

Abstract
Autophagy is a catabolic pathway in which normal or dysfunctional cellular components that accumulate during growth and differentiation are degraded via the lysosome and are recycled. During autophagy, cytoplasmic LC3 protein is lipidated and recruited to the autophagosomal membranes. The autophagosome then fuses with the lysosome to form the autolysosome, where the breakdown of the autophagosome vesicle and its contents occurs. The ubiquitin-associated protein p62, which binds to LC3, is also used to monitor autophagic flux. Cells undergoing autophagy should demonstrate the co-localization of p62, LC3, and lysosomal markers. Immunofluorescence microscopy has been used to visually identify LC3 puncta, p62, and/or lysosomes on a per-cell basis. However, an objective and statistically rigorous assessment can be difficult to obtain. To overcome these problems, multispectral imaging flow cytometry was used along with an analytical feature that compares the bright detail images from three autophagy markers (LC3, p62 and lysosomal LAMP1) and quantifies their co-localization, in combination with LC3 spot counting to measure autophagy in an objective, quantitative, and statistically robust manner.
Keywords: Cellular Biology, Issue 125, Autophagy, multispectral imaging flow cytometry (MIFC), ImageStream, autophagosome, autolysosome, LC3, p62, LAMP1
Introduction
Macroautophagy, hereafter referred to as autophagy, is a catabolic pathway in which damaged or dysfunctional components, long-lived proteins, and organelles are degraded via the lysosome and are recycled1. Autophagy is a dynamic, multistep process that involves the formation of an autophagosome; the fusion of the autophagosome with the lysosome, forming the autolysosome; and the degradation of the contents of the autolysosome2. The crucial biological marker used to identify autophagy in mammalian systems is the microtubule-associated protein 1A/1B-light chain 3 (LC3), which makes up the autophagosomal membrane. During autophagy, cytosolic LC3-1 is conjugated to phosphatidylethanolamine to form LC3-II; LC3-II is then incorporated into the autophagosomal membrane3. Another widely used marker for autophagic flux is the autophagy receptor sequestosome 1 (SQSTM1, p62) which physically links autophagic cargo to the autophagic membrane4,5.
Methods that have traditionally been used to measure autophagy are Western blots and fluorescence microscopy7. However, neither of these methods are considered to be the "gold standard,"2,6 as there are challenges associated with both of these techniques. Western blots only give an average because they use a homogenate sample, so observers cannot see what is happening in individual cells. On the other hand, fluorescence microscopy gives an observer information at the single-cell level, but it lacks throughput capabilities, making it difficult to obtain objective and statistically rigorous assessments. In recent years, the measurement of LC3 and p62 by multispectral imaging flow cytometry (MIFC, see the Table of Materials) has been gaining popularity due to its quantitative power, high throughput capabilities, multiplexing potential, and ability to image every cell. One of the most widely used methods to measure autophagy using MIFC, along with its companion analysis software (see the Table of Materials), is through the spot counting of LC3 puncta or autophagosomes8,9,10,11,12,13,14,15,16,17. However, an increase in autophagosomes does not necessarily mean that there is an increase in autophagy, as it could also represent a blockade in the process2. LC3-II turnover is a useful parameter to measure autophagic flux by analyzing cells in the presence and absence of a lysosomal degradation inhibitor, such as chloroquine. Chloroquine inhibits the fusion of the autophagosome to the lysosome, thereby permitting the quantitation of the autophagosome formation as a measure of the degree of autophagy by arresting autophagic flux before the lysosomal degradation can occur18. In addition, p62 is degraded primarily by autophagy, and if the lysosomal degradation of the autophagosome and its contents is blocked, an accumulation of p62 is expected17,19.
Autophagy is a multistep process, and the measurement of LC3 or p62 alone does not provide a complete picture of what is happening in the cells. Recent publications have emphasized the need to examine the concurrent formation of the autolysosome2,17,20,21. MIFC is uniquely able to measure the formation of the autolysosome by imaging the co-localization of the autophagosome to the lysosome15-17,20,21,22,23,24,25,26. In addition, the co-localization of p62 and LC3 using MIFC has also been explored to measure autophagic flux5. In the referenced analysis software, the "feature" that measures co-localization is called "Bright Detail Similarity R3" (BDS) and was specificallydesigned to compare the small, bright image details of two images. BDS is the log-transformed Pearson's correlation coefficient of the localized bright spots with a radius of 3 pixels or less; in other words, BDS calculates the degree of overlapping pixels from two different fluorescent channels. The bright spots are either correlated (i.e., same spatial location) or uncorrelated (i.e., different spatial locations). Therefore, the correlation coefficient varies between 0 (uncorrelated) and 1 (perfect correlation). The coefficient is log-transformed to increase the range to between zero and infinity26,27. BDS alone may not be sufficient; Rajan et al. found that using only BDS could lead to false-positive or false-negative results21. BDS evaluates the co-localization of two markers of autophagy but does not consider the number of autophagosomes. To account for the number of autophagosomes, Rajan et al. included spot-counting of the LC3 puncta21. Rajan et al. proposed using a bivariate scatter plot of LC3 spot count versus BDS. Using this bivariate plot, two populations were first identified: one with cells that have a high level of LC3 spots and one with cells that have a low level of LC3 spots. The high-LC3 spot population was further classified into two populations: cells with low co-localization (i.e., accumulation of autophagosomes) and cells with high co-localization (i.e., accumulation of autolysosomes). This bivariate plot allows one to distinguish between cells with very few autophagosomes and cells with an accumulation of autophagosomes and/or autolysosomes21.
Until now, the simultaneous co-localization of LC3, p62, and LAMP1 (lysosomal marker) was not possible using a single "Feature Type" in the MIFC companion analysis software (see the Table of Materials). However, a new feature, recently introduced in version 6.1, is called Bright Detail Colocalization 3 (BDC3). BDC3 compares the bright detail images from each of the three images (in this case, LC3, p62, and LAMP1) and quantifies the co-localization of the three probes (i.e., LC3, p62, and LAMP1). The BDC3 feature computes the Pearson's correlation coefficient modified to extend to three images. Since the bright spots in the three images are either correlated or uncorrelated, the correlation coefficient varies between 0 (uncorrelated) and 1 (perfect correlation). The coefficient is log-transformed to increase the dynamic range between 0 and infinity. By switching out the BDS feature with the BDC3 feature, the analysis method presented by Rajan et al. can now incorporate the three most-used markers to measure autophagy in one system at the same time. The ability to co-localize these three markers of autophagy in a single assay could lead to novel insights into the induction and regulation of autophagy. The following protocol outlines the steps to induce autophagy in Jurkat cells; label the cells with LC3, p62, and LAMP1 antibodies; acquire data on a multispectral imaging flow cytometer; and analyze the data to assess autophagic flux.
