

Abstract
Botulinum neurotoxin serotype A (BoNT/A) causes a debilitating and potentially fatal illness known as botulism. The toxin is also a bioterrorism threat, yet no pharmacological antagonist to counteract its effects has reached clinical approval. Existing strategies to negate BoNT/A intoxication have looked to antibodies, peptides or organic small molecules as potential therapeutics. In this work, a departure from the traditional drug discovery mindset was pursued, in which the enzyme’s susceptibility to metal ions was exploited. A screen of a series of metal salts showed marked inhibitory activity of group 11 and 12 metals against the BoNT/A light chain (LC) protease. Enzyme kinetics revealed that copper (I) and (II) cations displayed noncompetitive inhibition of the LC (Ki ≈ 1 μM), while mercury (II) cations were 10-fold more potent. Crystallographic and mutagenesis studies elucidated a key binding interaction between Cys165 on BoNT/A LC and the inhibitory metals. As potential copper prodrugs, ligand-copper complexes were examined in a cell-based model and were found to prevent BoNT/A cleavage of the endogenous protein substrate, SNAP-25, even at low μM concentrations of complexes. Further investigation of the complexes suggested a bioreductive mechanism causing intracellular release of copper, which directly inhibited the BoNT/A protease. In vivo experiments demonstrated that copper (II) dithiocarbamate and bis(thiosemicarbazone) complexes could delay BoNT/A-mediated lethality in a rodent model, indicating their potential for treating the harmful effects of BoNT/A intoxication. Our studies illustrate that metals can be therapeutically viable enzyme inhibitors; moreover, enzymes that share homology with BoNT LCs may be similarly targeted with metals.
Graphical Abstract
Introduction
Transition metals are well known for their medicinal properties especially against cancer. For example, cisplatin, a cis-diamminedichloro-platinum (II) complex, is one of the most successful metal-containing pharmaceuticals and is widely used in the clinic to treat a variety of cancers.1–2 The therapeutic properties of metals and metal complexes have typically been discovered serendipitously through phenotypic assays; moreover, their use in target-based screening campaigns has been limited. Metals have often been classified as undesirable medicinal agents due to their lack of target specificity and toxicity compared to drug-like, organic compounds. Yet, metal complexes are used against endogenous targets, e.g. auranofin an FDA-approved antirheumatic gold complex,3 and can possess a relatively high therapeutic index in the context of treating diseases caused by non-self biological entities such as cancers, parasites and infectious microbes.4 In the latter cases, the metal complex will exploit a biological “Achilles’ heel” unique to the disease-causing entity to induce selective killing. To highlight a few examples, the quickly replicating DNA found in cancer is a target for cisplatin;5–6 auranofin has been recently shown to inhibit the thioredoxin reductase (TrxR) found in the parasitic protozoan Entamoeba histolytica;7 and, the arsenic-containing drug melarsoprol targets a unique molecule trypanothione produced by Trypanosoma brucei.8 Although these metal complexes can have toxic side effects, their use is warranted because of the dire nature of the diseases that they can effectively treat.
Considering the largely untapped therapeutic potential of metal complexes, we posited that pursuing metals in our medicinal chemistry campaigns may be fruitful. In particular, botulinum neurotoxin serotype A (BoNT/A), one of seven toxin serotypes (A–G) produced by the bacterium Clostridium botulinum, presented an opportunity for metal-based therapy due to the failure of any existing small molecule BoNT/A inhibitors to reach clinical trials. Significant effort has been made to develop BoNT/A inhibitors because the toxin is the most common causative agent of human botulism (compared to the other serotypes)9 and is one of the most potent biological toxins with an estimated lethal dose by inhalation of under 1 μg.10 The deadly nature of BoNT/A has led some countries to weaponize the toxin throughout the 20th century, and today it is still classified by the CDC as a category A agent, and thus continues to be a bioterrorism threat.9, 11–12 Despite easy prevention by proper food storage techniques, foodborne botulism remains a human health risk and is a perennial concern in the food industry; in April of 2015, over 25 people suffered from botulism after eating improperly canned potatoes.13 Ironically, minute quantities of BoNT/A are used clinically under the trade names Botox and Dysport for cosmetic purposes or to ameliorate a myriad of neurological disorders including muscle spasms and dystonias.14–15 No cure exists for botulism and treatment options for botulism are limited, which include passive immunization of equine antitoxin and supportive therapy during prolonged paralysis. The key drawback of the antitoxin is it can only prevent further intoxication and cannot reverse preexisting paralysis because BoNTs act intracellularly.16–17 In the first step of the BoNT/A intoxication mechanism, the heavy chain (100 kDa) binds to the cell membrane of peripheral neurons and promotes endocytosis of the toxin. Next, the light chain (50 kDa) separates from the heavy chain by endosomal acidification and TrxR-mediated disulfide reduction18–19 and then translocates to the cytosol. Lastly, the BoNT/A zinc metalloprotease light chain (LC) cleaves synaptosomal associated protein 25 (SNAP-25), a component of the soluble NSF attachment protein receptor (SNARE) complex responsible for docking neurotransmitter-containing vesicles at the presynaptic membrane. Proteolysis of SNAP-25 halts the release of acetylcholine into the neuromuscular junction, thereby causing muscle paralysis.20 The BoNT/A LC persists in the neuronal cytosol for several months and continues to cleave newly synthesized SNAP-25.21 For this reason, neurons do not recover until the LC is slowly removed from the intracellular compartment.
The majority of anti-BoNT/A compounds have been designed to target the LC protease because of its key role in disrupting neurotransmission to muscles. The most common inhibition strategy has been to employ zinc chelator groups such as hydroxamates22–23 and quinolinols24–27 or SNAP-25 peptidomimetics28–30 with the goal of competing with SNAP-25 for the BoNT/A LC active site. Disruption of SNAP-25 binding at the LC exosites with small-molecule inhibitors has also been explored.31–34 More recently, translocation inhibitors targeting endogenous TrxR such as ebselen and auranofin have been shown to be effective at preventing BoNT/A intoxication in cell-based and in vivo models.35–36 The critical drawback to this therapeutic approach is that it provides minimal post-symptomatic relief because BoNT/A LC already translocated to the cytosol will continue to act unimpeded; therefore, direct LC inhibition remains an attractive, yet difficult strategy.
