDX, where 6 small pieces were subcutaneously transplanted into recipient mice. The leftover section of core 2 adjacent to section 1 was frozen in OCT and sectioned for direct PCR library preparation. B, Variant allele frequencies detected by sequencing in the pretreatment and postprogression biopsies, and in the generated PDX samples. Deep amplicon sequencing of RAD51C exon 4 (minimum coverage of 10,000×) was performed on these cores and on three recipient first passage (T1) mice. Although the first core analyzed contained all four RAD51C secondary mutations, only two of these were detected in the second core, which was used to generate the PDX. Spatial heterogeneity was even observed within the second core biopsy, with the c.577C>A mutation evident on one side of the core biopsy and at decreasing frequency toward the center of the core. The other side of the core biopsy predominantly contained the c.574_577delinsGGCG mutation, as did PDX tumors expanded in three T1 mice implanted with tissue from the same core. PDX T2 was treated with 450 mg/kg rucaparib for 2 weeks. C, Circos plot of the copy-number alterations detected by WGS in the postprogression biopsy (core 2 section 2) showing high levels of genomic instability. Losses are depicted in red, and gains in blue. D, Circos plot of copy-number alterations detected by WGS in the two analyzed PDX tumor samples obtained from the postprogression biopsy (core 2 section 2). The outer and inner copy-number tracks show the two analyzed tumor samples, and the middle track shows differences between them. E, RAD51C FISH assay of the postprogression biopsy (core 2 OCT block for serial sectioning) and the PDX sample treated with 450 mg/kg rucaparib for 2 weeks. Arrows point to the cells with three distinct signals visible for the postprogression biopsy and the PDX sample. H&E, hematoxylin and eosin.
To understand the context of the heterogeneity further, SNP array analysis (archival tissue and postprogression biopsies) and whole-genome sequencing (WGS) of the postprogression biopsy and two T1 PDXs were performed. Copy-number variation analysis revealed high levels of genomic instability in the archival sample and postprogression biopsies, with multiple amplifications and deletions detected throughout the genome. Little diversity was observed between the post-progression core biopsy that gave rise to the PDX, and the PDX itself; the two T1 PDXs analyzed were also highly concordant (Fig. 2C and D; Supplementary Table S4). Three copies of the RAD51C gene were observed in both archival and postprogression biopsies, which was confirmed by FISH analysis of postprogression and PDX tissue (Fig. 2E).
For case 6 with a germline RAD51D mutation (c.770_776del, p.G258Sfs*50), postprogression biopsy samples were collected from two different sites: a metastasis in the liver that was still responding to treatment and a growing metastasis in the spleen (Fig. 3A–C). The RAD51D secondary mutation (c.770_776delinsA) was found only in the splenic lesion that was progressing on rucaparib, suggesting that the detected secondary mutation conferred resistance (Table 1; Fig. 3D). To investigate the potential molecular basis of restored function observed with the RAD51D secondary mutation, we conducted molecular dynamics modeling of wild-type RAD51D, as well as RAD51D with both the primary and secondary mutations. Accurate simulation of the primary mutation was not possible because the frameshift-altered sequence diverged significantly from the sequence in the crystal structure. Molecular dynamics simulations indicated that in a DNA–RAD51D homoflament the S–G–R residues, which are replaced with lysine in the secondary mutation, were involved in double-stranded DNA (dsDNA) binding (Supplementary Fig. S8a–b and Supplementary Video), and modeling of the RAD51D with the secondary mutation indicated that the interaction with dsDNA was maintained (Supplementary Fig. S8c). Arginine-to-lysine substitutions are tolerated in evolutionary comparisons of this particular residue (Supplementary Fig. S8D); the interaction with dsDNA suggests that this secondary mutation can confer partial restoration of function.
Figure 3.
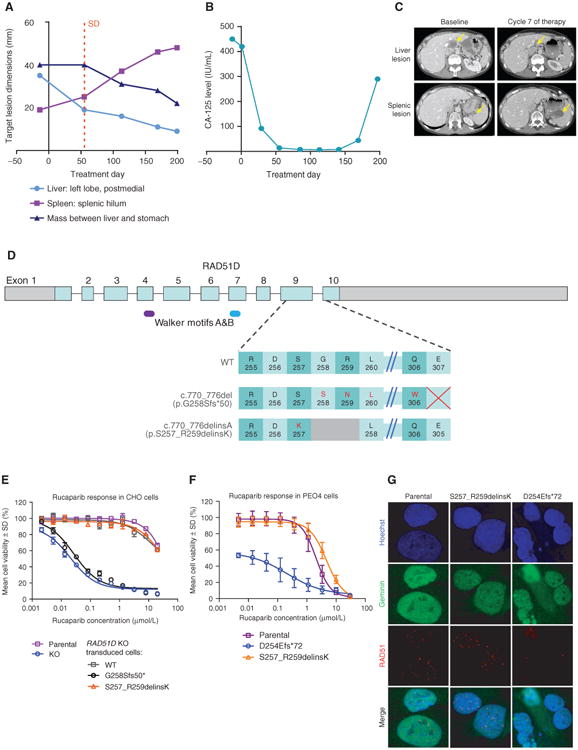
Identification and functional assessment of RAD51D secondary mutation identified in the postprogression biopsy in case 6. A, RECIST measurements of three tumor deposits in the patient with a germline RAD51D mutation (c.770_776del). The metastasis in the left lobe of the liver was biopsied prior to treatment. Surgery was performed following progression on rucaparib in order to remove the enlarging splenic lesion. The tumor deposit in the liver, which was still responding to rucaparib treatment, was also excised. B, Serum CA-125 levels monitored during the ARIEL2 Part 1 trial in the patient with the germline RAD51D mutation (c.770_776del). C, CT images obtained prior to (Baseline), during, and following (Cycle 7) treatment of the patient with the germline RAD51D mutation (c.770_776del). D, Diagram of the predicted RAD51D protein sequence changes caused by the primary mutation (c.770_776del) and the secondary mutation (c.770_776delinsA) detected in the patient with the germline RAD51D mutation. E, In vitro response to rucaparib in parental CHO cell line, CHO RAD51D KO clone, and CHO RAD51D KO clone transduced with WT, primary mutant, or secondary mutant RAD51D transcripts after treatment for 6 days. F, In vitro response to rucaparib in parental PEO4 cell line, PEO4 cells with the homozygous frameshift RAD51D mutation (c.762_763del), and PEO4 cells with the homozygous secondary RAD51D mutation (c.770_776delinsA) after treatment for 7 days. G, RAD51 foci formation 48 hours after rucaparib (10 μmol/L) exposure in geminin-positive cells in parental PEO4, PEO4 cells with the homozygous frameshift RAD51D mutation (c.762_763del), and PEO4 cells with the homozygous secondary RAD51D mutation (c.770_776delinsA). SD, stable disease.