Protocol
1. Preparation of Culture Medium and Lysosomal Degradation Inhibitor
1.1. Make ~500 mL of 1x RPMI culture medium. Add 5 mL of MEM non-essential amino acids (100x), 5 mL of sodium pyruvate (100 mM), 5 mL of penicillin-streptomycin-glutamine (100x), and 25 mL of fetal bovine serum (FBS) to a 500 mL bottle of RPMI – 1640 1x medium. The medium should be prepared in a biosafety cabinet. Store the RPMI culture medium at 2-8 °C. Heat the RPMI culture medium to 37 °C prior to adding it to the Jurkat cells.
Make 100 mM chloroquine, the lysosomal degradation inhibitor. Weigh out 0.05 g of chloroquine diphosphate salt and add it to 1 mL of cell culture-grade water in a 15-mL centrifuge tube. Vortex until the chloroquine has dissolved.
2. Culturing of Cells
Culture 80 mL of Jurkat cells clone E6-1 in RPMI culture medium at 37 °C and 5% CO2. NOTE: When culturing cells or working with the Jurkat cells prior to fixation, all work should be done in the biosafety cabinet.
Prior to inducing autophagy, count the Jurkat cells using a hemocytometer or another cell counting device to determine the cells/mL.
3. Inducing Autophagy in Cells
Take the Jurkat cells in exponential growth phase and split them into four samples of 20 mL each in 50-mL centrifuge tubes. Label the tubes as "Control," "Control + Chloroquine," "Starved," and "Starved + Chloroquine." NOTE: Jurkat cells should have a concentration of 2.5-5 x 105 cells/mL.
Wash the cells by adding 30 mL of Hank's Balanced Salt Solution without Ca2+ or Mg2+ (HBSS) to each sample. Centrifuge at 250 x g for 10 min. Pour off the supernatant into a waste beaker.
Resuspend the control samples in 20 mL of RPMI culture medium by pipetting up and down using a 25 mL sterile disposable pipette followed by performing light vortexing. Similarly, resuspend the starved samples in 20 mL of Earle's Balanced Salt Solution without Ca2+ or Mg2+ (EBSS).
Using a 25 mL sterile disposable pipette, transfer the control and starved samples to T75 flasks that have been labeled "Control," "Control + Chloroquine," "Starved," and "Starved + Chloroquine."
Add 20 µL of 100 mM chloroquine to the 20 mL of cell culture medium in the "Control + Chloroquine" and the "Starved + Chloroquine" flasks. Mix in the chloroquine by gently swirling the flask. Place all flasks in a 37 °C and 5% CO2 incubator for 2 h.
4. Preparation of Buffers for the Fixation and Labeling of LC3, LAMP1, and p62
Prepare 10 mL of 4% formalin fixation solution in PBS. Add 4 mL of 10% formalin stock to 1 mL of 10x Dulbecco's Phosphate-Buffered Saline solution without Ca2+ or Mg2+ (PBS) and 5 mL of ultrapure water in a 15 mL centrifuge tube. NOTE: CAUTION. Formalin/formaldehyde is toxic if inhaled or swallowed; is irritating to the eyes, respiratory system, and skin; and may cause sensitization by inhalation or skin contact. There is a risk of serious damage to eyes. It is a potential carcinogen.
Prepare 10 mL of 1% formalin final resuspension solution in PBS. Add 1 mL of 10% formalin stock to 9 mL of 1x PBS in a 15 mL centrifuge tube.
Prepare 500 mL of wash buffer by adding 10 mL of FBS to a 500 mL bottle of 1x PBS.
Prepare 50 mL of permeabilization buffer in a 50 mL centrifuge tube by adding 0.5 mL of 10% triton X-100 to the 49.5 mL wash buffer prepared in step 4.3.
Prepare anti-SQSTM1/p62-Alexa 488 (AF488) antibody/permeabilization buffer working solution by adding 5 µL of anti-SQSTM1/p62-AF488 to 495 µL of the permeabilization buffer made in step 4.4. Ensure that the final antibody concentration is 5 µg/mL. NOTE: Make antibody permeabilization buffer working solution just prior to adding it to the sample. Protect the solution from light.
Prepare mouse anti-LC3/permeabilization buffer working solution by adding 5 µL of mouse anti-LC3 to 495 µL of the permeabilization buffer made in step 4.4. Ensure that the final antibody concentration is 20 µg/mL. NOTE: Make antibody permeabilization buffer working solutions just prior to adding it to the sample. Protect the solution from light.
Prepare donkey anti-mouse-Alexa 647 (AF647)/permeabilization buffer working solution by adding 5 µL of AF647 labeled donkey anti-mouse – IgG to 495 µL of the permeabilization buffer made in step 4.4. Ensure that the final antibody concentration is 20 µg/mL. NOTE: Make the antibody permeabilization buffer working solution just prior to adding it to the sample. Protect the solution from light.
Prepare anti-human CD107a (LAMP1)-PE antibody/permeabilization buffer working solution by adding 25 µL of anti-CD107a (LAMP1)-PE to 475 µL of the permeabilization buffer made in step 4.4. Ensure that the final antibody concentration is 21 µg/mL. NOTE: Make the antibody permeabilization buffer working solution just prior to adding it to the sample. Protect the solution from light.
Prepare 10x DAPI nuclear stain by adding 4 µL of 5 mg/mL DAPI and 100 µL of 10% triton X-100 to 896 µL of 1x PBS.
5. Labeling the Jurkat Cells with LC3, LAMP1, and p62
- Remove 2 x 106 cells per sample (count the cells as in step 2.2). Place each sample in a 15-mL centrifuge tube that has been labeled with the appropriate sample name (i.e., Control, Control + Chloroquine, Starved, or Starved + Chloroquine).
- Add wash buffer so that there is a total volume of 15 mL in each 15-mL centrifuge tube. Centrifuge the samples at 250 x g for 10 min. Aspirate off the supernatant.
Add 100 µL of 4% formalin fixation solution to each of the cell pellets. Resuspend by pipetting up and down with a P200 pipette. Incubate for 10 min at room temperature. During the fixation step, transfer the samples to labeled, siliconized polypropylene microcentrifuge tubes. NOTE: Do not over-fix the cells; this might result in poor labeling results.
After the 10 min fixation step, add 1 mL of wash buffer to each sample. Pipette up and down with a P1000 pipette to mix the sample. Centrifuge at 250 x g for 5 min. Aspirate off the supernatant.
Add 100 µL of anti-SQSTM1/p62 - AF488 antibody/permeabilization buffer working solution to each sample. Resuspend by pipetting up and down with a P200 pipette. Incubate for 30 min in the dark at room temperature.
