

Abstract
Complement receptor 1 (CR1), a transmembrane glycoprotein that plays a key role in the innate immune system, is expressed on many cell types, but especially on red blood cells (RBCs). As a receptor for the complement components C3b and C4b, CR1 regulates the activation of the complement cascade and promotes the phagocytosis of immune complexes and cellular debris, as well as the amyloid-beta (Aβ) peptide in Alzheimer's disease (AD). Several studies have confirmed AD-associated single nucleotide polymorphisms (SNPs), as well as a copy-number variation (CNV) in the CR1 gene. Here, we describe an innovative method for determining the length polymorphism of the CR1 receptor. The receptor includes three domains, called long homologous repeats (LHR)-LHR-A, LHR-C, and LHR-D-and an n domain, LHR-B, where n is an integer between 0 and 3. Using a single pair of specific primers, the genetic material is used to amplify a first fragment of the LHR-B domain (the variant amplicon B) and a second fragment of the LHR-C domain (the invariant amplicon). The variant amplicon B and the invariant amplicon display differences at five nucleotides outside of the hybridization areas of said primers. The numbers of variant amplicons B and of invariant amplicons is deduced using a quantitative tool (high-resolution melting (HRM) curves), and the ratio of the variant amplicon B to the invariant amplicon differs according to the CR1 length polymorphism. This method provides several advantages over the canonical phenotype method, as it does not require fresh material and is cheaper, faster, and therefore applicable to larger populations. Thus, the use of this method should be helpful to better understand the role of CR1 isoforms in the pathogenesis of diseases such as AD.
Keywords: Genetics, Issue 125, CR1, CD35, complement C3b/C4b receptor, CR1 length polymorphism, complement, Alzheimer's disease, molecular biology, neurosciences, systemic lupus erythematosus, genetic risk
Introduction
AD, the most common cause of dementia, affects more than 30 million people around the world and is a major public health problem1. Clinically, AD is characterized by neurocognitive disorders leading to a progressive loss of autonomy2. AD is characterized by two neuropathological hallmarks, namely, extra-cellular amyloid deposits and intracellular neurofibrillary tangles3.
Traditionally, according to the age of onset of the disease, AD is classified into two forms. First is early-onset AD (EOAD), where onset most often occurs before the age of 65; this form accounts for less than 5% of AD cases. It is a rare, autosomal-dominant form of AD, which results in fully penetrant mutations either in the amyloid precursor protein (APP)4, presenilin 1 (PSEN1)5, or presenilin 2 (PSEN2)6 genes. Second, the more common form of the disease (>90% of AD cases) is called "sporadic" late-onset AD (LOAD) and most often occurs in individuals aged 65 years or older. It results from multiple genetic and environmental risk factors7. In LOAD, the 4 allele of the apolipoprotein E (APOE) gene is the major genetic risk factor8,9. Furthermore, more than 20 gene loci have been identified by genome-wide association studies (GWAS) as being associated with the risk of AD, one of which being the complement component (3b/4b) receptor 1 (CR1) gene10, located on chromosome 1q32 in a cluster of complement-related proteins. The CR1 gene encodes the complement receptor type 1 (CR1) protein, a component of the complement activity regulators.
CR1 (the C3b/C4b receptor, CD35), a transmembrane glycoprotein of approximately 200 kDa11, binds to the C3b, C4b, C3bi, C1q, mannan-binding lectin (MBL), and ficolin complement proteins12. The biological function of CR1 varies with the cell types in which it is expressed. In humans, 90% of the total circulating CR1 is found in red blood cells (RBCs)13. Present at the surface of RBCs, CR1 binds to C3b- or C4b-opsonized microorganisms or immune complexes, facilitating their clearance from circulation. Complexes bound to CR1 are indeed transferred to phagocytes when RBCs go through the liver and spleen11,14. By limiting the deposition of C3b and C4b, CR1 might prevent excessive complement activation. Therefore, the expression of CR1 on RBCs is considered an essential element in the protection of tissues, such as the cerebral nervous system, against immune complex deposition and the resulting diseases. The CR1 on RBCs is also known to play an important role in pathogenic infection15,16. In addition, CR1, as a key player in innate immunity, is involved in the regulation of the complement cascade and in the transport and clearance of immune complexes. CR1 exerts this activity by binding C3b and C4b fragments and dissociating classical and alternative convertases (dissociation of C2a from the C4b2a complex and dissociation of C3b from the C3bBb complex). As a cofactor of the plasma serine protease factor I (FI), CR1 inhibits the classical and alternative complement pathways by increasing the cleavage of C4b and C3b by FI, a property known as cofactor activity (CA), and by inhibiting the C3 amplification loop, in turn preventing further complement activation. Rogers and colleagues provide evidence that the Aβ peptide can bind and activate the complement pathway in the absence of antibodies17 and suggest that the Aβ peptide is cleared from circulation via complement-dependent adherence to the CR1 expressed on RBCs18.
