

Abstract
Peptides with deamidated asparagine residues and oxidized methionine residues are often not resolved sufficiently to allow quantitation of their native and modified forms using reversed phase (RP) chromatography. The accurate quantitation of these modifications is vital in protein biotherapeutic analysis because they can affect a protein’s function, activity, and stability. We demonstrate here that hydrophilic interaction liquid chromatography (HILIC) adequately and predictably separates peptides with these modifications from their native counterparts. Furthermore, coefficients describing the extent of the hydrophilicity of these modifications have been derived and were incorporated into a previously made peptide retention prediction model that is capable of predicting the retention times of peptides with and without these modifications.
Keywords: Deamidation of asparagine, Oxidation of methionine, Hydrophilic interaction liquid chromatography (HILIC), Mass spectrometry (MS), Separation, Quantitation, Modified peptides, Protein biotherapeutics, Peptide prediction retention model
Graphical abstract

Introduction
Many of the chemical modifications that accumulate in biotherapeutic agents during bioprocessing, purification, storage, or other stages increase the hydrophilicity of the amino acid side chain on the altered residue(s). These modifications can include the oxidation of methionine and the deamidation of asparagine, among others. The separation and quantitation of peptides that have these modifications is of paramount importance in protein biotherapeutics because the modifications can contribute to a loss of stability or activity [1–3].
Although little is known about the effects that deamidation of asparagine have on protein function, it is known that deamidation is involved in protein degradation and development [4–7]. This reaction is spontaneous and non-enzymatic, where asparagine residues undergo formation of a five-membered succinimide ring intermediate from an intramolecular attack, and subsequently hydrolyze under physiological conditions to form either aspartyl or isoaspartyl peptides, which can be found in both the D and L configurations (Figure 1). Deamidation occurs at a much faster rate (up to 70 times) when an unhindered amino acid residue such as glycine is on the C-terminal side of an asparagine in the primary sequence (XXX-Asn-Gly-XXX), but its rate is also affected by other conditions and characteristics such as temperature, pH, and protein structure [1, 4, 5, 8–12]. As deamidation changes the peptide/protein structure and conformation, it can significantly affect the function and stability of proteins. For example, deamidation of an Asn-Gly site in hemoglobin alters its affinity for oxygen, while the same modification alters the proteolytic cleavage of human growth hormone (hGH) [13, 14]. Deamidation has been studied using different analytical techniques, such as isoelectric focusing, capillary electrophoresis, and a variety of LC-MS/MS techniques, but they all have limitations that make analyzing deamidation a challenge [1, 7]. The greatest challenge for mass spectrometric analysis of deamidated proteins is that there is only a 1 Dalton mass shift between the modified and native forms, which causes the deamidated species to overlap with the mass-to-charge (m/z) ratios of the 13C isotopes of the unmodified species [15]. Without employing a separation technique that can fully distinguish the modified and unmodified versions, mass spectrometric analysis of deamidation can be highly challenging.
Figure 1.
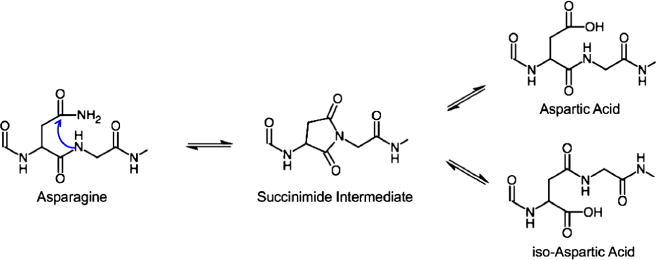
The deamidation of asparagine mechanism. Asparagine forms a five-membered succinimide ring intermediate from an intramolecular attack, and then hydrolyzes to form either aspartyl and isoaspartyl peptides (created using ChemDoodle by iChemLabs)
More is known about the oxidation of methionine compared with the deamidation of asparagine, presumably because the larger mass difference between modified and unmodified peptides makes it easier to study this post-translational modification (PTM) by mass spectrometry. The oxidation of methionine has been shown to affect the structure, stability, and biological functions of a variety of proteins, and is a major instability factor of protein pharmaceuticals, including monoclonal antibodies [16–18]. It is also associated with the development of several diseases, including Alzheimer’s disease, emphysema, and respiratory distress syndrome, among others [18–23]. Methionine (Met-S) can oxidize to form methionine sulfoxide (Met-SO) via a formal oxygen transfer, which can further oxidize to form methionine sulfone (Met-SO2) as shown in Figure 2. Met-SO can be reduced back to methionine using methionine sulfoxide reductase A (MsrA), which is found in most cells. MsrA has been shown to be important in Alzheimer’s disease, as the levels of MsrA in the brain of Alzheimer’s disease patients is significantly lower than in the brain of normal individuals, and this is reflected by increased levels of Met-SO in these regions [17, 21, 24–26]. Oxidation of methionine is similarly significant in “normal” aging, as a decline in MsrA activity leads to a 40% decrease in the maximum life span of mice, and overexpression of MsrA in Drosophila greatly extends their life span [26–28]. Just like deamidation of asparagine, oxidation of methionine affects protein structure, which in turn can lead to negative effects such as reduced protein activity or stability. Altered activity due to oxidation of methionine has been discovered in a plethora of different proteins including, but not limited to, chymotrypsin, ribonuclease B, lysozyme, and pepsin [29–33].
Figure 2.