Wash the cells by adding 1 mL of permeabilization buffer to each sample. Pipette up and down with a P1000 pipette to mix the sample. Centrifuge at 250 x g for 5 min. Aspirate off the supernatant.
Repeat steps 5.4-5.5 for each of the antibody working solutions. NOTE: The antibody working solutions are mouse anti-LC3/permeabilization buffer working solution, donkey anti-mouse - AF647/permeabilization buffer working solution, and anti-human CD107a (LAMP1) - PE antibody/permeabilization buffer working solution. Each antibody should be done separately and in the listed order. The order was chosen to minimize unwanted cross-reactivity between antibodies.
Add 100 µL of 1% formalin solution. Resuspend by pipetting up and down with a P200 pipette. Add 10 µL of the 10x DAPI nuclear stain to each sample. Lightly vortex each sample to mix. Incubate for 10 min in the dark at room temperature before running any of the samples on the instrument. NOTE: At this point, the sample can either be run on the MIFC or stored protected from light at 2-8 °C for up to a week.
6. Labeling of Single-color Controls
For each single-color control (i.e., AF488, PE, AF647, and DAPI), take 1 x 106 Jurkat cells, which can be from any of the sample populations. Put 1 x 106 cells in 15 mL centrifuge tubes labeled as either AF488, PE, AF647, or DAPI.
Add wash buffer so that there is a total volume of 15 mL in each 15 mL centrifuge tube. Centrifuge the samples at 250 x g for 10 min. Aspirate off the supernatant.
- Add 100 µL of wash buffer to the AF488, PE, and AF647 tubes. NOTE: For the DAPI tube, go to step 6.8.
- Resuspend by pipetting up and down with a P200 pipette. Transfer to 1.5-mL tubes.
Add 5 µL of either anti-CD45-AF488, anti-CD45-PE or anti-CD45-AF647 to the AF488, PE, or AF647 tubes, respectively. Mix by vortexing lightly.
Incubate the AF488, PE, and AF647 tubes for 30 min in the dark at room temperature. NOTE: Other markers can be used, such as LC3, p62, and LAMP-1 antibodies instead of anti-CD45 antibodies. However, CD45 is a reliable marker for single-color controls.
- Wash the AF488, PE, and AF647 cells by adding 1 mL of wash buffer to each sample.
- Pipette up and down with a P1000 pipette to mix the sample. Centrifuge at 250 x g for 5 min. Aspirate off the supernatant.
Add 100 µL of 1% formalin solution. Resuspend by pipetting up and down with a P200 pipette. NOTE: At this point, the sample can either be run on the MIFC or stored protected from light at 2-8 °C for up to a week.
- Add 100 µL of 1% formalin fixation solution to the DAPI tube.
- Resuspend by pipetting up and down with a P200 pipette. Incubate for 30 min at room temperature.
- Add 10 µL of 10x DAPI nuclear stain to the DAPI sample. Lightly vortex to mix. Incubate for 10 min in the dark at room temperature prior to running on the instrument. NOTE: At this point, the sample can either be run on the MIFC or stored protected from light at 2-8 °C for up to a week.
7. Starting and Calibrating the MIFC
Check to make sure the sheath, system calibration reagent (see the Table of Materials), debubbler, cleanser, and sterilizer containers are full and the waste tank is empty. If not, fill the containers and empty the waste tank.
Power up the system and double-click on the software (see the Table of Materials) icon to launch the application; this will start the MIFC software.
Click the Startup button and make sure that Start all calibrations and tests is selected. This will flush the system, load sheath, system calibration reagent, and calibrate the system.
8. Running the Samples and Single-color Controls on the MIFC
Load the default template by going to the File tab and selecting Load Default Template. NOTE: The default template contains the raw max pixel dot plots.
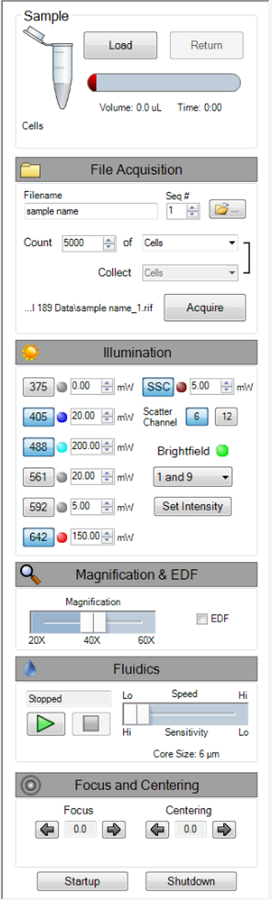
Turn on the 488 nm, 405 nm, 642 nm, and SSC lasers by clicking on their respective buttons. Set the laser power by entering the desired power next to each laser button. See Figure 1. NOTE: For this experiment, the laser powers are 200 mW, 20 mW, 150 mW, and 5 mW (Ch06) for the 488 nm, 405 nm, 642 nm, and SSC, respectively. The MIFC default template magnification it set to 40X, the default sensitivity is Hi, and all channels are enabled.
Confirm that the magnification slider is set to 40X, high-sensitivity mode is selected, and all channels are showing in the image gallery; Figure 1 shows the instruments settings used in this experiment.
Load the "Starved + Chloroquine" sample by clicking on the Load button and loading the 1.5 mL tube onto the instrument. NOTE: This experimental sample should have the highest signal of all samples for AF488, PE, and AF647 (the DAPI signal should have approximately the same signal strength for all experimental samples, including the "Starved + Chloroquine" sample).
Use the "Starved + Chloroquine" sample to confirm that the appropriate laser powers for the 488 nm, 405 nm, 642 nm, and SSC lasers are set. Use the raw max pixel dot plots from the default template to adjust the laser powers so that the signals are strong but do not reach a raw max pixel value of 4,096, which would saturate the CCD camera on the instrument. Use this laser setting for all experimental samples.
Click on the Create Dot Plots icon to create a new dot plot. Select "Area_M01" on the X-axis and "Aspect Ratio_M01" on the Y-axis for the "all" population. Click on the Create Polygon Region icon. Draw a region around the cells. Name this region "Cell." NOTE: Users can add additional plots and regions to narrow down the cells being collected. A common additional plot to add would be Gradient RMS to identify cells that are in focus.
Set the acquisition parameters. Specify the file name and destination folder, change the number of events to "5,000," and select the "Cell" collection population. Click the Acquire button to acquire the file. After collecting the file, return the sample by clicking the Return button. Remove the sample tube from the instrument.
For the next three samples ("Starved," "Control," and "Control + Chloroquine"), load the sample, enter the sample name, acquire the sample, and return the sample using the same settings as for the "Starved + Chloroquine" sample, above.
