

ABSTRACT
A variant within the gene locus encoding PTPN22 (protein tyrosine phosphatase, non-receptor type 22) emerged as an important risk factor for auto-inflammatory disorders, including rheumatoid arthritis, systemic lupus erythematosus and type 1 diabetes, but at the same time protects from Crohn disease, one of the 2 main forms of inflammatory bowel diseases. We have previously shown that loss of PTPN22 results in decreased NLRP3 (NLR family pyrin domain containing 3) activation and that this effect is mediated via enhanced NLRP3 phosphorylation. However, it is unclear how phosphorylation of NLRP3 mediates its inhibition. Here, we demonstrate that loss of macroautophagy/autophagy abrogates the inhibitory effect on NLRP3 activation observed upon loss of PTPN22. Phosphorylated, but not nonphosphorylated NLRP3 is found in autophagosomes, indicating that NLRP3 phosphorylation mediates its inactivation via promoting sequestration into phagophores, the precursors to autophagosomes. This finding shows that autophagy and NLRP3 inflammasome activation are connected, and that PTPN22 plays a key role in the regulation of those 2 pathways. Given its role in inflammatory disorders, PTPN22 might be an attractive therapeutic target, and understanding the cellular mechanisms modulated by PTPN22 is of crucial importance.
KEYWORDS: inflammasome, Lyp, NLRP3, NOD-like receptor protein, PEST-enriched phosphatase, SQSTM1, tyrosine phosphorylation
Introduction
Inflammasomes are multiprotein aggregates that form upon intracellular presence of damage-associated molecular patterns (DAMPs), and typically consist of 3 core components: (1) a receptor protein that initiates inflammasome assembly upon activation, (2) the adaptor protein PYCARD/ASC (PYD and CARD domain containing; not present in some inflammasome-complexes), and (3) the protease CASP1/caspase-1. Once assembled, inflammasomes mediate the cleavage of pro-CASP1 into its active form. Active CASP1 in turn cleaves the precursors of several inflammatory molecules, including IL1B (interleukin 1 β) and IL18 (interleukin 18) into their active forms.1 One of the best studied inflammasome receptors is NLRP3 (NLR family pyrin domain containing 3), which responds to several damage- and pathogen-associated molecules, including bacterial products, such as muramyl dipeptide (MDP)2 or Listeria toxin,3 particulate materials (e.g., silicium dioxide, titanium dioxide, monosodium crystals)4,5 as well as changes in intracellular potassium levels and increased levels of reactive oxygen species.6,7 Due to the potent pro-inflammatory response upon IL1B and IL18 release, inflammasome-inducing receptors and subsequent formation of inflammasome complexes is tightly regulated. Since NLRP3 responds to such a wide range of activators, control of NLRP3 activation is of special importance. In addition to its restricted expression, and polyubiquitination that mediates NLRP3 degradation via the proteasome,8 we have recently found that NLRP3 activation is negatively regulated by tyrosine phosphorylation.9 However the exact mechanism by which phosphorylation of NLRP3 interferes with its ability to form inflammasome-complexes is still unknown.
The phosphatase responsible for dephosphorylation and subsequently robust activation of NLRP3 upon the presence of danger molecules is PTPN22/PEP/Lyp (protein tyrosine phosphatase, nonreceptor type 22).9 While loss of PTPN22 results in decreased NLRP3-mediated IL1B secretion, presence of a variant in the gene encoding PTPN22 promotes inflammasome activity.9 This variant has been associated with increased risk to develop several chronic inflammatory disorders, including rheumatoid arthritis (RA)10,11 systemic lupus erythematosus (SLE)12-15 and type I diabetes, but at the same time is negatively associated with the onset of Crohn disease (CD), one subform of inflammatory bowel disease (IBD).16,17 PTPN22 is not only involved in inflammasome-activation, but also exerts other prominent functions such as regulating the response to type I interferons,18,19 and negative regulation of T cell receptor signaling cascades.20-22 We have further shown that PTPN22 expression is decreased in intestinal tissue of CD patients and interferes with MDP-induced autophagy.23,24
Autophagy is an important cellular process that mediates bulk degradation of damaged/dysfunctional proteins and organelles in the cytosol.25,26 In addition, autophagy is critically involved in degradation and removal of invading bacteria27-29 making it a pivotal player in intestinal homeostasis.27,30-32 Genetic variants in molecules involved in the autophagy machinery, such as ATG16L1 (autophagy-related 16 like 1)33 and IRGM (immunity-related GTPase M)17,34 increase the risk to develop IBD. Further, there is evidence that autophagy might be involved in the control of IL1B secretion.35-37
The aim of this study was to investigate whether PTPN22 interferes with cellular pathways influencing NLRP3 activation. Since PTPN22 controls autophagy, and autophagy controls IL1B secretion, the focus was to address whether PTPN22 mediates its control on NLRP3 activation in an autophagy-dependent manner.
Results
Inhibition of autophagy results in increased IL1B secretion in both PTPN22 competent and PTPN22-deficient cells
Because we have previously shown that loss of PTPN22 results in enhanced levels of autophagy,23 and autophagy has been implicated in the regulation of IL1B secretion35-37 we first addressed whether autophagy plays a role in the reduction of IL1B secretion observed upon loss of PTPN22. Therefore, THP-1 cells expressing either control or PTPN22-targeting shRNA, were treated with the autophagy inhibitors 3-methyladenine (3-MA), bafilomycin A1, or the phosphatidylinositol 3-kinase inhibitor wortmannin before treatment with MDP, a pathogen-associated molecular pattern (PAMP) that induces autophagy as well as the secretion of IL1B. As expected, loss of PTPN22 resulted in enhanced autophagy as observed by increased MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 β)-II accumulation, but impaired secretion of IL1B (Fig. 1A, B). Of note, loss of PTPN22 did not affect protein expression of pro-IL1B (Fig. 1A). Inhibition of autophagy using 3-MA, wortmannin, or bafilomycin A1 promoted secretion of IL1B in both PTPN22-competent and PTPN22-deficient cells (Fig. 1B). When autophagy was inhibited, PTPN22-deficient cells secreted as much IL1B as PTPN22-competent cells (Fig. 1B). The effects on IL1B secretion were due to differences in inflammasome activation: levels of cleaved CASP1 were decreased in PTPN22-deficient cells, but enhanced in cells where autophagy was inhibited (Fig. 1A). Similar results were obtained in bone marrow-derived dendritic cells (BMDC) from either wild-type (WT) or Ptpn22-deficient (ptpn22−/−) mice (Fig. 1C, D), and when monosodium urate (MSU) or ATP were used as inflammasome activators (Fig. S1): whereas PTPN22-deficient BMDC secreted reduced levels of IL1B compared with WT cells, this effect was abrogated upon inhibition of autophagy using 3-MA, wortmannin or bafilomycin A1 (Fig. 1C, D, Fig. S1).
Figure 1.
