

ABSTRACT
The mechanism underlying autophagic degradation of a protein aggregate remains largely unknown. A family of receptor proteins that simultaneously bind to the cargo and the Atg8 family of autophagy proteins (such as the MAP1LC3/LC3 subfamily) has been shown to confer cargo selectivity. The selectivity and efficiency of protein aggregate removal is also modulated by scaffold proteins that interact with receptor proteins and ATG proteins. During C. elegans embryogenesis, autophagic clearance of the cargoes PGL-1 and PGL-3 requires the receptor protein SEPA-1 and the scaffold protein EPG-2. SEPA-1 and EPG-2 also form aggregates that are degraded by autophagy. Here we investigated the effect of composition and organization of PGL granules on their autophagic degradation. We found that depletion of PGL-1 or PGL-3 facilitates the degradation of SEPA-1 and EPG-2. Removal of EPG-2 is also promoted when SEPA-1 is absent. Depletion of PGL-1 or PGL-3 renders the degradation of SEPA-1 independent of EPG-2. We further showed that overexpression of SEPA-1 or EPG-2 as well as SQST-1 or EPG-7 (scaffold protein), which belong to different classes of aggregate, has no evident effect on the degradation of the other type. Our results indicate that the composition and organization of protein aggregates provide another layer of regulation to modulate degradation efficiency.
KEYWORDS: aggrephagy, C. elegans, PGL granules, receptor protein, scaffold protein
Introduction
Autophagy involves the engulfment of a portion of the cytosolic contents in a double-membrane structure, called the autophagosome, and its subsequent delivery to the lysosome for degradation.1,2 Protein aggregates, formed by misfolded proteins, disease-related proteins or aggregate-prone proteins, can be selectively recognized and degraded by autophagy, a process known as aggrephagy.3,4 Loss of autophagy activity has been detected in a variety of human disorders that are associated with protein aggregate accumulation, including Lewy bodies (LBs) in Parkinson disease (PD) and polyglutamine-enriched inclusions in Huntington disease (HD).5-7 The mechanisms by which protein aggregates are selectively degraded by autophagy are not completely understood.
A family of proteins that bind to Atg8-family members has been shown to function as receptors that simultaneously interact with cargoes and the autophagosomal membrane-associated Atg8-family proteins, to mediate selective incorporation of cargoes into autophagosomes.8,9 Among these receptors, SQSTM1 mediates the autophagic degradation of ubiquitinated protein aggregates via its C-terminal ubiquitin-associating (UBA) domain.8,9 SQSTM1 also contains a self-oligomerization PB1 domain and an LC3-interacting region.10,11 Post-translational modifications of cargoes and receptor proteins regulate their binding affinities with each other and/or with Atg8-family proteins such as the LC3 subfamily, and thus modulate efficiency of aggrephagy.12-14 For example, acetylation of mutant HTT (huntingtin) protein promotes its interaction with SQSTM1 and its subsequent targeting into phagophores, the precursors to autophagosomes.12 Phosphorylation of OPTN by the protein kinase TBK1 enhances the LC3 binding affinity and autophagic clearance of cytosolic Salmonella enterica.14 Degradation efficiency of protein aggregates is also regulated by factors linking the cargo-receptor complexes with the autophagic assembly machinery. In mammalian cells, the phosphatidylinositol-3-phosphate-binding protein WDFY3 promotes degradation by binding simultaneously to SQSTM1-positive ubiquitinated protein aggregates and also to autophagy proteins such as those in the ATG12–ATG5-ATG16L1 complex and LC3.15,16 Each protein aggregate has a unique composition and contains distinct cargo proteins. Aggregates with the same set of cargoes can also vary in the amount, post-translational modification and organization of each component.6,7 It is largely unknown whether the distinct composition of a protein aggregate affects its degradation efficiency, and whether one type of protein aggregate affects the removal of another type.
A variety of protein aggregates are degraded by autophagy during C. elegans embryogenesis.4 Components of oocyte-derived P granules, PGL-1 and PGL-3, are degraded by autophagy in somatic cells in C. elegans embryos.17 The formation and degradation of PGL granules requires the self-oligomerized receptor protein SEPA-1, which interacts with PGL-3 and LGG-1 (a C. elegans ortholog of yeast Atg8 and the mammalian Atg8 family).17,18 SEPA-1 also forms aggregates and is degraded by autophagy, both of which are independent of PGL-1 and PGL-3.17 Degradation of PGL-1, PGL-3 and SEPA-1 requires the scaffold protein EPG-2, which directly interacts with SEPA-1 and also with LGG-1.18,19 Depletion of EPG-2 impairs the association of PGL granules with LGG-1 puncta.19 EPG-2 itself forms aggregates and is degraded by autophagy in a manner independent of PGL-1, PGL-3 and SEPA-1.19 Therefore, the removal of PGL-1, PGL-3, SEPA-1 and EPG-2 occurs in a linear hierarchical relationship in wild-type embryos. By altering the levels of each component of the PGL-1-PGL-3-SEPA-1-EPG-2 complex, we found that the degradation efficiency is significantly affected. We also found that accumulation of PGL granules has very little or no effect on the removal of SQST-1 and EPG-7 aggregates, and vice versa. Our results indicate that autophagic degradation efficiency is modulated by the composition of protein aggregates.