The functional capacity of the primary and secondary RAD51D mutations was further investigated in vitro using a previously described immortalized Chinese hamster (CHO) RAD51D KO cell line (29) and HR-competent human high-grade serous ovarian carcinoma cell line PEO4 (containing functional BRCA2). Introduction of RAD51D cDNA with the secondary mutation (c.770_776delinsA) conferred resistance to cisplatin, rucaparib, and other PARPis, whereas RAD51D cDNA containing the primary mutation did not (Fig. 3E; Supplementary Fig. S9). Two PEO4-derived clones, which contained either a homozygous frameshift mutation (c.762_763del, p.D254Efs*72) in the same exon as the primary frameshift mutation, or the homozygous secondary mutation (c.770_776delinsA, p.S257_R259delinsK) in endogenous RAD51D, were generated using CRISPR homology-directed repair. PEO4 with the frameshift mutation had increased cisplatin and rucaparib sensitivity compared with parental PEO4 or the cells with the secondary mutation (Fig. 3F; Supplementary Fig. S10). Furthermore, RAD51 foci formation assay confirmed proficient HR repair in parental PEO4 and cells expressing the secondary mutation, but not in cells with the frameshift mutation (Fig. 3G).
Discussion
In order to understand the development of secondary resistance to PARPi therapy, we analyzed 12 cases in which paired tumor biopsies were obtained both before treatment and following tumor progression, from patients with high-grade ovarian carcinoma receiving the PARPi rucaparib on the ARIEL2 Part 1 trial. Six of 12 cases were found to contain mutations in one of three DNA repair genes, BRCA1, RAD51C, or RAD51D, prior to therapy. Strikingly, in five of these six cases at progression we identified secondary mutations that restored the open reading frame by NGS of progressing lesions, including two cases that had two or more secondary mutations. In a seventh case, we detected a somatic frameshift mutation in CDK12, a reported regulator of HR, although no secondary mutations were observed in the progressing lesion.
Because secondary mutations in RAD51C and RAD51D have not been previously described in preclinical or clinical studies, we focused in detail on those mutations. Four distinct RAD51C secondary mutations were identified within one core biopsy, highlighting the selective pressure for the tumor cells to restore HR repair in the face of PARPi treatment. The observed microscopic spatial heterogeneity in the relapsed lymph node extends a previous report by Patch and colleagues (14), where 12 distinct secondary BRCA2 events were detected within one autopsy case, including five events, each identified at more than one metastatic site.
Functional analyses demonstrated that the RAD51C cDNA with the secondary mutations were able to restore RAD51C function in RAD51C KO ovarian cancer cells, as evidenced by increased HR, restored ability to bind the RAD51C binding partners RAD51B and XRCC3, and increased resistance to platinum and PARPi. Based on the observed variant frequencies reported for this case, including FISH and WGS analyses, we predict that each primary cancer cell contained three copies of the primary RAD51C mutation, whereas each cell with acquired resistance contained two copies of the primary mutation and one of four possible secondary mutations (Supplementary Fig. S11). We generated a PDX from the postprogression biopsy containing the RAD51C secondary mutations. However, with only one secondary mutation predominant in the biopsy material from which the PDX was generated and in the PDX itself, it was not possible to detect relative functional selection of the four secondary RAD51C mutations present in that lymph node under rucaparib pressure in vivo.
Molecular dynamics modeling of the RAD51D species with the secondary mutation revealed that Ser257–Gly258– Arg259 to Lys substitution maintains the interaction with dsDNA, which was observed in the wild-type RAD51D model. This was further supported by the presence of lysine at codon 259 in nonhuman RAD51D, suggesting that this secondary mutation could confer partial or full restoration of function. In vitro functional analyses of the primary and secondary RAD51D mutations provided additional evidence that the primary mutation sensitized cells to platinum and PARPi compounds, whereas the secondary mutation conferred resistance to both.
These observations provide evidence for restoration of functional HR under PARPi selection pressure in tumors with RAD51C or RAD51D mutations, supporting the view that mutations in these genes are synthetically lethal with PARP inhibition and demonstrating that secondary mutations are an important clinical mechanism of resistance in non-BRCA1/2 HR genes. Moreover, we observed secondary mutations at progression following PARPi therapy in five of six cases containing a primary mutation in a DNA repair gene at diagnosis. Collectively, these results identify the need for sequencing PARPi therapy early during a patient's disease course and highlight the urgent need for development of PARPi-containing combination or sequencing strategies capable of more robust cell killing, in order to circumvent or delay the development of PARPi resistance.
In summary, in vitro and in vivo analyses of primary and secondary mutations in RAD51C and RAD51D provided evidence for secondary mutations restoring most of the open reading frame beyond the primary mutation, thereby reinstituting HR function and contributing to development of clinical resistance to the PARPi rucaparib. Furthermore, these data support the role of primary mutations of RAD51C and RAD51D in conferring PARPi sensitivity and reveal secondary mutations in these genes as a mechanism of acquired PARPi resistance.
Methods
Patient Samples
Archival tumor and a tumor deposit suitable for attempted pre-treatment biopsy were required for all patients who enrolled in the ARIEL2 Part 1 trial (NCT01891344). A postprogression tumor biopsy was optional. RECIST imaging and cancer antigen 125 (CA-125) level monitoring was recorded for the duration of patients' enrollment in the trial.