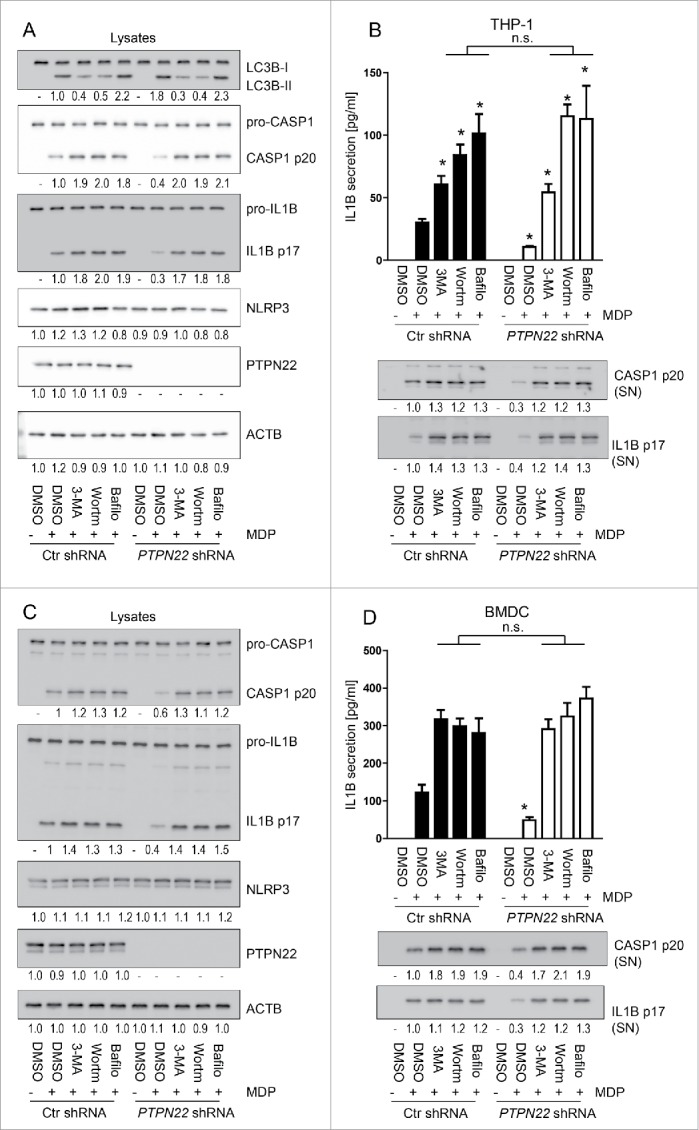
Reduction of IL1B secretion upon loss of PTPN22 is not observed in autophagy-deficient cells. PMA-differentiated THP-1 cells expressing either control, or PTPN22-specific shRNA (A and B), and BMDC from wild-type (WT) or PTPN22 deficient (ptpn22−/−) mice (C and D) were treated with 3-MA (1 mM), (Wortm; 10 uM), or bafilomycin A1 (Bafilo; 100 nM) to inhibit autophagy, 1 h before treatment with muramyl dipeptide (MDP, 100 ng/ml) for 24 h. The graphs show (A)and (C): representative western blot pictures of the indicated proteins and (B and D): results from IL1B ELISA. Data are representative for 1 out of 3 independent experiments with 3 replicates (n = 3). Numbers below the western blot pictures show results of densitometric measurements. * = p < 0.05, n.s. = not significant, one-way ANOVA with Bonferroni correction.
To confirm this finding, we next used nontargeting control siRNA or siRNA constructs specific for ATG16L1/Atg16l1, a molecule essential for autophagy induction. Again, inhibition of functional autophagy resulted in enhanced cleavage of CASP1 and enhanced IL1B secretion in both PTPN22-competent and PTPN22-deficient cells (Fig. 2A, B and Fig. S2A, B, Fig. S3A). Interestingly, the reduction in CASP1 cleavage and IL1B secretion observed upon loss of PTPN22 was no longer present when autophagy was blocked (Fig. 2A, B, Fig. S2A, B, Fig. S3A). Again, these effects were similar in cells treated with MSU (Fig. 2) or with MDP (Fig. S2, Fig. S3). Similar findings were obtained when we silenced LC3B/Lc3b (Fig. 2C, D, Fig. S2C, D, Fig. S3B), although cells lacking LC3B are not completely deficient in autophagy due to the presence of other LC3 isoforms. However, silencing of LC3B/Lc3b clearly reduced autophagy as indicated by enhanced levels of SQSTM1/p62 (sequestosome 1), a molecule degraded via autophagy. Taken together, these data strongly indicate a role for autophagy in the reduction of IL1B secretion and inflammasome activation observed in PTPN22-deficient cells.
Figure 2.
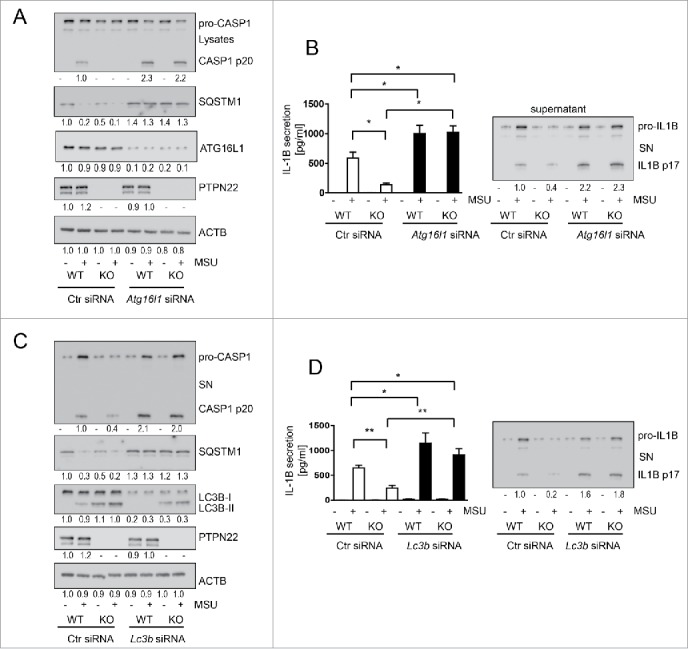
Knockdown of LC3B or ATG16L1 abrogates the reduction in IL1B secretion observed in PTPN22-deficient cells. BMDC from WT or ptpn22−/− mice were treated with nontargeting control siRNA or siRNA specific for Atg16l1 (A and B) or Lc3b (C and D) 48 h before priming with ultra-pure lipopolysaccharide (upLPS) (16 h) and subsequent activation with MSU (150 mg/ml) for 6 h. The graphs show (A and C): representative western blots from cell lysates for the indicated proteins; and (B and D): IL1B ELISA and western blot analysis from cell culture supernatants. Data are representative for 1 out of 3 independent experiments with 3 replicates (n = 3). Numbers below the western blot pictures show results of densitometric measurements. * = p < 0.05, ** = p < 0.01; one-way ANOVA with Bonferroni correction.
NLRP3 is found in autophagosomes upon inflammasome activation
We have previously shown that loss of PTPN22 results in enhanced phosphorylation of NLRP3, and that phosphorylation of NLRP3 results in decreased inflammasome activity. To test how autophagy might affect phosphorylated NLRP3, we analyzed whether NLRP3 is present in autophagosomes, and whether this is dependent on its phosphorylation status. To this end, we enriched autophagosomes37,38 from MDP- or MSU-treated THP-1 cells or BMDC and probed for NLRP3. While in untreated cells only a faint NLRP3 band was detectable in the autophagosome-enriched fraction, NLRP3 was readily detectable in autophagosomes of MDP- or MSU-treated cells (Fig. 3A, Fig. S4A, B). Of note, in cells lacking PTPN22, more NLRP3 was present in the autophagosome-enriched fraction upon NLRP3 activation (Fig. 3A). This was clearly an effect resulting from the lack of PTPN22 phosphatase function, because the same effect was observed in cells that express a loss-of-function variant in PTPN2213 (Fig. S4B). We also detected PYCARD/ASC, but not CASP1 or IL1B in autophagosomes upon inflammasome activation (Fig. 3A, Fig. S4B).
Figure 3.