Results
Degradation of PGL granules requires the direct interaction of SEPA-1 with EPG-2
sepa-1(bp456) contains a glutamine to produce a stop-codon mutation at amino acid 448. In sepa-1(bp456) mutants, and in autophagy mutants that carry the sepa-1(bp456) mutation, PGL-1 and PGL-3 are diffusely localized in the cytoplasm (Fig. 1A and B).17 We performed genetic screens of sepa-1(bp456) animals to identify mutants with accumulation of GFP::PGL-1 aggregates. One mutant allele, bp929, was cloned and found to contain a mutation in smg-1 (Figs. 1C, S1A and S1B). SMG-1 is an essential component of the nonsense mediated decay (NMD) pathway that surveys and degrades aberrant mRNAs containing a premature stop codon.20 Loss of function of other components in the NMD pathway such as smg-2 also caused the formation of PGL-1-PGL-3 granules in sepa-1(bp456) mutants (Fig. S1C). This suggests that sepa-1(bp456) mRNA, which is stabilized in smg mutants, is translated into a truncated SEPA-1(bp456) protein that mediates aggregation of PGL granules. The truncated SEPA-1(bp456) cannot be detected by the available antibody, as the anti-SEPA-1 antibody was raised against the C-terminal of SEPA-1 that is deleted at bp456. Our previous study showed that a SEPA-1 fragment containing amino acids 39 to 160 self-oligomerizes and also binds to PGL-3.17 We thus depleted SEPA-1 by RNAi to investigate the role of SEPA-1(bp456) in mediating aggregation of PGL-1 and PGL-3. RNAi depletion of sepa-1 abolished the formation of GFP::PGL-1 granules in sepa-1(bp456) smg-1 mutants (Fig. 1D). EPG-2 directly interacts with SEPA-1 but not with PGL-1 and PGL-3 in affinity isolation assays.21 However, SEPA-1(bp456) failed to interact with EPG-2 (Fig. 1E). Consistent with this, PGL-1 granules were separable from EPG-2 aggregates in sepa-1(bp456) smg-1 mutants and sepa-1(bp456) smg-1; lgg-1(RNAi) mutants (Fig. 1F-I). These results indicate that direct interaction of SEPA-1 and EPG-2 is required for efficient removal of PGL granules.
Figure 1.

Direct interaction of EPG-2 with SEPA-1 is essential for the degradation of PGL granules. (A) In wild-type embryos, GFP::PGL-1 is absent in somatic cells and is only observed in germline precursor cells (arrowhead). (B) In sepa-1(bp456) mutant embryos, GFP::PGL-1 is diffusely localized in somatic cells. (C) In sepa-1(bp456) smg-1 mutant embryos, GFP::PGL-1 aggregates are formed in somatic cells (examples are indicated by arrowheads). (D) Depletion of sepa-1 by RNAi in sepa-1(b456) smg-1 mutant embryos results in diffuse localization of GFP::PGL-1 in somatic cells. The phenotypes shown in (B to D) are fully penetrant. (E) MBP-SEPA-1(bp456), the truncated protein in sepa-1(bp456), fails to interact with His-EPG-2 in an affinity isolation assay. *indicates corresponding fusion proteins. Other bands may result from partial degradation of purified proteins. (F to I) GFP::PGL-1 granules are colocalized with EPG-2 in lgg-1(RNAi) mutant embryos (F), but are separable from EPG-2 aggregates in sepa-1 smg-1 mutant embryos (G) and in sepa-1 smg-1; lgg-1(RNAi) mutant embryos (H). Quantification of the colocalization of GFP::PGL-1 aggregates and EPG-2 puncta in lgg-1(RNAi), sepa-1 smg-1 and sepa-1 smg-1; lgg-1(RNAi) embryos at the ∼100 cell stage is shown in (I), n = 3 focal planes from 3 embryos, data are shown as mean ± SD, **P < 0.01, ns, no significant difference. DAPI images of the embryos are show in the left panels. Scale bar: 10 μm (A to D, F to H).
Degradation of PGL-3 is promoted by depleting PGL-1
The development of C. elegans embryos is independent of external nutrients. Degradation of protein aggregates such as PGL granules and SEPA-1 aggregates is mediated by basal autophagy activity.4 In wild-type and autophagy mutant embryos, the number of PGL granules and SEPA-1 aggregates is constant at a specific developmental stage, which greatly facilitates our study of the effect of composition and organization of PGL-1-PGL-3-SEPA-1-EPG-2 aggregates on the degradation of other components.
Formation of aggregates and autophagic removal of PGL-1 depend on PGL-3.17 PGL-1 directly associates with PGL-3.22 PGL-1 fails to be degraded and is diffusely localized in the somatic cells in pgl-3 mutants.17 In pgl-1 mutants, PGL-3, expressed from a GFP reporter, is degraded and shows no accumulation in somatic cells, resembling PGL-3 in wild-type animals (Fig. S1D and E).17 In sepa-1; pgl-1 double mutants, PGL-3::GFP is diffusely localized, as in sepa-1 single mutants (Fig. S1F and G). PGL-3 accumulates into a large number of aggregates in epg-2 mutants.19 We found that PGL-3 aggregates were smaller and less abundant in epg-2; pgl-1 double mutants (Fig. 2A-D). This suggests that in the absence of PGL-1, the degradation of PGL-3 is enhanced and becomes partially independent of EPG-2.
Figure 2.