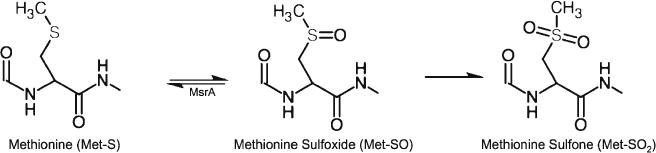
The oxidation of methionine mechanism. Methionine (Met-S) oxidizes to form methionine sulfoxide (Met-SO), which can further oxidize to form methionine sulfone (Met-SO2). Met-SO can be reduced back to methionine using methionine sulfoxide reductase A (MsrA) (created using ChemDoodle by iChemLabs)
Using conventional reversed-phase (RP) approaches, oxidized peptides can sometimes be separated from their native forms, but deamidated peptides are often not resolved from their unmodified counterparts. Hao et al. used a multidimensional RP-ERLIC-MS/MS approach to collect a triad of deamidated products together and subsequently separated them based on their pI to allow for identification [34]. High resolution hydrophilic interaction liquid chromatography (HILIC) can also be a solution to this problem, as the change in hydrophilicity of amino acid side chains resulting from these modifications may change the selectivity of the peptides that have these modifications sufficiently to allow for chromatographic separation, which could enable their quantitation.
Here, we demonstrate the capacity of HILIC-MS to separate and quantitate modified peptides and their native counterparts for the analysis of human immunoglobulin Gs (IgGs), and other standard proteins. Previously, we have created a peptide retention prediction model using HILIC that is based on the summation of amino acid coefficients [35]. Herein, the utility of this model is expanded by derivation of coefficients for the oxidation of methionine and for the deamidation of asparagine, which are now incorporated into the previous retention model. Modified and unmodified peptides can quickly and easily be identified by their predicted relative retention times in conjunction with their m/z ratio. This will provide an easier and consistent assessment of the extent of modifications in biotherapeutic agents, as well as allow for the separation, characterization, and potential isolation of peptides with these modifications.
Materials and Methods
Protein Digestion
Human IgGs were separated from human serum (Sigma-Aldrich, St. Louis, MO, USA) using a HiTrap Protein G column (General Electric Company, Fairfield, CT, USA). Cytochrome c, lysozyme, transferrin, and dextran were purchased from Sigma-Aldrich. Bovine serum albumin was purchased from Waters (Milford, MA, USA). These proteins as well as yeast and mosquito cuticular proteins were reduced using 10 mM dithiothreitol (DTT) and then alkylated using 55 mM iodoacetamide (IDA), both purchased from Sigma Aldrich. Sequencing-grade trypsin or chymotrypsin purchased from Promega (San Luis Obispo, CA, USA) was added at 50:1 (w/w, protein/trypsin) for incubation overnight in 50 mM ammonium bicarbonate (pH 7.0) at 37 °C. Three synthetic peptides with the same sequence of GFYPSDIAVE WESNGQPENNYK were purchased from Bachem (Bubendorf, Switzerland). One peptide was unmodified, one had a n-Asp modification at the 14th residue, and the last one had an isoAsp modification at the 14th residue.
LC-MS/MS Settings and Instrumentation
Data were acquired using a Finnegan LTQ (Thermo-Fisher, San Jose, CA, USA) in series with a 1100 Series Capillary LC system (Agilent Technologies, Palo Alto, CA, USA) with an ESI source that used spray tips made in-house. Samples were suspended in 25%H2O, 75% ACN, and 0.1% formic acid (Sigma-Aldrich) for direct injection into the LC system. Peptides were separated using a 200-μm × 150-mm HALO Penta-HILIC column packed with 2.7 μm diameter superficially porous particles that have a 90 Å pore diameter (Advanced Materials Technology, Wilmington, DE, USA) at room temperature. The gradient elution conditions employed a linear increase in aqueous solvent from 5%–70% over 90 min at a 2 μL/min flow rate, using the column at room temperature. The (strong) aqueous solvent contained 0.1% formic acid (Sigma Aldrich) with 50 mM ammonium formate (Thermo-Fisher) and the organic solvent was acetonitrile with 0.1% formic acid. The settings for the mass spectrometer included taking the five most intense ions from each full mass spectrum for fragmentation using collision-induced dissociation (CID), and the resulting MS/MS spectra were recorded.
To make sure that this model would be universal, some of the same digested proteins as well as the synthetic peptides were run on a 4000 Q Trap (AB Science, Chatham, NJ, USA). Peptides were separated by a 2.1 mm × 15 cm HALO Penta-HILIC column packed with 2.7-μ diameter superficially porous particles using a Nexera UFLC (Shimadzu, Columbia, MD, USA). The temperature of the column was 60 °C. The gradient used for each sample was 22%–52% water over 80 min at a 0.4-mL/min flow rate. Spectra were obtained using an ESI source.
For RP analysis using the Finnegan LTQ and 1100 Series Capillary LC system, samples were suspended in 95% H20, 5% ACN, and 0.1% FA (Sigma-Aldrich) and injected to the LC. Peptides were separated using a 200-μm × 150-mm HALO Peptide ES-C18 column packed with 5-μm diameter superficially porous particles (Advanced Materials Technology). The column was at room temperature. The gradient was 5%–75% ACN for 120 min at a 2 μL/min flow rate. The mobile phase contained 0.1% formic acid and 10 mM ammonium formate (Thermo-Fisher). The LC-MS/MS system and MS parameters were the same as the HILIC analysis.
