

Abstract
Glioblastoma (GBM) continues to carry an extremely poor clinical prognosis despite surgical, chemotherapeutic, and radiation therapy. Progressive tumor invasion into surrounding brain parenchyma represents an enduring therapeutic challenge. To develop anti-migration therapies for GBM, model systems that provide a physiologically relevant background for controlled experimentation are essential. Here, we present a protocol for generating slice cultures from human GBM tissue obtained during surgical resection. These cultures allow for ex vivo experimentation without passaging through animal xenografts or single cell cultures. Further, we describe the use of time-lapse laser scanning confocal microscopy in conjunction with cell tracking to quantitatively study the migratory behavior of tumor cells and associated response to therapeutics. Slices are reproducibly generated within 90 min of surgical tissue acquisition. Retrovirally-mediated fluorescent cell labeling, confocal imaging, and tumor cell migration analyses are subsequently completed within two weeks of culture. We have successfully used these slice cultures to uncover genetic factors associated with increased migratory behavior in human GBM. Further, we have validated the model's ability to detect patient-specific variation in response to anti-migration therapies. Moving forward, human GBM slice cultures are an attractive platform for rapid ex vivo assessment of tumor sensitivity to therapeutic agents, in order to advance personalized neuro-oncologic therapy.
Keywords: Medicine, Issue 125, slice culture, organotypic, glioblastoma, migration, invasion, time-lapse, retroviral labeling, confocal microscopy, personalized medicine, translational models, pre-clinical models
Introduction
The laboratory study of glioblastoma (GBM), is hindered by a lack of models that faithfully recapitulate the requisite pathologic characteristics of the human disease, namely tumor cell migration and invasion. Comparative studies of 2D and 3D in vitro invasion assays as well as 3D rodent slice culture models have uncovered mechanistically disparate cellular migration programs in these two contexts, potentially limiting the translatability of findings from 2D systems to the human disease1,2,3. The organotypic tumor slice culture and imaging paradigm described here allows for the study of tumor cell migration within slices of ex vivo human tumor tissue obtained from surgical resection. Thus, slice cultures of surgically resected tumor tissue in conjunction with time-lapse confocal microscopy provide a platform to study tumor cell migration in the native microenvironment without tissue dissolution or culture passaging.
There is extensive literature employing rodent brain slice culture models of GBM generated from human tumor xenografts, retroviral-induced tumors, and cellular overlays to study tumor invasion1,2,3,4,5. Recently, several groups have described the generation of organotypic slice cultures directly from human GBM tissue6,7,8,9,10. However, there is marked variation among published protocols with regards to slicing technique and culture media. Further, the use of organotypic slice cultures has focused on static experimental endpoints that have included changes in cell signaling, proliferation, and death. The protocol described herein expands upon prior slice culture paradigms by incorporating time-resolved observation of dynamic tumor cell behaviors through time-lapse laser scanning confocal microscopy. Recent discovery of inter11 and intratumoral12,13 genetic variation in human GBM underlines the importance of linking this heterogeneity with tumor cell behaviors and its implications on tumor response to therapy. Here, we report a streamlined and reproducible protocol for use of direct slice cultures from a human cancer tissue to visualize tumor cell migration in near real-time.
Protocol
Before collection of patient tissue samples is initiated, informed consent must be obtained from each patient under an approved Institutional Review Board (IRB) protocol. The authors of this protocol received consent for the work described under approved IRB protocols at the University of Colorado Hospital and Inova Fairfax Hospital. Data collected from these slice cultures were not used to direct patient care decisions.
1. Pre-slicing Preparation
Prepare "tissue processing" media and "slice culture maintenance" media the day before planned tumor resection and tissue collection (or utilize previously generated media within 2 weeks). Add 5 mL of Penicillin-Streptomycin solution (10,000 U/mL) and 5 mL of 1 M HEPES to 500 mL of a High-Glucose DMEM to generate the tissue processing medium.
Prepare 250 mL of slice culture maintenance media using a base of neuronal medium (e.g., Neurobasal) without phenol red. Supplement this medium with 10 mM HEPES, 1x B-27 supplement, 400 µM L-glutamine, 600 µM L-alanyl-L-glutamine dipeptide, 60 U/mL penicillin, 60 µg/mL streptomycin, and 6 U/mL nystatin.
Store all media at 4 °C for no longer than 2 weeks.
2. Day of Surgery: Tissue Acquisition
On the day of tissue acquisition, prepare 50 - 100 mL of 1% and 2% (wt/vol %) solutions of low melting temperature agarose in tissue processing media using autoclaved glassware and sterile technique in a laminar flow hood. Both concentrations of agarose are needed to accommodate unpredictable variation in tumor tissue consistency.
Heat the agarose suspension, using a microwave, until gentle boiling is observed. Place the agarose solution in a 37 °C water bath to maintain in a liquid state until use.
Place 1 mL of slice culture maintenance media in each well of a 6-well plate.
Use a sterile forceps to place empty PTFE culture inserts in each well. Place the plate in a humidified, water-jacketed, tissue culture incubator with a 5% CO2 atmosphere maintained at 37 °C.
Using a sterile Pasteur pipette in a laminar flow hood, bubble a mixture of 95% O2/5% CO2 gas into a flask containing ice cold tissue processing media for approximately 15 to 30 min, prior to tumor tissue acquisition. The use of supra-oxygenated media minimizes hypoxia in the bulk tissue during processing.