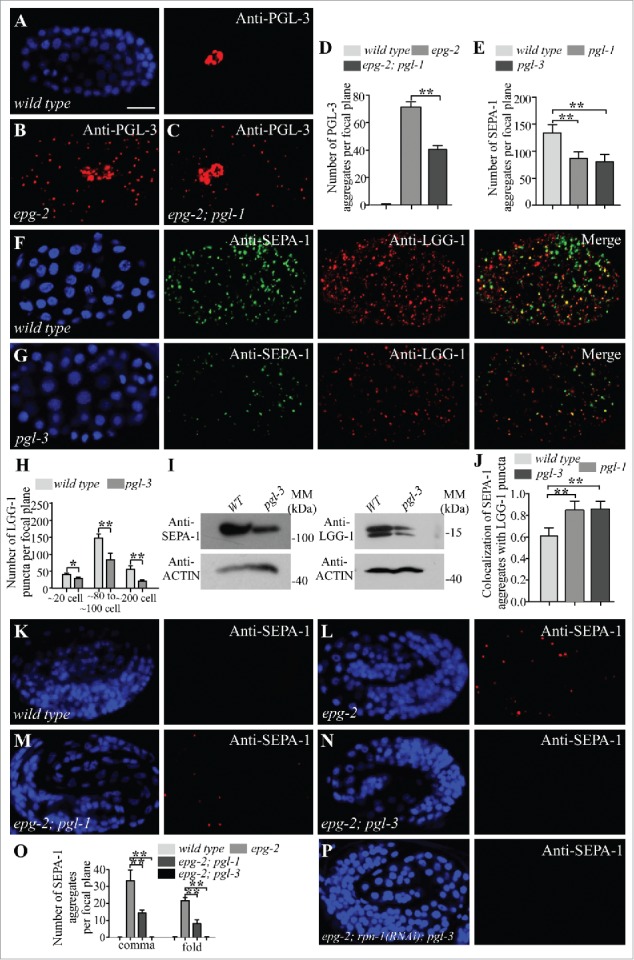
Depletion of PGL-1 and PGL-3 renders degradation of SEPA-1 independent of EPG-2. (A and B) In wild-type embryos, PGL-3 is not detected in somatic cells (A), but accumulates into a large number of granules in the somatic cells of epg-2 mutant embryos (B). (C) The number of PGL-3 aggregates in somatic cells is lower in epg-2; pgl-1 mutant embryos than epg-2 single mutants. Images were taken of embryos at the same developmental stage and using the same exposure time. (D) The number of PGL-3 aggregates per focal plane in wild-type, epg-2 and epg-2; pgl-1 embryos at the ∼100 cell stage. n = 3 focal planes from 3 embryos, data are shown as mean ± SD, **P < 0.01. (E to G) Compared to wild-type embryos (F), the number of SEPA-1 aggregates, detected by anti-SEPA-1, is decreased in pgl-3 mutants while the colocalization of SEPA-1 aggregates with LGG-1 puncta is increased. Quantification of the number of SEPA-1 aggregates in wild-type, pgl-1 and pgl-3 embryos at the ∼100 cell stage is shown in (E). n = 3 focal planes from 3 embryos, data are shown as mean ± SD, **P < 0.01. (H) The number of LGG-1 puncta per focal plane in wild-type and pgl-3 mutant embryos at the ∼20 cell stage, ∼80 to ∼100 cell stage and ∼200 cell stage. n = 3 focal planes from 3 embryos, data are shown as mean ± SD, *P < 0.05, **P < 0.01. (I) Immunoblotting assays revealed that protein levels of SEPA-1 and LGG-1 are decreased in extracts of pgl-3 mutant embryos compared with those of wild-type embryos. (J) The colocalization of SEPA-1 aggregates with LGG-1 puncta in wild-type, pgl-1 and pgl-3 embryos at the ∼100 cell stage. n = 3 focal planes from 3 embryos, data are shown as mean ± SD, **P < 0.01. (K to N) In wild-type embryos, no SEPA-1 aggregates are detected at the fold stage (K). SEPA-1 aggregates accumulate in epg-2 mutant embryos (L). Compared to epg-2 single mutants, the number of SEPA-1 aggregates is reduced in epg-2; pgl-1 mutant embryos (M) and in epg-2; pgl-3 mutant embryos (N). (O) Quantification of the number of SEPA-1 aggregates per focal plane in wild-type embryos and epg-2, epg-2; pgl-1 and epg-2; pgl-3 mutant embryos at the comma and the fold stage. n = 3 focal planes from 3 embryos, data are shown as mean ± SD, **P < 0.01. (P) Compared to epg-2; pgl-3 mutants, the number of SEPA-1 aggregates is not changed in epg-2; rpn-1(RNAi); pgl-3 mutant embryos. Scale bar: 10 μm (A to C, F, G, K to N, P).
Degradation of SEPA-1 is independent of EPG-2 when PGL-1 or PGL-3 is depleted
In wild-type embryos, the number and size of endogenous SEPA-1 aggregates are different at distinct embryonic stages.17 The number peaks at the ∼60 to ∼80 cell stage and decreases from the ∼200 cell stage, with no aggregates detectable in comma stage embryos.17 We determined whether loss of pgl-1 or pgl-3 has an effect on SEPA-1 removal. SEPA-1 aggregates, detected by anti-SEPA-1, were smaller and less abundant in pgl-1 and pgl-3 mutants than in wild-type embryos at the same developmental stage (Figs. 2E, F, G, S1H and S1L). The number of LGG-1 puncta in wild-type embryos is also dynamic; LGG-1 starts to form puncta at the ∼20 cell stage, which peak in number at the ∼80 to ∼100 cell stage and then decrease as development proceeds, becoming undetectable at the comma stage (Figs. 2F, H, S1I and S1K). The number of LGG-1 puncta in pgl-3 mutant is also reduced (Figs. 2G, H, S1J and S1L). Protein levels of SEPA-1 and LGG-1 were decreased in pgl-3 mutants compared with wild-type embryos (Fig. 2I). Consistent with the hypothesis that SEPA-1 is more quickly removed in the absence of PGL-1 or PGL-3, ∼90% of SEPA-1 aggregates colocalized with LGG-1 puncta in pgl-1 and pgl-3 mutants at the ∼80 to ∼100 cell stage, compared with ∼60% in wild-type embryos (Figs. 2F, G, J and S1H). Accumulation of SEPA-1 aggregates in lgg-1 mutants is not affected by loss of pgl-1 and pgl-3.21 These results indicate that depletion of PGL-1 or PGL-3 facilitates the removal of SEPA-1.