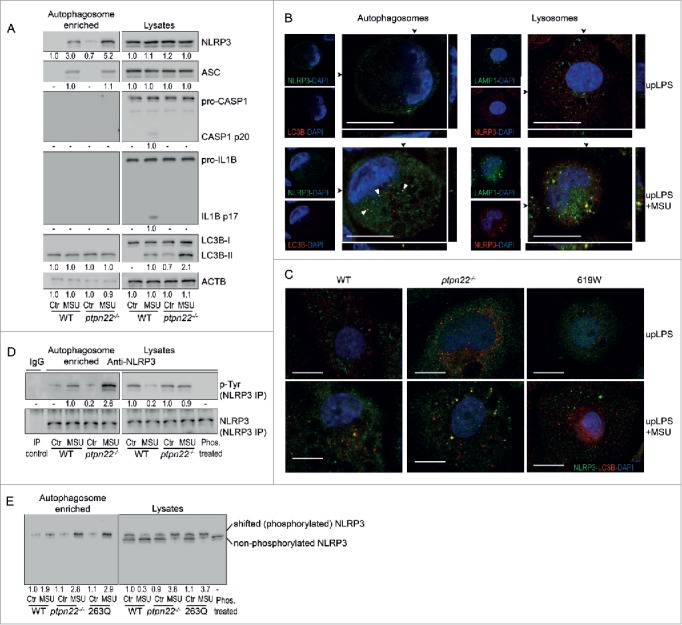
Phosphorylated NLRP3 is found in autophagosomes. (A) BMDC were primed for 16 h with upLPS to induce induction of NLRP3 and IL1B expression, before treatment with MSU for 6 h. The pictures show representative western blots from the indicated proteins in cell lysates and autophagosome-enriched fractions as indicated. (B) Confocal microscopy of THP-1 cells treated with LPS, or LPS and MSU, immunostained for NLRP3 (green) and LC3B (red; left side of the figure) or immunostained for NLRP3 (green) and the lysosome marker LAMP1 (red; right side of the figure); below and beside the image: z-stack at position of the arrows. scale bars: 10 μm. P-Tyr stands for phospho-tyrosine. (C) BMDC from WT mice, ptpn22−/− mice or mice expressing a gain-of-function variant in Ptpn22 (619W) were treated as in (A) and immunostained for NLRP3 (green) and LC3B (red). (D) BMDC from WT or ptpn22−/− mice were treated as in (A) and NLRP3 precipitated from either cell lysates or autophagosome-enriched fractions. The pictures show representative western blots for phospho-tyrosine (p-Tyr) and NLRP3 run in conventional SDS-PAGE. (E) control-transfected WT BMDC, control-transfected ptpn22−/− BMDC, or ptpn22−/− BMDC transfected with a vector expressing a loss-of-function variant in ptpn22 (263Q) were treated as in (A) and lysates run on Phos-tag gels, which allow the separation of phosphorylated from nonphosphorylated proteins due to slower migration of the phosphorylated forms. Data are representative for 1 out of 3 independent experiments with 3 replicates each (n = 3), except for confocal microscopy, where the experiment has been performed only twice with 3 replicates. Images are representative for at least 5 scanned areas for each depicted condition. Scale bars: 10 μm. P-Tyr stands for phospho-tyrosine. Numbers below the western blot pictures show results of densitometric measurements.
Localization of NLRP3 in autophagosomes was confirmed by confocal microscopy of cells costained for NLRP3 and LC3B. While in cells without inflammasome activation, NLRP3 was found all over the cytosol, activation of NLRP3 with MSU resulted in NLRP3 aggregation. Some of these NLRP3 aggregates colocalized with LC3B puncta, indicating that they were recruited to phagophores (Fig. 3B; left panel). NLRP3 was not only recruited into phagophores, but was also found in lysosomes upon NLRP3 activation, indicating that it is degraded in autolysosomes (Fig. 3B; right panel). In ptpn22−/− cells, or cells expressing the loss-of-function Ptpn22 variant, more LC3B puncta were observed in nontreated cells, and, upon activation with MSU, more NLRP3 aggregates colocalized with LC3B puncta. Conversely, cells expressing an altered function Ptpn22 variant, which results in a gain of function in terms of NLRP3 regulation,9 NLRP3 was not present in autophagosomes (Fig. 3C).
Only phosphorylated NLRP3 is detected in autophagosomes
We next precipitated NLRP3 from autophagosome-enriched fractions or whole cell lysates and probed for phospho-tyrosine (p-Tyr). Interestingly, in autophagosomes, p-Tyr was increased upon MSU-treatment, whereas in whole cell lysates it was decreased (Fig. 3D). In ptpn22−/− cells, we found no decrease in NLRP3 tyrosine phosphorylation upon inflammasome activation, and clearly more NLRP3 was present in autophagosomes (Fig. 3A). Since similar findings were observed in cells expressing a loss-of-function PTPN22 variant (Fig. S4C), this effect seems to be due to a loss of PTPN22 function, rather than absence of the protein per se. This finding strongly indicates that upon MDP- or MSU-treatment, phosphorylated NLRP3 is sequestered into phagophores. To address whether nonphosphorylated NLRP3 is also present in autophagosomes, we next ran whole cell lysate and autophagosome-enriched fractions on a Phos-tag gel (Fig. 3E). On these gels, phosphorylated proteins migrate at a slower speed than their nonphosphorylated forms.39 In the lysate of nontreated cells, NLRP3 appeared as a double band with approximately 50% appearing in the upper (phosphorylated) and 50% in the lower (nonphosphorylated) band. This is consistent with our previous findings that roughly half of NLRP3 is phosphorylated in nonactivated cells.9 In lysates, phosphorylation was decreased upon MDP treatment, where only a faint upper (phosphorylated) band was detectable. Of note, in ptpn22−/− cells or cells expressing a loss-of-function variant in Ptpn22 (263Q), MSU treatment did not decrease phosphorylation of NLRP3. Furthermore, in autophagosomes, the lower, nonphosphorylated band was not visible (Fig. 3E), indicating that only phosphorylated NLRP3 was sequestered into phagophores.
NLRP3 lacking the phosphorylation site is not recruited to phagophores
To further test the hypothesis that phosphorylation of NLRP3 is required for its recruitment into phagophores, we transfected BMDC from NLRP3-deficient mice with either wild-type NLRP3 or a mutated form of NLRP3 that lacks the phosphorylation site at tyrosine 859 (Y859F NLRP3). When analyzing autophagosome-enriched fractions for the presence of NLRP3, we only found NLRP3 in autophagosomes from BMDC transfected with WT NLRP3, but not in cells transfected with NLRP3Y859F, which lacks the phosphorylation site (Fig. 4A, Fig. S5A). In cells transfected with NLRP3Y859F, no colocalization of LC3B puncta with NLRP3 aggregates was observed (Fig. 4B). This further indicates that phosphorylated NLRP3—but not nonphosphorylated NLRP3—is sequestered to the phagophore upon inflammasome induction. Of interest, presence of the NLRP3Y859F construct resulted in increased CASP1 cleavage and enhanced IL1B secretion upon MSU and MDP treatment (Fig. 4C, Fig. S5). This observation indicates that phosphorylation of NLRP3 and subsequent degradation via autophagy is a regulatory mechanism of NLRP3 activation. In summary, these data indicate that NLRP3 phosphorylation results in sequestering of NLRP3 into phagophores and that this might be the mechanism for how phosphorylation of NLRP3 reduces its activation.
Figure 4.