In seeking an alternative method for disrupting the BoNT/A LC, tantalizing reports of a second metal binding site on the BoNT/A zinc metalloprotease LC have provided theoretical grounds for metal inhibition. The His-Glu-X-X-His motif is responsible for coordinating the zinc cofactor in the active site with ~70 nM affinity while a separate unidentified site can bind a second zinc atom with ~1 μM affinity.37–38 Intriguingly, transition metal cations such as silver, mercury and zinc were found to inhibit BoNT/A LC without displacing zinc at the active site.39–40 These studies coupled with our own observation of cationic copper as an inhibitor provided concrete rationale for the development of metal complexes as a novel class of BoNT/A LC inhibitors.
Results
Investigating metal salts as potential BoNT/A LC inhibitors
For screening purposes, the FRET-based SNAPtide assay has been reliably used for the discovery of small-molecule BoNT/A inhibitors.22, 41 The SNAPtide® substrate mimics SNAP-25 binding and Gln197-Arg198 cleavage at the LC active site, which can be measured by fluorescence. In screening a triazole-based ‘click’ library in the SNAPtide assay, we found molecules containing a copper chelator group, similar to ‘click’ ligands, e.g. TBTA, were highly active against the BoNT/A LC. Further investigation revealed that these compounds were only active because they released copper into the assay. To capitalize on this rather serendipitous discovery, development of anti-BoNT/A metal complexes was pursued. A logical starting point was selection of the most potent metal in a simple SNAPtide assay screen of water soluble metal salts. In the screen, at least one metal per group was tested from groups 1–15, using the metal species with the most stable oxidation state. Results from the screen revealed that group 11 and 12 transition metals, especially copper and mercury, showed significant inhibitory activity against the BoNT/A LC at 10 μM (Figure 1).
Figure 1. Screen of metals against BoNT/A LC identifies group 11 and 12 transition metals as inhibitors.

The water-soluble salt of each metal in its most stable oxidation state was tested at 10 μM in the FRET-based SNAPtide assay. Metals are arranged on the x-axis from left to right in order of increasing group number (1–15), and at least one metal was evaluated from each group. (*) represents activities below the 99.9% CI. Data represents mean ± SEM of two replicates.
BoNT/A LC inhibition kinetics in the presence of copper and mercury
In considering copper and mercury as the most potent metals for LC inhibition, kinetic studies were performed with these two metals in the SNAPtide assay. A plot of [S] versus V0 revealed classical Michaelis-Menten kinetics with clear enzyme saturation curves, and increasing copper and mercury concentrations had a marked inhibitory effect on Vmax (Figure 2A–C), which was reversible (Figure S2). Double reciprocal (Lineweaver-Burk) plots confirmed a Cu/Hg-mediated effect on Vmax with no apparent change in substrate Km at a full range of metal concentrations (Figure 2D–E). These data suggest a noncompetitive metal-mediated inhibition mechanism, implying that copper binds to the BoNT/A LC in a manner that does not interefere with substrate binding. A global fit of the SNAPtide kinetic data with a noncompetitive model revealed a similar Ki for both copper oxidation states (1.56 and 1.25 μM for Cu (II) and Cu (I), respectively) (Figure 2F). Hg showed more than 10-fold greater enzyme inhibition (Ki = 109 nM) relative to Cu, which is incredibly potent for an inhibitor containing one single atom (not including counter anions). For comparison, a tetrapeptide inhibitor of BoNT/A LC (RRGC) gave a similar Ki of 157 nM;28 however, RRGC contains 33 heavy atoms compared to only one heavy atom for the metals, thus demonstrating high ligand efficiency of Hg and Cu.
Figure 2. Enzyme kinetics demonstrate noncompetitive inhibition of BoNT/A LC by Cu/Hg.

Michaelis-Menten curves generated with varying concentrations of (A) CuCl2, (B) CuCl2 reduced to Cu (I) species with excess sodium ascorbate and (C) Hg(OAc)2. Lineweaver-Burk plots demonstrating noncompetitive inhibition of (D) CuCl2 and (E) Hg(OAc)2 data sets. (F) Calculated Ki values from a global fit of each data set. In panels AC, global fit curves (noncompetitive inhibition model) are overlaid with the raw data. In panels D and E, linear regressions were fit to the data. For panels A–E, data represents mean ± SEM of two replicates.
BoNT/A LC mutants, serotype B and E LCs and crystallographic analysis elucidate metal binding site
Following the observation of potent metal inhibition of BoNT/A LC, elucidation of the metal binding site on the enzyme was pursued. As sulfur-containing side chains of methionine and cysteine residues are known to chelate metals within enzymes,42–43 the only two cysteines in BoNT/A LC and two α-exosite methionines were mutated. Evaluation of the resulting mutants for copper inhibition revealed that swapping out Cys134, Met106 and Met344 for alanine had minimal effect on copper IC50, while mutation of Cys165 resulted in significant resistance to copper and mercury inhibition (Figure 3A). Furthermore mutation of adjacent residues Glu164 and Lys166 had almost no effect on the copper IC50. The C165S mutant LC has been previously characterized,44 showing no conformational departure from the wild type LC; therefore, we conclude that the reduced metal inhibition of the C165S mutant is due to a reduced capacity of the serine to chelate Cu/Hg. Similarly, serotype B and E LCs, bearing a threonine (171) and serine (161) in place of the active site cysteine, respectively, also demonstrated diminished sensitivity to metals (Figure 3A). X-ray crystallography was pursued to further investigate the interaction between Cys165 and Cu/Hg. Structure determination of metal-soaked crystalline BoNT/A LC (crystals were soaked with either CuCl2 or Hg(OAc)2) at the X-ray absorption edge of each respective metal revealed strong anomalous scattering localized to Cys165 (Figure 3B,C). Mercury binding appeared to be mediated entirely through this interaction (Figure 3B); however, copper binding occured through interactions with both Cys165 and the catalytic zinc binding site (Figure 3C). Coordination at the Cys165 site occured in a tetrahedral configuration with observed interactions between the Cys165 sidechain thiol (Cu-S distance: 2.3 Å), Cys165 backbone carbonyl (Cu-O distance: 2.5 Å) and an ordered water molecule (Cu-O distance: 1.9 Å). The coordination geometry and partial occupancy of copper at this site indicated that the observed species is likely Cu (I) produced through X-ray-mediated reduction during data collection. In the X-ray crystal structure, occupancy of the catalytic zinc binding site by Cu (II) is likely due to the high concentration of Cu (II) utilized (necessary to be equimolar with enzyme). Direct replacement of zinc by copper in solution is unlikely given that zinc titration was unable to rescue enzyme activity in the presence of inhibitory metals (Figure S3).