Degradation of PGL-1, PGL-3 and SEPA-1 is impaired and more PGL granules are detected in epg-2 mutants (Figs. 2B, L, S2B, S2D, S2L and S2M).19 Compared to autophagy mutants, such as atg-3, the number of SEPA-1 aggregates and PGL granules in epg-2 mutants was similar at the ∼100 cell stage (Figs. 2B, S2A to C and S2G), but was lower from the ∼200 cell and comma stages (Fig. S2D, E, H to J, M), indicating that PGL-1, PGL-3 and SEPA-1 may also be degraded by autophagy in the absence of EPG-2, but with reduced efficiency. Compared to epg-2 mutants, more PGL-1 aggregates are present in sepa-1(bp456) smg-1 mutants (Fig. S2I and K), indicating that the SEPA-1(bp456) protein also shows defects in mediating EPG-2-independent degradation of PGL-1 and PGL-3.
Compared to epg-2 single mutants, epg-2; pgl-1 mutants contained considerably fewer SEPA-1 aggregates at the comma stage and the 4-fold stage, while no SEPA-1 aggregates were detected in epg-2; pgl-3 (Figs. 2K-O and S2O-R). Degradation of SEPA-1 was not affected by inactivation of the proteasome components.17 Simultaneous depletion of rpn-1 by RNAi failed to increase the number of SEPA-1 aggregates in epg-2 mutant embryos (Fig. S2D and F) or in epg-2; pgl-3 mutant embryos (Fig. 2P). LGG-1 puncta and SEPA-1 aggregates are largely separable in epg-2 mutants. However, the colocalization of LGG-1 and SEPA-1 increased in epg-2; pgl-1 mutants at the ∼100 cell stage (Fig. S2N, S, T). These results indicate that in the absence of PGL-1 or PGL-3, degradation of SEPA-1 is largely independent of EPG-2, and may possibly occur by direct interaction with LGG-1.
Degradation of EPG-2 is facilitated in the absence of PGL-1, PGL-3 or SEPA-1
In wild-type embryos, the number of EPG-2 aggregates decreases when embryonic development proceeds.19 Compared to SEPA-1, EPG-2 aggregates are more numerous and persist to a later embryonic stage.21 In pgl-1, pgl-3 or sepa-1 mutants, EPG-2, detected by anti-EPG-2, still formed spherical aggregates in early stage embryos, but the number of aggregates was lower than in wild-type embryos (Figs. 3A to C and S3A to F). The number of EPG-2 aggregates in pgl-3 mutant embryos is reduced (Figs. 3D, G, H and S3C to F). SEPA-1 colocalized with EPG-2 aggregates in pgl-3 mutant embryos (Figs. 3G, H, S3C, and S3D). Protein levels of EPG-2 were decreased in pgl-3 mutants compared with WT embryos in an immunoblotting assay (Fig. 3E). Consistent with their enhanced autophagic degradation, EPG-2 aggregates colocalized more extensively with LGG-1 puncta in pgl-1, pgl-3 and sepa-1 mutants than in wild-type embryos (Figs. 3A, B, F, S3A, S3B). Taken together, these results indicate that depletion of PGL-1, PGL-3 or SEPA-1 promotes degradation of EPG-2.
Figure 3.
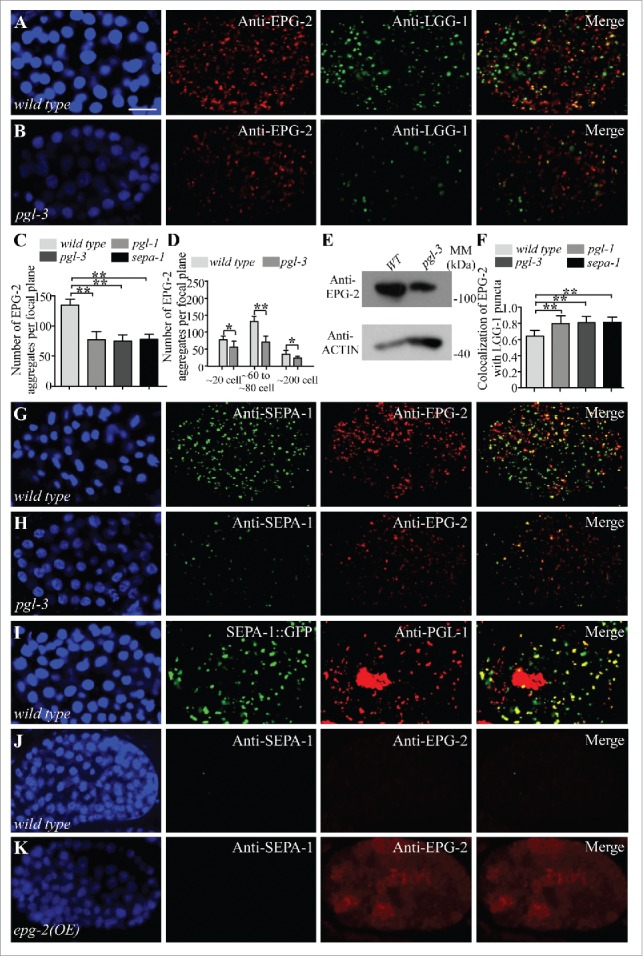
Depletion of PGL-1, PGL-3 and SEPA-1 facilitates the degradation of EPG-2. (A to C) Compared to wild-type embryos (A), colocalization of EPG-2 aggregates with LGG-1 puncta is increased in pgl-3 mutant embryos (B). Quantification of the number of EPG-2 aggregates per focal plane in wild-type embryos and pgl-1, pgl-3 and sepa-1 mutant embryos at the ∼100 cell stage is shown in (C). n = 3 focal planes from 3 embryos, data are shown as mean ± SD, **P < 0.01. (D) Quantification of the number of EPG-2 aggregates per focal plane in wild-type and pgl-3 embryos at the ∼20 cell stage, ∼60 to ∼80 cell stage and ∼200 cell stage. n = 3 focal planes from 3 embryos, data are shown as mean ± SD, *P < 0.05, **P < 0.01. (E) Immunoblotting assays reveal that protein levels of EPG-2 are decreased in extracts of pgl-3 mutant