Prepare labeled 50 mL conical tubes (enough for desired number of individual tumor regions to be isolated) containing 20 mL aliquots of supra-oxygenated ice-cold processing media to transport tissue between the operating room and slicing facility.
In conjunction with a neurosurgeon, pre-operatively plan the tumor region for sample acquisition. Select a tumor region with contrast enhancement as demonstrated on the patient's clinical imaging (T1 post-gadolinium magnetic resonance imaging (MRI) sequences). Previous experience suggests this area yields viable tumor tissue as opposed to necrotic tissue. NOTE: Generation of slices from the surrounding (peritumoral) white matter was attempted; however, background autofluorescence and decreased tumor cell density limited the experimental utility of these slices.
Obtain tumor tissue towards the beginning of tumor resection. Avoid collection of tumor tissue exposed to extensive bipolar cautery, which may compromise tissue viability secondary to thermal injury. Tissue samples acquired during piecemeal tumor resection demonstrate enhanced viability when compared to tissue acquired from lengthy en bloc resections. This observation may relate to differential tumor tissue deprivation of blood flow as a result of surgical resection and extended time to acquisition.
If desired, label and store tumor tissue in separate conical tubes to isolate samples from distinct tumor regions. (i.e. superficial vs. deep tumor tissue). Precise tissue locations can be recorded if/when intraoperative surgical navigation is available.
3. Slice Culture Preparation
NOTE: This protocol requires the use of fresh unfixed human tissue. All samples are presumed to be infectious, and should be handled according to universal blood borne pathogen protocols. Appropriate personal protective equipment should be donned at all times. Forceps and scalpels should be exposed to 15 min of UV light prior to use. During use, intermittently spray the tools with 70% ethanol (EtOH), allowing time for the liquid to evaporate before use. The slicing process is performed in a semi-sterile fashion utilizing a horizontal laminar flow hood with filtered air.
Place tumor tissue pieces in a Petri dish with ice-cold processing media. To wash the tumor tissue and minimize adherent red blood cells, use a pipette to gently exchange and discard the media in the Petri dish three times.
Using a scalpel, cut tumor pieces into rectangular box shapes. Trim tissue pieces to approximate 3 mm x 3 mm x 10 mm. Carefully remove any attached vessels through excision or gentle traction with fine forceps.
Pipette 5 - 7 mL of 37 °C agarose into a small cube-shaped (~ 2 cm3) plastic embedding mold. Confirm temperature of agarose solution is no greater than 37 °C with a sterile thermometer before use.
Allow agarose to sit for approximately 1 min on a bed of ice.
Place 2 - 4 tumor tissue "strips" into agarose with long axis oriented vertically. NOTE: Tissue strips will tend to "sink" in the agarose before solidification. To avoid this complication, temporarily hold the tissue strip in vertical orientation until agarose is further solidified.
Keep the tissue containing agarose mold on a bed of ice for 2 - 5 min to facilitate solidification.
Remove mold from ice and gently remove the agarose block by cutting the sides of the form with a scalpel. Avoid placing excessive force on the block, which may fracture the agarose.
Use a generous drop of cyanoacrylate glue to affix the agarose block to the vibratome specimen plate. Let glue set for approximately 1 - 2 min.
Fill the vibratome reservoir with ice-cold processing media to submerge the agarose block affixed to the specimen plate.
During slicing, bubble 95% O2 / 5% CO2 gas mixture into the vibratome reservoir through a trimmed sterile plastic pipette.
Set the vibratome slice thickness to 300 - 350 µm.
Adjust blade advance speed and blade amplitude according to tissue consistency. "Stiffer" GBM tissue requires slower blade advance speeds and higher amplitude. Exact blade speed and amplitude settings will vary according to vibratome specifications. NOTE: Several issues commonly arise during slicing. If the tissue strip dislodges from the agarose block, attempt embedding the tissue in a higher concentration agarose. If tumor slices are thicker than desired (machine set), or are asymmetric, increase the amplitude and decrease speed of vibratome blade. To ensure tumor tissue is not stuck to or "dragged" by the blade holder, as slicing occurs, carefully position the microspatula between the blade holder and slice, as the blade moves.
Transfer tissue slices to Petri dish with ice-cold processing media using a stainless steel microspatula.
Obtain 6-well plates containing PTFE inserts and equilibrated slice culture media from incubator (as prepared in section 2, step 4).
Plate tissue slices using a stainless steel microspatula. Minimize direct contact and manipulation of tissue by using a small fine bristle paintbrush to generate a "fluid wave" to gently push the slice off the spatula and onto the culture insert. NOTE: Minimize the amount of processing media introduced on top of the tissue culture insert. If excessive amounts of media are transferred causing the slices to float, use a sterile Pasteur pipette to remove media.
Return the 6-well plates containing tumor slices to an incubator maintained at 37 °C with a 5% CO2 atmosphere. NOTE: The entire embedding and slicing protocol should be completed within 90 min of tumor tissue acquisition from the operating room.
4. Slice Culture Maintenance
After 12 - 24 h, transfer the slice cultures by grasping each insert rim with a sterile forceps, and transfer to plates containing fresh slice culture maintenance media. Ensure the slice culture maintenance media aliquoted into each well equilibrates in the incubator for at least 15 min before transferring inserts.