CR1 exhibits three types of polymorphisms: structural or length polymorphisms, density polymorphisms, and Knops blood-group polymorphisms11,19. The structural polymorphism is related to a variation in the number of long homologous repeats (LHRs) and thus defines four isoforms. In fact, the extracellular domain of the CR1 protein is composed of a series of repeating units, called short consensus repeats (SCRs) or complement control repeats (CCPs). These SCRs have been demonstrated from the complement deoxyribonucleic acid (cDNA) encoding CR1. The SCRs are arranged in tandem groups of seven, known as LHRs. CR1 is arranged into four LHRs, designated as LHR-A, -B, -C, and -D, arising from the duplication of a seven-SCR unit19,20,21.
In increasing order of frequency, these CR1 isoforms determined by Western blot (WB) are CR1*1 (A/F) (fast migration on gel electrophoresis), CR1*2 (B/S) (slow migration on gel electrophoresis), CR1*3 (C/F`), and CR1*4 (D). The two most common isoforms, CR1*1 (A/F) and CR1*2 (B/S), are composed of four and five LHRs, respectively, while CR1*3 (C/F`) and CR1*4 (D) are composed of 3 and 6 LHRs, respectively. The most common isoform (CR1*1), composed of 30 SCRs, contains three C4b binding sites (SCRs 1-3; 8-10, and 15-17) and two C3b binding sites (SCRs 8-10 and 15-17), while SCRs 22-28 bind C1q, ficolins, and MBL12,20,21,22,23,24,25. Thus, CR1*2 contains one additional C3b/C4b binding site compared to CR1*1. Figure 1 illustrates the structures, nomenclatures, and molecular weights of the four different isoforms of CR1.
The density polymorphism corresponds to a stable phenotype that represents the level of constitutive expression of CR1 on RBCs. In healthy Caucasian subjects, it has been shown that the number of CR1 molecules per RBC can vary by up to a factor of ten (varying from 150 to 1,200 molecules per cell)26. RBCs of the Helgeson phenotype have a very low CR1 density, which was shown to be lower than 150 molecules per cell27,28. The CR1 density on RBCs is genetically associated with an autosomal codominant biallelic system on the CR1 gene, correlated with a HindIII restriction fragment length polymorphism (RFLP)29. A single-point mutation in Intron 27 of the CR1 gene, between the exons encoding the second SCR in LHR-D, results in the generation of a polymorphic HindIII site within this region30. Genomic HindIII fragments of 7.4 and 6.9 kDa identify alleles associated with high (H allele) or low (L allele) CR1 density on RBCs, respectively. However, no correlation was found between CR1 density on RBCs and HindIII polymorphisms in some West African populations31,32. The mechanism linking CR1 density regulation to a non-coding HindIII polymorphism remains unknown. Among several polymorphisms, Q981H in SCR16 and P1786R in SCR28 have been reported to be linked to the CR1 density on RBCs30,33.
The Knops (KN) polymorphism, according to the international nomenclature, is the 22nd blood group system to be indexed by the International Society of Blood Transfusion. It contains 9 antigenic specificities expressed by the CR1 on RBCs, including three antithetic antigenic pairs, KN1/KN2, KN3/KN6, and KN4/KN7, as well as 3 isolated antigens, KN5, KN8, KN9. The KN1, KN3, KN4, and KN5 antigens are high-frequency antigens of the KN system (i.e., expressed in more than 99% of the general population). However, the role of this polymorphism in AD remains to be determined13.
The protocol described in this work was designed to determine the CR1 length polymorphism genotypes involved in susceptibility to several diseases, such as AD, systemic lupus erythematosus, and malaria. Our method for CR1 length polymorphism determination takes advantage of the number of LHR-Bs comprising the CR1 isoforms and of the sequence differences between LHR-B and LHR-C (Figure 2).
Protocol
The protocol for human blood collection and handling was reviewed and approved by the regional ethics committee (CPP Est II), and the protocol number is 2011-A00594-37.
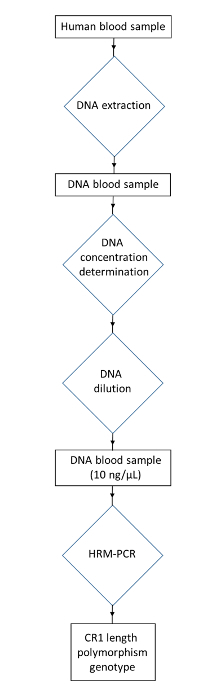
NOTE: The following protocol describes the handling of human blood. Please follow institutional guidelines while disposing of biohazardous material. Laboratory safety equipment, such as lab coats and gloves, should be worn. A flow chart describing the protocol is displayed in Figure 3.
1. Blood and Body Fluid DNA Extraction
Pipet 20 µL of proteinase K (15 µg/µL) into the bottom of a 1.5 mL tube.
Add 200 µL of sample to the 1.5-mL tube. Use up to 200 µL of whole blood, plasma, serum, buffy coat, or body fluids, or up to 5 x 106 lymphocytes in 200 µL of PBS.
Add 200 µL of lysis buffer to the sample. Mix by pulse-vortexing for 15 s.
Incubate at 56 °C for 10 min in a water bath.
Briefly centrifuge the 1.5 mL tube to remove the drops from the inside of the lid.
Add 200 µL of ethanol (96-100%) to the sample and mix again by pulse-vortexing for 15 s. After mixing, briefly centrifuge the 1.5 mL tube to remove the drops from the inside of the lid.