embryos compared with those of wild-type embryos. (F) Quantification of the colocalization of EPG-2 and LGG-1 in wild-type embryos and pgl-1, pgl-3 and sepa-1 mutant embryos at the ∼100 cell stage. n = 3 focal planes from 3 embryos, data are shown as mean ± SD, **P < 0.01. (G and H) Compared to wild-type embryos (G), the number of EPG-2 aggregates in pgl-3 mutant is reduced and the colocalization of SEPA-1 and EPG-2 aggregates is increased in pgl-3 mutant embryos (H). (I) In embryos expressing sepa-1::gfp, endogenous PGL-1, detected by anti-PGL-1, accumulates and colocalizes with SEPA-1::GFP aggregates at the ∼200 cell stage. (J and K) Compared to wild-type embryos (J), embryos overexpressing epg-2 display stronger diffuse EPG-2 signals, detected by anti-EPG-2, at the ∼200 cell stage, but no SEPA-1 aggregates are detected. Scale bar: 10 μm (A, B, G to K).
Overexpression of SEPA-1 impairs, while overexpression of EPG-2 facilitates, the removal of PGL granules
In wild-type embryos, PGL-1-PGL-3 granules are not detected in somatic cells from the ∼24 cell stage onwards.17 However, in embryos overexpressing SEPA-1::GFP, endogenous PGL-3 and PGL-1 accumulated and colocalized with SEPA-1::GFP aggregates in somatic cells, indicating that overexpression of SEPA-1 impairs the degradation of PGL-1 and PGL-3 (Fig. 3I and S3G). We examined whether overexpression of epg-2 affects the degradation of PGL granules in wild-type worms. Transgenic lines expressing epg-2 genomic DNA were analyzed. EPG-2, detected by anti-EPG-2, formed small aggregates at the ∼100 cell stage (Fig. S3H and I). From the ∼200 cell stage and onward, diffuse fluorescence was detected in embryos carrying the transgene (Fig. 3J and K). No ectopic accumulation of SEPA-1 was observed in embryos expressing high levels of epg-2 (Fig. 3J and K). In contrast, the number of SEPA-1::GFP aggregates at the late embryonic stage was decreased in embryos carrying a transgene expressing epg-2 (Fig. 4A to C), indicating that EPG-2 facilitates the removal of SEPA-1.
Figure 4.

Overexpression of PGL granules or SQST-1 aggregates has no effect on the degradation of the other. (A to C) A few SEPA-1::GFP aggregates are present in wild-type embryos at the 4-fold stage (A), while no aggregates are detected in embryos overexpressing epg-2 (the transgenic line is different from the one shown in [Figure 3K)]. Quantification of the numbers of SEPA-1::GFP aggregates per focal plane in wild-type and epg-2(OE) embryos at the fold stage is shown in (C). n = 3 focal planes from 3 embryos, data are shown as mean ± SD, *P < 0.05. (D and E) Compared to wild-type embryos (D), SQST-1::GFP accumulates into a large number of aggregates in embryos expressing sqst-1::gfp, but removal of EPG-7 is not affected (E). (F) In embryos expressing epg-7::gfp, EPG-7::GFP accumulates into a large number of aggregates and a large number of SQST-1 aggregates accumulate and colocalize with EPG-7::GFP. (G) Model for the composition of the PGL-1-PGL-3-SEPA-1-EPG-2 complex that modulates the efficiency of autophagic degradation. In wild-type embryos, PGL-1, PGL-3 and SEPA-1 are mainly degraded via the EPG-2-mediated degradation pathway. In the absence of PGL-1-PGL-3, SEPA-1 can be degraded in a manner independent of EPG-2. Depletion of SEPA-1 facilitates the removal of EPG-2. Overexpression of SEPA-1 traps PGL-1 and PGL-3 in the aggregates and impairs their EPG-2-mediated degradation. The density of LGG-1 proteins interacting with SEPA-1 and EPG-2 is proportional to the degradation efficiency of the complex. Scale bar: 10 μm (A, B, D to F).