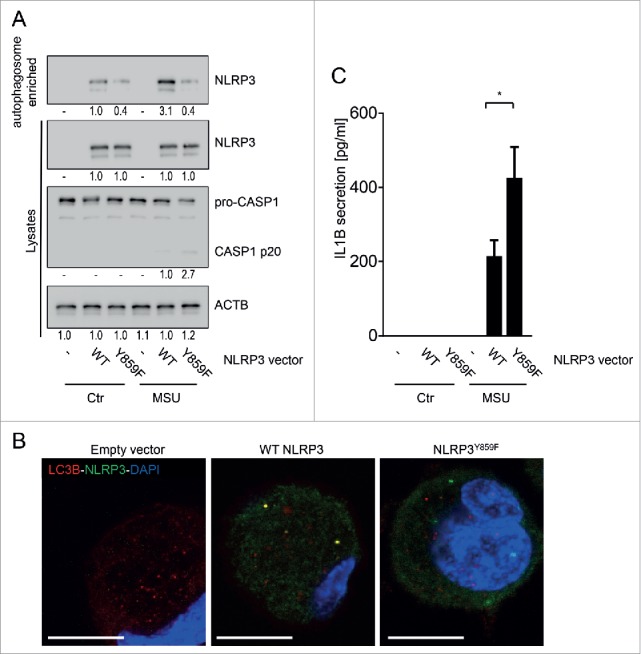
NLRP3 lacking the phosphorylation site is not recruited to phagophores. BMDC from nlrp3−/− mice were transfected with a wild-type (WT) Nlrp3 expression vector, or a Nlrp3 construct where Tyr859 in NLRP3 is replaced with a phenylalanine (Y859F; Y>F) to abolish NLRP3 phosphorylation. The cells were primed for 16 h with upLPS to induce NLRP3 and IL1B expression, before activation with MSU (150 µg/ml) for 6 h. Shown is (A) representative western blot pictures from cell lysates and autophagosome-enriched fractions, (B) confocal microscopy of LC3B (red) and NLRP3 (green) immunostained cells; blue: DNA stained with DAPI, scale bar: 10 μm; and (C) IL1B in the cell culture supernatant. Data are representative for 1 out of 3 independent experiments with 3 replicates each (n = 3), except for confocal microscopy where the experiment has been performed only twice with 3 replicates. Images are representative for at least 5 scanned areas for each depicted condition. Numbers below the western blot pictures show results of densitometric measurements. * = p < 0.05, one-way ANOVA with Bonferroni correction.
Phosphorylated NLRP3 interacts with SQSTM1 upon inflammasome activation
To understand how phosphorylated NLRP3 is recruited into phagophores we addressed whether NLRP3 interacts with molecules involved in targeting substrates to the phagophore. Indeed, we found that NLRP3 interacts with SQSTM1 upon inflammasome activation (Fig. 5A). Of note, this interaction was strictly dependent on phosphorylation of NLRP3, because in cells lacking the phosphorylation site no interaction between SQSTM1 and NLRP3 was observed. On the other hand, a phospho-mimetic variant of NLRP3 (NLRP3Y859E; reported in Ref. 9) resulted in increased interaction between SQSTM1 and NLRP3 (Fig. 5A). Further, loss of PTPN22 resulted in increased interaction of SQSTM1 and NLRP3 (Fig. 5B).
Figure 5.
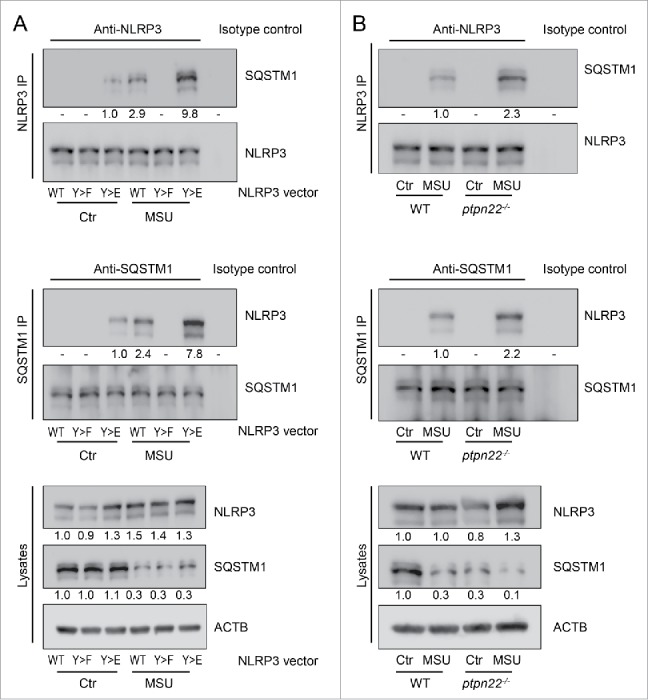
NLRP3 interacts with SQSTM1 upon its activation. (A) BMDC from nlrp3−/− mice were transfected with a wild-type (WT) Nlrp3 expression vector, a Nlrp3 construct where Tyr859 in NLRP3 is replaced with a phenylalanine (Y859F; Y>F) to abolish NLRP3 phosphorylation, or with a phsopho-mimetic NLRP3, where Tyr859 was replaced with a glutamate (Y859E; Y > E). The cells were primed for 16 h with upLPS to induce NLRP3 and IL1B expression, before activation with MSU (150 µg/ml) for 6 h. NLRP3 or SQSTM1 were precipitated from the lysate and probed for the indicated proteins. (B) BMDC from WT or ptpn22−/− cells were primed for 16 h with upLPS to induce NLRP3 and IL1B expression, before activation with MSU (150 µg/ml) for 6 h. NLRP3 or SQSTM1 were precipitated from the lysate and probed for the indicated proteins. Data is representative for 1 out of 3 independent experiments with 3 replicates each (n = 3). Numbers below the western blot pictures show results of densitometric measurements.
NLRP3 does not interact with SQSTM1 in PYCARD-deficient cells
So far we showed that phosphorylated NLRP3 interacts with SQSTM1; however, this is only the case when NLRP3 is activated. Because a portion of NLRP3 is phosphorylated in the steady-state, we wondered why phosphorylated NLRP3 is not recruited to phagophores in resting cells. Because NLRP3 activation results in its interaction with PYCARD, and we also found PYCARD in autophagosomes, we next addressed whether the interaction of NLRP3 with SQSTM1 is PYCARD dependent. When we treated PYCARD-deficient BMDC with MSU, we did not detect NLRP3 in autophagosomes (Fig. 6A, B). Immunoprecipitation of NLRP3 revealed that NLRP3 was phosphorylated in pycard−/− cells, but it did not interact with SQSTM1 (Fig. 6C). This result indicates that the interaction of NLRP3 with SQSTM1 was dependent on NLRP3-PYCARD interaction. In sum, our results indicate that phosphorylated NLRP3 binds SQSTM1 and is subsequently recruited into phagophores, once it interacts with PYCARD. Upon NLRP3 activation, PTPN22 dephosphorylates NLRP3 and thereby protects it from degradation, allowing robust inflammasome activity (summarized in Fig. S6).
Figure 6.
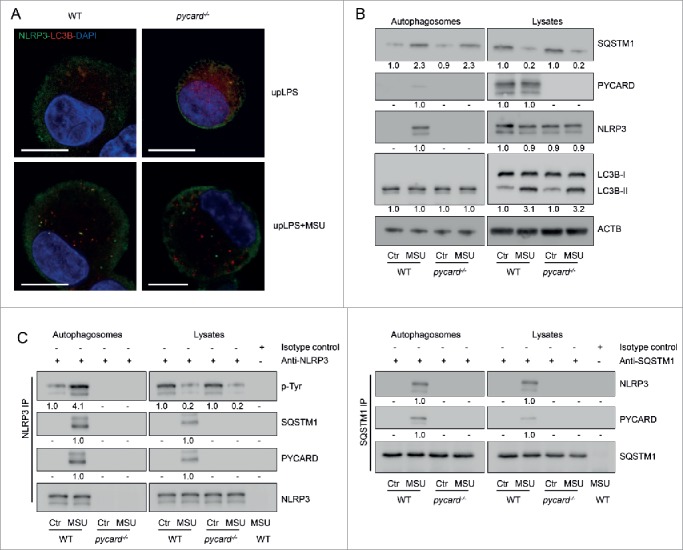
PYCARD is required for recruitment of NLRP3 into phagophores. BMDC from WT or pycard−/− cells were primed for 16 h with upLPS to induce NLRP3 and IL1B expression, before activation with MSU (150 μg/ml) for 6 h. (A) confocal microscopy of LC3B (red) and NLRP3 (green) immunostained cells; blue: DNA stained with DAPI, scale bar: 10 μm. (B) Western blots from autophagosome-enriched fractions and lysates were probed for the indicated proteins. (C) NLRP3 or SQSTM1 were precipitated from the lysate or from autophagosome-enriched fractions and probed for the indicated proteins. P-Tyr stands for phospho-tyrosine. Data are representative for 1 out of 3 independent experiments with 3 replicates each (n = 3), except for confocal microscopy where the experiment has been performed only once with 3 replicates. Images are representative for at least 5 scanned areas for each depicted condition. Numbers below the western blot pictures show results of densitometric measurements.