Cell Lines and Culture
The human ovarian carcinoma cell line OVCAR8 was obtained from the NCI. Early passages of the parental OVCAR8 and RAD51C KO 2-130 were banked, tested for Mycoplasma, and STR profiled; subsequent thaws were used within 6 months. The PEO4 cell line was obtained from F. Couch (Mayo Clinic) in 2013 and viably stored until 2016; subsequent thaws were used within 6 months. The PEO4 cells were routinely tested for Mycoplasma and were last authenticated by STR profiling in April 2017. The CHO cell lines parental and deficient for RAD51D were obtained from Dr. Larry H. Thompson (formerly of Lawrence Livermore National Laboratory) and Dr. Claudia Wiese (Colorado State University, Fort Collins, CO), tested for Mycoplasma, and passaged for 2 months. The MCF10A cells were provided by B.H. Park (John Hopkins University School of Medicine). Early-passage cells obtained were integrated with the DR-GFP reporter and viably stored; subsequent thaws were used within 4 months. These modified cells were used for generating RAD51C-conditional mutants and subsequent experiments. The RAD51C conditional MCF10A cell line tested negative for Mycoplasma on May 10, 2017, using the MycoAlert PLUS assay kit from Lonza.
The OVCAR8 cell line was cultured in RPMI-1640 (Corning) supplemented with 10% FBS (Peak Serum) and 1% penicillin and streptomycin (Corning) in a 5% CO2 atmosphere at 37°C The CHO cell line was cultured in MEM alpha (Corning) with 10% FBS (Peak) and 1% P/S (Corning) in 5% CO2 at 37°C The PEO4 and OVCAR8 cell lines (for RAD51 foci formation assays) were cultured in DMEM/F-12, GlutaMAX Supplement medium (Gibco) containing 5 μg/mL insulin, 50 ng/mL EGF, and 1 μg/mL hydrocortisone in a 5% CO2 atmosphere at 37°C The MCF10A cell line was cultured in DMEM HG/F-12 supplemented with 5% horse serum, 1% penicillin and streptomycin, 100 ng/mL cholera toxin, 20 ng/mL epidermal growth factor, 0.01 mg/mL insulin, and 500 ng/mL hydrocortisone in a 5% CO2 atmosphere at 37°C
Compounds
Rucaparib camsylate salt was manufactured by Lonza. Carboplatin, cisplatin, olaparib, niraparib, talazoparib, and veliparib were obtained from MedChem Express.
Generation of OVCAR8 RAD51C KO (Clone 2-130) and Overexpressing Cell Lines
To generate OVCAR8 RAD51C KO (clone 2-130), OVCAR8 cells were seeded at 0.2 × 106 cells per well in 6-well plates on day 1 in complete media. On day 2, cells were transfected with Fugene 6 (Pro-mega) according to the manufacturer's recommendation with 1 μg RAD51C CRISPR plasmid (Santa Cruz Biotechnology). Transfected cells were selected with 2 μg/mL puromycin (Thermo Fisher Scientific). Cells were single cell cloned by limiting dilution and expanded in complete media without puromycin. RAD51C KO was confirmed by genomic DNA sequencing. DNA was isolated using PureLink Genomic DNA mini kit (Thermo Fisher Scientific), and PCR was performed using KOD Hot Start Master Mix (EMD Millipore) with the follow primers: forward primer 5′-gcagaagccttagaaactctgc and reverse primer 5′-tgaataacgcagaaacttcctg, according to the manufacturer's recommendation. The PCR product was purified using a QIAquick PCR Purification kit (Qiagen) and Sanger sequencing was performed with primer 5′-tttcattaagggcactccacc. RAD51C clone 2–130 sequences showed 35-bp deletion RAD51C c.231_264 that generated nonsense mutation p.E80*.
To generate cells that transiently express wild-type and mutated RAD51C for RAD51 foci analysis, cells were transfected with commercially obtained, mutated RAD51C pcDNA3.1(+)/Hygro plasmids (OHu21400C, GenScript) using Lipofectamine 3000 Reagent (Thermo Fisher Scientific), following the manufacturer's instructions. After a 48-hour recovery period, transfected cells were selected with 200 μg/mL hygromycin (Thermo Fisher Scientific). To generate stable overexpressing RAD51C mutants, gene fragments containing specific mutations were synthesized (Integrated DNA Technologies) and cloned into the lentivector encoding RAD51C open reading frame under the control of the PGK promoter (Genecopoeia). Lentivirus vectors were packaged in HEK293 cells with third-generation lentiviral system (System Biosciences), and RAD51C 2-130 cells were transduced with 15 MOI plus polybrene 8 μg/mL for 24 hours. Cells were cultured in complete media for 48 hours before puromycin selection.
Generation of CHO Cell Lines Overexpressing Primary and Secondary RAD51D Mutations
To generate stable cell lines overexpressing RAD51D mutants, gene fragments containing specific mutations were synthesized (Integrated DNA Technologies) and cloned into the lentivector encoding RAD51D open reading frame (NM_002878.3) under the control of the PGK promoter (Genecopoeia). Lentivirus vectors were packaged in HEK293 cells with a third-generation lentiviral system (System Biosciences), and CHO RAD51D KO cells (29) were transduced with 15 MOI plus polybrene 8 μg/mL for 24 hours. Cells were cultured in complete media for 48 hours before puromycin selection.
Generation of PEO4 Cell Line with the Secondary RAD51D Mutation
To generate PEO4 cells with the secondary RAD51D mutation (c.770_776delinsA), PEO4 cells were transduced with lentiviral Cas9 vector (PUCas9Cherry) and doxycycline-inducible CRISPR guide with GFP vector (FgH1tUTG) with CRISPR guide 5′-CAACCACAT AACTCGAGACA (30). ssODN (40 pmol/L) containing the secondary mutation and a silent PAM mutation with 80-bp sequence overlap on each side (IDT) was transfected using Lipofectamine 3000 Reagent (Thermo Fisher Scientific), following the manufacturer's instructions. After overnight incubation, CRISPR guide was activated by doxycycline-supplemented medium (1 μg/mL) for 3 days. mCherry and GFP double-positive cells were single-cell plated using flow cytometry. After expanding for at least 2 weeks, single-cell colonies were sequenced for the presence of homozygous secondary mutation using a MiSeq platform. The colony with the frameshift RAD51D mutation (c.762_763del, p.D254Efs*72) was also selected from this process.