Carefully apply the mixture from step 1.6 to the membrane column (in a 2 mL collection tube). Without wetting the rim, close the cap and centrifuge at 6,000 x g for 1 min. Place the membrane column in a clean 2-mL collection tube and discard the tube containing the filtrate.
Carefully open the membrane column and add 500 µL of wash buffer 1 without wetting the rim. Close the cap and centrifuge at 6,000 x g for 1 min. Place the membrane column in a clean 2 mL collection tube and discard the tube containing the filtrate.
Carefully open the membrane column and add 500 µL of wash buffer 2 without wetting the rim. Close the cap and centrifuge at full speed (20,000 x g) for 3 min.
Place the membrane column in a new 2 mL collection tube and discard the collection tube with the filtrate. Centrifuge at 20,000 x g for 1 min.
Place the membrane column in a clean 1.5 mL tube and discard the collection tube containing the filtrate.
Carefully open the membrane column and add 200 µL of distilled water. Incubate at room temperature for 5 min and then centrifuge at 6,000 x g for 1 min.
Proceed to the determination of DNA concentration step or freeze the DNA samples at -20 °C
2. Determination of DNA Concentration
NOTE: See Figure 4.

Press 1 to select DNA mode on a spectrophotometer. Select a 5 mm path length using the left and right arrows. Select a dilution factor of 1 using the left and right arrows.
Select the unit (µg/mL) using the left and right arrows. Select a factor of 50 using the left and right arrows. Press OK.
Pipet 10 µL of distilled water into the cuvette. Put the cuvette in the spectrophotometer. Press the OA/100%T button.
Pipette 10 µL of the DNA sample (1/4 dilution in distilled water) into the cuvette. Put the cuvette in the spectrophotometer. Press the green button (read/start). Note the concentration.
Freeze the DNA sample at -20 °C.
3. HRM-PCR Protocol
Thaw the DNA samples. Dilute the DNA samples in 1.5 mL tubes with water to adjust them to a concentration of 10 ng/µL NOTE: The total volume of diluted DNA should be between 2 µL and 10 µL.
Thaw the primer solutions. Dilute the primer solutions in 1.5-mL tubes with water to adjust them to the same concentration of 6 µM. NOTE: The primer sequences and reaction conditions are provided in Table 1.
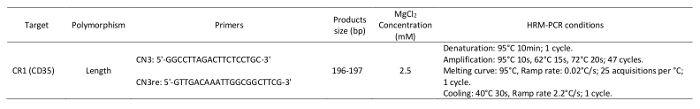 Table 1: Primers and parameters used in the high-resolution melting analysis.
Table 1: Primers and parameters used in the high-resolution melting analysis.
Thaw the HRM-PCR kit solutions and mix carefully by vortexing to ensure the recovery of all contents. Briefly spin the three vials containing the enzymatic mixture with DNA binding dye, MgCl2, and water in a microcentrifuge before opening them. Store them at room temperature.
- In a 1.5 mL tube at room temperature, prepare the PCR mix for one 20 µL reaction by adding the following components in the order listed below:
- 10 µL of enzymatic mixture with DNA binding dye;
- 2 µL of 25 mM MgCl2;
- 1 µL of primer 1, 6 µM (final concentration: 300 nM);
- 1 µL of primer 2, 6 µM (final concentration: 300 nM); and
- 5 µL of water. NOTE: To prepare the PCR mix for more than one reaction, multiply the volumes above by the number of reactions to be run, plus one additional reaction.
Mix carefully by vortexing.
Pipet 19 µL of PCR mix, prepared above, into each well of a white multiwell plate.
Add 1 µL of concentration-adjusted DNA template, prepared in step 3.1. NOTE: For control reactions, always run a negative control with the samples. To prepare a negative control, replace the template DNA with water.
Seal the white multiwell plate with sealing foil.
Place the white multiwell plate in the centrifuge and balance it with a suitable counterweight (i.e., another multiwell plate). Centrifuge for 1 min at 1,500 x g in a standard swing-bucket centrifuge containing a rotor for multiwell plates with suitable adaptors.
Load the white multiwell plate into the HRM-PCR instrument.
Start the HRM-PCR program with following PCR conditions: Denaturation: 95 °C for 10 min; 1 cycle. Amplification: 95 °C for 10 s, 62 °C for 15s, and 72 °C for 20s; 47 cycles. Melting curve: 95 °C; ramp rate: 0.02 °C/s; 25 acquisitions per °C; 1 cycle. Cooling: 40 °C for 30 s; ramp rate 2.2 °C/s; 1 cycle.
4. HRM Analysis to Determine the CR1 Length Polymorphism
NOTE: The methodology described (Figure 5) is specific to our software (See the Table of Materials), although other software packages may be used.