The relationship between degradation of PGL granules and SQST-1/EPG-7 aggregates
The C. elegans SQSTM1 ortholog SQST-1 is degraded by autophagy during embryogenesis.19 In wild-type embryos, SQST-1 is weak and diffuse in the cytoplasm. Degradation of SQST-1 requires the scaffold protein EPG-7, which interacts with SQST-1 and also with multiple ATG proteins.23 EPG-7 self-oligomerizes and is itself degraded by autophagy independent of SQST-1.23 PGL granules and SQST-1 aggregates are distinct types of aggregate, which are separate in various autophagy mutants.19 epg-2 mutants showed no accumulation of EPG-7 aggregates (Fig. S3J and K). The number of SQST-1 aggregates in epg-7 mutants was not evidently decreased by simultaneous depletion of pgl-1, pgl-3, sepa-1 or epg-2 (Fig. S4A-E). No SQST-1 aggregates were detected in pgl-1 and pgl-3 mutants (Figs. 4D, S4F and S4G). Loss of function of epg-7 causes no defect in removal of SEPA-1 and EPG-2 in late-stage embryos.23 The accumulation of PGL granules observed in epg-2 mutant embryos is not enhanced or suppressed by depletion of sqst-1, but it is slightly enhanced by loss of epg-7 function (Fig. S4H to J).
We determined whether overexpression of PGL granules or SQST-1/EPG-7 congests autophagic flux and consequently affects degradation of the other type of aggregate. Overexpression of SQST-1::GFP causes aggregate accumulation (Fig. 4D and E). However, removal of EPG-7, SEPA-1 and EPG-2 is not affected (Figs. 4D, E, S4K and S4L). In embryos carrying transgenic lines strongly expressing epg-7::gfp, numerous EPG-7::GFP aggregates were formed (Fig. 4F).23 The expression level and the number of EPG-7::GFP aggregates in those transgenic lines was further increased by loss of autophagy activity (Fig. S4M and N).23 No SQST-1 aggregates were detected in wild-type embryos, while a large number of SQST-1 aggregates accumulated and colocalized with EPG-7::GFP in embryos carrying the epg-7 overexpression (OE) transgene (Fig. 4F). The number of SQST-1 aggregates in epg-7::gfp (OE) animals, however, was much less than in lgg-1 mutant embryos (Fig. S4O to Q). The number of LGG-1 puncta is slightly increased in embryos overexpressing EPG-7 from the ∼20 cell stage to the comma stage; however, the abundance of LGG-1 puncta is not changed in SQST-1-overexpressing embryos (Fig. S4R to T). Degradation of PGL granules and EPG-2 aggregates was not affected in epg-7(OE) animals (Fig. S4U). In embryos carrying the epg-2(OE) transgene, no obvious SQST-1 aggregates and EPG-7 aggregates accumulated (Fig. S4V and W). Therefore, accumulation of PGL granules has no effect on the removal of SQST-1-EPG-7 aggregates, and vice versa.
Discussion
Here we demonstrated that autophagic degradation of protein aggregates is modulated by their composition. In wild-type embryos, degradation of SEPA-1 depends on the scaffold protein EPG-2.17,19 The number of SEPA-1 aggregates in epg-2 mutants, however, is lower than in autophagy mutants, indicating that SEPA-1 degradation also occurs slowly in an EPG-2-independent manner. In wild-type embryos, a portion of SEPA-1 aggregates colocalize with LGG-1 puncta. This suggests that SEPA-1 is in equilibrium between functional complexes that are degraded by autophagy and aggregates that are resistant to degradation or are slowly targeted for degradation. In epg-2 mutants, the presence of PGL-1 and PGL-3 in SEPA-1 aggregates may decrease direct interaction of the aggregates with the autophagic machinery, thus impairing the degradation of SEPA-1. In contrast, when the PGL-SEPA-1 interaction or stoichiometry is altered (like when PGL-3 is absent), SEPA-1 aggregates are recognized by the autophagic machinery independent of EPG-2. Accordingly, colocalization of SEPA-1 aggregates with LGG-1 puncta in epg-2 mutants is greatly enhanced by simultaneous depletion of PGL-1 or PGL-3. The removal of EPG-2 is also promoted in the absence of PGL-1, PGL-3 or SEPA-1. Therefore, the presence of PGL-1 and PGL-3 in the aggregates not only slows the removal of SEPA-1 and EPG-2 but also confers the requirement for EPG-2 on efficient SEPA-1 degradation.
Proteins degraded by autophagy possess distinct aggregation properties. Germline P granules that are partitioned into somatic founder cells are quickly disassembled. PGL-1 and PGL-3 are degraded during the first few cell divisions and no aggregates can be detected.17 Formation of PGL-1 and PGL-3 aggregates in autophagy mutants strictly depends on SEPA-1.17 SEPA-1 and EPG-2 form aggregates and gradually colocalize during embryogenesis.21 SQST-1 and EPG-7 are weakly expressed and diffusely localized in the cytoplasm.19,23 The aggregation of PGL-1 and PGL-3 is modulated by post-translational arginine methylation,21 which is performed by the arginine methyltransferase PRMT-1/EPG-11. Loss of function of prmt-1/epg-11 causes the incorporation of PGL-1 and PGL-3 into granules in a SEPA-1-independent manner, and thus alters the composition or organization of PGL granules.21 The PGL granules in prmt-1/epg-11 mutants show reduced association with EPG-2 and subsequently impaired degradation.21 When overexpressed, SEPA-1 and EPG-7 form big aggregates and trap endogenous PGL-1, PGL-3 and SQST-1, respectively, thus impairing their degradation (Fig. 4G). Overexpression of EPG-2, however, increases the diffuse fluorescent signal from the ∼200 cell stage and promotes the removal of SEPA-1 aggregates. Therefore, levels of aggregate-prone proteins and their propensity for aggregation modulate the degradation efficiency of protein aggregates.