RAD51 Foci Formation Assay
Cells were treated with DMSO or 10 μmol/L rucaparib for 24, 48, or 72 hours. Cells were fixed with either 4% paraformaldehyde or methanol, permeabilized with 0.2% TritonX-100, blocked with blocking buffer (1% BSA, 2% FCS or 1% BSA, 3% milk, 2% goat serum in PBS) and incubated with rabbit anti-RAD51 (ab133534 1:250; ab11055 1:400; Abcam) and either mouse anti-Geminin (ab104306 1:100; Abcam) or mouse anti-γH2AX (ab26350 1:400; Abcam) antibodies. For RAD51 foci formation with geminin staining, anti-rabbit 647, and anti-mouse 546 Alexa Fluor secondary antibodies (1:800; Invitrogen Molecular Probes) were used. Nuclei were counterstained with Hoechst 33342 (NucBlue Live ReadyProbes Reagent; Ther-mofisher Scientific). Cells were imaged using an LSM 780 inverse laser scanning microscope (Zeiss) and captured with a LSM T-PMT detector (Zeiss). At least 194 cells from four fields of view and two independent experiments were counted. Cells with ≥5 RAD51 foci/nucleus were scored using CellProfiler (version 2.2.0, Broad Institute). For RAD51 and γH2AX foci formation assay, anti-rabbit 488 and anti-mouse 594 Alexa Fluor secondary antibodies (A32731 1:500; A-11032 1:500; Invitrogen Molecular Probes) were used. Nuclei were counterstained with DAPI (Sigma). Cells were imaged using Leica DM 1000 LED at 40×.
Cell Viability Assays
Endpoint viability assays were performed using the CellTiter-Glo (Promega) assay according to the manufacturer's protocol. Briefly, cells were seeded at 600 to 800 cells per in 384-well plates or 2,000 cells in 96-well plates and allowed to establish overnight before adding treatments. Cells were treated for 6 to 7 days with compounds, over a range of concentrations, then the assay was terminated and viability assessed using luminescence detection on a Victor X4 plate reader (Perkin Elmer). Luminescence was normalized to DMSO control, and IC50 values were calculated using a sigmoidal dose–response curve fit analysis (Prism software, GraphPad).
HR Reporter Assay
The DR-GFP reporter was introduced into MCF10A cells as previously described (31). Cre recombinase was expressed in conditional RAD51C−/− MCF10A cells to remove an ectopic floxed RAD51C gene (R. Prakash and M. Jasin, unpublished) and in isogenic wild-type cells as a control. After Cre expression, cells were infected with an I-SceI– expressing lentivirus. GFP+ cells were measured by flow cytometry (BD FACScan) 48 hours after infection, and data were analyzed using FlowJo software. Without I-SceI expression, the number of GFP+ cells was ≤0.01.
Creation of Y2H and pWZL Expression Vectors
The RAD51C mutants were generated in the Y2H plasmids (pGAD-C1 and pGBD-C1) and pWZL plasmid using site-directed mutagenesis (Supplementary Table S5). RAD51C and RAD51B cDNA were subcloned into the pGAD-C1 and pGBD-C1 vectors using 5′-EcoRI and 3′-SalI restriction sites. The RAD51C cDNA and RAD51B cDNA were a gift from Jun Huang (32), and the pGAD-XRCC3 and pGBT-XRCC3 plasmids were a gift from David Schild (33).
Y2H Assay
The Y2H experiments were performed as previously described (34), except that the indicated GAL4 activating domain– and binding domain–expressing vectors were cotransformed into the YPJ694a yeast strain.
BRCA1 Colony PCR
Genomic DNA (10 ng) from postprogression biopsy (case 4) was PCR amplified with primers: forward 5′-gcatgtcgacGGGAACTAAC CAAACGGAGCA and reverse 5′-atgcaagcttGAGATCTTTGGGG TCTTCAGCA. Primers were designed with restriction sites for Sal1 and HindIII. Reaction was performed with KOD polymerase (EMD Millipore) according to the manufacturer's recommendation and cleaned by a QIAquick PCR kit (Qiagen). pUC19 (NEB) and PCR product were cut with Sal1 (NEB) and HindIII (NEB), and cleaned by a QIAquick PCR kit, before ligating with T4 ligase (NEB) and transforming Escherichia coli bacteria. Colonies were amplified by rolling circle amplification using bacteriophage phi29 DNA polymerase (NEB), before Sanger sequencing with M13 Forward-20 primer.
Western Blotting: OVCAR8, CHO, and PEO4
Whole-cell lysates were prepared using RIPA buffer (Sigma) containing protease inhibitor cocktail, and protein concentration was assessed by BCA colormetric protein determination (Pierce). Equal protein loads were resolved on precast 4% to 12% Bis-Tris gels under reducing conditions. Protein was transferred to nitrocellulose membranes using iBlot dry transfer method (Invitrogen), then probed with primary antibody anti-RAD51C (sc-398819 1:1,000; Santa Cruz Biotechnology), anti-RAD51D n-terminal (ab202063 1:1,000; Abcam), anti-RAD51D c-terminal (sc-398819 1:1,000; Santa Cruz Biotechnology), anti-GAPDH (#5174 1:1,000; Cell Signaling Technology), anti-tubulin (#2128 1:1,000; Cell Signaling Technology) or anti-actin (ab8229 1:1,000; Abcam) followed by peroxidase-labeled secondary antibody (sc-2020 1:5,000; Santa Cruz Biotechnology) and visualized by enhanced chemiluminescence (SuperSignal Chemiluminescent Substrate; Thermo Scientific) or IRDye-labeled secondary antibody (LI-COR Biotechnology) according to the manufacturer's instructions. Band volume analysis was conducted using Odyssey Fc (LI-COR Biotechnology).