Figure 3. Cys165 constitutes the putative metal binding site of BoNT/A LC.

(A) Normalized Cu and Hg IC50s of serotype B/E LCs and serotype A mutants relative to wild type BoNT/A LC IC50s. Bolded values indicate significant decreases in metal potency. NT = not tested. (B) X-ray crystal structures of BoNT/A LC soaked with Hg(OAc)2 or (C) CuCl2. Relevant side chains are displayed and labeled, including canonical zinc-binding residues. Anomalous difference maps are displayed as mesh in gray contoured at 5σ (B), or orange contoured at 4σ (C). Notably, Cys165 assumes two alternate rotamers, one of which is consistent with Cu (I) coordination geometry. Waters and acetate ions have been omitted for clarity.
Design and evaluation of copper complexes in a cell-based model for BoNT/A intoxication
Despite the intriguing discovery of group 11 and 12 metals as potent BoNT/A inhibitors, designing a practical metal-containing therapeutic presented a significant challenge. The major objective was to deliver the metal intracellularly to target cytosolic BoNT/A LC. Although mercury was a more potent LC inhibitor than copper, concerns regarding Hg toxicity and an extensive precedent for using copper for medicinal purposes shifted our focus to copper.45–47 Barring the action of transmembrane copper transporters such as Ctr1,48 copper salts show poor cell membrane permeability; therefore, well characterized copper ligands were investigated for their potential to shuttle the metal across the cell membrane. Fortunately, a wealth of previous studies exists on copper complexes as therapeutics for cancer and Alzheimer’s disease,46–47, 49–50 and in many cases, various ligands were employed as copper (II) ionophores to enhance copper bioavailability. Because the corresponding copper complexes of these ligands are known to increase intracellular concentrations of copper, we hypothesized that they would impede BoNT/A cleavage of SNAP-25 inside cells.
To test our hypothesis we employed a previously optimized cell-based model for BoNT/A intoxication in human induced pluripotent stem cell (hiPSC)-derived neurons.51 Following promising results in this assay with a variety of ligands, e.g., 2,2′-bipyridine (BIPY) and 5,7-dichloroquinolin-8-ol (DCOQ) in combination with CuCl2 (Figure S7, S8), 5,7-dichloro-2-[(dimethylamino)methyl]quinolin-8-ol (PBT2) (Figure 4A) was investigated because of its increased hydrophilicity and excellent copper affinity. Moreover, PBT2 possesses optimized physiochemical properties and has been used in clinical trials for Alzheimer’s disease.52–54 A dose-ranging evaluation of PBT2 complexed with copper showed efficacy in the low μM range for inhibiting BoNT/A-mediated cleavage of SNAP-25 (EC50 = 4.6 μM, Figure 4B,C). In examining the potential cytotoxicity of the PBT2-Cu complex, extracellular lactate dehydrogenase (LDH) was quantified and found to be comparable to non-treated controls, although morphological changes indicating early stages of cytotoxicity (cell rounding) were observed (Figure 4B).
Figure 4. Copper complexes protect human iPSC-derived neuronal cells from BoNT/A-induced SNAP-25 cleavage.

(A) Structures and names of active copper ligands and complexes in BoNT/A cellular assay. (B) Dose-responsive anti-BoNT/A activity of Cu(PBT2)2. +/− Tx C = Control lane with or without toxin. HiPSC-derived neurons were exposed to 200 U/well of BoNT/A1 and were treated at 1.5 post-toxin exposure with 2:1 PBT2/CuCl2 (preformed in DMSO at 100X concentration). Cells were harvested 7.5 h post-toxin and cell lysates were analyzed by Western blot for SNAP-25 cleavage. Morphology changes were graded on a scale of 0 (−) through 3 (+++) to indicate severity of cell rounding. Percent cytotoxicity was evaluated by LDH release from cells relative to a maximum LDH release control. Experiments were run in duplicate with one representative gel shown in the figure. (C) Densiometric representation of panel B gel for EC50 determination of Cu(PBT2)2. (D) Reduction of copper complex releases copper to inhibit BoNT/A LC. Sodium ascorbate (ASC) was added to BoNT/A LC in the presence of DCOQ-Cu complex and enzyme activity was measured by SNAPtide asssay. [DCOQ ligand] = 20 μM, [CuCl2] = 10 μM. ASC alone had no effect on activity while Cu alone gave <1% activity. For panels C/D, data represent mean ± SEM of two replicates.
Copper complex intracellular mechanism of action
Enzyme inhibition kinetics (Figure 2) and cell-based assay data (Figure 4) suggested that copper acts intracellularly as a direct inhibitor of the BoNT/A LC; however, an aspect that remained ambiguous is how complexed copper dissociates from the ligand to act on the LC. Multiple studies have shown that bis(thiosemicarbazone) (BTSC) complexes such as copper (II) glyoxal-bis(4-methylthiosemicarbazone) [Cu(GTSM)] (Figure 4A) cross the cell membrane and undergo reduction from Cu (II) to Cu (I) by a variety of intracellular reductants such as ascorbic acid (ASC).55–57 As a result, ligand affinity for copper is diminished, allowing for the uptake of copper by endogenous, cysteine-rich copper proteins.56 Similar to the reported mechanism of action of copper complexes, we hypothesized that the copper complexes demonstrating efficacy in the cell-based BoNT/A assay would release copper upon reduction, causing inhibition of the LC. Indeed, a robust reduction in enzyme SNAPtide assay activity was observed when increasing amounts of ASC were added to the copper complexes in the presence of the LC (Figure 4D, S4). Thiol-based reductants e.g. glutathione did not have the same effect as ASC because they readily chelate free copper, preventing enzyme inhibition (Figure S5).