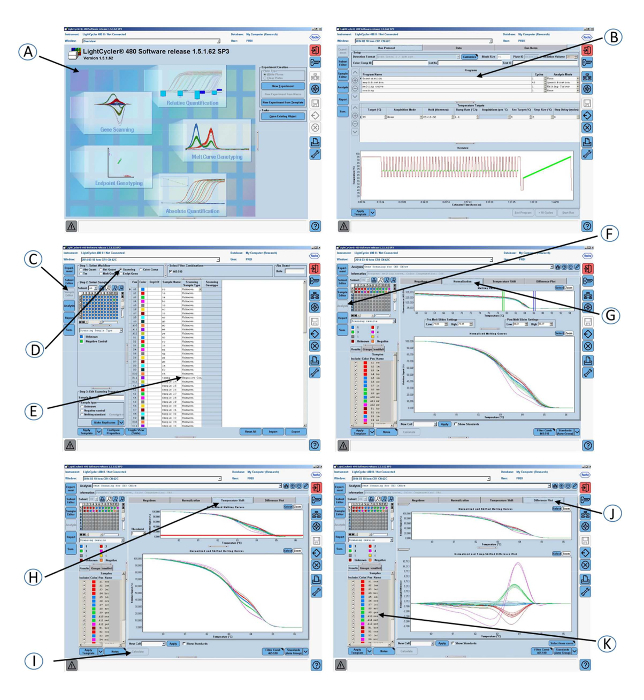
Open a gene scanning software to perform the CR1 length polymorphism scanning analysis.
Open the experiment containing the amplification program and the melting curve program.
Click Sample editor in the Module bar and then select the Scanning workflow.
Define the properties of the samples (i.e., name; unknown or negative control).
Click Analysis in the Module bar.
In the Create New Analysis list, select Gene Scanning.
Click the Normalization tab to normalize the melting curves.
Click the Temperature shift tab to reset the temperature axis (x-axis) of the melting curves. NOTE: The lower graph shows melting curves that are both normalized and temperature-shifted.
Click the Calculate button to analyze the results and determine the grouping.
Click the Difference plot tab in the charts area to view the Normalized and Shifted Melting Curves and the Normalized and Temperature Shifted Difference Plot.
Representative Results
Figure 6A displays the phenotyping of the CR1 length polymorphism by WB. Figure 6B and C displays the curves that are obtained during HRM-PCR analysis, enabling the determination of the CR1 length polymorphism. Analysis of the fusion curves using the software after the high-resolution melting of the PCR-derived amplicons from the genomic DNA of subjects with different CR1 isoforms produces curves that discriminate subjects according to their allotypic CR1 expression phenotypes.
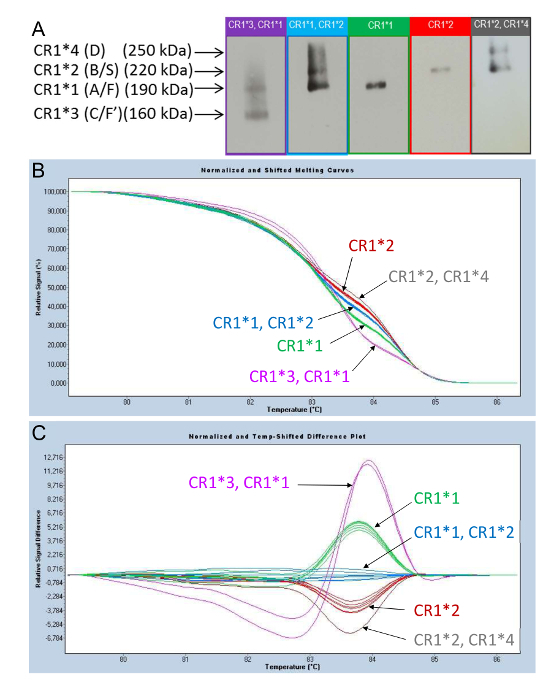
Figure 6B displays a first mode of presentation, called "Normalized and Shifted Melting Curves," while Figure 6C displays a second mode of presentation, called the "Normalized and Temp-Shifted Difference Plot."
The profiles of the curves are distributed according to the groups that represent the phenotypes displayed in Figure 6A: (CR1*3, CR1*1); (CR1*1, CR1*2); CR1*1; CR1*2; and (CR1*2, CR1*4). This enables the genotyping of the CR1 length polymorphism using this new molecular biology method.
Figure 1: Schematic representation of the structure and polymorphisms of the complement receptor 1 protein. Each short consensus repeat (SCR) or complement control protein (CCP) is represented by a circle. The SCRs are grouped into a repetitive, higher-order structures called LHRs. From the extracellular domain to the cell membrane, the LHRs are: LHR-A, -B, -C, -D, and -S (supplementary or additional LHR). In the most common CR1 isoform (CR1*1), the C4b/decay accelerating activity (DAA) binding site is located at LHR-A (SCRs 1-3, green circles), the C3b/C4b/cofactor activity (CA) binding site is located in the 3 N-terminal SCRs of LHR-B (SCRs 8-10, red circles) and LHR-C (SCRs 15-17, purple circles), and the binding site for C1q/MBL and ficolin is found in the LHR-D (SCRs 22-28, blue circles). The homologous structures are colored identically. The CR1*2 (S) isoform presents an additional LHR (LHR-S) and thus contains an additional C3b/C4b binding site. KDa, kilodalton. NRC, non-reduced conditions. RC, reduced conditions. TM, transmembrane domain. This figure was adapted from Brouwers et al.36 with permission from Nature Publishing Group. Please click here to view a larger version of this figure.