Western Blotting: MCF10A (Subcellular)
Nuclear extracts were collected from MCF10A cells using the cytoplasmic lysis buffer (10 mmol/L Tris-HCl, 0.34 mol/L sucrose, 3 mmol/L CaCl2, 2 mmol/L magnesium acetate, 0.1 mmol/L EDTA, 0.5% NP-40, protease inhibitor) and nuclear lysis buffer (20 mmol/L HEPES, 3 mmol/L EDTA, 10% glycerol, 150 mmol/L potassium acetate, 1.5 mmol/L MgCl2, 0.1% NP-40, protease inhibitor). Nuclear protein (30 μg) was used for detection. Protein was separated on 10% acrylamide gels and transferred to PVDF membranes, and protein was detected on a LiCor CLX scanner. RAD51C expression was detected with RAD51C antibody (ab55728 1:500; Abcam), and equal nuclear loading was detected using PCNA antibody (sc-56 1:250; Santa Cruz Biotechnology), and IR dye secondary antibodies from LiCor Biosciences. The image was adjusted for brightness and contrast using Photoshop (Adobe Systems Inc.).
Western Blotting: Y2H
Yeast expressing the indicated AD and BD constructs was grown overnight at 30°C in 5 mL YPD and then diluted to 0.2 OD600 in YPD for 90 minutes. Whole-cell lysates of equal cell numbers (0.2 OD600) were extracted by TCA precipitation. Protein was separated on 10% acrylamide gels and transferred to PVDF membranes, and protein was detected on a LiCor CLX scanner. RAD51C expression was detected with RAD51C antibody (ab55728 1:500; Abcam), equal loading was detected using a Kar2 antibody (sc-33630 1:2,000; Santa Cruz Biotechnology) and IR dye secondary antibodies (1:20,000) from LiCor Biosciences. The image was adjusted for brightness and contrast using Photoshop (Adobe Systems Inc.).
Generation and Treatment of PDX
All experiments involving animals were approved by the Walter and Eliza Hall Institute of Medical Research Animal Ethics Committee. A PDX was generated from the postprogression lymph node biopsy by transplanting fresh fragments subcutaneously into six NOD/SCID IL2Rγnull recipient mice (T1, passage 1; ref. 35), three of which developed tumors. The PDX tumors generated were transplanted into recipient mice (T2, passage 2), minced and cryopreserved in DMSO and snap-frozen for further analysis. A mouse harboring a T2 PDX tumor was treated with 450 mg/kg rucaparib for 2 weeks (oral gavage once daily Monday–Friday) at ≈0.1 cm3 in size. The tumor was harvested and snap-frozen 24 hours after treatment completion. DNA was extracted from the archival tumor sample and from the postprogression biopsy for sequencing analysis. SNP array analysis was performed on archival tissue and the postprogression biopsy, and WGS was performed on the postprogression biopsy and twoT1 PDXs.
Serial Section Analysis
A small fragment of one of the snap-frozen postprogression biopsy cores from patient 5 with the germline RAD51C mutation was embedded in Tissue-Tek optimal cutting temperature (OCT) compound (Sakura Finetek). Six serial sections were collected (40 μm each) with a 4-μm section cut for hematoxylin and eosin staining in between each section. Direct PCR was performed on each serial section using the Phusion Human Specimen Direct PCR Kit (Thermo Scientific) following the manufacturer's instructions. Briefy, 25 μL of buffer and 0.5 μL of DNA release reagent was added to each tube with section scrolls and incubated at 98°C for 2 minutes. Amplicon libraries were prepared using a two-step PCR approach: first internal PCR to amplify the region of interest (5′-tcgtcggcagcgtcagatgtgtataagagacag-tccaaaggagaacattttgtta forward primer and 5′-gtctcgtgggctcggagat gtgtataagagacag-tgtgtagtcacgacagcgaaa reverse primer), followed by second outer PCR to add the sequencing adaptors and indexes for multiplexing). Briefly, 2 μL of digested section sample for first PCR or inner product for second PCR was added to 5 μL of 5 × Q5 Reaction buffer (NEB), 1 μL of each primer (forward and reverse; 2 nmol/L each) for first PCR or 1 μL of each Nextera XT index (unique i5 and i7, Illumina), 0.5 μL of dNTPs (10 mmol/L each), 0.25 μL of Q5 Hot Start High-Fidelity DNA Polymerase (NEB), and 15.25 μL of dH2O. The PCR was performed as follows: 30 seconds at 98°C for initial denaturation, followed by 20 cycles of 10 seconds at 98°C, 15 seconds at 60°C for frst PCR or 63°C for second PCR, and 20 seconds at 72°C, followed 2 minutes at 72°C for final extension. The libraries were cleaned using standard Agencourt AmpureXP beads (Beckman Coulter) procedure with DNA to bead ratio of 1:0.9, and normalized to 1 nmol/L concentration (using the Qubit dsDNA HS Assay Kit for quantification). Libraries were sequenced using MiSeq Nano v2 300-cycle kit (Illumina) at 10 pmol/L final concentration to a minimum depth of 30,000×. Reads were aligned using bwa-mem to the Human GRCh38 genome, and visualized for further analysis using the IGV browser (36).
Genomic Analysis
All tumors were sequenced using Foundation Medicine's NGS-based T5 assay (19). Analyzed data were plotted using OncoPrint. Germline HRR gene mutations were confirmed by sequencing of DNA extracted from blood using the NGS-based BROCA assay (37).
WGS libraries were prepared for a postprogression biopsy of patient 5 and the two first-passage PDX tumors generated from this biopsy. The libraries were prepared using the TruSeq Nano library preparation kit (Illumina), and the sequencing was performed on the Illumina × Ten platform (Kinghorn Centre for Clinical Genomics, Darlinghurst).
Adaptor sequences were removed with Trimmomatic 0.36 (38) before mapping to the Human GRCh38 (GCA_000001405.15) and Mouse GRCm38 (GCF_000001635.25) genomes with Bowtie2 2.2.5 (39). Human reads were separated from mouse background with Xenomapper (40). Reads were sorted and indexed with Samtools 1.3.1 (41). Copy-number analysis was performed with HMMcopy 1.16 (42) and plotted with CIRCOS 0.67 (43). Coriell Cell Repository NA12878 reference cell line DNA previously processed with the TruSeq Nano kit at KCCG was used as an unrelated normal control.