Database Search Parameters
The resulting RAW files were converted using Trans-Proteomic Pipeline (Seattle Proteome Center, Seattle, WA, USA), and then the MS/MS spectra of each sample were searched using Mascot (Matrix Scientific, Boston, MA, USA) against corresponding protein databases of theoretical MS/MS spectra. The following parameters were utilized in Mascot: a peptide tolerance of 1000 ppm, a fragment tolerance of 0.6 Da, two max missed cleavages of trypsin, and a fixed modification of carbamidomethyl (C).
Selection of Peptides for Prediction Model and Post-Run Data Analysis
All peptides that had a higher Mascot score than 10 were considered. Peptide retention times were determined manually from. RAW files using the apex of the peaks displayed in Xcalibur software (Thermo-Fisher), and resulting MS/MS data were visually inspected for fragmentation that was consistent with peptide assignments. Chromatographic peaks for each peptide had to have a peak asymmetry value of between 0.25 and 4, and peptides exhibiting peak widths greater than 5.5 min were excluded from analysis. Peptide retention times in minutes were converted to glucose units based on dextran samples that were run immediately before. Linear regression analysis using StatPlus (AnalystSoft, Walnut, CA, USA) was used to find the incremental retention coefficients for each amino acid.
Results and Discussion
Resolving Modified and Unmodified Peaks
To evaluate the ability of RP chromatography to resolve oxidized and native peptides, samples were first run on a C18 RP column. Figure 3b shows the separation for the BSA peptide TVMENFVAFVDK, where the bold amino acid residue is the expected site of modification. In this figure, the oxidized and unoxidized versions are separated, which allows for the easy quantitation of these two species. However, this is not always the case with oxidation using RP chromatography, as shown by the chromatography of the BSA peptide ETYGDMADCCEK (Figure 3a), where the oxidized version and native version are not separated. These cases show that RP chromatography, using typical acidic mobile phase conditions, does not separate oxidized and unoxidized peptides with certainty, which can decrease detection relative sensitivity for modified versus unmodified peptides, leading to uncertainty about the significance of oxygen-driven degradation processes. The separation is also influenced by the composition of the neighboring residues to the methionine, which are different in the two peptides in terms of hydrophobicity.
Figure 3.
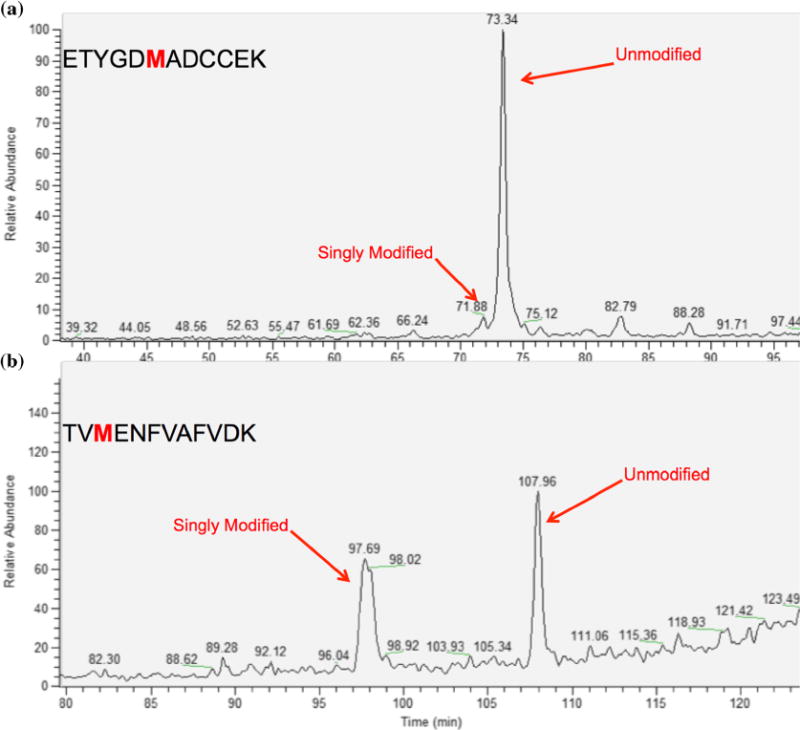
The separation of oxidized peptides and their native forms using a C18 column. Oxidized peptides are not consistently separating from their unmodified counterparts in a predictable fashion
Deamidation is a difficult modification to analyze because the 1 Da mass increase of the modification places the molecular ion for the deamidated peptide at the same nominal mass as the one 13C isotope of the unmodified species. Utilizing chromatography to resolve these two species decreases the complexity of identifying and quantitating the modified species. However, for the IgG peptide GFYPSDIAVEWESNGQPENNYK, the deamidated and non-deamidated peptides co-elute from a RP column, and this is shown in Figure 4. There were no cases using RP chromatography in any of the peptide samples in which the deamidated peptide and the native peptide had baseline separation. The majority of the peptides co-eluted and some had peak shoulders, but there was never enough separation to quantitate the peaks. The selectivity differences for the asparagine and iso-aspartic acid side chain functional groups are very small under typical low pH separation conditions, leading to minimal resolution capabilities. This separation problem is in addition to the similar masses that the modified and unmodified peptides possess, leading to significant errors in detection of the modified versus unmodified peptides. The mass difference between the unmodified 13C peptide and modified peptide is 0.0152 Da, which for the deamidated IgG peptide GFYPSDIAVEWESNGQPENNYK would require a mass spectrometer with a resolution of 167,377 to be able to resolve from its unmodified 13C version. In essence, using chromatographic techniques that are not able to separate deamidated peptides from their native forms requires very high-resolution mass spectrometers able to detect the minute mass difference. Even then, it still is difficult to identify the presence of a modified peptide with high certainty, and extremely difficult to determine relative abundance of the peptide pairs.