Development of second generation copper complexes
Given the evidence for a bioreductive mechanism that causes copper release for intracellular BoNT/A LC inhibition, we investigated cell-based activity of redox-active copper BTSC and dithiocarbamate (DTC) complexes, which inhibited BoNT/A LC in the SNAPtide assay (Figure S6). Similar to PBT2, the selected Cu(BTSC) and (DTC) complexes (Cu(GTSM) and copper (II) bis(2-methoxyethyl)dithiocarbamate [Cu(MEDTC)2], respectively, Figure 4A) showed excellent protection of BoNT/A-intoxicated neurons with EC50 values near 1 μM (Figure 5A–C). Even at 3 h post-toxin addition, both complexes were able to mitigate SNAP-25 cleavage. A key difference in experimental conditions for evaluating these complexes versus the other tested complexes is that Cu(GTSM) and (MEDTC)2 were synthesized as the complex while the other complexes were generated in situ; however, this condition did not appear to influence efficacy.
Figure 5. Cell-based activity of Cu(DTC)2 and (BTSC) complexes against BoNT/A.

Dose-response inhibition of (A) Cu(GTSM) and (B) Cu(MEDTC)2 on BoNT/A SNAP-25 cleavage. C) Densiometric representation of gel in panels A/B for EC50 determination of both complexes. +/− Tx C = Control lane with or without toxin. HiPSC-derived neurons were exposed to 200 U/well of BoNT/A1 and were treated with complexes (isolated as pure solids and dissolved at 100X concentration into DMSO) at 1.5 h or (*) 3 h post-toxin exposure. Cells were harvested 7.5 h after toxin addition and cell lysates were analyzed by Western blot for SNAP-25 cleavage. Experiments were run in duplicate with one representative gel shown in the figure. (D) Effect of additives on inhibition of 3 μM Cu(GTSM). NAC, BSO or ASC at 1 mM were incubated with neurons for 18 h prior to the assay as described for panel A/B. (E) Cu(GTSM) is a fast acting BoNT/A inhibitor. Control: cells were exposed to toxin and harvested at the indicated time points. Cu(GTSM): cells were exposed to toxin and treated with 3 μM Cu(GTSM) at the indicated time points then harvested at 7.5 h. For panels C/E, data represent mean ± SEM of two replicates.
To increase understanding of the intracellular action of the complexes, an attempt was made to modulate the inhibitory activity via addition of redox active compounds N-acetylcysteine (NAC) and ASC or glutathione synthesis inhibitor buthionine sulfoximine (BSO). Results indicated no observable effect on Cu(GTSM) inhibition of BoNT/A (Figure 5D) similar to what was observed for the Cu(PBT2)2 complex (Figure S9). Vitamin E analogue Trolox also had no effect on copper complex efficacy (Figure S7–9).
Further probing the cellular action of the Cu(GTSM) complex in a time-course experiment revealed that the complex acts very rapidly to halt BoNT/A cleaveage of SNAP-25. Addition of the complex almost immediately halted BoNT/A LC activity, as evidenced by a less than half hour delay between +complex and control curves of SNAP-25 cleavage versus time (Figure 5E). This result suggests that the complex acts directly on the LC. In contrast, BoNT/A translocation inhibitors act on indirect targets, e.g., TrxR, and display a delay in action of multiple hours.35
In vivo evaluation of second generation copper complexes
The promising results observed in the enzymatic and cell-based assays warranted further investigation of the copper complexes in vivo. Precedent for administration of PBT2 administration in mice54 and in clinical trials53 prompted us to perform a pilot study involving the coadministration of 2:1 PBT2/Cu in mice exposed to BoNT/A. An initial safety and pharmacokinetic study established that a 0.6 mg Cu/kg i.p. dose was non-toxic and produced adequate serum concentrations of compound (Figure S10A); unfortunately, at this dose, PBT2/Cu only slightly extended the time to death of BoNT/A intoxicated mice (Figure S10B).
BTSC and DTC complexes were also identified as promising candidates for in vivo experiments: Cu(BTSC) complexes have been tested in a variety of in vivo studies for cancer,58–59 Alzheimer’s60 and as a PET radiotracer.61–63 Cu(DTC)2 complexes have also been used as radiotracers,64 as well as a treatment for Menkes disease, which causes copper deficiency due to a dysfunctional ATP7A copper transport protein.65 Although in vivo half-lives of these complexes are relatively short, they are highly effective at facilitating biodistribution of copper. Following tissue uptake, the complex becomes trapped upon bioreduction, thus causing rapid intracellular copper release and decrease of the complex in the blood.61–65
Cu(GTSM) and Cu(MEDTC)2 were specifically selected for in vivo studies based on their established cell-based activity (Figure 5), but also because of their adequate water solubility. Isolated in solid form as the copper complexes, both could be formulated with aqueous vehicle containing 10% w/v or less 2-hydroxypropyl-β-cyclodextrin (β-CD); Cu(MEDTC)2 was especially easy to dissolve unlike related diethyl or pyrrolidine DTC complexes. Following intraperitoneal (i.p.) injection of BoNT/A, i.p. administration of Cu(GTSM) gave variable results with some mice surviving the toxin challenge, but statistically significant efficacy was not achieved (Figure S11). On the other hand, subcutaneous (s.c.) administration of the complex produced a marked increase in time to death post-toxin exposure (Figure S11). The failure of the i.p. route suggests that first-pass metabolism may greatly degrade the complex before adequate distribution can occur. In a second study, both complexes were evaluated at the dose of 1 mg/kg copper administered s.c., and both treatment groups showed an equally significant increase (P < 0.001) in time to death (Figure 6).
Figure 6. Copper complexes significantly extend time to death of mice exposed to BoNT/A.