Discussion
We have previously demonstrated that loss of PTPN22 reduces IL1B secretion via enhanced phosphorylation of the inflammasome receptor NLRP3.9 Here, we demonstrate that loss of functional autophagy abrogates the reduction of NLRP3 activation observed in PTPN22-deficient cells. This effect is explained by our observation that phosphorylation of NLRP3 mediates recruitment of NLRP3 into phagophores, which seems to be responsible for reduced NLRP3 activity. Our data clearly show that in autophagy-deficient cells phosphorylated NLRP3 is still able to efficiently form inflammasome complexes, most likely due to the lack of sequestration into phagophores. In turn, dephosphorylation of NLRP3—as observed upon robust inflammasome induction—protects NLRP3 from being sequestered into phagophores, and allows full NLRP3 activation.
Autophagy and inflammasome activation both play a crucial role in the development of several inflammatory disorders, including neurodegenerative diseases, cancer, rheumatoid arthritis, multiple sclerosis, IBD, and many more (reviewed in refs. 40-42); hence, understanding the mechanisms for how these pathways influence each other is of great relevance for understanding underlying pathomechanisms, and ultimately for the development of novel therapeutic strategies. In the intestines, autophagy plays a crucial role for tissue homeostasis and defense against invading bacteria.27,30 This role is highlighted by the fact that genetic variants resulting in defects in the autophagy machinery enhance the risk to develop IBD.17,33,43 Although rather protective in many cells and diseases, in some disorders autophagy might also drive pathologies: Enhanced autophagy promotes survival of inflammatory synovial fibroblasts in rheumatoid arthritis,44 and protects cancer cells from cell death.45 For the NLRP3 inflammasome the situation is a little different: in classical inflammatory disorders, such as arthritis, diabetes, or SLE, an increase of NLRP3 activation clearly promotes disease onset/progression.46-49 In the intestine, however, the role of NLRP3 is still a matter of dispute, with some reports showing protective effects upon loss of NLRP3, whereas others demonstrate that NLRP3-induced inflammasome activation is important for promoting wound healing and tissue repair.50-52
Although there is evidence that autophagy is involved in mediating unconventional secretion of inflammasome substrates,53 several reports show that loss of functional autophagy results in enhanced inflammasome activation.35-37 One mechanism proposed to mediate this effect is the accumulation of aged mitochondria upon loss of autophagy,54 and subsequently enhanced levels of reactive oxygen species in the cytosol, which induces inflammasome activity.55
Autophagy also controls inflammasome activation directly: ubiquitinated inflammasomes37 as well as IL1B35 can be found in autophagosomes. We demonstrate here an additional mechanism—how autophagy affects NLRP3-mediated IL1B secretion, which is dependent on the recruitment of phosphorylated NLRP3 into phagophores. Thereby, phosphorylation of NLRP3 mediates its interaction with SQSTM1 and subsequent accumulation in autophagosomes upon inflammasome activation; hence autophagy removes NLRP3 from the cytosol and thereby directly reduces NLRP3-mediated inflammasome assembly. Conversely, PTPN22-mediated dephosphorylation protects NLRP3 from recruitment into phagophores and thereby promotes inflammasome activation. Consistent with this, we only detected phosphorylated NLRP3 in autophagosomes, but not nonphosphorylated NLRP3. These findings are well in line with our previous results, where we observed that an increase in NLRP3 phosphorylation in PTPN22-deficient cells resulted in a reduction of inflammasome activation.9
Of note, treatment with the NLRP3 activators MSU and MDP did not promote protein expression levels of PTPN22; hence it might be surprising that PTPN22 has such a strong impact on downstream signaling events induced by these 2 molcules. Nevertheless, we previously showed that signals, which induce robust NLRP3 activation, also promote the interaction between NLRP3 and PTPN22,9 and increase the phosphatase activity of PTPN22.23
Further, we found that the recruitment of NLRP3 into phagophores was accompanied by the interaction of NLRP3 with SQSTM1, a molecule that targets its binding partners to autophagic degradation. We therefore speculate that the recruitment of NLRP3 into phagophores is a result of NLRP3-SQSTM1 interaction, which is induced upon inflammasome activation. We observed an increase in autophagy in both MDP- and MSU-treated cells; hence, sequestration of NLRP3 might also be the result of enhanced autophagy, and not the result of inflammasome activation. Of interest, however, neither the interaction of NLRP3 with SQSTM1 nor recruitment of NLRP3 into phagophores was observed in cells lacking PYCARD. Since NLRP3 only interacts with PYCARD upon inflammasome induction, this strongly indicates that inflammasome activation is necessary for the interaction of NLRP3 with SQSTM1, and subsequently also for NLRP3 sequestration into phagophores.
Upon its activation, NLRP3 forms complexes that serve as a platform for the induction of PYCARD aggregates, which then mediate CASP1 activation.56 We speculate that phosphorylated NLRP3 is less efficient in promoting the formation of PYCARD specks, but promotes binding with SQSTM1 in a PYCARD-dependent manner. In a broader view, this would also be in line with autophagy as a mechanism to remove damaged/nonfunctional proteins from the cytosol: if phosphorylated NLRP3 is less efficient in promoting PYCARD specks, it might be regarded as a nonfunctional protein.
Taken together, our results show that NLRP3 inflammasome activation and autophagy are closely interweaved pathways, and that PTPN22 crucially interferes with the function of these 2 pathways. In conclusion, our results explain the mechanism for how phosphorylation of NLRP3 reduces its activation and dampens secretion of IL1B, and why this effect is autophagy dependent. Further, this explains how loss of PTPN22 and subsequent enhanced NLRP3 phosphorylation mediate a decrease in NLRP3 inflammasome activation. Together with previous work from other groups, our findings in this and previous studies suggest that PTPN22 might be an interesting therapeutic target for a broad number of inflammatory diseases. However, given the important role of autophagy, it is obvious that understanding the molecular mechanism affected by PTPN22 in more detail is of important relevance before the development of possible treatment options.
Methods
Cell culture
THP-1 cells (DSMZ, ACC-16) were cultured in RPMI (Sigma-Aldrich, R8758) supplemented with 10% fetal calf serum (FCS; PAA Laboratories, P40–47100). Bone-marrow derived dendritic cells (BMDCs) were obtained as described previously.23 In brief, bone marrow was collected from femur and tibia of 8–10-wk-old mice and cultured for 7 d in RPMI supplemented with 10% FCS, 1% sodium pyruvate solution, 1% glutamine (Sigma-Aldrich, G7513), and 25% of conditioned medium from mouse- CSF2/GM-CSF-producing X63 hybridoma cells, corresponding to 100 IU CSF2/ml. X63-mCSF2 cells were a kind gift from E. Contassot (Clinic for Dermatology of the University Hospital Zurich, Zurich, Switzerland). ptpn22 heterozygous mice were obtained from Genentech and bred with each other to obtain WT and ptpn22−/− littermates, pycard−/− mice were a gift from E. Contassot.