Move inserts to new 6-well plates with fresh equilibrated slice culture media every 48 h.
If desired, aliquot old slice culture media with a pipette for immediate use in biochemical assays (i.e. ELISA) or freeze at -80 °C for future use.
5. Tumor Cell Labeling Via Green Fluorescence Protein Expressing Retrovirus
NOTE: Time-lapse microscopy for analysis of tumor cell migration requires stable, long-term fluorescent labeling of cells within the slice culture. Use of retrovirus is suggested because it selectively infects dividing cells, thereby enriching fluorescent labeling within the tumor cell population as opposed to microglia or other cell types present within the slice. Standardization of infection suggests that a viral titer of 104 CFUs/µL results in sufficient green fluorescent protein expression for the tracking and analysis of cell migration. Increased viral titer, use of non-selective virus (i.e. adenovirus, lentivirus), or other means of labeling all cells may preclude identification of clear cell boundaries during migration, thus complicating analysis. Use of alternative fluorescent markers can be utilized and optimized as needed.
Obtain retrovirus for infection of tumor slices either via standard protocols5,14 or from a commercially available source. Dilute the viral supernatant by adding the appropriate volume into unsupplemented neuronal medium to attain a viral titer of 104 CFUs/µL.
Between 7 - 10 days of culture infect the tumor slice cultures of interest with 5 - 10 µL of virus (104 CFUs/µL). Place the virus gently dropwise onto the surface of each tissue slice. Decrease supernatant volume added if the slice floats on the surface of the insert. Return plates containing slice cultures to incubator.
Assess the slices for labeled tumor cells beginning at 24 h after viral infection (see section 6). This time delay will vary dependent on viral incorporation and fluorescence gene expression kinetics. For the viral constructs used here, 72 h was required to observe robust fluorescent signal with a standard epifluorescence microscope. NOTE: Rarely, slices display a predominantly peripheral distribution of virally labeled cells, which can complicate confocal imaging. To avoid this complication, attempt to reduce slice thickness and thickness variation across the slice. If the slice culture has a thicker region near the center, this may prevent adequate nutrient penetration, thus limiting the population of actively dividing tumor cells. A qualitative assay to screen for this complication is the addition of a tetrazolium dye reagent (i.e. MTT) to the slice culture media. Areas of the slice that do not turn blue after adding the reagent indicate a lack of metabolic activity and compromised slice health.
6. Time-Lapse Single Photon Laser Scanning Confocal Imaging of Tumor Cell Migration
NOTE: After successful transduction and health of the culture is confirmed, cells may be imaged under control conditions, followed by an equal period of imaging under treatment conditions. Using this protocol, cells were successfully imaged and tracked for 12 hours in each condition. However, shorter or longer periods of imaging and environmental manipulation may also be informative.
- Loading the Microscope
- Before imaging, place 1 mL of fresh slice media into a glass bottom dish.
- Allow media in the glass bottom dish to equilibrate in an incubator for 15 min. NOTE: At this point soluble labeling agents, such as fluorescently conjugated lectins (i.e. Isolectin IB4 for microglia labeling) or ligand conjugated quantum dots, can be added to the slice culture media for identification of cellular subpopulations during imaging.
- Transfer the insert to be imaged to a glass bottom dish using sterile forceps in a laminar flow hood. Transport the dish to the microscope stage.
- Maintain slice cultures at 37 °C and 5% CO2 atmosphere in a sealed microscope stage-top incubator. Utilize sterile H2O humidification of the incubation chamber if available to prevent excessive media evaporation (especially important for longer imaging experiments).
- Utilize a confocal microscope with a long working distance 10X air objective and laser excitation for single-photon and/or multi-photon. NOTE: Ensure that the microscope objective lens provides adequate working distance. As a result of the added height from the stage-top incubator, glass bottom dish, and tissue culture insert, long working distance objectives are critical.
- Secure the glass bottom plate, remove the plastic Petri dish cover, and cover with a gas-permeable membrane.
- Image Acquisition
- Use the microscope to visually inspect the slice, and locate a suitable field with adequate density of fluorescently labeled tumor cells between the slice edge and the center. Avoid imaging fields at the edge of the slice, due to increased susceptibility of tissue shifts during imaging. Tissue slice harps may be employed to limit this shift.
- Use imaging software in multidimensional analysis mode to set the first (bottom) and last (top) Z-stack boundaries, such that all positions to be imaged contain visible fluorescent cellular signal. Imaging through 150 - 200 µm of the slice, with a constant Z-step of 10 µm provided adequate resolution for tracking cell paths (this can be adjusted for individual imaging needs).
- If the microscope has a motorized stage, ensure adequate time is allowed for each Z-stack acquisition. Set the time interval between acquisitions to equal scan time per position x number of positions to image (i.e. imaged tumor regions). NOTE: For simultaneous excitation of the green and red fluorophores, utilize a dual-line laser line-scanning program, with simultaneous 488 nm and 633 nm excitation. The wavelength of laser excitation selected will vary according to individual fluorescent proteins expressed.