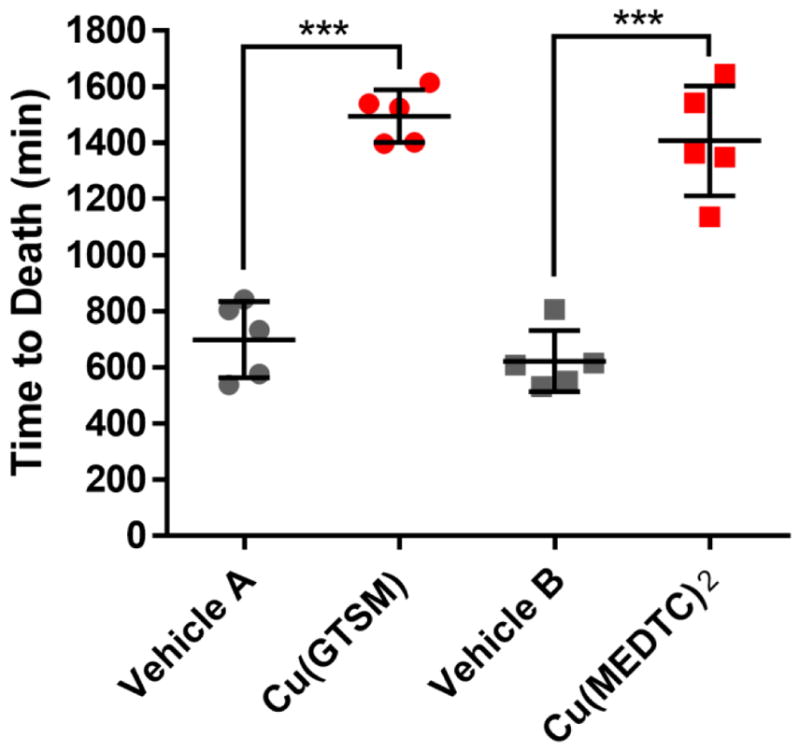
BoNT/A (5 LD50 units) was administered i.p. to mice (n = 5 per group) which were then treated 1 h post-toxin s.c. with vehicle A, vehicle B, vehicle A + Cu(GTSM) at 4.6 mg/kg and vehicle B + Cu(MEDTC)2 at 7.6 mg/kg. The doses for both complexes equated to a copper dose of 1 mg/kg. Vehicle A: citrate/phosphate buffer containing 10% w/v β-CD and 4% v/v DMSO. Vehicle B: saline containing 2.5% w/v β-CD, 2% v/v DMSO and 0.9% v/v benzyl alcohol. Data represent mean ± SEM with individual values shown. ***P < 0.001 by t test. Mice treated with complex only without toxin (n = 3 per complex) showed complete survival.
Discussion
Although copper complexes have been extensively investigated in preclinical models for cancer and Alzheimer’s,58–60, 66–70 the proposed mechanism of action of the complexes in these studies have been cryptic or nonspecific. Moreover, while metals have been reported to inhibit activity of certain enzymes,71–74 metals are not often viewed as therapeutically viable enzyme inhibitors in medicinal chemistry campaigns. On the contrary, we have identified copper and mercury as potent and specific inhibitors of BoNT/A LC protease, and copper complexes were effective in cell-based and in vivo models. Given the success of metals and the failure of many small molecules for inhibiting BoNT/A, other enzyme targets deemed “undruggable” may also be susceptible to metal-mediated inhibition, especially cysteine-containing targets that are homologous to BoNT/A LC. In elucidating a putative Cu/Hg binding site on the serotype A LC, our crystallographic studies have for the first time identified the direct involvement of Cys165. The observed noncompetitive inhibition mechanism which affects enzyme velocity but not substrate binding corroborates the crystal structures because Cys165 does not appear to interact with the SNAP-25 substrate. Mutagenesis studies indicated that the presence of this key cysteine residue was critical for inhibitory potency of Cu/Hg against BoNT/A LC, while serotypes B and E LCs lacking the cysteine could still be inhibited by metals, albeit more weakly. This result implicates the presence of a conserved metal binding site outside of Cys165, which could be investigated in future studies. Moreover, the therapeutic action of metals for ameliorating BoNT intoxication could extend to all BoNT serotypes.
To exploit the therapeutic potential of copper for inhibiting BoNT/A in a biological system, we utilized copper complexes as prodrugs. Our experiments suggest that intracellular bioreduction of these complexes releases copper to directly inhibit the BoNT protease. Pretreating neurons with antioxidants NAC, ASC and Trolox or a glutathione synthesis inhibitor BSO had little to no impact on copper complex efficacy, LDH release or cell rounding. This result shows that the anti-BoNT/A action of the complexes is not sensitive to changes in intracellular redox homeostasis, suggesting that the complexes could enter a highly-regulated copper transport system.56 We envision that upon reduction and release of copper from the complexes, endogenous copper chaperones could traffic the copper to the BoNT/A LC, causing inhibition. For example, chaperones CCS and Atox1 readily bind to Cu (I) and transport it to proteins SOD1 and ATP7A/B, respectively.42–43, 56, 75
In contrast to the anti-BoNT/A action of the examined copper complexes, the efficacy of the same complexes in cancer cells often depends on the generation of reactive oxygen species (ROS) and oxidative stress; anti-cancer activity was reported to decrease in the presence NAC and increase in the presence of BSO.66–69 Although our results show negligible effects of copper complexes on cytotoxicity, the observed cell rounding indicates the inevitable off-target effects of copper in inducing an unfolded-protein response through proteasome inhibition or by direct interference in protein folding.59, 70, 76–77 While ROS and proteasome inhibition play major roles in copper complexes as anticancer agents, they may still occur in our cell-based model but are unlikely to be involved in the copper-mediated inhibition of BoNT/A-induced SNAP-25 cleavage.