Receptor proteins and scaffold proteins directly interact with proteins of the Atg8 family, as well as other ATG proteins.17,23 Overexpression of one type of protein aggregate might sequestrate essential ATG proteins and consequently congest autophagic flux and affect autophagic degradation of other types of protein aggregate. However, we found that large protein aggregates resulting from overexpression of SEPA-1, as well as SQST-1 or EPG-7 cause no evident defect in the removal of the other type. Moreover, these aggregates are largely separable from LGG-1 puncta, indicating that additional factors are required for triggering aggrephagy initiation.
Efficient clearance of misfolded proteins and toxic aggregate-prone proteins is crucial for maintaining cellular homeostasis. Decreased solubility of specific disease-associated proteins and, concomitantly, increased aggregate formation is observed in various human diseases. Protein aggregates associated with different diseases differ in composition. For example, SNCA/α-synuclein accumulates in Parkinson disease, while β-amyloid accumulates in Alzheimer disease.5-7 SQSTM1 is generally present in ubiquitin-positive aggregates.24 Post-translational modification of these disease-related proteins modulates aggregation and thus affects the degradation efficiency. For example, sumoylated SNCA promotes its solubility and the sumoylation-defective mutant SNCA exhibits increased propensity for aggregation and cytotoxicity.25 Our study demonstrates that the composition and organization of protein aggregates, which are regulated by levels and post-translational modification of its components, have a great impact on autophagic degradation efficiency.
Materials and methods
Worm strains
The following strains were used in this work: N2 Bristol (wild-type), epg-2(bp287), atg-3(bp412), pgl-3(bp438), sepa-1(bp456), pgl-1(bn101), smg-1(bp929), epg-7(tm2508), sqst-1(ok2892), bpIs131(sepa-1::gfp, unc-76), bnIs1(Ppie-1::gfp::pgl-1, unc-119), bpIs241(epg-7::gfp(OE), rol-6), adIs2122(Plgg-1::gfp::lgg-1, rol-6), axIs1464(Ppie-1::gfp::pgl-3, unc-119), bpEx270(sqst-1::gfp(OE), unc-76) and bpEx213(epg-2(OE), unc-76).
Mapping and cloning of smg-1
bp929 was identified in screens for mutations that caused formation of GFP::PGL-1 aggregates in sepa-1(bp456) mutant embryos. Mapping with SNP markers placed bp929 between 0.04 and 2.8 on linkage group I. The molecular lesion in bp929 was then determined by whole genome sequencing. smg-1(bp929) failed to complement smg-1(e1228).
Indirect immunofluorescence
Embryos were collected, permeabilized by freeze-cracking, and fixed with methanol-acetone then blocked with PBS (140 mmol/L NaCl, 2.7 mmol/L KCl, 10 mmol/L Na2HPO4, 2 mmol/L KH2PO4, 1% BSA [Amresco, CAS: 9048–46–8]) for 1 h and incubated with primary antibodies for 1 to 2 h at room temperature. The embryos were then washed 3 times and incubated for 1 h with secondary antibodies. The following primary antibodies were generated in the lab: polyclonal rat anti-PGL-3, polyclonal rat anti-EPG-2, polyclonal rat anti-EPG-7, polyclonal rabbit anti-PGL-1, polyclonal rat anti-SQST-1, polyclonal rat or rabbit anti-LGG-1 and polyclonal rabbit anti-SEPA-1.
RNAi inactivation experiments in C. elegans
For RNAi injection experiments, single-stranded RNA (ssRNA) was transcribed from T7- and SP6-flanked PCR templates. ssRNAs were then annealed and injected into young adult animals carrying various reporters. Injected animals were placed on fresh plates overnight. The embryos laid during the next day were analyzed. All animals were cultured at 20°C. The DNA templates used for RNA synthesis were: lgg-1(C32D5: nt 35232–35666), smg-2(Y48G8AL: nt 45387–45406), sepa-1(T04D3: nt 66–931), and rpn-1(T22D1: nt 18247–25112).
epg-2 and epg-7::gfp Overexpression
Fosmid (WRM0614dc06) containing epg-2 genomic DNA or plasmid containing epg-7::gfp was injected at a concentration of 50 ng/ml with unc-76 expression vector (p76–16B) into unc-76(e911) animals. At least 3 stable transgenic lines were analyzed.
In vitro affinity isolation assays
cDNAs encoding full-length or truncated EPG-2 and SEPA-1 were cloned into pET-28a (for His tagging; Addgene, 45174) or pMal-C2X (for MBP fusion; Addgene, 75286). MBP fusion proteins were incubated with His-tagged proteins and amylose resin (for MBP fusion proteins; NEB, E8021). Bound proteins were analyzed by western blot using an anti-His antibody (Sigma, H1029). 20% of the fusion protein used for affinity isolation is shown as input.
Western blot assays
For western blot assays, embryonic extracts were subjected to SDS-PAGE and signals were detected using corresponding primary and secondary antibodies. As a gel loading control, anti-ACTIN monoclonal antibody (Sigma, A3853) was used.
Statistical analysis
All data are shown as mean ± SD. Unpaired Student t tests were performed for statistical analysis. At least 3 independent repeats were performed for each experiment. A P value less than 0.05 was considered significant (*); a P value less than 0.01 was considered extremely significant (**). In confocal images, 2 fluorescent markers were counted as colocalized when they overlapped partially or completely.