- Maintain uniform laser power and confocal pinhole settings between the imaged tumor regions. Utilize the lowest laser power setting needed to clearly demarcate the tumor cell bodies and processes to limit phototoxicity. Specific imaging parameter units (i.e. power and confocal pinhole settings) will vary according to microscope and laser source specifications.
- Compensate for potential media evaporation by adding 2 - 3 "buffer" Z-stack steps in the focal planes advancing towards the objective. This effectively prevents the tissue from leaving the range of Z-stack acquisitions in the vertical plane.
7. Image Post-Processing and Tumor Cell Tracking
NOTE: Many confocal imaging systems are equipped with proprietary image-processing software. The processing steps discussed below comprise a general protocol, which can be performed across software platforms. Specific instructions will be given for the open-source platforms, NIH ImageJ and MTrackJ15.
Open a Z-stack file. In ImageJ, click "Image→Stacks→Z Project." Chose the first and last Z-stack to include, and chose "Max Intensity" as the projection type. The result is a rendering called a maximum intensity projection (MIP). Create an MIP from each set of Z-stack images captured at each region imaged.
Concatenate the MIPs from each region to create a time series. Click "Image→Stacks→Images to Stack."
Manually identify the location of the cell body "centroid" by selecting the visually approximated center point of the tumor cell body. This is accomplished by clicking on the cell body using the "add" track functionality of MTrackJ (an open-source plugin-in for NIH ImageJ).
Click to demarcate the cell body location in each frame of the series of images. This creates a unique "track" for each cell. Sequentially, mark the cell body locations of the next cell. Repeat this process until all cell migration paths are tracked.
Once a population of cells is tracked in a given tumor micro region, export all cell track coordinates using the "measure" function of MTrackJ. Save the file as an .xls format so the raw data can be analyzed using spreadsheet or user-generated software.
Use the coordinates recorded for each point along a cell's migration track, to perform quantitative analyses including derivation of migration speed, directionality, and other migration metrics, as described in Parker et al16. NOTE: All calculated distances and speeds are under-estimates of actual values. This is inherent in the transformation of three-dimensional images (and migration paths) to two-dimensional data through generation of maximum intensity projections.
Representative Results
Our group has successfully generated slice cultures from over 50 patients undergoing initial GBM resection. This slice generation, culture, retroviral-labeling, imaging, and migration analysis protocol has been streamlined into a reproducible workflow (Figure 1). Critically, these organotypic GBM slices demonstrate concordance with originating tumor tissue throughout culture, including maintenance of pathologic hallmarks and microglia up to 15 days in culture (Figure 2). In addition, we have utilized this system to perform functional assays of tumor response to microenvironmental changes. As a metric of physiologic integrity, we examined how GBM slice cultures responded to hypoxia (1% O2) by measuring the production of vascular endothelial growth factor (VEGF), a process that occurs abundantly within the GBM microenvironment in vivo17,18. We demonstrated that by placing the slice cultures in hypoxic conditions, the slices mounted a rapid physiologic response, inducing VEGF release into the media (Figure 3).
To evaluate qualitative and quantitative aspects of tumor cell migration we utilized the time-lapse images to generate detailed migration maps. These maps demarcate all GBM cells tracked within the confines of tumor microregions (1 mm2), providing a static visualization of the dynamic migratory behavior of the tumor population. Quantitative measures of migration speed and directionality (cell displacement / total distance traveled) were calculated for each cell, allowing for investigation of changes in migration parameters across tumor regions, tumor samples, and in response to treatment (Figure 4).
The cell labeling and imaging protocol described here also provides sufficient spatial and temporal resolution to evaluate changes in cell morphology during migration through the native tumor microenvironment. We observed the presence of morphologically distinct motile tumor cells and microglial intermingled within the slice culture (Figure 5A). Tumor cell movement was characterized by a "search and burst" process, which involved repeated protrusion and retraction of filopodia from a static cell, followed by a short period of efficient movement. Imaging slice regions approximately every 10 min also provided adequate temporal resolution to record time-lapse images of tumor cells undergoing cell division. These dividing cells paused from migration, completed mitosis, and the daughter cells re-initiated migration without delay, all within a 3-h timeframe (Figure 5D). In contrast, microglia migrate at a higher and more consistent speed, with lower directionality than adjacent tumor cells, demonstrating their relatively inefficient migration (Figure 5C, 5E-G). Such observations may be important for gaining insight into the biology underlying patient- or cell-specific responses to treatment.
Finally, we used this protocol to demonstrated patient-to-patient variability in cell migration parameters at the population level, including a correlation of epidermal growth factor receptor (EGFR) genomic amplification with augmented migratory potential of tumor cells16. In addition, time-lapse microscopy of tumor slices before and after treatment with the anti-invasive drug, gefitinib, demonstrated a significant reduction in migration, which was specific to EGFR amplified tumor slices16.