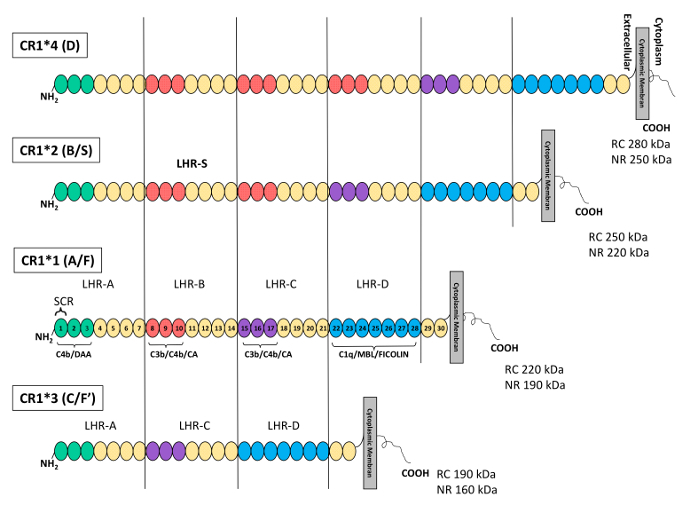
Figure 2: Schematic representation of the CR1 isoform structure with sequence amplicon positions obtained by HRM-PCR. Each box represents an SCR. The SCRs are grouped into LHRs: LHR-A, -B, -C and -D. The homologous structures are colored identically. The CR1*2 and CR1*4 isoforms present additional LHRs (LHR-B) and thus contain an additional amplicon area (amplicon B, in red). CR1*3 lacks LHR-B and contains less amplicon area (lacking amplicon B). Nucleotide differences between amplicon B (196 bp) and the invariant amplicon (197 bp) are depicted in red and blue, respectively. Differences in the ratio: (amplicon B, in red / invariant amplicon, in blue) between each CR1 isoform are determined by the HRM-PCR, enabling genotyping. The CN3 and CN3re primer sequences are underlined. TM, transmembrane domain. CYT, cytoplasmic tail. Please click here to view a larger version of this figure.
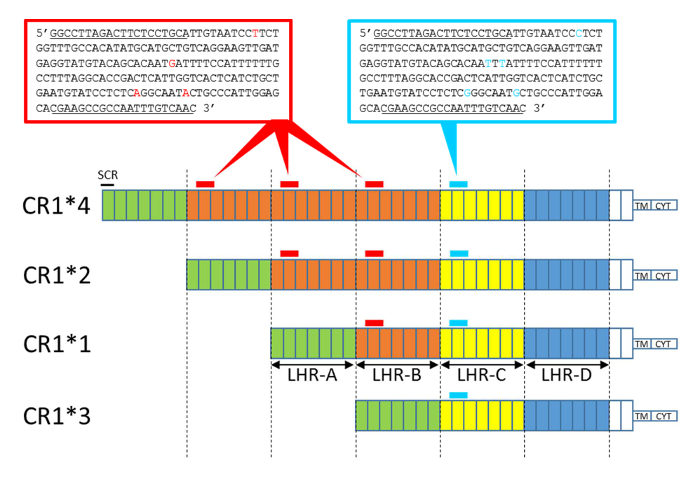
Figure 3: Flowchart of the protocol for genotyping the CR1 length polymorphism from human blood samples. Collect a human DNA sample. Extract the DNA to obtain a DNA blood sample. Determine the DNA concentration and dilute to obtain the DNA concentration at 10 ng/µL. Use HRM-PCR to obtain the CR1 length polymorphism genotype. Please click here to view a larger version of this figure.
Figure 4: Screenshots of the spectrophotometer used to determine the DNA concentration. Press 1 (A) to select DNA mode on the spectrophotometer. Select a 5 mm path length (B) using the left and right arrows (C). Select the unit (µg/mL) (D) using the left and right arrows (C). Select dilution factor 1 (E) using the left and right arrows (C). Select a factor of 50 (F) using the left and right arrows. Press OK (G). Pipet 10 µL of distilled water into the cuvette. Put the cuvette in the spectrophotometer (H). Press the OA/100%T button (I). Pipette 10 µL of DNA sample (1/4 dilution in distilled water) into the cuvette. Put the cuvette in the spectrophotometer (K). Press the green button (read/start) (L). Note the concentration (M). Please click here to view a larger version of this figure.
Figure 5: Screenshots of the graphical interface of the software used in step 4 of the protocol. (A) Open the gene scanning software. (B) Amplification program and melting curve program. (C) Click on Sample editor in the Module bar. (D) Select Scanning. (E) Define the properties of the samples. (F) Click on Analysis in the Module bar. (G) Click on the Normalization tab to normalize the melting curves. (H) Click on the Temperature shift tab to show the melting curves that are both normalized and temperature-shifted. (I) Click on the Calculate button to analyze the results and determine the grouping. (J) Click on the Difference plot tab to view the Normalized and Shifted Melting Curves and the Normalized and Temperature Shifted Difference Plot. (K) Colored grouping of the samples according to the CR1 length genotypes. Please click here to view a larger version of this figure.