After the samples have been acquired, run the single-color control samples and acquire data so that a compensation matrix can be made. Turn off the SSC laser by clicking on the SSC button. Turn off the brightfield channels by clicking on the Brightfield button and selecting OFF. NOTE: If channels were removed from the image gallery for experimental sample acquisition, re-enable all channels. Alternatively, single-color controls can be acquired by clicking on the Compensation tab and selecting Create Matrix; this opens the compensation wizard, which will walk through the step of collecting files and making a compensation matrix.
- Load the DAPI single-color control by clicking on the Load button and loading the 1.5 mL tube into the instrument. Enter the sample name (DAPI only), set the collection population to "All," and change the number of events to "500."
- Click the Acquire button to acquire the single-color control file.
- When the file collection is complete, return the sample by clicking the Return button. Remove the sample tube from the instrument.
- After the DAPI sample, load 10% bleach just like a sample by clicking on the Load button and loading 100 µL of 10% bleach in a 1.5 mL tube. Let the sample run for ~15 s. This will remove any residual DAPI from the tube.
- Run 100 µL of 1x PBS, as done in 8.10.3, to remove the residual 10% bleach. The instrument is now ready to run the AF488, PE, and AF647 single-color controls.
- Repeat steps 8.10-8.10.2 for each of the single-color controls (i.e., AF488, PE, and AF647). NOTE: There is no need to run 10% bleach or PBS between these samples, only after the DAPI single-color control.
9. Data Analysis in the MIFC Analysis Software
Launch the MIFC analysis software (see the Table of Materials).
- Click on Start Analysis to start the Open File Wizard. Select the data file to open by browsing to the "Starved + Chloroquine" raw image file (.rif). Select the file and click the Open button. Click Next.
- Apply compensation. Select a previously created compensation matrix and click Next. Alternatively, make a new compensation matrix by clicking the New Matrix button. NOTE: Steps 9.2.1.1-9.2.1.2 are the steps to create a new matrix.
- Add the single-color control files (i.e., DAPI only, AF488 only, PE only, and AF647 only) to the Control Files list by clicking on Add Files, selecting the files, and clicking the Open button. Click Next.
- The Create Compensation Matrix window will appear for this experiment. Ensure that Ch02, Ch03, Ch07, and Ch11 are selected and click Next. The compensation matrix will be created. Click Finish to save the compensation matrix. Add the compensation file to the Open File Wizard and click Next.
- Apply the analysis template. At this stage, there is no analysis template to apply, so click Next. NOTE: For the other experimental files, this "Starved + Chloroquine.daf" file can be used as a template.
- Name the file (the file names that correspond with the "rif" names are automatically generated) and click Next. Set the image display properties by selecting the "02," "03," "07," and "11" channels; channels 01, 06, and 09 are preselected. Click Next.
- At the end of the Open File Wizard, select the Apoptosis Wizard. Click on the Apoptosis Wizard icon and then on the Select Wizard button to open the Apoptosis Wizard.
- Select the nuclear image channel (Ch07) and click Next. Gate the cells in best focus. Click on the Create Line Region icon and draw a region on the Gradient RMS_M01_Ch01 from 60-90. Name this region "Focus," click OK, and click Next.
- Gate the single cells. Click on the Create Polygon Region icon. Draw a region around the single cells, name the region "Single," click OK, and click Next. Click No for "Do you want to analyze sub-populations?"
- Gate the nucleated cells. Click on the Create Line Region icon and draw a region that includes all nucleated cells. Name the region "Nucleated." Click OK and Next.
- Gate the apoptotic cells. Click on the Create Polygon Region icon. Draw a region around the apoptotic cells; these are cells with low nuclear Area_T50% and high brightfield contrast. Click Finish. Add a second region by clicking on the Create Polygon Region icon and drawing a region around the non-apoptotic cells; call this region "Cells."
- Select cells that are positive for LAMP1, p62, and LC3. Click on the Building Blocks icon, select Fluorescence Positives — One Color, and select the "Cells" population and Intensity_MC_Ch03. Label the X-Axis "Intensity LAMP1." Draw a region that includes cells with intensities greater than 10,000 by clicking on the Create Line Region icon and drawing the region. Label this region "LAMP1+." Follow the same steps as in 9.4 to select cells positive for p62 and LC3.
- For p62+ select the "LAMP1+" population and Intensity_MC_Ch02. Label the X-Axis "Intensity p62" and the region of p62+ cells "LAMP1+/p62+." For the LC3+, select the "LAMP1+/p62+" population and Intensity_MC_Ch11. Label the X-Axis "Intensity LC3" and the region of LC3+ cells "LAMP1+/p62+/LC3+." Use this "LAMP1+/p62+/LC3+" population for the rest of the analysis.
- Create a spot-count feature to count the LC3 spots. NOTE: This should be created using the "Starved + Chloroquine" sample.
- Click the Analysis tab and select Mask. Click the New and then the Function button. Under Function, select Peak; Mask, select M11; and Channel, select Ch11. Set the Spot to Cell Background Ratio to 4. Click OK to close the Define Mask Function window and OK to add it to the Mask list. Click Close to exit the Mask Manager.
- Click the Analysis tab and select Features. Click on the New button. Under Feature Type, selectSpot Count and under Mask, select Peak(M11, Ch11, Bright, 4). Type "Spot Count LC3" for the Name and click OK and Close to exit the Feature Manager. NOTE: The Spot wizard may also be used to create a spot-count feature. The spot-count feature used in this experiment may not be appropriate for other experiments and/or cell line. The user should test several spot-count masks/features when deciding on a mask/feature for a specific data set. Pugsley 2017 has further details on spot counting using the MIFC analysis software26.
- Click on the New Histogram icon. Select the "LAMP1+/p62+/LC3+" population. Select the Spot Count LC3 feature and click OK.
- Create the BDC3 feature.
- Click the Analysis tab and select Features. Click on the New button. Under Feature Type, select Bright Detail Colocalization 3; Mask, select MC; Image 1, select Ch02 (p62); Image 2, select Ch03 (LAMP1); and Image 3, select Ch11 (LC3). Type "BDC3 p62/LAMP1/LC3" for the Name and click OK and Close to exit theFeature Manager.
- Click on the New Histogram icon. Select the "LAMP1+/p62+/LC3+" population. Select the BDC3 p62/LAMP1/LC3 feature and click OK.
Click on the New Scatterplot icon. Select the "LAMP1+/p62+/LC3+" population. Select BDC3 p62/LAMP1/LC3 for the X-axis. Select the Spot Count LC3 feature for the Y-axis. Click OK.
Save the "Starved + Chloroquine" file. Click on File and select Save Data Analysis file (.daf). Click Save and Yes to replace the previous version of the file; this file can now be used as a template.