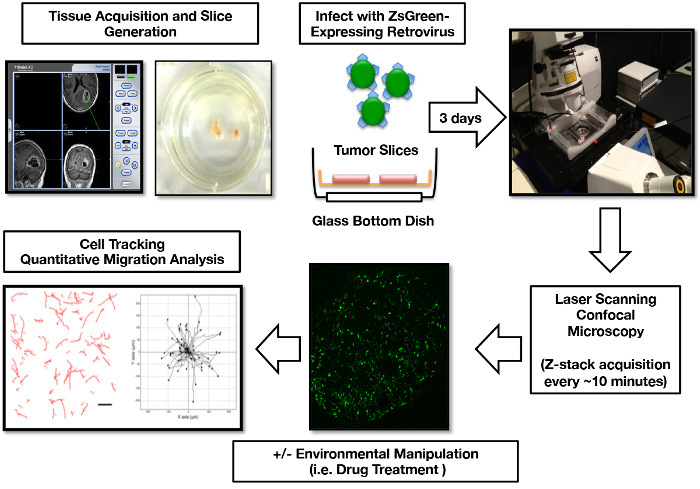 Figure 1:Human GBM Organotypic Slice Culture Infection, Imaging, and Cell Migration Analysis Workflow. Tumor tissue is localized to a specific region via intraoperative navigation equipment. One week post slicing, the ZsGreen expressing retrovirus is added to the slice cultures to label the mitotically active tumor cells. 3 days post-infection, slices are prepared for confocal imaging. The 3D imaging data is post-processed into 2D images for cell migration path tracking, generation of tumor cell migration maps, and calculation of tumor cell migration parameters. Portions of this figure were originally published in Parker et al, 201316 and reproduced with permission of Oxford University Press. Please click here to view a larger version of this figure.
Figure 1:Human GBM Organotypic Slice Culture Infection, Imaging, and Cell Migration Analysis Workflow. Tumor tissue is localized to a specific region via intraoperative navigation equipment. One week post slicing, the ZsGreen expressing retrovirus is added to the slice cultures to label the mitotically active tumor cells. 3 days post-infection, slices are prepared for confocal imaging. The 3D imaging data is post-processed into 2D images for cell migration path tracking, generation of tumor cell migration maps, and calculation of tumor cell migration parameters. Portions of this figure were originally published in Parker et al, 201316 and reproduced with permission of Oxford University Press. Please click here to view a larger version of this figure.
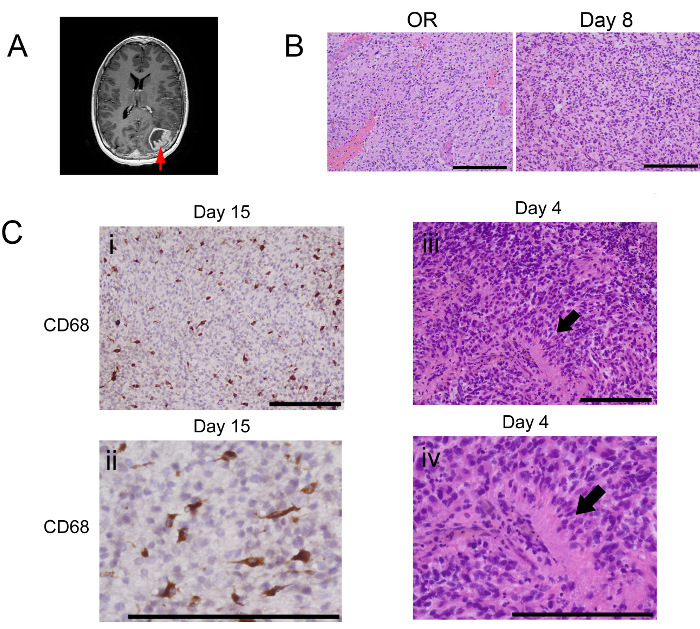 Figure 2. Human GBM Organotypic Slices Retain Histological Features Throughout Ex Vivo Culture. (A) T1 contrast enhanced MRI sequences were used to localize and document the region(s) of tissue acquisition (arrow). (B) H&E staining of initial donor tissue (OR) and slices at day 8 of culture from a slice culture generated from tissue obtained from the region highlighted in A. (C) Microenvironmental pathologic and cellular features of GBM, in vivo, are maintained throughout slice culture. (I, II) Immunohistochemistry for CD68, a microglia/macrophage marker, at low (top) and high (bottom) magnification, demonstrates microglial persistence in slices after 15 days of culture. H&E staining on day 4 of slice culture confirms maintenance of pseudopallisading necrosis, a pathologic hallmark of GBM, at both low (iii) and high (iv) magnification. Scale bars = 200 µm. Please click here to view a larger version of this figure.
Figure 2. Human GBM Organotypic Slices Retain Histological Features Throughout Ex Vivo Culture. (A) T1 contrast enhanced MRI sequences were used to localize and document the region(s) of tissue acquisition (arrow). (B) H&E staining of initial donor tissue (OR) and slices at day 8 of culture from a slice culture generated from tissue obtained from the region highlighted in A. (C) Microenvironmental pathologic and cellular features of GBM, in vivo, are maintained throughout slice culture. (I, II) Immunohistochemistry for CD68, a microglia/macrophage marker, at low (top) and high (bottom) magnification, demonstrates microglial persistence in slices after 15 days of culture. H&E staining on day 4 of slice culture confirms maintenance of pseudopallisading necrosis, a pathologic hallmark of GBM, at both low (iii) and high (iv) magnification. Scale bars = 200 µm. Please click here to view a larger version of this figure.