MDP, MSU, and ATP treatment
For experiments, THP-1 cells were seeded at a density of 1 × 106 cells/ml and differentiated into macrophages using 100 μg/ml PMA (Sigma-Aldrich, P1585) for 48 h. Medium was changed to serum-free medium 6 h before transfection with MDP (100 ng/ml; InvivoGen, tlrl-mdpc), or to serum-free medium containing ultrapure lipopolysaccharide (upLPS, 100 ng/ml; InvivoGen, tlrl-3pelps) 16 h before treatment with MSU (150 μg/ml; InvivoGen, tlrl-msu) or ATP (2 mM; InvivoGen tlrl-atp). For transfection, MDP (InvivoGen, tlrl-mdpc) was dissolved in DMSO and mixed with Lipofectamine® (InvivoGen, L3000–001) for 15 min before applying to the cell culture media. As a control, DSMO mixed with Lipofectamine® was used. MSU and ATP were applied onto the cells as suspensions. Likewise, BMDCs were collected at d 7 of BM cultures in differentiation medium, and seeded in new plates (0.5 × 106 cells/well in 12-well plates) in RPMI supplemented with 10% FCS. After 6 h, medium was changed for serum-free medium 6 h before transfection with MDP or treatment with MSU or ATP as described above.
Inhibitors, shRNA, and siRNA treatment
For experiments with inhibitors, cells were plated in serum-free medium 12 h before incubation with 3-MA (10 mM; Sigma-Aldrich, M9281), wortmannin (10 µM; Sigma-Aldrich, 95455), or bafilomycin A1 (100 nM; Sigma-Aldrich, B1793) for 1 h before treatment with MDP (100 ng/ml; InvivoGen, tlrl-mdpc), MSU (150 μg/ml; InvivoGen, tlrl-msu) or ATP (2 mM; InvivoGen, tlrl-atp). Knockdown of PTPN22 was induced in THP-1 cells using lentiviral particles containing PTPN22-specific shRNA as described previously.23 For knockdown of LC3B and ATG16L1 3 different silencer predesigned siRNA oligonucleotides targeting LC3B or ATG16L1 mRNA, respectively, were obtained from Life Technologies (AM16708A, IDs 44451, 44268, 44361 for ATG16L1 and IDs 30209, 30304, 130443 for LC3B). Per transfection, 100 pmol of each of the 3 gene-specific siRNA oligonucleotides were used. A nonspecific control siRNA SMARTpool (Life Technologies, AM16708A) at a concentration of 100 pmol per transfection was used as negative control. THP-1 cells were transfected using the Amaxa nucleofector® system (Lonza, AAB-1001) according to the manufacturer's instructions. After transfection, THP-1 cells were cultured in a 24-well plate for 48 h in RPMI supplemented with 10% FCS. For transfection of BMDC, siRNA was mixed with Lipofectamine® RNAiMAX (Life Technologies, 13778150) for 15 min before adding to the cell culture media. After 6 h, medium was changed and cells cultured for 24 h in RPMI supplemented with 10% FCS before activation as described above.
Reagents and antibodies
All reagents were commercially obtained and of analytical grade. The following antibodies were obtained from Cell Signaling Technology: anti-mouse/human LC3B (2775); anti-mouse/human ATG16L1 (8089); anti-phospho-tyrosine antibody (9411); anti-mouse/human SQSTM1 (5114). Anti-human CASP1 (sc514); and anti-human/mouse PTPN22 (sc48922) were obtained from Santa Cruz Biotechnology. Anti ACTB/actin β antibody was obtained from Merck/Millipore (MAB1501), anti-mouse NLRP3 antibody was from Adipogen (AG-20B-0014-C100), and anti-human NLRP3 antibody was from Enzo Life Sciences (ALX-804–819-C100).
NLRP3 vector transfections
pcDNA3.1 vectors expressing a FLAG-tagged WT mouse NLRP3 (NLRP3 WT) and mouse NLRP3 with Tyr859 substituted by a Phe (NLRP3Y859F) or by a glutamate (NLRP3Y859E) were obtained from Geneart (custom designed, described in Ref. 9). To overexpress WT and mutated NLRP3 vectors in BMDCs, the constructs were subcloned into a pLKO.1 vector, and together with pMD2.G and pMCV plasmids used to produce lentiviral particles in HEK293T cells as described previously.23
Western blot, immunoprecipitation, and Phos-tag gel electrophoresis
For western blotting, equal amounts of protein from each lysate were loaded on polyacrylamide gels and after separation by gel electrophoresis blotted onto nitrocellulose membranes. Membranes were incubated overnight with primary antibody, washed 3 times with washing buffer (Tris-buffered saline [0.2 M Tris base (Sigma-Aldrich, 10708976001) 1.5 M NaCl (Sigma-Aldrich, S3014)] 0.05% Tween (Sigma-Aldrich, P1379), 3% milk powder (Roth, T145.3) before incubation with horseradish peroxidase-coupled anti-mouse, anti-rabbit or anti-goat secondary antibodies (Santa Cruz Biotechnology, sc-2005, sc-2030, and sc-2020, respectively) for 2 h. Immunoreactive proteins were detected with a Fusion Solo S imager (Vilber Lourmat) using an enhanced chemiluminescence detection kit (Witec AG, K12045).
For immunoprecipitation, samples were pre-cleared with Sepharose G beads (GE Healthcare, 17–0618–01) and incubated on a rocker overnight at 4°C with 10 μg/ml anti-PTPN22, 10 μg/ml anti-mouse NLRP3, 15 μg/ml anti-human NLRP3 or 7 μg/ml anti-mouse/human SQSTM1 (Santa Cruz Biotechnology, sc-25575) antibodies, before precipitation with Sepharose G beads. After washing, the pellet was resuspended in 1x loading buffer, boiled for 10 min at 95°C, and the supernatants loaded on polyacrylamide gels. For detection of precipitated proteins, the same procedure was applied as used for western blotting.
To discriminate phosphorylated from nonphosphorylated proteins, Phos-tag gels were obtained from Wako Chemicals (304–93–521), and used according to the manufacturer's instructions and as described previously.39 In brief, cell lysates or autophagosome-enriched cell fractions were separated through 12% polyacrylamide gels containing 50 μM MnCl2 and 25 μM Phos-tag ligand. Gels were then blotted on nitrocellulose membranes and proteins detected using the same methods as for western blotting.
Autophagosome enrichment
Autophagosomes were enriched as described by Gao et al.38 In brief, cells were lysed in buffer B (0.25 M sucrose [Sigma-Aldrich, S9378], 1 mM EDTA, 20 mM HEPES [Siga-Aldrich, H0887], pH 7.4) by passing 15 times through a 22 G needle, and centrifuged for 10 min at 800 g to remove the nuclear fraction. The postnuclear fraction was incubated overnight with anti-LC3B antibody. Autophagosomes were then precipitated using Sepharose G beads, the pellets washed 3 times in ice-cold phosphate-buffered saline (Sigma-Aldrich, D8662) and subsequently lysed in M-PER lysis buffer (ThermoFisher Scientific, 78505) for further analysis.
RNA isolation and real-time PCR
Cells were suspended in 350 μl RLT buffer (Qiagen, 79216) and total RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, 74106) according to the manufacturer's instructions. RNA concentration was measured by absorbance at 260 and 280 nm. cDNA (cDNA) synthesis was performed using a High-Capacity cDNA Reverse Transcription Kit (Life Technologies, 4368813). Real-time polymerase chain reaction (PCR) was performed using FAST qPCR MasterMix for Taqman Assays (Life Technologies, 4352042) on a Fast HT7900 Real-Time PCR system using SDS Software (Life Technologies). Measurements were performed in triplicate, ACTB was used as an endogenous control, and results were analyzed using the ΔΔCT method. The real-time PCR contained an initial enzyme activation step (5 min, 95°C) followed by 45 cycles consisting of a denaturing (95°C, 15 sec) and an annealing/extending (60°C, 1 min) step. The used gene expression assays were all obtained from Life Technologies.