In vivo efficacy against BoNT/A was established for both Cu(MEDTC)2 and Cu(GTSM) complexes, and their success is likely attributed to a combination of strong stability under non-reducing conditions and ideal redox potentials that allow them to selectively release copper intracellularly. To our knowledge, copper in the form of these two complexes is one of the only direct BoNT/A LC inhibitors to demonstrate efficacy across enzymatic, cell-based and in vivo assays.78 Pharmacological blockade of BoNT/A internalization via dynamin inhibition79 and disruption of BoNT/A LC translocation via TrxR inhibition36 have been shown to prevent BoNT/A lethality in mice; however, these inhibitors must be administered prophylactically. In contrast, the copper complexes are fast acting and can provide protection of mice post-BoNT/A exposure. The approximate 13 h (2-fold) increase in mouse survival time indicates a significant complex-mediated reduction of BoNT/A intoxication. Complete survival was only observed in a few subjects (injected i.p.) – a result that was likely influenced by the relatively large dose of toxin (5 LD50 units) and relatively low dose of complex (1 mg Cu/kg). Future studies could improve efficacy of the complex via optimization of dosing, administration and physiochemical properties of the copper complexes; moreover, because BTSC and DTC copper complexes are known to have short half-lives, structural modification of the ligands to enhance in vivo stability and pharmacokinetic assessment of next generation complexes could be fruitful.
If the complexes (in the current state) are tested in humans, they would likely need to be administered intravenously (i.v.) to avoid metabolic stability problems; however, the currently approved antitoxin treatment for botulism must also be administered i.v. Side effects from the copper complex would be anticipated, however, it is reasonable to believe they would be no more adverse than the side effects of other metal-based therapeutics e.g. cisplatin. Because botulism is a temporary, yet life-threatening condition, we predict that the therapeutic index of copper DTC or BTSC complexes would be great enough to warrant clinical examination of the complexes as anti-botulism therapeutics, and in fact these copper complexes are already entering clinical trials for treatment of Alzheimer’s (NCT02870634) and cancer (NCT01777919, NCT02963051, NCT00742911).
Supplementary Material
Acknowledgments
We thank Dr. Jiajia Dong and Dr. Barry Sharpless for providing click library screening compounds, Dr. Amanda Roberts for performing preliminary mouse toxicology studies and NIAID (R01AI119564) for funding. This is manuscript # 29473 from The Scripps Research Institute.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01084. Experimental methods, supplementary text, additional enzymatic, cell-based and in vivo assay data, and original gels. Data and coordinates for the X-ray structures of BoNT/A LC soaked with Hg(OAc)2 or CuCl2 have been deposited in the Protein Data Bank with accession codes 5VGX and 5VGV, respectively.
References
- 1.Einhorn LH. J Clin Oncol. 1990;8:1777–81. doi: 10.1200/JCO.1990.8.11.1777. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Nature. 1969;222:385–6. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 3.Finkelstein AE, Walz DT, Batista V, Mizraji M, Roisman F, Misher A. Ann Rheum Dis. 1976;35:251–257. doi: 10.1136/ard.35.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lemire JA, Harrison JJ, Turner RJ. Nat Rev Microbiol. 2013;11:371–384. doi: 10.1038/nrmicro3028. [DOI] [PubMed] [Google Scholar]
- 5.Lee KB, Wang D, Lippard SJ, Sharp PA. Proc Natl Acad Sci U S A. 2002;99:4239–44. doi: 10.1073/pnas.072068399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinto AL, Lippard SJ. Proc Natl Acad Sci U S A. 1985;82:4616–4619. doi: 10.1073/pnas.82.14.4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debnath A, Parsonage D, Andrade RM, He C, Cobo ER, Hirata K, Chen S, Garcia-Rivera G, Orozco E, Martinez MB, Gunatilleke SS, Barrios AM, Arkin MR, Poole LB, McKerrow JH, Reed SL. Nat Med. 2012;18:956–60. doi: 10.1038/nm.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fairlamb AH, Henderson GB, Cerami A. Proc Natl Acad Sci U S A. 1989;86:2607–2611. doi: 10.1073/pnas.86.8.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. J Am Med Assoc. 2001;285:1059–1070. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 10.Herrero BA, Ecklund AE, Streett CS, Ford DF, King JK. Exp Mol Pathol. 1967;6:84. doi: 10.1016/0014-4800(67)90007-x. [DOI] [PubMed] [Google Scholar]
- 11.Shapiro RL, Hatheway C, Becher J, Swerdlow DL. J Am Med Assoc. 1997;278:433–435. [PubMed] [Google Scholar]
- 12.Hooper RR. Mil Med. 1983;148:901–902. [PubMed] [Google Scholar]
- 13.McCarty CL, Angelo K, Beer KD, Cibulskas-White K, Quinn K, de Fijter S, Bokanyi R, Germain ES, Baransi K, Barlow K, Shafer G, Hanna L, Spindler K, Walz E, DiOrio M, Jackson BR, Luquez C, Mahon BE, Basler C, Curran K, Matanock A, Walsh K, Slifka KJ, Rao AK. Morb Mortal Wk Rep. 2015;64:802–803. doi: 10.15585/mmwr.mm6429a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bihari K. Curr Med Res Opin. 2005;21:433–438. doi: 10.1185/030079905X36396. [DOI] [PubMed] [Google Scholar]
- 15.Carruthers A, Carruthers J. Dermatol Surg. 1998;24:1168–1170. doi: 10.1111/j.1524-4725.1998.tb04092.x. [DOI] [PubMed] [Google Scholar]
- 16.Tacket CO, Shandera WX, Mann JM, Hargrett NT, Blake PA. Am J Med. 1984;76:794–798. doi: 10.1016/0002-9343(84)90988-4. [DOI] [PubMed] [Google Scholar]
- 17.Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Nat Struct Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- 18.Pirazzini M, Tehran DA, Zanetti G, Lista F, Binz T, Shone CC, Rossetto O, Montecucco C. Toxicon. 2015;107:32–6. doi: 10.1016/j.toxicon.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 19.Pirazzini M, Bordin F, Rossetto O, Shone CC, Binz T, Montecucco C. FEBS Lett. 2013;587:150–155. doi: 10.1016/j.febslet.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 20.Schiavo G, Rossetto O, Catsicas S, Delaureto PP, Dasgupta BR, Benfenati F, Montecucco C. J Biol Chem. 1993;268:23784–23787. [PubMed] [Google Scholar]