Figure 6: Phenotyping of the CR1 length polymorphisms observed in the WB and their corresponding profile curves obtained by HRM-PCR. (A) Phenotyping of the CR1 length polymorphisms using WB. (B) and (C) Genotyping of CR1 length polymorphisms using HRM-PCR analysis. HRM curve analysis of PCR amplicons obtained from the genomic DNA of subjects displaying different CR1 isoforms led to the identification of specific curve profiles: (CR1*3, CR1*1) (purple); (CR1*1, CR1*2) (blue); CR1*1 (green); CR1*2 (red); and (CR1*2, CR1*4) (gray). These correspond to the CR1 length polymorphism alleles. As shown by the two modes of presentation-"Normalized and Shifted Melting Curves" (B) and "Normalized and Temp-Shifted Difference Plot" (C)-the curve profiles are distributed according to the (CR1*3, CR1*1); (CR1*1, CR1*2); CR1*1; CR1*2; and (CR1*2, CR1*4) groups. This figure was adapted from Mahmoudi et al.38 with permission from Elsevier. Please click here to view a larger version of this figure.
Discussion
Here, we describe a widely accessible methodology to study CR1 length polymorphisms. The molecular biology techniques for amplification or segmental hybridization have never been able to give satisfactory results that allow for the determination of CR1 length polymorphisms in all individuals. This is because of the repetitive structure of the CR1 gene (i.e., the highly repetitive SCRs of CR1). The molecular biology method described here exploits the quantitative distribution of LHR-B in the CR1 molecule. The CR1 molecule includes n LHR-B domains, where n is a number between 0 and 3, and only one LHR-C domain, as described and illustrated in Figure 2. Quantitative domain B detection makes it possible to perfectly discriminate one additional or missing domain B.
From the extracted genetic material, a first DNA fragment from LHR-B is amplified by PCR using a single pair of specific primers, representing a characteristic variant of LHR-B called "variant amplicon B." A second DNA fragment, called "invariant amplicon" and belonging to LHR-C, is also amplified. This second DNA fragment is a fragment of LHR-C, but another invariant fragment of LHR-A or -D could also be used.
The two DNA fragments, variant amplicon B and the invariant amplicon, are amplified by the same primers and exhibit five differences in nucleotide sequences. This characteristic makes it possible in the subsequent step to determine the number of variant amplicon B and the number of invariant amplicons using a quantitative molecular biology tool, HRM, which was adapted for this purpose. The ratio between the number of amplicon B and the invariant amplicon (ratio: amplicon B / invariant amplicon) varies according to the length of the CR1 molecule. Indeed, CR1*4 displays 3 amplicon B units to 1 invariant amplicon, CR1*2 displays 2 amplicon B units to 1 invariant amplicon, CR1*1 displays 1 amplicon B to 1 invariant amplicon, and CR1*3 displays 0 amplicon B units to 1 invariant amplicon (Figure 2). Thus, this HRM-PCR method enables the genotyping of the CR1 length polymorphism.
Nevertheless, at the start of the study, this technique requires the use of reference subjects whose CR1 phenotypes are already known (i.e., previously established by WB) in order to be able to establish the genotyping of CR1 according to the reference profile of curves obtained by HRM-PCR. Two types of representation, namely, "Normalized and Shifted Melting Curves" and "Normalized and Temp-Shifted Difference Plot," are available. They display distinct groups of curve profiles colored according to the CR1 length polymorphism phenotyping obtained by WB (Figure 6). From the "Normalized and Shifted Melting Curves" step, our method makes it possible to discriminate the distinct groups of curves corresponding to the isoforms of CR1, grouped by color. However, the "Normalized and Temp Shifted Difference Plot," which corresponds to a different mathematical representation, allows for the easier visualization of the distinct groups of curves. Although the manufacturer's software was routinely used in this study, other software (such as uANALYSE) could be used as a data extraction program for curve analysis.
To date, the WB analysis technique is the original technique that enables the identification of the different CR1 length polymorphisms at the level of the protein. However, this technique has a number of disadvantages. In particular, protein analysis by WB can only be done after the extraction of the cell membranes. As a result, it is complex and very time-consuming. Moreover, this technique requires obtaining a fresh blood sample in appropriate conditions at the beginning of the procedure to achieve useful results. On the contrary, HRM-PCR performed on DNA makes it possible to use even dry blood spots or samples after long-term storage. HMR-PCR requires a specialized DNA melt instrument, either a dedicated instrument or an HMR-capacity thermocycler with HRM software. SNP detection is dependent on several factors: i) SNPs included in large PCR fragments are more difficult to detect than the same SNPs in small PCR fragments; ii) SNPs belonging to classes III and IV are more difficult to detect than those of classes I and II34; and iii) SNPs that are flanked by nearest-neighbor base symmetry (e.g., 5'-G(G/C) C-3') cannot be sorted by melting, regardless of the resolution power of the HMR platform. It may be useful to test the predicted PCR fragments containing the SNP of interest with the uMELT software35 to assess the validity of the assay. Finally, HMR-PCR is an analogic method, meaning that the similarity of melting curves between a reference and a template does not necessarily imply that the template sequence is identical to the reference, as the template sequence may be different from the reference but thermodynamically equivalent. However, these limitations do not apply to the method described here, which is based on the melting of a mix of amplicons that differ in terms of 5 nucleotide sequences.