Confocal microscopy
For confocal microscopy, THP-1 cells were differentiated into macrophages on cover slides using 100 μg/ml PMA (Sigma-Aldrich, P1585) for 48 h, before activation with MSU for 6 h. Antibodies used for detection of LC3B and NLRP3 were the same as described above, goat anti-mouse Alexa Fluor 647 (ThermoFisher Scientific; A-21235) and goat anti-rabbit Alexa Fluor 564 (ThermoFisher Scientific; A-11035) were used as secondary antibodies. Immunofluorescence confocal microscopy was performed using a CSLM SP5 Leica microscope and image analyses performed using the LAS lite imaging software.
Statistical analyses
Data are presented as means ± Standard Deviation for a series of n experiments. Data are expressed as relative values of the respective control groups. Statistical analysis was performed by analysis of variance (ANOVA) followed by Student–Newman–Keuls post hoc test and bonferroni correction where multiple groups were compared with each other. P values < 0.05 were considered significant.
Supplementary Material
Abbreviations
- ATG16L1
autophagy-related 16 like 1
- BMDC
bone-marrow derived dendritic cells
- CD
Crohn disease
- DAMPS
damage-associated molecular patterns
- IBD
inflammatory bowel disease
- IRGM
immunity related GTPase M
- MAP1LC3B/LC3B
microtubule associated protein 1 light chain 3 β
- MDP
muramyl dipeptide
- MSU
monosodium urate
- NLRP3
NLR family pyrin domain containing 3
- PTPN22
protein tyrosine phosphatase, non-receptor type 22
- PYCARD
PYD and CARD domain containing
- RA
rheumatoid arthritis
- SLE
systemic lupus erythematosus
- SQSTM1
sequestosome 1
- 3-MA
3-methyladenene
Disclosure of potential conflicts of interest
The authors declare no financial conflict of interests. ACC is an employee of Genentech, South San Francisco, CA.
Funding
This research was supported by research grants from the Swiss National Science Foundation to MS (Grant No. 314730–146204, Grant No. 314730_166381 and Grant No. CRSII3_154488/1) and the Swiss IBD Cohort (Grant No. 3347CO-108792) to GR; a grant from the Holcim Foundation for Research to MRS, and a grant from the European Crohn and Colitis Foundation to MRS. The funding institutions had no role in study design and data interpretation.
References
- [1].Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229-65. https://doi.org/ 10.1146/annurev.immunol.021908.132715. PMID:19302040 [DOI] [PubMed] [Google Scholar]
- [2].Martinon F, Agostini L, Meylan E, Tschopp J. Identification of bacterial muramyl dipeptide as activator of the NALP3/cryopyrin inflammasome. Curr Biol. 2004;14:1929-34. https://doi.org/ 10.1016/j.cub.2004.10.027. PMID:15530394 [DOI] [PubMed] [Google Scholar]
- [3].Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228-32. https://doi.org/ 10.1038/nature04515. PMID:16407890 [DOI] [PubMed] [Google Scholar]
- [4].Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol. 2007;178:5271-6. https://doi.org/ 10.4049/jimmunol.178.8.5271. PMID:17404311 [DOI] [PubMed] [Google Scholar]
- [5].Ruiz PA, Moron B, Becker HM, Lang S, Atrott K, Spalinger MR, Scharl M, Wojtal KA, Fischbeck-Terhalle A, Frey-Wagner I, et al.. Titanium dioxide nanoparticles exacerbate DSS-induced colitis: role of the NLRP3 inflammasome. Gut. 2016;66(7):1216-24. https://doi.org/ 10.1136/gutjnl-2015-310297. PMID:26848183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142-53. https://doi.org/ 10.1016/j.immuni.2013.05.016. PMID:23809161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Martinon F. Signaling by ROS drives inflammasome activation. Eur J Immunol. 2010;40:616-9. https://doi.org/ 10.1002/eji.200940168. PMID:20201014 [DOI] [PubMed] [Google Scholar]
- [8].Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL, Gibson KF, Zou C, Chen BB, Mallampalli RK. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J Biol Chem. 2015;290:18124-33. https://doi.org/ 10.1074/jbc.M115.645549. PMID:26037928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, Scharl S, Gutte PM, Grütter MG, Beer HD, et al.. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest. 2016;126:1783-800. https://doi.org/ 10.1172/JCI83669. PMID:27043286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Begovich AB, Carlton VE, Honigberg LA, Schrodi SJ, Chokkalingam AP, Alexander HC, Ardlie KG, Huang Q, Smith AM, Spoerke JM, et al.. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet. 2004;75:330-7. https://doi.org/ 10.1086/422827. PMID:15208781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Michou L, Lasbleiz S, Rat A-C, Migliorini P, Balsa A, Westhovens R, Barrera P, Alves H, Pierlot C, Glikmans E, et al.. Linkage proof for PTPN22, a rheumatoid arthritis susceptibility gene and a human autoimmunity gene. Proc Natl Acad Sci U S A. 2007;104:1649-54. https://doi.org/ 10.1073/pnas.0610250104. PMID:17237219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Eliopoulos E, Zervou MI, Andreou A, Dimopoulou K, Cosmidis N, Voloudakis G, Mysirlaki H, Vazgiourakis V, Sidiropoulos P, Niewold TB, et al.. Association of the PTPN22 R620W polymorphism with increased risk for SLE in the genetically homogeneous population of Crete. Lupus. 2011;20:501-6. https://doi.org/ 10.1177/0961203310392423. PMID:21543514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Orru V, Tsai SJ, Rueda B, Fiorillo E, Stanford SM, Dasgupta J, Hartiala J, Zhao L, Ortego-Centeno N, D'Alfonso S, et al.. A loss-of-function variant of PTPN22 is associated with reduced risk of systemic lupus erythematosus. Hum Mol Genet. 2009;18:569-79. https://doi.org/ 10.1093/hmg/ddn363. PMID:18981062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ramirez M, Quintana G, Diaz-Gallo LM, Caminos J, Garces M, Cepeda L, Rondón F, Restrepo JF, Egea E, Garavito G, et al.. The PTPN22 C1858T variant as a risk factor for rheumatoid arthritis and systemic lupus erythematosus but not for systemic sclerosis in the Colombian population. Clin Exp Rheumatol. 2012;30:520-4. PMID:22704547 [PubMed] [Google Scholar]
- [15].Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, MacMurray J, Meloni GF, Lucarelli P, Pellecchia M, et al.. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet. 2004;36:337-8. https://doi.org/ 10.1038/ng1323. PMID:15004560 [DOI] [PubMed] [Google Scholar]
- [16].Diaz-Gallo LM, Espino-Paisan L, Fransen K, Gomez-Garcia M, van Sommeren S, Cardena C, Rodrigo L, Mendoza JL, Taxonera C, Nieto A, et al.. Differential association of two PTPN22 coding variants with Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2287-94. https://doi.org/ 10.1002/ibd.21630. PMID:21287672 [DOI] [PubMed] [Google Scholar]
- [17].WelcomeTrustConsortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661-78. https://doi.org/ 10.1038/nature05911. PMID:17554300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang Y, Ewart D, Crabtree JN, Yamamoto A, Baechler EC, Fazeli P, Peterson EJ. PTPN22 variant R620W is associated with reduced Toll-like receptor 7-induced Type I interferon in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67:2403-14. https://doi.org/ 10.1002/art.39211. PMID:26018863 [DOI] [PubMed] [Google Scholar]
- [19].Wang Y, Shaked I, Stanford SM, Zhou W, Curtsinger JM, Mikulski Z, Shaheen ZR, Cheng G, Sawatzke K, Campbell AM, et al.. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type 1 interferon-dependent immunity. Immunity. 2013;39:111-22. https://doi.org/ 10.1016/j.immuni.2013.06.013. PMID:23871208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Vang T, Liu WH, Delacroix L, Wu S, Vasile S, Dahl R, Yang L, Musumeci L, Francis D, Landskron J, et al.. LYP inhibits T-cell activation when dissociated from CSK. Nat Chem Biol. 2012;8:437-46. https://doi.org/ 10.1038/nchembio.916. PMID:22426112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yu X, Sun JP, He Y, Guo X, Liu S, Zhou B, Hudmon A, Zhang ZY. Structure, inhibitor, and regulatory mechanism of Lyp, a lymphoid-specific tyrosine phosphatase implicated in autoimmune diseases. Proc Natl Acad Sci U S A. 2007;104:19767-72. https://doi.org/ 10.1073/pnas.0706233104. PMID:18056643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hasegawa K, Martin F, Huang G, Tumas D, Diehl L, Chan AC. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science. 2004;303:685-9. https://doi.org/ 10.1126/science.1092138. PMID:14752163 [DOI] [PubMed] [Google Scholar]