Figure 4.
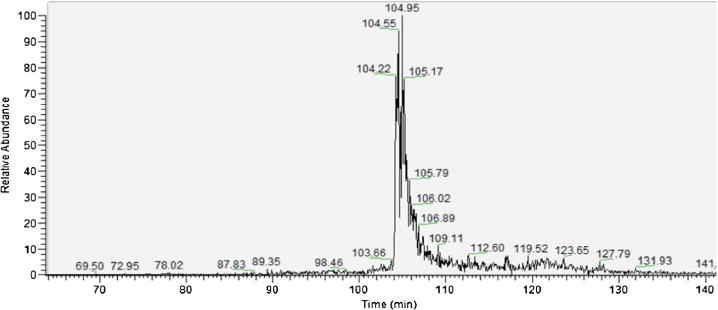
The chromatography of the IgG deamidated peptide GFYPSDIAVEWESNGQPENNYK and its native forms using a C18 column. Both modified and native forms co-eluted around 105 min
From the HILIC peptide retention model that we previously created, there is a substantial difference between asparagine and aspartic acid coefficients, which indicates that deamidated peptides should be separated from their non-deamidated forms. There is also a significant difference between alanine and serine that implies adding an oxygen to methionine should be enough to effectively separate peptides with oxidized methionine residues from their native counterparts [35]. To test this, the same samples were analyzed using HILIC separation conditions. Figure 5 shows baseline separation for the IgG oxidized peptide KDSGFQMNQLR (Figure 5a) and the IgG deamidated peptide GFYPSDIAVEWESNGQPENNYK (Figure 5b). Both modified peptides and their native forms exhibited baseline separation on the HILIC column, allowing for confident quantitation of the peak areas or heights even with low-resolution mass spectrometers. These modifications increase the hydrophilicity of their respective peptides, which in turn increases the retention time. Using HILIC to analyze deamidation negates the requirement to employ a highresolution mass spectrometer, as necessitated by the overlap of the unmodified peptide 13C isotope envelope, and the modified peptide mass. The separation of the modified peptides from the native structure is predictable, and occurs regardless of peptide sequence, as a variety of different types of residues were adjacent to the site of deamidation in the peptides that were used in the study. Separation selectivity factors (α) and resolution values for unmodified and modified peptide pairs that were separated and identified together are shown in Table 1.
Figure 5.
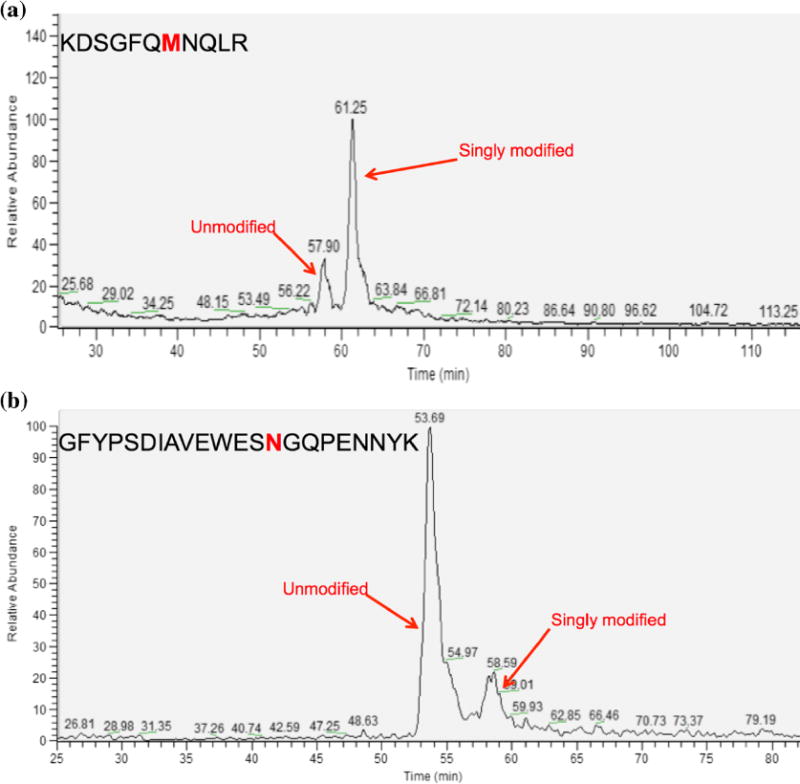
The separation of oxidized and deamidated peptides from their native forms using a HILIC column. Deamidated and oxidized peptides are adequately separated enough from their unmodified counterparts to allow for quantitation
Table 1.