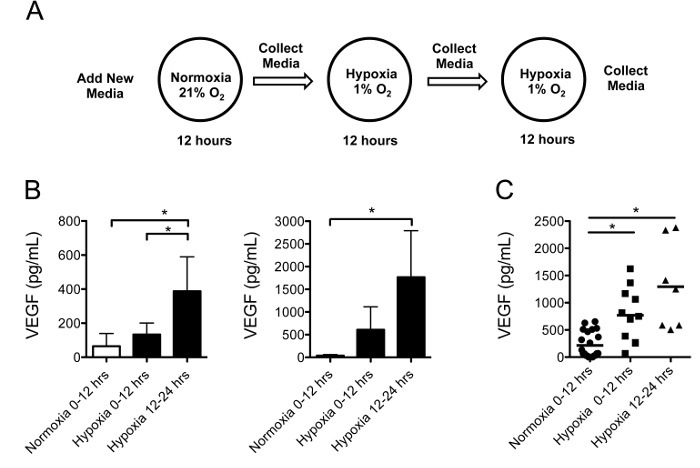 Figure 3:Human GBM Slice Cultures Secrete VEGF in Response to Hypoxia. (A) The experiment utilized individual culture inserts containing 3 similar sized tumor slices generated from the same tumor region. The slices were maintained in normoxia for 12 h, followed by two 12 h intervals of hypoxia, with new media added before each interval. (B) VEGF secretion into the media measured by ELISA (mean ± standard deviation) from slice cultures generated from two representative tumors, was significantly increased under hypoxia than normoxia (p <0.05). (C) A pooled analysis of slice cultures from 4 different tumors demonstrated increased VEGF secretion after sequential 12 h intervals of hypoxia compared to normoxia (p <0.05). Please click here to view a larger version of this figure.
Figure 3:Human GBM Slice Cultures Secrete VEGF in Response to Hypoxia. (A) The experiment utilized individual culture inserts containing 3 similar sized tumor slices generated from the same tumor region. The slices were maintained in normoxia for 12 h, followed by two 12 h intervals of hypoxia, with new media added before each interval. (B) VEGF secretion into the media measured by ELISA (mean ± standard deviation) from slice cultures generated from two representative tumors, was significantly increased under hypoxia than normoxia (p <0.05). (C) A pooled analysis of slice cultures from 4 different tumors demonstrated increased VEGF secretion after sequential 12 h intervals of hypoxia compared to normoxia (p <0.05). Please click here to view a larger version of this figure.
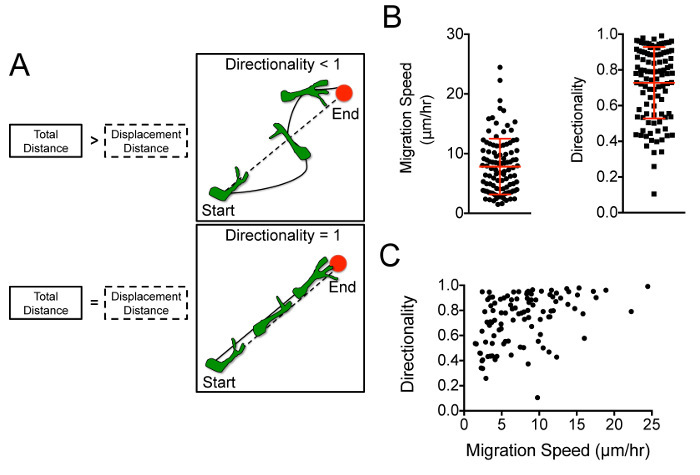 Figure 4:Tumor Cell Path Track Data Allows Quantitative Determination of Cell Speed and Directionality. (A) Tumor cells with a directionality of 1 represent perfect efficiency along a straight vector, whereas those with lower directionality engage in inefficient meandering paths, as represented in this schematic16. Reprinted with permission from Oxford University Press. (B) Analysis of representative path track data ("low" resolution ~55 min cell tracking intervals) demonstrates cellular variability in speed and directionality. (C) Directionality versus migration speed for each cell track is plotted to visualize migration behavior across the cell population (each dot represents an individual cell). Please click here to view a larger version of this figure.
Figure 4:Tumor Cell Path Track Data Allows Quantitative Determination of Cell Speed and Directionality. (A) Tumor cells with a directionality of 1 represent perfect efficiency along a straight vector, whereas those with lower directionality engage in inefficient meandering paths, as represented in this schematic16. Reprinted with permission from Oxford University Press. (B) Analysis of representative path track data ("low" resolution ~55 min cell tracking intervals) demonstrates cellular variability in speed and directionality. (C) Directionality versus migration speed for each cell track is plotted to visualize migration behavior across the cell population (each dot represents an individual cell). Please click here to view a larger version of this figure.