- 21.Whitemarsh RC, Tepp WH, Johnson EA, Pellett S. PloS One. 2014;9:e90252. doi: 10.1371/journal.pone.0090252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boldt GE, Kennedy JP, Janda KD. Org Lett. 2006;8:1729–32. doi: 10.1021/ol0603211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silvaggi NR, Boldt GE, Hixon MS, Kennedy JP, Tzipori S, Janda KD, Allen KN. Chem Biol. 2007;14:533–42. doi: 10.1016/j.chembiol.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 24.Caglic D, Krutein MC, Bompiani KM, Barlow DJ, Benoni G, Pelletier JC, Reitz AB, Lairson LL, Houseknecht KL, Smith GR, Dickerson TJ. J Med Chem. 2014;57:669–676. doi: 10.1021/jm4012164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roxas-Duncan V, Enyedy I, Montgomery VA, Eccard VS, Carrington MA, Lai HG, Gul N, Yang DCH, Smith LA. Antimicrob Agents Chemother. 2009;53:3478–3486. doi: 10.1128/AAC.00141-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai HG, Feng MH, Roxas-Duncan V, Dakshanamurthy S, Smith LA, Yang DCH. Arch Biochem Biophys. 2009;491:75–84. doi: 10.1016/j.abb.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Bremer PT, Adler M, Phung CH, Singh AK, Janda KD. J Med Chem. 2017;60:338–348. doi: 10.1021/acs.jmedchem.6b01393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumaran D, Rawat R, Ludivico ML, Ahmed SA, Swaminathan S. J Biol Chem. 2008;283:18883–91. doi: 10.1074/jbc.M801240200. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt JJ, Stafford RG. FEBS Lett. 2002;532:423–426. doi: 10.1016/s0014-5793(02)03738-9. [DOI] [PubMed] [Google Scholar]
- 30.Zuniga JE, Schmidt JJ, Fenn T, Burnett JC, Arac D, Gussio R, Stafford RG, Badie SS, Bavari S, Brunger AT. Structure. 2008;16:1588–1597. doi: 10.1016/j.str.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bremer PT, Xue S, Janda KD. Chem Commun. 2016;52:12521–12524. doi: 10.1039/c6cc06749b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silhar P, Capkova K, Salzameda NT, Barbieri JT, Hixon MS, Janda KD. J Am Chem Soc. 2010;132:2868. doi: 10.1021/ja910761y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu X, Legler PM, Southall N, Maloney DJ, Simeonov A, Jadhav A. J Comput Aid Mol Des. 2014;28:765–778. doi: 10.1007/s10822-014-9758-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eubanks LM, Silhar P, Salzameda NT, Zakhari JS, Feng XC, Barbieri JT, Shoemaker CB, Hixon MS, Janda KD. ACS Med Chem Lett. 2010;1:268–272. doi: 10.1021/ml100074s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seki H, Xue S, Pellett S, Šilhár P, Johnson EA, Janda KD. J Am Chem Soc. 2016;138:5568–5575. doi: 10.1021/jacs.5b12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pirazzini M, Tehran DA, Zanetti G, Megighian A, Scorzeto M, Fillo S, Shone CC, Binz T, Rossetto O, Lista F, Montecucco C. Cell Rep. 2014;8:1870–1878. doi: 10.1016/j.celrep.2014.08.017. [DOI] [PubMed] [Google Scholar]
- 37.Fu FN, Lomneth RB, Cai SW, Singh BR. Biochemistry. 1998;37:5267–5278. doi: 10.1021/bi9723966. [DOI] [PubMed] [Google Scholar]
- 38.Schiavo G, Rossetto O, Santucci A, Dasgupta BR, Montecucco C. J Biol Chem. 1992;267:23479–23483. [PubMed] [Google Scholar]
- 39.Burnett JC, Schmidt JJ, Stafford RG, Panchal RG, Nguyen TL, Hermone AR, Vennerstrom JL, McGrath CF, Lane DJ, Sausville EA, Zaharevitz DW, Gussio R, Bavari S. Biochem Biophys Res Comm. 2003;310:84–93. doi: 10.1016/j.bbrc.2003.08.112. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed SA, Smith LA. J Protein Chem. 2000;19:475–487. doi: 10.1023/a:1026549431380. [DOI] [PubMed] [Google Scholar]
- 41.Boldt GE, Kennedy JP, Hixon MS, McAllister LA, Barbieri JT, Tzipori S, Janda KD. J Comb Chem. 2006;8:513–521. doi: 10.1021/cc060010h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N, Pierattelli R, Rosato A, Voulgaris P. Nat Chem Biol. 2006;2:367–368. doi: 10.1038/nchembio797. [DOI] [PubMed] [Google Scholar]
- 43.Pufahl RA, Singer CP, Peariso KL, Lin SJ, Schmidt PJ, Fahrni CJ, Culotta VC, PennerHahn JE, OHalloran TV. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]
- 44.Stura EA, Le Roux L, Guitot K, Garcia S, Bregant S, Beau F, Vera L, Collet G, Ptchelkine D, Bakirci H, Dive V. J Biol Chem. 2012;287:33607–14. doi: 10.1074/jbc.M112.396697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santini C, Pellei M, Gandin V, Porchia M, Tisato F, Marzano C. Chem Rev. 2014;114:815–862. doi: 10.1021/cr400135x. [DOI] [PubMed] [Google Scholar]
- 46.Tegoni M, Valensin D, Toso L, Remelli M. Curr Med Chem. 2014;21:3785–3818. doi: 10.2174/0929867321666140601161939. [DOI] [PubMed] [Google Scholar]
- 47.Helsel ME, Franz KJ. Dalton Trans. 2015;44:8760–8770. doi: 10.1039/c5dt00634a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J, Pena MMO, Nose Y, Thiele DJ. J Biol Chem. 2002;277:4380–4387. doi: 10.1074/jbc.M104728200. [DOI] [PubMed] [Google Scholar]
- 49.Duncan C, White AR. Metallomics. 2012;4:127–138. doi: 10.1039/c2mt00174h. [DOI] [PubMed] [Google Scholar]
- 50.Louie AY, Meade TJ. Chem Rev. 1999;99:2711–2734. doi: 10.1021/cr9804285. [DOI] [PubMed] [Google Scholar]
- 51.Whitemarsh RCM, Strathman MJ, Chase LG, Stankewicz C, Tepp WH, Johnson EA, Pellett S. Toxicol Sci. 2012;126:426–435. doi: 10.1093/toxsci/kfr354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adlard PA, Bica L, White AR, Nurjono M, Filiz G, Crouch PJ, Donnelly PS, Cappai R, Finkelstein DI, Bush AI. PloS One. 2011;6 doi: 10.1371/journal.pone.0017669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Hrrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW, Grp PES. Lancet Neurol. 2008;7:779–786. doi: 10.1016/S1474-4422(08)70167-4. [DOI] [PubMed] [Google Scholar]
- 54.Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI. Neuron. 2008;59:43–55. doi: 10.1016/j.neuron.2008.06.018. [DOI] [PubMed] [Google Scholar]
- 55.Kraker A, Krezoski S, Schneider J, Minkel D, Petering DH. J Biol Chem. 1985;260:3710–3718. [PubMed] [Google Scholar]
- 56.Xiao ZG, Donnelly PS, Zimmermann M, Wedd AG. Inorg Chem. 2008;47:4338–4347. doi: 10.1021/ic702440e. [DOI] [PubMed] [Google Scholar]
- 57.Dearling JLJ, Lewis JS, McCarthy DW, Welch MJ, Blower PJ. Chem Commun. 1998:2531–2532.