Selectivity Factor (α) and Resolution Values for Modified and Unmodified Peptide Pairs. Modified Residues are Underlined in Red
| Sequence | Modification | Selectivity (α) | Resolution |
|---|---|---|---|
| GFYPSDIAVEWES GQPENNYK | Asn/(Iso)Asp14 | 1.099 | 1.928 |
| G PTVEVELTTEKGVFR | Asn/(Iso)Asp2 | 1.087 | 2.006 |
| LLGVAGGQAFEGAPT VEIAR | Asn/(Iso)Asp16 | 1.134 | 2.088 |
| PVILADACCSR | Asn/(Iso)Asp1 | 1.098 | 1.431 |
| TVDYTADDV GFNAVVSK | Asn/(Iso)Asp10 | 1.062 | 1.424 |
| VVEEYTADPV GFNAVVHR | Asn/(Iso)Asp11 | 1.073 | 1.348 |
| VVSVLTVLHQDWL GK | Asn/(lso)Asp14 | 1.119 | 1.579 |
| VVSVLTVVHQDWL GK | Asn/(Iso)Asp14 | 1.123 | 1.874 |
| IET R | Met/MetS04 | 1.097 | 1.364 |
| KDSGFQ NQLR | Met/MetS07 | 1.058 | 1.288 |
| PCTEDYLSLILNR | Met/MetS01 | 1.198 | 1.990 |
| TV ENFVAFVDK | Met/MetS03 | 1.139 | 1.932 |
From the deamidation mechanism, it is clear that two potential, n-Asp and isoAsp, modified products can be present. To deduce the form of the deamidated products, synthetic peptides with the sequence GFYPSDIAVEWESNGQPENNYK were run on the 4000 Q Trap LC-MS system. These synthetic peptides had three versions: unmodified, n-Asp at the 14th residue, and isoAsp at the 14th residue, and Figure 6 shows a run with all of the versions separated. The least retained peak is the unmodified form of the peptide, whereas the peak in the middle is the aspartyl version, and the peak most retained is the isoaspartyl version. Comparing these results to the same deamidated peptide in Figure 7 shows that the deamidated peak at 37 min is the isoaspartyl version of the peptide, and peak at 35 min corresponds to the aspartyl version of the peptide. There is another set of three peaks around this peptide that are not labeled, most likely indicating a second deamidation site at the first asparagine in the “NN” motif in the peptide sequence. However, the separation of this peptide is consistent with the first two peptides shown in Figure 7, which only differ in the residue in the 8th position (the earlier eluting peptide has a leucine where the later eluting peptide has a valine). These peptides share the same elution order as the synthetic peptide. For all the samples run on the LTQ, it was ambiguous as to which deamidation product was present before the synthetic peptides were run because only one deamidated peak would appear. It is known that isoAsp is two to three times more abundant than the n-Asp, so the deamidation that has been seen for the peptides of the current study actually corresponds to isoAsp, and the ratio of peak abundances of n-Asp to isoAsp in Figure 7 is analogous to this statement [4, 5, 8, 12, 34]. Due to the different physical properties of these two deamidation products, such as pKa (n-Asp: 3.9, isoAsp: 3.2), the retention times will be different in the mildly acidic conditions of separation used during the current study. A higher percentage of isoAsp will be charged at this pH, increasing the retention in comparison to n-Asp. There was no indication that the D and L configurations could be resolved.
Figure 6.
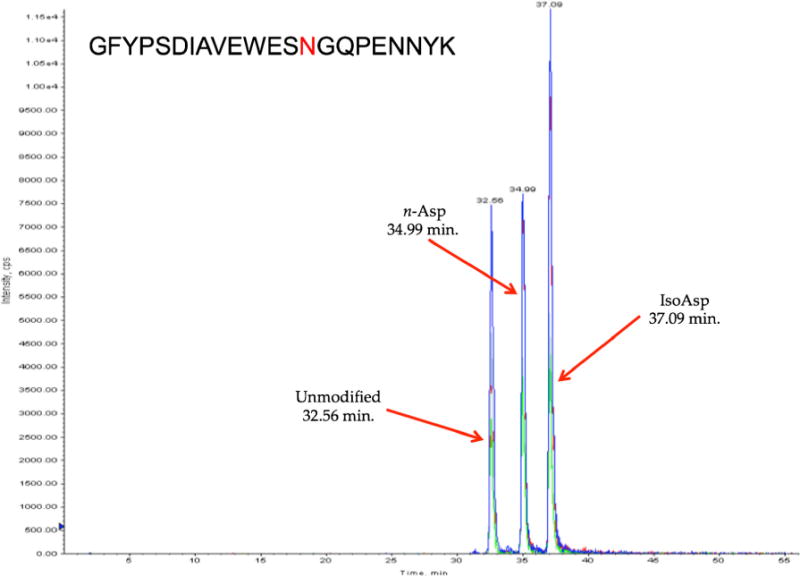
The separation of the unmodified, n-Asp, and isoAsp versions of the synthetic peptide GFYPSDIAVEWESNGQPENNYK, with the site of modification at the residue in red. The unmodified form eluted first, followed by the n-Asp form, and finally the isoAsp form
Figure 7.