Open the "Control" file. Click on File and then Open. Select the" Control.rif" file and click Open. Select the compensation matrix from step 9.2.1 and the "Starved + Chloroquine.daf" file as the template. Click OK.
- Create regions for autophagosome accumulation and autolysosome accumulation using the "Control" sample to set the regions.
- Click on the Create Rectangle Region icon. Draw a region from X-coordinates of -0.1 to 3 and Y-coordinates of 1.5 to -0.3. Name this region "Low Spots." Click on theCreate Rectangle Region icon. Draw a region from X-coordinates of -0.1 to 1 and Y-coordinates of 17.5 to 1.5. Name this region "High Spot/Low BDC3."
- Click on the Create Rectangle Region icon. Draw a region from X-coordinates of 1 to 3 and Y-coordinates of 17.5 to 1.5. Name this region "High Spots/High BDC3."
Save the "Control" file. Click on File and select Save Data Analysis file (.daf). Click Save and Yes to replace the previous version of the file; this file can now be used as a template.
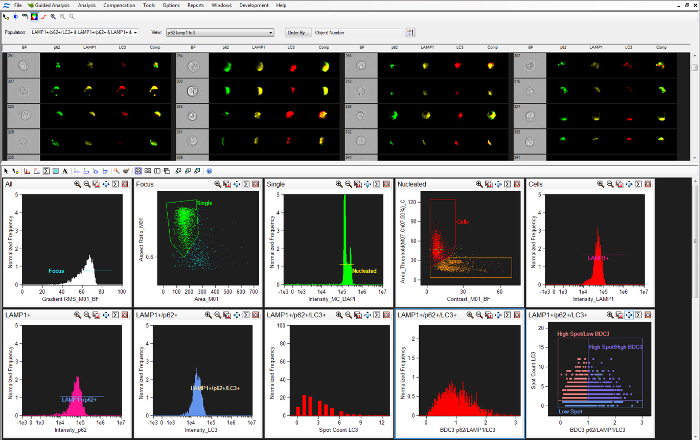
Open the "Control + Chloroquine" file by clicking on File and then Open. Select the "Control + Chloroquine.rif" file and click Open. Select the compensation matrix from step 9.2.1 and the "Control.daf" file as the template. Click OK. Repeat this for the "Starved" and "Starved + Chloroquine" files. NOTE: The batch function in the analysis software may also be used to apply the compensation matrix and/or template to the other files. The final gating strategy is shown in Figure 2.
Representative Results
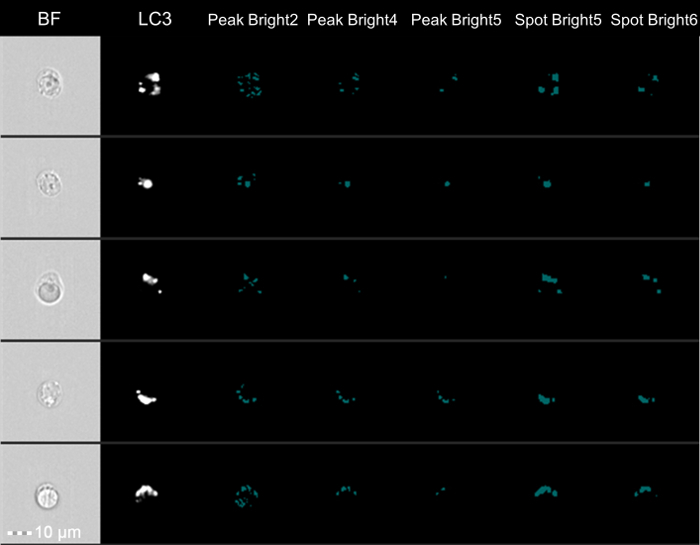
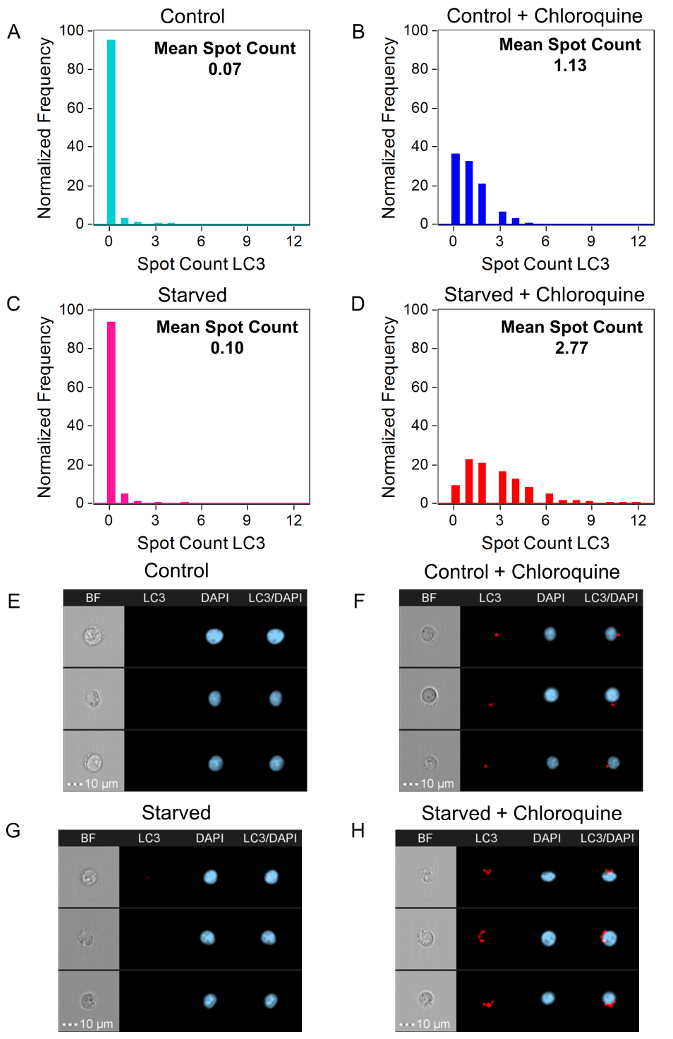
This analysis method uses multiple features to asses autophagic flux. In order to fully understand the final bivariate plot, the individual analysis features must first be investigated. The counting of autophagosomes is a logical way to measure autophagy; however, the size/shape/brightness of LC3 puncta can vary drastically between cells. Variation can make it difficult to count autophagosomes manually or using a spot-count feature in the analysis software. Therefore, no spot-count feature will be perfect due to this large variability in autophagosomes. However, a good spot-count feature will work on most cells26. Figure 3 shows examples of spot masking of LC3 puncta in Jurkat cells using different spot masks. The spot-count feature selected for this data set is Spot Count_Peak(M11, Ch11-LC3-AF647, Bright, 4)_4, meaning that the spot-count feature counted the spots that the peak mask identified within the default channel 11 mask (M11) on channel 11 (Ch11-LC3-AF647) with a spot-to-cell background ratio of 4 (Bright, 4). Figure 4 shows spot-count histograms and representative images for the mean spot count for the Control, Control + Chloroquine, Starved, and Starved + Chloroquine Jurkat cells labeled with anti-LC3-AF647. The Control and Starved mean spot counts are not significantly different; with the addition of chloroquine, there is a large difference in the mean of the Control + Chloroquine compared to the Starved + Chloroquine.