In addition, the technique described here, based on the analysis of HRM, has many advantages. First, the CR1 length polymorphisms are determined more rapidly, since the results are obtained in about 2 h once the extraction of the genetic material has been performed. Conversely, traditional biochemical techniques based on WB protein analysis require steps that are relatively long: the electrophoresis of cell membrane extracts through an acrylamide gel, a step to transfer proteins to a nitrocellulose or polyvinylidene fluoride (PVDF) membrane, and a step to identify the isoforms of the CR1 molecule by immunochemiluminescence. Second, the HRM-PCR technique also has the advantage of not being too expensive, which is particularly important for a method likely to be applied to many individuals (i.e., largescale) to determine their susceptibility to the development of pathologies such as Alzheimer's disease, lupus, or malaria.
Recently, we and others have shown that the CR1*2 isoform, which contains one additional C3b/C4b binding site, was associated with AD36,37,38. In our previously published study, the CR1 length polymorphisms at the level of the gene and the protein were consistent in 98.9% of subjects38. The discordant results obtained when comparing length polymorphisms obtained by HRM-PCR with those obtained by WB corresponded to subjects with a (CR1*1, CR1*2) genotype profile determined by HRM (gene) but expressing only the CR1*1 isoform at the surface of the erythrocyte according to WB (protein). In our study, the lack of CR1*2 isoform expression (individuals bearing a silent allele) was reproducible when the WB was performed with a longer exposure time and could be explained by the existence of a silent CR1 allele (CR1*2), described in the literature by Helgeson39. Nevertheless, further studies on larger populations are needed to support this hypothesis.
It should be noted that private SNPs (SNPs only occurring in a small family group or even in a single individual) led to distinct curve profiles that should be deciphered using reference phenotype determination (WB) but that cannot be confused with the canonical profile to avoid any misinterpretation of the CR1 length polymorphism genotype.
Disclosures
The authors are the inventors of a patent owned by the University of Reims Champagne-Ardenne (URCA) (patent number WO 2015166194)
Acknowledgments
We thank all the members of the Plateforme Régionale de Biologie Innovante, the staff of the Department of Immunology, and the staff of the Department of Internal Medicine and Geriatrics, who contributed to optimizing and validating the protocol. This work was funded by the Reims University Hospitals (grant number AOL11UF9156). We also thank Fiona Ecarnot (EA3920, University Hospital Besancon, France) for editorial assistance.
References
- Prince M, et al. World Alzheimer Report 2015, The Global Impact of Dementia: An analysis of prevalence, incidence, cost and trends. Alzheimer's Disease International. 2015.
- McKhann GM, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349(6311):704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375(6534):754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, et al. A familial Alzheimer's disease locus on chromosome 1. Science. 1995;269(5226):970–973. doi: 10.1126/science.7638621. [DOI] [PubMed] [Google Scholar]
- Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8) doi: 10.1101/cshperspect.a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu Rev Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- Lambert JC, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41(10):1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Liu D, Niu ZX. The structure, genetic polymorphisms, expression and biological functions of complement receptor type 1 (CR1/CD35) Immunopharmacol Immunotoxicol. 2009;31(4):524–535. doi: 10.3109/08923970902845768. [DOI] [PubMed] [Google Scholar]
- Jacquet M, et al. Deciphering complement receptor type 1 interactions with recognition proteins of the lectin complement pathway. J Immunol. 2013;190(7):3721–3731. doi: 10.4049/jimmunol.1202451. [DOI] [PubMed] [Google Scholar]
- Pham BN, et al. Analysis of complement receptor type 1 expression on red blood cells in negative phenotypes of the Knops blood group system, according to CR1 gene allotype polymorphisms. Transfusion. 2010;50(7):1435–1443. doi: 10.1111/j.1537-2995.2010.02599.x. [DOI] [PubMed] [Google Scholar]
- Cosio FG, Shen XP, Birmingham DJ, Van Aman M, Hebert LA. Evaluation of the mechanisms responsible for the reduction in erythrocyte complement receptors when immune complexes form in vivo in primates. J Immunol. 1990;145(12):4198–4206. [PubMed] [Google Scholar]
- Krych-Goldberg M, Moulds JM, Atkinson JP. Human complement receptor type 1 (CR1) binds to a major malarial adhesin. Trends Mol Med. 2002;8(11):531–537. doi: 10.1016/s1471-4914(02)02419-x. [DOI] [PubMed] [Google Scholar]
- Cohen JH, Geffriaud C, Caudwell V, Kazatchkine MD. Genetic analysis of CR1 (the C3b complement receptor, CD35) expression on erythrocytes of HIV-infected individuals. Aids. 1989;3(6):397–399. doi: 10.1097/00002030-198906000-00011. [DOI] [PubMed] [Google Scholar]