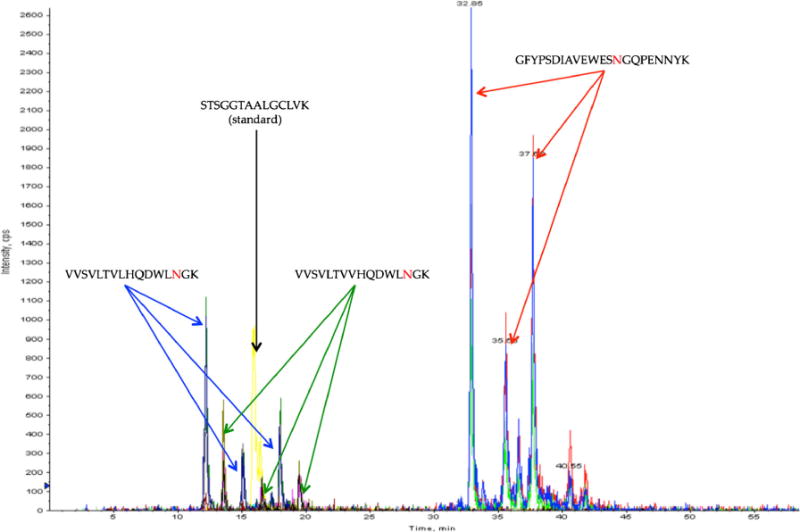
Analysis of peptides for detecting deamidation by LCMS with the QTrap 4000. For the three peptides that have deamidation sites, there are peaks corresponding to the unmodified form, the n-Asp form, and the isoAsp form of the asparagine residue in red. The unmodified form of each peptide elutes first, followed by the n-Asp form, and finally the isoAsp form. The first two deamidated peptides differ at the 8th position, where the first peptide has a leucine and the second peptide has a valine
Finally, the extent of the hydrophilic retention shifts for both modifications is consistent with the HILIC column, but was shown to be inconsistent with the RP system employed. This is due to the hydrophilic modifications having a greater selectivity difference with the HILIC stationary phase than the RP stationary phase, allowing for the prediction of the retention to be heightened for HILIC [36]. It is also due to secondary effects in RP, such as amino acid location or neighboring residue composition, as the two peptides, TVMENFVAFVDK and ETYGDMADCCEK, exhibited different retention behavior. The first peptide has a hydrophobic neighboring residue and exhibits baseline separation between modified and unmodified forms, whereas the second peptide has hydrophilic residues on either side and the oxidized version is not fully separated from the native peptide. RP resolution is driven by hydrophobicity differences in analytes, so the change in polarity of the first peptide due to oxidation could affect the surface interactions of the hydrophobic residue next to the methionine, leading to a better separation than obtained for the second peptide. Whatever underlies a mechanistic interpretation of the selectivity differences in separation, it is clear that RP is much more sensitive to sequence effects than HILIC, for which was observed a consistent retention shift regardless of neighboring residues.
Peptide Prediction Model Coefficients
We have previously created a model that predicts peptide retention based on amino acid composition [35]. In this model, coefficients for each amino acid were derived using linear regression analysis of 50 unmodified peptides, and the retention time of a peptide can be predicted by using Equation 1 shown below, where RT is the predicted retention time, Li is the amount of residue i in the peptide, AAi is the amino acid coefficient of residue i, and b0 is the intercept of the model:
| (1) |
We have recently expanded this model using data from 297 unmodified peptides, and it has a very high correlation coefficient (0.94553), indicating accurate prediction.
The amino acid coefficients are expressed in glucose units (GU) from procainamide-labeled dextran samples that were run before each sample. This approach allows the model to be used on any LC-MS system as long as a dextran standard ladder is run before the protein sample of interest, and the retention times of peptides are then converted from minutes to GU based on the logarithmic fit for the dextran samples. Dextran elutes in order of increasing monosaccharide linkage and provides a useful reference for the retention times of peptides. Excluding the actual stationary phase and mobile phase composition, this approach also allows for modifications to a LC-MS system to occur, such as the changing of the length of a capillary line or detector configuration, which would not affect the conversion of a peptide’s retention time to GU. To ensure that dextran would be a suitable retention time calibrant, peptide standards were run on two different LC-MS systems over the course of a month, and data analysis indicated that the retention times of the standards had minimal changes. These two systems had differing column lengths, column temperatures, gradients, and flow rates, yet the retention times of peptides that were run on both systems were within 3.73% of each other and only differed by an average of 0.52 GU (2.29 min).
Two new coefficients were created for the isoAsp form of the deamidated asparagine residues and oxidized methionine residues to be able to predict the retention of peptides with these modifications. Twelve deamidated peptides and 27 Metoxidized peptides were discovered and incorporated into the model. These modified peptides were from some of the samples used to create the unmodified peptide retention model (IgGs, mosquito cuticular proteins, yeast proteins, BSA, cytochrome c, transferrin, and lysozyme), and regression analysis was used to derive these coefficients. The deamidation coefficient corresponding to the isoaspartyl form that was derived had a value of 1.409 (R-squared = 0.94186), indicating that the modification is very hydrophilic and will increase the retention time of peptides with this modification. This coefficient was on the higher end of all coefficients in the unmodified peptide retention model, only less than the three most hydrophilic residues: lysine, arginine, and histidine. The large deamidation coefficient (1.409) that was derived supports the claim that the deamidated peaks are indeed isoAsp. This coefficient is much larger than the difference between the asparagine coefficient and the aspartic acid coefficient, which would correspond to the formation of the n-Asp product.