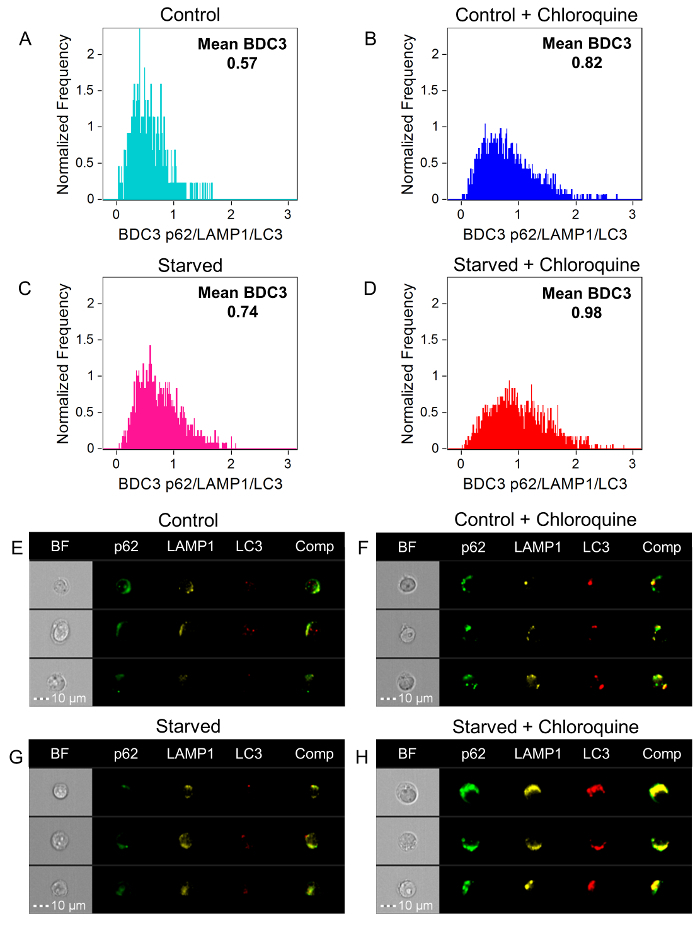
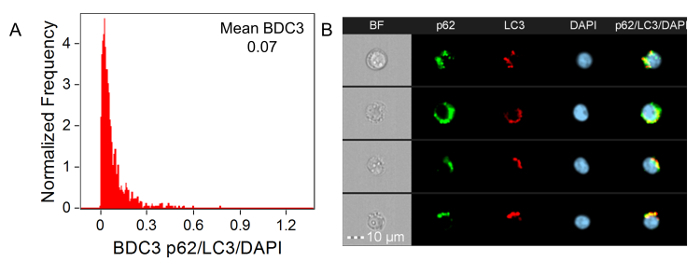
The next feature to investigate is BDC3. BDC3 is a measurement of the co-localization of three markers/probes, in this case, LC3, p62, and LAMP1. Figure 5A-5D shows BDC3 histograms for the Control, Control + Chloroquine, Starved, and Starved + Chloroquine Jurkat cells labeled with anti-p62-AF488, anti-LAMP1-PE, and anti-LC3-AF647. There is a shift between the Control mean to the Starved mean, as well as the Control + Chloroquine mean to the Starved + Chloroquine mean. However, looking at the images of cells from the mean BDC3 scores in Figure 5E-5H, there is a greater difference between the samples than the histograms may lead one to believe. This is because BDC3 does not consider the number of autophagy organelles that co-localize, resulting in a large degree of variability in the number of autophagosomes for the same BDC3 score. In most cases, there is overlap between all three probes because, even at basal levels, p62, LAMP1, and LC3 should, to a certain extent, co-localize or reside in similar regions in the cells. As a contrast, an example of three probes that should not co-localize are anti-p62-AF488, anti-LC3-AF647, and DAPI nuclear dye for the Starved + Chloroquine sample, shown in Figure 6.
When the spot-count feature and the BDC3 feature are combined, the presence of different subpopulations that improve the ability to distinguish between the various samples/conditions are evident. Figure 7 shows the bivariate plot of spot count of LC3 versus BDC3 p62/LAMP1/LC3 for the four samples: Control, Control + Chloroquine, Starved, and Starved + Chloroquine. The Control sample was used to set the gating strategy for three populations: Low Spots, High Spots/Low BDC3, and High Spots/High BDC3. The Control samples demonstrated that greater than 98% of the cells had 1 or fewer spots. The boundary between the High Spots/Low BDC3 and High Spots/High BDC3 was set to a BDC3 score of 1 because more than 91% of the Control sample had a BDC3 score of less than 1. A summary of the results for the bivariate plots is shown in Table 1.
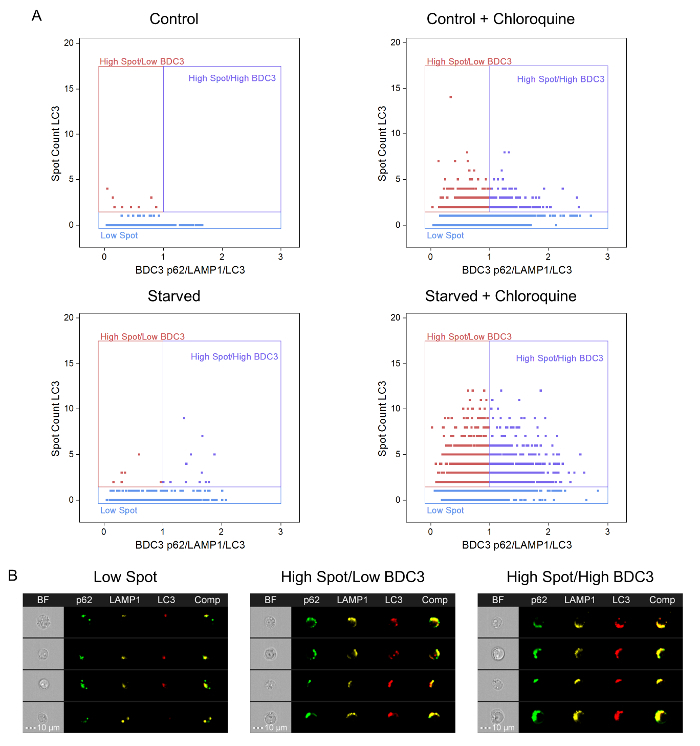
Figure 1: MIFC Instrument Setting. A screenshot of the MIFC instrument settings used for this experiment, outlined in step 8 of the protocol. Please click here to view a larger version of this figure.