High-depth amplicon analysis of RAD51C exon 4 was performed on the multiple samples from patient 5 with germline RAD51C mutation (pretreatment biopsy, postprogression biopsy, and 4 PDX tumors) in triplicate. DNA was extracted using a QIAamp DNA mini kit (Qiagen). Amplicon libraries were prepared using a two-step PCR approach, as described for serial section analysis, with 20 ng DNA input in the first PCR. The PCR was performed as follows: 30 seconds at 98°C for initial denaturation, followed by 16 cycles for first PCR or 12 cycles for second PCR of 10 seconds at 98°C, 15 seconds at 60°C for first PCR or 63°C for second PCR, and 20 seconds at 72°C, followed by 2 minutes at 72°C for final extension. The libraries were cleaned and sequenced as described for serial section analysis to a minimum depth of 10,000×. Reads were aligned using bwa-mem to the Human GRCh38 genome and visualized for further analysis using IGV browser (36).
RAD51C FISH
Frozen OCT sections from the postprogression biopsy (Core 2 OCT block for serial sectioning) and the PDX sample treated with 450 mg/kg rucaparib for 2 weeks, both from patient 5 with the germline RAD51C mutation, as well as an unrelated control PDX were thawed, fixed in 4% paraformaldehyde, and pretreated with a SPOT-Light Tissue Pretreatment kit (Invitrogen). Briefly, sections were incubated in pretreatment solution for 15 minutes at 95°C, washed in PBS, and incubated with enzyme for 10 minutes at room temperature. Sections were dehydrated, incubated with denaturation buffer (70% formamide, 2× SSC, pH 7.0–8.0) for 5 minutes at 73°C, dehydrated, and incubated with prepared RAD51C probe (as per the manufacturer's instructions; Empire Genomics) for 24 hours at 37°C. Sections were then washed with WS1 (0.4× SSC/0.3% NP-40) for 2 minutes at 73°C, followed by a wash with WS2 (2× SSC/0.1% NP-40) for 1 minute at room temperature. Nuclei were counterstained with Hoechst 33342 (NucBlue Live ReadyProbes Reagent; Thermofisher Scientific) and coverslipped with fluoromount G (SouthernBiotech). Sections were imaged using an LSM 780 inverse laser scanning microscope (Zeiss) and captured with an LSM T-PMT detector (Zeiss).
Molecular Dynamics Modeling
Molecular models were made of RAD51D to test the effects of deletions and mutations using NAMD (44). Initial models were constructed from primary sequence using the Swiss-Model web server (45), which constructed a model using the Saccharomyces cerevisiae 1SZP pdb structure as a template (46). The model was constructed as a 6 monomer helical complex with an additional 58-bp double-stranded DNA loosely positioned along the central helical axis. A homomeric model was constructed, as appropriate crystal structures were not available to allow modeling of the heteromeric RAD51B/C/D and XRCC2 complex. This model provides general information on the interaction between the subunits and between the protein and DNA. RAD51 paralogues and RecA have been shown to bind both ssDNA in an ATP-catalyzed reaction and dsDNA in a filament structure. As a computational simplification, we chose to model only the interaction with dsDNA. A wild-type model and two mutant models were constructed (p.S257_R259delinsK and p.G258Sfs*50, see Fig. 3D for annotation) using VMD (47) and the psfgen module. All models were solvated and ionized with sodium chloride to approximately 0.15 mol/L and electrical neutrality. The initial dimensions of the wild-type system were 108 × 108 × 200 Angstroms, with a total of 222,339 atoms for the WT, and 230,572 or 226,326 atoms for the primary and secondary mutants, respectively. Each model was equilibrated for 1 ns before performing production runs. Production runs were performed using NAMD 2.10 at 310K using a NPT ensemble (constant pressure and temperature). Long-range Coulomb forces were computed with the Particle Mesh Ewald method with a grid spacing of 1 Å. 2 fs time steps were used with nonbonded interactions calculated every 2 fs and full electrostatics every 4 fs while hydrogens were constrained with the SHAKE algorithm. The cutoff distance was 12 Å with a switching distance of 10 Å and a pair-list distance of 14 Å. Pressure was controlled to 1 atmosphere using the Nosé-Hoover Langevin piston method using a piston period of 100 fs and a piston decay of 50 fs. Trajectory frames were captured every 100 ps. Simulation trajectories were viewed with VMD (47).
Supplementary Material
Acknowledgments
We thank D. Schild for XRCC3 Y2H plasmids, J. Huang for the cDNA used to make the RAD51C and RAD51B Y2H vectors, Drs. Larry Thompson and Claudia Wiese for their generous gift of the RAD51D CHO cell lines, Dr. Marco Herald for the Cas9/CRISPR vectors, and T. Phuong and E. Lieschke for technical assistance. We acknowledge Elaina Mann and Lan-Thanh Vo (Clovis Oncology) for tumor specimen shipment and processing. We thank Kyle Gowen and Joel Greenbowe (Foundation Medicine) for assistance in modeling of tumor clonal fractions with secondary RAD51C mutations. On behalf of the Australian Ovarian Cancer Study, David Bowtell gratefully acknowledges the cooperation of the participating institutions in Australia and also acknowledges the contribution of the study nurses, research assistants, and all clinical and scientific collaborators to the study. We would like to thank all of the women who participated in these research programs.
Grant Support: This work was supported by fellowships and grants from the National Health and Medical Research Council (NHMRC Australia; Project grant 1062702 to C.L. Scott); the Cancer Council Victoria (Sir Edward Dunlop Fellowship in Cancer Research to C.L. Scott); the Victorian Cancer Agency (Clinical Fellowships CRF10-20 and CRF16014 to C.L. Scott); CRC for Cancer Therapeutics (PhD top-up scholarship to G.-Y. Ho); the Stafford Fox Medical Research Foundation including a WEHI Centenary Stafford Fox Fellowship to H. Barker; the National Institutes of Health (2P50CA083636 to E.M. Swisher); and the Wendy Feuer Ovarian Cancer Research Fund (to E.M. Swisher). This work was made possible through the Australian Cancer Research Foundation, the Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS. We thank Clovis Oncology for funding Foundation Medicine's analyses and providing rucaparib for in vivo experiments. K.A. Bernstein is supported by a National Institutes of Health Grant (ES024872), the V Foundation for Cancer Research, and a Stand Up To Cancer Innovative Research Grant (Grant Number SU2C-AACR-IRG-02-16). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. R. Prakash was supported by NIH F32GM110978. M. Jasin is supported by NIH R01CA185660 and the MSK Cancer Center Support Grant/Core Grant (NIH P30CA008748). E.M. Kass, E.M. Swisher, M. Jasin, R.L. Coleman and S.H. Kaufmann are supported by a Stand Up To Cancer–Ovarian Cancer Research Fund–Ovarian Cancer National Alliance–National Ovarian Cancer Coalition Dream Team Translational Research Grant (Grant Number: SU2C-AACR-DT16-15). MD modeling was supported by Victorian Life Sciences Computation Initiative grant LTU0011 on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government, Australia. The Australian Ovarian Cancer Study was supported by the U.S. Army Medical Research and Materiel Command under DAMD17-01-1-0729, The Cancer Council Tasmania, The Cancer Foundation of Western Australia, and the National Health and Medical Research Council of Australia (NHMRC; ID400413 and ID400281).