- 58.Palanimuthu D, Shinde SV, Somasundaram K, Samuelson AG. J Med Chem. 2013;56:722–734. doi: 10.1021/jm300938r. [DOI] [PubMed] [Google Scholar]
- 59.Cater MA, Pearson HB, Wolyniec K, Klaver P, Bilandzic M, Paterson BM, Bush AI, Humbert PO, La Fontaine S, Donnelly PS, Haupt Y. ACS Chem Biol. 2013;8:1621–1631. doi: 10.1021/cb400198p. [DOI] [PubMed] [Google Scholar]
- 60.Crouch PJ, Hung LW, Adlard PA, Cortes M, Lal V, Filiz G, Perez KA, Nurjono M, Caragounis A, Du T, Laughton K, Volitakis I, Bush AI, Li QX, Masters CL, Cappai R, Cherny RA, Donnelly PS, White AR, Barnham KJ. Proc Natl Acad Sci U S A. 2009;106:381–386. doi: 10.1073/pnas.0809057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.John EK, Green MA. J Med Chem. 1990;33:1764–1770. doi: 10.1021/jm00168a035. [DOI] [PubMed] [Google Scholar]
- 62.Green MA, Klippenstein DL, Tennison JR. J Nucl Med. 1988;29:1549–1557. [PubMed] [Google Scholar]
- 63.Torres JB, Andreozzi EM, Dunn JT, Siddique M, Szanda I, Howlett DR, Sunassee K, Blower PJ. J Nucl Med. 2016;57:109–114. doi: 10.2967/jnumed.115.162370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Matsumoto K, Fujibayashi Y, Konishi J, Yokoyama A. Radioisotopes. 1990;39:482–6. doi: 10.3769/radioisotopes.39.11_482. [DOI] [PubMed] [Google Scholar]
- 65.Nomura S, Nozaki S, Hamazaki T, Takeda T, Ninomiya E, Kudo S, Hayashinaka E, Wada Y, Hiroki T, Fujisawa C, Kodama H, Shintaku H, Watanabe Y. J Nucl Med. 2014;55:845–851. doi: 10.2967/jnumed.113.131797. [DOI] [PubMed] [Google Scholar]
- 66.Barilli A, Atzeri C, Bassanetti I, Ingoglia F, Dall’Asta V, Bussolati O, Maffini M, Mucchino C, Marchio L. Mol Pharm. 2014;11:1151–1163. doi: 10.1021/mp400592n. [DOI] [PubMed] [Google Scholar]
- 67.Stefani C, Al-Eisawi Z, Jansson PJ, Kalinowski DS, Richardson DR. J Inorg Biochem. 2015;152:20–37. doi: 10.1016/j.jinorgbio.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 68.Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P, Richardson DR. Cancer Res. 2011;71:5871–5880. doi: 10.1158/0008-5472.CAN-11-1218. [DOI] [PubMed] [Google Scholar]
- 69.Tardito S, Bussolati O, Maffini M, Tegoni M, Giannetto M, Dall’Asta V, Franchi-Gazzola R, Lanfranchi M, Pellinghelli MA, Mucchino C, Mori G, Marchio L. J Med Chem. 2007;50:1916–1924. doi: 10.1021/jm061174f. [DOI] [PubMed] [Google Scholar]
- 70.Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Breast Cancer Res. 2005;7:R897–R908. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maret W, Jacob C, Vallee BL, Fischer EH. Proc Natl Acad Sci U S A. 1999;96:1936–1940. doi: 10.1073/pnas.96.5.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Holmquist B, Vallee BL. J Biol Chem. 1974;249:4601–4607. [PubMed] [Google Scholar]
- 73.Wilson M, Hogstrand C, Maret W. J Biol Chem. 2012;287:9322–9326. doi: 10.1074/jbc.C111.320796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shaw WHR. J Am Chem Soc. 1954;76:2160–2163. [Google Scholar]
- 75.Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 76.Daniel KG, Gupta P, Harbach RH, Guida WC, Dou QP. Biochem Pharmacol. 2004;67:1139–1151. doi: 10.1016/j.bcp.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 77.Tardito S, Bassanetti I, Bignardi C, Elviri L, Tegoni M, Mucchino C, Bussolati O, Franchi-Gazzola R, Marchio L. J Am Chem Soc. 2011;133:6235–42. doi: 10.1021/ja109413c. [DOI] [PubMed] [Google Scholar]
- 78.Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Baldwin MR, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. Proc Natl Acad Sci U S A. 2007;104:2602–2607. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harper CB, Martin S, Nguyen TH, Daniels SJ, Lavidis NA, Popoff MR, Hadzic G, Mariana A, Chau N, McCluskey A, Robinson PJ, Meunier FA. J Biol Chem. 2011;286:35966–35976. doi: 10.1074/jbc.M111.283879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.