The oxidized Met coefficient was also found to be hydrophilic, with a value of 0.633. This is a large difference from the unoxidized methionine coefficient (−0.337), and it was shown that this difference is sufficient for ready separation of the unmodified peptides from the modified ones. As with the value of the deamidation coefficient, the oxidized methionine coefficient is greater than expected, based on the modest difference in coefficients between alanine and serine. This comparison was made because both cases differ by the addition of an oxygen atom, so the expected difference between the alanine/serine and oxidized/unoxidized deviations should be minimal. The oxidized samples slightly increased the R-squared of the model to 0.94637, whereas the deamidated samples did the opposite, slightly decreasing the R-squared to 0.94186. However, the incorporation of both of the coefficients into the previous model barely affected the overall R-squared value, and this is because the total amount of modified peptides (39) was significantly less than the amount of unmodified peptides used to create the original model (297). The coefficients that were derived for the hydrophilic modifications do not affect the values for the unmodified amino acid coefficients because we are more concerned with the separation of the modified and unmodified peptides rather than their actual retention times. In time there will be more instances of these modifications and we can gather a better understanding of the impact the coefficients have to the overall fit of the model. For now, we have found that both of these modifications are hydrophilic, with deamidation being one of the most hydrophilic coefficients in the model, and the coefficients explain why we are able to see sufficient separation between the modified and unmodified peptides using the HILIC mode of separation.
Conclusion
Deamidated asparagine residues and oxidized methionine residues were shown to be resolvable from their native forms using HILIC chromatography, which allows for individual peak quantitation. This is particularly useful for deamidation, where the mass differences between a peptide containing an unmodified 13C isotope and a deamidated asparagine residue are too small to resolve from one another without using a highresolution mass spectrometer. In the current examples, analyses were conducted using an LTQ instrument, with only limited mass resolution capabilities. By being able to fully separate peptides with and without these modifications, the identification process can be heightened and peptides with modifications can be more easily quantitated, which is vital in protein biotherapeutics where the quantitation of analytes with modifications needs to be known. Additionally, coefficients describing each modification’s hydrophilicity were derived and incorporated into a peptide retention prediction model that was previously presented. Both coefficients were shown to be very hydrophilic and did not affect the already high R-squared value of the original model by a significant amount.
Acknowledgments
Support for this work comes from NIH grant GM0 93747 to B.B. The authors thank Clayton Seigel for his help with the data processing.
References
- 1.Gervais D. Protein deamidation in biopharmaceutical manufacture: understanding, control, and impact. J Chem Technol Biotechnol. 2016;91:569–575. [Google Scholar]
- 2.Jenkins N, Murphy L, Tyther R. Post-translational modifications of recombinant proteins: significance for biopharmaceuticals. Mol Biotechnol. 2008;39:113–118. doi: 10.1007/s12033-008-9049-4. [DOI] [PubMed] [Google Scholar]
- 3.Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, Yao Z, Sreedhara A, Cano T, Tesar D, Nijem I, Allison DE, Wong PY, Kao YH, Quan C, Joshi A, Harris RJ, Motchnik P. Charge variants in IgG1: isolation, characterization, in vitro binding properties and pharmacokinetics in rats. MAbs. 2010;6:613–624. doi: 10.4161/mabs.2.6.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bischoff R, Kolbe HVJ. Deamidation of asparagine and glutamine residues in proteins and peptides: structural determinants and analytical methodology. J Chromatogr B. 1994;662:261–278. doi: 10.1016/0378-4347(94)00203-7. [DOI] [PubMed] [Google Scholar]
- 5.Catak S, Monard G, Aviyente V, Ruiz-Lopez MF. Deamidation of asparagine residues: direct hydrolysis versus succinimide-mediated deamidation mechanisms. J Phys Chem A. 2009;113:1111–1120. doi: 10.1021/jp808597v. [DOI] [PubMed] [Google Scholar]
- 6.Liu DT. Deamidation: a source of miscroheterogeneity in pharmaceutical proteins. Trends Biotechnol. 1992;10:364–369. doi: 10.1016/0167-7799(92)90269-2. [DOI] [PubMed] [Google Scholar]
- 7.Yang H, Zubarev RA. Mass spectrometric analysis of asparagine deamidation and aspartate isomerization in polypeptides. Electrophoresis. 2010;31(11):1764–1772. doi: 10.1002/elps.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geiger T, Clarke S. Deamidation, isomerization, and racemization at asparaginyl and aspartyl residues in peptides. J Biol Chem. 1987;262(2):785–794. [PubMed] [Google Scholar]
- 9.Patel K, Borchardt RT. Chemical pathways of peptide degradation. II. Kinetics of deamidation of an asparaginyl residue in a model hexapeptide. Pharm Res. 1990;7(7):703–711. doi: 10.1023/a:1015807303766. [DOI] [PubMed] [Google Scholar]