- [23].Spalinger MR, Lang S, Vavricka SR, Fried M, Rogler G, Scharl M. Protein tyrosine phosphatase non-receptor type 22 modulates NOD2-induced cytokine release and autophagy. PLoS One. 2013;8:e72384. https://doi.org/ 10.1371/journal.pone.0072384. PMID:23991106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Spalinger MR, Lang S, Weber A, Frei P, Fried M, Rogler G, Scharl M. Loss of protein tyrosine phosphatase nonreceptor type 22 regulates interferon-gamma-induced signaling in human monocytes. Gastroenterology. 2013;144:978–88 e10. https://doi.org/ 10.1053/j.gastro.2013.01.048. PMID:23380085 [DOI] [PubMed] [Google Scholar]
- [25].Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741-52. https://doi.org/ 10.1038/nrm2239. PMID:17717517 [DOI] [PubMed] [Google Scholar]
- [26].Ohsumi Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211-6. https://doi.org/ 10.1038/35056522. PMID:11265251 [DOI] [PubMed] [Google Scholar]
- [27].Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al.. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11:55-62. https://doi.org/ 10.1038/ni.1823. PMID:19898471 [DOI] [PubMed] [Google Scholar]
- [28].Deretic V. Autophagy as an innate immunity paradigm: Expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012;24:21-31. https://doi.org/ 10.1016/j.coi.2011.10.006. PMID:22118953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol. 2007;7:767-77. https://doi.org/ 10.1038/nri2161. PMID:17767194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med. 2010;16:90-7. https://doi.org/ 10.1038/nm.2069. PMID:19966812 [DOI] [PubMed] [Google Scholar]
- [31].Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al.. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259-63. https://doi.org/ 10.1038/nature07416. PMID:18849966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Inoue J, Nishiumi S, Fujishima Y, Masuda A, Shiomi H, Yamamoto K, Nishida M, Azuma T, Yoshida M. Autophagy in the intestinal epithelium regulates Citrobacter rodentium infection. Arch Biochem Biophys. 2012;521:95-101. https://doi.org/ 10.1016/j.abb.2012.03.019. PMID:22475450 [DOI] [PubMed] [Google Scholar]
- [33].Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, et al.. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207-11. https://doi.org/ 10.1038/ng1954. PMID:17200669 [DOI] [PubMed] [Google Scholar]
- [34].Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, et al.. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn's disease susceptibility. Nat Genet. 2007;39:830-2. https://doi.org/ 10.1038/ng2061. PMID:17554261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, Lambe EM, Creagh EM, Golenbock DT, Tschopp J, et al.. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286:9587-97. https://doi.org/ 10.1074/jbc.M110.202911. PMID:21228274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al.. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264-8. https://doi.org/ 10.1038/nature07383. PMID:18849965 [DOI] [PubMed] [Google Scholar]
- [37].Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255-63. https://doi.org/ 10.1038/ni.2215. PMID:22286270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gao W, Kang JH, Liao Y, Ding WX, Gambotto AA, Watkins SC, Liu YJ, Stolz DB, Yin XM. Biochemical isolation and characterization of the tubulovesicular LC3-positive autophagosomal compartment. J Biol Chem. 2010;285:1371-83. https://doi.org/ 10.1074/jbc.M109.054197. PMID:19910472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, Mizuguchi J, Schweighoffer E, Tybulewicz V, Mitsuyama M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol. 2013;14:1247-55. https://doi.org/ 10.1038/ni.2749. PMID:24185614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Freeman LC, Ting JP. The pathogenic role of the inflammasome in neurodegenerative diseases. J Neurochem. 2016;136(Suppl 1):29-38. https://doi.org/ 10.1111/jnc.13217. PMID:26119245 [DOI] [PubMed] [Google Scholar]
- [41].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42. https://doi.org/ 10.1016/j.cell.2007.12.018. PMID:18191218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Netea-Maier RT, Plantinga TS, van de Veerdonk FL, Smit JW, Netea MG. Modulation of inflammation by autophagy: Consequences for human disease. Autophagy. 2016;12:245-60. https://doi.org/ 10.1080/15548627.2015.1071759. PMID:26222012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, et al.. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39:596-604. https://doi.org/ 10.1038/ng2032. PMID:17435756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dai Y, Hu S. Recent insights into the role of autophagy in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2016;55:403-10. https://doi.org/ 10.1093/rheumatology/kev337. PMID:26342228 [DOI] [PubMed] [Google Scholar]
- [45].Huang YH, Al-Aidaroos AQ, Yuen HF, Zhang SD, Shen HM, Rozycka E, McCrudden CM, Tergaonkar V, Gupta A, Lin YB, et al.. A role of autophagy in PTP4A3-driven cancer progression. Autophagy. 2014;10:1787-800. https://doi.org/ 10.4161/auto.29989. PMID:25136802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Biologics in Rheumatoid Arthritis Genetics and Genomics Study Syndicate (BRAGGSS), Eyre S, Churchman SM, Wilson AG, Isaacs JD, et al.. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis. 2014;73:1202-10. https://doi.org/ 10.1136/annrheumdis-2013-203276. PMID:23687262 [DOI] [PubMed] [Google Scholar]
- [47].Choi AM, Nakahira K. Dampening insulin signaling by an NLRP3 ‘meta-flammasome'. Nat Immunol. 2011;12:379-80. https://doi.org/ 10.1038/ni.2028. PMID:21502990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yang Q, Yu C, Yang Z, Wei Q, Mu K, Zhang Y, Zhao W, Wang X, Huai W, Han L. Deregulated NLRP3 and NLRP1 inflammasomes and their correlations with disease activity in systemic lupus erythematosus. J Rheumatol. 2014;41:444-52. https://doi.org/ 10.3899/jrheum.130310. PMID:24334646 [DOI] [PubMed] [Google Scholar]
- [49].Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278-86. https://doi.org/ 10.1038/nature10759. PMID:22258606 [DOI] [PubMed] [Google Scholar]
- [50].Zaki MH, Boyd KL, Vogel P, Kastan MB, Lamkanfi M, Kanneganti TD. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379-91. https://doi.org/ 10.1016/j.immuni.2010.03.003. PMID:20303296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zhang J, Fu S, Sun S, Li Z, Guo B. Inflammasome activation has an important role in the development of spontaneous colitis. Mucosal Immunol. 2014;7:1139-50. https://doi.org/ 10.1038/mi.2014.1. PMID:24472848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Chen GY, Nunez G. Inflammasomes in intestinal inflammation and cancer. Gastroenterology. 2011;141:1986-99. https://doi.org/ 10.1053/j.gastro.2011.10.002. PMID:22005480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011;30:4701-11. https://doi.org/ 10.1038/emboj.2011.398. PMID:22068051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci U S A. 2010;107:832-7. https://doi.org/ 10.1073/pnas.0913170107. PMID:20080761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221-5. https://doi.org/ 10.1038/nature09663. PMID:21124315 [DOI] [PubMed] [Google Scholar]
- [56].Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schröder GF, Fitzgerald KA, Wu H, Egelman EH. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193-206. https://doi.org/ 10.1016/j.cell.2014.02.008. PMID:24630722 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.