The management group of the AOCS group included D. Bowtell, G. Chenevix-Trench, A. Green, P. Webb, A. DeFazio, and D. Gertig. The project and data managers included N. Trafcante, S. Fereday, S. Moore, J. Hung, K. Harrap, T. Sadkowsky, and N. Pandeya. The Research Nurses and Assistants of the AOCS group included M. Malt, A. Mellon, R. Robertson, T. Vanden Bergh, M. Jones, P. Mackenzie, J. Maidens, K. Nattress, Y.E. Chiew, A. Stenlake, H. Sullivan, B. Alexander, P. Ashover, S. Brown, T. Corrish, L. Green, L. Jackman, K. Ferguson, K. Martin, A. Martyn, B. Ranieri, J. White, V. Jayde, P. Mamers, L. Bowes, L. Galletta, D. Giles, J. Hendley, K. Alsop, T. Schmidt, H. Shirley, C. Ball, C. Young, S. Viduka, H. Tran, S. Bilic, L. Glavinas, and J. Brooks. The Clinical and Scientific Collaborators of the AOCS group included R. Stuart-Harris, F. Kirsten, J. Rutovitz, P. Clingan, A. Glasgow, A. Proietto, S. Braye, G. Otton, J. Shannon, T. Bonaventura, J. Stewart, S. Begbie, M. Friedlander, D. Bell, S. Baron-Hay, A. Ferrier (dec.), G. Gard, D. Nevell, N. Pavlakis, S. Valmadre, B. Young, C. Camaris, R. Crouch, L. Edwards, N. Hacker, D. Marsden, G. Robertson, P. Beale, J. Beith, J. Carter, C. Dalrymple, R. Houghton, P. Russell, M. Links, J. Grygiel, J. Hill, A. Brand, K. Byth, R. Jaworski, P. Harnett, R. Sharma, G. Wain, B. Ward, D. Papadimos, A. Crandon, M. Cummings, K. Horwood, A. Obermair, L. Perrin, D. Wyld, J. Nicklin, M. Davy, M.K. Oehler, C. Hall, T. Dodd, T. Healy, K. Pittman, D. Henderson, J. Miller, J. Pierdes, P. Blomfield, D. Challis, R. McIntosh, A. Parker, B. Brown, R. Rome, D. Allen, P. Grant, S. Hyde, R. Laurie, M.Robbie, D. Healy, T. Jobling, T. Manolitsas, J. McNealage, P. Rogers, B. Susil, E. Sumithran, I. Simpson, K. Phillips, D. Rischin, S. Fox, D.Johnson, S. Lade, M. Loughrey, N. O'Callaghan, W. Murray, P. Waring, V. Billson, J. Pyman, D. Neesham, M. Quinn, C. Underhill, R. Bell, L.F.Ng, R. Blum, V. Ganju, I. Hammond, Y. Leung, A. McCartney (dec.), M.Buck, I. Haviv, D. Purdie, D. Whiteman, and N. Zeps.
Footnotes
note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Disclosure of Potential Conficts of Interest: A.V. Tinker reports receiving commercial research support from AstraZeneca. M. Friedlander is a consultant/advisory board member for AstraZeneca. D.M. O'Malley is a consultant/advisory board member for Clovis, AstraZeneca, Tesaro, Novocure, Genentech/Roche, Janssen, and Eisai, and is on the steering committee for Amgen. T.C. Harding has ownership interest (including patents) in Clovis Oncology. I.A. McNeish is a consultant/advisory board member for Clovis Oncology. C.L. Scott has received speakers bureau honoraria from Prime Oncology and is a consultant/advisory board member for Clovis Oncology and AstraZeneca. No potential conficts of interest were disclosed by the other authors.