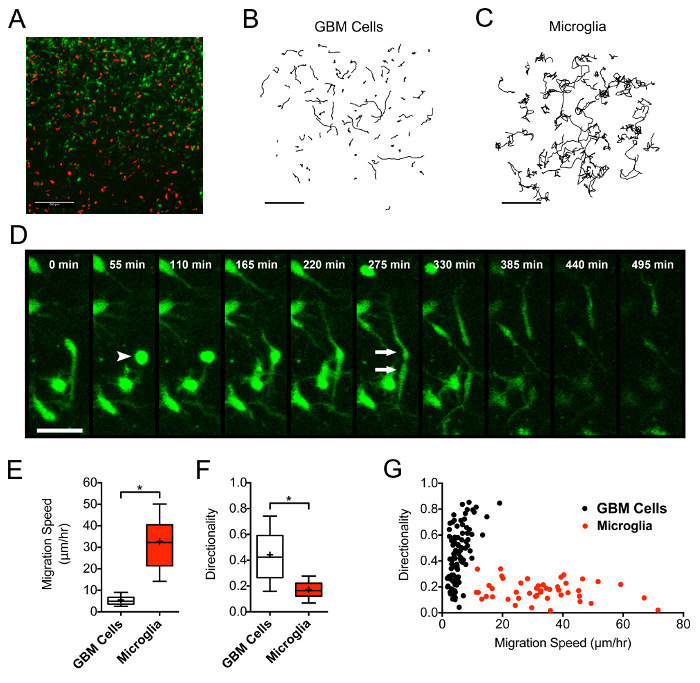 Figure 5:Microglia within GBM Slice Cultures are Characterized by High Migration Speed and Low Directionality Relative to Tumor Cells. (A) Replicating tumor cells selectively expressing ZsGreen retrovirus and microglia tagged with an Isolectin-IB4-647 conjugate exist intermingled in a representative tumor micro-region from a representative slice culture. (B-C) The paths of individually tracked GBM (B) and microglia (C) in the same tumor micro-region demonstrate discordant migration behaviors. Scale bars = 200 µm. (D) An actively migrating tumor cell pauses, retracts its processes (arrowhead), undergoes cell division, and two daughter cells migrate away in opposing directions (arrows). Scale bars = 50 µm. (E and F) Microglia demonstrate increased migration speed (p <0.0001) and decreased directionality (p <0.0001) when compared to tumor cells in the same region. (G) The distribution of tumor and microglial cells based on speed and directionality demonstrates the unique migratory phenotypes of the two cell populations. Please click here to view a larger version of this figure.
Figure 5:Microglia within GBM Slice Cultures are Characterized by High Migration Speed and Low Directionality Relative to Tumor Cells. (A) Replicating tumor cells selectively expressing ZsGreen retrovirus and microglia tagged with an Isolectin-IB4-647 conjugate exist intermingled in a representative tumor micro-region from a representative slice culture. (B-C) The paths of individually tracked GBM (B) and microglia (C) in the same tumor micro-region demonstrate discordant migration behaviors. Scale bars = 200 µm. (D) An actively migrating tumor cell pauses, retracts its processes (arrowhead), undergoes cell division, and two daughter cells migrate away in opposing directions (arrows). Scale bars = 50 µm. (E and F) Microglia demonstrate increased migration speed (p <0.0001) and decreased directionality (p <0.0001) when compared to tumor cells in the same region. (G) The distribution of tumor and microglial cells based on speed and directionality demonstrates the unique migratory phenotypes of the two cell populations. Please click here to view a larger version of this figure.
Discussion
Organotypic slice cultures from human cancer tissue provide an attractive and underutilized platform for pre-clinical translational experimentation. Understanding of population-level behaviors of tumor cells with regards to migration, proliferation, and cell death in the native tumor microenvironment is lacking. Critically, studying tumor response to therapy in a dynamic, time-resolved fashion at the level of cell behavior may shed light on novel mechanisms of treatment resistance. Human tumor slice cultures provide a link between the human disease process and current ex vivo and in vivo modeling techniques19. We recently validated the technique described here as a method to study GBM migration, reporting for the first time measurable inter-tumoral variations in cell migration behavior related to EGFR amplification and signaling16. This study also utilized the slice culture model to test the patient-specific effectiveness of the EGFR inhibitor, gefitinib, as a potential anti-invasive therapy for GBM 16.
Several of the common pitfalls during tissue slicing, retroviral infection, imaging, and image analysis paradigm have been discussed above. However, the retroviral infection protocol warrants further attention. Given patient-to-patient variation, it may prove challenging to titrate the density of virally labeled tumor cells within each slice. If insufficient numbers of cells are labeled by the viral construct, add additional 5-10 µL aliquots of retroviral supernatant to the surface of each slice daily until desired concentration of labeled tumor cells is achieved. In primary slice cultures, the percent of replicating cells is generally lower than in transformed cell lines, thus limiting the subset of cells permissive to retroviral incorporation at any time point. Alternatively, if too many cells are labeled, preventing accurate demarcation of cell migration paths, dilute the viral supernatant with neuronal media to achieve a lower effective titer. Tumor associated microglia incorporated the fluorescent protein expressing retrovirus at a frequency of approximately of 1% of all labeled cells. We were able to visually isolate these cells for separate analysis by the use of a microglia binding lectin for post-imaging data analyses (Figure 5).
The tumor microenvironment including aspects of nutrient delivery, cell-cell interactions, and extracellular matrix all play a role in the pathogenesis of GBM20. Direct human GBM slice cultures eliminate the need for passaging within small animal models or disseminated cell culture, while providing a close recapitulation of the human tumor microenvironment. Further, slice cultures provide uniform access to nutrients across samples, while maintaining cell-cell and cell-ECM interactions. By reducing variations in cellular access to nutrients which are known to occur within tumors, we propose observed differences in the cultures shed light on intrinsic differences among tumor cell behaviors (i.e. migration) at the population level. However, interpreting data collected across slice cultures generated from human tumors is complicated by inherent inter- and intra-tumoral heterogeneity. Critically, further study is needed to characterize the potential genetic and epigenetic shifts that may occur during ex vivo maintenance of human tumor slice cultures.