Supplementary Material
Abbreviations
- ATG
autophagy related
- epg
ectopic PGL granules
- GFP
green fluorescent protein
- LC3
light chain 3
- LGG-1LC3
LC3GABARAP and GATE-16 family
- OE
overexpression
- PGL
P granule abnormality protein
- PRMT
protein arginine methyltransferase
- rpn
proteasome regulatory particle non-ATPase-like
- SEPA-1
suppressor of ectopic P granules in autophagy mutant
- smg
suppressor with morphological effect on genitalia
- SNCA
synuclein α
- SQST-1
sequestosome related
- WT
wild type
Acknowledgments
We are grateful to Dr. Isabel Hanson for editing work.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC) (31630048, 31421002, 31561143001), a grant from the National Basic Research Program of China (2013CB910100) to H.Z. The research of Hong Zhang was supported in part by an International Early Career Scientist grant from the Howard Hughes Medical Institute.
References
- [1].Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat Rev Mol Cell Biol 2009; 10:458-67; PMID: 19491929; https://doi.org/ 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- [2].Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID: 24366339; https://doi.org/ 10.1038/cr.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID: 18191218; https://doi.org/ 10.1016/j.cell.2007.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lu Q, Wu F, Zhang H. Aggrephagy: Lessons from C. elegans. Biochem J 2013; 452:381-90; PMID: 23725457; https://doi.org/ 10.1042/BJ20121721 [DOI] [PubMed] [Google Scholar]
- [5].Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 2003; 4:49-60; PMID: 12511861; https://doi.org/ 10.1038/nrn1007 [DOI] [PubMed] [Google Scholar]
- [6].Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl) 2003; 81:678-99; PMID: 12942175; https://doi.org/ 10.1007/s00109-003-0464-5 [DOI] [PubMed] [Google Scholar]
- [7].Aguzzi A, O'Connor T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat Rev Drug Discov 2010; 9:237-48; PMID: 20190788; https://doi.org/ 10.1038/nrd3050 [DOI] [PubMed] [Google Scholar]
- [8].Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci 2013; 126:3237-47; PMID: 23908376. [DOI] [PubMed] [Google Scholar]
- [9].Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID: 21189453; https://doi.org/ 10.4161/auto.7.3.14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID: 17580304; https://doi.org/ 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [11].Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283:22847-57; PMID: 18524774; https://doi.org/ 10.1074/jbc.M802182200 [DOI] [PubMed] [Google Scholar]
- [12].Jeong H, Then F, Melia TJ, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, et al.. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009; 137:60-72; PMID: 19345187; https://doi.org/ 10.1016/j.cell.2009.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44:279-89; PMID: 22017874; https://doi.org/ 10.1016/j.molcel.2011.07.039 [DOI] [PubMed] [Google Scholar]
- [14].Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al.. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333:228-33; PMID: 21617041; https://doi.org/ 10.1126/science.1205405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HC, Lamark T, et al.. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 2010; 38:265-79; PMID: 20417604; https://doi.org/ 10.1016/j.molcel.2010.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Øvervatn A, Stenmark H, Bjørkøy G, Simonsen A, et al.. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010; 6:330-44; PMID: 20168092; https://doi.org/ 10.4161/auto.6.3.11226 [DOI] [PubMed] [Google Scholar]
- [17].Zhang Y, Yan L, Zhou Z, Yang P, Tian E, Zhang K, Zhao Y, Li Z, Song B, Han J, et al.. SEPA-1 mediates the specific recognition and degradation of P granule components by autophagy in C. elegans. Cell 2009; 136:308-21; PMID: 19167332; https://doi.org/ 10.1016/j.cell.2008.12.022 [DOI] [PubMed] [Google Scholar]
- [18].Wu F, Watanabe Y, Guo XY, Qi X, Wang P, Zhao HY, Wang Z, Fujioka Y, Zhang H, Ren JQ, et al.. Structural basis of the differential function of the two C. elegans Atg8 homologs, LGG-1 and LGG-2, in autophagy. Mol Cell 2015; 60:914-29; PMID: 26687600; https://doi.org/ 10.1016/j.molcel.2015.11.019 [DOI] [PubMed] [Google Scholar]
- [19].Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al.. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010; 141:1042-55; PMID: 20550938; https://doi.org/ 10.1016/j.cell.2010.04.034 [DOI] [PubMed] [Google Scholar]
- [20].Kervestin S, Jacobson A. NMD: A multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 2012; 13:700-12; PMID: 23072888; https://doi.org/ 10.1038/nrm3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li S, Yang P, Tian E, Zhang H. Arginine methylation modulates autophagic degradation of PGL granules in C. elegans. Mol Cell 2013; 52:421-33; PMID: 24140420; https://doi.org/ 10.1016/j.molcel.2013.09.014 [DOI] [PubMed] [Google Scholar]
- [22].Kawasaki I, Amiri A, Fan Y, Meyer N, Dunkelbarger S, Motohashi T, Karashima T, Bossinger O, Strome S. The PGL family proteins associate with germ granules and function redundantly in Caenorhabditis elegans germline development. Genetics 2004; 167:645-61; PMID: 15238518; https://doi.org/ 10.1534/genetics.103.023093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lin L, Yang P, Huang X, Zhang H, Lu Q, Zhang H. The scaffold protein EPG-7 links cargo-receptor complexes with the autophagic assembly machinery. J Cell Biol 2013; 201:113-29; PMID: 23530068; https://doi.org/ 10.1083/jcb.201209098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 2009; 34:259-69; PMID: 19450525; https://doi.org/ 10.1016/j.molcel.2009.04.026 [DOI] [PubMed] [Google Scholar]
- [25].Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, Urlaub H, Zweckstetter M, Kügler S, Melchior F, et al.. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol 2011; 194:49-60; PMID: 21746851; https://doi.org/ 10.1083/jcb.201010117 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.