- Rogers J, et al. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1992;89(21):10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, et al. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006;27(12):1733–1739. doi: 10.1016/j.neurobiolaging.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Krych-Goldberg M, Atkinson JP. Structure-function relationships of complement receptor type 1. Immunol Rev. 2001;180:112–122. doi: 10.1034/j.1600-065x.2001.1800110.x. [DOI] [PubMed] [Google Scholar]
- Klickstein LB, et al. Human C3b/C4b receptor (CR1). Demonstration of long homologous repeating domains that are composed of the short consensus repeats characteristics of C3/C4 binding proteins. J Exp Med. 1987;165(4):1095–1112. doi: 10.1084/jem.165.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourcade D, Miesner DR, Atkinson JP, Holers VM. Identification of an alternative polyadenylation site in the human C3b/C4b receptor (complement receptor type 1) transcriptional unit and prediction of a secreted form of complement receptor type 1. J Exp Med. 1988;168(4):1255–1270. doi: 10.1084/jem.168.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krych M, Hourcade D, Atkinson JP. Sites within the complement C3b/C4b receptor important for the specificity of ligand binding. Proc Natl Acad Sci USA. 1991;88(10):4353–4357. doi: 10.1073/pnas.88.10.4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krych-Goldberg M, et al. Decay accelerating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J Biol Chem. 1999;274(44):31160–31168. doi: 10.1074/jbc.274.44.31160. [DOI] [PubMed] [Google Scholar]
- Klickstein LB, Barbashov SF, Liu T, Jack RM, Nicholson-Weller A. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity. 1997;7(3):345–355. doi: 10.1016/s1074-7613(00)80356-8. [DOI] [PubMed] [Google Scholar]
- Ghiran I, et al. Complement receptor 1/CD35 is a receptor for mannan-binding lectin. J Exp Med. 2000;192(12):1797–1808. doi: 10.1084/jem.192.12.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornillet P, Philbert F, Kazatchkine MD, Cohen JH. Genomic determination of the CR1 (CD35) density polymorphism on erythrocytes using polymerase chain reaction amplification and HindIII restriction enzyme digestion. J Immunol Methods. 1991;136(2):193–197. doi: 10.1016/0022-1759(91)90006-2. [DOI] [PubMed] [Google Scholar]
- Moulds JM, Nickells MW, Moulds JJ, Brown MC, Atkinson JP. The C3b/C4b receptor is recognized by the Knops, McCoy, Swain-langley, and York blood group antisera. J Exp Med. 1991;173(5):1159–1163. doi: 10.1084/jem.173.5.1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulds JM, Moulds JJ, Brown M, Atkinson JP. Antiglobulin testing for CR1-related (Knops/McCoy/Swain-Langley/York) blood group antigens: negative and weak reactions are caused by variable expression of CR1. Vox Sang. 1992;62(4):230–235. doi: 10.1111/j.1423-0410.1992.tb01204.x. [DOI] [PubMed] [Google Scholar]
- Wilson JG, et al. Identification of a restriction fragment length polymorphism by a CR1 cDNA that correlates with the number of CR1 on erythrocytes. J Exp Med. 1986;164(1):50–59. doi: 10.1084/jem.164.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong WW, et al. Structure of the human CR1 gene. Molecular basis of the structural and quantitative polymorphisms and identification of a new CR1-like allele. J Exp Med. 1989;169(3):847–863. doi: 10.1084/jem.169.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera AH, Xiang L, Martin SG, Lewis J, Wilson JG. Analysis of complement receptor type 1 (CR1) expression on erythrocytes and of CR1 allelic markers in Caucasian and African American populations. Clin Immunol Immunopathol. 1998;87(2):176–183. doi: 10.1006/clin.1998.4529. [DOI] [PubMed] [Google Scholar]
- Rowe JA, et al. Erythrocyte CR1 expression level does not correlate with a HindIII restriction fragment length polymorphism in Africans; implications for studies on malaria susceptibility. Genes Immun. 2002;3(8):497–500. doi: 10.1038/sj.gene.6363899. [DOI] [PubMed] [Google Scholar]
- Birmingham DJ, et al. A polymorphism in the type one complement receptor (CR1) involves an additional cysteine within the C3b/C4b binding domain that inhibits ligand binding. Mol Immunol. 2007;44(14):3510–3516. doi: 10.1016/j.molimm.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venter JC, et al. The sequence of the human genome. Science. 2001;291(5507):1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Dwight Z, Palais R, Wittwer CT. uMELT: prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics. 2011;27(7):1019–1020. doi: 10.1093/bioinformatics/btr065. [DOI] [PubMed] [Google Scholar]
- Brouwers N, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17(2):223–233. doi: 10.1038/mp.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazrati LN, et al. Genetic association of CR1 with Alzheimer's disease: a tentative disease mechanism. Neurobiol Aging. 2012;33(12):2945–2949. doi: 10.1016/j.neurobiolaging.2012.07.001. [DOI] [PubMed] [Google Scholar]
- Mahmoudi R, et al. Alzheimer's disease is associated with low density of the long CR1 isoform. Neurobiol. Aging. 2015;36:1712–1765. doi: 10.1016/j.neurobiolaging.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Helgeson M, Swanson J, Polesky HF. Knops-Helgeson (Kna), a high-frequency erythrocyte antigen. Transfusion. 1970;10(3):137–138. doi: 10.1111/j.1537-2995.1970.tb00720.x. [DOI] [PubMed] [Google Scholar]