Figure 2: Analysis Software Gating Strategy. A screenshot of the analysis software gating scheme, outlined in step 9 of the protocol. Please click here to view a larger version of this figure.
Figure 3: LC3 Spot Masking. Jurkat cell images and masks used to create the spot-count feature. Shown are BrightField (BF), LC3-AF647, Peak(M11, Ch11-LC3-AF647, Bright,2), Peak(M11, Ch11-LC3-AF647, Bright,4), Peak(M11, Ch11-LC3-AF647, Bright,5), Spot(M11, Ch11-LC3-AF647, Bright,5,3,1), and Spot(M11, Ch11-LC3-AF647, Bright,6,2,1). The mask that worked best for all cells shown was Peak(M11, Ch11-LC3-AF647, Bright,4). Please click here to view a larger version of this figure.
Figure 4: LC3 Spot-count Histograms. Using the spot-count feature Spot Count_Peak(M11, Ch11-LC3-AF647, Bright, 4)_4 and the LC3-AF647 spot-count histograms for Control (A, light blue), Control + Chloroquine (B, blue), Starved (C, pink), and Starved + Chloroquine (D, red). The mean spot counts for Control, Control + Chloroquine, Starved, and Starved + Chloroquine are 0.07, 1.13, 0.10, and 2.77, respectively. BF, LC3-AF647 (red), DAPI nuclear dye (blue), and a composite of the LC3-AF647 and DAPI images of representative cells for the mean spot count are shown for Control (E, 0 spots), Control + Chloroquine (F, 1 spot), Starved (G, 0 spots), and Starved + Chloroquine (H, 3 spots). Please click here to view a larger version of this figure.
Figure 5: BDC3 Histograms of p62/LAMP1/LC3. BDC3 p62/LAMP1/LC3 histograms for Control (A, light blue), Control + Chloroquine (B, blue), Starved (C, pink), and Starved + Chloroquine (D, red). The mean BDC3 score for Control, Control + Chloroquine, Starved, and Starved + Chloroquine are 0.57, 0.82, 0.74, and 0.98, respectively. BF; p62-AF488 (green); LAMP1-PE (yellow); LC3-AF647 (red); and a composite of the p62-AF488, LAMP1-PE, and LC3-AF647 images of representative cells for the mean BDC3 are shown for Control (E), Control + Chloroquine (F), Starved (G), and Starved + Chloroquine (H). Please click here to view a larger version of this figure.
Figure 6: BDC3 Histograms of p62/LC3/DAPI for the Starved + Chloroquine Sample. (A) BDC3 p62/LC3/DAPI histogram for the Starved + Chloroquine sample is shown. The mean BDC3 score is 0.07. (B) BF; p62-AF488 (green); LC3-AF647 (red); DAPI (blue); and a composite of the p62-AF488, LC3-AF647, and DAPI images of representative cells for the mean BDC3 is shown for the Starved + Chloroquine sample. Please click here to view a larger version of this figure.
Figure 7: Bivariate Plots of LC3 Spot Count versus BDC3 p62/LAMP1/LC3. (A) Bivariate plots of LC3 spot count versus BDC3 p62/LAMP1/LC3 for Control, Control + Chloroquine, Starved, and Starved + Chloroquine Jurkat cells. (B) BF; p62-AF488 (green); LAMP1-PE (yellow); LC3-AF647 (red); and a composite of the p62-AF488, LAMP1-PE, and LC3-AF647 images from three regions (i.e., Low Spots, High Spots/Low BDC3, and High Spots/High BDC3) are shown for the Starved + Chloroquine sample. Please click here to view a larger version of this figure.
| Cell Count | Bright Detail Colocalization 3 Mean | Spot Count LC3 Mean | % Low Spot | % High Spot/Low BDC3 | % High Spot/High BDC3 | |
| Control | 439 | 0.57 | 0.07 | 98.2 | 1.8 | 0.0 |
| Control + Chloroquine | 1432 | 0.82 | 1.13 | 68.9 | 19.5 | 11.7 |
| Starved | 1204 | 0.74 | 0.10 | 98.4 | 0.6 | 1.0 |
| Starved + Chloroquine | 1811 | 0.98 | 2.77 | 32.1 | 36.9 | 31.0 |
Table 1: Summary of Jurkat LC3 Spot Count versus BDC3 p62/LAMP1/LC3 Bivariate Plot
Discussion
When performing this analysis, there are a few things to consider. It is important to have appropriate control samples. For this experiment, two different controls were used: Control and Control + Chloroquine. The Control sample was used to set the threshold for the regions. It represents the basal level of autophagy without stopping lysosomal degradation and is the control for the Starved sample. However, the Control sample is not an appropriate control to compare with the results from the Starved + Chloroquine sample because chloroquine itself influences the control sample. Therefore, it was necessary to have a Control + Chloroquine sample.
Prior to performing any analysis for autophagy, it is important to only analyze/gate on single cells that are in focus (i.e., gradient RMS greater than 60), as the results could be affected if there is more than one cell or the images are out of focus. It is crucial that the autophagosomes and lysosomes are in focus for proper spot counting and BDC3 analysis. Apoptotic cells must be removed prior to autophagy analysis, as they may skew results and should not be included. There should be appropriate staining for all three markers (i.e., LC3, p62, and LAMP1). For example, all three markers are intracellular and should have a punctate signal; if the signal is not punctate, or if it is on the surface of the cell, the staining would not be appropriate and the experiment/staining should be redone. In addition, for BDC3 to be accurate, it is essential to gate on cells that are bright for all three fluorescent markers of interest. Gating on positive events is needed to prevent the measurement of BDC3 on non-specific binding, imaging artifacts, and/or noise. This is crucial, as BDC3 can potentially amplify artifacts that are not true signals. Since BDC3 can only be performed on bright positive events, many cells may not be included in the final analysis; this is especially true for control cells that do not have any accumulation of LC3, p62, and/or LAMP1. Thus, a limitation of this assay is that BDC3 only examines cells that are positive for all three markers, which might not be appropriate for all autophagy experiments.
Like BDS, which has been previously reported15,16,17,20,21,22,23,24,25,26, BDC3 alone does not take into account the number of autophagy organelles that co-localize, resulting in a large degree of variability in the number of autophagosomes. Therefore, the bivariate approach presented by Rajan et al. was employed. Looking at the bivariate plots, one might ask if the cells must be positive for LC3, p62, and LAMP1; why are there so many cells that have 0 spots? This is because the LC3 signal might be bright enough for co-localization, but it might not meet the threshold set by the peak mask (Peak (M11, Ch11-LC3-AF647, Bright, 4)) necessary for it to be considered a spot.