- 10.Robinson NE, Robinson AB. Molecular clocks. Proc Natl Acad Sci. 2001;98(3):944–949. doi: 10.1073/pnas.98.3.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scotchler JW, Robinson AB. Deamidation of glutaminyl residues: dependence on pH, temperature and ionic strength. Anal Biochem. 1974;59:319–322. doi: 10.1016/0003-2697(74)90040-2. [DOI] [PubMed] [Google Scholar]
- 12.Tyler-Cross R, Schirch V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J Biol Chem. 1991;266(33):22549–22556. [PubMed] [Google Scholar]
- 13.Charache S, Fox J, McCurdy P, Kazazian H, Jr, Winslow R, Hathaway P, van Beneden R, Jessop M. Post-synthetic deamidation of hemoglobin providence (β 82 Lys → Asn, Asp) and its effect on oxygen transport. J Clin Invest. 1977;59:652–658. doi: 10.1172/JCI108683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis UJ, Singh RNP, Bonewald LF, Seavey BK. Altered proteolytic cleavage of human growth hormone as a result of deamidation. J Biol Chem. 1981;256(22):11645–11650. [PubMed] [Google Scholar]
- 15.Robinson NE, Zabrouskov V, Zhang J, Lampi KJ, Robinson AB. Measurement of deamidation of intact proteins by isotopic envelope and mass defect with ion cyclotron resonance Fourier transform mass spectrometry. Rapid Commun Mass Spectrom. 2006;20(23):3535–3541. doi: 10.1002/rcm.2767. [DOI] [PubMed] [Google Scholar]
- 16.Gaza-Bulseco G, Faldu S, Hurkmans K, Chumsae C, Liu H. Effect of methionine oxidation of a recombinant monoclonal antibody on the binding affinity to protein A and protein G. J Chromatogr B. 2008;870:55–62. doi: 10.1016/j.jchromb.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 17.Stadtman ER, Moskovitz J, Levine RL. Oxidation of methionine residues of proteins: biological consequences. Anitoxid Redox Signal. 2003;5(5):577–582. doi: 10.1089/152308603770310239. [DOI] [PubMed] [Google Scholar]
- 18.Vogt W. Oxidation of methionyl residues in proteins: tools, targets, and reversal. Free Radic Biol Med. 1995;18(1):93–105. doi: 10.1016/0891-5849(94)00158-g. [DOI] [PubMed] [Google Scholar]
- 19.Carney JM, Smith CD, Carney AM, Butterfield DA. Aging- and oxygen-induced modifications in brain chemistry and behavior. Ann NY Acad Sci. 1994;738:44–53. doi: 10.1111/j.1749-6632.1994.tb21788.x. [DOI] [PubMed] [Google Scholar]
- 20.Cochrane CG, Spragg R, Revak SD. Pathogenesis of the adult respiratory distress syndrome. J Clin Invest. 1983;71:754–761. doi: 10.1172/JCI110823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gabbita SP, Aksenov MY, Lovell MA, Markesbery WR. Decrease in peptide methionine sulfoxide reductase in Alzheimer’s disease brain. J Neurochem. 1999;73(4):1660–1666. doi: 10.1046/j.1471-4159.1999.0731660.x. [DOI] [PubMed] [Google Scholar]
- 22.Gladstone IM, Jr, Levine RL. Oxidation of proteins in neonatal lungs. Pediatrics. 1993;93:764–768. [PubMed] [Google Scholar]
- 23.Pryor WA, Dooley MM, Church DF. Inactivation of human α-1-proteinase inhibitor by gas-phase cigarette smoke. Biochem Biophys Res Commun. 1984;122(2):676–681. doi: 10.1016/s0006-291x(84)80086-8. [DOI] [PubMed] [Google Scholar]
- 24.Liang X, Kaya A, Zhang Y, Le DT, Hua D, Gladyshev VN. Characterization of methionine oxidation and methionine sulfoxide reduction using methionine-rich cysteine-free proteins. BMC Biochem. 2012;13(21):1–10. doi: 10.1186/1471-2091-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 26.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci. 2001;98(23):12920–12925. doi: 10.1073/pnas.231472998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruan H, Tang XD, Chen ML, Joiner MLA, Sun G, Brot N, Weissbach H, Heinemann SH, Iverson L, Wu CF, Hoshi T. High-quality life extension by the enzyme peptide methionine sulfoxide reductase. Proc Natl Acad Sci. 2002;999(5):2748–2753. doi: 10.1073/pnas.032671199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine RL, Moskovitz J, Stadtman ER. Oxidation of methionine in proteins: roles in antioxidant defense and cellular regulation. Life. 2000;50:301–307. doi: 10.1080/713803735. [DOI] [PubMed] [Google Scholar]
- 30.Jori G, Galiazzo G, Marzotto A, Scoffone E. Selective and reversible photo-oxidation of the methionyl residues in lysozyme. J Biol Chem. 1968;243(16):4272–278. [PubMed] [Google Scholar]
- 31.Kido K, Kassell B. Oxidation of methionine residues of porcine and bovine pepsins. Biochemistry. 1975;14(3):631–635. doi: 10.1021/bi00674a026. [DOI] [PubMed] [Google Scholar]
- 32.Ray WJ, Jr, Latham HG, Jr, Katsoulis M, Koshland DE., Jr Evidence for involvement of a methionine residue in the enzymatic action phosphoglucomutase and chymotrypsin. J Am Chem Soc. 1960;82(17):4743–4744. [Google Scholar]
- 33.Schachter H, Dixon GH. Identification of the methionine involved in the active center of chymotrypsin. Biochem Biophys Res Commun. 1962;9(2):132–137. doi: 10.1016/0006-291x(62)90101-8. [DOI] [PubMed] [Google Scholar]
- 34.Hao P, Qian J, Dutta B, Cheow ESH, Sim KH, Meng W, Adav SS, Alpert A, Sze SK. Enhanced separation and characterization of deamidated peptides with RP-ERLIC-based multidimensional chromatography coupled with tandem mass spectrometry. J Proteome Res. 2012;11:1804–1811. doi: 10.1021/pr201048c. [DOI] [PubMed] [Google Scholar]
- 35.Badgett MJ, Boyes B, Orlando R. Prediction of peptide retention times in hydrophilic liquid interaction chromatography (HILIC) based on amino acid composition. Chromatogr Today. 2015;8(4):39–42. [Google Scholar]
- 36.Alpert AJ. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids, and other polar compounds. J Chromatogr. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]