The use of human tumor slice cultures in parallel with Phase I/II clinical trials is a promising strategy to correlate slice parameters with patient clinical outcomes. Validation of these potential predictive/prognostic parameters is necessary before slice cultures can be used to personalize oncologic therapy. Our work, as well as that of others, demonstrates feasibility of biomarker validation21, as well as rapid ex vivo testing of therapeutic agents in slice cultures from GBM9,16. Similar human slice culture techniques using lung22, colon22, head and neck23, breast24, and prostate cancer25 tissues suggest this approach is generalizable across human cancers.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We would like to thank Dr. Lee Niswander and Dr. Rada Massarwa for their technical expertise and contributions to the slice culture confocal imaging protocol described here. Further thanks to Dr. Kalen Dionne who provided expertise regarding optimizing brain tumor tissue slicing and culture parameters.
References
- Beadle C, et al. The role of myosin II in glioma invasion of the brain. Mol Biol Cell. 2008;19:3357–3368. doi: 10.1091/mbc.E08-03-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farin A, et al. Transplanted glioma cells migrate and proliferate on host brain vasculature: a dynamic analysis. Glia. 2006;53:799–808. doi: 10.1002/glia.20334. [DOI] [PubMed] [Google Scholar]
- Panopoulos A, Howell M, Fotedar R, Margolis RL. Glioblastoma motility occurs in the absence of actin polymer. Mol Biol Cell. 2011;22:2212–2220. doi: 10.1091/mbc.E10-10-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivkovic S, et al. Direct inhibition of myosin II effectively blocks glioma invasion in the presence of multiple motogens. Mol Biol Cell. 2012;23:533–542. doi: 10.1091/mbc.E11-01-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assanah M, et al. Glial progenitors in adult white matter are driven to form malignant gliomas by platelet-derived growth factor-expressing retroviruses. J Neurosci. 2006;26:6781–6790. doi: 10.1523/JNEUROSCI.0514-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaichana KL, et al. Preservation of glial cytoarchitecture from ex vivo human tumor and non-tumor cerebral cortical explants: A human model to study neurological diseases. J Neurosci Methods. 2007;164:261–270. doi: 10.1016/j.jneumeth.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grube S, et al. Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis. J Neurooncol. 2014;118:277–287. doi: 10.1007/s11060-014-1452-z. [DOI] [PubMed] [Google Scholar]
- Hovinga KE, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells. 2010;28:1019–1029. doi: 10.1002/stem.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz F, et al. Organotypic slice cultures of human glioblastoma reveal different susceptibilities to treatments. Neurooncol. 2013;15:670–681. doi: 10.1093/neuonc/not003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, et al. Vorinostat modulates cell cycle regulatory proteins in glioma cells and human glioma slice cultures. J Neurooncol. 2011;105:241–251. doi: 10.1007/s11060-011-0604-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities. in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill BJ, et al. MRI-localized biopsies reveal subtype-specific differences in molecular and cellular composition at the margins of glioblastoma. Proc Natl Acad Sci USA. 2014;111:12550–12555. doi: 10.1073/pnas.1405839111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snuderl M, et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer cell. 2011;20:810–817. doi: 10.1016/j.ccr.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Kakita A, Goldman JE. Patterns and dynamics of SVZ cell migration in the postnatal forebrain: monitoring living progenitors in slice preparations. Neuron. 1999;23:461–472. doi: 10.1016/s0896-6273(00)80800-4. [DOI] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I, van Cappellen WA. Tracking in cell and developmental biology. Sem Cell Dev Biol. 2009;20:894–902. doi: 10.1016/j.semcdb.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Parker JJ, et al. Gefitinib selectively inhibits tumor cell migration in EGFR-amplified human glioblastoma. Neurooncol. 2013;15:1048–1057. doi: 10.1093/neuonc/not053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat DJ, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64:920–927. doi: 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Shamir ER, Ewald AJ. Three-dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol. 2014;15:647–664. doi: 10.1038/nrm3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60:502–514. doi: 10.1002/glia.21264. [DOI] [PubMed] [Google Scholar]
- Di Cristofori A, et al. The vacuolar H+ ATPase is a novel therapeutic target for glioblastoma. Oncotarget. 2015;6:17514–17513. doi: 10.18632/oncotarget.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaira V, et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc Natl Acad Sci USA. 2010. pp. 8352–8356. [DOI] [PMC free article] [PubMed]
- Gerlach MM, et al. Slice cultures from head and neck squamous cell carcinoma: a novel test system for drug susceptibility and mechanisms of resistance. Br J Cancer. 2014;110:479–488. doi: 10.1038/bjc.2013.700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday DL, et al. The practicalities of using tissue slices as preclinical organotypic breast cancer models. J Clin Pathol. 2013;66:253–255. doi: 10.1136/jclinpath-2012-201147. [DOI] [PubMed] [Google Scholar]
- Maund SL, Nolley R, Peehl DM. Optimization and comprehensive characterization of a faithful tissue culture model of the benign and malignant human prostate. Lab Invest. 2014;94:208–221. doi: 10.1038/labinvest.2013.141. [DOI] [PMC free article] [PubMed] [Google Scholar]