The protocol presented here used MIFC to count LC3 puncta and the co-localization of three autophagy markers to measure autophagic flux. Under basal conditions (Control sample), the number of autophagosomes was low, and few cells were found with "High Spots." With the addition of chloroquine, which inhibits autophagosome/lysosome fusion, there was an increase in the number of LC3 spots. Since the lysosome is unable to break down the autophagosome and the p62, which resides in the autophagosome, this leads to an increase in the co-localization of the LC3, p62, and LAMP1 autolysosome accumulation. This effect was amplified under nutrient starvation, which induces autophagy. However, without the addition of chloroquine, there was not a significant increase in the number of autophagosomes, most likely due to an increase in the rate of autophagic turnover. When cells were starved in the presence of chloroquine, there was an increase in the co-localization and number of autophagosomes, which supports the conclusion that starvation increases autophagic flux. EBSS is known to be a powerful inducer of autophagy. Therefore, it is expected that the increase would be large. If another method, such as drug induction, is used to induce autophagy, the difference between the control and treated samples may be subtler.
This particular protocol was designed to measure autophagy in human cell lines, but the assay could be adapted to other species by switching to antibodies for that particular species. In addition, the analysis method could be used for any intracellular application that requires the co-localization of three probes/markers.
Disclosures
I am employed by MilliporeSigma, the maker of the Amnis brand ImageStream, which was used in this study.
Acknowledgments
I would like to thank my coworkers at MilliporeSigma, Philip J. Morrissey and Sherree L. Friend, for their support and guidance over the years. I would also like to thank Vidya Venkatachalam and Bryan R. Davidson for their help with the BDC3 feature in the IDEAS software, Ryan P. Jessup for help editing this manuscript, Raymond Kong for help on the day of shooting, and a special thank you to makeup artist Cynthia Xamonthiene.
References
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Developmental Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Zhang XJ, Chen S, Huang KX, Le WD. Why should autophagic flux be assessed? Acta Pharmacologica Sinica. 2013;34:595–599. doi: 10.1038/aps.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Takashi U, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- Larsen KB, Lamark T, Øvervatn A, Harneshaug I, Johansen T, Bjørkøy G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy. 2010;6(6):784–793. doi: 10.4161/auto.6.6.12510. [DOI] [PubMed] [Google Scholar]
- Demishtein A, Porat Z, Elazar Z, Shvets E. Applications of flow cytometry for measurement of autophagy. Methods. 2015;75:87–95. doi: 10.1016/j.ymeth.2014.12.020. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsov I, et al. BAC-mediated transgenic expression of fluorescent autophagic protein Beclin 1 reveals a role for Beclin 1 in lymphocyte development. Cell Death Differ. 2008;15(9):1385–1395. doi: 10.1038/cdd.2008.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloyan A, Sayegh J, Osinska H, Chua BH, Robbins J. Manipulation of death pathways in desmin-related cardiomyopathy. Circ Res. 2010;106(9):1524–1532. doi: 10.1161/CIRCRESAHA.109.212639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman BJ, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene. 2011;30(16):1855–1867. doi: 10.1038/onc.2010.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez AL, Kong R, George T, He L, Yue Z, van Dyk LF. Gammaherpesvirus 68 infection of endothelial cells requires both host autophagy genes and viral oncogenes for optimal survival and persistence. J Virol. 2011;85(13):6293–6308. doi: 10.1128/JVI.00001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tra T, et al. Autophagy in human embryonic stem cells. PLoS One. 2011;6(11) doi: 10.1371/journal.pone.0027485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan N, et al. Bafilomycin A1 targets both autophagy and apoptosis pathways in pediatric B-cell acute lymphoblastic leukemia. Haematologica. 2015;100(3):345–356. doi: 10.3324/haematol.2014.113324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leveque-El Mouttie L, et al. Autophagy is required for stem cell mobilization by G-CSF. Blood. 2015;125(19):2933–2936. doi: 10.1182/blood-2014-03-562660. [DOI] [PubMed] [Google Scholar]
- Watson AS, et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015;1:15008. doi: 10.1038/cddiscovery.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranks AJ, et al. Autophagy Controls Acquisition of Aging Features in Macrophages. J Innate Immun. 2015;7(4):375–391. doi: 10.1159/000370112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke AJ, et al. Autophagy is activated in systemic lupus erythematosus and required for plasmablast development. Ann Rheum Dis. 2015;74(5):912–920. doi: 10.1136/annrheumdis-2013-204343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes G. Flow cytometric assays for the study of autophagy. Methods. 2015;82:21–28. doi: 10.1016/j.ymeth.2015.03.027. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadwal K, et al. A novel method for autophagy detection in primary cells: Impaired levels of macroautophagy in immunosenescent T cells. Autophagy. 2012;8(4):677–689. doi: 10.4161/auto.18935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan R, Karbowniczek M, Pugsley HR, Sabnani MK, Astrinidis A, La-Beck NM. Quantifying autophagosomes and autolysosomes in cells using imaging flow cytometry. Cytometry A. 2015;87(5):451–458. doi: 10.1002/cyto.a.22652. [DOI] [PubMed] [Google Scholar]
- Kwok AS, et al. HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery of autophagosomes. J Neurochem. 2011;119(6):1155–1161. doi: 10.1111/j.1471-4159.2011.07521.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Herrera G, et al. Deleterious Mutations in LRBA Are Associated with a Syndrome of Immune Deficiency and Autoimmunity. Am J Hum Genet. 2012;90(6):986–1001. doi: 10.1016/j.ajhg.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LR, Phadwal K, Simon AK, Harris AL. ATF4 orchestrates a program of BH3-only protein expression in severe hypoxia. Mol Biol Rep. 2012;39(12):10811–10822. doi: 10.1007/s11033-012-1975-3. [DOI] [PubMed] [Google Scholar]
- Pike LR, et al. Transcriptional up-regulation of ULK1 by ATF4 contributes to cancer cell survival. Biochem J. 2013;449(2):389–400. doi: 10.1042/BJ20120972. [DOI] [PubMed] [Google Scholar]
- Pugsley HR. Quantifying autophagy: Measuring LC3 puncta and autolysosome formation in cells using multispectral imaging flow cytometry. Methods. 2017;112:147–156. doi: 10.1016/j.ymeth.2016.05.022. [DOI] [PubMed] [Google Scholar]
- IDEAS. Image Data Exploration and Analysis Software User's Manual Version 6.2. 2015.