Authors' Contributions: Conception and design: M. Nguyen, M. Jasin, R.L. Coleman, A. Oza, M.J. Wakefeld, S.H. Kaufmann, A.D. Simmons, T.C. Harding, M. Raponi, E.M. Swisher, K.K. Lin, C.L. Scott
Development of methodology: O. Kondrashova, M. Nguyen, K. Shield-Artin, M.I. Harrell, H. Barker, R. Prakash, E.M. Kass, M.R. Sullivan, G.J. Brunette, K.A. Bernstein, R.L. Coleman, A. Oza, J. Sun, M.J. Wakefeld, T.C. Harding
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): O. Kondrashova, M. Nguyen, K. Shield-Artin, A.V. Tinker, N.N.H. Teng, M.I. Harrell, G.-Y. Ho, H. Barker, M. Jasin, R. Prakash, E.M. Kass, M.R. Sullivan, G.J. Brunette, R.L. Coleman, A. Floquet, M. Friedlander, G. Kichenadasse, D.M. O'Malley, A. Oza, J. Sun, L. Maloney, D. Bowtell, I.A. McNeish, E.M. Swisher, C.L. Scott
Analysis and interpretation of data (e.g., statistical analysis, bio-statistics, computational analysis): O. Kondrashova, M. Nguyen, K. Shield-Artin, M.I. Harrell, M.J. Kuiper, H. Barker, M. Jasin, R. Prakash, E.M. Kass, M.R. Sullivan, G.J. Brunette, K.A. Bernstein, R.L. Coleman, A. Oza, J. Sun, L. Maloney, M.J. Wakefeld, A.D. Simmons, T.C. Harding, E.M. Swisher, K.K. Lin, C.L. Scott
Writing, review, and/or revision of the manuscript: O. Kondrashova, M. Nguyen, K. Shield-Artin, A.V. Tinker, N.N.H. Teng, M.I. Harrell, H. Barker, M. Jasin, R. Prakash, E.M. Kass, M.R. Sullivan, G.J. Brunette, K.A. Bernstein, R.L. Coleman, A. Floquet, M. Friedlander, G. Kichenadasse, D.M. O'Malley, A. Oza, L. Robillard, L. Maloney, D. Bowtell, H. Giordano, M.J. Wakefeld, S.H. Kaufmann, A.D. Simmons, T.C. Harding, M. Raponi, I.A. McNeish, E.M. Swisher, K.K. Lin, C.L. Scott
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M. Nguyen, M.J. Kuiper, K.A. Bernstein, L. Robillard, L. Maloney, H. Giordano, M. Raponi, E.M. Swisher
Study supervision: N.N.H. Teng, M. Jasin, K.A. Bernstein, R.L. Coleman, A. Oza, L. Maloney, T.C. Harding, M. Raponi, I.A. McNeish, E.M. Swisher, K.K. Lin, C.L. Scott
Other (conception and design specifically to one portion of the study on the analysis of RAD51C mutations): M. Jasin
References
- 1.Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-Ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol. 2015;33:1397–406. doi: 10.1200/JCO.2014.58.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–17. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 3.Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–54. doi: 10.1158/2159-8290.CD-15-0714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y, O'Connor MJ, De Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8:362ps17. doi: 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- 5.Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–8. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alsop K, Fereday S, Meldrum C, deFazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30:2654–63. doi: 10.1200/JCO.2011.39.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852–61. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- 8.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375:2154–64. doi: 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 9.Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87. doi: 10.1016/S1470-2045(16)30559-9. [DOI] [PubMed] [Google Scholar]
- 10.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 12.Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29:3008–15. doi: 10.1200/JCO.2010.34.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stover EH, Konstantinopoulos PA, Matulonis UA, Swisher EM. Bio-markers of response and resistance to DNA repair targeted therapies. Clin Cancer Res. 2016;22:5651–60. doi: 10.1158/1078-0432.CCR-16-0247. [DOI] [PubMed] [Google Scholar]
- 14.Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489–94. doi: 10.1038/nature14410. [DOI] [PubMed] [Google Scholar]
- 15.Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229:422–9. doi: 10.1002/path.4140. [DOI] [PubMed] [Google Scholar]
- 16.Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20:764–75. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 19.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profl-ing test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song H, Dicks E, Ramus SJ, Tyrer JP, Intermaggio MP, Hayward J, et al. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol. 2015;33:2901–7. doi: 10.1200/JCO.2015.61.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prakash R, Zhang Y, Feng W, Jasin M. Homologous recombination and human health: the roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb Perspect Biol. 2015;7:a016600. doi: 10.1101/cshperspect.a016600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2:482–90. doi: 10.1001/jamaoncol.2015.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42:410–4. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 24.Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43:879–82. doi: 10.1038/ng.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J Biol Chem. 2014;289:9247–53. doi: 10.1074/jbc.M114.551143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ekumi KM, Paculova H, Lenasi T, Pospichalova V, Bosken CA, Rybarikova J, et al. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015;43:2575–89. doi: 10.1093/nar/gkv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonatti F, Pepe C, Tancredi M, Lombardi G, Aretini P, Sensi E, et al. RNA-based analysis of BRCA1 and BRCA2 gene alterations. Cancer Genet Cytogenet. 2006;170:93–101. doi: 10.1016/j.cancergencyto.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, et al. The BRCA1- 11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76:2778–90. doi: 10.1158/0008-5472.CAN-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinz JM, Tebbs RS, Wilson PF, Nham PB, Salazar EP, Nagasawa H, et al. Repression of mutagenesis by Rad51D-mediated homologous recombination. Nucleic Acids Res. 2006;34:1358–68. doi: 10.1093/nar/gkl020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aubrey BJ, Kelly GL, Kueh AJ, Brennan MS, O'Connor L, Milla L, et al. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Reports. 2015;10:1422–32. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 31.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–8. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu T, Wan L, Wu Y, Chen J, Huang J. hSWS1•SWSAP1 is an evolutionarily conserved complex required for efficient homologous recombination repair. J Biol Chem. 2011;286:41758–66. doi: 10.1074/jbc.M111.271080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schild D, Lio YC, Collins DW, Tsomondo T, Chen DJ. Evidence for simultaneous protein interactions between human Rad51 paralogs. J Biol Chem. 2000;275:16443–9. doi: 10.1074/jbc.M001473200. [DOI] [PubMed] [Google Scholar]
- 34.Godin SK, Meslin C, Kabbinavar F, Bratton-Palmer DS, Hornack C, Mihalevic MJ, et al. Evolutionary and functional analysis of the invariant SWIM domain in the conserved Shu2/SWS1 protein family from Saccharomyces cerevisiae to Homo sapiens. Genetics. 2015;199:1023–33. doi: 10.1534/genetics.114.173518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Topp MD, Hartley L, Cook M, Heong V, Boehm E, McShane L, et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol Oncol. 2014;8:656–68. doi: 10.1016/j.molonc.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci. 2011;108:18032–7. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wakefield MJ. Xenomapper: mapping reads in a mixed species context. JOSS. 2016;1:18. [Google Scholar]
- 41.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ha G, Roth A, Lai D, Bashashati A, Ding J, Goya R, et al. Integrative analysis of genome-wide loss of heterozygosity and monoallelic expression at nucleotide resolution reveals disrupted pathways in triple-negative breast cancer. Genome Res. 2012;22:1995–2007. doi: 10.1101/gr.137570.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Research. 2009;19:1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protocols. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 46.Conway AB, Lynch TW, Zhang Y, Fortin GS, Fung CW, Symington LS, et al. Crystal structure of a Rad51 filament. Nat Struct Mol Biol. 2004;11:791–6. doi: 10.1038/nsmb795. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.